Chapter 164
Atheromatous Embolization
Jeffrey W. Olin, John R. Bartholomew
Atheromatous embolization is a poorly recognized and underdiagnosed multisystem disorder that is associated with high risk of all-cause and cardiovascular mortality.1 There are a myriad of clinical manifestations that may occur across all specialties, making differential diagnoses broad and diagnosis difficult. In addition, atheromatous embolization is a confusing entity to many physicians because it is known by many different names: cholesterol embolization, cholesterol crystal embolization, blue toe syndrome, purple toe syndrome, atheroembolism, and pseudovasculitis. For the purposes of this chapter, these terms will be used interchangeably. Once atheroembolism occurs, therapy involves three major strategies: treat the end organ that is undergoing embolization, prevent further embolization from occurring, and prevent future cardiovascular morbidity and mortality. Therefore, it is critical for clinicians to have a high index of suspicion and to recognize the clinical manifestations of this syndrome.
Incidence
The incidence of cholesterol embolism syndrome varies based on population characteristics, diagnostic criteria, and study design. Among unselected series of autopsy studies, the incidence of cholesterol crystal embolism ranges from 0.18% to 2.4%.2 However, autopsy studies performed in selected populations with atherosclerosis and in those who have undergone aortic manipulation have reported a greater prevalence of cholesterol crystal embolism, ranging from 12% to 77%.3,4 In autopsy studies of patients with known advanced atherosclerosis who recently had an arteriogram or a cardiac or vascular surgical procedure, the incidence is also high.5 Blauth et al5 identified atheroemboli in 22% of their postcardiac surgery autopsy cases, whereas Ramirez et al6 reported a 27% incidence in patients who had arteriography performed before their deaths. In general, retrospective autopsy studies may overestimate the frequency of the disease because of the detection of subclinical cases and the selection bias inherent in obtaining information after necropsy. In contrast, clinically significant atheroembolic disease in clinical studies may be missed because of short-term follow-up. The prevalence of atheroembolic disease in clinical studies has been estimated to be between 1% and 4%.7–11 There are two factors that may be associated with either an increased prevalence of atheromatous embolization (the continuing increasing age of the population, and thus, more advanced atherosclerosis) or a decreased prevalence (better guide wires, catheters, and endovascular and surgical techniques). However, there have been no recent studies on current prevalence rates.
Pathogenesis
The first description of atheroembolism was published more than a century ago by the German pathologist, Panum.12 However, Flory13 is credited for accurately describing the syndrome in 1945. Among 267 consecutive autopsies, he observed 9 instances of cholesterol crystal embolism: none in 63 cases in which aortic plaque ulceration was absent; 2 instances in 147 (1.4%) cases with moderate aortic plaque erosion; and 7 instances in 57 (12.8%) cases with severe aortic plaque ulceration. Since that seminal paper, it has become quite clear that the risk of atheroembolism is directly related to the severity of aortic atherosclerosis.
Atherosclerotic plaques consist of a fibrous cap, under which are macrophages, necrotic debris, and cholesterol crystals. The vulnerable plaque, or the plaque at the highest risk of rupture, are those with a thin fibrous cap surrounding a large lipid-rich core.14 Kronzon and Saric15 have stated that there are six key elements required for the development of “cholesterol embolization syndrome”: (1) plaque in large arteries, such as the aorta, iliac, or carotid arteries; (2) spontaneous, traumatic, or iatrogenic plaque rupture; (3) embolization of cholesterol crystals, platelets, fibrin, and other detritus; (4) lodging of emboli in small arteries with a diameter of 100 to 200 µm, leading to occlusion of vessels; (5) foreign body inflammatory response to the atheromatous emboli; and (6) end organ damage to physical and inflammatory occlusion of multiple small arteries. Cholesterol crystals are white and rhomboidal or rectangular in shape. They can also be elongated, biconvex, and needle-shaped, and range in size from 250 µm to less than 10 µm in diameter.8 The cholesterol crystals dissolve in paraffin-fixed sections, leaving needle-like clefts (Fig. 164-1). Frozen or wet formalin-fixed sections reveal doubly refractile cholesterol crystals; with the Schultz histochemical stain, these crystals stain blue-green.16
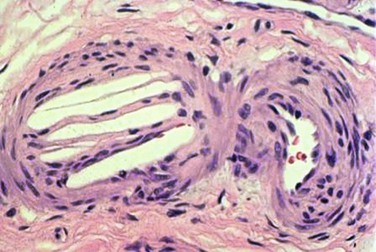
Figure 164-1 Typical appearance of the cholesterol clefts in a specimen from a kidney biopsy. The convex-shaped crystals within the small arterioles of the kidney dissolve during the fixation process leaving the ghosts (left side), which is what is seen in the histopathology (hematoxlyin & eosin stain, 200 times original magnification). Note the three red blood cells in the arteriole (right side).
Because they are light in weight and hydrophobic, they pass quickly through blood vessels until they are stopped by arterial bifurcations, a narrowing of the vessel lumen, or when they reach the end of the arterial circulation.8 Cholesterol emboli tend to be diffuse and lodge in arteries 100 to 200 µm in size,10 although small crystals have been observed in capillaries in the end-arterial circulation.
Cholesterol crystals that lodge in the arterioles immediately incite an inflammatory response characterized by varying degrees of polymorphonuclear and eosinophilic infiltration.8,13,17,18 By 2 to 4 weeks, a more chronic inflammatory infiltrate is seen. Cholesterol crystals become embedded in multinucleated giant cells and smooth muscle cells.17 Endothelial proliferation and fibrous tissue can be found surrounding the crystals, ultimately leading to luminal obliteration.17,18 At 1 to 2 months, crystals may extrude out of the vessel lumen and be buried in the adventitia, or remain in the lumen, embedded within organized thrombus that may re-canalize.17 The crystals are resistant to breakdown by macrophages and have been shown to persist in tissue for up to 9 months.19,20 Arterial lumina are eventually occluded by the accumulation of cells and fibrous material. These pathologic changes result in tissue effects distal to the cholesterol crystal emboli, including ischemia, and rarely, infarction, depending on the extent of organ involvement. This type of foreign body reaction is the reason it may take weeks to months for serum creatinine to rise in patients with atheroembolic renal disease and illustrates why renal function does not usually recover.19,21–23
Risk Factors and Simple Preventive Strategies
The most important risk factor for atheroembolism is established atherosclerosis. In 1945, Flory13 was the first person to hypothesize such a relationship between cholesterol crystal embolism and a diseased atherosclerotic aorta. Forty-seven years later, Blauth et al5 reported on 46 patient autopsies in which severe atherosclerosis of the ascending aorta was accompanied by evidence of atheroemboli in other vascular beds. Significant risk factors for atheroembolism included peripheral arterial disease, hypertension, older age, and coronary artery disease.5
There are several precipitating factors that have been implicated in the occurrence of plaque instability and consequent atheroembolism, including trauma, vascular surgery,24,25 angiographic and endovascular procedures,1,19,22,26 anticoagulation,7,27,28 and thrombolysis.29–32 Although spontaneous atheroembolism was once the most common presentation of atheromatous embolization, endovascular revascularization techniques are now the most frequent precipitating cause of the atheromatous embolization syndrome.21,22,33–35 In 354 patients who were followed for an average of 2 years, Scolari et al22 found that atheroembolic disease was spontaneous in only 23.5% of cases.
Coronary Interventions
Manipulation of the aorta with catheters or guide wires can cause mechanical trauma, consequently dislodging atheromatous material from the arterial wall.36 One study that evaluated the frequency of atherothrombotic material retrieved during placement of coronary catheters found 0.5% of 7621 patients had macroscopically visible atherothrombotic debris.37 None of these patients, however, had clinically apparent atheroembolic disease. In a review of 4587 cardiac catheterizations, Drost et al38 found 7 cases of clinical cholesterol embolization (0.002%). Colt et al39 found 8 cases after heart catheterization, percutaneous transluminal coronary angioplasty, and intra-aortic balloon pump insertion involving 3733 procedures (0.002%). Coronary angiography with angioplasty and stenting is considered the most common arteriographic procedure to incite atheroembolism.10,22 Saklayen et al,40 in a prospective analysis of 267 patients who underwent coronary angiography, found the incidence of cholesterol embolism to be less than 2%. Similar statistics were found in a more recent prospective analysis by Fukumoto et al,41 who reported the occurrence of clinically apparent cholesterol embolism (livedo pattern on the feet, blue toe syndrome, digital gangrene, or renal failure) in 1.4% of 1786 patients who underwent left heart catheterization.
Although it is impossible to predict the risk of atheroembolism in a given patient, the presence of severe peripheral artery disease, aortic aneurysm, and protruding mobile atheroma by transesophageal echocardiography (TEE),42 or aortic plaque more than 4 mm in thickness43 increase the risk of distal embolization and should therefore influence the vascular approach.40,44 Utilization of long guide wire (260 cm) exchanges is recommended, and back bleeding from guiding catheters (once the wire is removed) allows for removal of debris. Advancement and removal of catheters should occur over a guide wire to straighten the catheter and minimize contact with the aortic wall. Brachial and radial access may minimize embolization from the abdominal aorta, but not from the ascending aorta or arch. However, a prospective study of 1579 patients who underwent coronary angioplasty did not find significant differences between the brachial and femoral approaches.45 In contrast, in another study that involved 3733 procedures, there were no cases of cholesterol embolization after cardiac catheterization when the brachial artery was used.39
Aortography
Catheter manipulation of the aorta is frequent in the diagnosis and evaluation of patients with vascular disease before revascularization, and as with coronary angiography, the risk of atheroembolism is a serious concern. Ramirez et al,6 in a retrospective study of 71 autopsy cases, reported a 27% incidence of cholesterol embolization in patients who had arteriography performed compared with a 4.3% incidence of spontaneous cholesterol emboli in an age- and disease-matched control group who did not undergo arteriography.6 The rigidity of the catheter used and the force of the contrast injection appeared to contribute to the risk of embolization. Although some have advocated the use of a softer, more flexible catheter to avoid such a complication,6 the most important factor determining risk remains the severity of the atherosclerotic disease in the aorta. It should be understood, however, that the studies that reported a significant incidence of cholesterol embolization after angiography were conducted nearly 25 years ago. With the advent of better, smaller and more flexible catheters, guide wires and balloons, as well as superior operator technique and the use of the “no touch” technique,46 the incidence of atheromatous embolization following angiography is much less than it was 3 decades ago.
Endovascular Therapy
Endovascular therapy for patients with peripheral vascular disease has become widely utilized as an alternative to surgical revascularization. Clinically important atheroembolism appears to be a relatively infrequent, but not absent, complication of endovascular therapy. In a retrospective analysis of 493 patients who underwent a total of 565 aortoiliac stent placements, Lin et al47 found the incidence of atheroembolism to be 1.6%. This percentage is comparable to the findings of previous clinical studies that noted that atheroembolism ranged between 1.3% and 3.6%.48,49 In a study that utilized duplex ultrasound at the time of renal artery stenting, microembolic signals were detected in the renal parenchyma in every single case.1,50 In addition, when emboli protection devices were utilized in carotid stent procedures51 or renal artery stent procedures,52–58 visible atherosclerotic debris was frequently retrieved. Several studies showed that distal protection devices used at the time of renal artery stenting were associated with less deterioration in renal function compared with stenting without the use of these devices (Fig. 164-2).53–55 In a small prospective randomized trial, Cooper et al54 demonstrated that during renal artery stenting, the combination of distal embolic protection and the use of the glycoprotein IIb/IIIa inhibitor abciximab resulted in an improvement in renal function compared with the use of either of these modalities alone.54 Additionally, Paul et al59 showed that there is a much greater improvement in renal blood flow (as measured by the renal frame count) when a distal protection device was used during renal artery stent implantation compared with renal artery stenting without a distal protection device. Although distal protection devices are routinely used in carotid artery stenting, this is not the case in renal artery stenting. In the initial design of the CORAL trial, a distal protection device was used in every patient who was randomized to stenting. However, due to slow recruitment, this requirement was dropped from the protocol, and the decision was left up to the discretion of the interventionalist.60
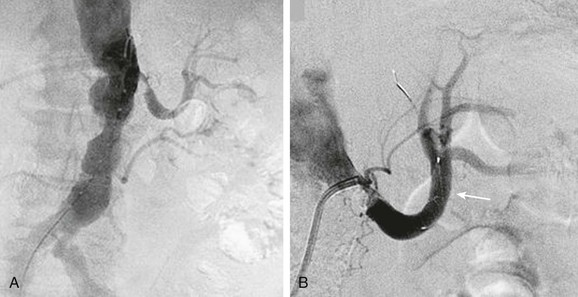
Figure 164-2 A, Catheter-based angiogram demonstrating a severely atherosclerotic aorta with a severe stenosis of the left renal artery and an occluded right renal artery (left panel). A selective angiogram of the left renal artery shows the severity of stenosis. B, There was a 7-mm Spider embolic protection device (arrow) (ev3 Endovascular, Inc., Plymouth, Minn.) placed before renal artery stenting (right panel).
The presence of a shaggy aorta and diffuse soft ulcerative plaque on TEE studies clearly identify a high-risk population.43 Hence, heightened awareness, proper patient selection, utilization of the most advanced catheters, guide wires, and balloons, use of emboli protection devices, and greater operator expertise may have a favorable impact on the incidence of atheroembolism during endovascular interventions. There are times in which endovascular procedures using covered stent grafts may be quite effective in patients who experience embolization, as is discussed in the Treatment section.
Vascular Surgery
The effect of atheroembolism after major vascular surgery was first recognized by Thurlbeck et al in 1957.61 In their series, atheroembolism was present at autopsy in more than 75% of the patients who died after aortic aneurysm surgery. Atheromatous embolization was the cause of death or significantly contributed to mortality in nearly half of the patients in this series. Subsequently, numerous studies confirmed the importance of vascular surgery as a precipitator of atheroembolism. Vascular surgery procedures may disrupt plaque when the vessel is manipulated, cross-clamped, or incised during surgery. Other vascular surgery procedures known to precipitate cholesterol embolization include aortoiliac and aortofemoral bypass, carotid endarterectomy, and renal artery revascularization.10 In a retrospective series of 1011 patients who underwent infrarenal aortic surgery or infrainguinal surgery, the diagnosis of cholesterol embolization was 2.9%.62 Due to the advent of better surgical techniques and an awareness of the risk of surgery precipitating atheromatous embolization, this complication has become much less common than reported in the older literature.63
Cardiac Surgery
Atheroembolization is a recognized complication of cardiac surgery and has profound medical and economic consequences. Doty et al,25 in a retrospective analysis of 18,402 patients who underwent cardiac surgery, found evidence of atheroembolism in 0.2% of patients at autopsy. The clinical presentation of atheroembolism in this study was broad and included five distinct organ systems: heart, central nervous system, gastrointestinal (GI) tract, kidneys, and the lower extremities. In 21% of these cases, death was directly attributable to atheroembolism.25 Kolh et al64 documented a significant increase in intensive care unit stay, overall hospital stay, and total hospital cost in patients with documented atheroembolism after cardiac surgery. TEE can identify significant aortic plaque preoperatively with sensitivity and specificity in excess of 90%.65 Multidetector computed tomographic (CT) angiography may also be a valuable imaging tool to image the aorta and iliac arteries. Alteration of the cannulation site and avoidance of aortic manipulation for coronary artery bypass based on such findings may reduce the incidence of atheroembolism.
It is clear that the most effective strategy for the management of atheroemboli in vascular surgery is prevention. In high-risk surgical patients, noninvasive procedures, such as magnetic resonance imaging, TEE, or CT angiography are excellent techniques to screen for the presence of aortic atherosclerosis preoperatively. When a severely atherosclerotic aorta is visualized, alternative surgical procedures should be considered to minimize aortic manipulation. Appropriate surgical techniques to prevent atheroemboli during operation are now well recognized and documented.63
Anticoagulation
An increased risk of cholesterol embolization with anticoagulation and clinical improvement when anticoagulation was removed has been reported in case reports for nearly half a century.7,10,27 One hypothesis is that anticoagulation may prevent thrombus formation over unstable atherosclerotic plaque, thus allowing exposed cholesterol crystals to embolize. Another hypothesis is that these agents may initiate the disruption of a complex plaque by causing intraplaque hemorrhage.66–69 Based on these small case series and case reports, some investigators have recommended that warfarin be discontinued, when feasible, in patients who have had an episode of cholesterol embolization for which no other precipitant can be identified.
However, the data are not entirely clear in assessing anticoagulation safety in patients with a large amount of aortic plaque. The assumption that anticoagulation precipitates cholesterol emboli syndrome was not confirmed by the SPAF-3 trial, in which patients with documented aortic plaque identified by TEE and who were assigned to adjusted-dose warfarin therapy had a low annual rate of cholesterol embolization (0.7% per patient-year; 95% confidence interval, 0.1%-5.3%).70 Furthermore, cholesterol embolization was not seen in patients with documented aortic arch plaque of more than 1 mm in thickness treated with warfarin in The French Study of Aortic Plaques in Stroke Group.43 In a prospective evaluation of 25 patients with cholesterol emboli syndrome after cardiac catheterization, Fukumoto et al41 failed to show any association between the use of anticoagulants and cholesterol embolism. In addition, anticoagulation has been advocated for patients with crescendo transient ischemic attacks (TIAs), a syndrome caused by athermatous embolization to the eye or brain.
Whether the use of anticoagulation is associated with a higher incidence of atheroemboli remains controversial, but the current literature suggests that it is safe to continue anticoagulation therapy in patients with a compelling reason to do so, such as those with atrial fibrillation or venous thromboembolism.
Thrombolysis
Atheromatous emboli have also been associated with thrombolytic therapy in case reports and small series,30,32 but again this is controversial. Thrombolytic agents act by converting plasminogen to plasmin; plasmin directly degrades fibrin. Theoretically, any therapy that causes the thrombus to undergo lysis may leave atherosclerotic plaque uncovered, placing the patient at risk for embolization. In one small prospective study, no relationship between the administration of thrombolytic therapy and cholesterol emboli syndrome was found.71
Clinical Features
Epidemiology
The syndrome of atheroembolism usually affects elderly persons who have multiple risk factors for atherosclerosis, but may occur in younger individuals with advanced atheroscleosis.2 In a recent prospective study to identify risk factors for cholesterol embolism in patients who underwent cardiac catheterization, Fukumoto et al41 confirmed that cholesterol emboli syndrome occurred more frequently in patients with generalized atherosclerosis (e.g., multivessel coronary disease, cerebrovascular disease). In addition, the authors found a significant relationship between C-reactive protein and cholesterol embolism (odds ratio 4.6; P = .01, using multivariate analysis), indicating an important possible association between systemic inflammation and cholesterol emboli syndrome.41
The increased frequency of this disease in men72 may be explained by a difference in the prevalence of atherosclerosis between the sexes. In addition, atheroemboli occurs almost exclusively in patients older than 50 years of age.72 A race predilection has also been reported, because atheroemboli are less likely to occur in African American patients (32 : 1 ratio).72,73 However, because African Americans appear to have an increased prevalence of atherosclerosis, it has been suggested by some investigators that this may be a failure to recognize the classic features of this syndrome because of skin pigmentation and the propensity of African American patients to develop renal failure secondary to poorly controlled blood pressure.
Clinical Findings
Patients almost always have symptomatic atherosclerosis manifested clinically by angina, myocardial infarction, TIA, stroke, renal artery disease, mesenteric ischemia, or peripheral arterial disease and claudication.74 Atheroembolism can present with a myriad of symptoms (Box 164-1).75–77 In general, the organs affected by cholesterol embolism depend on the location of the embolic source. Atheroemboli from the ascending aorta and proximal aortic arch usually manifest with central nervous system or retinal pathology, whereas cholesterol crystal emboli originating from the descending thoracic or abdominal aortas affect the visceral organs and extremities. In general, bilateral lower extremity atheroembolism signifies a source proximal to the aortic bifurcation, whereas unilateral emboli may originate either proximally or in any artery distal to the aortic bifurcation. Patients with one or more large atheromatous plaques in the aorta may present with a catastrophic event, such as an acutely ischemic limb, or renal or mesenteric infarction.33,78 Conversely, patients with microemboli may have milder localized signs or a clinical picture that suggests a systemic illness. There may be a temporal delay in clinical findings (especially for renal failure) after the inciting event of up to 8 weeks.76
Cutaneous Manifestations
Skin manifestations are among the most common clinical manifestations of atheroembolism, and the common cutaneous features are livedo reticularis and blue toes.79,80 The appearance of cutaneous signs can be delayed, with 50% of patients in one series showing skin signs of atheroembolism more than 30 days after their procedure or other inciting event.79
Livedo Reticularis.
Livedo reticularis is a blue-red mottling or discoloration of the skin that occurs in a netlike pattern, most commonly seen on the buttocks, thighs, or legs (Fig. 164-3A). A detailed skin examination performed in both the supine and upright posture is necessary because livedo reticularis has been shown to be more readily demonstrable in the upright position.81 Livedo reticularis is caused by obstruction of small arteries, capillaries, or venules in the deep dermis.10,22 When the skin is biopsied in patients with atheromatous embolization, cholesterol crystals may be seen in the dermal blood vessels. Livedo reticularis, however, is not pathognomonic of atheroemboli and has an extensive differential diagnosis including, but not limited to, other causes of intravascular obstruction (i.e., antiphospholipid antibody syndrome, cryoglobulinemia, endocarditis, left atrial myxoma), vasculitis, or be drug-induced (i.e., quinidine, quinine, amantidine, catecholamines).82 Furthermore, there are also physiologic (Cutis marmorata) and idiopathic (livedoid vasculitis) forms of livedo reticularis. Livedo can occur in young healthy women and appears to be related to abnormal sensitivity of the dermal blood vessels to cold (primary livedo reticularis). This livedo reticularis pattern usually disappears on re-warming. Patients who exhibit this should be reassured that there is no serious circulatory abnormality present.
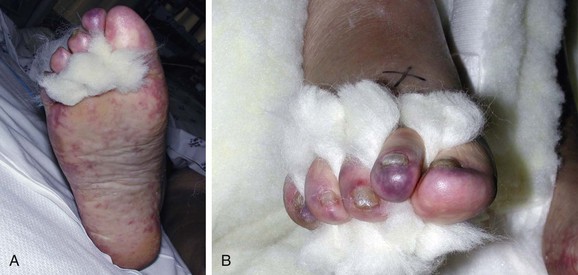
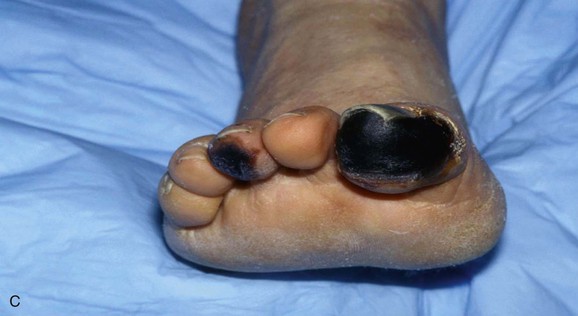
Figure 164-3 A, Characteristic appearance of the foot in a patient with atheromatous embolization. There is a patchy distribution of livedo reticularis on the lateral aspect, plantar surface, and heel of the right foot. Note the purple second toe. B, Typical appearance of blue or purple toes that may occur in atheromatous embolization. C, More severe cases may progress to gangrene.
Blue Toe Syndrome.
In its classic presentation, the blue toe syndrome presents as the sudden appearance of a cool, cyanotic, and painful toe in the presence of palpable distal pulses (Fig. 164-3B).66–68,79 Discoloration may also be seen on the sole of the foot. The discoloration may be patchy (see Fig. 164-3A), and comparison of both feet shows that the distribution is not symmetric. These lesions may progress to ulceration, necrosis, and frank gangrene (Fig. 164-3C).10 Accessory lesions may be present on the lateral and posterior aspects of the heels, which later develop into linear fissures with skin edge gangrene and a dark, necrotic base.
Other Skin Manifestations.
Other skin manifestations include splinter hemorrhages, petichiae, purpura, ulcers,83 and raised nodules that appear as the result of subepidermal inflammation surrounding cholesterol crystals,10,79 These nodules are painful, violaceous in appearance with a necrotic center, and may mimic a necrotizing vasculitits, such as polyarteritis nodosa or leukocytoclastic vasculitis. Ulceration of the penis and scrotum have also been described.84
Renal Involvement
The kidneys are a prime target for cholesterol crystal embolization due to the enormous amount of blood that flows through the kidney and the close proximity of the proximal renal arteries to the plaque-bearing abdominal aorta.21–23
Incidence.
Mayo et al,9 in a review of 402 nephrology consultation charts, found that the incidence of clinically detectable atheroembolism amounted to at least 4% of all inpatients examined, representing approximately 5% to 10% of the acute renal failure patients encountered. Scolari et al10 estimated encountering at least one case per month with atheroembolic renal disease. Since this report, these investigators have seen 354 patients who have been followed for an average of 2 years.22 Most investigators believe that this condition is significantly underdiagnosed.1,21,22
Pathology.
Pathologically, the classic lesion of atheroembolic renal disease is the occlusion of medium-sized arterioles (150-200 µm in diameter) and glomerular capillaries with cholesterol emboli.10 In addition to the ischemic obstructive mechanical phenomenon produced at the onset, this pathologic condition produces an inflammatory reaction within the arterioles. The initial stages, which occur several days to weeks after the inciting event, are characterized by an inflammatory infiltration consisting of polymorphonuclear leukocytes, macrophages, and multinucleated giant cells. Because the cellular infiltrate leads to thickening and fibrosis of the arterioles, later stages are characterized by glomerular sclerosis, tubular atrophy, and interstitial fibrosis.21,76,85 A kidney biopsy specimen from an individual patient may reveal different stages of histologic evolution because dislodged atheromatous debris may be showered into the circulation at different time intervals.85 Furthermore, the involvement tends to be patchy, and therefore, a renal biopsy may not always show the classic pathologic lesions of this disease.85
Clinical Features.
The net effects of this pathogenic process, when combined with varying amounts of cholesterol embolization, have several different clinical presentations (Fig. 164-4).10,21–23 It is important to recognize the differences in the patient with acute tubular necrosis (ATN) (Fig. 164-4A) from those with atheroembolic renal disease (Figs. 164-4B-D). Marked renal impairment with acute onset is the easiest form of atheroembolic renal disease to recognize. It has the closest temporal relationship to the inciting event and is generally considered the consequence of massive embolization, often resulting in catastrophic consequences. The subacute form of atheroembolic renal disease, the most frequently observed, is more insidious in onset, occurring a few weeks after the inciting event. Renal impairment may worsen over weeks to months due to an inflammatory response and foreign-body reaction, or the cyclic occurrence of cholesterol crystal embolic showers (Fig. 164-4B). Some patients with advanced renal failure come to the attention of physicians with few clues as to the exact onset of renal impairment. Under this circumstance, atheroembolic renal disease can only be diagnosed by performing a renal biopsy. Atheroembolic renal disease may also be present with chronic stable renal impairment (Fig. 164-4C) in an asymptomatic patient. Clinical features tend to be similar to those of ischemic nephropathy and nephrosclerosis. The role of cholesterol embolization in this setting is somewhat unclear. Many patients are misdiagnosed either because a renal biopsy is not performed or cholesterol embolization is missed on biopsy due the patchy distribution of the emboli. Recovery of dialysis-dependent renal failure (Fig. 164-4D) may occur in 21% to 39% of patients who initially require renal replacement therapy.23 Atheroembolic renal disease is often associated with severe and poorly controlled hypertension.68,76,86,87 When large segments of small arterioles are occluded, ischemic atrophy of substantial portions of the kidney occurs. As glomerular filtration declines, the renin-angiotensin-aldosterone system is activated, causing hypertension.76 Severe, accelerated, labile, and malignant hypertension have all been reported, and atheroembolic renal disease should be strongly considered in patients who present with resistant hypertension.87,88




Figure 164-4 Patterns of renal failure in patients with acute tubular necrosis (ATN) and atheroembolic renal disease. A, ATN: the serum creatinine rises within 24 hours of the inciting event and progresses to a peak over a period of days to weeks. Under most circumstances, the creatinine slowly returns to normal or near normal. B, There is a delay of one to several weeks from the inciting event until the creatinine begins to rise. This may progress to end-stage renal disease over a period of weeks to months. C, Delay from the inciting event, followed by a rise in the serum creatinine over a period of weeks to months eventually leveling out and leaving the patient with chronic stable renal disease. D, There have been an increasing number of reports in which the patient requires renal replacement therapy and after a period of time, the renal function improves to the point where the patient no longer requires dialysis. (From Bartholomew JR, et al: Atheromatous embolization. In Young JR, et al, editors: Peripheral vascular diseases, ed 2, St. Louis, 1996, C.V. Mosby Co., pp 266).
Prognosis.
The renal outcome for patients with atheroembolic renal disease is quite variable. In early reports, the renal outcome was uniformly dismal, with progression over weeks or months to end-stage renal failure.67,86 Over the past decade, however, spontaneous recovery of renal function in patients with atheroembolic renal disease have appeared in the literature, even after variable periods of dialytic support.7,26,89,90 The improvement in renal function may be related to reversal of inflammation, resolution of ATN in ischemic areas, and hypertrophy of surviving nephrons.10,91 Despite these promising case reports, most patients with atheroembolic renal disease continue to have either advanced chronic renal insufficiency or progress to end-stage renal disease requiring dialytic support. In the large study by Scolari et al,22 of the 354 patients followed for an average of 2 years, 116 (32.7%) patients required dialysis therapy. Eighty-three patients remained on maintenance dialysis therapy, whereas 33 were able to discontinue dialysis. Five patients re-started dialysis within 2 to 6 months of stopping therapy. Cumulative renal survival probability was reduced by the presence of heart failure (P < .001), baseline chronic kidney disease categories (P < .001), age older than 70 years (P < .039), iatrogenic atheromatous embolization (P < .001), acute and/or subacute onset (P < .001), and leg (P < .007), or GI involvement (P < .001). Statin treatment was associated with protective effects both when already in place at the time of diagnosis and when initiated after diagnosis (P < .001). Multivariable analysis showed that the same factors plus diabetes were associated with a significant increased risk for end-stage renal disease.
Gastrointestinal Involvement
Although this site is frequently overlooked, cholesterol embolization commonly occurs in the GI tract.
Incidence.
Sites of GI involvement include the colon, which is involved in up to 42% of cases, with the small bowel in 33%, and the stomach in 12%.92–94 The preferential involvement of the GI tract is probably a result of its rich vascular supply. Other areas of digestive system involvement that have been reported include the pancreas, liver, gallbladder, and less commonly, the spleen.92,93,95 The pancreas and liver are frequent sites of cholesterol embolization as indicated by autopsy reports, although clinically overt pancreatitis and hepatitis are an exceedingly rare presentation of cholesterol embolization.95 In contrast, cholesterol embolization to the gallbladder, while even rarer, tends to be clinically significant when present, with a clinical presentation ranging from chronic acalculous cholecystitis to acute gangrenous cholecystitis.93
Clinical Features.
The most common manifestations of GI tract involvement are abdominal pain, diarrhea, and GI bleeding.92 Abdominal pain may be caused by bowel ischemia with or without infarction, or by fibrous stricture with bowel obstruction as a consequence of tissue repair after repeated showers of atheroembolism.10 The pathogenesis of diarrhea may be related to multiple mechanisms, including mucosal inflammation, accumulation of luminal blood, and malabsorption.92 GI bleeding is caused by superficial mucosal ulceration, erosions, and microinfarcts.94
The diagnosis of cholesterol embolism is rarely made by endoscopy alone because most endoscopic findings are nonspecific, including congestive or erythematous mucosa, erosions, ulcerations, necrosis, inflammatory polyps, or strictures.2,7,94,95 Mucosal punch biopsies from the stomach, duodenum, or colon may be helpful in making the diagnosis occasionally, demonstrating the typical appearance of cholesterol crystals.94
Prognosis.
The prognosis of patients with GI involvement tends to be poor; the overall death rate is high. Patients with GI involvement commonly have a multisystem cholesterol embolization syndrome. In a retrospective review of 10 patients with histologically proven cholesterol crystal emboli diagnosed by endoscopic GI biopsy, death due to atherosclerotic complications occurred in 5 patients (multisystem failure [3], stroke [1], and ruptured abdominal aortic aneurysm [1]) within 3 months after diagnosis. All of these patients also had cutaneous manifestations and end-stage renal disease.94
Central Nervous System and Eye Involvement
Cholesterol embolization commonly occurs in the brain and eye and causes significant morbidity and mortality.2
Retinal.
The culprit atherosclerotic plaques are located in the ascending aorta, aortic arch, and/or carotid arteries.96,97 Patients may develop visual disturbances (e.g., amaurosis fugax) or variable degrees of blindness caused by central or branch retinal artery occlusion.8 Retinal cholesterol embolization is evident as yellow, highly refractile plaques (Hollenhorst’s plaques) at arterial bifurcations on ophthalmoscopic examination (Fig. 164-5).8,98 From 2000 to 2005, 130 patients were analyzed with either Hollenhorst’s plaques or branch retinal artery occlusions.99 This study found a low rate of significant extracranial carotid artery disease among these patients (<30% in 68%, 30%-60% in 22%, and >60% in 8%). Only six patients underwent a carotid endarterectomy or received a stent. With a median follow-up of 22 months, no stroke or TIA occurred, and overall survival was 94% in this group of patients.99 Among the 3654 survivors of the Blue Mountain Eye Study, the cumulative incidence of retinal emboli was only 2.9%.100 The authors believed that this was an underestimation of the true incidence due to the transient nature of emboli and differential loss to follow-up. The same authors101 assessed the relationship between retinal emboli and mortality in elderly patients. Of the 8384 patients with retinal photographs available, 2506 (30%) patients died over a 10- to 12-year period of time. The cumulative mortality was higher in patients with than without emboli (all-cause mortality: 56% versus 30%; stroke-related mortality: 12% versus 4%; and cardiovascular mortality: 30% versus 16%).

Figure 164-5 This 59-year-old man underwent stenting of the left internal carotid artery. A, Two months later, retinal examination of the left eye showed multiple, tiny refractile retinal arteriolar cholesterol emboli and a saddle embolus superior to the optic nerve (arrow). B, Two months later, repeat examination showed an increase in the number of cholesterol emboli. C, Four weeks later, a sudden, painless loss of the left superior visual field occurred. Examination revealed whitening in the inferior macular region (arrow), a finding that was consistent with an occlusion at the second major bifurcation of the inferior temporal branch of the retinal artery. (From Colucciello M: Images in clinical medicine. Retinal arteriolar cholesterol emboli. N Engl J Med 358:826, 2008.)
Cerebral.
Cerebral cholesterol embolism may be manifested as TIA, stroke, altered mental status, headache, dizziness, or organic brain syndrome.7,96,102 In a retrospective review of 29 patients with autopsy proven brain cholesterol emboli, encephalopathy was the predominant finding on neurologic examination.103 This was most likely due to the diffuse and bi-hemispheric nature of cholesterol embolization. Involvement of the spinal cord artery, which can lead to lower extremity paralysis, has been rarely reported.75 A case history that is comprised of a procedure that involves the ascending thoracic aorta, acute renal failure, and encephalopathy in an elderly patient should raise the suspicion of cholesterol emboli to the brain. Radiologic studies can be helpful by showing multiple small ischemic lesions or border zone infarcts.
Other Areas
Cholesterol emboli can occur in virtually any organ. Cardiac manifestations include angina pectoris and myocardial infarction. The usual source in these circumstances is the aortic root or proximal coronary artery.104 Pulmonary involvement has rarely been reported in the context of cholesterol embolism.105,106 Hemoptysis and dyspnea are the most common respiratory symptoms described. The pathogenesis of pulmonary involvement in cholesterol embolism may be related to direct deposition of atheroemboli in the lungs107 or de novo production of pulmonary lesions as a result of systemic inflammation associated with cholesterol embolism.108 Atheromatous emboli have also been demonstrated in the spleen, bone marrow,109 muscle, prostate, thyroid, and adrenal glands in autopsy studies.2,66,67,86
Nonspecific findings such as fever, weight loss, headaches, and myalgias have been reported and may suggest a multisystem illness (see Box 164-1).7,26,110
Diagnosis
The diagnosis of cholesterol embolization syndrome remains a significant challenge for physicians. The symptoms and signs are nonspecific and diverse, which is why this disease is sometimes referred to as the “great masquerader.”11,110 For this reason, a high index of suspicion and a thorough understanding of the various clinical manifestations are needed to correctly make the diagnosis antemortem. The diagnosis can often be made on clinical grounds alone, without histologic evaluation, in a patient who has a precipitating event, acute or subacute renal failure, difficult to control hypertension, and evidence of peripheral embolization.7
In the absence of obvious clinical clues, a definitive diagnosis may require a biopsy. The highest yield for histologic confirmation is in the skin in patients exhibiting livedo reticularis or in an affected organ, such as an amputated extremity in a patient with gangrenous toes, the kidney in a patient with new-onset renal failure, or in the GI tract in a patient with abdominal pain and GI bleeding.
Laboratory Tests
There are no specific laboratory tests that are diagnostic of atheromatous embolization. Eosinophilia can be found in up to 80% of cases and is probably related to the generation of complement C5, which has chemotactic properties for eosinophils.111 Eosinophilia, however, tends to be transient and short-lived.2,20 Laboratory markers of inflammation, including C-reactive protein, fibrinogen, and the erythrocyte sedimentation rate, have also been found to be elevated in many patients.67 Other reported laboratory findings include leukocytosis, anemia, thrombocytopenia, and decreased complement levels.66,67,112 Laboratory data may also reflect specific organ involvement. Elevations in serum levels of amylase, hepatic transaminases, blood urea nitrogen, creatinine, and creatinine phosphokinase may be seen with involvement of the pancreas, liver, kidney, and muscles, respectively. Mild proteinuria, microhematuria, and hyaline or granular casts are the most common urinary findings in patients with confirmed cholesterol embolism.67,113 Nephrotic range proteinuria, and eosinophiluria have also been reported less commonly.19,113,114
Imaging Modalities
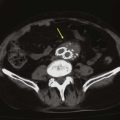
Invasive vascular procedures requiring aortic instrumentation should be avoided as a diagnostic modality due to the potential risk for producing recurrent atheroembolism. Noninvasive imaging studies, such as multidetector CT angiography, magnetic resonance angiography, and TEE, can assist in confirming the diagnosis if a markedly irregular and shaggy aorta is demonstrated (Figs. 164-6 and 164-7) It should be emphasized that these imaging modalities only demonstrate that the patient has significant underlying atherosclerosis, but cannot discern whether the atherosclerotic lesions are responsible for the embolic events.


Figure 164-6 Multidetector CT angiogram demonstrating marked calcified atherosclerotic disease in the infrarenal abdominal aorta in a reformatted view (A) and axial view (B). C, Multidector CT angiogram (axial view) of the infrarenal aorta showing an ectatic aorta with calcified plaque and extensive thrombus formation.


Differential Diagnosis
Diseases that should be considered in the differential diagnosis include, but are not limited to, contrast nephropathy, ATN from ischemic injury, necrotizing vasculitis, leukocytoclastic vasculitis, thrombotic thrombocytopenic purpura, antiphospholipid antibody syndrome, and multiple myeloma. Thromboembolism from the heart or aneurysms, and other cardiac sources of emboli, such as an atrial myxoma, nonbacterial thrombotic endocarditis, and infective endocarditis115 should always be excluded.
No laboratory test uniformly helps in the diagnosis of atheroembolic disease. The peripheral blood eosinophilia, hypocomplementemia, elevated sedimentation rate, and increased level of C-reactive protein seen in these patients are nonspecific findings that can also be found in patients with systemic or renal vasculitis110,116 Atheroembolic disease should be distinguished from vasculitis on the basis of other clinical findings and histology. The urine sediment in patients with atheroembolic renal disease is usually benign or shows only microhematuria. Rarely, eosinophiluria may be present.117 In contrast, the urine sediment in patients with ATN often demonstrates pigmented casts (dirty brown casts) and renal tubular cells. Atheroembolic renal disease can further be differentiated from ATN or contrast nephropathy based on the time frame of renal impairment. In contrast nephropathy and ATN, renal failure occurs within 48 to 72 hours after the inciting event, whereas in patients with atherombolic renal disease, the rise in creatinine is often delayed for 7 to 10 days.21,22,75,76 In addition, full recovery of renal function is the rule for contrast nephropathy and ATN if the underlying precipitating factor is corrected, whereas it is the exception in atheroembolic renal disease.10,76 There are, however, examples of late recovery of renal function in patients with atheroemboli.118 ATN is further characterized by normal blood pressure levels as opposed to the severe and refractory hypertension present in many patients with atheroembolic renal disease. The other previously listed disorders that may mimic cholesterol embolization due to their multisystem involvement should be excluded by appropriate diagnostic studies.
Treatment
There have been no randomized controlled trials of any therapeutic intervention for patients with cholesterol embolization, and no agent has been strongly correlated with favorable outcomes in case series. Clearly, the most important aspect of therapy is prevention (see section on Risk, Precipitating Factors, and Simple Preventive Strategies). Once atheromatous embolization has occurred, therapy is mostly supportive. Avoiding further inciting events such as aortic manipulation, good control of hypertension and heart failure, dialytic support, and adequate nutrition are the mainstays of treatment.7,68 Symptomatic care of the end organ where the emboli are located and risk factor modification to prevent myocardial infarction, stroke, and the progression of atherosclerotic disease are important treatment goals.75,119
Medical Therapy
Pathologic descriptions of cholesterol embolism highlight the severe inflammatory reaction that contributes to vascular obstruction. Although the inflammatory process caused by cholesterol embolism may suggest a role for anti-inflammatory agents,20 the use of corticosteroids has had conflicting results. Dahlberg et al,69 in a report of two patients given prednisone (60 mg daily) or methylprednisolone (80 mg daily) for 5 days, found a rapid and dramatic improvement in the manifestations of peripheral embolization.69 More recent case reports described improvement in renal function in patients with suspected atheroembolism when corticosteroid therapy was used.120–125 Corticosteroid administration has also been shown to be helpful in relieving symptoms related to mesenteric ischemia, such as abdominal pain and food intolerance, and for ischemic leg pain.7 In contrast, in a series of 67 patients, of whom 18 were treated with corticosteroids, no survival benefit was attributed directly to this therapy.7 Furthermore, in large retrospective series, Falanga et al79 found that corticosteroid use was associated with 100% mortality. Based upon the available literature, corticosteroid use cannot be recommended on a routine basis for this patient population, although some authorities have recommended corticosteroids in patients with multisystem atheroembolic disease.23
Statins have been reported to be beneficial in the treatment of livedo reticularis caused by cholesterol embolization126 and for treatment of atheroembolic renal disease.127 Furthermore, in a retrospective analysis of 519 patients with severe thoracic aortic plaque visualized on TEE, multivariate analysis showed that statin use was independently protective against recurrent embolic events (P = .0001).128 The mechanism of the beneficial effects of statin therapy is most likely related to the plaque-stabilizing activity of these drugs.
Iloprost (Schering AG, France), a prostacyclin analogue, has been reported to be beneficial for treatment of atheromatous embolization. It is a potent vasodilator that inhibits platelet aggregation and also has cytoprotective properties. It has been used to treat pain at rest and nonhealing ulcers in patients with critical limb ischemia, as well as severe Raynaud’s phenomenon and thromboangiitis obliterans. The dosage reported varies between 1.5 and 2 ng/kg/min. Side effects include headache and flushing. At higher doses, GI distress and hypotension are commonly reported. Lowering the dosage generally improves these side effects. In the largest reported series, four patients treated by Elinav et al129 received intravenous iloprost continuously for 10 to 14 days, followed by another 2 to 3 weeks of 8-hour infusions 2 or 3 times a week and once a week thereafter. The patients all had ischemia of their distal extremities, and one patient had renal failure. At follow-up, all patients reported a decrease in pain and improvement in their nongangrenous skin lesions, and there was also improvement in renal function in the one patient.129 There are several other individual case reports that suggest similar results with ischemic lesions130 and renal involvement.131 Previous experimental studies of renal injury have shown that iloprost protects the kidney by increasing renal plasma flow and fractional excretion of sodium without affecting the glomerular filtration rate or plasma rennin activity.132,133 Anecdotally, the prostaglandin analogues appear to be beneficial in the healing of ischemic ulcerations associated with atheroemboli. It is not known whether this class of drug is effective in patients with atheroembolic renal failure.
Low-density lipoprotein (LDL) apheresis with or without the addition of steroids was shown to be effective in improving skin manifestations. Hasegawa et al134 showed that combining corticosteroids and LDL apheresis improved atheroembolism related to blue toe syndrome. The mechanism is unknown, although it was postulated to improve blood viscosity by removing large molecular weight substances and decreasing total and oxidized LDL, thus improving endothelial function and reducing circulating inflammatory cytokines and chemokines.134
Other forms of therapy that have been advocated include antiplatelet agents such as aspirin66,67 and dipyridimole, low-molecular-weight dextran, intra-arterial papavarine, and the use of platelet infusions to help stabilize the source of atheroemboli. However, no controlled trials have shown that any of these forms of therapy are of benefit.
All patients should receive optimal risk factor modification with an antiplatelet agent (aspirin, clopidogrel, or both), a statin, and an angiotensin-converting enzyme inhibitor or angiotensin receptor blocking agent, all of which have been found to improve mortality in patients with underlying atherosclerotic disease.75,135–140
Surgical and Endovascular Therapy
Surgical or endovascular treatment is a viable option when a source of cholesterol emboli can be identified, and the source is surgically or endovascularly accessible. However, it is often difficult to identify which of the many atherosclerotic plaques is the culprit lesion.
Surgical Therapy
The aims of surgical therapy for atheroembolic disease are, first and foremost, the elimination of the embolic source, and second, arterial reconstruction of any hemodynamically significant proximal occlusive disease to encourage healing through improved end-arterial bed perfusion. Given the frequent instability of these lesions, surgical treatment has been perceived to be safer than endovascular approaches because the surgeon can clamp the artery proximal and distal to the lesion before manipulating the diseased vessel in an attempt to decrease the risk of recurrent embolization.141 Thromboendarterectomy or resection and graft replacement have been the surgical approaches most commonly used.142 Two prospective series reported favorable outcomes with vascular resection of atherosclerotic segments of large arteries identified as being the source of previous cholesterol crystal embolism.143,144 However, when the suprarenal aorta was involved, greater mortality rates were observed, likely related to the risk for visceral and renal ischemia and/or atheroemboli.143 Thus, according to some investigators, surgical elimination of the presumed source of cholesterol embolization should be reserved for patients with lower limb ischemia and an infrarenal source of embolization.10 In patients who are too weak for a major surgical intervention, ligation of the external iliac arteries or common femoral arteries, followed by an extra anatomic bypass (e.g., axillobifemoral bypass) has been advocated.142,145 The ligation prevents further embolization from reaching the legs, although embolization to the kidneys and intestines may still occur.
Endovascular Therapy
There are a few reports of intra-arterial treatment of embolizing lesions, including thrombolytic administration,62,146 percutaneous atherectomy,147,148 balloon angioplasty,62,146 and stent implantation.149,150 Intra-arterial thrombolytic administration in isolation is controversial. By destroying the platelet-fibrin thrombus that covers the atheromatous ulcerated plaques, thrombolysis may allow for the liberation of the cholesterol crystals into the arterial circulation with consequent microembolization. There are no data currently on the adjunctive use of tissue plasminogen activator in treating lesions believed to be responsible for atheroembolic events.
In percutaneous transluminal angioplasty, the intima is fractured and remolded, which could increase the risk of distal embolization. However, anecdotal reports utilizing this approach have shown symptomatic improvement in leg pain, re-establishment of peripheral pulses, and no evidence of recurrent embolization. Stent placement, in conjunction with angioplasty, may provide a protective scaffold to help secure these lesions. However, a potential risk of recurrent atheroembolism may exist due to either plaque dislodgement or extrusion of atheromatous material through stent interstices at the time of stent placement.
The possibility of procedural-related distal embolization due to stent placement was highlighted in a recent study by Ohki et al,151 that demonstrated a significant risk of distal embolization secondary to intra-arterial stent placement in an ex vivo carotid endarterectomy model. In contrast, Matchett et al,149 in a retrospective report of 15 patients treated with stent placement for blue toe syndrome, found no procedure-related embolization and only one recurrent embolization during follow-up. Renshaw et al152 reported successful angioplasty with stenting in eight patients with unilateral blue toe syndrome. Symptoms resolved in all eight patients over the ensuing month, and there were no recurrences with a mean follow-up of 18.5 months.152
The short-term results in studies utilizing percutaneous atherectomy, in which the plaque is shaved off the wall of the vessel and removed through a collection device, are similar to those of patients undergoing percutaneous transluminal angioplasty or surgery.147,148
The availability of covered stent grafts in recent years has raised their potential utility in the management of patients with distal atheroembolic lesions. Covered stents offer the added advantage of completely excluding the diseased segment, preventing the escape of thrombus or plaque debris. Kumins et al153 reported on the successful use of the Wallgraft endoprosthesis in two patients with distal microembolism from common iliac artery pathology.
Carroccio et al154 recently reported on endovascular stent graft repair for abdominal aortic aneurysms in 16 patients with atheromatous embolization syndrome. The 30-day mortality was 0%, and the abdominal aortic aneurysms were successfully excluded in 88% of patients. Resolution of foot ischemia and prevention of further atheromatous embolization occurred in 89% of the patients still alive at 1 year. Six patients died during a mean follow-up of 26 months, further illustrating the very high mortality in this patient population.
Distal embolic protection devices, especially filters, were originally designed to minimize atheroembolism during percutaneous coronary or carotid interventions. An off-label use of these devices is to protect the outflow arteries from atheroembolism during peripheral interventions. It would not be cost effective to utilize distal embolic protection for all peripheral interventions, and the arterial diameters compatible with these devices are limited. Nonetheless, distal embolic protection may be a useful adjunct for endovascular treatment of lesions deemed to be a high risk for embolization during the procedure, especially in patients who have already experienced an atheroembolic event.
Because of the limited studies available in the literature regarding the role of endovascular therapy for atheromatous embolization, its clinical efficacy is difficult to compare with operative treatment strategies. Undoubtedly, further clinical evaluation is warranted to further validate endovascular therapy in the treatment of atheroembolism.
Pain Control
Pain control is a critical aspect of the management of peripheral cholesterol embolism. The degree of pain associated with lower extremity ischemic and necrotic lesions secondary to cholesterol embolism is generally severe and disproportionate to the extent of the tissue involved. Sympathectomy has received attention as a surgical measure for the palliation of atheroembolic lesions. Lee et al155 demonstrated that adjunctive sympathectomy resulted in improved healing of distal digital ischemic ulcers. Sympathectomy is easily performed during aortic procedures, or it can be achieved postoperatively through lumbar sympathetic block or laparoscopic techniques.
More recently, Ghilardi et al156 reported on two cases of inferior limb ischemia secondary to cholesterol embolism treated with the temporary surgical implantation of spinal cord stimulation devices. Spinal cord stimulation in this study was found to provide rapid and effective pain control in the reported cases and improvement in peripheral microcirculation manifested by the rapid resolution of the necrotic lesions within 4 to 6 weeks.156 These surgical techniques may not only be an adjunct to direct surgical treatment of the offending arterial segment, but also may be useful to control the pain of severe atheroembolic lower extremity lesions in patients who are not candidates for direct reconstruction of the embolic source. It is often helpful to ask a pain specialist to help in the management of the severe pain that may occur in these patients.
Outcome
In general, the prognosis of patients with atheroembolic disease is poor and most likely related to the severe and diffuse atherosclerosis that is present in this patient population. The course, however, varies depending on type of clinical presentation. Patients with symptoms limited to an extremity tend to have a better prognosis compared with patients with disseminated cholesterol crystal embolization, particularly when there is evidence of visceral and renal involvement. The reported 1-year mortality in four different reports varied from 64% to 81% (Table 164-1).19,67–69 Causes of death were multifactorial and included cardiac, central nervous system, and GI ischemia.

Special Considerations
Atheroembolism Arising from the Thoracic Aorta
The importance of the thoracic aorta as a source of cerebral and peripheral vascular emboli has been ascertained only recently.
Epidemiology
Tunick et al,157 in a retrospective study that compared 122 patients with a history of stroke, TIA, or peripheral emboli with 122 age- and gender-matched controls, found protruding atheromas to be an independent risk factor for embolic symptoms. Plaques located proximal to the ostium of the left subclavian artery were found in 60% of patients 60 years of age or older with ischemic stroke, with plaques more than 4 mm in thickness having the strongest association with protruding atheromas.158 Similar results were found in an autopsy study that showed that ulcerated plaques in the ascending aorta and aortic arch, which included both atheromatous material and thrombus, were significantly more prevalent in those who experienced cerebral embolic events compared with those without such plaques.159
A separate issue is the role of aortic plaque as a predictor of subsequent stroke and other embolic events, which has been evaluated in prospective studies.160,161 Three recent studies, in which the majority of patients who sustained a recent stroke were followed prospectively, showed an association between aortic arch atherosclerosis and cerebral and/or peripheral embolic events.162–164 Tunick et al163 found an annual event rate of vascular events of 33% in patients who had protruding plaques more than 5 mm thick in the thoracic aorta compared with 7% in matched control individuals. In a similar study, Mitusch et al164 found a significantly higher rate of vascular events in patients who were found to have complex plaque (>5 mm in thickness or plaques with mobile components) on echocardiographic examination compared with those found to have only moderate atherosclerosis (13.7% versus 4.1%/100 person-years, respectively). Davila-Roman et al,162 in a prospective long-term, follow-up study of 1957 patients who underwent cardiac surgery, found atherosclerosis of the ascending aorta to be an independent predictor of long-term neurologic events and mortality. Based on these recently published echocardiographic and pathologic studies, the overall vascular risk arising from advanced atherosclerosis of the thoracic aorta may be as high as that of established sources of embolism, including nonvalvular atrial fibrillation, left atrial thrombi, or severe stenosis of the internal carotid artery origin.158
Risk Profile
The vascular risks attributable to complex atherosclerosis of the thoracic aorta appear to be mainly correlated with the thickness of the plaques and the morphologic parameters associated with plaques. TEE of the ascending aorta and aortic arch has been used to identify plaque size and morphology as risk factors for embolic events (see Figs. 164-7A and B). A review from the French Study of Aortic Plaques in Stroke Group43 included 331 patients with an initial ischemic stroke who were followed for 2 to 4 years. Patients were divided into groups based on their degree of aortic plaque thickness(>4, 1-3.9, and <1 mm). At follow-up, the patients with plaque thickness more than 4 mm had a significantly greater incidence of recurrent stroke and vascular events (Table 164-2). An analysis of 788 person-year follow-up to determine the effect of plaque morphology on the risk of ischemic disease158 demonstrated that the only plaque morphology that increased the risk of ischemic events was the absence of plaque calcification. Ulceration and hypoechoic plaques had no predictive value in evaluating vascular events. Overall, it was determined that aortic plaques that were more than 4 mm in thickness increased the risk of vascular events, and this risk was further increased by the lack of plaque calcification. These authors hypothesized that the noncalcified plaques were probably lipid-laden plaques with a thin fibrous cap, which are unstable and prone to ulceration, rupture, and thrombosis.165 Pedunculated, mobile plaques have also been associated with an increased risk of recurrent embolization.157,166,167
Table 164-2
Incidence of Events According to Plaque Thickness in the Aortic Arch Proximal to the Ostium of the Left Subclavian Artery

* Includes brain infarction, myocardial infarction, peripheral embolism, and death from vascular causes.
(From The French Study of Aortic Plaques in Stroke Group: Atherosclerotic disease of the aortic arch as a risk factor for recurrent ischemic stroke. N Engl J Med 334:1216-1221, 1996.)
Treatment
Complex thoracic aortic plaques are not only valuable “markers” of severe widespread atherosclerosis,158,159 but also identify individuals at a high risk for cardiovascular events.43
The data concerning the efficacy of anticoagulation in patients with complex thoracic aortic plaques are conflicting. Ferrari et al168 used TEE in a prospective cohort to compare antiplatelet therapy to anticoagulation therapy and found that patients treated with antiplatelet agents had more combined vascular events and a higher mortality rate than patients treated with oral anticoagulants. Similar results were reported by Dressler et al,167 who found that patients with mobile aortic atheroma who did not receive warfarin had a higher incidence of vascular events than those who received warfarin treatment (27% had strokes vs. 0%). Conversely, a retrospective study showed no significant effect on the risk of events with the use of warfarin.128 In the 2010 American College of Cardiology/American Heart Association Guidelines on thoracic aortic disease, the section on treatment of aortic atheromatous embolization states, “There is no definitive therapeutic regimen for this high-risk patient group because no randomized trial has been completed.”169
Lipid-lowering therapy, primarily with a statin, is warranted in all patients with symptomatic atherosclerotic vascular disease.139 In a recent retrospective analysis of patients with complex aortic arch plaques, statin therapy significantly and independently reduced the risk of embolic events.163
Finally, surgical treatment of thoracic aorta atherosclerosis may be considered. Surgical therapy cannot be recommended routinely for the asymptomatic patient because the risks inherent in such a complex procedure outweigh the benefits.170 However, performing aortic arch endarterectomy or aortic resection along with the planned cardiac procedure, if severe atheromatous disease is discovered, has been addressed. Stern et al171 reported a large increase in intraoperative stroke and mortality when surgery was performed to limit the risk of stroke after cardiopulmonary bypass. At this time, in the absence of randomized controlled trials, surgical indications for aortic endarterectomy should be restricted to highly selected patients who have multiple and documented embolic events with a low operative risk, despite optimal medical treatment.166
Selected Key References
Belenfant X, Meyrier A, Jacquot C. Supportive treatment improves survival in multivisceral cholesterol crystal embolism. Am J Kidney Dis. 1999;33(5):840–850.
Falanga V, Fine MJ, Kapoor WN. The cutaneous manifestations of cholesterol crystal embolization. Arch Dermatol. 1986;122(10):1194–1198.
The most detailed review of the skin findings in patients with atheromatous embolization..
The French Study of Aortic Plaques in Stroke Group. Atherosclerotic disease of the aortic arch as a risk factor for recurrent ischemic stroke. N Engl J Med. 1996;334(19):1216–1221.
Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE Jr, Eagle KA, Hermann LK, Isselbacher EM, Kazerooni EA, Kouchoukos NT, Lytle BW, Milewicz DM, Reich DL, Sen S, Shinn JA, Svensson LG, Williams DM. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. [American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine] Circulation. 2010;121:e266–e369.
Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation. 2010;122:631–641.
A comprehensive and up to date review of the cholesterol embolization syndrome..
Scolari F, Ravani P. Atheroembolic renal disease. Lancet. 2010;375:1650–1660.
The most recent and comprehensive review of the effects of atheromatous embolization on the kidney..
Smith SC Jr, Benjamin EJ, Bonow RO, Braun LT, Creager MA, Franklin BA, Gibbons RJ, Grundy SM, Hiratzka LF, Jones DW, Lloyd-Jones DM, Minissian M, Mosca L, Peterson ED, Sacco RL, Spertus J, Stein JH, Taubert KA. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol. 2011;58:2432–2446.
Tunick PA, Nayar AC, Goodkin GM, Mirchandani S, Francescone S, Rosenzweig BP, Freedberg RS, Katz ES, Applebaum RM, Kronzon I. Effect of treatment on the incidence of stroke and other emboli in 519 patients with severe thoracic aortic plaque. [NYU Atheroma Group] Am J Cardiol. 2002;90(12):1320–1325.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Olin JW. Atheroembolic renal disease: underdiagnosed and misunderstood. Catheter Cardiovasc Interv. 2007;70(6):789–790.
2. Moolenaar W, et al. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156(6):653–657.
3. Rhodes JM. Cholesterol crystal embolism: an important “new” diagnosis for the general physician. Lancet. 1996;347:1641.
4. Moolenaar W, et al. Gastrointestinal blood loss due to cholesterol crystal embolization. J Clin Gastroenterol. 1995;21(3):220–223.
5. Blauth CI, et al. Atheroembolism from the ascending aorta. An emerging problem in cardiac surgery. J Thorac Cardiovasc Surg. 1992;103(6):1104–1111.
6. Ramirez G, et al. Cholesterol embolization: a complication of angiography. Arch Intern Med. 1978;138(9):1430–1432.
7. Belenfant X, et al. Supportive treatment improves survival in multivisceral cholesterol crystal embolism. Am J Kidney Dis. 1999;33(5):840–850.
8. Hollenhorst RW. Vascular status of patients who have cholesterol emboli in the retina. Am J Opthamol. 1966;61:1159–1165.
9. Mayo RR, et al. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100(5):524–529.
10. Scolari F, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36(6):1089–1109.
11. Lie JT. Cholesterol atheromatous embolism. The great masquerader revisited. Pathol Annu. 1992;27(Pt 2):17–50.
12. Panum P. Experimentelle Beitrage Zur Lehre Von der Embolie. Arch Pathol Anat Physiol Klin Med. 1862;25:308–310.
13. Flory C. Arterial occlusions produced by emboli from eroded aortic atheromatous plaques. Am J Pathol. 1945;21:549–565.
14. Arroyo LH, et al. Mechanisms of plaque rupture: mechanical and biologic interactions. Cardiovasc Res. 1999;41(2):369–375.
15. Kronzon I, et al. Cholesterol embolization syndrome. Circulation. 2010;122:631–641.
16. Pennington M, et al. Cholesterol embolization syndrome: cutaneous histopathological features and the variable onset of symptoms in patients with different risk factors. Br J Dermatol. 2002;146(3):511–517.
17. Jones DB, et al. Atheromatous emboli in renal biopsies. An ultrastructural study. Am J Pathol. 1975;78(2):261–276.
18. Kang K, et al. Subtle clues to the diagnosis of cholesterol embolism. Am J Dermatopathol. 1996;18(4):380–384.
19. Thadhani RI, et al. Atheroembolic renal failure after invasive procedures. Natural history based on 52 histologically proven cases. Medicine (Baltimore). 1995;74(6):350–358.
20. Fabbian F, et al. A possible role of corticosteroids in cholesterol crystal embolization. Nephron. 1999;83(2):189–190.
21. Mittal BV, et al. Atheroembolic renal disease: a silent masquerader. Kidney Int. 2008;73(1):126–130.
22. Scolari F, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116(3):298–304.
23. Scolari F, et al. Atheroembolic renal disease. Lancet. 2010;375:1650–1660.
24. Piriou V, et al. Severe systemic cholesterol embolization after open heart surgery. Br J Anaesth. 1996;77(2):277–280.
25. Doty JR, et al. Atheroembolism in cardiac surgery. Ann Thorac Surg. 2003;75(4):1221–1226.
26. Scolari F, et al. Cholesterol atheromatous embolism: an increasingly recognized cause of acute renal failure. Nephrol Dial Transplant. 1996;11(8):1607–1612.
27. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 24-1998. A 76-year-old woman with cardiac and renal failure and gastrointestinal bleeding. N Engl J Med. 1998;339(5):329–337.
28. Moll S, et al. Cholesterol emboli associated with warfarin treatment. Am J Hematol. 2004;77(2):194–195.
29. Turnbull RG, et al. Multiple spontaneous intestinal perforations from atheroembolism after thrombolytic therapy: a case report. Can J Surg. 1994;37:325–328.
30. Rivera-Manrique E, et al. Cholesterol embolism: a fatal complication after thrombolytic therapy for acute myocardial infarction. Arch Intern Med. 1998;158(14):1575.
31. Izumi C, et al. Clinical evaluation of cholesterol embolization syndrome after cardiac catheterization. J Cardiol. 1998;31(4):201–206.
32. Adorati M, et al. Cholesterol embolism and acute interstitial nephritis: two adverse effects of streptokinase thrombolytic therapy in the same patient. Nephrol Dial Transplant. 1998;13(5):1262–1264.
33. Sarwar S, et al. Catastrophic cholesterol crystal embolization after endovascular stent placement for peripheral vascular disease. Am J Med Sci. 2008;335(5):403–406.
34. Edwards MS, et al. Atheroembolism during percutaneous renal artery revascularization. J Vasc Surg. 2007;46(1):55–61.
35. Hiramoto J, et al. Atheroemboli during renal artery angioplasty: an ex vivo study. J Vasc Surg. 2005;41(6):1026–1030.
36. Keeley EC, et al. Scraping of aortic debris by coronary guiding catheters: a prospective evaluation of 1,000 cases. J Am Coll Cardiol. 1998;32(7):1861–1865.
37. Eggebrecht H, et al. Potential embolization by atherosclerotic debris dislodged from aortic wall during cardiac catheterization: histological and clinical findings in 7,621 patients. Catheter Cardiovasc Interv. 2000;49(4):389–394.
38. Drost H, et al. Cholesterol embolism as a complication of left heart catheterisation. Report of seven cases. Br Heart J. 1984;52(3):339–342.
39. Colt HG, et al. Cholesterol emboli after cardiac catheterization. Eight cases and a review of the literature. Medicine (Baltimore). 1988;67(6):389–400.
40. Saklayen MG, et al. Incidence of atheroembolic renal failure after coronary angiography. A prospective study. Angiology. 1997;48(7):609–613.
41. Fukumoto Y, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42(2):211–216.
42. Shmuely H, et al. Acute stroke after coronary angiography associated with protruding mobile thoracic aortic atheromas. Neurology. 1997;49(6):1689–1691.
43. The French Study of Aortic Plaques in Stroke Group. Atherosclerotic disease of the aortic arch as a risk factor for recurrent ischemic stroke. N Engl J Med. 1996;334(19):1216–1221.
44. Kiemeneij MD, et al. A randomized comparison of percutaneous transluminal coronary angioplasty by the radial, brachial and femoral approaches: the Access Study. J Am Coll Cardiol. 1997;29(6):1269–1275.
45. Johnson LW, et al. Peripheral vascular complications of coronary angioplasty by the femoral and brachial techniques. Cathet Cardiovasc Diagn. 1994;31(3):165–172.
46. Feldman RL, et al. No-touch technique for reducing aortic wall trauma during renal artery stenting. Catheter Cardiovasc Interv. 1999;46(2):245–248.
47. Lin PH, et al. Late complication of aortoiliac stent placement- atheroembolization of the lower extremities. J Surg Res. 2002;103(2):153–159.
48. Toogood GJ, et al. Early experience with stenting for iliac occlusive disease. Eur J Vasc Endovasc Surg. 1998;15(2):165–168.
49. Ballard J, et al. Aortoiliac stent deployment versus surgical reconstruction: analysis of outcome and cost. J Vasc Surg. 1998;28(1):94–103.
50. Kawarada O, et al. The characteristics of dissemination of embolic materials during renal artery stenting. Catheter Cardiovasc Interv. 2007;70(6):784–788.
51. Roubin GS, et al. Immediate and late clinical outcomes of carotid artery stenting in patients with symptomatic and asymptomatic carotid artery stenosis: a 5-year prospective analysis. Circulation. 2001;103(4):532–537.
52. Henry M, et al. Protected renal stenting with the PercuSurge GuardWire device: a pilot study. J Endovasc Ther. 2001;8(3):227–237.
53. Holden A, et al. Renal artery stent revascularization with embolic protection in patients with ischemic nephropathy. Kidney Int. 2006;70(5):948–955.
55. Henry M, et al. Endovascular treatment of a renal artery stenosis: techniques, indications, results. Role of embolic protection devices (part 4). Angiol Sosud Khir. 2007;13(4):57–66.
56. Henry M, et al. Recent advances in renal artery stenting. J Cardiovasc Surg (Torino). 2007;48(4):411–442.
57. Henry M, et al. New distal embolic protection device the FiberNet 3 dimensional filter: first carotid human study. Catheter Cardiovasc Interv. 2007;69(7):1026–1035.
58. Dubel GJ, et al. Distal embolic protection for renal arterial interventions. Cardiovasc Intervent Radiol. 2008;31(1):14–22.
59. Paul TK, et al. Renal embolic protection devices improve blood flow after stenting for atherosclerotic renal artery stenosis. Cath Cardiovasc Interv. 2012;80:1019–1022.
60. Cooper CJ, et al. Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart Journal. 2006;152:59–66.
61. Thurlbeck W, et al. Atheromatous emboli to the kidneys after aortic surgery. N Engl J Med. 1957;257:442–447.
62. Sharma PV, et al. Changing patterns of atheroembolism. Cardiovasc Surg. 1996;4(5):573–579.
63. Kaufman JL. Atheroembolism and microthromboembolic syndromes (blue toe syndrome and disseminated atheroembolism). Rutherford RB. Vascular surgery. ed 5. WB Saunders Company: Philadelphia; 2000:836–845.
64. Kolh PH, et al. Atheroembolization in cardiac surgery. The need for preoperative diagnosis. J Cardiovasc Surg (Torino). 1999;40(1):77–81.
65. Davila-Roman VG, et al. Transesophageal echocardiography in the detection of cardiovascular sources of peripheral vascular embolism. Ann Vasc Surg. 1995;9(3):252–260.
66. Kassirer JP. Atheroembolic renal disease. N Engl J Med. 1969;280(15):812–818.
67. Fine MJ, et al. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38(10):769–784.
68. Saleem S, et al. Atheroembolic renal disease. Semin Nephrol. 1996;16(4):309–318.
69. Dahlberg PJ, et al. Cholesterol embolism: experience with 22 histologically proven cases. Surgery. 1989;105(6):737–746.
70. Blackshear JL, et al. Making the diagnosis when the patient has ‘blue toes.’. Geriatrics. 1994;49(12):37–45.
71. Blankenship JC, et al. Prospective assessment of cholesterol embolization in patients with acute myocardial infarction treated with thrombolytic vs conservative therapy. Chest. 1995;107(3):662–668.
72. Donohue K, et al. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol. 2003;17(5):504–511.
73. Saklayen MG. Atheroembolic renal disease: preferential occurrence in whites only. Am J Nephrol. 1989;9(1):87–88.
74. Meyrier A. Cholesterol crystal embolism: diagnosis and treatment. Kidney Int. 2006;69(8):1308–1312.
75. Rose R, et al. Atheromatous embolization syndrome. Rutherford RB. Vascular surgery. ed 6. WB Saunders Co.: Philadelphia; 2005:986–999.
76. Bartholomew JR, et al. Atheromatous embolization. Young JR, et al. Peripheral vascular diseases. ed 2. C.V. Mosby Co.: St. Louis; 1996.
77. Quinones A, et al. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
78. Faria B, et al. Atheroembolic renal disease with rapid progression and fatal outcome. Clin Exp Nephrol. 2011;15:159–163.
79. Falanga V, et al. The cutaneous manifestations of cholesterol crystal embolization. Arch Dermatol. 1986;122(10):1194–1198.
80. Jucgla A, et al. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol. 2006;55(5):786–793.
81. Chaudhary K, et al. Livedo reticularis: an underutilized diagnostic clue in cholesterol embolization syndrome. Am J Med Sci. 2001;321(5):348–351.
82. Callen JP, et al. Color atlas of dermatology. ed 2. WB Saunders: Philadelphia; 2000.
83. Schanz S, et al. Cholesterol embolism: an often unrecognized cause of leg ulcers. Br J Dermatol. 2002;146(6):1107–1108.
84. Rosansky SJ. Multiple cholesterol emboli syndrome. South Med J. 1982;75:677–680.
85. Modi KS, et al. Atheroembolic renal disease. J Am SocNephrol. 2001;12(8):1781–1787.
86. Lye WC, et al. Renal cholesterol embolic disease. Case report and review of the literature. Am J Nephrol. 1993;13:489–493.
87. Ripple MG, et al. Cholesterol embolization in renal allografts. Transplantation. 2000;69(10):2221–2225.
88. Dalakos TG, et al. Malignant hypertension resulting from atheromatous embolization predominantly of one kidney. Am J Med. 1974;57(1):135–138.
89. Theriault J, et al. Atheroembolic renal failure requiring dialysis: potential for renal recovery? A review of 43 cases. Nephron Clin Pract. 2003;94(1):c11–c18.
90. Gorriz JL, et al. Recovery of renal function after renal failure due to cholesterol crystal embolism. Nephrol Dial Transplant. 1999;14(9):2261–2262.
91. Scolari F, et al. Predictors of renal and patient outcomes in atheroembolic renal disease: a prospective study. J Am Soc Nephrol. 2003;14(6):1584–1590.
92. Moolenar W, et al. Cholesterol crystal embolisation to the alimentary tract. Gut. 1996;38(2):196–200.
93. Ben Horin S, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98(7):1471–1479.
94. Paraf F, et al. Cholesterol crystal embolization demonstrated on GI biopsy. Am J Gastroenterol. 2001;96(12):3301–3304.
95. Lawson JM. Cholesterol crystal embolization: more common than we thought? Am J Gastroenterol. 2001;96(12):3230–3232.
96. Masuda J, et al. Atheromatous embolism in the brain: a clinicopathologic analysis of 15 autopsy cases. Neurology. 1994;44(7):1231–1237.
97. Beal MF, et al. Cholesterol embolism as a cause of transient ischemic attacks and cerebral infarction. Neurology. 1981;31(7):860–865.
98. Colucciello M. Images in clinical medicine. Retinal arteriolar cholesterol emboli. N Engl J Med. 2008;358(8):826.
99. Dunlap AB, et al. The fate of patients with retinal artery occlusion and Hollenhorst plaque. J Vasc Surg. 2007;46(6):1125–1129.
100. Cugati S, et al. Ten-year incidence of retinal emboli in an older population. Stroke. 2006;37(3):908–910.
101. Wang JJ, et al. Retinal arteriolar emboli and long-term mortality: pooled data analysis from two older populations. Stroke. 2006;37(7):1833–1836.
102. Laloux P, et al. Lacunar infarctions due to cholesterol emboli. Stroke. 1991;22(11):1440–1444.
103. Ezzeddine MA, et al. Clinical characteristics of pathologically proved cholesterol emboli to the brain. Neurology. 2000;54(8):1681–1683.
104. Teja K, et al. Intramural coronary arteritis from cholesterol emboli: a rare cause of unstable angina preceding sudden death. Am Heart J. 1985;110(1 Pt 1):168–170.
105. Sabatine MS, et al. Pulmonary cholesterol crystal embolization. Chest. 1997;112(6):1687–1692.
106. Walton TJ, et al. Systemic cholesterol crystal embolisation with pulmonary involvement: a fatal combination after coronary angiography. Postgrad Med J. 2002;78(919):288–289.
107. Hillion D, et al. Syndrome of alveolar hemorrhage associated with systemic cholesterol embolism. Ann Med Interne (Paris). 1986;137(8):660–662.
108. Sijpkens Y, et al. Vasculitis due to cholesterol embolism. Am J Med. 1997;102(3):302–303.
109. Reuter MD, et al. Spontaneous cholesterol crystal embolization to bone marrow. South Med J. 2007;100(5):533–536.
110. Olin JW. Syndromes that mimic vasculitis. Curr Opin Cardiol. 1991;6:768–774.
111. Cecioni I, et al. Eosinophilia in cholesterol atheroembolic disease. J Allergy Clin Immunol. 2007;120(6):1470–1471.
112. Low complement in atheroembolic disease. Lancet. 1985;2(8447):136.
113. Haqqie SS, et al. Nephrotic-range proteinuria in renal atheroembolic disease: report of four cases. Am J Kidney Dis. 1996;28(4):493–501.
114. Greenberg A, et al. Focal segmental glomerulosclerosis associated with nephrotic syndrome in cholesterol atheroembolism: clinicopathological correlations. Am J Kidney Dis. 1997;29(3):334–344.
115. Mieszczanska H, et al. Cholesterol emboli mimicking acute bacterial endocarditis. Heart Lung. 2002;31(6):452–454.
116. Peat DS, et al. Cholesterol emboli may mimic systemic vasculitis. BMJ. 1996;313(7056):546–547.
117. Wilson DM, et al. Eosinophiluria in atheroembolic renal disease. Am J Med. 1991;91(2):186–189.
118. Siddiqui S, et al. Recovery of renal function after 90 d on dialysis: implications for transplantation in patients with potentially reversible causes of renal failure. Clin Transplant. 2008;22(2):136–140.
119. Liew YP, et al. Atheromatous embolization. Vasc Med. 2005;10(4):309–326.
120. Mann SJ, et al. Treatment of atheroembolization with corticosteroids. Am J Hypertens. 2001;14(8 Pt 1):831–834.
121. Nakahama H, et al. Small dose oral corticosteroid treatment rapidly improved renal function in a patient with an acute aggravation of chronic renal failure due to cholesterol embolism. Nephrol Dial Transplant. 2001;16(4):872–873.
122. Matsumura T, et al. A case of cholesterol embolism confirmed by skin biopsy and successfully treated with statins and steroids. Am J Med Sci. 2006;331(5):280–283.
123. Yucel AE, et al. Cholesterol crystal embolization mimicking vasculitis: success with corticosteroid and cyclophosphamide therapy in two cases. Rheumatol Int. 2006;26(5):454–460.
124. Koga J, et al. Cholesterol embolization treated with corticosteroids–two case reports. Angiology. 2005;56(4):497–501.
125. Sharma A, et al. Favorable outcome in atheroembolic renal disease with pulse steroid therapy. Indian J Nephrol. 2012;22:473–476.
126. Finch TM, et al. Livedo reticularis caused by cholesterol embolization may improve with simvastatin. Br J Dermatol. 2000;143(6):1319–1320.
127. Woolfson RG, et al. Improvement in renal cholesterol emboli syndrome after simvastatin. Lancet. 1998;351(9112):1331–1332.
128. Tunick PA, et al. Effect of treatment on the incidence of stroke and other emboli in 519 patients with severe thoracic aortic plaque. Am J Cardiol. 2002;90(12):1320–1325.
129. Elinav E, et al. Improvement in cholesterol emboli syndrome after iloprost therapy. BMJ. 2002;324(7332):268–269.
130. Minatohara K. Renal failure associated with blue toe syndrome: effective treatment with intravenous prostaglandin E-1. Acta Derm Venereol. 2006;86(4):364–365.
131. Radauceanu A, et al. Use of a prostacyclin analogue in cholesterol crystal embolism. Diabet Med. 1998;15:262–263.
132. Lifschitz MD, et al. Prostaglandin I2 attenuates ischemic acute renal failure in the rat. Am J Physiol. 1984;247(5 Pt 2):F714–F717.
133. Neumayer HH, et al. Amelioration of postischemic acute renal failure by prostacyclin analogue (iloprost): long-term studies with chronically instrumented conscious dogs. J Cardiovasc Pharmacol. 1986;8(4):785–790.
134. Hasegawa M, et al. The evaluation of corticosteroid therapy in conjunction with plasma exchange in the treatment of renal cholesterol embolic disease. A report of 5 cases. Am J Nephrol. 2000;20(4):263–267.
135. Hankey GJ. Angiotensin-converting enzyme inhibitors for stroke prevention: is there HOPE for PROGRESS After LIFE? Stroke. 2003;34(2):354–356.
136. The Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342(3):145–153.
137. Cannon CP. Effectiveness of clopidogrel versus aspirin in preventing acute myocardial infarction in patients with symptomatic atherothrombosis (CAPRIE trial). Am J Cardiol. 2002;90(7):760–762.
138. Collaboration, Antithrombotic Trialists. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324(7329):71–86.
139. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):7–22.
140. Smith SC Jr, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation. J Am Coll Cardiol. 2011;58:2432–2446.
141. Dougherty MJ, et al. Endovascular treatment of embolization of aortic plaque with covered stents. J Vasc Surg. 2002;36(4):727–731.
142. Friedman SG, et al. External iliac ligation and axillary-bifemoral bypass for blue toe syndrome. Surgery. 1994;115(1):27–30.
143. Keen RR, et al. Surgical management of atheroembolization. J Vasc Surg. 1995;21(5):773–780.
144. Baumann DS, et al. An institutional experience with arterial atheroembolism. Ann Vasc Surg. 1994;8(3):258–265.
145. Kaufman JL, et al. The role of extraanatomic exclusion bypass in the treatment of disseminated atheroembolism syndrome. Ann Vasc Surg. 1990;4(3):260–263.
146. Brewer ML, et al. Blue toe syndrome: treatment with anticoagulants and delayed percutaneous transluminal angioplasty. Radiology. 1988;166(1 Pt 1):31–36.
147. Clugston RA, et al. Atherectomy of the distal aorta using a kissing-balloon technique for the treatment of blue toe syndrome. AJR Am J Roentgenol. 1992;159(1):125–127.
148. Dolmatch BL, et al. Blue toe syndrome: treatment with percutaneous atherectomy. Radiology. 1989;173(3):799–804.
149. Matchett WJ, et al. Blue toe syndrome: treatment with intra-arterial stents and review of therapies. J Vasc Interv Radiol. 2000;11(5):585–592.
150. Murphy KD, et al. Iliac artery stent placement with the Palmaz stent: follow-up study. J Vasc Interv Radiol. 1995;6(3):321–329.
151. Ohki T, et al. Ex vivo human carotid artery bifurcation stenting correlation of lesion characteristics with embolic potential. J Vasc Surg. 1998;27(3):463–471.
152. Renshaw A, et al. Angioplasty with stenting is effective in treating blue toe syndrome. Vasc Endovascular Surg. 2002;36(2):155–159.
153. Kumins NH, et al. Early experience using the Wallgraft in the management of distal microembolism from common iliac artery patholology. Ann Vasc Surg. 2002;16(2):181–186.
154. Carroccio A, et al. The role of aortic stent grafting in atheromatous embolization syndrome: results after a mean of 15 months follow-up. J Vasc Surg. 2004;40:424–429.
155. Lee BY, et al. Lumbar sympathectomy for toe gangrene. Long-term follow-up. Am J Surg. 1983;145(3):398–401.
156. Ghilardi G, et al. Temporary spinal cord stimulation for peripheral cholesterol embolism. J Cardiovasc Surg (Torino). 2002;43(2):255–258.
157. Tunick PA, et al. Protruding atheromas in the thoracic aorta and systemic emobolization. Ann Intern Med. 1991;115(6):423–427.
158. Amarenco P, et al. Atherosclerotic disease of the aortic arch and the risk of ischemic stroke. N Engl J Med. 1994;331(22):1474–1479.
159. Amarenco P, et al. The prevalence of ulcerated plaques in the aortic arch in patients with stroke. N Engl J Med. 1992;326(4):221–225.
160. Kronzon I, et al. Aortic atherosclerotic disease and stroke. Circulation. 2006;114(1):63–75.
161. Molisse TA, et al. Complications of aortic atherosclerosis: atheroemboli and thromboemboli. Curr Treat Options Cardiovasc Med. 2007;9(2):137–147.
162. Davila-Roman VG, et al. Atherosclerosis of the ascending aorta is an independent predictor of long-term neurologic events and mortality. J Am Coll Cardiol. 1999;33(5):1308–1316.
164. Mitusch R, et al. Vascular events during follow-up in patients with aortic arch atherosclerosis. Stroke. 1997;28(1):36–39.
165. Cohen A, et al. Atherosclerosis of the thoracic aorta: from risk stratification to treatment. Am J Cardiol. 2002;90(12):1333–1335.
166. Laperche T, et al. Mobile thromboses of the aortic arch without aortic debris: a transesophageal echocardiographic finding associated with unexplained arterial embolism. Circulation. 1997;96(1):288–294.
167. Dressler MD, et al. Mobile aortic atheroma and systemic emboli: efficacy of anticoagulation and influence of plaque morphology on recurrent stroke. J Am Coll Cardiol. 1998;31(1):134–138.
168. Ferrari E, et al. Atherosclerosis of the thoracic aorta and aortic debris as a marker of poor prognosis: benefit of oral anticoagulants. J Am Coll Cardiol. 1999;33(5):1317–1322.
169. Hiratzka LF, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121(13):e266–e369.
170. Bojar RM, et al. Surgical treatment of systemic atheroembolism from the thoracic aorta. Ann Thorac Surg. 1996;61(5):1389–1393.
171. Stern A, et al. Protruding aortic arch atheromas: risk of stroke during heart surgery with and without aortic arch endarterectomy. Am Heart J. 1999;138(4 (Pt 1)):746–752.