CHAPTER 84 Alcoholic Liver Disease
EPIDEMIOLOGY
Although two thirds of American adults drink alcohol, only a minority are problem drinkers. Nevertheless, the number of alcoholics in the United States is estimated to be 14 million.1 The total costs of alcohol abuse amount to $185 billion annually, most of which are related to lost productivity and motor vehicle collisions. Alcoholic liver disease also is a major health care problem, accounting for 40% of deaths from cirrhosis and more than 30% of cases of hepatocellular carcinoma in the United States.1,2 In both Europe and the United States, alcoholic liver disease and its complications account for 50,000 deaths annually.3
Numerous studies have shown that alcoholic liver disease develops in women after a shorter duration of drinking and with a lower daily alcohol intake than in men.4,5 Population-based surveys have documented that men usually must drink 40 to 80 g of alcohol daily and women 20 to 40 g daily for 10 to 12 years to achieve a significant risk of liver disease.4–6 Table 84-1 illustrates the alcohol content of various beverages, their typical serving sizes, and the daily alcohol intake, for at least 10 years, that puts both men and women at risk for the development of alcoholic liver disease.

SPECTRUM OF DISEASE
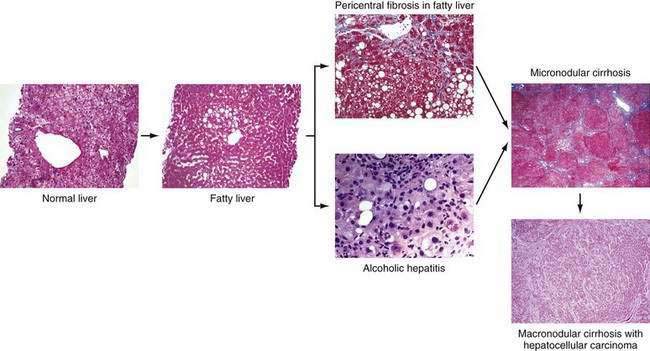
Chronic alcohol abuse can result in a spectrum of liver injury that ranges from mild fatty infiltration to cirrhosis and hepatocellular carcinoma (Fig. 84-1).7,8 Fat accumulation in liver cells, which is the earliest and most predictable response to alcohol ingestion, is seen in 90% of heavy drinkers. Although fatty liver generally is a benign condition that usually reverses quickly with abstinence, cirrhosis can develop within five years in 10% of patients who continue to drink heavily.9 Cirrhosis is particularly likely to develop if the steatosis manifests as a mixed micro- and macrovesicular pattern rather than the macrovesicular pattern seen in most alcoholics.8 Much more important than steatosis alone is the development of necroinflammation, with or without fat infiltration, and fibrosis (alcoholic hepatitis) that occurs in approximately 10% to 35% of heavy drinkers. Alcoholic hepatitis is an important clinical entity for the following two reasons: (1) patients with severe alcoholic hepatitis have extremely high short-term mortality rates, and (2) alcoholic hepatitis is a well-documented precursor of cirrhosis, with an associated long-term risk of cirrhosis that is nine times higher than that for patients with fatty liver alone.9 With continued alcohol abuse, a fine mesh-like pattern of cirrhosis with prominent involvement of the central vein (micronodular, or Laennec’s, cirrhosis) develops in 8% to 20% of heavy drinkers. Over time, and especially with continued abstinence, this lesion can evolve to include broad bands of fibrosis that separate large nodules of liver tissue (macronodular cirrhosis). Hepatocellular carcinoma typically develops in this setting.
PATHOGENESIS
ETHANOL METABOLISM AND TOXIC METABOLITES
The liver is the main organ responsible for ethanol metabolism; other organs such as the stomach contribute to much lesser degrees. Ethanol is metabolized by three major systems in the liver: alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP2E1), and, of least importance, catalase.10 ADH is actually a set of cytoplasmic enzymes with multiple isoforms. ADH is the primary enzyme system responsible for metabolism of ethanol at low concentrations, whereas CYP2E1 contributes at higher concentrations of ethanol (greater than 10 mM) and is induced by exposure to ethanol. Both ADH and CYP2E1 convert ethanol to acetaldehyde, which is then converted to acetate by aldehyde dehydrogenase (ALDH). Acetaldehyde is a highly reactive and potentially toxic compound that is responsible for many of the systemic toxic effects of alcohol, such as nausea, headaches, and flushing. The “Oriental flush syndrome” results from impaired metabolism of acetaldehyde caused by inheritance of the ALDH22 allele, which encodes an inactive form of ALDH2. Persons from East Asia who are homozygous for this mutation rarely drink ethanol because they invariably experience toxic systemic effects, such as flushing and tachycardia, when they do drink.
Acetaldehyde also is postulated to play an etiologic role in alcoholic liver disease. Acetaldehyde can form adducts with reactive residues on proteins or small molecules (e.g., cysteines). These chemical modifications can alter or interfere with normal biologic processes and can be directly toxic to the cell. Modified molecules also may stimulate the host’s immune response and cause autoimmune-like manifestations. Antibodies against such oxidatively modified proteins have been reported in both human and animal models of alcoholic liver disease.11 An example is the hybrid adduct of malondialdehyde and acetaldehyde (MAA), unique to alcohol exposure, which induces an immune reaction in human alcoholics and in animal models of alcoholic liver disease.11 Acetaldehyde also has been shown to impair mitochondrial glutathione transport and to sensitize hepatocytes to tumor necrosis factor (TNF)-mediated killing.12
In addition to forming cytotoxic metabolites such as acetaldehyde, ethanol metabolism can alter the cellular oxidation-reduction (redox) state, thereby modulating liver injury. Specifically, the oxidation of ethanol uses nicotinamide-adenine dinucleotide (NAD+) as an electron acceptor and thereby causes a shift in the ratio of reduced NAD (NADH) to NAD+ to a more reduced state.10 This change in the redox state can impair normal carbohydrate and lipid metabolism; multiple effects ensue, including a decrease in the supply of adenosine triphosphate (ATP) to the cell and an increase in hepatic steatosis.
OTHER METABOLIC MECHANISMS
Oxidative Stress
Oxidative stress is an imbalance between pro-oxidants and antioxidants. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are products of normal metabolism and can be beneficial to the host (e.g., by contributing to bacterial killing).13 Overproduction of ROS and RNS or inadequate antioxidant defenses (e.g., low levels of vitamins, selenium, mitochondrial glutathione), or both, can lead to liver injury. Oxidative stress is well documented in alcoholic liver disease.14 Studies in normal volunteers have shown that acute alcohol consumption causes a dose-related increase in urinary isoprostane levels (a marker of lipid peroxidation, which is an indirect marker of oxidative stress), and patients with alcoholic hepatitis exhibit high isoprostane levels.15 Major ROS and RNS include superoxide anion, hydrogen peroxide, and hydroxyl radical (ROS) and nitric oxide, peroxynitrite (RNS), and hypohalous acid. Oxidative stress usually is documented by detection of one of several indirect markers (1) protein oxidation (e.g., protein thiol or carbonyl products); (2) lipid oxidation (e.g., isoprostanes, malondialdehyde); (3) DNA oxidation (e.g., oxodeoxyguanosine); or (4) depletion or induction of antioxidant defenses (e.g., vitamin E, glutathione, thioredoxin).
The stimulus for oxidative stress in the liver comes from multiple sources. In hepatocytes, CYP2E1 activity increases after alcohol consumption—in part because of stabilization of messenger RNA (mRNA). The CYP2E1 system leaks electrons to initiate oxidative stress.13 CYP2E1 is localized in the hepatic lobule in areas of alcohol-induced liver injury. Moreover, overexpression of CYP2E1 in mice and in HepG2 cells (a hepatocyte cell line) in vitro leads to enhanced alcohol hepatotoxicity.16,17 On the other hand, alcohol-induced liver injury still develops in CYP2E1-knockout mice. These findings suggest that increased CYP2E1 probably plays a role in alcoholic liver injury but is not the sole or dominant factor. In the CYP2E1-knockout mice, other compensatory mechanisms may be operational, such as induction of other microsomal enzymes. Nonparenchymal cells and infiltrating inflammatory cells (e.g., polymorphonuclear neutrophils) are another major source of pro-oxidants that are used for normal cellular processes, such as killing invading organisms. Major enzyme systems for pro-oxidant production in Kupffer cells include nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase and inducible nitric oxide synthase (iNOS). Mice deficient in NADPH oxidase or mice treated with the drug diphenyleneiodonium sulfate, which blocks NADPH oxidase, are resistant to ethanol-induced liver injury.18,19 Infiltrating neutrophils use enzyme systems such as myeloperoxidase to generate hypochlorus acid (HOCl−, a halide species that causes oxidative stress) and RNS.
The critical role of oxidative stress in the development of alcoholic liver disease has been validated in multiple studies in rats and mice fed alcohol and treated with various antioxidants, ranging from ebselen to green tea polyphenols, that overexpress both superoxide dismutase I and II. Various antioxidants have been shown to block or attenuate the development of alcoholic liver disease in rodents.13
Mitochondrial Dysfunction
Mitochondria are the major consumers of molecular oxygen and major generators of ROS in the liver. Mitochondrial dysfunction is well documented in alcoholic liver disease and contributes to oxidative stress.20,21
Mitochondrial abnormalities in alcoholic liver disease include megamitochondria observed on light and electron microscopy and functional mitochondrial abnormalities as documented by an abnormal 13C ketoacid breath test result (ketoacids are metabolized by mitochondria).22 Short-term administration of alcohol causes increased hepatic superoxide generation in liver mitochondria, with an increased flow of electrons along the respiratory electron transport chain. The increased NADH/NAD+ ratio caused by ethanol intake favors superoxide generation.13 Because hepatic mitochondria lack catalase, glutathione plays a critical role in protecting mitochondria against oxidative stress. Mitochondria do not make glutathione but instead import it from the cytosol. In alcoholic liver disease, the transport of glutathione into mitochondria is impaired, and selective mitochondrial glutathione depletion is observed.23 Glutathione depletion also sensitizes the liver to the toxic effects of TNF, and TNF also impairs mitochondrial function. Finally, an increase in mitochondrial membrane depolarization and permeability leads to hepatocyte death, especially apoptotic (programmed), rather than necrotic, cell death.
Hypoxia
The centrilobular area of the hepatic lobule (the functional unit of the liver) has the lowest oxygen tension and greatest susceptibility to hypoxia. Chronic alcohol intake increases oxygen uptake by the liver and increases the lobular oxygen gradient. A chronic intragastric feeding model in rats has been used to define the mechanisms underlying hepatic hypoxia and the association of these mechanisms with cycling of urinary alcohol levels (UALs).24 At high UALs, hepatic hypoxia is observed, along with reduced ATP levels; the NADH/NAD+ ratio is shifted to the reduced state; and the hypoxia-inducible factor (HIF) 1 and 2 genes are up-regulated. When UALs fall, reperfusion injury occurs, with free radical formation and peak liver enzyme release from hepatocytes. Stimuli for this cycle of events include catecholamines (levels of which coincide with peak UALs) and modulators of vascular tone (e.g., nitric oxide, endothelin-1); modulation of these stimuli offers future therapeutic possibilities.
Impaired Proteasome Function
The 26S ubiquitin-proteasome pathway is the primary proteolytic pathway of eukaryotic cells (see Chapter 72). It controls the levels of numerous proteins involved in gene regulation, cell division, and surface receptor expression, as well as stress response and inflammation. The proteasome system is now considered a cellular defense mechanism because it also removes irregular and damaged proteins generated by mutations, translational errors, or oxidative stress.25 This pathway involves two major steps: (1) covalent attachment of multiple ubiquitin molecules to the protein substrate and (2) degradation of the targeted protein by the 26S proteasome complex. The 26S ubiquitin-proteasome pathway may play a pathogenic role in the development of alcoholic liver disease.26
Early clinical studies showed that hepatomegaly caused by chronic alcohol consumption resulted in part from accumulation of protein in the liver.27 Animal studies have demonstrated that chronic ethanol feeding results in a significant decrease in proteolytic activity of the proteasome; this decreased activity can lead to abnormal protein accumulation, including accumulation of oxidized proteins.28,29 The decrease in proteasome function correlates significantly with the level of hepatic oxidative stress. Patients with alcoholic cirrhosis have increased serum ubiquitin levels, suggesting damaged proteasome function.30 Also, hepatocytes from alcoholics contain large amounts of ubiquitin in the form of cellular inclusions, or Mallory (or Mallory-Denk) bodies, which accumulate because they are not degraded efficiently by the proteasome.31 The formation of Mallory bodies, which occurs when the ubiquitin-proteasome system is inhibited or overwhelmed, is a pathway of liver injury caused by diverse toxins, including alcohol.31 As hepatocytes die as a result of proteasome inhibition, they inappropriately release cytokines such as interleukin (IL)-8 and IL-18. IL-8 recruits neutrophils and probably plays a role in neutrophil infiltration in alcoholic hepatitis, and IL-18 sustains inflammation in the liver.32
Abnormal Metabolism of Methionine, S-adenosylmethionine, and Folate
In mammals, the liver plays a central role in methionine metabolism; nearly one half of the daily intake of methionine is metabolized in the liver (Fig. 84-2). The first step in methionine metabolism is the formation of S-adenosylmethionine (SAM) in a reaction catalyzed by methionine adenosyltransferase (MAT). Activity of this enzyme is depressed in alcoholic liver disease.33 SAM is the principal biological methyl donor through the transmethylation pathway; the precursor of aminopropyl groups used in polyamine biosynthesis; and a precursor of glutathione through its conversion to cysteine along the transsulfuration pathway. Under normal conditions, most of the SAM generated daily is used in transmethylation reactions, in which methyl groups are added to a large number of molecules by means of specific methyltransferases.34 These compounds include DNA, RNA, biogenic amines, phospholipids, histones, and other proteins; methylation of these compounds may modulate cellular functions and integrity. In this process, SAM is converted to S-adenosylhomocysteine (SAH), which is a potent competitive inhibitor of most methyltransferases. Both an increase in SAH and a decrease in the SAM/SAH ratio are known to inhibit transmethylation reactions.34–36
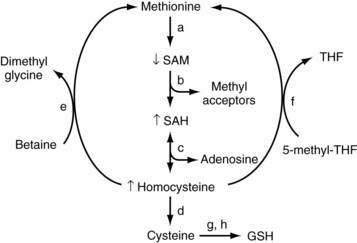
Deficiency of SAM in patients with alcoholic liver disease was first noted in the early 1980s, when it was observed that alcoholic subjects had delayed clearance of an oral bolus of methionine (presumably because of blocked conversion of methionine to SAM).37 Functional MAT activity was subsequently shown to be subnormal in liver biopsy specimens from alcoholic subjects.38 Subnormal hepatic SAM levels also are noted in various experimental models of liver injury. Exogenous administration of SAM corrects the deficiency and attenuates the severity of these experimental forms of liver injury.33,39
Because SAM is a precursor of glutathione, SAM deficiency results in glutathione deficiency, which is observed in many forms of liver disease.40 In animal studies, exogenous SAM corrects hepatic deficiencies of both SAM and glutathione.41 Because glutathione is required for optimal expression of MAT activity in liver, hepatic deficiency of MAT may be caused in part by glutathione deficiency. Also, hepatic MAT is sensitive to oxidative stress, and the subnormal hepatic MAT activity in patients with alcoholic liver disease could result from oxidation of MAT.42
In models of alcohol-induced hepatotoxicity, SAM has been shown to maintain mitochondrial glutathione levels. Depletion of mitochondrial glutathione is thought to be one pathogenic factor in the development of alcoholic liver disease, and SAM, but not other glutathione prodrugs, prevents mitochondrial glutathione depletion in experimental alcoholic liver disease (possibly by protecting mitochondrial glutathione transport systems).43 SAM also decreases lipopolysaccharide (LPS)-stimulated TNF release and increases IL-10 release in a monocyte cell line.33 Similarly, in rats fed a diet to induce SAM deficiency, serum TNF levels increase, and sensitivity to endotoxin-induced hepatotoxicity, which can be blocked by injection of SAM, increases markedly.44 These data support the concept that SAM may have direct hepatoprotective functions and may modify LPS-stimulated cytokine production.
Although serum SAM levels are decreased in patients with alcoholic liver disease, levels of the downstream products SAH and homocysteine are elevated. Homocysteine has been postulated to play a role in the pathogenesis of fatty liver seen with alcoholic liver disease, and in experimental animals reduction of homocysteine levels with administration of betaine (to convert homocysteine to methionine) attenuates the severity of alcoholic liver disease.45 Elevated SAH levels have been shown to sensitize hepatocytes to TNF-mediated destruction, and SAH may be a critical physiologic sensitizer of TNF-mediated killing in liver injury.36 Homocysteine and SAH can be removed by giving betaine, which facilitates regeneration of methionine from homocysteine. Folic acid also can play a critical role in the regeneration of homocysteine to methionine by means of 5-methyltetrahydrofolate (5-MTHF).46 Fatty liver develops in mice that lack the gene for MTHF reductase (MTHFR), and steatohepatitis develops in MAT1A–knockout mice; these findings further support a role for this critical pathway in the development of steatosis and steatohepatitis. Halsted and colleagues have shown that folic acid deficiency enhances the development of alcohol-induced liver injury in micropigs and that alcohol feeding interferes with normal folic acid metabolism in multiple different pathways—from impaired intestinal uptake to increased renal excretion.46 Collectively, the data support a role for altered methionine-transmethylation-transsulfuration metabolism in alcoholic liver disease and link these pathways to TNF hepatotoxicity.35
IMMUNE AND INFLAMMATORY MECHANISMS
Kupffer Cell Activation and Dysregulated Cytokine Production
Cytokines are low-molecular-weight mediators of cellular communication (see Chapter 2).47 Multiple cell types in the liver are potential sources of the increased release of proinflammatory cytokines observed in alcoholic liver disease. Kupffer cells are prominent producers of proinflammatory cytokines such as TNF-α, as well as certain anti-inflammatory cytokines such as IL-10. Sinusoidal endothelial cells express adhesion molecules, which modulate white blood cell adhesion and transmigration. Activated stellate cells produce collagen in response to signals such as the profibrotic cytokine transforming growth factor-β (TGF-β). Hepatocytes are a relatively newly recognized source of cytokine production, including IL-8, a major neutrophil chemotactic peptide and angiogenesis factor.
Major stimuli for the observed increase in proinflammatory cytokine production in alcoholic liver disease are believed to be ROS and intestine-derived LPS.47 In alcoholic liver disease, intestinal permeability is increased and the frequency of endotoxemia is high. LPS activates the redox-sensitive transcription factor NF-κB in Kupffer cells, thereby resulting in the production of certain cytokines such as TNF (Fig. 84-3). TNF can increase intestinal permeability, induce oxidative stress, and perpetuate this cycle. Generation of ROS through the metabolism of alcohol also can activate NF-κB and stimulate proinflammatory cytokine production.48 Although necrosis has traditionally been thought to be the major mechanism of hepatocyte cell death in alcoholic liver disease, increased apoptosis also has been documented. As hepatocytes die of apoptosis, they can be taken up by Kupffer cells and stimulate TNF production.49 Moreover, when alcohol-fed rodents are treated with caspase inhibitors to block apoptosis, liver inflammation and injury are markedly attenuated.50 When hepatocytes die of proteasome inhibition-mediated apoptosis, the dying hepatocytes release IL-8 and IL-18, which cause sustained inflammation.32 Therefore, in alcoholic liver disease, hepatocyte apoptosis may sustain proinflammatory cytokine production and cell injury or death.
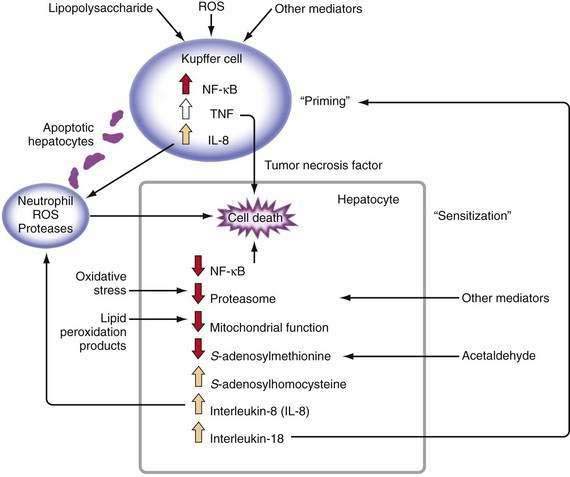
TNF metabolism is dysregulated in alcoholic hepatitis, as suggested by the observation that cultured monocytes (which produce substantial amounts of TNF) from patients with alcoholic hepatitis produce significantly increased amounts of TNF in response to LPS stimulation.51 Increased serum TNF concentrations in patients with alcoholic hepatitis show a strong correlation with disease severity and risk of mortality.47 Serum concentrations of TNF-inducible cytokines and chemokines, such as IL-6, IL-8, IL-18, monocyte chemoattractant protein 1 (MCP-1), and others, are elevated in patients with alcoholic hepatitis or cirrhosis, and the levels often correlate with markers of the acute-phase response, reduced liver function, and poor clinical outcomes.47
This enhanced cytokine response to a physiologic stimulus such as LPS is termed priming. Increased serum or urinary levels of neopterin and other markers indicate that monocytes and Kupffer cells are primed in alcoholic liver disease. This priming for LPS-stimulated TNF production has been reproduced in vitro by culturing monocyte cell lines with relevant concentrations of alcohol. This response appears to be mediated, at least in part, by induction of CYP2E1 and oxidative stress.52
Studies in rats, mice, and tissue culture have validated a pathogenic role for cytokines (especially TNF) in the development of alcoholic liver disease.47 Rats chronically fed alcohol are more sensitive to the hepatotoxic effects of injected LPS and have much higher LPS-stimulated plasma levels of TNF in comparison with control rats. Liver injury can be attenuated by giving a prostaglandin analog to down-regulate TNF production.53 Because rats have a natural aversion to alcohol, an intragastric route is often used to deliver high amounts of alcohol. Studies using the intragastric alcohol-feeding model have demonstrated that the development of liver injury coincides with an increase in TNF mRNA expression in the liver and in isolated Kupffer cells.54,55 Rats fed ethanol intragastrically also have high blood LPS levels and increased expression of CYP2E1, as well as markers of oxidative stress and lipid peroxidation.
Not only are levels of proinflammatory cytotoxic cytokines increased in alcoholic liver disease, but also monocyte and Kupffer cell production of protective anti-inflammatory cytokines such as IL-10 is decreased.56 The importance of this observation for humans has been confirmed using IL-10–knockout mice, in which more severe ethanol-induced hepatotoxicity develops and increased levels of pro-inflammatory cytokines such as TNF are seen.57
Several strategies have been devised to decrease cytokine production or activity in an attempt to block or attenuate liver injury. Examples include antibiotics to modulate intestinal flora and LPS, gadolinium chloride to destroy Kupffer cells, and antioxidants such as glutathione prodrugs to inhibit cytokine production. Each of these strategies has been successful in attenuating alcohol-induced liver injury in rats.47 Prebiotics such as oat bran also have been shown to decrease endotoxemia in experimentally induced alcoholic liver injury. Perhaps the most compelling data that relate TNF to alcohol-induced liver injury are from studies in which anti-TNF antibody has been used to prevent liver injury in alcohol-fed rats.58 Similarly, alcoholic liver injury does not develop in mice that lack the TNF type I receptor.59
Hepatocytes normally are resistant to TNF killing. Hepatocytes from rats fed alcohol or hepatocytes incubated in alcohol are sensitized to TNF killing, however.16,60 Some potentially relevant mechanisms for this sensitization include mitochondrial depletion of glutathione, accumulation of SAH, and inhibition of proteasomes, among others. Therefore, in alcoholic liver disease, monocytes and Kupffer cells are primed to increase production of TNF and to sensitize hepatocytes to TNF killing. These processes are closely intertwined with previously described mechanisms such as oxidative stress, mitochondrial dysfunction, abnormal metabolism of methionine, and dysfunction of proteasomes.
Immune Responses to Altered Hepatocellular Proteins
Alcoholic hepatitis may persist histologically for many months after exposure to ethanol has ceased, suggesting an ongoing immune or autoimmune response. Autoimmune reactions are now well documented in patients with alcoholic liver disease, with autoantibodies directed against phospholipids, alcohol dehydrogenase, heat shock protein, and other potential antigens.61 Patients with alcoholic liver disease are at increased risk for the development of immune responses directed at neoantigens generated from the interactions of metabolites of alcohol (e.g., acetaldehyde or hydroxyethyl radicals) with liver proteins.62 Studies have linked genetic susceptibility and autoimmunity in alcoholic liver disease. Some humans have a genetic mutation in the cytotoxic T lymphocyte–associated antigen 4G (CTLA-4G) allele that leads to inappropriately activated T cell function. One of the breakdown products of alcohol metabolism, the hydroxyethyl radical, can modify CYP2E1 and, in the presence of the CTLA-4G mutation, increase the risk that anti-CYP2E1 autoantibodies will develop.63 This is one pathway whereby alcohol abuse may break “self-tolerance” in the liver.
GENDER AND GENETIC FACTORS
Female gender is now a well-accepted risk factor for the development and rapid progression of alcoholic liver disease.4,5 Although rates of metabolism and elimination of alcohol have been reported to be more rapid in women than in men, when adjusted for liver volume, elimination rates are similar between genders.64 Studies in rats or mice fed alcohol chronically have demonstrated that females are more susceptible than males to liver injury. Risk factors for the development of liver disease in females appear to include increased endotoxemia, lipid peroxidation, and chemokine (e.g., monocyte chemotactic protein-1) mRNA levels and activation of the critical transcription factor NF-κB.65 These risk factors are critical for determining “safe” levels of alcohol consumption in women. Many authorities consider any amount of alcohol above 20 g a day to be a risk factor for the development of liver disease in women; however, differences in levels of alcohol dehydrogenase in gastric mucosa between men and women are not thought to play a major role in the greater susceptibility of women to alcoholic liver injury.
Genetic polymorphisms in alcohol-metabolizing systems such as CYP2E1 and ADH have been postulated to play a role in the development of alcoholic liver disease.66 None of these polymorphisms, however, adequately explains the diverse pathologic responses seen among patients with alcoholic liver disease. Polymorphisms in the promoter regions of cytokines TNF and IL-10 also have been reported to predispose affected persons to the development of alcoholic liver disease and are under active study.67,68
EMERGING MECHANISMS
Three areas deserve recognition as emerging mechanisms of liver injury. The first is the area of epigenetics, which refers to changes in gene expression that do not involve DNA coding sequence modifications. Epigenetic mechanisms typically involve DNA methylation and a variety of modifications of histones. Epigenetic modifications can be transient or long term and can be tissue or organ specific. The critical steps and enzymes (e.g., histone acetyltransferase, DNA methyltransferase) responsible for epigenetic modifications are under active investigation by multiple laboratories.69
A second area is the endoplasmic reticulum (ER) stress response, which is induced by the accumulation of unfolded or misfolded proteins. To deal with the ER stress response, cells activate a series of signaling pathways termed the unfolded protein response (UPR), which can be either protective (usually in the short term) or detrimental (usually in the long term). One of the effects of a prolonged UPR can be increased production of triglycerides and cholesterol, leading to fatty liver. Some potential inducers of the ER stress in alcoholic liver disease include elevated homocysteine levels, acetaldehyde adducts, and oxidative stress.70
A third area is the endogenous cannabinoids, which are ubiquitous lipid signaling molecules that mediate their effects by specific cannabinoid receptors, CB1 and CB2. Studies have demonstrated that inhibition of CB1 receptors can cause weight loss and attenuate fatty liver and hyperlipidemia in animal models of obesity and steatohepatitis. Moreover, CB1 blockade reduces hepatic fibrosis in a variety of animal models of cirrhosis.71
FIBROSIS
The development of hepatic fibrosis, leading to cirrhosis, indicates major progression of alcoholic liver disease and represents a maladaptive wound healing response. The development of fibrosis is a dynamic state, with constant remodeling of scar tissue; fibrosis may regress with discontinuation of exposure to alcohol. The stellate cell is the major source of collagen production in the liver. It normally exists in a quiescent state and serves as a major storehouse for vitamin A. Following activation, the stellate cell assumes a myofibroblast-like contractile phenotype and produces collagen. The cytokine TGF-β is a major stimulus for stellate cell activation and collagen production. Other cytokines implicated in activation of stellate cells include platelet-derived growth factor and connective tissue growth factor (see Chapter 90). Oxidative stress plays a major role in stellate cell activation, and a variety of antioxidants can block both stellate cell activation and collagen production in vitro. Serum levels of 4-hydroxy-nonenal, a specific product of lipid peroxidation, are elevated in patients with alcoholic liver disease and up-regulate both procollagen type I and tissue inhibitor of metalloproteinase-1 (TIMP-1) gene expression. Matrix metalloproteinase-1 plays a major role in degrading type I collagen. TIMP-1 levels also are elevated in alcoholic liver disease. The result appears to be an increase in stellate cell activation and collagen production on the one hand and a decrease in matrix degradation on the other hand.72–74
DIAGNOSIS OF ALCOHOL ABUSE
The diagnosis of alcohol abuse is based on a history of heavy alcohol intake and the presence of other organ system damage or an excessive frequency of falls, lacerations, and fractures. Physicians typically identify only 50% of patients with drinking problems because of both inadequate questioning by physicians and denial by patients.75 Underdiagnosis is particularly common in older patients.76 The uniform application of screening tools such as the four-item AUDIT-C (Alcohol Use Disorders Identification Test) consumption questions and the four-item CAGE (need to cut down, annoyed by criticism, guilty about drinking, need for an eye-opener in the morning) questionnaire dramatically improves the recognition of patients with problem drinking in primary care clinics.77 Even the use of a single question: “When was the last time you had more than x drinks in one day?,” where x equals four for women and five for men, dramatically improves the diagnosis of alcoholism in primary care settings.78
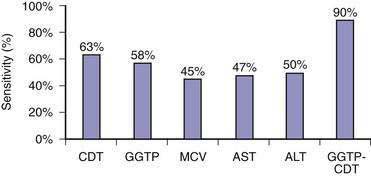
Because of the reluctance of many patients to share their drinking histories candidly, continued interest has been directed toward laboratory measures that can reliably identify patients with problem drinking. Blood or breath alcohol measurements are the most sensitive and specific indicators of recent alcohol abuse, particularly among binge drinkers.79 The major limitation of these tests is the short half-life of ethanol in blood, urine, and breath. As a result, continued efforts have focused on developing biomarkers of alcohol abuse that are detectable over longer periods of time. The most specific of these biomarkers is carbohydrate-deficient transferrin (CDT).80 CDT levels increase in serum with ingestion of 50 to 80 g/day of ethanol for two to three weeks and decline gradually during abstinence, with a half-life of approximately 15 days; however, the sensitivity of this test is only 35% to 40% among alcoholics who consume 100 g of ethanol daily.80 Conditions that can influence the CDT level include gender, age, body mass index, smoking, anorexia, pregnancy, and acute trauma with blood loss.80 These issues, as well as a lack of methodologic standardization, has limited the use of CDT measurement in clinical practice. Mean corpuscular erythrocyte volume (MCV), serum gamma glutamyl transpeptidase (GGTP) levels, and the ratio of mitochondrial aspartate aminotransferase (AST) to total AST (mitochondrial AST/total AST) and combinations of these markers have been touted as more accurate measures of alcohol abuse. A mathematically formulated equation incorporating serum GGTP and CDT levels appears to have the best diagnostic accuracy of any currently available assays for chronic alcohol abuse (Fig. 84-4).81 Measurement of two alcohol metabolites, phosphatidylethanol and ethyl glucuronide, also shows promise as an innovative way to detect recent alcohol use.80
DIAGNOSIS OF ALCOHOLIC LIVER DISEASE
HISTORY
Most patients with fatty liver are asymptomatic. Although patients with alcoholic hepatitis and cirrhosis may be asymptomatic, many present with a variety of complaints including anorexia, nausea and vomiting, weakness, jaundice, weight loss, abdominal pain, fever, and diarrhea.82
PHYSICAL EXAMINATION
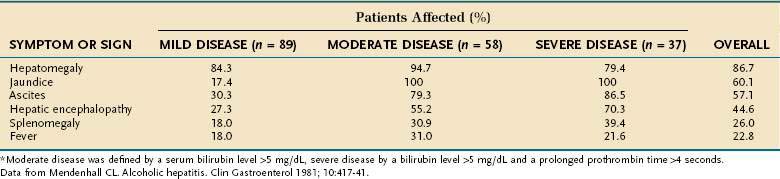
The most extensive demographic information on alcoholic liver disease in the United States comes from studies of hospitalized patients who were assigned the diagnosis on the basis of clinical and histologic parameters.82,83 Although these studies included few asymptomatic patients, they provide a useful guide to diagnosis. The most common physical finding in patients with fatty liver and alcoholic hepatitis is hepatomegaly, which is detectable in more than 75% of patients, regardless of disease severity. Patients with alcoholic hepatitis and cirrhosis also may have hepatic tenderness, an audible bruit over the liver, spider angiomata, splenomegaly, and peripheral edema. Jaundice and ascites, which are found in approximately 60% of patients, are more frequent in patients with severe disease (Table 84-2). Various degrees of hepatic encephalopathy can be seen, usually in the most severely ill patients. Some patients with alcoholic hepatitis have a fever, with temperatures as high as 104°F, that can persist for weeks.
In patients with well-compensated cirrhosis, findings on the physical examination can be normal; however, most patients have obvious hepatomegaly and splenomegaly. As the disease progresses, the liver decreases in size and has a hard and nodular consistency. Patients with decompensated cirrhosis typically have muscle wasting, ascites, spider angiomata, palmar erythema, and Dupuytren’s contractures. Enlarged parotid and lacrimal glands often are seen, and severely ill patients may have Muercke’s lines or white nails. Patients with hepatopulmonary syndrome often have digital clubbing (see Chapter 92).
LABORATORY FEATURES
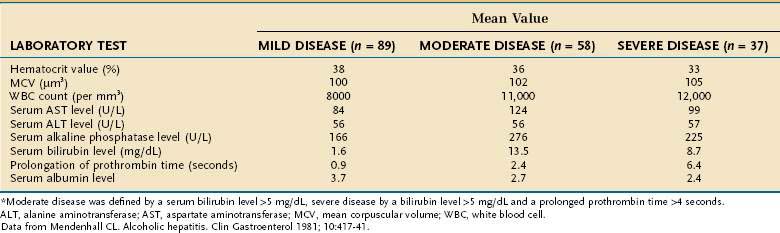
Only one third of hospitalized patients with fatty liver have laboratory abnormalities, which usually consist of mild increases in serum AST and alanine aminotransferase (ALT) levels. As illustrated in Table 84-3, surprisingly modest elevations of serum aminotransferase levels are seen in patients with alcoholic hepatitis and cirrhosis, even when the disease is severe.82,83 Serum AST levels are almost always less than 300 to 500 U/L and typically are associated with trivial elevation of serum ALT levels, resulting in an AST/ALT ratio greater than 2, which is characteristic of alcoholic liver disease, in part because of deficiency of pyridoxal 5′ phosphate (a cofactor of aminotransferases) in alcoholic patients (see Chapter 73). Serum alkaline phosphatase levels can range from normal to values greater than 1000 U/L. Serum bilirubin levels range from normal to 20 to 40 mg/dL, and serum albumin levels may be normal or depressed to a value as low as 1.0 to 1.5 g/dL. Most patients with alcoholic liver disease are anemic and have some degree of thrombocytopenia. By contrast, the white blood cell count usually is normal or elevated, occasionally to levels consistent with a leukemoid state. Severely ill patients usually have marked prolongation of the prothrombin time—often expressed as the international normalized ratio (INR)—and often have elevated serum creatinine values.82–85
HISTOPATHOLOGY
The clinical diagnosis of alcoholic liver disease is quite sensitive and specific; therefore, liver biopsy is rarely needed to establish the diagnosis.86 A liver biopsy is essential for determining precisely the severity of hepatic injury, however, and for clarifying the diagnosis in atypical cases (see Fig. 84-1). Centrilobular and perivenular fatty infiltration in the liver is seen in most persons who drink more than 60 g of alcohol daily. Classic histologic features of alcoholic hepatitis include ballooning degeneration of hepatocytes, alcoholic hyaline (Mallory, or Mallory-Denk, bodies) within damaged hepatocytes, and a surrounding infiltrate composed of polymorphonuclear leukocytes. Most patients have moderate to severe fatty infiltration. Varying degrees of fibrosis may be present, and many patients exhibit an unusual perisinusoidal distribution of fibrosis, at times with partial or complete obliteration of the terminal hepatic venules (sclerosing hyaline necrosis). Cirrhosis can be identified by the presence of nodules of hepatic tissue that are completely surrounded by fibrous tissue.7,8
Alcoholic cirrhosis typically is micronodular or mixed micro- and macronodular. In patients with coexisting alcoholic hepatitis, alcoholic hyaline is almost universal, and sclerosing hyaline necrosis and moderate-to-severe fatty infiltration are common.7,8 In patients with alcoholic cirrhosis who abstain from alcohol for long periods, a frequent finding is a gradual transformation to macronodular cirrhosis, which is indistinguishable from cirrhosis caused by other forms of liver disease.
CONDITIONS THAT MAY RESEMBLE ALCOHOLIC LIVER DISEASE
Nonalcoholic Fatty Liver Disease
The condition that is most challenging to differentiate from alcoholic liver disease is NAFLD. The two conditions are histologically indistinguishable. As a consequence, the differentiation between alcoholic liver disease and NAFLD has to be made on clinical grounds. The strongest evidence in support of a diagnosis of NAFLD rather than alcoholic liver disease is a history of daily alcohol intake less than 20 g/day. When a patient’s alcohol intake is questionable, differentiating the two conditions can be difficult, if not impossible. Patients with NAFLD are more likely than patients with alcoholic liver disease to be asymptomatic and often have peripheral insulin resistance, obesity, hypertension, and dyslipidemia.87,88 A model that incorporates the MCV, AST/ALT ratio, body mass index, and gender shows promise in more clearly differentiating patients with alcoholic liver disease from those with NAFLD.89 The serum CDT level can be useful for distinguishing heavy drinkers from abstinent patients with NAFLD; however, the accuracy of this test for detecting moderate but clinically significant levels of alcohol intake is less clear (see earlier and Chapter 85).80
Hereditary Hemochromatosis
On occasion, distinguishing patients with alcoholic liver disease and secondary iron overload from those with liver disease caused by hereditary hemochromatosis, particularly those with decompensated cirrhosis, can be difficult. Patients with end-stage liver disease from alcoholic cirrhosis may have elevated serum iron and ferritin levels and increased hepatic iron levels suggestive of hereditary hemochromatosis.90 In fact, more than 20% of patients with end-stage alcoholic cirrhosis have clinically important hepatic siderosis.91 To complicate matters further, 15% to 40% of patients with hereditary hemochromatosis consume more than 80 g of alcohol daily.92
The overlapping clinical features of hereditary hemochromatosis and alcoholic liver disease include hepatomegaly, testicular atrophy, cardiomyopathy, and glucose intolerance. Testing for mutations in the gene for hereditary hemochromatosis, HFE, is the best method for differentiating the two conditions among whites. Few patients with alcoholic cirrhosis and iron overload are homozygous for C282Y or heterozygous for the C282Y and H63D HFE genes, whereas some have a hepatic iron index value greater than 1.9 that might otherwise suggest hereditary hemochromatosis (see Chapter 74).90,91
Amiodarone Hepatotoxicity
Much less common and less difficult than NAFLD to distinguish from alcoholic liver disease is amiodarone hepatotoxicity. Although the hepatic histologic features of this condition may be similar to those of alcoholic hepatitis with or without cirrhosis, the clinical setting usually distinguishes amiodarone hepatotoxicity from alcoholic liver disease (see Chapter 86).8,93
Budd-Chiari Syndrome
Occasional patients with severe alcoholic liver disease can be misdiagnosed as having acute Budd-Chiari syndrome (hepatic vein thrombosis) on the basis of rapid clinical deterioration, marked hepatomegaly, caudate lobe hypertrophy, and failure to visualize the hepatic veins by Doppler ultrasonography.94 Careful evaluation of these patients usually reveals clinical and biochemical features typical of severe alcoholic hepatitis. Patent hepatic veins usually can be demonstrated by venography. Liver biopsy is particularly useful in distinguishing the characteristic histologic features of alcoholic liver disease from those of Budd-Chiari syndrome. Failure to recognize alcoholic hepatitis as the underlying cause of the liver disease before initiating anticoagulation or performing portacaval shunt surgery can result in high mortality rates (see Chapter 83).94
DIFFERENTIAL DIAGNOSIS OF CLINICAL DETERIORATION
Acetaminophen Hepatotoxicity
The most common cause of severe drug-induced liver injury encountered in the United States is acetaminophen hepatotoxicity (see Chapter 86). Two clinical patterns of liver injury have been identified: (1) suicidal or accidental ingestion of large quantities of acetaminophen sufficient to cause hepatic injury and (2) ingestion of lesser quantities of acetaminophen by patients predisposed to injury because of up-regulation of the hepatic enzymes that convert acetaminophen to a hepatotoxic metabolite. The latter type of toxicity is seen most commonly in chronic alcoholics who take excessive acetaminophen over a period of days to weeks for relief of a headache, toothache, or other minor pain.95,96 The clinical features in these patients are indistinguishable from those of alcoholic liver disease, with one obvious exception: AST values are typically more than 1000 U/L, much higher than those in patients with alcoholic liver disease. Because liver injury typically has occurred by the time of hospitalization, acetaminophen levels are not helpful for diagnosis or management. Recognition of the cause of the unusually elevated serum aminotransferase levels comes from careful questioning of the patient and family about acetaminophen ingestion in the days to weeks before hospitalization. The morbidity and mortality associated with this condition are considerable.95,96 Because many of these patients have a history of recent heavy alcohol use, few are candidates for liver transplantation.
Acute Viral Illness
Patients with alcoholic cirrhosis are vulnerable to decompensation from a variety of viral illnesses. Acute viral hepatitis can result in the sudden onset of liver failure, with extremely high mortality rates (see Chapters 77 through 81).97,98 Sudden decompensation and liver failure also have been reported during infection with influenza A virus.99 These dramatic cases illustrate the importance of routine immunization against hepatitis A and B and influenza in patients with alcoholic cirrhosis.
Hepatocellular Carcinoma
Occasional patients with alcoholic cirrhosis who have been abstinent for many years decompensate suddenly, with the abrupt onset of hepatic encephalopathy, variceal bleeding, or ascites. Not infrequently, the underlying cause is hepatocellular carcinoma. Unfortunately, the sudden onset of symptoms frequently results from tumor invasion of the portal or hepatic veins; as a result, the prognosis for these patients is dismal.3 The risk of hepatocellular carcinoma in patients with alcoholic cirrhosis underscores the need for surveillance for this neoplasm in these patients, especially those who abstain from alcohol and in whom the long-term prognosis is otherwise good (see Chapter 94).
COFACTORS THAT MAY INFLUENCE PROGRESSION OF ALCOHOLIC LIVER DISEASE
CHRONIC HEPATITIS C
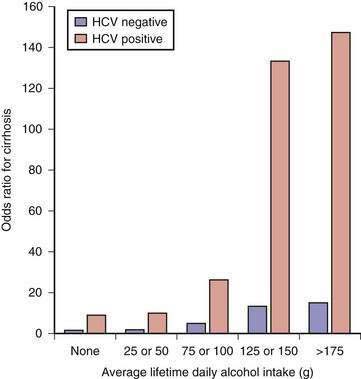
The cofactor that influences progression of alcoholic liver disease most profoundly is HCV infection. Between one fourth and one third of patients with alcoholic liver disease have serologic or virologic evidence (or both) of HCV infection.100 The prevalence of HCV infection is highest in patients who have used injection drugs; however, the risk is high even among those who deny drug use. Histologic features of focal lymphoid aggregates, portal inflammation, and periportal or bridging fibrosis are common in liver biopsy specimens from alcoholics with HCV infection.101 Of greater importance, liver disease is more severe, advanced disease develops at a younger age, and survival is shorter in patients with both alcoholic liver disease and HCV infection than in patients with alcoholic liver disease and no evidence of HCV infection.100 In one of the more striking examples of the interaction between alcohol abuse and hepatitis C, Corrao and colleagues found that the .relative risk of cirrhosis was 10-fold higher among heavy drinkers with chronic hepatitis C than among those who had no evidence of HCV infection (Fig. 84-5).102 In addition, alcohol and HCV act synergistically in the development of hepatocellular carcinoma (see Chapter 79).103–105
OBESITY AND SMOKING
The risk of liver disease is two to three times higher in drinkers who are obese than in drinkers who have a normal body mass index.106 Although an increased risk of fatty liver is not surprising in obese persons (see Chapter 85), obesity also appears to be an independent risk factor for both alcoholic hepatitis and cirrhosis.106,107 Cigarette smoking also has been shown to accelerate the progression of fibrosis in patients with alcoholic liver disease,108,109 and smoking appears to accelerate disease progression in patients with HCV infection who drink heavily.110
PROGNOSIS
The prognosis for an individual patient with alcoholic liver disease depends on the degree of pathologic injury, patient’s nutritional status, presence of complications of advanced liver disease, presence of other comorbid conditions such as HCV infection, and patient’s ability to discontinue destructive patterns of drinking. In studies that have examined the natural history of alcoholic liver disease on the basis of histologic characteristics at diagnosis, patients with fatty liver have had the best outcome (70% to 80% survival rate at four to five years); those with alcoholic hepatitis or cirrhosis have had an intermediate outcome (50% to 75% survival rate at four to five years); and those with cirrhosis combined with alcoholic hepatitis have had the worst outcome (30% to 50% survival rate at four to five years) (Fig. 84-6).111 Among all patients with alcoholic liver disease, the average one-year and five-year survival rates are approximately 80% and 50%, respectively.80 Alcoholic cirrhosis also appears to be an independent risk factor for hepatocellular carcinoma.2,3,100–102 Among alcoholics, men older than 50 years of age appear to be most vulnerable to the development of hepatocellular carcinoma (see Chapter 94).112
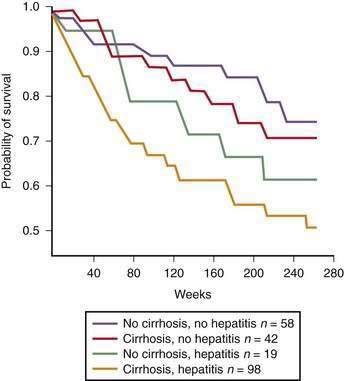
Figure 84-6. Survival of patients with alcoholic liver disease stratified by histologic severity of disease.
(From Orrego H, Black JE, Blendis LM, Medline A. Prognosis of alcoholic cirrhosis in the presence or absence of alcoholic hepatitis. Gastroenterology 1987; 92:208-14, with permission.)
ALCOHOLIC HEPATITIS
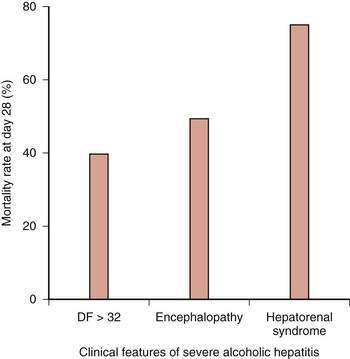
The prognosis among patients with alcoholic hepatitis can vary dramatically. In patients with severe disease, the mortality rate is high, approaching that for patients with fulminant hepatic failure. Clinical features associated with severe disease include hepatic encephalopathy, marked prolongation of the prothrombin time, elevation of the serum bilirubin level above 25 mg/dL, depression of the serum albumin level, an elevated serum creatinine level, and older age. Other important prognostic variables in patients with severe alcoholic hepatitis are spontaneous hepatic encephalopathy and hepatorenal syndrome (Fig. 84-7).79,81,82 The one-month mortality rate in patients with spontaneous hepatic encephalopathy is approximately 50%, and the rate in those with hepatorenal syndrome is 75%.79,81,82,113–115
Three models have been shown to predict short-term prognosis in these often critically ill patients. Maddrey and Boitnott discovered a simple formula they called the discriminant function (DF), calculated as [4.6 × prothrombin time − control value (seconds)] + serum bilirubin (mg/dL). The DF has proved useful for identifying patients with poor short-term survival rates.113 Three prospective studies have demonstrated that patients with a DF value of 32 or more have a poor prognosis, with one-month mortality rates of 35% to 45%.82,114,115 By contrast, patients with a DF value less than 32 have short-term survival rates of 90% to 100%.113,116 The Model for End-stage Liver Disease (MELD) score (which includes the serum bilirubin level, INR, and serum creatinine level) and the Glasgow alcoholic hepatitis score (which includes age, WBC count, blood urea nitrogen level, prothrombin time ratio [ratio of the patient’s prothrombin time to the control value], and serum bilirubin level) also have been shown to predict survival in patients with severe alcoholic hepatitis.117,118 Because both the MELD and Glasgow scores include measures of renal function, they appear to be more accurate than the DF in determining the prognosis of patients with concomitant kidney injury.
ALCOHOLIC CIRRHOSIS
The clinical tool used most widely to determine prognosis in patients with alcoholic cirrhosis is the Child-Turcotte-Pugh (CTP) score and Child (or Child-Pugh) classification (see Chapter 90). This simple classification system, which was designed specifically to assess the risk of mortality following portacaval shunt surgery in cirrhotic patients with variceal bleeding, has gained favor as a rapid method for determining the prognosis of patients with various chronic liver diseases. The Child classification is as effective as quantitative liver function tests (see Chapter 73) and disease-specific prognostic models for determining short-term prognosis in groups of patients awaiting liver transplantation (see Chapter 95).119 Despite its limitations, the Child classification has been adopted widely for risk-stratifying patients with cirrhosis because of its simplicity and ease of use. Five-year survival rates for patients with alcoholic cirrhosis decrease dramatically as the CTP score and Child’s class become higher at the time of clinical presentation (Fig. 84-8).117
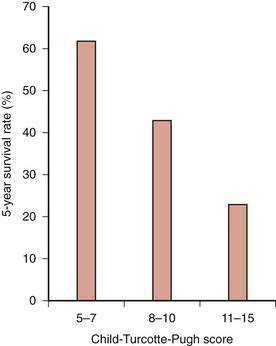
Figure 84-8. Five-year survival rates in patients with alcoholic cirrhosis according to their Child-Turcotte-Pugh scores.
(Data from Poynard T, Naveau S, Doffoel M, et al. Evaluation of efficacy of liver transplantation in alcoholic cirrhosis using matched and simulated controls: Five-year survival. Multi-centre group. J Hepatol 1999; 30:1130-7.)
The development of ascites, variceal bleeding, hepatic encephalopathy, spontaneous bacterial peritonitis, or hepatorenal syndrome also has a significant impact on the prognosis of patients with alcoholic cirrhosis. The five-year survival rate for persons in whom any of these complications develop is only 20% to 50% of that for patients with compensated cirrhosis.120 The most ominous complications are spontaneous bacterial peritonitis and rapid-onset hepatorenal syndrome (see Chapters 91 and 92). Fewer than half of the patients in whom spontaneous bacterial peritonitis develops can be expected to survive one year; the median survival of patients with hepatorenal syndrome is less than two weeks.121,122
Other models that have been used to predict prognosis in patients with alcoholic cirrhosis are the Beclere model, a proportional hazards model developed by Poynard and colleagues, and the MELD score.119,123 The Beclere model, which was developed from a database of 818 patients with alcoholic cirrhosis who were followed prospectively for four years, includes the serum bilirubin level, serum albumin level, patient’s age, and presence or absence of hepatic encephalopathy.119 The MELD model was developed at the Mayo Clinic to assess short-term prognosis in patients undergoing transjugular intrahepatic portosystemic shunt placement.123 This model has been shown to be useful for predicting short-term survival in groups of patients with various liver diseases (see Chapter 90).
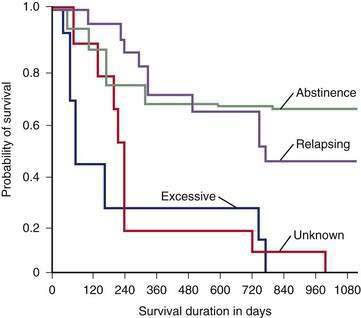
Abstinence from continued excessive drinking is the most important predictor of survival in patients who survive an initial hospitalization for alcoholic cirrhosis.124 The rate of survival over the ensuing two years is 70% to 80% among patients who abstain or dramatically reduce their excessive drinking, compared with only 20% to 30% in those who continue to drink heavily (Fig. 84-9).124
TREATMENT
ABSTINENCE AND LIFESTYLE MODIFICATION
Virtually every study of abstinence in alcoholic liver disease shows beneficial effects on patient survival, even in patients with decompensated cirrhosis (see Fig. 84-9). Reducing but not completely stopping alcohol consumption also has been shown to improve projected survival in patients with alcoholic liver disease.125,126 Heavy drinkers who receive so-called brief interventions, which are less than 1 hour in duration and incorporate simple motivational counseling techniques, are twice as likely as control patients to moderate or stop their drinking 6 to 12 months later.126 Regular meetings with a nurse or other health care professional to emphasize abstinence and adherence to medication appear to be more effective than intense counseling by alcohol treatment specialists.124,127 Abstinence invariably causes resolution of hepatic steatosis. An additional goal of abstinence is to prevent ongoing injury, fibrosis, and the possible development of hepatocellular carcinoma, but few studies have addressed the effects of abstinence on disease progression. Short-term treatment with naltrexone, an opioid antagonist, decreases the chance of relapse in one third of heavy drinkers; however, this agent is contraindicated in patients with liver disease because of its extensive hepatic metabolism and potential hepatotoxicity.127,129 Baclofen, a gamma aminobutyric acid (GABA) B-receptor agonist, shows promise as the first safe and effective agent to improve abstinence and decrease the likelihood of relapse in patients with alcoholic cirrhosis.130
As discussed earlier, obesity, which is increasing in frequency among alcoholics as well as the general population, is associated with the development of fatty liver, steatohepatitis, and cirrhosis and appears to be a major risk factor for progression of alcoholic liver disease (see Chapter 85).41,42 The majority of alcoholics smoke cigarettes, another risk factor for more severe alcoholic liver disease. Therefore, lifestyle modifications including significant reduction or cessation of alcohol consumption, weight control, and elimination of cigarette smoking, are important initial approaches to the treatment of alcoholic liver disease.
NUTRITIONAL SUPPORT
Nutritional abnormalities are pervasive in alcoholics. Two divergent patterns are common: obesity and malnutrition. Persons who combine alcohol abuse with a high calorie diet frequently develop truncal obesity, which can accelerate the progression of the underlying alcoholic liver disease.41,42,125 The high frequency of obesity is not surprising given the high caloric content (7.1 kcal/g) of alcohol.
By contrast, malnutrition is a widespread clinical problem among patients with alcoholic liver disease when a substantial proportion of nutrient-rich dietary calories are replaced with alcohol.131 Every patient with moderate to severe alcoholic hepatitis or cirrhosis shows some signs of malnutrition, with up to 50% of total energy intake derived from alcohol.132 The frequency of malnutrition increases dramatically with the severity of liver disease.128 For example, the frequency of profound malnutrition increases from 20% in patients with Child’s class A cirrhosis to 60% in those with Child’s C cirrhosis.127 Furthermore, a strong association exists between protein-calorie malnutrition and complications of alcoholic disease such as infections, encephalopathy, ascites, and variceal bleeding.131,132
Levels of folate, vitamin B6, thiamine, and vitamin A, as well as those of trace elements such as selenium, zinc, copper, and magnesium, are often severely reduced in patients with alcoholic liver disease.125 The mechanisms underlying the profound malnutrition frequently observed in patients with moderate to severe alcoholic liver disease are complex and multifactorial and include (1) anorexia induced by increased proinflammatory cytokines such as TNF and leptin; (2) intestinal fat and protein malabsorption; and (3) a catabolic state that promotes gluconeogenesis from skeletal and visceral proteins.131–133 Evaluating malnutrition in patients with liver disease can be difficult because the tests most commonly used to assess nutritional status (e.g., serum albumin concentration, anthropometry, immune status) often are affected by the liver disease. The creatinine-height index appears to be the most reliable indicator of loss of muscle mass in patients with moderate-to-severe liver disease.133,134
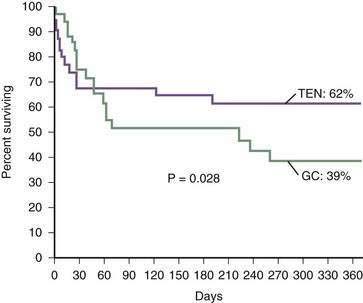
These difficulties have stimulated research into various forms of nutritional support for patients with severe alcoholic hepatitis and cirrhosis.135–138 In the first large study of intensive nutritional support in patients with moderate to severe alcoholic hepatitis, Mendenhall and colleagues demonstrated increased survival in patients provided aggressive nutritional support.132 In a pivotal multicenter study by Cabre and coworkers, patients were randomized to receive prednisone 40 mg daily (see later) or a liver-specific formula containing 2000 calories per day through a feeding tube.136 The one-month mortality rates were similar in both groups, but the one-year mortality rate was significantly lower in the patients who received the enteral nutrition, in great part because of reduced infectious complications, in comparison with patients who received glucocorticoids (Fig. 84-10). This study clearly demonstrates the important role of enteral nutrition in hospitalized patients with severe alcoholic liver disease. By contrast, no study has, to date, demonstrated a survival benefit for treatment with parenteral nutrition.131
Studies of nutritional support in outpatients are limited. Hirsch and colleagues demonstrated that patients attending an outpatient liver clinic who took an enteral nutritional support product that contained 1000 kcal and 34 g of protein had significantly improved protein intake and fewer hospitalizations in comparison with those not receiving the supplement.137 Cirrhotics have decreased hepatic glycogen stores and an accelerated metabolic reaction to short-term fasting. Studies have shown that nighttime snacks (710 kcal = 2 cans of Ensure Plus) attenuate this catabolic response and significantly improve lean tissue over a one-year period of supplementation.138
ANTI-INFLAMMATORY AND ANTICYTOKINE DRUGS
Glucocorticoids
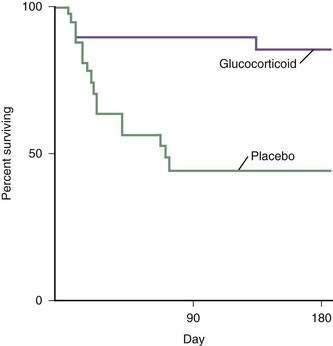
Glucocorticoid therapy has been the most extensively studied and the most controversial treatment for patients with alcoholic hepatitis. A total of 10 small, single-center, placebo-controlled randomized trials of glucocorticoid therapy were published from 1971 to 1984, and only two showed a benefit.114 Helman and colleagues demonstrated improved survival only in patients who had hepatic encephalopathy within the first 10 days after hospital admission.17 Maddrey and colleagues confirmed the prognostic importance of encephalopathy and found that a DF value greater than 32 (see earlier) was as effective as detecting encephalopathy in selecting patients at high risk for early mortality and that these patients appeared to benefit from glucocorticoid therapy.113 These two prognostic tools—hepatic encephalopathy and an elevated DF—were used to select patients for entry into a subsequent multicenter study that demonstrated a dramatic improvement in short-term survival with glucocorticoid therapy.114 The cumulative 28-day mortality rate for this severely ill group of patients was 35% in the placebo recipients, compared with only 6% in patients who received methylprednisolone. Using the same selection criteria for study entry, Ramond and colleagues confirmed the improvement in short-term survival and also demonstrated a continued survival benefit for up to six months after treatment with glucocorticoids (Fig. 84-11).115 Additional follow-up of these patients revealed that the survival benefit of glucocorticoid therapy persisted for one but not two years after treatment.133 No major complications were associated with glucocorticoid therapy in these studies.114,115 These two clinical trials included only patients with severe disease; patients with gastrointestinal bleeding requiring transfusions and active infection were excluded. Furthermore, none of the patients had evidence of hepatorenal syndrome before entry into the studies.
Glucocorticoids should not be used in patients with mild alcoholic hepatitis; however, a short course of glucocorticoids (e.g., prednisone, 40 mg daily for 28 days, followed by 20 mg daily for 7 days and 10 mg daily for 7 days) may be beneficial in patients with severe disease. The DF, MELD score, and Glasgow index can each be used to select patients for treatment. Glucocorticoids should not be used in patients with gastrointestinal bleeding requiring blood transfusions or with evidence of active infection and probably are not effective in patients with hepatorenal syndrome.19,139 The response to glucocorticoids can be determined within seven days (primarily by a reduction in serum bilirubin levels), and treatment can be discontinued at that time if a response has not been achieved.140 Figure 84-12 illustrates the factors that should be taken into account when glucocorticoid therapy is considered in patients with severe alcoholic hepatitis.
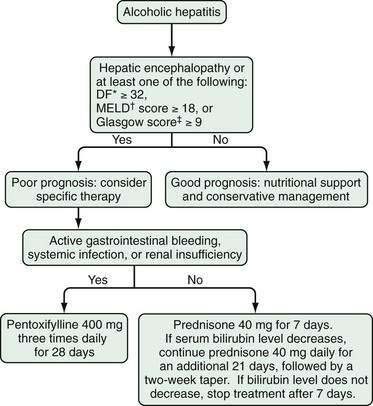
Figure 84-12. Algorithm for the management of patients with alcoholic hepatitis. *The DF is calculated as follows: 4.6 (prothrombin time of patient − prothrombin time of control) + serum bilirubin level (in mg/dL). †The Model for End-stage Liver Disease (MELD) score is based on the serum bilirubin level, international normalized ratio, and serum creatinine level (see Chapter 90).‡The Glasgow alcoholic hepatitis score is based on the patient’s age, white blood cell count, blood urea nitrogen level, ratio of prothrombin time to a control value, and serum bilirubin level. DF, discriminant function.
Pentoxifylline
Pentoxifylline is a nonselective phosphodiesterase inhibitor that increases intracellular concentrations of adenosine 3′,5′-cyclic monophosphate (cAMP) and guanosine 3′,5′-cyclic monophosphate (cGMP) and may thereby inhibit TNF production. Pentoxifylline also has been shown to decrease gene transcription and to affect multiple steps in the cytokine/chemokine inflammatory pathway, either directly or indirectly by inhibiting TNF.141 Selected effects of pentoxifylline include inhibition of cytokine/chemokine synthesis (e.g., MCP-1, IL-8, macrophage inflammatory protein [MIP]-1α and MIP-1β), decreased expression of adhesion molecules on endothelial cells, decreased activation of neutrophils, decreased proliferation of lymphocytes and monocytes, and decreased binding and transmigration of leukocytes. Pentoxifylline also reduces fibroblast proliferation and secretion of collagen and other interstitial matrix proteins.
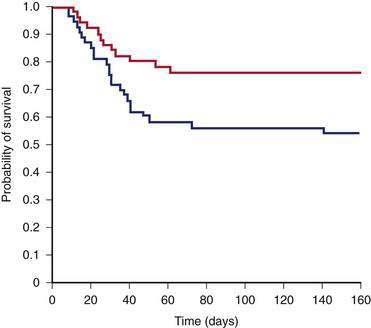
Akriviadis and colleagues performed a prospective, randomized, double-blind clinical trial of pentoxifylline in patients with severe alcoholic hepatitis (DF greater than 32).20 Forty-nine patients received pentoxifylline, 400 mg orally three times daily, and 52 received placebo (vitamin B12) for four weeks. Only 12 patients (24.5%) who received pentoxifylline died, compared with 24 (46%) who received placebo (Fig. 84-13). Pentoxifylline therapy was associated with a significant decrease in the frequency of hepatorenal syndrome as a cause of death and was well tolerated with no major side effects. On the basis of this single trial, pentoxifylline appears to be a viable alternative to glucocorticoids, particularly in patients with clinically important renal dysfunction.
Glucocorticoids Followed by Pentoxifylline
Louvet and his colleagues in France explored the possibility of switching patients to pentoxifylline if they failed to demonstrate a response to the first seven days of corticosteroid therapy.142 Unfortunately, these patients did not obtain any benefit from the early switch. Therefore, the optimal approach to the management of nonresponders to glucocorticoid therapy remains unresolved.
Specific Anti-Tumor Necrosis Factor Therapy
Dysregulated cytokine metabolism was described in alcoholic hepatitis long before it was recognized in inflammatory bowel disease and rheumatoid arthritis. An initial concern in alcoholic liver disease arose from early observations that low (“basal”) amounts of TNF were important for liver regeneration.143 Therefore, many investigators suggested that down-regulating, without totally blocking, TNF activity would be a preferred therapeutic intervention. Indeed, many therapies used in alcoholic liver disease (e.g., glucocorticoids, pentoxifylline, S-adenosylmethionine) decrease but do not abolish TNF activity.
Because therapy with anti-TNF antibodies has been shown to block development of alcohol-induced liver injury in rats, it was initially studied in small clinical trials in patients with alcoholic hepatitis, with apparent success.144 A large, double-blind, randomized controlled trial in France in which patients with acute alcoholic hepatitis were treated with prednisolone or prednisolone plus high-dose infliximab was terminated, however, because of an increased rate of infectious complications in the patients who received combined therapy.145 Etanercept was postulated to be more appropriate than infliximab in patients with alcoholic liver disease because of its shorter duration of action, but a National Institutes of Health (NIH)-sponsored multicenter trial reported similar one-month mortality rates and significantly worse six-month mortality rates in patients with moderate-to-severe alcoholic hepatitis treated with etanercept compared with those treated with placebo.146 Therefore, at the present time, agents that inhibit, but do not totally block, inflammatory mediators such as TNF appear to be preferable to those that totally block these mediators in patients with alcoholic hepatitis.
ANTIOXIDANTS
S-adenosylmethionine
As discussed earlier, alcoholic liver disease is characterized by elevated plasma methionine concentrations and decreased clearance of intravenously or orally administered methionine. Decreased MAT activity results in decreased plasma (and presumably intrahepatic) SAM levels.147 Administration of SAM has been reported to protect against experimental liver injury caused by alcohol, acetaminophen, carbon tetrachloride, and galactosamine.69,75 Theoretical benefits of SAM in alcoholic liver disease include its roles as an antioxidant and a critical methyl donor and its actions in maintaining mitochondrial function, decreasing TNF levels, and producing glutathione. A multicenter clinical study reported that SAM in a daily dose of 1200 mg significantly reduced the mortality rate and decreased the need for liver transplantation in patients with Child’s A and B alcoholic cirrhosis.148
Silymarin
Silymarin, the active ingredient extracted from Silybum marianum (also known as milk thistle), has been shown in experimental animals to protect against various hepatotoxins, including carbon tetrachloride, acetaminophen, iron (in iron overload), and poisonous mushrooms (see Chapter 87).149 It has antioxidant properties, protects against lipid peroxidation, and exerts anti-inflammatory and antifibrotic effects. Despite these properties, insufficient data are available from well-conducted clinical trials to demonstrate improvement in mortality, complications of cirrhosis, or histology in patients with alcoholic liver disease.150,151 Nevertheless, silymarin has become the most popular form of complementary and alternative medicine therapy for patients with liver disease because of its good safety profile, and ongoing NIH studies should answer questions concerning its efficacy (see Chapter 127).
Vitamin E
Vitamin E deficiency has been well documented in patients with alcoholic liver disease.152 Vitamin E has hepatoprotective effects in experimental liver injury, with potentially beneficial effects that include membrane stabilization, reduced NF-κB activation and TNF production, and inhibition of hepatic stellate cell activation and collagen production.152–154 Unfortunately, the largest randomized study of vitamin E supplementation in patients with alcoholic liver disease did not show a significant benefit, possibly because a relatively low dose was used.154
Combination Antioxidant Therapy
Oxidative stress plays an etiologic role in the development of alcoholic liver disease (see earlier), and antioxidants block the development of alcoholic liver disease in experimental animals. An initial study of a combination of antioxidants in patients with alcoholic hepatitis reported beneficial effects,155 whereas a subsequent trial in which glucocorticoids were compared with an antioxidant cocktail was stopped after an interim analysis found a significant benefit in the glucocorticoid-treated group.156 These inconsistent results led to a third trial in which patients with acute alcoholic hepatitis were randomized to antioxidant therapy alone or with glucocorticoids; neither treatment improved six-month survival.157 None of these studies evaluated whether or not antioxidant therapy actually decreases oxidative stress. At this time, antioxidants should not be used as sole therapy for alcoholic hepatitis; whether they are of benefit as adjunctive therapy in patients with alcoholic hepatitis or alcoholic cirrhosis and which antioxidant should be used remain unclear.
DRUGS OF UNLIKELY BENEFIT
Colchicine
Colchicine has many potential therapeutic mechanisms of action in alcoholic liver disease, including inhibition of collagen production, enhancement of collagenase activity, and anti-inflammatory activity. Initial positive studies158 led to a large Veterans Administration (VA) Cooperative Study of colchicine therapy in patients with alcoholic cirrhosis that showed no beneficial effects on overall or liver-related mortality.159 A smaller study from Europe also showed no beneficial effects of colchicine therapy in patients with alcoholic liver disease.160
Propylthiouracil
Chronic alcohol feeding in animal models can induce a hypermetabolic state with increased oxygen consumption similar to the hypermetabolic state associated with hyperthyroidism. This hypermetabolic state may lead to relative hypoxia in the centrilobular area of hepatic lobules. Propylthiouracil has been postulated to attenuate the hypermetabolic state, function as an antioxidant, and improve portal blood flow. Nevertheless, a Cochrane review of six randomized trials involving more than 700 patients found no beneficial effect of propylthiouracil therapy in patients with alcoholic liver disease.161
Anabolic Steroids
Anabolic steroids have been shown to decrease fatty infiltration in the liver and are hepatoprotective. As noted earlier, patients with end-stage liver disease frequently are malnourished and often have low circulating levels of the anabolic hormone insulin-like growth factor-1. These observations provide a rationale for using anabolic steroids to treat alcoholic liver disease.132 Nevertheless, a Cochrane review was not able to demonstrate efficacy for anabolic steroids (specifically oxandrolone) in patients with alcoholic liver disease, although such therapy did appear to be safe.162
Ursodeoxycholic Acid
Urosodeoxycholic acid, an agent used for a variety of cholestatic liver disorders (see Chapter 89), was evaluated in one large multicenter controlled clinical trial in patients with severe alcoholic liver disease and found to have no beneficial effect on six-month survival.163
Polyenylphosphatidylcholine
Polyenylphosphatidylcholine, or lecithin, a lipid extract obtained from soybeans, has been shown to prevent septal fibrosis and cirrhosis in alcohol-fed baboons and to stimulate the release of collagenase activity by cultured hepatic stellate cells. It also has antioxidant effects and decreases TNF production.164 Multiple positive studies of polyenylphosphatidylcholine in animal models of liver disease led to a VA Cooperative Study that evaluated the effects of this drug in humans with early alcoholic liver disease.165 Results of this study were negative; however, patients decreased their alcohol use markedly during the trial, thus decreasing the likelihood that a beneficial effect of polyenylphosphatidylcholine could be demonstrated.
LIVER TRANSPLANTATION
Liver transplantation for alcoholic liver disease remains one of the most contentious and controversial areas in transplantation medicine. Short-term outcomes following liver transplantation in patients with alcoholic liver disease are comparable to those for patients who receive transplants for most other conditions, with seven-year survival rates of 60% (see also Chapter 95).166 Profound confusion in the early postoperative period is more likely to develop in patients with alcoholic cirrhosis, however, than in those undergoing transplantation for other liver diseases.167 The result can be a prolonged hospitalization and an increase in the cost of transplantation. A number of transplant centers have reported distinctly lower long-term survival rates among patients with alcoholic liver disease, particularly among those who return to heavy drinking.168 In addition, patients with alcoholic liver disease have an increased risk of pharyngeal, esophageal, and gastric malignancies after transplantation.169 This risk is particularly high among persons who begin smoking heavily again after the operation.170
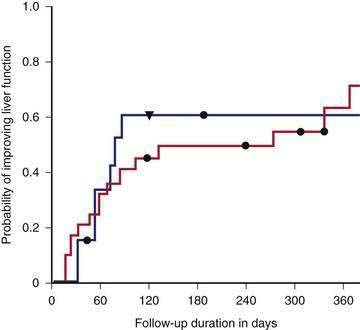
Many patients with apparently advanced alcoholic liver disease can recover to the degree that transplantation is not required if they reduce their alcohol intake significantly or abstain completely (Fig. 84-14).124 Because the benefits of abstinence can be so dramatic, requiring a period of abstinence before proceeding with transplantation is reasonable for patients with alcoholic liver disease. Patients who have a CTP score of 11 or greater despite at least six months of abstinence have improved survival with liver transplantation compared with predicted survival based on the Beclere model (Fig. 84-15).119 Similar, although less impressive, results have been shown using other prognostic models.171 The optimal length of pretransplant abstinence remains controversial.40 Some experts have argued that patients with severe alcoholic hepatitis should be abstinent for one year before being considered for transplantation, whereas others have argued that patients should be considered for transplantation if they continue to have CTP scores of 11 or greater after only three months of abstinence.124,139
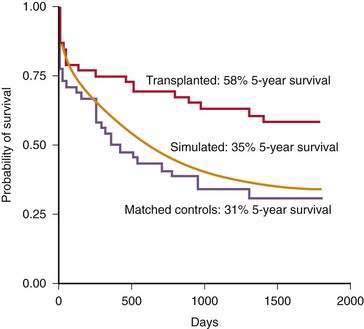
Evidence of a survival benefit following transplantation is less clear for patients with milder alcoholic liver disease, unless they have hepatocellular carcinoma. Patients with CTP scores of 5 to 7 do not benefit from liver transplantation.119 The survival benefit from transplantation for patients with a CTP score of 8 to 10 after 6 months of abstinence is minimal compared with predicted survival using the Beclere and MELD models.119,171 Furthermore, a trial in which patients with a CTP score of 8 to 10 were randomized to receive immediate transplantation or to be observed expectantly showed a lower two-year survival rate among the patients randomized to undergo immediate transplantation (73% versus 80%), primarily because of a high risk of postoperative malignancy.172
OPTIMAL MANAGEMENT
Alcoholic liver disease accounts for 50,000 deaths annually in the United States and Europe. The optimal management of patients with alcoholic liver disease begins with a dramatic reduction in or elimination of alcohol intake, which often can be accomplished successfully using “brief interventions” by a nurse, primary care physician, or gastroenterologist. Abstinence can have a profound impact on survival even in patients with decompensated cirrhosis. The next important step is to eliminate other factors, such as cigarette smoking and obesity, which can enhance disease progression. Treatment of concomitant HCV infection may be an important aspect of management in some patients (see Chapter 79).
For patients with alcoholic cirrhosis, no drugs available in the United States have documented efficacy. Many patients already are taking agents such as milk thistle or SAM that may be of benefit and appear to be safe. These agents are not covered by insurance, however, and access to such agents often depends on the socioeconomic status of the patient. Because of the risk of decompensation with superimposed infections, all patients with alcoholic cirrhosis should receive vaccinations for hepatitis A and B and annual vaccinations for influenza. In addition, they should undergo regular screening for hepatocellular carcinoma (see Chapter 94). Screening is particularly important in older patients with cirrhosis who have been abstinent for sustained periods. Finally, liver transplantation has been shown to be effective in carefully selected patients who have discontinued drinking (see Chapter 95).
Akriviadis E, Botla R, Briggs W, et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: A double-blind, placebo-controlled trial. Gastroenterology. 2000;119:1637-48. (Ref 85.)
Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 2003;124:778-90. (Ref 13.)
Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial Etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135:1953-60. (Ref 146.)
Cabre E, Rodriguez-Iglesias P, Caballeria J, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: A multicenter randomized trial. Hepatology. 2000;32:36-42. (Ref 136.)
Ji C. Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J Gastroenterol Hepatol. 2008;23(Suppl 1):S16-24. (Ref 70.)
Kunos G, Osei-Hyiaman D, Batkai S, Gao B. Cannabanoids hurt, heal in cirrhosis. Nat Med. 2006;12:608-10. (Ref 71.)
Mathurin P, Mendenhall CL, Carithers RL, et al. Glucocorticoids improve short-term survival in patients with severe alcoholic hepatitis (AH): Individual data analysis of the last three randomized placebo controlled double blind trials of glucocorticoids in severe AH. J Hepatol. 2002;36:480-7. (Ref 116.)
Niemelä O. Biomarkers in alcoholism. Clin Chimica Acta. 2007;377:39-49. (Ref 80.)
Pfitzmann R, Schwenzer J, Rayes N, et al. Long-term survival and predictors of relapse after orthotopic liver transplantation for alcoholic liver disease. Liver Transplant. 2007;13:197-205. (Ref 168.)
Plank LD, Gane EJ, Peng S, et al. Nocturnal nutritional supplementation improves total body protein status of patients with liver cirrhosis: A randomized 12-month trial. Hepatology. 2008;48:557-66. (Ref 138.)
Purohit V, Abdelmalek MF, Barve S, et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am J Clin Nutr. 2007;86:14-24. (Ref 35.)
Shukla SD, Velazquez J, French SW, et al. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32:1525-34. (Ref 69.)
Stewart S, Prince M, Bassendine M, et al. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J Hepatol. 2007;47:277-83. (Ref 157.)
Veldt BJ, Laine F, Guillygomarc’h A, et al. Indication of liver transplantation in severe alcoholic liver cirrhosis: Quantitative evaluation and optimal timing. J Hepatol. 2002;36:93-8. (Ref 124.)
Yip WW, Burt AD. Alcoholic liver disease. Semin Diagn Pathol. 2006;23:149-60. (Ref 8.)
1. Kim WR, Brown RSJr, Terrault NA, El Serag H. Burden of liver disease in the United States: Summary of a workshop. Hepatology. 2002;36:227-42.
2. Hassan MM, Hwang LY, Hatten CJ, et al. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206-13.
3. Bosetti C, Levi F, Lucchini F, et al. Worldwide mortality from cirrhosis: An update to 2002. J Hepatol. 2007;46:827-39.
4. Fuchs CS, Stampfer MJ, Colditz GA, et al. Alcohol consumption and mortality among women. N Engl J Med. 1995;332:1245-50.
5. Becker U, Deis A, Sorensen TI, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: A prospective population study. Hepatology. 1996;23:1025-9.
6. Thun MJ, Peto R, Lopez AD, et al. Alcohol consumption and mortality among middle-aged and elderly U.S. adults. N Engl J Med. 1997;337:1705-14.
7. Lefkowitch JH. Morphology of alcoholic liver disease. Clin Liver Dis. 2005;9:37-53.
8. Yip WW, Burt AD. Alcoholic liver disease. Semin Diagn Pathol. 2006;23:149-60.
9. Mathurin P, Beuzin F, Louvet A, et al. Fibrosis progression occurs in a subgroup of heavy drinkers with typical histological features. Alimen Pharm Ther. 2007;25:1047-54.
10. Arteel G, Marsano L, Mendez C, et al. Advances in alcoholic liver disease. Best Pract Res Clin Gastroenterol. 2003;17:625-47.
11. Thiele GM, Worrall S, Tuma DJ, et al. The chemistry and biological effects of malondialdehyde-acetaldehyde adducts. Alcohol Clin Exp Res. 2001;25(5 Suppl ISBRA):218S-224S.
12. Lluis JM, Colell A, Garcia-Ruiz C, et al. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124:708-24.
13. Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 2003;124:778-90.
14. Reinke LA, Lai EK, DuBose CM, McCay PB. Reactive free radical generation in vivo in heart and liver of ethanol-fed rats: Correlation with radical formation in vitro. Proc Natl Acad Sci USA. 1987;84:9223-7.
15. Meagher EA, Barry OP, Burke A, et al. Alcohol-induced generation of lipid peroxidation products in humans. J Clin Invest. 1999;104:805-13.
16. Wu D, Cederbaum AI. Ethanol cytotoxicity to a transfected HepG2 cell line expressing human cytochrome P4502E1. J Biol Chem. 1996;271:23914-19.
17. Morgan K, French SW, Morgan TR. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology. 2002;36:122-34.
18. Kono H, Rusyn I, Uesugi T, et al. Diphenyleneiodonium sulfate, an NADPH oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1005-12.
19. Kono H, Rusyn I, Yin M, et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867-72.
20. Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology. 2002;122:2049-63.
21. Adachi M, Ishii H. Role of mitochondria in alcoholic liver injury. Free Radic Biol Med. 2002;32:487-91.
22. Witschi A, Mossi S, Meyer B, et al. Mitochondrial function reflected by the decarboxylation of [13C]ketoisocaproate is impaired in alcoholics. Alcohol Clin Exp Res. 1994;18:951-5.
23. Coll O, Colell A, Garcia-Ruiz C, et al. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology. 2003;38:692-702.
24. French SW. The role of hypoxia in the pathogenesis of alcoholic liver disease. Hepatol Res. 2004;29:69-74.
25. Yang Y, Yu X. Regulation of apoptosis: The ubiquitous way. FASEB J. 2003;17:790-9.
26. Donohue TMJr. The ubiquitin-proteasome system and its role in ethanol-induced disorders. Addict Biol. 2002;7:15-28.
27. Baraona E, Leo MA, Borowsky SA, Lieber CS. Pathogenesis of alcohol-induced accumulation of protein in the liver. J Clin Invest. 1977;60:546-54.
28. Fataccioli V, Andraud E, Gentil M, et al. Effects of chronic ethanol administration on rat liver proteasome activities: Relationship with oxidative stress. Hepatology. 1999;29:14-20.
29. Osna NA, Donohue TMJr. Implication of altered proteasome function in alcoholic liver injury. World J Gastroenterol. 2007;13:4931-7.
30. Takagi M, Yamauchi M, Toda G, et al. Serum ubiquitin levels in patients with alcoholic liver disease. Alcohol Clin Exp Res. 1999;23(4 Suppl):76S-80S.
31. Bardag-Gorce F, van Leeuwen FW, Nguyen V, et al. The role of the ubiquitin-proteasome pathway in the formation of Mallory bodies. Exp Mol Pathol. 2002;73:75-83.
32. Joshi-Barve S, Barve SS, Butt W, et al. Inhibition of proteasome function leads to NF-kappaB-independent IL-8 expression in human hepatocytes. Hepatology. 2003;38:1178-87.
33. McClain CJ, Hill DB, Song Z, et al. S-adenosylmethionine, cytokines, and alcoholic liver disease. Alcohol. 2002;27:185-92.
34. Mato JM, Alvarez L, Ortiz P, Pajares MA. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol Ther. 1997;73:265-80.
35. Purohit V, Abdelmalek MF, Barve S, et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am J Clin Nutr. 2007;86:14-24.
36. Song Z, Zhou Z, Uriarte S, et al. S-adenosylhomocysteine sensitizes to TNF hepatotoxicity in mice and liver cells: A possible etiological factor in alcoholic liver disease. Hepatology. 2004;40:989-97.
37. Horowitz JH, Rypins EB, Henderson JM, et al. Evidence for impairment of transsulfuration pathway in cirrhosis. Gastroenterology. 1981;81:668-75.
38. Cabrero C, Duce AM, Ortiz P, et al. Specific loss of the high-molecular-weight form of S-adenosyl-L-methionine synthetase in human liver cirrhosis. Hepatology. 1988;8:1530-4.
39. Song Z, McClain CJ, Chen T. S-adenosylmethionine protects against acetaminophen-induced hepatotoxicity in mice. Pharmacology. 2004;71:199-208.
40. Chawla RK, Lewis FW, Kutner MH, et al. Plasma cysteine, cystine, and glutathione in cirrhosis. Gastroenterology. 1984;87:770-6.
41. Lieber CS, Casini A, DeCarli LM, et al. S-adenosyl-l-methionine attenuates alcohol-induced liver injury in the baboon. Hepatology. 1990;11:165-72.
42. Sanchez-Gongora E, Ruiz F, Mingorance J, et al. Interaction of liver methionine adenosyltransferase with hydroxyl radical. FASEB J. 1997;11:1013-19.
43. Colell A, Garcia-Ruiz C, Miranda M, et al. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;115:1541-51.
44. Chawla RK, Watson WH, Eastin CE, et al. S-adenosylmethionine deficiency and TNF-alpha in lipopolysaccharide-induced hepatic injury. Am J Physiol. 1998;275(1 Pt 1):G125-9.
45. Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J Gastroenterol. 2004;10:1699-708.
46. Halsted CH, Villanueva JA, Devlin AM, Chandler CJ. Metabolic interactions of alcohol and folate. J Nutr. 2002;132(8 Suppl):2367S-2372S.
47. McClain CJ, Song Z, Barve S, et al. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G497-502.
48. Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10:709-20.
49. Canbay A, Feldstein AE, Higuchi H, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188-98.
50. Deaciuc IV, D’Souza NB, de Villiers WJ, et al. Inhibition of caspases in vivo protects the rat liver against alcohol-induced sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res. 2001;25:935-43.
51. McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9:349-51.
52. Zhang Z, Bagby GJ, Stoltz D, et al. Prolonged ethanol treatment enhances lipopolysaccharide/phorbol myristate acetate-induced tumor necrosis factor-alpha production in human monocytic cells. Alcohol Clin Exp Res. 2001;25:444-9.
53. Honchel R, Ray MB, Marsano L, et al. Tumor necrosis factor in alcohol enhanced endotoxin liver injury. Alcohol Clin Exp Res. 1992;16:665-9.
54. Kamimura S, Tsukamoto H. Cytokine gene expression by Kupffer cells in experimental alcoholic liver disease. Hepatology. 1995;22(4 Pt 1):1304-9.
55. Nanji AA, Zhao S, Sadrzadeh SM, Waxman DJ. Use of reverse transcription-polymerase chain reaction to evaluate in vivo cytokine gene expression in rats fed ethanol for long periods. Hepatology. 1994;19:1483-7.
56. Le Moine O, Marchant A, De Groote D, et al. Role of defective monocyte interleukin-10 release in tumor necrosis factor-alpha overproduction in alcoholic cirrhosis. Hepatology. 1995;22:1436-9.
57. Hill DB, D’Souza NB, Lee EY, et al. A role for interleukin-10 in alcohol-induced liver sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res. 2002;26:74-82.
58. Iimuro Y, Gallucci RM, Luster MI, et al. Antibodies to tumor necrosis factor alpha attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology. 1997;26:1530-7.
59. Yin M, Wheeler MD, Kono H, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942-52.
60. Pastorino JG, Hoek JB. Ethanol potentiates tumor necrosis factor-alpha cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology. 2000;31:1141-52.
61. Chedid A, Chadalawada KR, Morgan TR, et al. Phospholipid antibodies in alcoholic liver disease. Hepatology. 1994;20:1465-71.
62. Clot P, Bellomo G, Tabone M, et al. Detection of antibodies against proteins modified by hydroxyethyl free radicals in patients with alcoholic cirrhosis. Gastroenterology. 1995;108:201-7.
63. Vidali M, Stewart SF, Rolla R, et al. Genetic and epigenetic factors in autoimmune reactions toward cytochrome P4502E1 in alcoholic liver disease. Hepatology. 2003;37:410-19.
64. Kwo PY, Ramchandani VA, O’Connor S, et al. Gender differences in alcohol metabolism: Relationship to liver volume and effect of adjusting for body mass. Gastroenterology. 1998;115:1552-7.
65. Nanji AA, Jokelainen K, Fotouhinia M, et al. Increased severity of alcoholic liver injury in female rats: Role of oxidative stress, endotoxin, and chemokines. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1348-56.
66. Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63:49-63.
67. Grove J, Daly AK, Bassendine MF, Day CP. Association of a tumor necrosis factor promoter polymorphism with susceptibility to alcoholic steatohepatitis. Hepatology. 1997;26:143.
68. Grove J, Daly AK, Bassendine MF, et al. Interleukin 10 promoter region polymorphisms and susceptibility to advanced alcoholic liver disease. Gut. 2000;46:540-5.
69. Shukla SD, Velazquez J, French SW, et al. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32:1525-34.
70. Ji C. Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J Gastroenterol Hepatol. 2008;23(Suppl 1):S16-24.
71. Kunos G, Osei-Hyiaman D, Batkai S, Gao B. Cannabanoids hurt, heal in cirrhosis. Nat Med. 2006;12:608-10.
72. Friedman SL. Liver fibrosis-from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38-53.
73. Zamara E, Novo E, Marra F, et al. 4-Hydroxynonenal as a selective pro-fibrogenic stimulus for activated human hepatic stellate cells. J Hepatol. 2004;40:60-8.
74. Reeves HL, Friedman SL. Activation of hepatic stellate cells—a key issue in liver fibrosis. Front Biosci. 2002;7:d808-26.
75. Saitz R, Mulvey KP, Plough A, Samet JH. Physician unawareness of serious substance abuse. Am J Drug Alcohol Abuse. 1997;23:343-54.
76. Khan N, Davis P, Wilkinson TJ, et al. Drinking patterns among older people in the community: Hidden from medical attention? NZ Med J. 2002;115:72-5.
77. Bradley KA, DeBenedetti AG, Volk RJ, et al. AUDIT-C as a brief screen for alcohol misuse in primary care. Alcohol Clin Exp Res. 2007;31:1208-17.
78. Seale JP, Boltri JM, Shellenberger S. Primary care validation of a single screening question for drinkers. J Stud Alcohol. 2006;67:778-84.
79. Savola O, Niemelä O, Hillbom M. Blood alcohol is the best indicator of hazardous alcohol drinking in young adults and working-age patients with trauma. Alcohol Alcohol. 2004;39:340-5.
80. Niemelä O. Biomarkers in alcoholism. Clin Chimica Acta. 2007;377:39-49.
81. Hietala J, Koivisto H, Anttila P, et al. Comparison of the combined marker GGT-CDT and the conventional laboratory markers of alcoholic abuse in heavy drinkers, moderate drinkers and abstainers. Alcohol Alcohol. 2006;41:528-33.
82. Helman RA, Temko MH, Nye SW, Fallon HJ. Alcoholic hepatitis: Natural history and evaluation of prednisolone therapy. Ann Intern Med. 1971;74:311-21.
83. Mendenhall CL. Alcoholic hepatitis. Clin Gastroenterol. 1981;10:417-41.
84. Depew W, Boyer T, Omata M, et al. Double-blind controlled trial of prednisolone therapy in patients with severe acute alcoholic hepatitis and spontaneous encephalopathy. Gastroenterology. 1980;78:524-9.
85. Akriviadis E, Botla R, Briggs W, et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: A double-blind, placebo-controlled trial. Gastroenterology. 2000;119:1637-48.
86. Elphick DA, Dube AK, McFarlane E, et al. Spectrum of liver histology is presumed decompensated alcoholic liver disease. Am J Gastroenterol. 2007;102:780-8.
87. Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917-23.
88. Falck-Ytter U, Younossi ZM, Marchesini G, McCullough AJ. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis. 2001;21:17-26.
89. Dunn W, Angulo P, Sanderson S, et al. Utility of a new model to diagnose an alcohol basis for steatohepatitis. Gastroenterology. 2006;131:1057-63.
90. Cotler SJ, Bronner MP, Press RD, et al. End-stage liver disease without hemochromatosis associated with elevated hepatic iron index. J Hepatol. 1998;29:257-62.
91. Gleeson D, Evans S, Bradley M, et al. HFE genotypes in decompensated alcoholic liver disease: Phenotypic expression and comparison with heavy drinking and normal controls. Am J Gastroenterol. 2006;101:304-10.
92. Fletcher LM, Powell LW. Hemochromatosis and alcoholic liver disease. Alcohol. 2003;30:131-6.
93. Simon JB, Manley PN, Brien JF, Armstrong PW. Amiodarone hepatotoxicity simulating alcoholic liver disease. N Engl J Med. 1984;311:16772.
94. Janssen HL, Tan AC, Tilanus HW, et al. Pseudo-Budd-Chiari syndrome: Decompensated alcoholic liver disease mimicking hepatic venous outflow obstruction. Hepatogastroenterology. 2002;49:810-12.
95. Seeff LB, Cuccherine BA, Zimmerman HJ, et al. Acetaminophen hepatotoxicity in alcoholics: A therapeutic misadventure. Ann Intern Med. 1986;105:399-404.
96. Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology. 2005;42:1364-72.
97. Akriviadis EA, Redeker AG. Fulminant hepatitis A in intravenous drug users with chronic liver disease. Ann Intern Med. 1989;110:838-9.
98. Feller A, Uchida T, Rakela J. Acute viral hepatitis superimposed on alcoholic liver cirrhosis: Clinical and histopathologic features. Liver. 1985;5:239-46.
99. Duchini A, Hendry RM, Redfield DC, Pockros PJ. Influenza infection in patients before and after liver transplantation. Liver Transpl. 2000;6:531-42.
100. Koff RS, Dienstag JL. Extrahepatic manifestations of hepatitis C and the association with alcoholic liver disease. Semin Liver Dis. 1995;15:101-9.
101. Fong T-L, Kanel GC, Conrad A, et al. Clinical significance of concomitant hepatitis C infection in patients with alcoholic liver disease. Hepatology. 1994;19:554-7.
102. Corrao G, Arico S. Independent and combined action of hepatitis C virus infection and alcohol consumption on the risk of symptomatic liver cirrhosis. Hepatology. 1998;27:914-19.
103. Hassan MM, Wuang L-Y, Hatten CJ, et al. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206-13.
104. Yamauchi M, Nakahara M, Maezawa Y, et al. Prevalence of hepatocellular carcinoma in patients with alcoholic cirrhosis and prior exposure to hepatitis C. Am J Gastroenterol. 1993;88:39-43.
105. Neuberger J, Schulz KH, Day C, et al. Transplantation for alcoholic liver disease. J Hepatol. 2002;36:130-7.
106. Naveau S, Giraud V, Borotto E, et al. Excess weight risk factor for alcoholic liver disease. Hepatology. 1997;25:108-11.
107. Raynard B, Balian A, Fallik D, et al. Risk factors of fibrosis in alcohol-induced liver disease. Hepatology. 2002;35:635-8.
108. Klatsky AL, Armstrong MA. Alcohol, smoking, coffee, and cirrhosis. Am J Epidemiol. 1992;136:1248-57.
109. Corrao G, Lepore AR, Torchio P, et al. The effect of drinking coffee and smoking cigarettes on the risk of cirrhosis associated with alcohol consumption. A case-control study. Provincial Group for the Study of Chronic Liver Disease. Eur J Epidemiol. 1994;10:657-64.
110. Pessione F, Ramond MJ, Njapoum C, et al. Cigarette smoking and hepatic lesions in patients with chronic hepatitis C. Hepatology. 2001;34:121-5.
111. Orrego H, Blake JE, Blendis LM, Medline A. Prognosis of alcoholic cirrhosis in the presence and absence of alcoholic hepatitis. Gastroenterology. 1987;92:208-14.
112. Poynard T, Aubert A, Lazizi Y, et al. Independent risk factors for hepatocellular carcinoma in French drinkers. Hepatology. 1991;13:896-901.
113. Maddrey WC, Boitnott JK, Bedine MS, et al. Glucocorticoid therapy of alcoholic hepatitis. Gastroenterology. 1978;75:193-9.
114. Carithers RLJr, Herlong HF, Diehl AM, et al. Methylprednisolone therapy in patients with severe alcoholic hepatitis: A randomized multicenter trial. Ann Intern Med. 1989;110:685-90.
115. Ramond MJ, Poynard T, Rueff B, et al. A randomized trial of prednisolone in patients with severe alcoholic hepatitis. N Engl J Med. 1992;326:507-12.
116. Mathurin P, Mendenhall CL, Carithers RL, et al. Glucocorticoids improve short-term survival in patients with severe alcoholic hepatitis (AH): Individual data analysis of the last three randomized placebo controlled double blind trials of glucocorticoids in severe AH. J Hepatol. 2002;36:480-7.
117. Srikureja W, Kyulo NL, Runyon BA, et al. MELD score is a better prognostic model that Child-Turcotte-Pugh score or discriminant function score in patients with alcoholic hepatitis. J Hepatol. 2005;42:700-6.
118. Forrest EH, Evans CDJ, Stewart S, et al. Analysis of factors predictive of mortality in alcoholic hepatitis and derivation and validation of the Glasgow alcoholic hepatitis score. Gut. 2005;54:1174-9.
119. Poynard T, Naveau S, Doffoel M, et al. Evaluation of efficacy of liver transplantation in alcoholic cirrhosis using matched and simulated controls: 5-year survival. Multi-centre group. J Hepatol. 1999;30:1130-7.
120. Ginès P, Quintero E, Arroyo V, et al. Compensated cirrhosis: Natural history and prognosis factors. Hepatology. 1987;7:122-8.
121. Andreu M, Sola R, Sitges SA, et al. Risk factors for spontaneous bacterial peritonitis in cirrhotic patients with ascites. Gastroenterology. 1993;104:1133-8.
122. Ginès A, Escorsell A, Ginès P, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology. 1993;105:229-36.
123. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33:464-70.
124. Veldt BJ, Laine F, Guillygomarc’h A, et al. Indication of liver transplantation in severe alcoholic liver cirrhosis: Quantitative evaluation and optimal timing. J Hepatol. 2002;36:93-8.
125. Powell WJ, Klatskin G. Duration and survival in patients with Laennec’s cirrhosis. Am J Med. 1968;44:406-20.
126. Wilk AI, Jensen NM, Havighurst MS. Meta-analysis of randomized control trials addressing brief interventions in heavy alcohol drinkers. J Gen Intern Med. 1997;12:274-83.
127. Anton RF, O’Malley SS, Ciraulo DA, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence. JAMA. 2006;295:2003-17.
128. Lieber CS, Weiss DG, Groszmann R, et al. I. Veterans Affairs Cooperative Study of polyenylphosphatidylcholine in alcoholic liver disease: Effects on drinking behavior by nurse/physician teams. Alcohol Clin Exp Res. 2003;27:1757-64.
129. Srisurapanont M, Jarusuraisin N. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev 2005; CD001867.
130. Addolorato G, Leggio L, Ferrulli A, et al. Effectiveness and safety of baclofen for maintenance of alcohol abstinence in alcohol-dependent patients with liver cirrhosis: Randomized, double-blind controlled study. Lancet. 2007;370:1915-22.
131. Stickel G, Hoehn B, Schuppan D, Seitz HK. Review article: Nutritional therapy in alcoholic liver disease. Aliment Pharmacol Ther. 2003;18:357-73.
132. Mendenhall C, Roselle GA, Gartside P, Moritz T. Relationship of protein calorie malnutrition to alcoholic liver disease: A reexamination of data from two Veterans Administration Cooperative Studies. Alcohol Clin Exp Res. 1995;19:635-41.
133. Muller MJ, Lautz HU, Plogmann B, et al. Energy expenditure and substrate oxidation in patients with cirrhosis: The impact of cause, clinical staging and nutritional state. Hepatology. 1992;15:782-94.
134. Halsted CH. Nutrition and alcoholic liver disease. Semin Liver Dis. 2004;24:289-304.
135. Kearns PJ, Young H, Garcia G. Accelerated improvement of alcoholic liver disease with enteral nutrition. Gastroenterology. 1992;102:200-5.
136. Cabre E, Rodriguez-Iglesias P, Caballeria J, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: A multicenter randomized trial. Hepatology. 2000;32:36-42.
137. Hirsch S, Bunout D, de la Maza P, et al. Controlled trial on nutrition supplementation in outpatients with symptomatic alcoholic cirrhosis. JPEN J Parenter Enteral Nutr. 1993;17:119-24.
138. Plank LD, Gane EJ, Peng S, et al. Nocturnal nutritional supplementation improves total body protein status of patients with liver cirrhosis: A randomized 12-month trial. Hepatology. 2008;48:557-66.
139. Mathurin P, Duchatelle V, Ramond MJ, et al. Survival and prognostic factors in patients with severe alcoholic hepatitis treated with prednisone. Gastroenterology. 1996;110:1847-53.
140. Louvet A, Naveau S, Abdelnour M, et al. The Lille model: A new tool for therapeutic strategy in patient with severe alcoholic hepatitis treated with steroids. Hepatology. 2007;45:1348-54.
141. Strieter RM, Remick DG, Ward PA, et al. Cellular and molecular regulation of tumor necrosis factor-alpha production by pentoxifylline. Biochem Biophys Res Commun. 1988;155:1230-6.
142. Louvet A, Diaz E, Dharancy S, et al. Early switch to pentoxyfylline in patients with severe alcoholic hepatitis is inefficient in non-responders to corticosteroids. J Hepatol. 2008;48:465-70.
143. Akerman PA, Cote PM, Yang SQ, et al. Long-term ethanol consumption alters the hepatic response to the regenerative effects of tumor necrosis factor-alpha. Hepatology. 1993;17:1066-73.
144. Tilg H, Jalan R, Kaser A, et al. Anti-tumor necrosis factor-alpha monoclonal antibody therapy in severe alcoholic hepatitis. J Hepatol. 2003;38:419-25.
145. Naveau S, Chollet-Martin S, Dharancy S, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology. 2004;39:1390-7.
146. Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial Etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135:1953-60.
147. Lee TD, Sadda MR, Mendler MH, et al. Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2004;28:173-81.
148. Mato JM, Camara J, Fernandez DP, et al. S-adenosylmethionine in alcoholic liver cirrhosis: A randomized, placebo-controlled, double-blind, multicenter clinical trial. J Hepatol. 1999;30:1081-9.
149. Luper S. A review of plants used in the treatment of liver disease: Part 1. Altern Med Rev. 1998;3:410-21.
150. Rambaldi A, Jacobs BP, Iaquinto G, Gluud C. Milk thistle for alcoholic and/or hepatitis B or C virus liver diseases. Cochrane Database Syst Rev 2007; CD003620.
151. Ball KR, Kowdley KV. A review of silybum marianum (milk thistle) as a treatment for alcoholic liver disease. J Clin Gastroenterol. 2005;39:520-8.
152. McClain C, Hill DB, Marsano L. Nutrition and liver disease. In: Bowman BA, Russell RM, Russell R, editors. Present Knowledge in Nutrition. 8th ed. Washington, DC: International Life Sciences Institute; 2006:747-763.
153. Hill DB, Devalaraja R, Joshi-Barve S, et al. Antioxidants attenuate nuclear factor-kappa B activation and tumor necrosis factor-alpha production in alcoholic hepatitis patient monocytes and rat Kupffer cells, in vitro. Clin Biochem. 1999;32:563-70.
154. de la Maza MP, Petermann M, Bunout D, Hirsch S. Effects of long-term vitamin E supplementation in alcoholic cirrhotics. J Am Coll Nutr. 1995;14:192-6.
155. Wenzel G, Kuklinski B, Ruhlman C, Ehrhardt D. Alcohol-induced toxic hepatitis—a “free radical” associated disease. Lower fatality by adjuvant antioxidant therapy. Z Gesamte Int Med. 1993;48:490-6.
156. Phillips M, Curtis H, Portmann B, et al. Antioxidants versus corticosteroids in the treatment of severe alcoholic hepatitis—a randomized clinical trial. J Hepatol. 2006;44:784-90.
157. Stewart S, Prince M, Bassendine M, et al. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J Hepatol. 2007;47:277-83.
158. Kershenobich D, Vargas F, Garcia-Tsao G, et al. Colchicine in the treatment of cirrhosis of the liver. N Engl J Med. 1988;318:1709-13.
159. Morgan TR, Weiss DG, Nemchausky N, et al. Colchicine treatment of alcoholic cirrhosis: A randomized, placebo-controlled clinical trial of patient survival. Gastroenterology. 2005;128:882-90.
160. Cortez-Pinto H, Alexandrino P, Camilo ME, et al. Lack of effect of colchicine in alcoholic cirrhosis: Final results of a double blind randomized trial. Eur J Gastroenterol Hepatol. 2002;14:377-81.
161. Rambaldi A, Gluud C. Propylthiouracil for alcoholic liver disease. Cochrane Database Syst Rev 2005; CD002800.
162. Rambaldi A, Iaquinto G, Gluud C. Anabolic-androgenic steroids for alcoholic liver disease: A Cochrane review. Am J Gastroenterol. 2002;97:1674-81.
163. Pelletier G, Roulot D, Davion T, et al. A randomized controlled trial of ursodeoxycholic acid in patients with alcohol-induced cirrhosis and jaundice. Hepatology. 2003;37:887-92.
164. Cao Q, Mak KM, Lieber CS. Dilinoleoylphosphatidylcholine decreases acetaldehyde-induced TNF-alpha generation in Kupffer cells of ethanol-fed rats. Biochem Biophys Res Commun. 2002;299:459-64.
165. Lieber CS, Weiss DG, Groszmann R, et al. II. Veterans Affairs Cooperative Study of polyenylphosphatidylcholine in alcoholic liver disease. Alcohol Clin Exp Res. 2003;27:1765-72.
166. Roberts MS, Angus DC, Bryce CL, et al. Survival after liver transplantation in the United States: A disease-specific analysis of the UNOS database. Liver Transpl. 2004;10:886-97.
167. Buis CI, Wiesner RH, Krom RA, et al. Acute confusional state following liver transplantation for alcoholic liver disease. Neurology. 2002;59:601-5.
168. Pfitzmann R, Schwenzer J, Rayes N, et al. Long-term survival and predictors of relapse after orthotopic liver transplantation for alcoholic liver disease. Liver Transplant. 2007;13:197-205.
169. Jain A, DiMartini A, Kashyap R, et al. Long-term follow-up after liver transplantation for alcoholic liver disease under tacrolimus. Transplantation. 2000;70:1335-42.
170. Dimartini A, Javed L, Russell S, et al. Tobacco use following liver transplantation for alcoholic liver disease: An underestimated problem. Liver Transplant. 2005;11:679-83.
171. Young TA, Neuberger J, Longworth L, et al. Survival gain after liver transplantation for patients with alcoholic liver disease: A comparison across models and centers. Transplantation. 2003;76:1479-86.
172. Miguet JP, Vanlemmens C, Milan C, et al. Impact of liver transplantation on survival in Pugh B alcoholic cirrhotic patients: A multicenter randomized trial. J Hepatol. 2003;38(Suppl 1):90A.