[level-membership-for-basic-science-category]58
Zinc and copper
Zinc
Zinc physiology
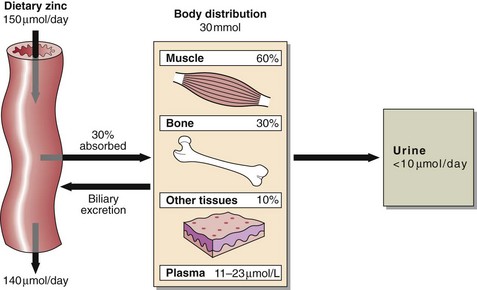
Zinc deficiency is a major health problem in the poorer nations of the world. The daily requirement, which varies with age, sex and during pregnancy is about 150 µmol (10 mg) per day (Fig 58.1). Zinc is present in all protein-rich foods and approximately 30% is absorbed. Phytates are indigestible in man, bind calcium, iron and zinc, and reduce their absorption. In the liver, zinc is incorporated into metalloenzymes, while in blood the majority of the zinc is contained in the erythrocytes. In plasma, 90% of zinc is bound to albumin and 10% to α2-macroglobulin. Zinc reserves in the body are small and are located mainly in muscle and bone. Zinc is excreted in urine, in bile, in pancreatic fluid and in milk in lactating mothers.
Zinc deficiency
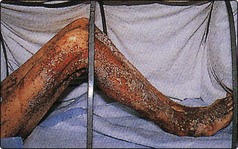
In children, the rate of growth during rehabilitation from famine has been clearly related to the dietary supply of bioavailable zinc. Zinc deficiency is known to occur in patients on intraveneous nutrition and causes a characteristic skin rash (Fig 58.2) and hair loss. Wound breakdown and delayed healing are other complications. Acrodermatitis enteropathica, a rare inherited disorder of zinc metabolism, manifests itself in infancy as skin rash. Untreated, the prognosis is poor, but oral zinc therapy leads to complete remission. Cadmium displaces zinc from metalloproteins, and zinc deficiency can be a consequence of chronic cadmium poisoning.
Copper
Copper physiology
About 50% of the average daily dietary copper of around 25 µmol (1.5 mg) is absorbed from the stomach and the small intestine (Fig 58.3). There is evidence that not all modern diets contain sufficient copper, especially when large amounts of refined carbohydrate are consumed. Copper absorption is facilitated by cation transport enzymes in the mucosal cells. A high zinc intake will block the absorption of copper by inducing metallothionine in the mucosal cells. Copper has high affinity for metallothionine and is lost when the mucosal cells are shed in the faeces. Absorbed copper is transported to the liver bound to albumin where it is incorporated into caeruloplasmin, that contains 6 copper atoms per molecule, and exported into the circulation. (Fig 58.4).
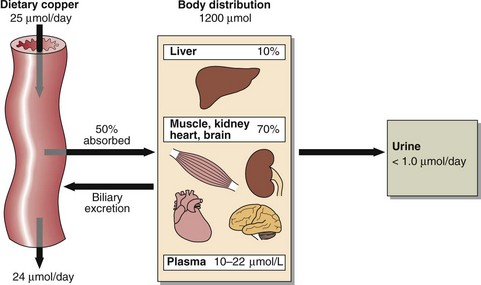
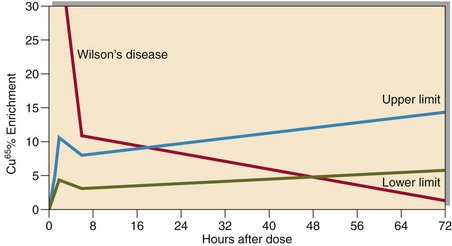
Fig 58.4 Cu65 uptake test. The most abundant stable isotope of copper is Cu63. A standard dose of Cu65, another naturally occurring stable isotope, is given orally. The initial increase in enrichment is followed by a decrease at 6 hours representing liver uptake. The later increase represents the export of caeruloplasmin from the liver. In Wilson’s disease the initial increase is exaggerated – typically 5 times normal with subsequent diminished export from the liver. In Cu malabsorption, due to disease or Zn induced mucosal block, the initial increase is blunted but the subsequent export from the liver is normal.
Laboratory assessment
 Serum copper. 90% is bound to caeruloplasmin. Total copper concentration may vary either due to changes in copper itself or to changes in the concentration of caeruloplasmin.
Serum copper. 90% is bound to caeruloplasmin. Total copper concentration may vary either due to changes in copper itself or to changes in the concentration of caeruloplasmin.

 Urinary copper. Normal excretion is <1.0 µmol/24 hours.
Urinary copper. Normal excretion is <1.0 µmol/24 hours.
 The oral Cu65 absorption test (Fig 58.4) is a powerful tool in the investigation of patients found to have a low plasma copper.
The oral Cu65 absorption test (Fig 58.4) is a powerful tool in the investigation of patients found to have a low plasma copper.
Inborn errors of copper metabolism
Wilson’s disease
Wilson’s disease is caused by a mutation in the gene ATP7B that codes for a cation transporting enzyme involved in copper transport. Urinary free copper excretion is high and total serum concentrations low (Table 58.1). Confirmation is by measurement of copper in a liver biopsy, which is usually greater than 250 µg/g dry weight in patients with the disease. A non invasive 65Cu-oral uptake test is a reliable test for the diagnosis of Wilson’s disease and available in some specialist laboratories.
Table 58.1
Biochemistry determinations in patients with Wilson’s disease
| Investigation | Normal adult | Wilson’s disease |
| Serum copper µmol/L | 10–22 | <10 |
| Caeruloplasmin g/L | 0.15–0.6 | <0.15 |
| Urinary copper µmol/24h | <1 | 5–15 |
| Liver copper µg/g dry weight | 20–50 | >250 |
 Clinical note
Clinical noteZinc and copper
 Adequate zinc is needed for growth in children.
Adequate zinc is needed for growth in children.
 Symptomatic zinc deficiency in the adult causes dermatitis, hair loss and poor wound healing.
Symptomatic zinc deficiency in the adult causes dermatitis, hair loss and poor wound healing.
 Serum zinc concentrations persistently below 5 µmol/L warn of impending clinical deficiency.
Serum zinc concentrations persistently below 5 µmol/L warn of impending clinical deficiency.

 The major inborn error of copper metabolism is Wilson’s disease.
The major inborn error of copper metabolism is Wilson’s disease.
 Wilson’s disease is treatable and requires prompt diagnosis.
Wilson’s disease is treatable and requires prompt diagnosis.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]58
Zinc and copper
Zinc
Zinc physiology
Zinc deficiency is a major health problem in the poorer nations of the world. The daily requirement, which varies with age, sex and during pregnancy is about 150 µmol (10 mg) per day (Fig 58.1). Zinc is present in all protein-rich foods and approximately 30% is absorbed. Phytates are indigestible in man, bind calcium, iron and zinc, and reduce their absorption. In the liver, zinc is incorporated into metalloenzymes, while in blood the majority of the zinc is contained in the erythrocytes. In plasma, 90% of zinc is bound to albumin and 10% to α2-macroglobulin. Zinc reserves in the body are small and are located mainly in muscle and bone. Zinc is excreted in urine, in bile, in pancreatic fluid and in milk in lactating mothers.
Zinc deficiency
In children, the rate of growth during rehabilitation from famine has been clearly related to the dietary supply of bioavailable zinc. Zinc deficiency is known to occur in patients on intraveneous nutrition and causes a characteristic skin rash (Fig 58.2) and hair loss. Wound breakdown and delayed healing are other complications. Acrodermatitis enteropathica, a rare inherited disorder of zinc metabolism, manifests itself in infancy as skin rash. Untreated, the prognosis is poor, but oral zinc therapy leads to complete remission. Cadmium displaces zinc from metalloproteins, and zinc deficiency can be a consequence of chronic cadmium poisoning.
