[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 75 Wilson Disease
Copper, a component of several essential enzymes, is toxic to tissues when present in excess. Dietary intake of copper generally exceeds the trace amount required, and mechanisms to control influx and efflux from cells must maintain an appropriate balance. Two disorders of copper transport are known: Menkes disease, an X-linked defect in transport of copper from the intestine, and Wilson disease, an autosomal recessive disorder of copper overload. Wilson disease was first described, in 1912 by Kinnear Wilson, as a familial disease characterized by progressive, lethal neurologic dysfunction with chronic liver disease and a corneal abnormality, the Kayser-Fleischer (KF) ring.1 Wilson also observed that some younger siblings of typical patients died of severe liver disease without experiencing neurologic abnormalities. In this disease, inadequate hepatic copper excretion leads to copper accumulation in the liver, brain, kidney, and cornea. The incidence in most populations is on the order of 1 in 30,000.
THE COPPER PATHWAY
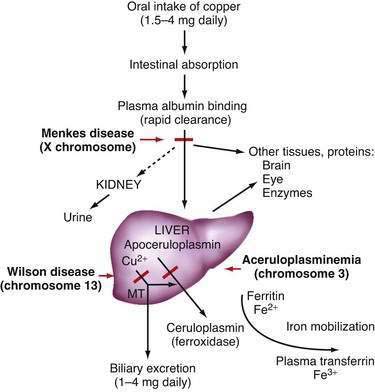
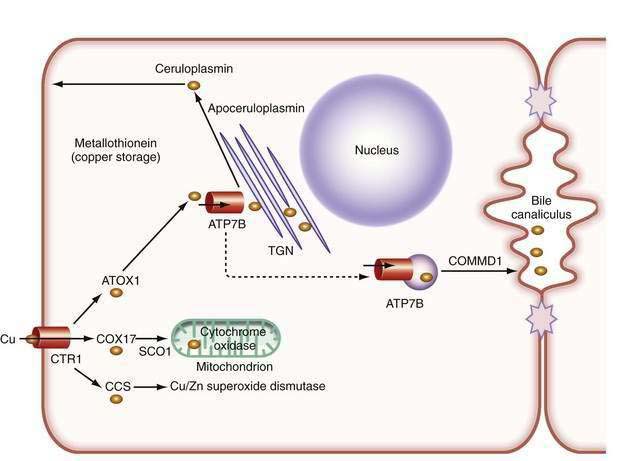
In the liver, copper is incorporated into the protein apoceruloplasmin to produce ceruloplasmin (also called holoceruloplasmin). More than 90% of the copper in plasma is an integral part of ceruloplasmin, an α2-glycoprotein that contains six molecules of copper and has a molecular weight of 132 kd. The normal serum concentration of ceruloplasmin in adults, as measured by immunochemical or enzymatic techniques, is 200 to 400 mg/L, rising from a very low level at birth to 300 to 500 mg/L in the first years of life. Ceruloplasmin is an acute-phase reactant that is elevated by inflammation (including inflammatory hepatic disease), pregnancy, and the use of exogenous estrogen. The majority of ingested copper is excreted via the bile; a small fraction is excreted in urine. When intestinal or liver cells are overloaded with copper, the metallothioneins, a class of low-molecular-weight cysteine-rich proteins, are induced and sequester copper in a nontoxic form. The normal pathways of copper transport in the body and in the hepatocyte are shown in Figures 75-1 and 75-2.
THE BASIC MOLECULAR DEFECT
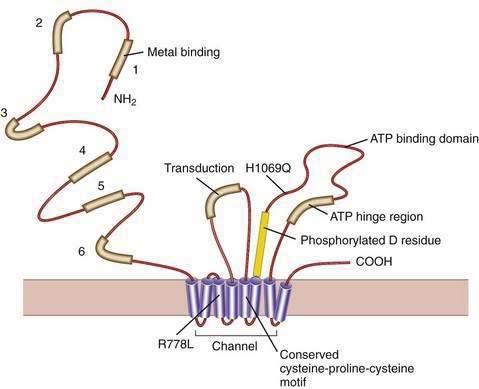
Our understanding of the basic defect in Wilson disease increased dramatically with the cloning of the genes, first for X-linked Menkes disease and then for Wilson disease. The gene for Menkes disease (ATP7A), which was cloned by using a chromosomal breakpoint in an affected female patient, was found to be related to bacterial copper resistance genes. Cloning of the Wilson disease gene (ATP7B) was accomplished by a combination of linkage analysis, physical mapping of the relevant region of chromosome 13q14, and recognition of its extensive homology with the Menkes disease gene.2,3 The coding region of the gene is 4.1 kilobases in length, with messenger RNA (mRNA) of about 8 kilobases. The product, ATP7B (or the Wilson ATPase), is a membrane P-type adenosine triphosphatase (ATPase) that consists of 1443 amino acid residues and has a molecular mass of 160 kd. The predicted structure,2 as confirmed by structural studies,4 has six copper binding domains, a phosphorylation domain, an ATP-binding region, and eight transmembrane domains. (Fig. 75-3). All functionally important regions of the gene are conserved between bacteria and yeast. Mutations in the ATP7B gene result in retention of copper in the liver as well as impaired incorporation of copper into ceruloplasmin. The Long-Evans cinnamon (LEC) rat and the toxic milk (tx) mouse have mutations in their homologous ATP7B genes and are thus suitable models for the study of Wilson disease mechanisms and therapy.5,6
Although the gene for Menkes disease is expressed in many tissues, including muscle, kidney, heart, and intestine, the gene for Wilson disease is expressed predominantly in the liver and kidney, with minor expression in brain, lungs, and placenta.2 Studies in cultured cells show localization of the Wilson ATPase in the trans-Golgi network, with trafficking from the trans-Golgi network to cytoplasmic vesicles in the presence of increased copper.7 When intracellular copper concentrations are elevated, the Wilson ATPase is found near the apical (bile canalicular) membrane in hepatocytes, consistent with its proposed function of facilitating excretion of copper via bile.8
Additional proteins are involved in the intracellular transport of copper. Molecular copper is not free in the cell but is transported to specific proteins by copper chaperones.9 The chaperone ATOX1 transports copper to the Wilson ATPase. Other chaperones deliver copper to superoxide dismutase in the cytoplasm and various copper-containing proteins in mitochondria.
The study of inherited hepatic copper toxicosis in Bedlington terriers has identified a new component of the copper transport system. Affected dogs show clinical variability that ranges from death or hepatic disease at two to three years of age to less severe chronic disease to a high level of hepatic copper. The proposed defective canine gene was identified by positional cloning through the use of markers to identify a region containing the COMMD1 (initially called the MURR1) gene. The gene has a deletion of one exon in some,10 but not all,11 affected dogs. Although the gene responsible for the excess copper remains to be identified, the disease in Bedlington terriers has highlighted other genes that may be involved in response to excess copper; COMMD1 interacts not only with the Wilson ATPase,12 but also with the X-linked inhibitor of apoptosis (XIAP), which can also bind copper.13 COMMD1 could be involved in response to, and facilitate elimination of, excess copper.
CLINICAL FEATURES
The clinical presentation of Wilson disease is extremely variable. The age at onset of symptoms is much broader than previously thought, ranging from 3 to 55 years. Wilson disease with hepatic involvement has been identified in one- to two-year-old patients and in patients older than age 60.14 Patients may present with chronic or fulminant liver disease, a progressive neurologic disorder without clinically prominent hepatic dysfunction, isolated acute hemolysis, or psychiatric illness. The clinical variability often makes confirmation of the diagnosis difficult. Further description of the clinical features and treatment has been published.14,15
HEPATIC PRESENTATION
Wilson disease may present in children and young adults with clinical liver disease indistinguishable from autoimmune hepatitis.16 As in autoimmune hepatitis, the onset may be acute. Fatigue, malaise, arthropathy, and rashes may occur; laboratory findings include elevated aminotransferase levels, a greatly increased serum immunoglobulin (IgG) concentration, and detectable nonspecific autoantibodies such as antinuclear antibodies and anti-smooth muscle (anti-actin) antibodies. Wilson disease must be specifically ruled out because the treatment of the two diseases is entirely different. With appropriate treatment, the long-term outlook for patients with Wilson disease that manifests as autoimmune hepatitis appears to be favorable, even if cirrhosis is present.
Wilson disease may present as fulminant hepatic failure, with severe coagulopathy and encephalopathy. Acute intravascular hemolysis is usually present, and renal failure may develop. Because the patient typically has not been suspected of having underlying liver disease, fulminant viral hepatitis is usually the working diagnosis. Unlike fulminant viral hepatitis, fulminant Wilson disease is typically characterized by disproportionately low aminotransferase levels (usually much less than 1500 U/L) at the onset of clinically apparent disease. The serum alkaline phosphatase level is in the normal range or even low for the patient’s age, and the bilirubin level is often disproportionately elevated as a result of hemolysis. In adults who present with Wilsonian fulminant liver failure, the combination of a ratio of the alkaline phosphatase level to the total bilirubin level of less than 4 and a ratio of the aspartate aminotransferase (AST) to alanine aminotransferase (ALT) level of greater than 2.2 can be extremely helpful for making the diagnosis, whereas the serum ceruloplasmin level is not (see later).17 Slit-lamp examination may reveal Kayser-Fleischer rings (see later). Urinary copper excretion is greatly elevated. These patients require urgent liver transplantation because they do not respond well to chelation treatment; albumin dialysis and related techniques may serve as temporary procedures until liver transplantation can be performed (see later).18 This presentation of Wilson disease is not rare, and affected patients account for a substantial proportion of persons transplanted for fulminant hepatic failure (see Chapters 93 and 95).
Recurrent bouts of hemolysis may predispose to the development of gallstones. Cirrhosis, if present, may be a further predisposing factor. Children with unexplained cholelithiasis, particularly with small bilirubinate stones, should be tested for Wilson disease. Compared with other chronic liver diseases, Wilson disease is rarely complicated by hepatocellular carcinoma; however, patients may have an increased propensity to abdominal malignancies,19 and hepatocellular carcinoma has been reported in a child with Wilson disease.
PATHOLOGY
Changes in hepatocellular mitochondria, identified with electron microscopy, are an important feature in Wilson disease.20 The mitochondria vary in size, and the numbers of dense bodies in mitochondria may be increased. The most striking change is dilatation of the tips of the mitochondrial cristae as a result of separation of the inner and outer membranes of the cristae, with widening of the intercristal space until the appearance is irregularly cystic. The crista resembles a tennis racquet if only the tip is dilated. This finding, although not entirely specific for Wilson disease, can be helpful diagnostically, even in young and minimally affected patients. Involvement of hepatocytes may be not uniform, and abnormalities may be found in some hepatocytes in some lobules and not in others. The mitochondrial changes are probably a consequence of oxidative damage from excessive copper in hepatocytes.21
DIAGNOSIS
The patient with the classic combination of chronic liver disease, tremor or dystonia, and Kayser-Fleischer rings is readily diagnosed on clinical grounds, but such patients are uncommon. A diagnostic scoring system has been proposed22 but has been evaluated only in children. Suggestive clinical symptoms are often the main prerequisite for diagnosing Wilson disease, and laboratory investigations may provide confirmation. Kayser-Fleischer rings should be sought through a careful slit-lamp examination and repeated if necessary. Lack of Kayser-Fleischer rings does not exclude the diagnosis of Wilson disease. Routine liver biochemical test levels are usually abnormal, with mild-to-moderate elevations of the serum aminotransferase levels. Serum levels of the ALT may be much lower than those of AST, possibly reflecting damage to hepatocellular mitochondria.
Two major disturbances of copper disposition in Wilson disease are defective incorporation of copper into ceruloplasmin and decreased biliary excretion of copper. A summary of biochemical features in Wilson disease in comparison with normal persons is shown in Table 75-1. The classic feature of a low ceruloplasmin concentration has proved less typical than previously thought because hepatic inflammation may be sufficient to elevate serum ceruloplasmin levels. Also, the normal range for serum ceruloplasmin is increased in very young children. The method of measuring ceruloplasmin may account in part for finding normal ceruloplasmin levels in patients with Wilson disease. Immunologic methods, which are commonly used in most laboratories, measure both apoceruloplasmin and holoceruloplasmin and typically overestimate the true amount of functional ceruloplasmin in plasma. The oxidase assay, although technically less convenient for laboratories that perform automated testing, provides a more reliable measure of ceruloplasmin for diagnosis because the assay measures enzymatically active, copper-containing ceruloplasmin. This method permits an accurate estimate of non–ceruloplasmin-bound copper23 and can also indicate possible early copper deficiency in treated patients.24
Table 75-1 Biochemical Parameters in Patients with Wilson Disease and in Normal Adults
| NORMAL ADULTS | WILSON DISEASE* | |
|---|---|---|
| Serum ceruloplasmin (mg/L) | 200-350 | 0-200 |
| Serum copper (µg/L) | 700-1520 | 190-640 |
| (µmol/L) | 11-24 | 3-10 |
| Urinary copper (µg/d) | <40 | 100-1000 |
| (µmol/d) | <0.6 | >1.6 |
| Liver copper (µg/g dry weight) | 20-50 | >200 |
* For all the assays, results in homozygotes and heterozygotes may overlap in some cases.
Serum ceruloplasmin measurement by itself is not a sufficient diagnostic test for Wilson disease. A low serum level of ceruloplasmin is not unique to Wilson disease; synthesis of ceruloplasmin may be reduced in other types of chronic liver disease, intestinal malabsorption, nephrosis, and malnutrition. Furthermore, a low ceruloplasmin concentration is found in at least 10% of heterozygotes for Wilson disease. Almost complete absence of ceruloplasmin is found in hereditary aceruloplasminemia, a rare autosomal recessive condition that is associated with neurologic, retinal, and pancreatic degeneration caused by iron accumulation in the brain, retina, and pancreas.25,26 Anemia and an increase in the plasma ferritin level are observed. Aceruloplasminemia has confirmed the important function of ceruloplasmin as a ferroxidase that oxidizes iron for transport from ferritin to transferrin, a function proposed in the late 1960s. Targeted disruption of the ceruloplasmin gene in a mouse model has confirmed the critical role of ceruloplasmin in transporting iron out of cells.27 Rarely, patients with Wilson disease who undergo rigorous, prolonged chelation therapy may resemble those with clinical aceruloplasminemia if ceruloplasmin oxidase activity is reduced to undetectable levels.28 The concurrence of Wilson disease and mutation-confirmed hereditary hemochromatosis has been reported.29
Studies of urinary copper excretion, preferably with three separate 24-hour collections, have proved useful for diagnosis. Urinary copper excretion reflects the non–ceruloplasmin-bound copper concentration in plasma. The collection must be complete, and the volume and total creatinine excretion should be measured; precautions against contamination with copper in the collection process are essential. The basal 24-hour urinary copper excretion is elevated at least two to three times normal in the vast majority of patients. The conventional cutoff criterion of 100 µg/day, although typical, is not sufficiently sensitive. A patient with a basal 24-hour urinary copper excretion of greater than 40 µg/day merits further investigation for Wilson disease. Heterozygotes usually have a normal 24-hour urinary copper excretion, although the value may be borderline abnormal in some cases. Although a normal person may excrete as much as 20 times the baseline level of copper after administration of penicillamine, a patient with symptomatic Wilson disease will excrete considerably more. Urinary copper excretion of 2.5 µmol (1600 µg) or more per 24 hours is diagnostic of Wilson disease. This provocative test lacks sensitivity for identifying presymptomatic affected siblings.30
Hepatic tissue copper concentration, which usually is measured by neutron activation analysis or atomic absorption spectrometry, may provide important diagnostic information. A hepatic copper content greater than 250 µg per g dry weight of liver is considered diagnostic of Wilson disease. On the basis of a large series of genetically-diagnosed patients, a value of greater than 70 µg per g dry weight has been proposed as a better diagnostic threshold, although some specificity is lost.31 Hepatic parenchymal concentrations of less than 40 µg per g dry weight are regarded as strong evidence against a diagnosis of Wilson disease. Liver biopsy samples must be collected without extraneous copper contamination, but in general ordinary disposable liver biopsy needles can be used. The sample submitted must be adequate in size. In the early stages of Wilson disease, when copper is distributed diffusely in the liver cell cytoplasm, this measurement may clearly indicate hepatic copper overload. In later stages of hepatic Wilson disease, the measurement of hepatic copper is less reliable because copper is distributed unequally in the liver. Moreover, liver biopsy may not be safe in such patients because of coagulopathy or ascites—therefore this diagnostic parameter may not be available. Some heterozygotes have minor elevations of liver tissue copper. An elevated hepatic copper concentration is not specific for Wilson disease; patients with chronic cholestasis or diseases such as Indian childhood cirrhosis may have elevated hepatic copper levels.
In view of the numerous available diagnostic tests, a methodical approach is required. The classic patient with Wilson disease, whether displaying hepatic or neurologic findings, may be considered to be someone between 6 and 40 years of age with serum ceruloplasmin level less than 5 mg/dL (<50 mg/L) and definite Kayser-Fleischer rings. Otherwise, in the presence of chronic liver disease (indicated by hepatomegaly or biochemical abnormalities) or typical neurologic symptoms, the combination of a low serum ceruloplasmin level (less than 140 mg/dL32) and elevated basal 24-hour urinary copper excretion (>40 µg/day) is highly suggestive of Wilson disease. The measurement of 24-hour urinary copper excretion after administration of penicillamine may be so definitive that the diagnosis is no longer in doubt. Typical ocular findings complete the clinical diagnosis but may be lacking. A percutaneous liver biopsy is useful for assessing the severity of liver damage and measuring parenchymal copper concentration, which is regarded by some to be the sine qua non for the diagnosis of Wilson disease. This procedure, however, may have to be delayed in patients with severe liver dysfunction. Other clinical entities in the differential diagnosis must be appropriately excluded. In the patient who does not have classic manifestations, extensive studies must be pursued meticulously; ultimately a gene mutation analysis may be the only convincing and reliable diagnostic procedure.
MUTATION ANALYSIS
More than 500 reported mutations in the ATP7B gene have been detected in many different populations since the original mutations were described. These mutations are recorded in the Wilson Disease Mutation Database (http://www.wilsondisease.med.ualberta.ca/database.asp), as described by Kenney and Cox.33 This database includes references and population sources. The usual approach to detection of a mutation is high-throughput sequencing of either selected or all exons of the gene. Although identification of a mutation is technically straightforward, care must be taken that the change detected causes disease and is not a rare normal variant, particularly for missense mutations, in which a single amino acid is substituted. Because of the similarity between yeast and mammalian copper transport systems, yeast and cell assay systems have been developed for the functional assessment of variants.34–36 The identification of one mutation may be adequate to confirm the diagnosis, if characteristic clinical symptoms and at least some of the biochemical features are present and if the one mutation detected is clearly established as a disease-causing mutation.
The majority of mutations in ATP7B identified to date are missense mutations. Small deletions, insertions, nonsense, and splice-site mutations occur throughout the gene (Cox, Mutation Database). Large gene deletions, which are found in about 20% of patients with Menkes disease, are rare, and the mutation spectrum for the Menkes gene, ATP7A, is different from that for ATP7B.37 Various ethnic groups have different specific mutations. The common histidine1069glutamine (His1069Gln) mutation3 is present, at least in the heterozygous state, in 35% to 75% of Europeans with Wilson disease; the higher number is relevant only for eastern Europe.38 Exon 8 of the gene is particularly rich in mutations in European populations. The mutation arginine778leucine is common in Chinese populations.39 Because no mutation is present in high frequency in Japanese and Mediterranean populations, mutation detection is more challenging in such populations. In populations with ethnic homogeneity or in which the spectrum of mutations is established, testing strategies can identify the mutations in more than 90% of patients, as in Sardinians, in whom the disease frequency is 1 in 7000 live births.40 In populations with a limited number of mutations, use of a “Wilson disease chip”41 may be cost-effective. If the patient is clinically normal, has only mild signs of the disease, or has a late age of onset, the possibility exists that the patient could actually be a heterozygote. Up to now, however, heterozygotes have not been known to become clinically affected or to require treatment.
Gene deletions, duplications, nonsense mutations, and splice-site mutations would be predicted to prevent almost completely the formation of the gene product and thus to produce a severe defect. This prediction is true to some extent,39,42 and the onset of liver disease at three years of age has been reported in a patient with a severe gene defect that was predicted to abolish the gene product.43 The common His1069Gln mutation tends to be associated with neurologic disease and later onset44; however, this mutation has been reported in homozygotes as young as nine years of age with hepatic disease. The positions of this and other missense mutations (amino acid substitutions) are shown in Figure 75-4. Most patients are compound heterozygotes, carrying two different mutations for the gene. Because homozygotes for a single mutation are relatively infrequent, correlation of clinical features with specific mutations is difficult.
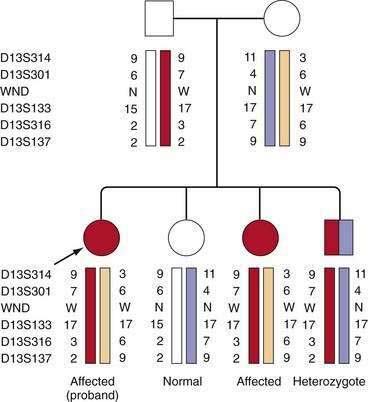
Presymptomatic Diagnosis of Siblings
If mutations have been identified in a patient, mutational analysis is easily carried out in siblings (sibs) by direct testing for the mutations found in the patient. If mutations have not been identified, accurate diagnosis can be achieved using markers flanking the gene. The most useful genetic markers are stretches of dinucleotides or trinucleotides, which show such extensive variability in the normal population so that parents within any one family will carry different alleles of these markers. This variability allows the tracking of the disease gene as it segregates within families, as shown in Figure 75-4. It is important that informative markers flank the gene because an erroneous diagnosis could rarely result if markers on only one side of the gene are informative and a recombinant event has occurred close to the gene. The combination of markers, or haplotype, reliably indicates the genetic status within the family. According to marker studies, an occasional person considered, as a result of biochemical testing, to have a high probability of being a presymptomatic patient has been shown to be a heterozygote. Therefore, confirmation of the genotype is highly recommended before treatment is initiated. If a heterozygote is found to accumulate copper stores in later decades of life, he or she may need to be followed for signs of copper overload.
TREATMENT
Three treatments for Wilson disease are generally recognized: d-penicillamine, trientine, and zinc (Table 75-2). Chelation with tetrathiomolybdate is a relatively new and still experimental option. With effective lifelong chelation treatment, most patients live normal, healthy lives. Starting treatment early is critical, and the outcome is best for patients in whom the disease is diagnosed and treatment begun when the patient is presymptomatic. Whether routine institution of chelation therapy in infancy (<2 years old) is advantageous or deleterious remains unknown. Likewise, the potential role of gene transfer therapy remains uncertain.45 Dietary treatment alone is ineffective, but most patients should eliminate copper-rich foods from the diet. These foods include organ meats, shellfish, nuts, chocolate, and mushrooms. Vegetarians require specific dietary counseling. If the concentration of copper in the patient’s drinking water is believed to be high, the water should be analyzed and a copper-removing device should be installed in the plumbing system.
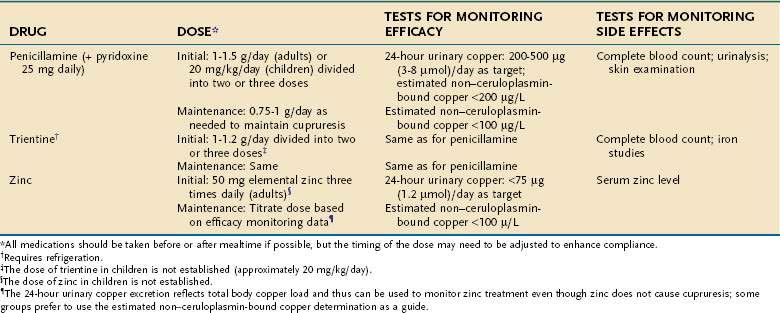
Penicillamine, introduced in 1956 by J. M. Walshe, is effective in most patients with Wilson disease. Penicillamine, which is the sulfhydryl-containing amino acid cysteine substituted with two methyl groups, greatly increases urinary excretion of copper. Only the d-penicillamine form is used clinically. Studies in the LEC rat model indicate that penicillamine inhibits the accumulation of copper in hepatocellular lysosomes and solubilizes copper for mobilization from these particles, but not from cytoplasmic metallothioneins.46 In addition to its chelating action, penicillamine inhibits collagen cross-linking and has some immunosuppressive properties. The neurologic status of patients with mainly neurologic symptoms may worsen initially after treatment with penicillamine is started47; most, but not all, recover with continued use of penicillamine. A febrile reaction with rash and proteinuria develops in some patients within 7 to 10 days of beginning treatment. Penicillamine can be restarted slowly, along with glucocorticoids, but changing to an alternative chelator is preferred.
Trientine, or triethylene tetramine dihydrochloride (2,2,2-tetramine), the official short name of which is “trien,” is the usual second-line treatment for patients who are intolerant of penicillamine. Trientine differs chemically from penicillamine in lacking sulfhydryl groups. Copper is chelated by forming a stable complex with the four constituent nitrogens in a planar ring. Trientine increases urinary copper excretion and may interfere with intestinal absorption of copper. Trientine is a less potent chelator than penicillamine, but the difference is not clinically important. Trientine produces little significant toxicity in patients with Wilson disease—apart from causing occasional gastritis and inducing iron deficiency, apparently by chelating dietary iron. Bone marrow suppression is extremely rare. Adverse effects of penicillamine resolve and do not recur during treatment with trientine.48 Neurologic worsening after treatment with trientine is begun has rarely been reported. Trientine is highly effective, even in patients with advanced liver fibrosis or as initial treatment in children.49
Oral zinc, used in Europe since the 1970s,50 has been investigated extensively as a treatment modality in North America. Its mechanism of action is entirely different from that of the chelators. In pharmacologic doses, zinc interferes with the absorption of copper from the gastrointestinal tract and increases the excretion of copper in the stool. The postulated mechanism of action is through the induction of metallothionein in enterocytes. The metallothionein has a greater affinity for copper than for zinc and preferentially binds copper from the intestinal contents. Once bound, the copper is not absorbed but is lost in the feces as enterocytes are shed during normal turnover.51 Additionally, zinc may interfere with lipid peroxidation and enhance the availability of glutathione.51
Problems with zinc therapy include gastritis, which is a common side effect, and uncertainty about dosing. Using a zinc salt other than zinc sulfate may minimize gastritis; any zinc salt is equally acceptable for the treatment of Wilson disease. Food interferes with the effectiveness of zinc, and some investigators recommend that no food be eaten for one hour before or after a dose of zinc is taken. This dosing regimen tends to increase the severity of gastritis and may be sufficiently inconvenient to compromise compliance, as in adolescents. An alternative approach is to be less rigorous about avoiding zinc at mealtimes and to titrate the dose according to the urinary copper excretion or serum non–ceruloplasmin-bound copper concentration. Treatment with zinc appears to have few side effects52 but may not be effective in all patients. Rare patients experience a deterioration in hepatic Wilson disease when started on zinc. Zinc may have immunosuppressant effects and reduce leukocyte chemotaxis. Studies in rats suggest a possible interference with bone formation. The long-term effectiveness of zinc requires further investigation, but current data indicate that zinc is effective as maintenance therapy and has low toxicity.
For patients who present with decompensated chronic liver disease, combining zinc with a conventional chelator (preferably trientine) has become a popular treatment strategy despite a lack of extensive validation. The two types of treatments must be temporally dispersed through the day, with at least four to five hours between administration of the two drugs, or else they may neutralize each other. This intensive induction regimen is best suited to patients with severe hepatic or neurologic disease53 and remains investigational. Some patients with severe hepatic Wilson disease may fail on this regimen and require urgent liver transplantation.
Ammonium tetrathiomolybdate may be especially suitable for treatment of severe neurologic Wilson disease because, unlike penicillamine, it is not associated with early neurologic deterioration.54 Tetrathiomolybdate interferes with the absorption of copper from the intestine and binds to plasma copper with high affinity. Unlike penicillamine, tetrathiomolybdate has been found in LEC rats to remove copper from metallothionein at low doses; at higher doses, an insoluble copper complex is deposited in the liver.55 Although tetrathiomolybdate is regarded as non-toxic, bone marrow suppression and hepatotoxicity are noteworthy adverse side effects. Little is known about where the mobilized copper and molybdate may be deposited. The optimal dose and length of treatment, as well as long-term side effects, require further study. Such a potent copper-binding drug could produce copper deficiency.
Antioxidants may be a useful adjunct for preventing tissue damage. Studies in copper-loaded animals and in patients with Wilson disease indicate that copper enhances free radical production in tissues and that this effect may be an important cause of liver damage.56 Oxidative damage may be reflected in mutations of the TP53 tumor suppressor gene, increased activity of nitric oxide synthase in patients with hepatic Wilson disease,55 and DNA strand breaks in the brains of LEC rats.57 Oxidative effects of excess copper may contribute to development and progression of liver damage.58 Alpha-tocopherol may be beneficial adjunctive treatment for patients with severe hepatic decompensation.
For pregnant patients with Wilson disease, treatment must be continued throughout pregnancy. Postpartum hepatic decompensation is a risk if treatment is stopped completely during pregnancy. Although many successful pregnancies have occurred during treatment with penicillamine, the drug is officially classified as a teratogen. Occasional reports of severe collagen defects in the offspring of a patient treated with penicillamine may be caused in part by copper deficiency as a result of prolonged aggressive treatment as well as the teratogenic effects of penicillamine.59 Treatment with zinc may be less likely to produce adverse effects on developing collagen in the fetus. The safety of trientine during pregnancy is unknown, apart from favorable anecdotal reports. Judicious reduction of the dose of penicillamine or trientine by approximately 25% of the pre-pregnancy dose is advisable, especially if delivery by cesarean section is anticipated.
PROGNOSIS
The role of liver transplantation in Wilson disease is limited (see Chapters 93 and 95). Fulminant liver failure in a patient with Wilson disease necessitates liver transplantation (see earlier). Some patients with severe liver disease that is unresponsive to drug therapy may also proceed to early transplantation. The outcome is favorable, with one-year survival rates of 80% to 90% and excellent survival beyond one year.60,61 Severe neurologic disease may improve after liver transplantation, but experience is conflicting14 and liver transplantation cannot be recommended for neurologic Wilson disease. Patients with neurologic or psychiatric manifestations of Wilson disease appear to have poor outcomes after liver transplantation and adhere poorly to medical regimens.62 Therefore, liver transplantation should be reserved for patients who present with severe, decompensated liver disease that is unresponsive to therapy or with fulminant liver failure. Live donor-related liver transplantations, even when the graft is from a family member who may be a heterozygote, have been found to yield adequately functioning grafts.63,64 If the donor is the brother or sister of the patient, genetic analysis of ATP7B should be included in the assessment of the donor.
Brewer GJ, Dick RD, Johnson VD, et al. Treatment of Wilson’s disease with zinc: XV long-term follow-up studies. J Lab Clin Med. 1998;132:264-78. (Ref 52.)
Bull PC, Thomas GR, Rommens JM, et al. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327-37. (Ref 2.)
Emre S, Atillasoy EO, Ozdemir S, et al. Orthotopic liver transplantation for Wilson’s disease: A single-center experience. Transplantation. 2001;72:1232-6. (Ref 61.)
Ferenci P, Steindl-Munda P, Vogel W, et al. Diagnostic value of quantitative hepatic copper determination in patients with Wilson’s disease. Clin Gastroenterol Hepatol. 2005;3:811-18. (Ref 31.)
Harris ZL, Takahashi Y, Miyajima H, et al. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci USA. 1995;92:2539-43. (Ref 25.)
Hsi G, Cullen LM, Macintyre G, et al. Sequence variation in the ATP-binding domain of the Wilson disease transporter, ATP7B, affects copper transport in a yeast model system. Hum Mutat. 2008;29:491-501. (Ref 36.)
Kenney SM, Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat. 2007;28:1171-7. (Ref 33.)
Korman JD, Volenberg I, Balko J, et al. Screening for Wilson disease in acute liver failure: A comparison of currently available diagnostic tests. Hepatology. 2008;48:1167-74. (Ref 17.)
Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut. 2007;56:115-20. (Ref 47.)
Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: An update. Hepatology. 2008;47:2089-111. (Ref 14.)
Wilson DC, Phillips MJ, Cox DW, Roberts EA. Severe hepatic Wilson’s disease in preschool-aged children. J Pediatr. 2000;137:719-22. (Ref 43.)
1. Wilson SAK. Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of the liver. Brain. 1912;34:295-509.
2. Bull PC, Thomas GR, Rommens JM, et al. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327-37.
3. Tanzi RE, Petrukhin KE, Chernov I, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344-50.
4. Fatemi N, Sarkar B. Structural and functional insights of Wilson disease copper-transporting ATPase. J Bioenerg Biomembr. 2002;34:339-49.
5. Wu J, Forbes JR, Chen HS, Cox DW. The LEC rat has a deletion in the copper transporting ATPase gene homologous to the Wilson disease gene. Nat Genet. 1994;7:541-5.
6. Theophilos MB, Cox DW, Mercer JF. The toxic milk mouse is a murine model of Wilson disease. Hum Mol Genet. 1996;5:1619-24.
7. Hung IH, Suzuki M, Yamaguchi Y, et al. Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J Biol Chem. 1997;272:21461-6.
8. La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: Role in copper homeostasis. Arch Biochem Biophys. 2007;463:149-67.
9. Field LS, Luk E, Culotta VC. Copper chaperones: Personal escorts for metal ions. J Bioenerg Biomembr. 2002;34:373-39.
10. van de Sluis B, Rothuizen J, Pearson PL, et al. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet. 2002;11:165-73.
11. Coronado VA, Damaraju D, Kohijoki R, Cox DW. New haplotypes in the Bedlington terrier indicate complexity in copper toxicosis. Mamm Genome. 2003;14:483-91.
12. Tao TY, Liu F, Klomp L, et al. The copper toxicosis gene product Murr1 directly interacts with the Wilson disease protein. J Biol Chem. 2003;278:41593-6.
13. Mufti AR, Burstein E, Csomos RA, et al. XIAP Is a copper binding protein deregulated in Wilson’s disease and other copper toxicosis disorders. Mol Cell. 2006;21:775-85.
14. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: An update. Hepatology. 2008;47:2089-111.
15. Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology. 2003;37:1475-92.
16. Milkiewicz P, Saksena S, Hubscher SG, Elias E. Wilson’s disease with superimposed autoimmune features: Report of two cases and review. J Gastroenterol Hepatol. 2000;15:570-4.
17. Korman JD, Volenberg I, Balko J, et al. Screening for Wilson disease in acute liver failure: A comparison of currently available diagnostic tests. Hepatology. 2008;48:1167-74.
18. Kreymann B, Seige M, Schweigart U, et al. Albumin dialysis: Effective removal of copper in a patient with fulminant Wilson disease and successful bridging to liver transplantation: A new possibility for the elimination of protein-bound toxins. J Hepatol. 1999;31:1080-5.
19. Walshe JM, Waldenstrom E, Sams V, et al. Abdominal malignancies in patients with Wilson’s disease. QJM. 2003;96:657-62.
20. Sternlieb I. Mitochondrial and fatty changes in hepatocytes of patients with Wilson’s disease. Gastroenterology. 1968;5:354-67.
21. Roberts EA, Robinson BH, Yang S. Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol Genet Metab. 2008;93:54-65.
22. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139-42.
23. Walshe JM. Wilson’s disease: The importance of measuring serum caeruloplasmin non-immunologically. Ann Clin Biochem. 2003;40:115-21.
24. Macintyre G, Gutfreund KS, Martin WR, et al. Value of an enzymatic assay for the determination of serum ceruloplasmin. J Lab Clin Med. 2004;144:294-301.
25. Harris ZL, Takahashi Y, Miyajima H, et al. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci USA. 1995;92:2539-43.
26. Yoshida K, Furihata K, Takeda S, et al. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat Genet. 1995;9:267-72.
27. Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. 1999;96:10812-17.
28. Hayashi H, Yano M, Fujita Y, Wakusawa S. Compound overload of copper and iron in patients with Wilson’s disease. Med Mol Morphol. 2006;39:121-6.
29. Dib N, Valsesia E, Malinge MC, et al. Late onset of Wilson’s disease in a family with genetic haemochromatosis. Eur J Gastroenterol Hepatol. 2006;18:43-7.
30. Muller T, Koppikar S, Taylor RM, et al. Re-evaluation of the penicillamine challenge test in the diagnosis of Wilson’s disease in children. J Hepatol. 2007;47:270-6.
31. Ferenci P, Steindl-Munda P, Vogel W, et al. Diagnostic value of quantitative hepatic copper determination in patients with Wilson’s disease. Clin Gastroenterol Hepatol. 2005;3:811-18.
32. Mak CM, Lam CW, Tam S. Diagnostic accuracy of serum ceruloplasmin in Wilson disease: Determination of sensitivity and specificity by ROC curve analysis among ATP7B-genotyped subjects. Clin Chem. 2008;54:1356-62.
33. Kenney SM, Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat. 2007;28:1171-7.
34. Forbes JR, Cox DW. Functional characterization of missense mutations in ATP7B: Wilson disease mutation or normal variant? Am J Hum Genet. 1998;63:1663-74.
35. Hsi G, Cullen LM, Moira Glerum D, Cox DW. Functional assessment of the carboxy-terminus of the Wilson disease copper-transporting ATPase, ATP7B. Genomics. 2004;83:473-81.
36. Hsi G, Cullen LM, Macintyre G, et al. Sequence variation in the ATP-binding domain of the Wilson disease transporter, ATP7B, affects copper transport in a yeast model system. Hum Mutat. 2008;29:491-501.
37. Hsi G, Cox DW. A comparison of the mutation spectra of Menkes disease and Wilson disease. Hum Genet. 2003;114:165-72.
38. Caca K, Ferenci P, Kuhn HJ, et al. High prevalence of the H1069Q mutation in East German patients with Wilson disease: Rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol. 2001;35:575-81.
39. Thomas GR, Forbes JR, Roberts EA, et al. The Wilson disease gene: Spectrum of mutations and their consequences. Nat Genet. 1995;9:210-17.
40. Lovicu M, Dessi V, Zappu A, et al. Efficient strategy for molecular diagnosis of Wilson disease in the Sardinian population. Clin Chem. 2003;49:496-8.
41. Gojova L, Jansova E, Kulm M, et al. Genotyping microarray as a novel approach for the detection of ATP7B gene mutations in patients with Wilson disease. Clin Genet. 2008;73:441-52.
42. Panagiotakaki E, Tzetis M, Manolaki N, et al. Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B). Am J Med Genet A. 2004;131A:168-73.
43. Wilson DC, Phillips MJ, Cox DW, Roberts EA. Severe hepatic Wilson’s disease in preschool-aged children. J Pediatr. 2000;137:719-22.
44. Stapelbroek JM, Bollen CW, van Amstel JK, et al. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: Results of a meta-analysis. J Hepatol. 2004;41:758-63.
45. Merle U, Stremmel W, Encke J. Perspectives for gene therapy of Wilson disease. Curr Gene Ther. 2007;7:217-20.
46. Klein D, Lichtmannegger J, Heinzmann U, Summer KH. Dissolution of copper-rich granules in hepatic lysosomes by d-penicillamine prevents the development of fulminant hepatitis in Long-Evans cinnamon rats. J Hepatol. 2000;32:193-201.
47. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut. 2007;56:115-20.
48. Scheinberg IH, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson’s disease. N Engl J Med. 1987;317:209-13.
49. Arnon R, Calderon JF, Schilsky M, et al. Wilson disease in children: Serum aminotransferases and urinary copper on triethylene tetramine dihydrochloride (trientine) treatment. J Pediatr Gastroenterol Nutr. 2007;44:596-602.
50. Hoogenraad TU, Van Haltum J, Van der Hamer CJA. Management of Wilson’s disease with zinc sulphate. J Neurol Sci. 1987;77:137-46.
51. Yuzbasiyan-Gurkan V, Grider A, Nostrant T, et al. Treatment of Wilson’s disease with zinc: X. Intestinal metallothionein induction. J Lab Clin Med. 1992;120:380-6.
52. Brewer GJ, Dick RD, Johnson VD, et al. Treatment of Wilson’s disease with zinc: XV long-term follow-up studies. J Lab Clin Med. 1998;132:264-78.
53. Askari FK, Greenson J, Dick RD, et al. Treatment of Wilson’s disease with zinc. XVIII. Initial treatment of the hepatic decompensation presentation with trientine and zinc. J Lab Clin Med. 2003;142:385-90.
54. Brewer GJ, Askari F, Lorincz MT, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63:521-7.
55. Ogra Y, Suzuki KT. Targeting of tetrathiomolybdate on the copper accumulating in the liver of LEC rats. J Inorg Biochem. 1998;70:49-55.
56. Sokol RJ, Twedt D, McKim JM, et al. Oxidant injury to hepatic mitochondria in patients with Wilson’s disease and Bedlington terriers with copper toxicosis. Gastroenterology. 1994;107:1788-98.
57. Hayashi M, Fuse S, Endoh D, et al. Accumulation of copper induces DNA strand breaks in brain cells of Long-Evans Cinnamon (LEC) rats, an animal model for human Wilson disease. Exp Anim. 2006;55:419-26.
58. Nagasaka H, Inoue I, Inui A, et al. Relationship between oxidative stress and antioxidant systems in the liver of patients with Wilson disease: Hepatic manifestation in Wilson disease as a consequence of augmented oxidative stress. Pediatr Res. 2006;60:472-7.
59. Pinter R, Hogge WA, McPherson E. Infant with severe penicillamine embryopathy born to a woman with Wilson disease. Am J Med Genet. 2004;128A:294-8.
60. Eghtesad B, Nezakatgoo N, Geraci LC, et al. Liver transplantation for Wilson’s disease: A single-center experience. Liver Transpl Surg. 1999;5:467-74.
61. Emre S, Atillasoy EO, Ozdemir S, et al. Orthotopic liver transplantation for Wilson’s disease: A single-center experience. Transplantation. 2001;72:1232-6.
62. Medici V, Mirante VG, Fassati LR, et al. Liver transplantation for Wilson’s disease: The burden of neurological and psychiatric disorders. Liver Transpl. 2005;11:1056-63.
63. Asonuma K, Inomata Y, Kasahara M, et al. Living related liver transplantation from heterozygote genetic carriers to children with Wilson’s disease. Pediatr Transplant. 1999;3:201-5.
64. Tamura S, Sugawara Y, Kishi Y, et al. Living-related liver transplantation for Wilson’s disease. Clin Transplant. 2005;19:483-6.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 75 Wilson Disease
Copper, a component of several essential enzymes, is toxic to tissues when present in excess. Dietary intake of copper generally exceeds the trace amount required, and mechanisms to control influx and efflux from cells must maintain an appropriate balance. Two disorders of copper transport are known: Menkes disease, an X-linked defect in transport of copper from the intestine, and Wilson disease, an autosomal recessive disorder of copper overload. Wilson disease was first described, in 1912 by Kinnear Wilson, as a familial disease characterized by progressive, lethal neurologic dysfunction with chronic liver disease and a corneal abnormality, the Kayser-Fleischer (KF) ring.1 Wilson also observed that some younger siblings of typical patients died of severe liver disease without experiencing neurologic abnormalities. In this disease, inadequate hepatic copper excretion leads to copper accumulation in the liver, brain, kidney, and cornea. The incidence in most populations is on the order of 1 in 30,000.
THE COPPER PATHWAY
In the liver, copper is incorporated into the protein apoceruloplasmin to produce ceruloplasmin (also called holoceruloplasmin). More than 90% of the copper in plasma is an integral part of ceruloplasmin, an α2-glycoprotein that contains six molecules of copper and has a molecular weight of 132 kd. The normal serum concentration of ceruloplasmin in adults, as measured by immunochemical or enzymatic techniques, is 200 to 400 mg/L, rising from a very low level at birth to 300 to 500 mg/L in the first years of life. Ceruloplasmin is an acute-phase reactant that is elevated by inflammation (including inflammatory hepatic disease), pregnancy, and the use of exogenous estrogen. The majority of ingested copper is excreted via the bile; a small fraction is excreted in urine. When intestinal or liver cells are overloaded with copper, the metallothioneins, a class of low-molecular-weight cysteine-rich proteins, are induced and sequester copper in a nontoxic form. The normal pathways of copper transport in the body and in the hepatocyte are shown in Figures 75-1 and 75-2.
THE BASIC MOLECULAR DEFECT
Our understanding of the basic defect in Wilson disease increased dramatically with the cloning of the genes, first for X-linked Menkes disease and then for Wilson disease. The gene for Menkes disease (ATP7A), which was cloned by using a chromosomal breakpoint in an affected female patient, was found to be related to bacterial copper resistance genes. Cloning of the Wilson disease gene (ATP7B) was accomplished by a combination of linkage analysis, physical mapping of the relevant region of chromosome 13q14, and recognition of its extensive homology with the Menkes disease gene.2,3 The coding region of the gene is 4.1 kilobases in length, with messenger RNA (mRNA) of about 8 kilobases. The product, ATP7B (or the Wilson ATPase), is a membrane P-type adenosine triphosphatase (ATPase) that consists of 1443 amino acid residues and has a molecular mass of 160 kd. The predicted structure,2 as confirmed by structural studies,4 has six copper binding domains, a phosphorylation domain, an ATP-binding region, and eight transmembrane domains. (Fig. 75-3). All functionally important regions of the gene are conserved between bacteria and yeast. Mutations in the ATP7B gene result in retention of copper in the liver as well as impaired incorporation of copper into ceruloplasmin. The Long-Evans cinnamon (LEC) rat and the toxic milk (tx) mouse have mutations in their homologous ATP7B genes and are thus suitable models for the study of Wilson disease mechanisms and therapy.5,6
Although the gene for Menkes disease is expressed in many tissues, including muscle, kidney, heart, and intestine, the gene for Wilson disease is expressed predominantly in the liver and kidney, with minor expression in brain, lungs, and placenta.2 Studies in cultured cells show localization of the Wilson ATPase in the trans-Golgi network, with trafficking from the trans-Golgi network to cytoplasmic vesicles in the presence of increased copper.7 When intracellular copper concentrations are elevated, the Wilson ATPase is found near the apical (bile canalicular) membrane in hepatocytes, consistent with its proposed function of facilitating excretion of copper via bile.8
Additional proteins are involved in the intracellular transport of copper. Molecular copper is not free in the cell but is transported to specific proteins by copper chaperones.9 The chaperone ATOX1 transports copper to the Wilson ATPase. Other chaperones deliver copper to superoxide dismutase in the cytoplasm and various copper-containing proteins in mitochondria.
The study of inherited hepatic copper toxicosis in Bedlington terriers has identified a new component of the copper transport system. Affected dogs show clinical variability that ranges from death or hepatic disease at two to three years of age to less severe chronic disease to a high level of hepatic copper. The proposed defective canine gene was identified by positional cloning through the use of markers to identify a region containing the COMMD1 (initially called the MURR1) gene. The gene has a deletion of one exon in some,10 but not all,11 affected dogs. Although the gene responsible for the excess copper remains to be identified, the disease in Bedlington terriers has highlighted other genes that may be involved in response to excess copper; COMMD1 interacts not only with the Wilson ATPase,12 but also with the X-linked inhibitor of apoptosis (XIAP), which can also bind copper.13 COMMD1 could be involved in response to, and facilitate elimination of, excess copper.
CLINICAL FEATURES
The clinical presentation of Wilson disease is extremely variable. The age at onset of symptoms is much broader than previously thought, ranging from 3 to 55 years. Wilson disease with hepatic involvement has been identified in one- to two-year-old patients and in patients older than age 60.14 Patients may present with chronic or fulminant liver disease, a progressive neurologic disorder without clinically prominent hepatic dysfunction, isolated acute hemolysis, or psychiatric illness. The clinical variability often makes confirmation of the diagnosis difficult. Further description of the clinical features and treatment has been published.14,15
HEPATIC PRESENTATION
Wilson disease may present in children and young adults with clinical liver disease indistinguishable from autoimmune hepatitis.16 As in autoimmune hepatitis, the onset may be acute. Fatigue, malaise, arthropathy, and rashes may occur; laboratory findings include elevated aminotransferase levels, a greatly increased serum immunoglobulin (IgG) concentration, and detectable nonspecific autoantibodies such as antinuclear antibodies and anti-smooth muscle (anti-actin) antibodies. Wilson disease must be specifically ruled out because the treatment of the two diseases is entirely different. With appropriate treatment, the long-term outlook for patients with Wilson disease that manifests as autoimmune hepatitis appears to be favorable, even if cirrhosis is present.
Wilson disease may present as fulminant hepatic failure, with severe coagulopathy and encephalopathy. Acute intravascular hemolysis is usually present, and renal failure may develop. Because the patient typically has not been suspected of having underlying liver disease, fulminant viral hepatitis is usually the working diagnosis. Unlike fulminant viral hepatitis, fulminant Wilson disease is typically characterized by disproportionately low aminotransferase levels (usually much less than 1500 U/L) at the onset of clinically apparent disease. The serum alkaline phosphatase level is in the normal range or even low for the patient’s age, and the bilirubin level is often disproportionately elevated as a result of hemolysis. In adults who present with Wilsonian fulminant liver failure, the combination of a ratio of the alkaline phosphatase level to the total bilirubin level of less than 4 and a ratio of the aspartate aminotransferase (AST) to alanine aminotransferase (ALT) level of greater than 2.2 can be extremely helpful for making the diagnosis, whereas the serum ceruloplasmin level is not (see later).17 Slit-lamp examination may reveal Kayser-Fleischer rings (see later). Urinary copper excretion is greatly elevated. These patients require urgent liver transplantation because they do not respond well to chelation treatment; albumin dialysis and related techniques may serve as temporary procedures until liver transplantation can be performed (see later).18 This presentation of Wilson disease is not rare, and affected patients account for a substantial proportion of persons transplanted for fulminant hepatic failure (see Chapters 93 and 95).
Recurrent bouts of hemolysis may predispose to the development of gallstones. Cirrhosis, if present, may be a further predisposing factor. Children with unexplained cholelithiasis, particularly with small bilirubinate stones, should be tested for Wilson disease. Compared with other chronic liver diseases, Wilson disease is rarely complicated by hepatocellular carcinoma; however, patients may have an increased propensity to abdominal malignancies,19 and hepatocellular carcinoma has been reported in a child with Wilson disease.
PATHOLOGY
Changes in hepatocellular mitochondria, identified with electron microscopy, are an important feature in Wilson disease.20 The mitochondria vary in size, and the numbers of dense bodies in mitochondria may be increased. The most striking change is dilatation of the tips of the mitochondrial cristae as a result of separation of the inner and outer membranes of the cristae, with widening of the intercristal space until the appearance is irregularly cystic. The crista resembles a tennis racquet if only the tip is dilated. This finding, although not entirely specific for Wilson disease, can be helpful diagnostically, even in young and minimally affected patients. Involvement of hepatocytes may be not uniform, and abnormalities may be found in some hepatocytes in some lobules and not in others. The mitochondrial changes are probably a consequence of oxidative damage from excessive copper in hepatocytes.21
DIAGNOSIS
The patient with the classic combination of chronic liver disease, tremor or dystonia, and Kayser-Fleischer rings is readily diagnosed on clinical grounds, but such patients are uncommon. A diagnostic scoring system has been proposed22 but has been evaluated only in children. Suggestive clinical symptoms are often the main prerequisite for diagnosing Wilson disease, and laboratory investigations may provide confirmation. Kayser-Fleischer rings should be sought through a careful slit-lamp examination and repeated if necessary. Lack of Kayser-Fleischer rings does not exclude the diagnosis of Wilson disease. Routine liver biochemical test levels are usually abnormal, with mild-to-moderate elevations of the serum aminotransferase levels. Serum levels of the ALT may be much lower than those of AST, possibly reflecting damage to hepatocellular mitochondria.
Two major disturbances of copper disposition in Wilson disease are defective incorporation of copper into ceruloplasmin and decreased biliary excretion of copper. A summary of biochemical features in Wilson disease in comparison with normal persons is shown in Table 75-1
[/not-level-membership-for-gastroenterology-and-hepatology-category]
