[level-membership-for-internal-medicine-category]
Chapter 13 Water, electrolytes and acid–base balance
Distribution and composition of body water
 Intracellular fluid (28 L, about 35% of lean bodyweight)
Intracellular fluid (28 L, about 35% of lean bodyweight)
 Extracellular – the interstitial fluid that bathes the cells (9.4 L, about 12%)
Extracellular – the interstitial fluid that bathes the cells (9.4 L, about 12%)

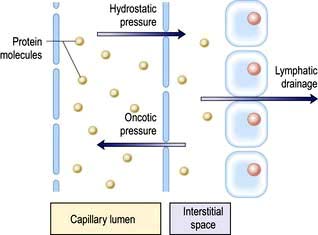
The intracellular and interstitial fluids are separated by the cell membrane; the interstitial fluid and plasma are separated by the capillary wall (Fig. 13.1). In the absence of solute, water molecules move randomly and in equal numbers in either direction across a semi-permeable membrane. However, if solutes are added to one side of the membrane, the intermolecular cohesive forces reduce the activity of the water molecules. As a result, water tends to stay in the solute-containing compartment because there is less free diffusion across the membrane. This ability to hold water in the compartment can be measured as the osmotic pressure.
Osmotic pressure
 The intracellular fluid contains mainly potassium (K+) (most of the cell Mg2+ is bound and osmotically inactive)
The intracellular fluid contains mainly potassium (K+) (most of the cell Mg2+ is bound and osmotically inactive)

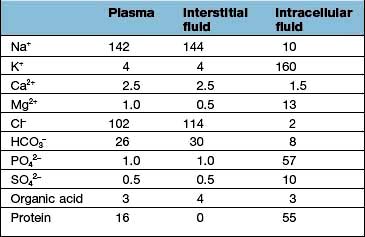
Regulation of the plasma volume is somewhat more complicated because of the tendency of the plasma proteins to hold water in the vascular space by an oncotic effect which is, in part, counterbalanced by the hydrostatic pressure in the capillaries that is generated by cardiac contraction (Fig. 13.1). The composition of intracellular and extracellular fluids is shown in Table 13.1.
Distribution of different types of replacement fluids
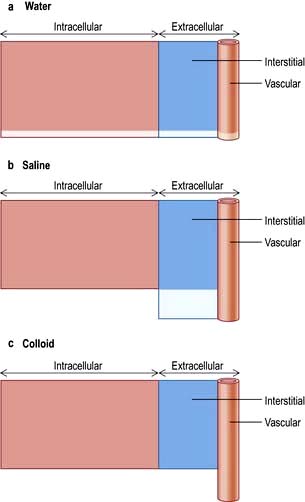
Figure 13.2 shows the relative effects on the compartments of the addition of identical volumes of water, saline and colloid solutions. Thus, 1 L of water given intravenously as 5% glucose is distributed equally into all compartments, whereas the same amount of 0.9% saline remains in the extracellular compartment. The latter is thus the correct treatment for extracellular water depletion – sodium keeping the water in this compartment. The addition of 1 L of colloid with its high oncotic pressure stays in the vascular compartment and is a treatment for hypovolaemia.
Regulation of extracellular volume (Fig. 13.3)
 Plasma fluid constitutes one-third of extracellular volume (4.6 L), and of this,
Plasma fluid constitutes one-third of extracellular volume (4.6 L), and of this,
 85% (3.9 L) lies in the venous side and only 15% (0.7 L) resides in the arterial circulation.
85% (3.9 L) lies in the venous side and only 15% (0.7 L) resides in the arterial circulation.
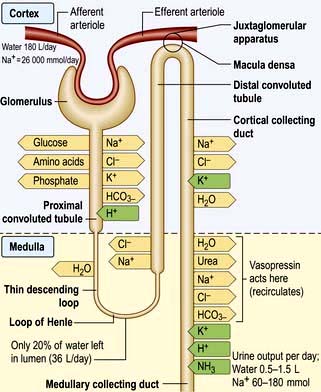
These changes in Na+ excretion can result from alterations both in the filtered load, determined primarily by the glomerular filtration rate (GFR), and in tubular reabsorption, which is affected by multiple factors. In general, it is changes in tubular reabsorption that constitute the main adaptive response to fluctuations in the effective circulating volume. How this occurs can be appreciated from Table 13.2 and Figure 13.4 and Figure 12.2 (see p. 563), which depicts the sites and determinants of segmental Na+ reabsorption. Although the loop of Henle and distal tubules make a major overall contribution to net Na+ handling, transport in these segments primarily varies with the amount of Na+ delivered; that is, reabsorption is flow-dependent. In comparison, the neurohumoral regulation of Na+ reabsorption according to body needs occurs primarily in the proximal tubules and collecting ducts.

Neurohumoral regulation of extracellular volume
 Intrarenal receptors. Receptors in the walls of the afferent glomerular arterioles respond, via the juxtaglomerular apparatus, to changes in renal perfusion, and control the activity of the renin-angiotensin-aldosterone system (see p. 1006). In addition, sodium concentration in the distal tubule and sympathetic nerve activity alter renin release from the juxtaglomerular cells. Prostaglandins I2 and E2 are also generated within the kidney in response to angiotensin II, acting to maintain glomerular filtration rate and sodium and water excretion, modulating the sodium-retaining effect of this hormone.
Intrarenal receptors. Receptors in the walls of the afferent glomerular arterioles respond, via the juxtaglomerular apparatus, to changes in renal perfusion, and control the activity of the renin-angiotensin-aldosterone system (see p. 1006). In addition, sodium concentration in the distal tubule and sympathetic nerve activity alter renin release from the juxtaglomerular cells. Prostaglandins I2 and E2 are also generated within the kidney in response to angiotensin II, acting to maintain glomerular filtration rate and sodium and water excretion, modulating the sodium-retaining effect of this hormone.
 Extrarenal receptors. These are located in the vascular tree in the left atrium and major thoracic veins, and in the carotid sinus body and aortic arch. These volume receptors respond to a slight reduction in effective circulating volume and result in increased sympathetic nerve activity and a rise in catecholamines. In addition, volume receptors in the cardiac atria control the release of a powerful natriuretic hormone – atrial natriuretic peptide (ANP) – from granules located in the atrial walls (see p. 943).
Extrarenal receptors. These are located in the vascular tree in the left atrium and major thoracic veins, and in the carotid sinus body and aortic arch. These volume receptors respond to a slight reduction in effective circulating volume and result in increased sympathetic nerve activity and a rise in catecholamines. In addition, volume receptors in the cardiac atria control the release of a powerful natriuretic hormone – atrial natriuretic peptide (ANP) – from granules located in the atrial walls (see p. 943).
A salt load, for example, leads to an increase in the effective circulatory and extracellular volume, raising both renal perfusion pressure, and atrial and arterial filling pressure. The increase in the renal perfusion pressure reduces the secretion of renin, and subsequently that of angiotensin II and aldosterone (see Fig. 12.5), whereas the rise in atrial and arterial filling pressure increases the release of ANP. These factors combine to reduce Na+ reabsorption in the collecting duct, thereby promoting excretion of excess Na+.
With more marked hypovolaemia, a decrease in GFR leads to an increase in proximal and thin ascending limb Na+ reabsorption which contributes to Na+ retention. This is brought about by enhanced sympathetic activity acting directly on the kidneys and indirectly by stimulating the secretion of renin/angiotensin II (see Fig. 13.3b) and non-osmotic release of antidiuretic hormone (ADH), also called vasopressin. The pressure natriuresis phenomenon may be the final defence against changes in the effective circulating volume. Marked persistent hypovolaemia leads to systemic hypotension and increased salt and water absorption in the proximal tubules and ascending limb of Henle. This process is partly mediated by changes in renal interstitial hydrostatic pressure and local prostaglandin and nitric oxide production.
Volume regulation in oedematous conditions
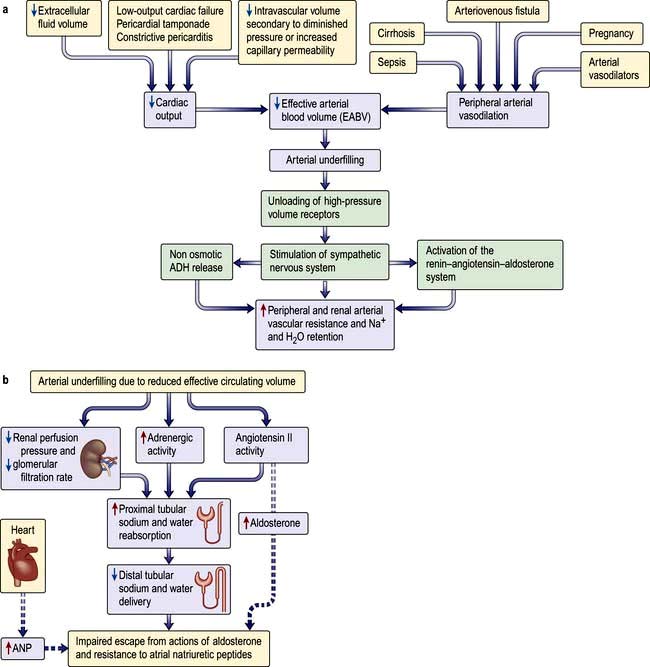
Sodium and water are retained despite increased extracellular volume in oedematous conditions such as cardiac failure, hepatic cirrhosis and hypoalbuminaemia. Here the principal mediator of salt and water retention is the concept of arterial underfilling due either to reduced cardiac output or diminished peripheral arterial resistance. Arterial underfilling in these settings leads to reduction of pressure or stretch (i.e. ‘unloading’ of arterial volume receptors), which results in activation of the sympathetic nervous system, activation of the renin-angiotensin-aldosterone system and non-osmotic release of ADH. These neurohumoral mediators promote salt and water retention in the face of increased extracellular volume. The common nature of the degree of arterial fullness and neurohumoral pathway in the regulation of extracellular volume in health and disease states forms the basis of Schrier’s unifying hypothesis of volume homeostasis (Fig. 13.3a).
Mechanism of impaired escape from actions of aldosterone and resistance to ANP
Not only is the activity of the renin-angiotensin-aldosterone system increased in oedematous conditions such as cardiac failure, hepatic cirrhosis and hypoalbuminaemia, but also the action of aldosterone is more persistent than in normal subjects and patients with Conn’s syndrome, who have increased aldosterone secretion (see p. 989).
In patients with the above oedematous conditions, e.g. heart failure, escape from the sodium-retaining actions of aldosterone does not occur and therefore they continue to retain sodium in response to aldosterone. Accordingly they have substantial natriuresis when given spironolactone, which blocks mineralocorticoid receptors. Alpha-adrenergic stimulation and elevated angiotensin II increase sodium transport in the proximal tubule, and reduced renal perfusion and GFR further increases sodium absorption from the proximal tubules by presenting less sodium and water in the tubular fluid. Sodium delivery to the distal portion of the nephron, and thus the collecting duct, is reduced. Similarly, increased cardiac ANP release in these conditions requires optimum sodium concentration at the site of its action in the collecting duct for its desired natriuretic effects. Decreased sodium delivery to the collecting duct is therefore the most likely explanation for the persistent aldosterone-mediated sodium retention, absence of escape phenomenon and resistance to natriuretic peptides in these patients (Fig. 13.3b).
Regulation of water excretion



The cortical collecting duct has two cell types (see also p. 597) with very different functions:
 Principal cells (about 65%) have sodium and potassium channels in the apical membrane and, as in all sodium-reabsorbing cells, Na+/K+-ATPase pumps in the basolateral membrane.
Principal cells (about 65%) have sodium and potassium channels in the apical membrane and, as in all sodium-reabsorbing cells, Na+/K+-ATPase pumps in the basolateral membrane.

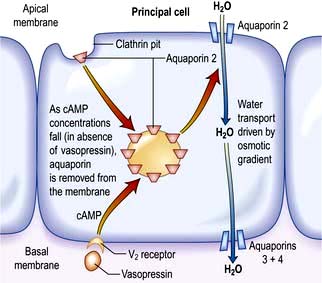
The ADH-induced increase in collecting duct water permeability occurs primarily in the principal cells. ADH acts on V2 (vasopressin) receptors located on the basolateral surface of principal cells, resulting in the activation of adenyl cyclase. This leads to protein kinase activation and to preformed cytoplasmic vesicles that contain unique water channels (called aquaporins) moving to and then being inserted into the luminal membrane. Four renal aquaporins have been well characterized and are localized in different areas of the cells of the collecting duct. The water channels span the luminal membrane and permit water movement into the cells down a favourable osmotic gradient (Fig. 13.5). This water is then rapidly returned to the systemic circulation across the basolateral membrane. When the ADH effect has worn off, the water channels aggregate within clathrin-coated pits, from which they are removed from the luminal membrane by endocytosis and returned to the cytoplasm. A defect in any step in this pathway, such as in attachment of ADH to its receptor or the function of the water channel, can cause resistance to the action of ADH and an increase in urine output. This disorder is called nephrogenic diabetes insipidus.
Plasma osmolality
1. Ingestion of a water load leads to an initial reduction in the plasma osmolality, thereby diminishing the release of ADH. The ensuing reduction in water reabsorption in the collecting ducts allows the excess water to be excreted in a dilute urine.
2. Water loss resulting from sweating is followed by, in sequence, a rise in both plasma osmolality and ADH secretion, enhanced water reabsorption, and the appropriate excretion of a small volume of concentrated urine. This renal effect of ADH minimizes further water loss but does not replace the existing water deficit. Thus, optimal osmoregulation requires an increase in water intake, which is mediated by a concurrent stimulation of thirst. The importance of thirst can also be illustrated by studies in patients with central diabetes insipidus, who are deficient in ADH. These patients often complain of marked polyuria, which is caused by the decline in water reabsorption in the collecting ducts. However, they do not typically become hypernatraemic, because urinary water loss is offset by the thirst mechanism.
Osmoregulation versus volume regulation
The roles of these two pathways should be considered separately when evaluating patients.
 A water load is rapidly excreted (in 4–6 h) by inhibition of ADH release so that there is little or no water reabsorption in the collecting ducts. This process is normally so efficient that volume regulation is not affected and there is no change in ANP release or in the activity of the renin-angiotensin-aldosterone system. Thus, a dilute urine is excreted, and there is little alteration in the excretion of Na+.
A water load is rapidly excreted (in 4–6 h) by inhibition of ADH release so that there is little or no water reabsorption in the collecting ducts. This process is normally so efficient that volume regulation is not affected and there is no change in ANP release or in the activity of the renin-angiotensin-aldosterone system. Thus, a dilute urine is excreted, and there is little alteration in the excretion of Na+.

This principle of separate volume and osmoregulatory pathways is also evident in the syndrome of inappropriate ADH secretion (SIADH). Patients with SIADH (see p. 993) have impaired water excretion and hyponatraemia (dilutional) caused by the persistent presence of ADH. However, the release of ANP and aldosterone is not impaired and, thus, Na+ handling remains intact. These findings have implications for the correction of the hyponatraemia in this setting which initially requires restriction of water intake.
Increased extracellular volume
 Venous tone, which determines the capacitance of the blood compartment and thus hydrostatic pressure
Venous tone, which determines the capacitance of the blood compartment and thus hydrostatic pressure



Causes
Heart failure
Reduction in cardiac output and the consequent fall in effective circulatory volume and arterial filling lead to activation of the renin-angiotensin-aldosterone system, non-osmotic release of ADH, and increased activity of the renal sympathetic nerves via volume receptors and baroreceptors (Fig. 13.3a). Sympathetic overdrive also indirectly augments ADH and renin-angiotensin-aldosterone response in these conditions. The cumulative effect of these mediators results in increased peripheral and renal arteriolar resistance and water and sodium retention. These factors result in extracellular volume expansion and increased venous pressure, causing oedema formation.
Hepatic cirrhosis
The mechanism is complex, but involves peripheral vasodilatation (possibly owing to increased nitric oxide generation) resulting in reduced effective arterial blood volume (EABV) and arterial filling. This leads to an activation of a chain of events common to cardiac failure and other conditions with marked peripheral vasodilatation (Fig. 13.3). The cumulative effect results in increased peripheral and renal resistance, water and sodium retention, and oedema formation.
Nephrotic syndrome
Interstitial oedema is a common clinical finding with hypoalbuminaemia, particularly in the nephrotic syndrome. Expansion of the interstitial compartment is secondary to the accumulation of sodium in the extracellular compartment. This is due to an imbalance between oral (or parenteral) sodium intake and urinary sodium output, as well as alterations of fluid transfer across capillary walls. The intrarenal site of sodium retention is the cortical collecting duct (CCD) where Na+/K+-ATPase expression and activity are increased threefold along the basolateral surface (Fig. 13.4). In addition, amiloride-sensitive epithelial sodium channel activity is also increased in the CCD. The renal sodium retention should normally be counterbalanced by increased secretion of sodium in the inner medullary collecting duct, brought about by the release of ANP. This regulatory pathway is altered in patients with nephrotic syndrome by enhanced kidney specific catabolism of cyclic GMP (the second messenger for ANP) following phosphodiesterase activation.
Oedema generation was classically attributed to the decrease in the plasma oncotic pressure and the subsequent increase in the transcapillary oncotic gradient. However, the oncotic pressure and transcapillary oncotic gradient remain unchanged and the transcapillary hydrostatic pressure gradient is not altered. Conversely, capillary hydraulic conductivity (a measure of permeability) is increased. This is determined by intercellular macromolecular complexes between the endothelial cells consisting of tight junctions (made of occludins, claudins and ZO proteins) and adherens junctions (made of cadherin, catenins and actin cytoskeleton). Elevated TNF-α levels in nephrotic syndrome activate protein kinase C, which changes phosphorylation of occludin and capillary permeability. In addition, increased circulating ANP can increase capillary hydraulic conductivity by altering the permeability of intercellular junctional complexes. Furthermore, reduction in effective circulatory volume and the consequent fall in cardiac output and arterial filling can lead to a chain of events as in cardiac failure and cirrhosis (see above and Fig. 13.3). These factors result in extracellular volume expansion and oedema formation.
Sodium retention
A decreased GFR decreases the renal capacity to excrete sodium. This may be acute, as in the acute nephritic syndrome (see p. 582), or may occur as part of the presentation of chronic kidney disease. In end-stage renal failure, extracellular volume is controlled by the balance between salt intake and its removal by dialysis.
 Oestrogens cause mild sodium retention, due to a weak aldosterone-like effect. This is the cause of weight gain in the premenstrual phase.
Oestrogens cause mild sodium retention, due to a weak aldosterone-like effect. This is the cause of weight gain in the premenstrual phase.


 Thiazolidinediones (TZD) (see p. 1011) are widely used to treat type 2 diabetes. Their mechanism of action is attributed to binding and activation of the PPAR-γ system. PPARs are nuclear transcription factors essential to the control of energy metabolism that are modulated via binding with tissue-specific fatty acid metabolites. Of the three PPAR isoforms, γ has been extensively studied and is expressed at high levels in adipose and liver tissues, macrophages, pancreatic-β cells and principal cells of the collecting duct. These drugs have been asociated with salt and water retention and are contraindicated in patients with heart failure. Recent evidence suggests that TZD-induced oedema (like insulin) is also due to upregulation of epithelial Na transporter channel (ENaC) but by different pathways. Diuretics of choice for TZD-induced oedema are amiloride and triamterene.
Thiazolidinediones (TZD) (see p. 1011) are widely used to treat type 2 diabetes. Their mechanism of action is attributed to binding and activation of the PPAR-γ system. PPARs are nuclear transcription factors essential to the control of energy metabolism that are modulated via binding with tissue-specific fatty acid metabolites. Of the three PPAR isoforms, γ has been extensively studied and is expressed at high levels in adipose and liver tissues, macrophages, pancreatic-β cells and principal cells of the collecting duct. These drugs have been asociated with salt and water retention and are contraindicated in patients with heart failure. Recent evidence suggests that TZD-induced oedema (like insulin) is also due to upregulation of epithelial Na transporter channel (ENaC) but by different pathways. Diuretics of choice for TZD-induced oedema are amiloride and triamterene.
Other causes of oedema
 Initiation of insulin treatment for type 1 diabetes and refeeding after malnutrition are both associated with the development of transient oedema. The mechanism is complex but involves upregulation of ENaC in the principal cell of the collecting duct. This transporter is amiloride sensitive which makes amiloride or triamterene the diuretic of choice in insulin-induced oedema.
Initiation of insulin treatment for type 1 diabetes and refeeding after malnutrition are both associated with the development of transient oedema. The mechanism is complex but involves upregulation of ENaC in the principal cell of the collecting duct. This transporter is amiloride sensitive which makes amiloride or triamterene the diuretic of choice in insulin-induced oedema.

 Oedema is also caused by increased interstitial oncotic pressure as a result of increased capillary permeability to proteins. This can occur as part of a rare complement-deficiency syndrome; with therapeutic use of interleukin 2 in cancer chemotherapy; or in ovarian hyperstimulation syndrome (see p. 981).
Oedema is also caused by increased interstitial oncotic pressure as a result of increased capillary permeability to proteins. This can occur as part of a rare complement-deficiency syndrome; with therapeutic use of interleukin 2 in cancer chemotherapy; or in ovarian hyperstimulation syndrome (see p. 981).
Treatment
The mainstay of treatment is the use of diuretic agents, which increase sodium, chloride and water excretion in the kidney (Table 13.3). These agents act by interfering with membrane ion pumps which are present on numerous cell types; they mostly achieve specificity for the kidney by being secreted into the proximal tubule, resulting in much higher concentrations in the tubular fluid than in other parts of the body.
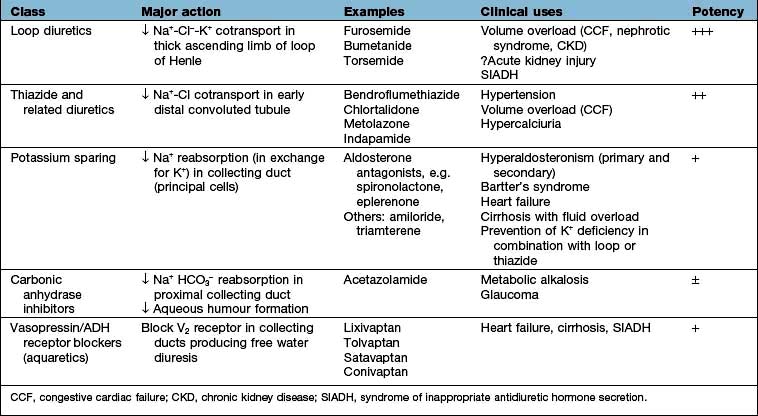
Clinical use of diuretics
Loop diuretics
These potent diuretics are useful in the treatment of any cause of systemic extracellular volume overload. They stimulate excretion of both sodium chloride and water by blocking the sodium-potassium-2-chloride (NKCC2) channel in the thick ascending limb of Henle (Fig. 13.6) and are useful in stimulating water excretion in states of relative water overload. They also act by causing increased venous capacitance, resulting in rapid clinical improvement in patients with left ventricular failure, preceding the diuresis. Unwanted effects include:




 Allergic tubulointerstitial nephritis and other allergic reactions
Allergic tubulointerstitial nephritis and other allergic reactions
 Myalgia – especially with high-dose bumetanide
Myalgia – especially with high-dose bumetanide

 Interference with excretion of lithium, resulting in toxicity.
Interference with excretion of lithium, resulting in toxicity.
Thiazide diuretics (see p. 719)
These are less potent than loop diuretics. They act by blocking a sodium chloride channel in the distal convoluted tubule (Fig. 13.7). They cause relatively more urate retention, glucose intolerance and hypokalaemia than loop diuretics. They interfere with water excretion and may cause hyponatraemia, particularly if combined with amiloride or triamterene. This effect is clinically useful in diabetes insipidus. Thiazides reduce peripheral vascular resistance by mechanisms that are not completely understood but do not appear to depend on their diuretic action, and are widely used in the treatment of essential hypertension. They are also used extensively in mild to moderate cardiac failure. Thiazides reduce calcium excretion. This effect is useful in patients with idiopathic hypercalciuria, but may cause hypercalcaemia. Numerous agents are available, with varying half-lives but little else to choose between them. Metolazone is not dependent for its action on glomerular filtration, and therefore retains its potency in renal impairment.
Potassium-sparing diuretics (see Fig. 13.8)
 Aldosterone antagonists, which compete with aldosterone in the collecting ducts and reduce sodium absorption, e.g. spironolactone and eplerenone (which has a shorter half-life). Spironolactone is used in patients with heart failure because it significantly reduces the mortality in these patients by antagonizing the fibrotic effect of aldosterone on the heart. Eplerenone is devoid of antiandrogenic or antiprogesterone properties.
Aldosterone antagonists, which compete with aldosterone in the collecting ducts and reduce sodium absorption, e.g. spironolactone and eplerenone (which has a shorter half-life). Spironolactone is used in patients with heart failure because it significantly reduces the mortality in these patients by antagonizing the fibrotic effect of aldosterone on the heart. Eplerenone is devoid of antiandrogenic or antiprogesterone properties.

Aquaretics (vasopressin or antidiuretic hormone antagonists)
Vasopressin V2 receptor antagonists are very useful agents in the treatment of conditions associated with elevated levels of vasopressin, such as heart failure, cirrhosis and SIADH (see p. 993). Non-peptide vasopressin V2 receptor antagonists are efficacious in producing free water diuresis in humans. Studies in patients with heart failure and cirrhosis suggest that such agents will allow normalization of serum osmolality with less water restriction (see p. 650).
Resistance to diuretics






Decreased extracellular volume
Clinical features
 Loss of interstitial fluid leads to loss of skin elasticity (’turgor’) – the rapidity with which the skin recoils to normal after being pinched. Skin turgor decreases with age, particularly at the peripheries. The turgor over the anterior triangle of the neck or on the forehead is a very useful sign in all ages.
Loss of interstitial fluid leads to loss of skin elasticity (’turgor’) – the rapidity with which the skin recoils to normal after being pinched. Skin turgor decreases with age, particularly at the peripheries. The turgor over the anterior triangle of the neck or on the forehead is a very useful sign in all ages.

Loss of more than this causes the following:
Postural hypotension
Normally the blood pressure rises if a subject stands up, as a result of increased venous return due to venoconstriction (this maintains cerebral perfusion). Loss of extracellular fluid (underfill) prevents this and causes a fall in blood pressure. This is one of the earliest and most reliable signs of volume depletion, as long as the other causes of postural hypotension are excluded (Table 13.4).
Table 13.4 Postural hypotension: some causes of a fall in blood pressure from lying to standing



Investigations
 Box 13.1
Box 13.1





Treatment
The overriding principle is to replace what is missing.
Loss of plasma
Loss of plasma, as occurs in burns or severe peritonitis, should be treated with human plasma or a plasma substitute (see p. 390).
Loss of water and electrolytes
Sodium bicarbonate (500 mg, 6 mmol each of Na+ and HCO3− per tablet) is used in doses of 6–12 tablets/day with 2–3 L of water. This is used in milder chronic sodium depletion with acidosis (e.g. chronic kidney disease, post-obstructive renal failure, renal tubular acidosis). Sodium bicarbonate is less effective than sodium chloride in causing positive sodium balance. Oral rehydration solutions are described in Box 4.10.
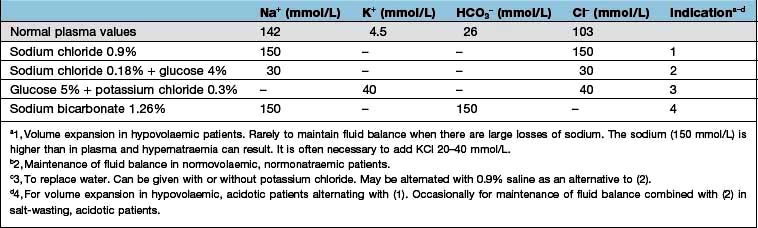
Intravenous fluids are sometimes required (Table 13.6). Rapid infusion (e.g. 1000 mL per hour or even faster) is necessary if there is hypotension and evidence of impaired organ perfusion (e.g. oliguria, confusion); in these situations, plasma expanders (colloids) are often used in the first instance to restore an adequate circulating volume (see p. 887). Repeated clinical assessments are vital in this situation, usually complemented by frequent measurements of central venous pressure (see p. 872, for the management of shock). Severe hypovolaemia induces venoconstriction, which maintains venous return; over-rapid correction does not give time for this to reverse, resulting in signs of circulatory overload (e.g. pulmonary oedema) even if a total body extracellular fluid (ECF) deficit remains. In less severe ECF depletion (such as in a patient with postural hypotension complicating acute tubular necrosis), the fluid should be replaced at a rate of 1000 mL every 4–6 h, again with repeated clinical assessment. If all that is required is avoidance of fluid depletion during surgery, 1–2 L can be given over 24 h, remembering that surgery is a stimulus to sodium and water retention and that over-replacement may be as dangerous as under-replacement. Regular monitoring by fluid balance charts, bodyweight and plasma biochemistry is mandatory.
Loss of water alone
FURTHER READING
Ahmed MS, Wong CF, Pai P. Cardiorenal syndrome – a new classification and current evidence on its management. Clin Nephrol 2010; 74(4):245–257.
Bie P. Blood volume, blood pressure and total body sodium: internal signaling and output control. Acta Physiol (Oxford) 2009; 195(1):187–196.
Bie P, Damkjaer M. Renin secretion and total body sodium: pathways of integrative control. Clin Exp Pharmacol Physiol 2010; 37(2):e34–42.
Schrier RW. Molecular mechanisms of clinical concentrating and diluting disorders. Prog Brain Res 2008; 170:539–550.
Wakil A, Atkin SL. Serum sodium disorders: safe management. Clin Med 2010; 10:79–82.
Disorders of sodium concentration



Table 13.8 Causes of hyponatraemia with normal extracellular volume (euvolaemia)
Table 13.9 Causes of hyponatraemia with increased extracellular volume (hypervolaemia)
|
Heart failure |
Oliguric kidney injury |
|
Liver failure |
Hypoalbuminaemia |
Hyponatraemia with hypovolaemia
This is due to salt loss in excess of water loss; the causes are listed in Table 13.7. In this situation, ADH secretion is initially suppressed (via the hypothalamic osmoreceptors); but as fluid volume is lost, volume receptors override the osmoreceptors and stimulate both thirst and the release of ADH. This is an attempt by the body to defend circulating volume at the expense of osmolality.
Clinical features
With sodium depletion the clinical picture is usually dominated by features of volume depletion (see p. 638). The diagnosis is usually obvious where there is a history of gut losses, diabetes mellitus or diuretic abuse. Examination of the patient is often more helpful than the biochemical investigations, which include plasma and urine electrolytes and osmolality.
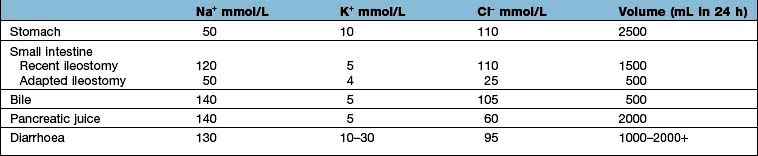
Table 13.10 shows the potential daily losses of water and electrolytes from the gut. Losses due to renal or adrenocortical disease may be less easily identified but a urinary sodium concentration of >20 mmol/L, in the presence of clinically evident volume depletion, suggests a renal loss.




Hyponatraemia with euvolaemia (see Table 13.8)
 With normal kidney function, dilution hyponatraemia is uncommon even if a patient drinks approximately 1 L per hour.
With normal kidney function, dilution hyponatraemia is uncommon even if a patient drinks approximately 1 L per hour.


 Marathon runners drinking excess water and ‘sports drinks’ can become hyponatraemic.
Marathon runners drinking excess water and ‘sports drinks’ can become hyponatraemic.

Clinical features



Investigations
The cause of hyponatraemia with apparently normal extracellular volume requires investigation:
 Plasma and urine electrolytes and osmolalities. The plasma concentrations of sodium, chloride and urea are low, giving a low osmolality. The urine sodium concentration is usually high and the urine osmolality is typically higher than the plasma osmolality. However, maximal dilution (<50 mosmol/kg) is not always present.
Plasma and urine electrolytes and osmolalities. The plasma concentrations of sodium, chloride and urea are low, giving a low osmolality. The urine sodium concentration is usually high and the urine osmolality is typically higher than the plasma osmolality. However, maximal dilution (<50 mosmol/kg) is not always present.

Treatment




Osmotic demyelination syndrome (ODS)
Avoiding ODS
In the absence of vasopressin, it is generally assumed that the total urine volume is equal to the volume of filtrate delivered to the distal nephron, which is the GFR minus the volume reabsorbed in the proximal convoluted tubule (PCT). Approximately 80% of the GFR is reabsorbed in PCT under normal circumstance (increases even more in the presence of intravascular volume depletion). However, in real life water excretion will be less than the volume of distal delivery of filtrate, even in the absence of vasopressin, because a significant degree of water is reabsorbed in the inner medullary collecting duct through its residual water permeability, prompted by a very high osmotic force in the interstitium (see Fig. 12.2).
Antidiuretic hormone antagonists (vasopressin antagonists)
Vasopressin V2 receptor antagonists (see p. 645), which produce a free water diuresis, are being used in clinical trials for the treatment of hyponatraemic encephalopathy. Three oral agents, lixivaptan, tolvaptan and satavaptan, are selective for the V2 (antidiuretic) receptor, while conivaptan blocks both the V1A and V2 receptors.
FURTHER READING
Nemerovski C, Hutchinson DJ. Treatment of hypervolemic or euvolemic hyponatremia associated with heart failure, cirrhosis, or the syndrome of inappropriate antidiuretic hormone with tolvaptan: a clinical review. Clin Ther 2010; 32(6):1015–1032.
Rozen-Zvi B, Yahav D, Gheorghiade M et al. Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta-analysis. Am J Kidney Dis 2010; 56(2):325–337.
Hyponatraemia with hypervolaemia
The common causes of hyponatraemia due to water excess are shown in Table 13.9. In all these conditions, there is usually an element of reduced glomerular filtration rate with avid reabsorption of sodium and chloride in the proximal tubule. This leads to reduced delivery of chloride to the ‘diluting’ ascending limb of Henle’s loop and a reduced ability to generate ‘free water’, with a consequent inability to excrete dilute urine. This is commonly compounded by the administration of diuretics that block chloride reabsorption and interfere with the dilution of filtrate either in Henle’s loop (loop diuretics) or distally (thiazides).
Hypernatraemia
This is much rarer than hyponatraemia and nearly always indicates a water deficit. Causes are listed in Table 13.11).
|
|
Treatment
Treatment is that of the underlying cause, e.g.


If there is clinical evidence of volume depletion (see p. 646), this implies that there is a sodium deficit as well as a water deficit. Treatment of this is discussed on page 647.
Disorders of potassium concentration
Regulation of serum potassium concentration







 acidosis – K+ exchanged for H+ across cell membrane
acidosis – K+ exchanged for H+ across cell membrane
 cell damage or cell death – resulting in massive K+ release.
cell damage or cell death – resulting in massive K+ release.
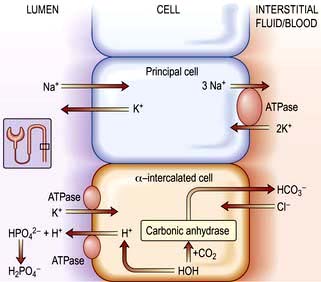
Renal excretion of potassium is increased by aldosterone, which stimulates K+ and H+ secretion in exchange for Na+ in the principal cells of the collecting duct (Fig. 13.8). Because H+ and K+ are interchangeable in the exchange mechanism, acidosis decreases and alkalosis increases the secretion of K+. Aldosterone secretion is stimulated by hyperkalaemia and increased angiotensin II levels, as well as by some drugs, and this acts to protect the body against hyperkalaemia and against extracellular volume depletion. The body adapts to dietary deficiency of potassium by reducing aldosterone secretion. However, because aldosterone is also influenced by volume status, conservation of potassium is relatively inefficient, and significant potassium depletion may therefore result from prolonged dietary deficiency.
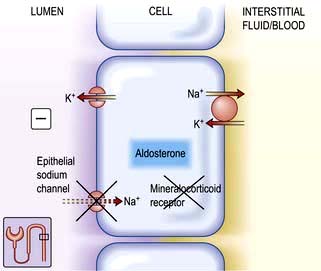
Figure 13.8 Aldosterone-regulated transport in the cortical collecting ducts. Under normal conditions, the epithelial sodium channel is the rate-limiting barrier for the normal entry of sodium from the lumen into the cell. The resulting lumen-negative transepithelial voltage (indicated by the minus sign) drives potassium secretion from the principal cells and proton secretion from the α-intercalated cells (see Fig. 13.11). In Liddle’s syndrome, a mutation in the gene encoding the epithelial sodium channel results in persistent unregulated reabsorption of sodium and increased secretion of potassium (not shown). In pseudohypoaldosteronism type I autosomal recessive, loss of function mutations (X) in this gene inactivate the channel. In the autosomal dominant variety, the mutation is in the gene encoding the mineralocorticoid regulation of the activity of the epithelial sodium channel. Either mechanism reduces the activity of the epithelial sodium channel, thus causing salt wasting and decreasing the secretion of potassium and protons.
Hypokalaemia
Common causes
Causes
The most common causes of chronic hypokalaemia are diuretic treatment (particularly thiazides) and hyperaldosteronism. Acute hypokalaemia is often caused by intravenous fluids without potassium and redistribution into cells. The common causes are shown in Table 13.12.
Rare causes
Bartter’s syndrome (clinically similar to loop diuretics)
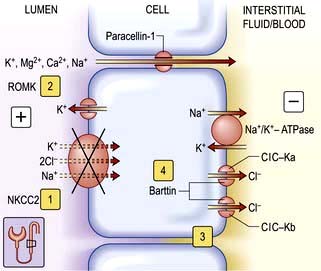
This consists of metabolic alkalosis, hypokalaemia, hypercalciuria, occasionally hypomagnesaemia (see p. 657), normal blood pressure, and an elevated plasma renin and aldosterone. The primary defect in this disorder is an impairment in sodium and chloride reabsorption in the thick ascending limb of the loop of Henle (Fig. 13.6). Mutation in the genes encoding either the sodium-potassium-2-chloride cotransporter (NKCC2), the ATP-regulated renal outer medullary potassium channel (ROMK) or kidney-specific basolateral chloride channels (ClC-Kb) – Bartter types I, II and III, respectively – causes loss of function of these channels, with consequent impairment of sodium and chloride reabsorption. There is also an increased intrarenal production of prostaglandin E2 which is secondary to sodium and volume depletion, hypokalaemia and the consequent neurohumoral response rather than a primary defect. PGE2 causes vasodilatation and may explain why the blood pressure remains normal.
Treatment is with combinations of potassium supplements, amiloride and indomethacin.
Gitelman’s syndrome (similar to thiazide diuretics)
Gitelman’s syndrome is a phenotype variant of Bartter’s syndrome characterized by hypokalaemia, metabolic alkalosis, hypocalciuria, hypomagnesaemia, normal blood pressure, and elevated plasma renin and aldosterone. There are striking similarities between the Gitelman’s syndrome and the biochemical abnormalities induced by chronic thiazide diuretic administration. Thiazides act in the distal convoluted tubule to inhibit the function of the apical sodium-chloride cotransporter (NCCT) (Fig. 13.7). Analysis of the gene encoding the NCCT has identified loss of function mutations in Gitelman’s syndrome.
Like Bartter’s syndrome, defective NCCT function leads to increased solute delivery to the collecting duct, with resultant solute wasting, volume contraction and an aldosterone-mediated increase in potassium and hydrogen secretion. Unlike Bartter’s syndrome, the degree of volume depletion and hypokalaemia is not sufficient to stimulate prostaglandin E2 production. Impaired function of NCCT is predicted to cause hypocalciuria, as does thiazide administration. Impaired sodium reabsorption across the apical membrane, coupled with continued intracellular chloride efflux across the basolateral membrane, causes the cell to become hyperpolarized. This in turn stimulates calcium reabsorption via apical, voltage-activated calcium channels. Decreased intracellular sodium also facilitates calcium efflux via the basolateral sodium-calcium exchanger. The mechanism for urinary magnesium losses is described on page 656.
Liddle’s syndrome
This is characterized by potassium wasting, hypokalaemia and alkalosis, but is associated with low renin and aldosterone production, and high blood pressure. There is a mutation in the gene encoding for the amiloride-sensitive epithelial sodium channel in the distal tubule/collecting duct. This leads to constitutive activation of the epithelial sodium channel, resulting in excessive sodium reabsorption with coupled potassium and hydrogen secretion. Unregulated sodium reabsorption across the collecting tubule results in volume expansion, inhibition of renin and aldosterone secretion and development of low renin hypertension (Fig. 13.8).
Clinical features
Hypokalaemia is usually asymptomatic, but severe hypokalaemia (<2.5 mmol) causes muscle weakness. Potassium depletion may also cause symptomatic hyponatraemia (see p. 648).
Treatment
The underlying cause should be identified and treated where possible. Table 13.13 shows some examples.
| Cause | Treatment |
|---|---|
|
Dietary deficiency |
Increase intake of fresh fruit/vegetables or oral potassium supplements (20–40 mmol daily). (Potassium supplements can cause gastrointestinal irritation) |
|
Hyperaldosteronism, e.g. cirrhosis, thiazide therapy |
Spironolactone/eplerenone. Co-prescription of a potassium-sparing diuretic with a similar onset and duration of action |
|
Intravenous fluid replacement |
Add 20 mmol of K+/L of fluid with monitoring |
The treatment of adrenal disorders is described on page 958.
Hyperkalaemia
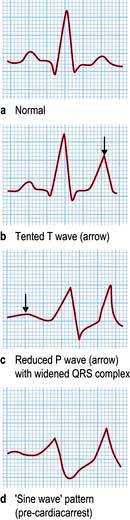
Causes
Acute self-limiting hyperkalaemia occurs normally after vigorous exercise and is of no pathological significance. Hyperkalaemia in all other situations is due either to increased release from cells or to failure of excretion (Table 13.14). The most common causes are renal impairment and drug interference with potassium excretion. The combination of ACE inhibitors with potassium-sparing diuretics or NSAIDs is particularly dangerous.
Rare causes
Hyporeninaemic hypoaldosteronism
This is also known as type 4 renal tubular acidosis (see p. 664). Hyperkalaemia occurs because of acidosis and hypoaldosteronism.
Pseudohypoaldosteronism type 1 (autosomal recessive and dominant types)
This is a disease of infancy, apparently due to resistance to the action of aldosterone. It is characterized by hyperkalaemia and evidence of sodium wasting (hyponatraemia, extracellular volume depletion). Autosomal recessive forms result from loss of function because of mutations in the gene for epithelial sodium channel activity (opposite to Liddle’s syndrome). This disorder involves multiple organ systems and is especially marked in the neonatal period. With aggressive salt replacement and control of hyperkalaemia, these children can survive and the disorder appears to become less severe with age. The autosomal dominant type is due to mutations affecting the mineralocorticoid receptor (Fig. 13.8). These patients present with salt wasting and hyperkalaemia but do not have other organ-system involvement.
Gordon’s syndrome (familial hyperkalaemic hypertension, pseudohypoaldosteronism type 2)
This appears to be a mirror image of Gitelman’s syndrome (see p. 653), in which primary renal retention of sodium causes hypertension, volume expansion, low renin/aldosterone, hyperkalaemia and metabolic acidosis. There is also an increased sensitivity of sodium reabsorption to thiazide diuretics, suggesting that the thiazide-sensitive sodium-chloride cotransporter (NCCT) is involved. Genetic analyses, however, have excluded abnormalities in NCCT. The involvement of two loci on chromosomes 1 and 12 and further genetic heterogeneity has also been found. These genes do not correspond to ionic transporters but to unexpected proteins, WNK (With No lysine Kinase) 1 and WNK 4, which are two closely related members of a novel serine–threonine kinase family. WNK 4 normally inhibits NCCT by preventing its membrane translocation from the cytoplasm. Loss of function mutation in WNK 4 results in escape of NCCT from normal inhibition and its overactivity as seen from the patient’s phenotype. WNK 1 is an inhibitor of WNK 4 and in some patients with Gordon’s syndrome, gain of function mutation in WNK 1 results in functional deficiency of WNK 4 and overactivity of NCCT.
Treatment
Treatment for severe hyperkalaemia requires both urgent measures to save lives and maintenance therapy to keep potassium down, as summarized in Emergency Box 13.1. The cause of the hyperkalaemia should be found and treated.
 Emergency Box 13.1
Emergency Box 13.1
Correction of severe hyperkalaemia


FURTHER READING
Bubien JK. Epithelial Na+ channel (ENaC), hormones, and hypertension. J Biol Chem 2010; 285(31):23527–23531.
Flatman PW. Co-transporters, WNKs and hypertension: an update. Curr Opin Nephrol Hypertens 2008; 17(2):186–192.
Furgeson SB, Linas S. Mechanisms of type I and type II pseudohypoaldosteronism. J Am Soc Nephrol 2010; 21(11):1842–1845.
Wang WH, Yue P, Sun P et al. Regulation and function of potassium channels in aldosterone-sensitive distal nephron. Curr Opin Nephrol Hypertens 2010; 19(5):463–470.
Disorders of magnesium concentration
Renal handling of magnesium
Cortical thick ascending limb of Henle (cTAL)
Approximately 30% of Mg2+ is bound to plasma proteins but the remaining fraction is freely filterable. The major site of magnesium transport is the cortical thick ascending limb (cTAL) of the loop of Henle, where 65–70% of the filtered load is reabsorbed with only 10–20% being reabsorbed in the proximal tubule (Fig. 13.6). This transport is passive, paracellular and carried out by tight junction proteins (paracellin-1 and claudins). This process is driven by the lumen-positive electrochemical gradient, characteristic of this segment. This voltage gradient is created by the apical disproportionate net transport of two Cl− to one Na+ (by the bumetanide-sensitive sodium-potassium-2-chloride transporter) and the secretion of K+ (via the ROMK) (see Fig. 13.6). Loss of function mutations in these key reabsorptive processes lead to hypomagnesaemia as part of distinctive clinical syndromes described below.
Bartter’s syndrome (p. 652). Hypomagnesaemia is not present in all patients with Bartter’s syndrome because expected dissipation of the luminal positive voltage gradient is prevented by lack of dilution of tubular fluid which maintains transepithelial voltage in the normal range. Compensatory increased absorption of Mg2+ in the distal convoluted tubule (DCT) can also partly prevent hypomagnesaemia in this condition.
Familial hypomagnesaemia, hypercalciuria and nephrocalcinosis (FHHNC) is characterized by excessive renal magnesium and calcium wasting. Individuals develop bilateral nephrocalcinosis and progressive chronic kidney disease. Patients also have elevated PTH levels, which precedes any reduction in GFR. A substantial proportion of patients show incomplete distal renal tubular acidosis, hypocitraturia and hyperuricaemia. Extrarenal involvement such as myopia, nystagmus, chorioretinitis has been reported. The main defect in magnesium and calcium reabsorption lies in cTAL. Ten different mutations have been identified in a novel gene which encodes for paracellin-1 and claudins 16/19 complex, members of the claudin family of tight junction proteins (see p. 23).
Distal convoluted tubule (DCT)
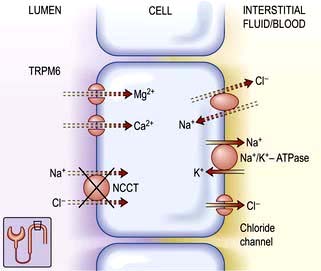
The reabsorption rate in the DCT (10%) is much lower than in the cTAL, but it defines the final urinary excretion, as there is no significant reabsorption in the collecting duct: 3–5% of filtered magnesium is finally excreted in the urine. Magnesium reabsorption in the DCT is transcellular and active (Fig. 13.7). The DCT has a slight lumen-negative voltage of approximately −5 mV. The luminal Mg2+ concentration in the DCT ranges between 0.2 and 0.7 mmol/L, whereas the intracellular concentration of Mg2+ is estimated to be maintained around 0.2–1.0 mmol/L. Therefore the voltage difference across the apical membrane plays a key role in Mg2+ transport within the DCT. Magnesiotropic proteins, including the transient receptor potential channel melastatin member 6 (TRPM6), the pro-epidermal growth factor (EGF), the potassium channels Kir4.1 as well as the hepatocyte nuclear factor 1B (HNF1B) are situated in DCT.
Thiazide-sensitive Na+–Cl− cotransporter in the DCT plays a role in sodium and chloride absorption and maintenance of lumen-negative voltage. Loss of function mutation in this cotransporter results in Gitelman’s syndrome (p. 645; see Fig. 13.7). Hypomagnesaemia is likely due to a reduced abundance of TRPM6. The observed hypocalciuria is caused by an increased proximal tubular reabsorption, a process that occurs in response to the mild volume depletion.
The HNF1b gene encodes a transcription factor linked to the regulation of the FXYD2 gene. Defects in HNF1b gene have been implicated in genetic defects of beta-cell function (p. 1007). Interestingly, almost half of the carriers of a mutation in the HNF1b gene display hypomagnesaemia (<0.65 mmol/L) due to renal wasting of Mg2+. As in patients with FXYD2 mutations, hypocalciuria is present.
Hypomagnesaemia
In addition to the familial causes above, hypomagnesaemia most often develops as a result of deficient intake, defective gut absorption, or excessive gut or urinary loss (Table 13.15). It can also occur with acute pancreatitis, possibly owing to the formation of magnesium soaps in the areas of fat necrosis. The serum magnesium is usually <0.7 mmol/L (1.4 mEq/L). The phenotypes can in many cases be mimicked by drug treatment such as aminoglycosides and cisplatinum compounds. Due to the severe effects of hypomagnesaemia, routine measurements of serum Mg2+ should be conducted in the critically ill as well as in patients who are exposed to drugs and other conditions associated with Mg2+ deficiency.
SIADH, syndrome of inappropriate antidiuretic hormone secretion.
Relationship between hypomagnesaemia and plasma calcium



Hypermagnesaemia
This primarily occurs in patients with acute or chronic kidney disease given magnesium-containing laxatives or antacids. It can also be induced by magnesium-containing enemas. Mild hypermagnesaemia may occur in patients with adrenal insufficiency. Causes are given in Table 13.16.
Treatment
FURTHER READING
Glaudemans B, van der Wiist J, Scola RH et al. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest 2009; 119:936–942.
San-Cristobal P, Dimke H, Hoenderop JG et al. Novel molecular pathways in renal Mg2+ transport: a guided tour along the nephron. Curr Opin Nephrol Hypertens 2010; 19(5):456–462.
Yang L, Frindt G, Palmer LG. Magnesium modulates ROMK channel–mediated potassium secretion. J Am Soc Nephrol 2010; 21:2109–2116.
Disorders of phosphate concentration
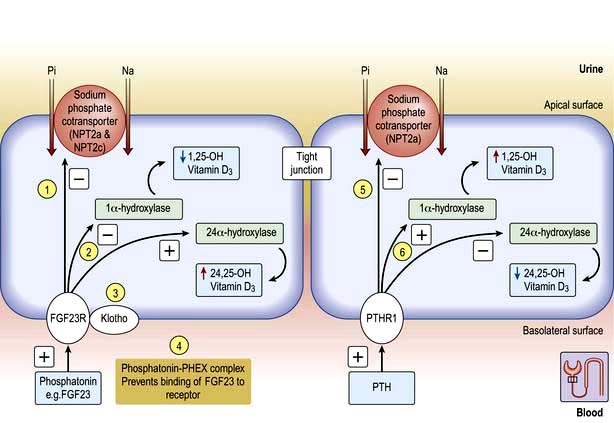
Osteoblast-secreted phosphaturic factors (phosphatonins) such as fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein (MEPG) and frizzled related protein 4 (FRP-4) play a role in phosphate homeostasis. FGF23 is the most extensively investigated phosphatonin which binds to its receptors FGFR1 in the kidney and causes phosphaturia and also regulates vitamin D by inactivation of 1α-hydroxylase (CYP27B1) and upregulation of 24 hydroxylase (CYP24A1) enzymes with the net result of low 1,25 vitamin D synthesis. Moreover, FGF23 requires Klotho (p. 652) to act as a coreceptor with FGFR1 for its activity. Loss of function mutation in either FGF23 or Klotho results in a similar phenotype of shortened lifespan, premature ageing (p. 37) including hyperphosphataemia and as expected from mode of action increased 1–25 vitamin D levels. Klotho can also inhibit phosphate absorption directly in the absence of FGF23 or PTH.
Phosphate absorption is an active process carried out by a family of sodium-phosphate cotransporters (NPT) in the gut and kidneys. NPT2a and NPT2c are expressed in the brush border of the renal proximal tubule whilst NPT2b is expressed in lungs and intestine. NPT2a plays a central role in the renal reabsorption of phosphate but essentially requires a companion protein called sodium hydrogen exchanger regulatory factor 1 (NHERF1) for membrane sorting. Intestinal absorption is carried out by NPT2b but its mutation does not cause any phosphate abnormalities because of the compensation taking place by the renal expression of NPT2a and possibly NPT2c. Under normal circumstances plasma phosphate levels are kept constant, e.g. after a phosphate-rich meal, the intestinal bone axis releases FGF23 from bone which inhibits NPT2a and causes phosphate excretion. Moreover, phosphate in the plasma either directly or indirectly by lowering ionized calcium causes the release of PTH which also inhibits NPT2a and NHERF1 resulting in phosphaturia (Fig. 13.9). These two principal mechanisms keep plasma phosphate levels within normal limits on a daily basis.
 Muscle weakness, e.g. diaphragmatic weakness, decreased cardiac contractility, skeletal muscle rhabdomyolysis
Muscle weakness, e.g. diaphragmatic weakness, decreased cardiac contractility, skeletal muscle rhabdomyolysis


Causes
Decreased renal reabsorption of phosphate
Excessive phosphatonins (FGF23)
Normal adaptive increases in 1,25-dihydroxyvitamin D3 synthesis in response to low phosphate levels do not occur in TIO, ADHR and XLR, aggravating phosphaturia (Fig. 13.9).
Hyperphosphataemia
Hyperphosphataemia is common in patients with CKD (see p. 618 and Table 13.18). Hyperphosphataemia is usually asymptomatic but may result in precipitation of calcium phosphate, particularly in the presence of a normal or raised calcium or of alkalosis. Uraemic itching may be caused by a raised calcium phosphate product. Prolonged hyperphosphataemia causes hyperparathyroidism, and periarticular and vascular calcification.
Usually, no treatment is required for acute hyperphosphataemia, as the causes are self-limiting. Treatment of chronic hyperphosphataemia is with gut phosphate binders and dialysis (see p. 622).
FURTHER READING
Markadieu N, Bindels RJ, Hoenderop JG. The renal connecting tubule: resolved and unresolved issues in Ca(2+) transport. Intl J Biochem Cell Biol 2011; 43(1):1–4.
Nakatani T, Sarraj B, Ohnishi M et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J 2009; 23:433–441.
Prié D, Friedlander G. Genetic disorders of renal phosphate transport. N Engl J Med 2010; 362:2399–2409.
Tiosano D, Hochberg Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab 2009; 27:392–401.
Acid–base disorders
The concentration of hydrogen ions in both extracellular and intracellular compartments is extremely tightly controlled, and very small changes lead to major cell dysfunction. The blood pH is tightly regulated and is normally maintained at between 7.38 and 7.42. Any deviation from this range indicates a change in the hydrogen ion concentration [H+] because blood pH is the negative logarithm of [H+] (Table 13.19). The [H+] at a physiological blood pH of 7.40 is 40 nmol/L. An increase in the [H+] – a fall in pH – is termed acidaemia. A decrease in [H+] – a rise in the blood pH – is termed alkalaemia. The disorders that cause these changes in the blood pH are acidosis and alkalosis, respectively.
| pH | [H+] (nmol/L) |
|---|---|
|
6.9 |
126 |
|
7.0 |
100 |
|
7.1 |
79 |
|
7.2 |
63 |
|
7.3 |
50 |
|
7.4 |
40 |
|
7.5 |
32 |
|
7.6 |
25 |
Normal acid–base physiology
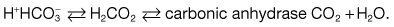
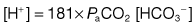
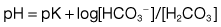
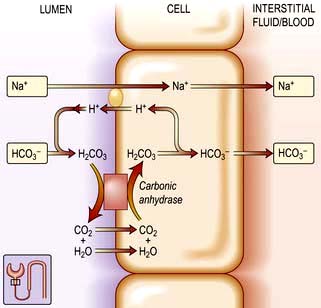
Renal reabsorption of bicarbonate
The plasma [HCO3−] is normally maintained at approximately 25 mmol/L. In individuals with a normal glomerular filtration rate (120 mL/min), about 4500 mmol of bicarbonate is filtered each day. If this filtered bicarbonate were not reabsorbed, the plasma [HCO3−] would fall, along with blood pH. Thus, maintenance of normal plasma [HCO3−] requires that essentially all of the bicarbonate in the glomerular filtrate be reabsorbed (Fig. 13.10).
Renal excretion of [H+] (Fig. 13.11)
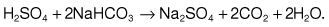



Buffer systems in acid excretion
Two buffer systems are involved in acid excretion: the titratable acids such as phosphate, and the ammonia system. Each system is responsible for excreting about half of the daily acid load of 50–100 mmol under physiological conditions (Fig. 13.11).
Titratable acid
A titratable acid is a filtered buffer substance having a conjugate anion that can be titrated within the pH range occurring physiologically in the urine. Phosphoric acid (pKa 6.8) is the usual titratable urinary buffer. Hydrogen ions bind to the conjugate anions of the titratable acids and are excreted in the urine. For each hydrogen ion excreted in this form, a bicarbonate ion is regenerated within the cell and returned to the blood (Fig. 13.11).
Ammonium (NH4+)
In the setting of metabolic acidosis, titratable acids cannot increase significantly because the availability of titratable acid is fixed by the plasma concentration of the buffer and by the GFR. The ammonia buffer system, by contrast, can increase several hundred-fold when necessary. Consequently, impaired renal excretion of hydrogen ions is always associated with a defect in ammonium excretion (Fig. 13.12).
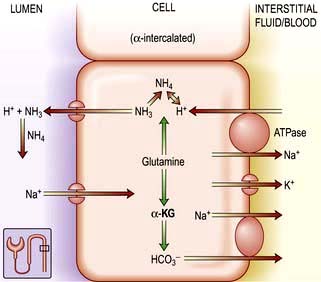
All ammonia used to buffer urinary hydrogen ions in the collecting tubule is synthesized in the proximal convoluted tubule. Glutamine is the primary source of ammonia. It undergoes deamination catalysed by glutaminase, resulting in α-ketoglutaric acid (Fig. 13.12) and ammonia. Once formed, ammonia can diffuse into the proximal tubule lumen and become acidified, forming ammonium. Once in the proximal tubule lumen, ammonium flows along the tubule to the thick ascending limb of Henle’s loop. Here, it is transported out of the tubule into the medullary interstitium. Ammonium then dissociates to ammonia, leading to a high interstitial ammonia concentration. The notion that ammonia diffuses down its concentration gradient into the lumen of the collecting tubule has recently been challenged by the discovery of rhesus (Rh) associated glycoproteins acting as ammonia transport proteins also called RhCG/Rhcg which are expressed in the basolateral and apical surfaces of DCT, inner medullary collecting duct and type A intercalated cells. These proteins play a fundamental role in renal ammonia excretion under both basal and acidosis states. Once secreted, NH3 reacts with the hydrogen ions secreted by the collecting tubular cells to form ammonium. Because ammonium (NH4) is not lipid-soluble, it is trapped in the lumen and excreted in the urine as ammonium chloride. Two conditions predominantly promote ammonia synthesis by the proximal tubular cell: systemic acidosis and hypokalaemia.
Causes of acid–base disturbance
Acid–base disturbance may be caused by:


Both may, and usually do, co-exist. For instance, metabolic acidosis causes hyperventilation (via medullary chemoreceptors, see p. 794), leading to increased removal of CO2 in the lungs and partial compensation for the acidosis. Conversely, respiratory acidosis is accompanied by renal bicarbonate retention, which could be mistaken for primary metabolic alkalosis. The situation is even more complex if a patient has both respiratory disease and a metabolic disturbance.
Diagnosis
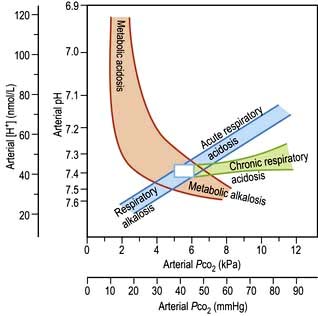
Clinical history and examination usually point to the correct diagnosis. Table 13.20 shows the typical blood changes, but in complicated patients the acid–base nomogram (Fig. 13.13) is invaluable. The [H+] and PaCO2 are measured in arterial blood (for precautions see p. 659) as well as the bicarbonate. If the values from a patient lie in one of the bands in the diagram, it is likely that only one abnormality is present. If the [H+] is high (pH low) but the PaCO2 is normal, the intercept lies between two bands: the patient has respiratory dysfunction, leading to failure of CO2 elimination, but this is partly compensated for by metabolic acidosis, stimulating respiration and CO2 removal (this is the most common ‘combined’ abnormality in practice).
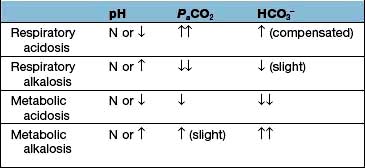
Respiratory acidosis and alkalosis
Metabolic acidosis




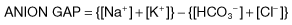
Metabolic acidosis with a normal anion gap
If the anion gap is normal in the presence of acidosis, this suggests that H+Cl− is being retained or that Na+HCO3− is being lost. Causes of a normal-anion-gap acidosis are given in Table 13.21. In these conditions, plasma bicarbonate decreases and is replaced by chloride to maintain electroneutrality. Consequently, these disorders are sometimes referred to collectively as hyperchloraemic acidoses.
Table 13.21 Causes of metabolic acidosis with a normal anion gap
Renal tubular acidosis (RTA)
Type 4 renal tubular acidosis
Also called ‘hyporeninaemic hypoaldosteronism’, this is probably the most common of these disorders. The cardinal features are hyperkalaemia and acidosis occurring in a patient with mild chronic kidney disease, usually caused by tubulo-interstitial disease (e.g. reflux nephropathy) or diabetes. Gordon’s syndrome (see p. 655) shares biochemical abnormalities but differs in having normal GFR and hypertension. Plasma renin and aldosterone are found to be low, even after measures which would normally stimulate their secretion. The features for the diagnosis are shown in Table 13.22. An identical syndrome is caused by chronic ingestion of NSAIDs, which impair renin and aldosterone secretion. In the presence of acidosis, urine pH may be low. Treatment is with fludrocortisone, sodium bicarbonate, diuretics, or ion exchange resins to remove potassium, or a combination of these. Dietary potassium restriction alone is ineffective.
Table 13.22 Features of hyporeninaemic hypoaldosteronism (type 4 renal tubular acidosis)
|
Low plasma bicarbonate and hyperchloraemia Normal ACTH stimulation test (p. 944) Low basal 24-hour urinary aldosterone Subnormal response of plasma renin and plasma aldosterone to stimulation |
Type 2 (‘proximal’) renal tubular acidosis
This is very rare in adult practice. It is caused by failure of sodium bicarbonate reabsorption in the proximal tubule. The cardinal features are acidosis, hypokalaemia, an inability to lower the urine pH below 5.5 despite systemic acidosis, and the appearance of bicarbonate in the urine despite a subnormal plasma bicarbonate. This disorder normally occurs as part of a generalized tubular defect, together with other features such as glycosuria and amino-aciduria. Inherited forms of isolated type 2 RTA are described as both autosomal dominant and recessive patterns of inheritance, where putative mutations are in the Na+-H+ antiporter in the apical membrane and Na+-HCO3− cotransporter in the basolateral membrane of proximal tubular cells respectively (see Fig. 13.10). Treatment is with sodium bicarbonate: massive doses may be required to overcome the renal ‘leak’.
Type 1 (’distal’) renal tubular acidosis
This is due to a failure of H+ excretion in the distal tubule (Table 13.23). It consists of:




Table 13.23 Causes of distal renal tubular acidosis (type 1 RTA)
a May also cause proximal renal tubular acidosis.
These features may be present only in the face of increased acid production; hence the need for an acid load test in diagnosis (Practical Box 13.1). Other features include:
 Low urinary citrate (owing to increased citrate absorption in the proximal tubule where it can be converted to bicarbonate)
Low urinary citrate (owing to increased citrate absorption in the proximal tubule where it can be converted to bicarbonate)

 Practical Box 13.1
Practical Box 13.1



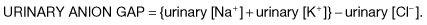
Metabolic acidosis with a high anion gap
If the anion gap is increased, there is an unmeasured anion present in increased quantities. This is either one of the acids normally present in small, but unmeasured quantities, such as lactate, or an exogenous acid. Causes of a high-anion-gap acidosis are given in Table 13.24.
Table 13.24 Causes of metabolic acidosis with an increased anion gap
|
Kidney failure (serum sulphate and phosphate) Accumulation of organic acids Type B – decreased hepatic lactate metabolism deficiency (decreased pyruvate dehydrogenase activity)
|
Lactic acidosis
Increased lactic acid production occurs when cellular respiration is abnormal, because of either a lack of oxygen in the tissues (‘type A’) or a metabolic abnormality, such as drug-induced (‘type B’) (Table 13.24). The most common cause in clinical practice is type A lactic acidosis, occurring in septic or cardiogenic shock. Significant acidosis can occur despite a normal blood pressure and PaCO2, owing to splanchnic and peripheral vasoconstriction. Acidosis worsens cardiac function and vasoconstriction further, contributing to a downward spiral and fulminant production of lactic acid.
Mixed metabolic acidosis
Clinical features
Acidosis increases delivery of oxygen to the tissues by shifting the oxyhaemoglobin dissociation curve to the right, but it also leads to inhibition of 2,3-BPG production, which returns the curve towards normal (see p. 870). Cardiovascular dysfunction is common in acidotic patients, although it is often difficult to dissociate the numerous possible causes of this. Acidosis is negatively inotropic. Severe acidosis also causes venoconstriction, resulting in redistribution of blood from the peripheries to the central circulation, and increased systemic venous pressure, which may worsen pulmonary oedema caused by myocardial depression. Arteriolar vasodilatation also occurs, further contributing to hypotension.







Metabolic alkalosis
Classification and definitions
Metabolic alkalosis has been classified on the basis of underlying pathophysiology (Table 13.25).
Chloride may be lost from the gut, kidney or skin. The loss of gastric fluid rich in acid results in alkalosis because bicarbonate generated during the production of gastric acid returns to the circulation. In Zollinger–Ellison syndrome (see p. 370) or gastric outflow obstruction these losses can be massive. Although sodium and potassium loss in the gastric juice is variable, the obligate urinary loss of these cations is intensified by bicarbonaturia, which occurs during disequilibrium.




Clinical features
The symptoms of metabolic alkalosis per se are difficult to separate from those of chloride, volume or potassium depletion. Tetany (see p. 997), apathy, confusion, drowsiness, cardiac arrhythmias and neuromuscular irritability are common when alkalosis is severe. The oxyhaemoglobin dissociation curve is shifted to the left. Respiration may be depressed.
Treatment
Chloride-responsive metabolic alkalosis
Although replacement of the chloride deficit is essential in chloride depletion states, selection of the accompanying cation – sodium, potassium or proton – is dependent on the assessment of extracellular fluid volume status (see p. 646), the presence or absence of associated potassium depletion, and the degree and reversibility of any depression of GFR. If kidney function is normal, bicarbonate and base equivalents will be excreted with sodium or potassium, and metabolic alkalosis will be rapidly corrected as chloride is made available.
Chloride-resistant metabolic alkalosis
Metabolic alkalosis due to potassium depletion is managed by the correction of the underlying cause (see hypokalaemia). Mild to moderate alkalosis requires oral potassium chloride administration. However, the presence of cardiac arrhythmia or generalized weakness requires intravenous potassium chloride.
Greenlee MM, Lynch IJ, Gumz ML, et al. The renal H,K-ATPases. Curr Opin Nephrol Hypertens. 2010;19(5):478–482.
Kaplan LJ, Kellum JA. Fluids, pH, ions and electrolytes. Curr Opin Crit Care. 2010;16(4):323–331.
Karet FE. Disorders of water and acid-base homeostasis. Nephron Physiol. 2011;118(1):28–34.
Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol. 2007;2(1):162–174.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 13 Water, electrolytes and acid–base balance
Distribution and composition of body water
Intracellular fluid (28 L, about 35% of lean bodyweight)
Extracellular – the interstitial fluid that bathes the cells (9.4 L, about 12%)
The intracellular and interstitial fluids are separated by the cell membrane; the interstitial fluid and plasma are separated by the capillary wall (Fig. 13.1). In the absence of solute, water molecules move randomly and in equal numbers in either direction across a semi-permeable membrane. However, if solutes are added to one side of the membrane, the intermolecular cohesive forces reduce the activity of the water molecules. As a result, water tends to stay in the solute-containing compartment because there is less free diffusion across the membrane. This ability to hold water in the compartment can be measured as the osmotic pressure.
Osmotic pressure
The intracellular fluid contains mainly potassium (K+) (most of the cell Mg2+ is bound and osmotically inactive)
Regulation of the plasma volume is somewhat more complicated because of the tendency of the plasma proteins to hold water in the vascular space by an oncotic effect which is, in part, counterbalanced by the hydrostatic pressure in the capillaries that is generated by cardiac contraction (Fig. 13.1). The composition of intracellular and extracellular fluids is shown in Table 13.1.
Distribution of different types of replacement fluids
Figure 13.2 shows the relative effects on the compartments of the addition of identical volumes of water, saline and colloid solutions. Thus, 1 L of water given intravenously as 5% glucose is distributed equally into all compartments, whereas the same amount of 0.9% saline remains in the extracellular compartment. The latter is thus the correct treatment for extracellular water depletion – sodium keeping the water in this compartment. The addition of 1 L of colloid with its high oncotic pressure stays in the vascular compartment and is a treatment for hypovolaemia.
Regulation of extracellular volume (Fig. 13.3)
Plasma fluid constitutes one-third of extracellular volume (4.6 L), and of this,
85% (3.9 L) lies in the venous side and only 15% (0.7 L) resides in the arterial circulation.
These changes in Na+ excretion can result from alterations both in the filtered load, determined primarily by the glomerular filtration rate (GFR), and in tubular reabsorption, which is affected by multiple factors. In general, it is changes in tubular reabsorption that constitute the main adaptive response to fluctuations in the effective circulating volume. How this occurs can be appreciated from Table 13.2 and Figure 13.4 and Figure 12.2 (see p. 563), which depicts the sites and determinants of segmental Na+ reabsorption. Although the loop of Henle and distal tubules make a major overall contribution to net Na+ handling, transport in these segments primarily varies with the amount of Na+ delivered; that is, reabsorption is flow-dependent. In comparison, the neurohumoral regulation of Na+ reabsorption according to body needs occurs primarily in the proximal tubules and collecting ducts.
Neurohumoral regulation of extracellular volume
Intrarenal receptors. Receptors in the walls of the afferent glomerular arterioles respond, via the juxtaglomerular apparatus, to changes in renal perfusion, and control the activity of the renin-angiotensin-aldosterone system (see p. 1006). In addition, sodium concentration in the distal tubule and sympathetic nerve activity alter renin release from the juxtaglomerular cells. Prostaglandins I2 and E2 are also generated within the kidney in response to angiotensin II, acting to maintain glomerular filtration rate and sodium and water excretion, modulating the sodium-retaining effect of this hormone.
Extrarenal receptors. These are located in the vascular tree in the left atrium and major thoracic veins, and in the carotid sinus body and aortic arch. These volume receptors respond to a slight reduction in effective circulating volume and result in increased sympathetic nerve activity and a rise in catecholamines. In addition, volume receptors in the cardiac atria control the release of a powerful natriuretic hormone – atrial natriuretic peptide (ANP) – from granules located in the atrial walls (see p. 943).
A salt load, for example, leads to an increase in the effective circulatory and extracellular volume, raising both renal perfusion pressure, and atrial and arterial filling pressure. The increase in the renal perfusion pressure reduces the secretion of renin, and subsequently that of angiotensin II and aldosterone (see Fig. 12.5), whereas the rise in atrial and arterial filling pressure increases the release of ANP. These factors combine to reduce Na+ reabsorption in the collecting duct, thereby promoting excretion of excess Na+.
With more marked hypovolaemia, a decrease in GFR leads to an increase in proximal and thin ascending limb Na+ reabsorption which contributes to Na+ retention. This is brought about by enhanced sympathetic activity acting directly on the kidneys and indirectly by stimulating the secretion of renin/angiotensin II (see Fig. 13.3b) and non-osmotic release of antidiuretic hormone (ADH), also called vasopressin. The pressure natriuresis phenomenon may be the final defence against changes in the effective circulating volume. Marked persistent hypovolaemia leads to systemic hypotension and increased salt and water absorption in the proximal tubules and ascending limb of Henle. This process is partly mediated by changes in renal interstitial hydrostatic pressure and local prostaglandin and nitric oxide production.
Volume regulation in oedematous conditions
Sodium and water are retained despite increased extracellular volume in oedematous conditions such as cardiac failure, hepatic cirrhosis and hypoalbuminaemia. Here the principal mediator of salt and water retention is the concept of arterial underfilling due either to reduced cardiac output or diminished peripheral arterial resistance. Arterial underfilling in these settings leads to reduction of pressure or stretch (i.e. ‘unloading’ of arterial volume receptors), which results in activation of the sympathetic nervous system, activation of the renin-angiotensin-aldosterone system and non-osmotic release of ADH. These neurohumoral mediators promote salt and water retention in the face of increased extracellular volume. The common nature of the degree of arterial fullness and neurohumoral pathway in the regulation of extracellular volume in health and disease states forms the basis of Schrier’s unifying hypothesis of volume homeostasis (Fig. 13.3a).
Mechanism of impaired escape from actions of aldosterone and resistance to ANP
Not only is the activity of the renin-angiotensin-aldosterone system increased in oedematous conditions such as cardiac failure, hepatic cirrhosis and hypoalbuminaemia, but also the action of aldosterone is more persistent than in normal subjects and patients with Conn’s syndrome, who have increased aldosterone secretion (see p. 989).
In patients with the above oedematous conditions, e.g. heart failure, escape from the sodium-retaining actions of aldosterone does not occur and therefore they continue to retain sodium in response to aldosterone. Accordingly they have substantial natriuresis when given spironolactone, which blocks mineralocorticoid receptors. Alpha-adrenergic stimulation and elevated angiotensin II increase sodium transport in the proximal tubule, and reduced renal perfusion and GFR further increases sodium absorption from the proximal tubules by presenting less sodium and water in the tubular fluid. Sodium delivery to the distal portion of the nephron, and thus the collecting duct, is reduced. Similarly, increased cardiac ANP release in these conditions requires optimum sodium concentration at the site of its action in the collecting duct for its desired natriuretic effects. Decreased sodium delivery to the collecting duct is therefore the most likely explanation for the persistent aldosterone-mediated sodium retention, absence of escape phenomenon and resistance to natriuretic peptides in these patients (Fig. 13.3b).
Regulation of water excretion
The cortical collecting duct has two cell types (see also p. 597) with very different functions:
Principal cells (about 65%) have sodium and potassium channels in the apical membrane and, as in all sodium-reabsorbing cells, Na+/K+-ATPase pumps in the basolateral membrane.
The ADH-induced increase in collecting duct water permeability occurs primarily in the principal cells. ADH acts on V2 (vasopressin) receptors located on the basolateral surface of principal cells, resulting in the activation of adenyl cyclase. This leads to protein kinase activation and to preformed cytoplasmic vesicles that contain unique water channels (called aquaporins) moving to and then being inserted into the luminal membrane. Four renal aquaporins have been well characterized and are localized in different areas of the cells of the collecting duct. The water channels span the luminal membrane and permit water movement into the cells down a favourable osmotic gradient (Fig. 13.5). This water is then rapidly returned to the systemic circulation across the basolateral membrane. When the ADH effect has worn off, the water channels aggregate within clathrin-coated pits, from which they are removed from the luminal membrane by endocytosis and returned to the cytoplasm. A defect in any step in this pathway, such as in attachment of ADH to its receptor or the function of the water channel, can cause resistance to the action of ADH and an increase in urine output. This disorder is called nephrogenic diabetes insipidus.
Plasma osmolality
1. Ingestion of a water load leads to an initial reduction in the plasma osmolality, thereby diminishing the release of ADH. The ensuing reduction in water reabsorption in the collecting ducts allows the excess water to be excreted in a dilute urine.
2. Water loss resulting from sweating is followed by, in sequence, a rise in both plasma osmolality and ADH secretion, enhanced water reabsorption, and the appropriate excretion of a small volume of concentrated urine. This renal effect of ADH minimizes further water loss but does not replace the existing water deficit. Thus, optimal osmoregulation requires an increase in water intake, which is mediated by a concurrent stimulation of thirst. The importance of thirst can also be illustrated by studies in patients with central diabetes insipidus, who are deficient in ADH. These patients often complain of marked polyuria, which is caused by the decline in water reabsorption in the collecting ducts. However, they do not typically become hypernatraemic, because urinary water loss is offset by the thirst mechanism.
Osmoregulation versus volume regulation
The roles of these two pathways should be considered separately when evaluating patients.
A water load is rapidly excreted (in 4–6 h) by inhibition of ADH release so that there is little or no water reabsorption in the collecting ducts. This process is normally so efficient that volume regulation is not affected and there is no change in ANP release or in the activity of the renin-angiotensin-aldosterone system. Thus, a dilute urine is excreted, and there is little alteration in the excretion of Na+.
This principle of separate volume and osmoregulatory pathways is also evident in the syndrome of inappropriate ADH secretion (SIADH). Patients with SIADH (see p. 993) have impaired water excretion and hyponatraemia (dilutional) caused by the persistent presence of ADH. However, the release of ANP and aldosterone is not impaired and, thus, Na+ handling remains intact. These findings have implications for the correction of the hyponatraemia in this setting which initially requires restriction of water intake.
Increased extracellular volume
Venous tone, which determines the capacitance of the blood compartment and thus hydrostatic pressure
Causes
Heart failure
Reduction in cardiac output and the consequent fall in effective circulatory volume and arterial filling lead to activation of the renin-angiotensin-aldosterone system, non-osmotic release of ADH, and increased activity of the renal sympathetic nerves via volume receptors and baroreceptors (Fig. 13.3a). Sympathetic overdrive also indirectly augments ADH and renin-angiotensin-aldosterone response in these conditions. The cumulative effect of these mediators results in increased peripheral and renal arteriolar resistance and water and sodium retention. These factors result in extracellular volume expansion and increased venous pressure, causing oedema formation.
Hepatic cirrhosis
The mechanism is complex, but involves peripheral vasodilatation (possibly owing to increased nitric oxide generation) resulting in reduced effective arterial blood volume (EABV) and arterial filling. This leads to an activation of a chain of events common to cardiac failure and other conditions with marked peripheral vasodilatation (Fig. 13.3). The cumulative effect results in increased peripheral and renal resistance, water and sodium retention, and oedema formation.
Nephrotic syndrome
Interstitial oedema is a common clinical finding with hypoalbuminaemia, particularly in the nephrotic syndrome. Expansion of the interstitial compartment is secondary to the accumulation of sodium in the extracellular compartment. This is due to an imbalance between oral (or parenteral) sodium intake and urinary sodium output, as well as alterations of fluid transfer across capillary walls. The intrarenal site of sodium retention is the cortical collecting duct (CCD) where Na+/K+-ATPase expression and activity are increased threefold along the basolateral surface (Fig. 13.4). In addition, amiloride-sensitive epithelial sodium channel activity is also increased in the CCD. The renal sodium retention should normally be counterbalanced by increased secretion of sodium in the inner medullary collecting duct, brought about by the release of ANP. This regulatory pathway is altered in patients with nephrotic syndrome by enhanced kidney specific catabolism of cyclic GMP (the second messenger for ANP) following phosphodiesterase activation.
Oedema generation was classically attributed to the decrease in the plasma oncotic pressure and the subsequent increase in the transcapillary oncotic gradient. However, the oncotic pressure and transcapillary oncotic gradient remain unchanged and the transcapillary hydrostatic pressure gradient is not altered. Conversely, capillary hydraulic conductivity (a measure of permeability) is increased. This is determined by intercellular macromolecular complexes between the endothelial cells consisting of tight junctions (made of occludins, claudins and ZO proteins) and adherens junctions (made of cadherin, catenins and actin cytoskeleton). Elevated TNF-α levels in nephrotic syndrome activate protein kinase C, which changes phosphorylation of occludin and capillary permeability. In addition, increased circulating ANP can increase capillary hydraulic conductivity by altering the permeability of intercellular junctional complexes. Furthermore, reduction in effective circulatory volume and the consequent fall in cardiac output and arterial filling can lead to a chain of events as in cardiac failure and cirrhosis (see above and Fig. 13.3). These factors result in extracellular volume expansion and oedema formation.
Sodium retention
A decreased GFR decreases the renal capacity to excrete sodium. This may be acute, as in the acute nephritic syndrome (see p. 582), or may occur as part of the presentation of chronic kidney disease. In end-stage renal failure, extracellular volume is controlled by the balance between salt intake and its removal by dialysis.
Oestrogens cause mild sodium retention, due to a weak aldosterone-like effect. This is the cause of weight gain in the premenstrual phase.
Thiazolidinediones (TZD) (see p. 1011) are widely used to treat type 2 diabetes. Their mechanism of action is attributed to binding and activation of the PPAR-γ system. PPARs are nuclear transcription factors essential to the control of energy metabolism that are modulated via binding with tissue-specific fatty acid metabolites. Of the three PPAR isoforms, γ has been extensively studied and is expressed at high levels in adipose and liver tissues, macrophages, pancreatic-β cells and principal cells of the collecting duct. These drugs have been asociated with salt and water retention and are contraindicated in patients with heart failure. Recent evidence suggests that TZD-induced oedema (like insulin) is also due to upregulation of epithelial Na transporter channel (ENaC) but by different pathways. Diuretics of choice for TZD-induced oedema are amiloride and triamterene.
Other causes of oedema
Initiation of insulin treatment for type 1 diabetes and refeeding after malnutrition are both associated with the development of transient oedema. The mechanism is complex but involves upregulation of ENaC in the principal cell of the collecting duct. This transporter is amiloride sensitive which makes amiloride or triamterene the diuretic of choice in insulin-induced oedema.
Oedema is also caused by increased interstitial oncotic pressure as a result of increased capillary permeability to proteins. This can occur as part of a rare complement-deficiency syndrome; with therapeutic use of interleukin 2 in cancer chemotherapy; or in ovarian hyperstimulation syndrome (see p. 981).
Treatment
The mainstay of treatment is the use of diuretic agents, which increase sodium, chloride and water excretion in the kidney (Table 13.3). These agents act by interfering with membrane ion pumps which are present on numerous cell types; they mostly achieve specificity for the kidney by being secreted into the proximal tubule, resulting in much higher concentrations in the tubular fluid than in other parts of the body.
Clinical use of diuretics
Loop diuretics
These potent diuretics are useful in the treatment of any cause of systemic extracellular volume overload. They stimulate excretion of both sodium chloride and water by blocking the sodium-potassium-2-chloride (NKCC2) channel in the thick ascending limb of Henle (Fig. 13.6) and are useful in stimulating water excretion in states of relative water overload. They also act by causing increased venous capacitance, resulting in rapid clinical improvement in patients with left ventricular failure, preceding the diuresis. Unwanted effects include:
Allergic tubulointerstitial nephritis and other allergic reactions
Myalgia – especially with high-dose bumetanide
Interference with excretion of lithium, resulting in toxicity.
Thiazide diuretics (see p. 719)
These are less potent than loop diuretics. They act by blocking a sodium chloride channel in the distal convoluted tubule (Fig. 13.7). They cause relatively more urate retention, glucose intolerance and hypokalaemia than loop diuretics. They interfere with water excretion and may cause hyponatraemia, particularly if combined with amiloride or triamterene. This effect is clinically useful in diabetes insipidus. Thiazides reduce peripheral vascular resistance by mechanisms that are not completely understood but do not appear to depend on their diuretic action, and are widely used in the treatment of essential hypertension. They are also used extensively in mild to moderate cardiac failure. Thiazides reduce calcium excretion. This effect is useful in patients with idiopathic hypercalciuria, but may cause hypercalcaemia. Numerous agents are available, with varying half-lives but little else to choose between them. Metolazone is not dependent for its action on glomerular filtration, and therefore retains its potency in renal impairment.
Potassium-sparing diuretics (see Fig. 13.8)
Aldosterone antagonists, which compete with aldosterone in the collecting ducts and reduce sodium absorption, e.g. spironolactone and eplerenone (which has a shorter half-life). Spironolactone is used in patients with heart failure because it significantly reduces the mortality in these patients by antagonizing the fibrotic effect of aldosterone on the heart. Eplerenone is devoid of antiandrogenic or antiprogesterone properties.
Aquaretics (vasopressin or antidiuretic hormone antagonists)
Vasopressin V2 receptor antagonists are very useful agents in the treatment of conditions associated with elevated levels of vasopressin, such as heart failure, cirrhosis and SIADH (see p. 993). Non-peptide vasopressin V2 receptor antagonists are efficacious in producing free water diuresis in humans. Studies in patients with heart failure and cirrhosis suggest that such agents will allow normalization of serum osmolality with less water restriction (see p. 650).
Resistance to diuretics
Decreased extracellular volume
Clinical features
Loss of interstitial fluid leads to loss of skin elasticity (’turgor’) – the rapidity with which the skin recoils to normal after being pinched. Skin turgor decreases with age, particularly at the peripheries. The turgor over the anterior triangle of the neck or on the forehead is a very useful sign in all ages.
Loss of more than this causes the following:
Postural hypotension
Normally the blood pressure rises if a subject stands up, as a result of increased venous return due to venoconstriction (this maintains cerebral perfusion). Loss of extracellular fluid (underfill) prevents this and causes a fall in blood pressure. This is one of the earliest and most reliable signs of volume depletion, as long as the other causes of postural hypotension are excluded (Table 13.4).
Table 13.4 Postural hypotension: some causes of a fall in blood pressure from lying to standing
Investigations
Treatment
The overriding principle is to replace what is missing.
Loss of plasma
Loss of plasma, as occurs in burns or severe peritonitis, should be treated with human plasma or a plasma substitute (see p. 390).
Loss of water and electrolytes
Sodium bicarbonate (500 mg, 6 mmol each of Na+ and HCO3− per tablet) is used in doses of 6–12 tablets/day with 2–3 L of water. This is used in milder chronic sodium depletion with acidosis (e.g. chronic kidney disease, post-obstructive renal failure, renal tubular acidosis). Sodium bicarbonate is less effective than sodium chloride in causing positive sodium balance. Oral rehydration solutions are described in Box 4.10.
Intravenous fluids are sometimes required (Table 13.6). Rapid infusion (e.g. 1000 mL per hour or even faster) is necessary if there is hypotension and evidence of impaired organ perfusion (e.g. oliguria, confusion); in these situations, plasma expanders (colloids) are often used in the first instance to restore an adequate circulating volume (see p. 887). Repeated clinical assessments are vital in this situation, usually complemented by frequent measurements of central venous pressure (see p. 872, for the management of shock). Severe hypovolaemia induces venoconstriction, which maintains venous return; over-rapid correction does not give time for this to reverse, resulting in signs of circulatory overload (e.g. pulmonary oedema) even if a total body extracellular fluid (ECF) deficit remains. In less severe ECF depletion (such as in a patient with postural hypotension complicating acute tubular necrosis), the fluid should be replaced at a rate of 1000 mL every 4–6 h, again with repeated clinical assessment. If all that is required is avoidance of fluid depletion during surgery, 1–2 L can be given over 24 h, remembering that surgery is a stimulus to sodium and water retention and that over-replacement may be as dangerous as under-replacement. Regular monitoring by fluid balance charts, bodyweight and plasma biochemistry is mandatory.
Loss of water alone
FURTHER READING
Ahmed MS, Wong CF, Pai P. Cardiorenal syndrome – a new classification and current evidence on its management. Clin Nephrol 2010; 74(4):245–257.
Bie P. Blood volume, blood pressure and total body sodium: internal signaling and output control. Acta Physiol (Oxford) 2009; 195(1):187–196.
Bie P, Damkjaer M. Renin secretion and total body sodium: pathways of integrative control. Clin Exp Pharmacol Physiol 2010; 37(2):e34–42.
Schrier RW. Molecular mechanisms of clinical concentrating and diluting disorders. Prog Brain Res 2008; 170:539–550.
Wakil A, Atkin SL. Serum sodium disorders: safe management. Clin Med 2010; 10:79–82.
Disorders of sodium concentration
Table 13.8 Causes of hyponatraemia with normal extracellular volume (euvolaemia)
Table 13.9 Causes of hyponatraemia with increased extracellular volume (hypervolaemia)
|
Heart failure |
Oliguric kidney injury |
|
Liver failure |
Hypoalbuminaemia |
[/not-level-membership-for-internal-medicine-category]
