Von Willebrand disease and other inherited coagulation disorders
Von Willebrand disease
Von Willebrand disease (vWD) is the most common inherited bleeding disorder. The prevalence of symptomatic vWD is approximately 0.01%. Identification of milder forms is complicated by the broad range of von Willebrand factor (vWF) levels in the normal population; it is important to recognize that the diagnosis of vWD requires the presence of bleeding symptoms (see Fig 37.1).
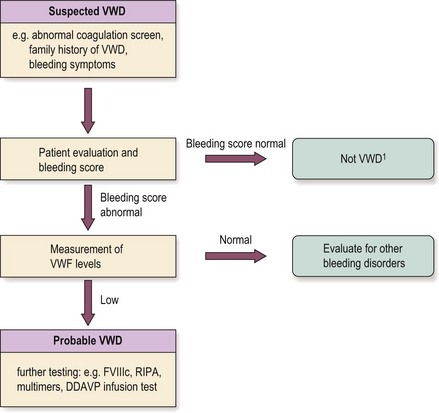
Fig 37.1 One approach to vWD diagnosis.
In practice, this has to be individualised (see text). vWD, von Willebrand disease; vWF, von Willebrand factor; FVIIIc, factor VIII; RIPA, ristocetin-induced platelet aggregation.
1In children should have lower threshold for measuring vWF levels.
All vWD is caused by mutations in the gene for vWF. vWF is an adhesive glycoprotein secreted by endothelium and megakaryocytes (see also p. 12). It is a multimeric protein with a characteristic normal distribution of multimer sizes in plasma. vWF has two key functions: promotion of platelet adhesion to damaged endothelium and other platelets (Fig 37.2) and the transport and stabilisation of factor VIII. Thus, the clinical disorder of vWD is associated with excessive bleeding due to abnormal platelet function and low factor VIII activity. The relationship between the risk of bleeding and vWF level is not strong until the level is very low. The clinical and laboratory heterogeneity of vWD necessitates the definition of several subtypes.
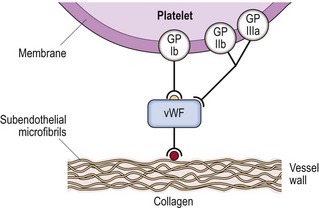
Fig 37.2 The role of von Willebrand factor in platelet adhesion.
Following vessel wall injury, large multimers of vWF bind to subendothelial microfibrils and also to glycoprotein (Gp) Ib on the platelet membrane thus mediating platelet adhesion. A secondary binding site with platelet Gp IIb/IIIa promotes further adhesion.
Classification (Table 37.1 and Fig 37.3)
Table 37.1
Summary of classification of vWD
 Type 1 vWD is a partial quantitative deficiency of vWF
Type 1 vWD is a partial quantitative deficiency of vWF
 Type 2 vWD is a qualitative deficiency of vWF
Type 2 vWD is a qualitative deficiency of vWF
 Type 3 vWD is a virtually complete deficiency of vWF
Type 3 vWD is a virtually complete deficiency of vWF
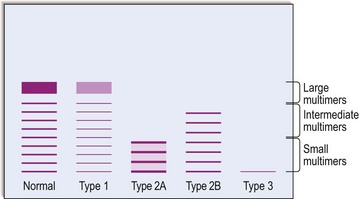
 Type 2A vWD is a qualitative variant with an absence of high molecular weight vWF multimers
Type 2A vWD is a qualitative variant with an absence of high molecular weight vWF multimers

 Type 2M vWD is a qualitative variant not caused by absence of high molecular weight multimers
Type 2M vWD is a qualitative variant not caused by absence of high molecular weight multimers
 Type 2N vWD is a qualitative variant with reduced affinity of vWF for factor VIII
Type 2N vWD is a qualitative variant with reduced affinity of vWF for factor VIII
Note: Mixed phenotypes may be caused by compound heterozygosity.
Clinical features
Laboratory diagnosis
1. Blood count. The platelet count is normal except for a moderate reduction in some cases of type 2B disease.
2. Activated partial thromboplastin time (APTT). Usually prolonged due to low factor VIII : C levels. The prothrombin time (PT) is normal.
3. Quantitative immunoassay for vWF antigen.
4. Functional assay of vWF. The commonest methodology is the ‘ristocetin cofactor assay’. Collagen binding assays are also used.
5. Factor VIII : C assay. Often low. May be borderline or normal in mild type 1 disease.
6. Multimer analysis. The multimer composition of circulating VWF is assessed by either crossed immunoelectrophoresis or sodium dodecyl sulphate electrophoresis (see Fig 37.3).
7. Platelet aggregation studies. Ristocetin (an obsolete antibiotic) induces platelet aggregation in normal plasma but not in severe vWD. An exception is the type 2B variant where platelets aggregate at unusually low concentrations of ristocetin.
8. Blood group. Normal plasma vWF levels tend to be lower in group O individuals.
9. General tests of primary haemostasis. The bleeding time is now little used. The PFA-100 (see p. 70) is a useful screening test but is also abnormal in other platelet disorders.
Other inherited coagulation disorders
Abnormalities of fibrinogen
Von Willebrand disease and other inherited coagulation disorders





