[level-membership-for-cardiovascular-category]1
Voltage-Gated Sodium Channels and Electrical Excitability of the Heart
Voltage-gated sodium channels initiate action potentials in cardiac myocytes and other excitable cells, and they are responsible for propagation of action potentials through the atria, conduction system, and ventricles of the heart. As shown in Figure 1-1, action potentials in atrial and ventricular muscle fibers rise rapidly from a resting potential near −80 mV and reach their peak within 1 ms. During this brief interval, cardiac sodium channels respond to the change in pacemaker potential as it reaches threshold and open to allow rapid Na+ entry. Sodium channels begin to inactivate as soon as they open and inactivate to 98% or 99% completion within a few milliseconds. The plateau phase of the cardiac action potential is generated by opening of voltage-gated calcium channels (see Chapter 2), and the cell is finally repolarized by slower opening of voltage-gated potassium channels (see Chapter 3). The rate of conduction of the action potential through the cardiac tissue depends directly on the rate of rise of the cardiac action potential and therefore on the density of sodium channels and their rate of activation.
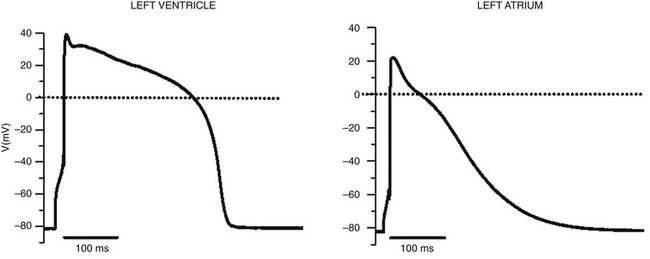
Figure 1-1 Cardiac action potential in sheep heart. Cardiac myocytes in left ventricle or left atrium were impaled with a microelectrode, and the cardiac action potential was recorded. (Courtesy J. Jalife.)
Subunit Structure of Sodium Channels
Sodium channel proteins purified from excitable cells are complexes composed of an approximately 260-kD α-subunit in association with one or two auxiliary β-subunits of approximately 33 to 39 kD.1 Purified sodium channel complexes of α- and β-subunits are sufficient for voltage-dependent gating and ion conduction in artificial lipid membranes and expression of the α-subunit alone is sufficient for physiologic function in recipient nonexcitable cells, indicating that this subunit has all the necessary structural elements for voltage-dependent gating and ion conduction.1–3 The primary sequence predicts that the sodium channel α-subunit folds into four internally repeated domains (I-IV), each of which contains six α-helical transmembrane segments (S1-S61,3–6; Figure 1-2). In each domain, the S1-S4 segments serve as the voltage-sensing module, and the S5 and S6 segments and the reentrant P loop between them serve as the pore-forming module. Extracellular loops connect the S5 and S6 transmembrane segments to the P loop in each domain, whereas the other extracellular loops are small. Large intracellular loops link the four homologous domains, and the large N-terminal and C-terminal domains also contribute substantially to the mass of the internal face of sodium channels. This view of sodium channel architecture, originally derived from hydrophobicity analysis of the amino acid sequence,4 has been largely confirmed by biochemical, electrophysiologic, and structural experiments.6
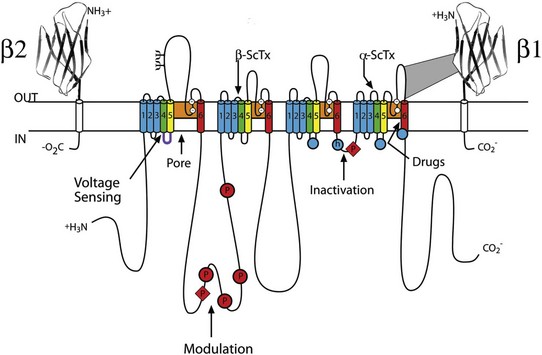
Figure 1-2 Transmembrane organization of sodium channel subunits. The primary structures of the subunits of the voltage-gated ion channels are illustrated as transmembrane folding diagrams. Cylinders represent probable alpha helical segments: blue, S1-S3; green, S4; yellow, S5; red, S6; shaded orange area, outer pore loop; purple, intracellular S4-S5 helix. Bold lines represent the polypeptide chains of each subunit with length approximately proportional to the number of amino acid residues in the brain sodium channel subtypes. The extracellular domains of the β1- and β2-subunits are shown as immunoglobulin-like folds Ψ, sites of probable N-linked glycosylation. P, sites of demonstrated protein phosphorylation by PKA (circles) and PKC (diamonds); white circles, the outer (EEDD) and inner (DEKA) rings of amino residues that form the ion selectivity filter and the tetrodotoxin binding site; ++, S4 voltage sensors; h in shaded circle, inactivation particle in the inactivation gate loop; open shaded circles, sites implicated in forming the inactivation gate receptor. The structure of the extracellular domain of the β-subunits is illustrated as an immunoglobulin-like fold based on amino acid sequence homology to the myelin P0 protein.8,22 Sites of binding of α- and β-scorpion toxins and a site of interaction between α- and β1-subunits are also shown. (Adapted from Catterall WA: From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26:13–25, 2000.)
The auxiliary β subunits were identified in the initial purification studies of sodium channels.1 These subunits have a single transmembrane segment, a large N-terminal extracellular domain that is homologous in structure to a variable chain (V-type) immunoglobulin-like fold, and a short C-terminal intracellular segment (see Figure 1-2).7–10 The β-subunits interact with α-subunits through their extracellular immunoglobulin (Ig)-fold domains, modulate α-subunit function, and enhance their cell surface expression.11 Like other proteins with an extracellular Ig-fold, they also serve as cell adhesion molecules by interacting with extracellular matrix proteins, cell adhesion molecules, and cytoskeletal linker proteins.12–17 These interactions are thought to localize and stabilize sodium channels in specific subcellular compartments and to bring crucial signaling molecules to the sodium channel to regulate it. Deletion of the genes encoding β-subunits causes alterations in sodium channel function; reduced action potential conduction and abnormal development of myelin folds in axons; hyperexcitability and epilepsy in the brain; and arrhythmias in the heart.18–20
Three-Dimensional Structure of Sodium Channels
Sodium channel architecture has been revealed in three dimensions by determination of the crystal structure of the bacterial sodium channel NavAb at high resolution (2.7 Å (0.27 nm); Figure 1-3). This ancient sodium channel has a simple structure—four identical subunits that are each similar to one homologous domain of a mammalian sodium channel without the large intracellular and extracellular loops of the mammalian protein.21 This structure has revealed a wealth of new information about the structural basis for sodium selectivity and conductance, the mechanism for block of the channel by therapeutically important drugs, and the mechanism of voltage-dependent gating. As viewed from the top, NavAb has a central pore surrounded by four pore-forming modules composed of S5 and S6 segments and the intervening pore loop (see Figure 1-3, A). Four voltage-sensing modules composed of S1-S4 segments are symmetrically positioned around the outer rim of the pore module (see Figure 1-3, A). The transmembrane architecture of NavAb shows that the adjacent subunits have swapped their functional domains such that each voltage-sensing module is most closely positional around with the pore-forming module of its neighbor (see Figure 1-3, B). It is likely that this domain-swapped arrangement enforces concerted gating of the four subunits or domains of sodium channels.
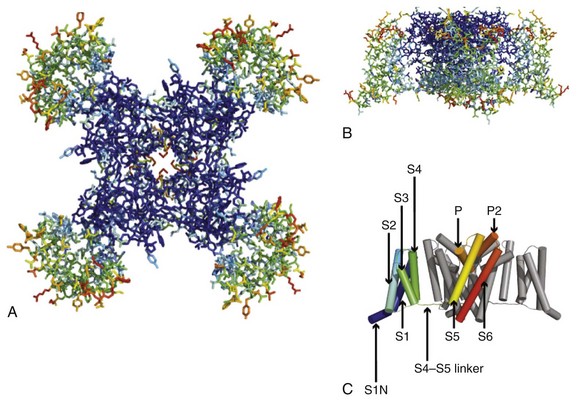
Figure 1-3 Three-dimensional structure of sodium channels. A, Top view of NavAb channels colored according to crystallographic temperature factors of the main-chain (blue < 50 Å2 to red > 150 Å2). B, Side view of NavAb. C, Structural elements in NavAb. The structural components of one subunit are highlighted. 1-6, Transmembrane segments S1-S6. (Adapted from Payandeh J, Scheuer T, Zheng N, et al: The crystal structure of a voltage-gated sodium channel. Nature 475:353–358, 2011.)
Comparison of the primary structures of the auxiliary β-subunits to those of other proteins revealed a close structural relationship to the family of proteins that contain Ig-like folds, which include many cell-adhesion molecules.8 The extracellular domains of these type-I single-membrane-spanning proteins are predicted to fold in a similar manner as myelin protein P0, whose Ig-like fold is known to be formed by a sandwich of two β-sheets held together by hydrophobic interactions (see Figure 1-222). Myelin P0 protein is a cell adhesion molecule involved in tight wrapping of myelin sheets, and many related cell adhesion molecules with extracellular Ig-folds and a single membrane-spanning segment are involved in cell-cell interactions among neurons and glia.22 As expected from their structure, NaVβ subunits interact with extracellular matrix molecules, other cell adhesion molecules, and intracellular cytoskeletal proteins and signaling proteins.12–16,23
Sodium Channel Structure and Function
Hodgkin and Huxley24 defined the three key functions of sodium channels: (1) voltage-dependent activation, (2) fast inactivation, and (3) selective ion conductance. Building on this foundation, detailed biophysical studies revealed the ion selectivity of the channel pore, detected the movement of the voltage sensors as gating current, and developed mechanistic models for these essential channel functions.25,26 Recent structure-function studies using molecular, biochemical, structural, and electrophysiologic techniques have provided clear understanding of the molecular and structural basis for these sodium channel functions.
Outer Pore and Selectivity Filter
Voltage clamp studies showed that sodium channels are highly selective for sodium versus potassium and other monovalent cations.26,27 Analysis of ion selectivity and block by tetrodotoxin and saxitoxin led to a model of tetrodotoxin and saxitoxin as plugs of the selectivity filter in the outer pore of sodium channels.26 Mutational analysis identified a key glutamate residue in the membrane-reentrant loop in domain I as a crucial residue for tetrodotoxin and saxitoxin binding.28 Additional studies revealed a pair of important amino acid residues, mostly negatively charged, in analogous positions in all four domains (see Figure 1-2, B, small white circles).29–31 Mutation of a set of four residues in analogous positions in each domain (aspartate in domain I, glutamate in domain II, lysine in domain III, and alanine in domain IV, DEKA) to glutamates confers calcium selectivity,32 indicating that the side chains of these amino acid residues are likely to interact with sodium ions as they are conducted through the ion selectivity filter of the pore. These results showed that the narrow selectivity filter in the outer pore is formed by the reentrant P loops between transmembrane segments S5 and S6 of each domain (see Figure 1-2). Mutations in this postulated ring of four amino acid residues have strong effects on selectivity for organic and inorganic monovalent cations, in agreement with the idea that they form the selectivity filter, and structure-function studies suggest specific structural interactions and functional roles for the P loops from the four domains.33–36
In the bacterial sodium channel NavAb, the overall pore architecture includes a large external vestibule, a narrow ion selectivity filter containing the amino acid residues shown to determine ion selectivity in vertebrate sodium and calcium channels, a large central cavity that is lined by the S6 segments and is filled with water, and an intracellular activation gate formed at the crossing of the S6 segments at the intracellular surface of the membrane (Figure 1-4, A). The activation gate is tightly closed in the NavAb structure (see Figure 1-4, B), and there is no space for ions or water to move through it. This general architecture resembles voltage-gated potassium channels (see Chapter 3). Although the overall pore architecture of sodium and potassium channels is similar, the structures of their ion selectivity filters and their mechanisms of ion selectivity and conductance are completely different. Potassium channels select K+ by direct interaction with a series of four ion coordination sites formed by the backbone carbonyls of the amino acid residues that comprise the ion selectivity filter (Chapter 3). No charged amino acid residues are involved, and no water molecules intervene between K+ and its interacting backbone carbonyls in the ion selectivity filter of potassium channels. In contrast, the NavAb ion selectivity filter has a high field strength site at its extracellular end (see Figure 1-4, C; Glu177 = ε177), which is formed by amino acid residues that are highly conserved and are key determinants of ion selectivity in vertebrate sodium and calcium channels (see Figure 1-2). Considering its dimensions of approximately 4.6 Å2, Na+ with two planar waters of hydration could fit in this high–field-strength site. This outer site is followed by two ion coordination sites formed by backbone carbonyls (see Figure 1-4, D). These two carbonyl sites are perfectly designed to bind Na+ with four planar waters of hydration, but would be much too large to bind Na+ directly. In fact, the NavAb selectivity filter is large enough to fit the entire K+ channel ion selectivity filter inside it. Thus, the chemistry of Na+ selectivity and conductance is opposite to that of K+: negatively charged residues interact with Na+ to remove most (but not all) of its waters of hydration, and Na+ is conducted as a hydrated ion interacting with the pore through its inner shell of bound waters. Theoretical considerations of sodium selectivity and conductance predicted an outer high–field-strength site that would only partially dehydrate the permeating ion and two inner sites that would conduct and rehydrate the permeant Na+ ion because of the high energy of hydration of Na+ (see Figure 1-4, C, D).37
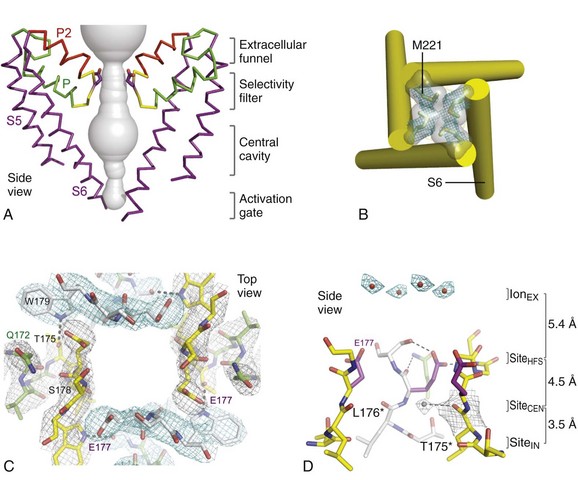
Figure 1-4 NavAb pore and selectivity filter. A, Architecture of the NavAb pore. Purple, Glu177 side-chains; gray, pore volume. B, The closed activation gate at the intracellular end of the pore illustrating the close interaction of Met221 residues in closing the pore. C, Top view of the ion selectivity filter. Symmetry-related molecules are colored white and yellow; P-helix residues are colored green. Hydrogen bonds between Thr175 and Trp179 are indicated by gray dashes. Electron densities from Fo-Fc omit maps are contoured at 4.0 σ (blue and gray), and subtle differences can be appreciated (small arrows). D, Side view of the selectivity filter. Glu177 (purple) interactions with Gln172, Ser178, and the backbone of Ser180 are shown in the far subunit. Fo-Fc omit map, 4.75 σ (blue); putative cations or water molecules (red spheres, IonEX). Electron-density around Leu176 (gray; Fo-Fc omit map at 1.75 σ) and a putative water molecule are shown (grey sphere). Na+-coordination sites: SiteHFS, SiteCEN and SiteIN. (Adapted from Payandeh J, Scheuer T, Zheng N, et al: The crystal structure of a voltage-gated sodium channel. Nature 475:353–358, 2011.)
Voltage-Dependent Activation
The voltage dependence of the activation of sodium channels derives from outward movement of approximately 12 gating charges as a consequence of depolarization of the membrane and reduction of the membrane electric field.25,38 The S4 segments of each homologous domain serve as the primary voltage sensors for activation.4,5 They contain repeated motifs of a positively charged amino acid residue followed by two hydrophobic residues creating a transmembrane spiral of positive charges. Upon depolarization, outward movement and rotation of S4 is thought to initiate a conformational change that opens the sodium channel pore.4,5 This “sliding helix” or “helical screw” model is supported by strong evidence. For example, neutralization of the positively charged residues in S4 reduce the voltage-dependence of gating.39 The outward and rotational gating movement of S4 segment has been detected directly by reaction of substituted cysteine residues in S4 segments with extracellular sulfhydryl reagents following channel activation and by analysis of the movement of fluorescent probes incorporated into these substituted cysteine residues.40,41 Other support for this mechanism derives from a wide range of structure and function studies.42
In the structure of the bacterial sodium channel NavAb, the S4 segment is in a transmembrane position in its activated state and its positive charges are thought to be neutralized by negative charges in the nearby S1 and S2 segments (Figure 1-5, A).21 At the resting membrane potential, the force of the electric field, which is negative inside the cell, would pull the positive charges inward. Depolarization would abolish this force and allow an outward movement of the S4 helix and its gating charges, catalyzed by exchange of ion pair partners (see Figure 1-5, B; Movie 1-1). After conformational changes have occurred in all four domains, the transmembrane pore can open and conduct ions (see Movie 1-1). This structural model shows that the S4 segment and its gating charges move through a narrow gating pore that focuses the transmembrane electric field to a distance of approximately 5 Å normal to the membrane and allows the gating charges to move from an intracellular aqueous vestibule to an extracellular aqueous vestibule with a short transit through the channel protein (see Video 1-1).
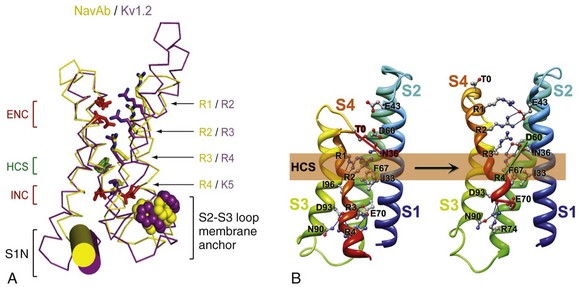
Figure 1-5 The voltage-sensing domain (VSD). A, Side view of the VSD illustrating the extracellular negative charge-cluster (red; ENC), the intracellular negative charge-cluster (red; INC), hydrophobic constriction site (green; HCS), residues of the S1N helix (cyan), and phenylalanine of the S2-S3 loop (purple). S4 segment and gating charges (R1-R4) are in yellow. B, Transmembrane view of the lowest-energy Rosetta models of the VSD of NaChBac in Resting State 1 (left) and Activated State 3 (right). Side chains of the gating-charge-carrying arginines in S4 and key residues in S1, S2, and S3 segments are shown in stick representation and labeled. Gray, blue, and red atoms are C, N, and O, respectively. The HCS is highlighted by orange bars. The lowest-energy models of Resting State 1 predict that R1 forms hydrogen bonds with the backbone carbonyl of I96 (in S3) at the extracellular edge of the HCS. On the intracellular side of the HCS, R3 makes ionic interactions with the amino acid residues of the intracellular negatively charged cluster, including E70 (in S2) and D93 (in S3), and R4 forms an ion pair with D93 (in S3). The lowest-energy models for Activated State 3 predict that R1 forms an ion pair with E43 (in S1), R2 forms an ion pair with E43 (in S1), R3 forms hydrogen bond with Y156 (in S5) and makes ionic interactions with D60 (in S2) and E43 (in S1), and R4 forms an ion pair with D60 (in S2). See Videos 1-1 and 1-2. (Adapted from Payandeh J, Scheuer T, Zheng N, et al: The crystal structure of a voltage-gated sodium channel. Nature 475:353–358, 2011; and Yarov-Yarovoy V, DeCaen PG, Westenbroek RE, et al: Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci U S A 109:E93–102, 2012.)
Pore Opening
From these structural models, one can visualize the steps in the gating of a voltage-gated ion channel. In the closed state, the negative internal membrane potential of −70 to −90 mV pulls the S4 gating charges inward by electrostatic force. The inward position of the S4 segment exerts a force on the S4-S5 linker, twists and straightens the S6 segment, and closes the pore at its inner mouth. When the cell is depolarized, the electrostatic force pulling the S4 segment inward is relieved. In response to the change in electrostatic force, the S4 segment moves outward with each positive gating charge interacting with charged amino acid side chains in turn to ease their movement through the voltage sensor module. When the R3 and R4 gating charges pass the hydrophobic constriction site in the center of the voltage sensor module, the force on the S4-S5 linker is sufficient to pull on the pore-forming module and twist and bend the S6 segment, resulting in opening of the pore at its intracellular end. During activation of the voltage sensors of sodium channels, the S4-S5 intracellular linkers in each domain (see Figure 1-2, B, purple) exert a force on the adjacent S6 segments, and the pore opens by bending the S6 segment (see Figure 1-2, B, red). In bacterial voltage-gated potassium channels and sodium channels, bending of the S6 segment occurs at a critical hinge glycine residue about one-third down the S6 segment,43–45 and this bending motion allows the opening of the inner mouth of the pore (Videos 1 and 2; Figure 1-5) and ion rapid movement across the membrane.
Fast Inactivation
Fast inactivation of the sodium channel is a critical process that occurs within milliseconds of channel opening. The generally accepted model of this process involves a conserved inactivation gate formed by the intracellular loop connecting domains III and IV (see Figure 1-2), which serves as a hinged lid that binds to the intracellular end of the pore and blocks it (Figure 1-6). Intracellular perfusion of proteases prevents fast inactivation.25 Site-directed antipeptide antibodies against the short, highly conserved intracellular loop connecting domains III and IV of the sodium channel α-subunit (see Figure 1-2), but not antibodies directed to other intracellular domains, were found to prevent fast sodium channel inactivation.46,47 Moreover, the accessibility of this site for antibody binding was reduced when the membrane was depolarized to induce inactivation, suggesting that the loop connecting domains III and IV forms an inactivation gate that folds into the channel structure during inactivation.46,47 Cutting the loop between domains III and IV by expression of the sodium channel in two pieces greatly slows inactivation.39 Mutagenesis studies of this region revealed a hydrophobic triad of isoleucine, phenylalanine, and methionine (IFM) that is critical for fast inactivation (see Figure 1-2, B, blue circle with h),48 and peptides containing this motif can serve as pore blockers and can restore inactivation to sodium channels having a mutated inactivation gate.49 The latch of this fast inactivation gate is formed by three key hydrophobic residues, IFM, and an adjacent threonine (T). These results support a model in which the IFM motif serves as a tethered pore blocker that binds to a receptor in the intracellular mouth of the pore. Inactivation is impaired in proportion to the hydrophilicity of amino acid substitutions for the key phenylalanine residue (F1489), suggesting that it forms a hydrophobic interaction with an inactivation gate receptor during inactivation.50 Voltage-dependent movement of the inactivation gate has been detected by measuring the accessibility of a cysteine residue substituted for F1489.51 This substituted cysteine residue becomes inaccessible to reaction with sulfhydryl reagents as the inactivation gate closes. Glycine and proline residues that flank the IFM motif may serve as molecular hinges to allow closure of the inactivation gate like a hinged lid (see Figure 1-6, A).52
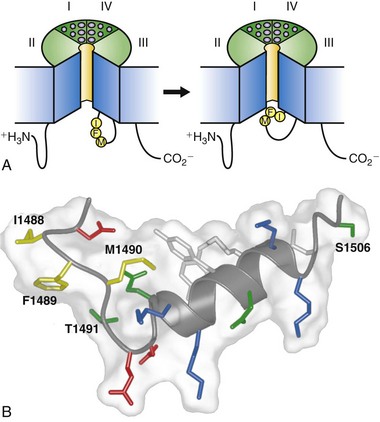
Figure 1-6 The molecular mechanism of fast sodium channel inactivation. A, The hinged-lid mechanism. The intracellular loop connecting domains III and IV of the sodium channel is depicted as forming a hinged lid with the critical phenylalanine (Phe1489) within the IFM motif shown occluding the mouth of the pore during the inactivation process. The circles represent the transmembrane helices. B, Three-dimensional structure of the central segment of the inactivation gate as determined by multidimensional NMR. Isoleucine 1488, phenylalanine 1489, and methionine 1490 (IFM) are illustrated in yellow. Threonine 1491, which is important for inactivation, and serine 1506, which is a site of phosphorylation and modulation by protein kinase C, are also indicated. (Adapted from Catterall WA: From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26:13–25, 2000.)
The three-dimensional structure of the central portion of the inactivation gate has been determined by expression as a separate peptide and analysis by multidimensional NMR methods.53 These experiments reveal a rigid α-helix flanked on its N-terminal side by two turns, the second of which contains the IFM motif (see Figure 1-6, B). The fold of the inactivation gate peptide projects F1489 into the solvent away from the core of the peptide, an unusual position for a hydrophobic residue in a short peptide. In this position, F1489 is poised to serve as a tethered ligand that occludes the pore. The nearby threonine (T1491), which is an important residue for inactivation,50 also is in position to interact with the inactivation gate receptor in the pore. In contrast, the methionine of the IFM motif (M1490) is buried in the core of the peptide, interacting with two tyrosine residues in the alpha helix. This hydrophobic interaction stabilizes the fold of the peptide and forces F1489 into its exposed position. The structure of the inactivation gate peptide in solution suggests that the rigid α-helix serves as a scaffold to present the IFM motif and T1491 to a receptor in the mouth of the pore as the gate closes.
Scanning mutagenesis experiments have revealed multiple amino acid residues that can form the inactivation gate receptor within and near the intracellular mouth of the pore (see Figure 1-1, B, blue circles), including hydrophobic residues at the intracellular end of transmembrane segment IVS654 and amino acid residues in intracellular loops IIIS4-S555 and IVS4-S5.56–59 Mutations of residues in each of these positions impair inactivation by destabilizing the inactivated state, as expected for disruption of the inactivation gate receptor. In addition, mutations in intracellular loop IVS4-S5 impair closed channel block by IFM-containing peptides, consistent with function as the inactivation gate receptor,57 and paired insertions of charged residues in the IIIS4-S5 loop and in the IFM motif indicate that these peptide segments interact during inactivation.55 Evidently, multiple peptide segments form a complex inactivation gate receptor into which the inactivation gate closes to occlude the inner pore.
Coupling of Activation to Fast Inactivation
Sodium channel inactivation derives most or all of its voltage dependence from coupling to the activation process driven by transmembrane movements of the S4 voltage sensors.25 Increasingly strong evidence implicates the S4 segment in domain IV in this process. Mutations of charged amino acid residues at the extracellular end of the IVS4 segment have strong and selective effects on inactivation.60 α-Scorpion toxins and sea anemone toxins uncouple activation from inactivation by binding to a receptor site at the extracellular end of the IVS4 segment and preventing its normal gating movement,61,62 evidently trapping it in a position that is permissive for activation but not for fast inactivation. The IIIS4 and IVS4 segments, detected by covalently incorporated fluorescent probes, are specifically immobilized in the outward position by fast inactivation, arguing that their movement is coupled to the inactivation process.63 Together, these results provide strong evidence that outward movement of the S4 segment in domain IV is the signal to initiate fast inactivation of the sodium channel by closure of the intracellular inactivation gate. The molecular mechanism for coupling of this movement of IVS4 to inactivation gate closure is an interesting subject for further investigation.
Slow Inactivation
In addition to the fast inactivation process discovered by Hodgkin and Huxley in their classic work, a separate slow inactivation process operating on the time scale of 100 ms to seconds also terminates the Na+ influx through Na+ channels. This process is engaged during repetitive generation of action potentials in nerve and muscle cells and limits the length of trains of repetitive action potentials. Bacterial Na+ channels whose structure has been determined have a slow inactivation process, although their homotetrameric structure means that they do not have a structural component analogous to the intracellular loop connecting domains III and IV of vertebrate channels, which mediates fast inactivation. The structure of the slow-inactivated bacterial Na+ channel64 reveals that the pore has partially collapsed by movement of two opposing S6 segments toward the central axis of the pore and corresponding movement of the two adjacent pairs of S6 segments away from the axis (Figure 1-7). This movement is observed at the selectivity filter at the extracellular end of the pore, in the central cavity, and at the activation gate at the intracellular end of the pore (see Figure 1-7). This asymmetric collapse of the pore is accompanied by subtle rotation of the voltage-sensing domain around the cylindrical exterior surface of the pore domain (see Figure 1-7). It is likely that the pore collapse is important for stabilization of the sodium channel in the inactivated state, which requires strong, long-duration hyperpolarization for recovery to the resting state.
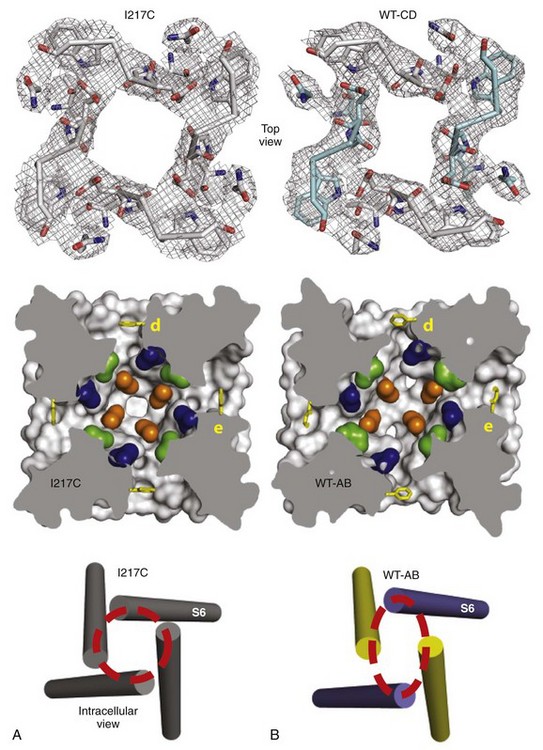
Figure 1-7 Structural basis for slow inactivation. A, Structure of the functional elements of the pore in the pre-open state. Top, Selectivity filter; middle, central cavity with amino acid residues of the drug-binding site shown in color; bottom, activation gate. B, Structure of the functional elements of the pore in the slow-inactivated state shown as in A. (Adapted from Payandeh J, Gamal El-Din TM, Scheuer T, et al: Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486:135–139, 2012.)
Inner Pore and Receptor Site for Local Anesthetic and Antiarrhythmic Drugs
Sodium channels are the molecular targets for drugs used in control of cardiac arrhythmias, local anesthesia, prevention of acute pain, and treatment of epilepsy and bipolar disorder. Sodium channel–blocking drugs are also in development for treatment of chronic pain. Sodium channel blocking drugs bind to a specific receptor site within the pore of sodium channels, formed by the S6 segments in domains I, III, and IV21,65–68 (Figure 1-8). Their binding blocks ion movement through the pore and stabilizes the inactivated state of sodium channels. Antiarrhythmic drugs and antiepileptic drugs share similar, overlapping receptor sites. Complete block of sodium channels would be lethal; however, these drugs selectively block sodium channels in depolarized or rapidly firing cells, such as axons carrying high-intensity pain information and rapidly firing nerve and cardiac muscle cells that drive epileptic seizures or cardiac arrhythmias.69–71 This selective block arises because the drugs can reach their binding site in the pore of the sodium channel more rapidly when the pore is repetitively opened, and they bind with high affinity to inactivated sodium channels that are generated in rapidly firing or depolarized cells. The conformational change observed in the central cavity during slow inactivation may be responsible for the increased affinity for drug block of the inactivated state (see Figure 1-7). The use-dependent action of the sodium channel–blocking drugs is essential for their therapeutic efficacy.
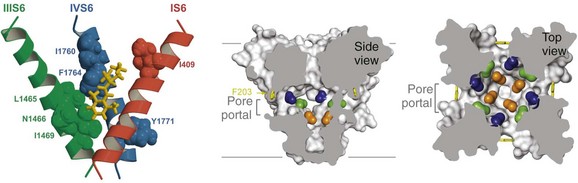
Figure 1-8 Drug binding in the central cavity. A, Three-dimensional model of proposed orientation of amino acid residues within the Na+ channel pore with respect to the local anesthetic etidocaine. Only transmembrane segments IS6 (red), IIIS6 (green), and IVS6 (blue) are shown. Residues important for etidocaine binding are shown in space-filling representation. B, Side-view through the pore module illustrating fenestrations (portals) and hydrophobic access to central cavity. Phe203 side-chains, yellow sticks. Surface representations of NavAb residues aligning with those implicated in drug binding and block. Blue, Thr206; green, Met209; orange, Val213; gray lines, membrane boundaries. Electron-density from an Fo-Fc omit map is contoured at 2.0 σ. C, Top-view sectioned below the selectivity filter, colored as in B. (Adapted from Payandeh J, Scheuer T, Zheng N, et al: The crystal structure of a voltage-gated sodium channel. Nature 475:353–358, 2011.)
High-affinity binding of local anesthetics to the inactivated state of sodium channels requires two critical amino acid residues, phe1764 and tyr1771 in brain type IIA channels, which are located on the same side of the IVS6 transmembrane segment two α-helical turns apart (see Figure 1-8, A; phe1764 and tyr1771 in blue, etidocaine in red).65–68 It is likely that the tertiary amino group of local anesthetics interacts with phe1764, which is located more deeply in the pore, and that the aromatic moiety of the local anesthetics interacts with tyr1771, which is located nearer to the intracellular end of the pore (see Figure 1-8, blue residues). Subsequent work has shown that sodium channel–blocking drugs of diverse structure that are used as antiarrhythmic drugs and as anticonvulsants also interact with the same site as local anesthetics, but also create additional interactions with other nearby amino acid residues.72–76
The amino acid residues that form the receptor sites for Na+ channel blockers line the inner surface of the S6 segments and create a three-dimensional drug receptor site whose occupancy would block the pore (see Figure 1-8, B). Remarkably, fenestrations lead from the lipid phase of the membrane sideways into the drug receptor site, providing a hydrophobic access pathway for drug binding (see Figure 1-8, B). This form of drug binding from the membrane phase was predicted in early studies of the mechanism of block of Na+ channels by different local anesthetics.26 Access to the drug binding site in NavAb channels from the membrane phospholipid bilayer is limited by the side chain of a single amino acid residue (see Figure 1-8, B), which could control drug access and egress from the drug receptor site and possibly entry and egress of physiologic lipid modulators.
In addition to these important pharmaceutical agents, sodium channels are the molecular targets for a large number of neurotoxins that paralyze prey by preventing neuromuscular function.77,78 These toxins act at five or more distinct receptor sites and either block the pore of the channel or alter the kinetics or voltage dependence of gating. The pore blockers tetrodotoxin and saxitoxin bind to neurotoxin receptor site 1, which is formed by the P loops in the four domains (see Figure 1-2).
Sodium Channel Genes
Sodium channels are the founding members of ion channel superfamily that includes voltage-gated calcium channels, TRP channels, voltage-gated, inward rectifying, and two-pore-domain potassium channels, and cyclic nucleotide-regulated CNG and HCN channels.79 In evolution, the four-domain sodium channel was last among the voltage-gated ion channels to appear, and it is only found in multicellular organisms. It is thought that sodium channels evolved by two rounds of gene duplication from ancestral single-domain bacterial sodium channels. Voltage-gated sodium channel genes are present in a variety of metazoan species, including the fly, leech, squid, and jellyfish. The biophysical properties, pharmacology, gene organization, and even intron-splice sites of these invertebrate sodium channels are largely similar to the mammalian sodium channels.
Ten related sodium channel genes are found in vertebrates, and nine encode voltage-gated sodium channels (Figure 1-9).79,80 More than 20 exons comprise each of the sodium channel α-subunit genes in mammals. Genes encoding sodium channels Nav1 1, Nav1 2, Nav1 3, and Nav1 7 are localized on chromosome 2 in humans, and these channels share similarities in sequence, biophysical characteristics, block by nanomolar concentrations of tetrodotoxin, and broad expression in neurons. A second cluster of genes encoding Nav1 5, Nav1 8, and Nav1 9 channels is localized to human chromosome 3p21-24. Although they are more than 75% identical in sequence to the group of channels on chromosome 2, these sodium channels all contain amino acids substitutions that confer varying degrees of resistance to the pore blocker tetrodotoxin. In Nav1 5, the principal cardiac isoform,81 a single amino acid change from phenylalanine to cysteine in the pore region of domain I, is responsible for a 200-fold reduction in tetrodotoxin (TTX) sensitivity compared with those channels on chromosome 2.82 At the identical position in Nav1 8 and Nav1 9, the amino acid residue is serine, and this change results in even greater resistance to TTX.83 These two channels are primarily expressed in peripheral sensory neurons. Compared with the sodium channels on chromosomes 2 and 3, Nav1 4 (which is expressed in skeletal muscle) and Nav1 6 (which is highly abundant in the central nervous system) have greater than 85% sequence identity and similar functional properties, including TTX-sensitivity in the nanomolar concentration range. A tenth sodium channel, Nax, whose gene is located near the sodium channels of chromosome 2, is evolutionarily more distant.80 Key differences in functionally important regions of voltage sensor and inactivation gate and lack of functional expression of voltage-gated sodium currents in heterologous cells suggest that Nax might not function as a voltage-dependent sodium channel. Consistent with this conclusion, targeted deletion of the Nax gene in mice causes functional deficits in sensing plasma salt levels.84
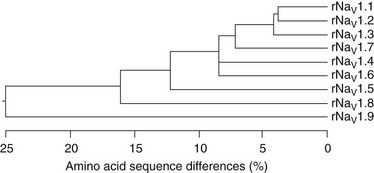
Figure 1-9 Amino acid sequence similarity of voltage-gated sodium channel α-subunits. Amino acid sequence similarity of voltage-gated sodium channel α-subunits. A comparison of amino acid identity for rat sodium channels Nav1.1 to Nav1.9. The comparison was performed with Megalign in the program DNAStar (using the Clustal method) for the four domains and the cytoplasmic linker connecting domains III and IV. (Adapted from Catterall WA: From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26:13–25, 2010.)
The NaVβ subunits are encoded by four distinct genes in mammals.7–10 The gene encoding β1 maps to chromosome 19q13, whereas β2 and β4 are located on chromosome 11q22-23 and β3 is located nearby on chromosome 11q24. The β1 and β3 subunits associate noncovalently with the α-subunits, whereas β2 and β4 are covalently linked by a disulfide bond. These distinct modes of association and the corresponding similarities in amino acid sequence suggest that the similar pairs of β-subunits (β1 and β3 vs. β2 and β4) may be able to substitute for each other in interaction with sodium channel α-subunits.
Although NaV1.5 channels are primarily expressed in the heart and are often termed the cardiac sodium channel,81 several of the brain sodium channel subtypes are also expressed at lower levels in the heart, where they are differentially localized in subcellular compartments in a cell-specific and species-specific manner. In human atrial myocytes, NaV1.5 channels are localized in a striated pattern on the cell surface at the z-lines in each sarcomere. In contrast, NaV1.2 channels are localized primarily at the intercalated discs, but at a lower concentration, and NaV1.1 channels are localized at low density in a scattered punctate pattern over the cell surface. Because all the “brain” and “skeletal muscle” sodium channel subtypes are inhibited by nanomolar concentrations of TTX and NaV1.5 channels require micromolar concentrations of TTX for inhibition, the contribution of the TTX-sensitive sodium channels to cardiac contractility can be tested in careful does-response experiments. Such experiments indicate that 12% to 27% of sodium current is conducted by the TTX-sensitive sodium channels, and block of these channels reduces the amplitude and velocity of contraction of atrial muscle strips approximately 15%.85 The differential localization of these subtypes suggests specific functions for the NaV1.2 channels in the initiation of the action potential at intercalated discs and the NaV1.5 channels in depolarizing the atrial membrane near the voltage-gated calcium channels at the z-lines in order to efficiently initiate excitation-contraction coupling.
In contrast to this distribution of sodium channel subtypes in human cardiac myocytes, the distribution of sodium channels in mouse ventricular myocytes is rather different.86–88 In the mouse heart, NaV1.5 channels are localized in high density in the intercalated discs, whereas NaV1.1 channels are localized in lower density at the z-lines.86 The high concentration of sodium channels at the ends of mouse ventricular myocytes suggests that they conduct action potentials in a salutatory manner, like a myelinated nerve; that is, the large sodium current entering at the intercalated disc instantaneously depolarizes the entire myocyte and directly activates the high density of sodium channels at the intercalated discs at the other end of the cell, rather than being conducted progressively across the cell. The instantaneous depolarization of the entire cell surface would then initiate action potentials in the T-tubules conducted by the TTX-sensitive sodium channels there. This altered mode of initiation and conduction of the cardiac action potential could be a specialization that supports the rapid beating rate of the mouse heart (600 bpm) without loss of synchrony and force of contraction.
References
1. Catterall, WA. The molecular basis of neuronal excitability. Science. 1984; 223:653–661.
2. Hartshorne, RP, Keller, BU, Talvenheimo, JA, et al. Functional reconstitution of the purified brain sodium channel in planar lipid bilayers. Proc Natl Acad Sci USA. 1985; 82:240–244.
3. Numa, S, Noda, M. Molecular structure of sodium channels. Ann N Y Acad Sci. 1986; 479:338–355.
4. Guy, HR, Seetharamulu, P. Molecular model of the action potential sodium channel. Proc Natl Acad Sci USA. 1986; 508:508–512.
5. Catterall, WA. Molecular properties of voltage-sensitive sodium channels. Annu Rev Biochem. 1986; 55:953–985.
6. Catterall, WA. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 2000; 26:13–25.
7. Isom, LL, De Jongh, KS, Patton, DE, et al. Primary structure and functional expression of the b1 subunit of the rat brain sodium channel. Science. 1992; 256:839–842.
8. Isom, LL, Ragsdale, DS, De Jongh, KS, et al. Structure and function of the beta-2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM-motif. Cell. 1995; 83:433–442.
9. Morgan, K, Stevens, EB, Shah, B, et al. Beta-3: An additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci U S A. 2000; 97:2308–2313.
10. Yu, FH, Westenbroek, RE, Silos-Santiago, I, et al. Sodium channel beta-4, a new disulfide-linked auxiliary subunit with similarity to beta-2. J Neurosci. 2003; 23:7577–7585.
11. McCormick, KA, Srinivasan, J, White, K, et al. The extracellular domain of the b1 subunit is both necessary and sufficient for b1-like modulation of sodium channel gating. J Biol Chem. 1999; 274:32638–32646.
12. Ratcliffe, CF, Qu, Y, McCormick, KA, et al. A sodium channel signaling complex: Modulation by associated receptor protein tyrosine phosphatase b. Nat Neurosci. 2000; 3:437–444.
13. Srinivasan, J, Schachner, M, Catterall, WA. Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proc Natl Acad Sci USA. 1998; 95:15753–15757.
14. Ratcliffe, CF, Westenbroek, RE, Curtis, R, et al. Sodium channel beta-1and beta-3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol. 2001; 154:427–434.
15. Kazarinova-Noyes, K, Malhotra, JD, McEwen, DP, et al. Contactin associates with sodium channels and increases their functional expression. J Neurosci. 2001; 21:7517–7525.
16. Malhotra, JD, Koopmann, MC, Kazen-Gillespie, KA, et al. Structural requirements for interaction of sodium channel beta 1 subunits with ankyrin. J Biol Chem. 2002; 277:26681–26688.
17. Xiao, ZC, Ragsdale, DS, Malhotra, JD, et al. Tenascin-R is a functional modulator of sodium channel b subunits. J Biol Chem. 1999; 274:26511–26517.
18. Chen, C, Bharucha, V, Chen, Y, et al. Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci U S A. 2002; 99:17072–17077.
19. Chen, C, Westenbroek, RE, Xu, X, et al. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci. 2004; 24:4030–4042.
20. Lopez-Santiago, LF, Meadows, LS, Ernst, SJ, et al. Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J Mol Cell Cardiol. 2007; 43:636–647.
21. Payandeh, J, Scheuer, T, Zheng, N, et al. The crystal structure of a voltage-gated sodium channel. Nature. 2011; 475:353–358.
22. Shapiro, L, Doyle, JP, Hensley, P, et al. Crystal structure of the extracellular domain from Po, the major structural protein of peripheral nerve myelin. Neuron. 1996; 17:435–449.
23. Isom, LL. Sodium channel b-subunits: Anything but auxiliary. Neuroscientist. 2001; 7:42–54.
24. Hodgkin, AL, Huxley, AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952; 117:500–544.
25. Armstrong, CM. Sodium channels and gating currents. Physiol Rev. 1981; 61:644–682.
26. Hille, B. Ionic Channels of Excitable Membranes, ed 3. Sunderland, MA: Sinauer Associates Inc; 2001.
27. Hille, B. The permeability of the sodium channel to metal cations in myelinated nerve. J Gen Physiol. 1972; 59:637–658.
28. Noda, M, Suzuki, H, Numa, S, et al. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989; 259:213–216.
29. Terlau, H, Heinemann, SH, Stühmer, W, et al. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991; 293:93–96.
30. Lipkind, GM, Fozzard, HA. A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophys J. 1994; 66:1–13.
31. Penzotti, JL, Fozzard, HA, Lipkind, GM, et al. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys J. 1998; 75:2647–2657.
32. Heinemann, SH, Terlau, H, Stühmer, W, et al. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992; 356:441–443.
33. Chiamvimonvat, N, O’Rourke, B, Kamp, TJ, et al. Functional consequences of sulfhydryl modification in the pore-forming subunits of cardiovascular Ca2+ and Na+ channels. Circ Res. 1995; 76:325–334.
34. Pérez-García, MT, Chiamvimonvat, N, Marban, E, et al. Structure of the sodium channel pore revealed by serial cysteine mutagenesis. Proc Natl Acad Sci USA. 1996; 93:300–304.
35. Schlief, T, Schönherr, R, Imoto, K, et al. Pore properties of rat brain II sodium channels mutated in the selectivity filter domain. Eur Biophys J. 1996; 25:75–91.
36. Sun, YM, Favre, I, Schild, L, et al. On the structural basis for size-selective permeation of organic cations through the voltage-gated sodium channel—Effect of alanine mutations at the DEKA locus on selectivity, inhibition by Ca2+ and H+, and molecular sieving. J Gen Physiol. 1997; 110:693–715.
37. Hille, B. Ionic selectivity, saturation, and block in sodium channels. A four-barrier model. J Gen Physiol. 1975; 66:535–560.
38. Hirschberg, B, Rovner, A, Lieberman, M, et al. Transfer of twelve charges is needed to open skeletal muscle Na+ channels. J Gen Physiol. 1995; 106:1053–1068.
39. Stuhmer, W, Conti, F, Suzuki, H, et al. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989; 339:597–603.
40. Yang, N, Horn, R. Evidence for voltage-dependent S4 movement in sodium channel. Neuron. 1995; 15:213–218.
41. Yang, NB, George, AL, Jr., Horn, R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996; 16:113–122.
42. Catterall, WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010; 67:915–928.
43. Jiang, Y, Lee, A, Chen, J, et al. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002; 417:515–522.
44. Jiang, Y, Lee, A, Chen, J, et al. The open pore conformation of potassium channels. Nature. 2002; 417:523–526.
45. Zhao, Y, Yarov-Yarovoy, V, Scheuer, T, et al. A gating hinge in Na+ channels; a molecular switch for electrical signaling. Neuron. 2004; 41:859–865.
46. Vassilev, PM, Scheuer, T, Catterall, WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988; 241:1658–1661.
47. Vassilev, P, Scheuer, T, Catterall, WA. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc Natl Acad Sci USA. 1989; 86:8147–8151.
48. West, JW, Patton, DE, Scheuer, T, et al. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc Natl Acad Sci USA. 1992; 89:10910–10914.
49. Eaholtz, G, Scheuer, T, Catterall, WA. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994; 12:1041–1048.
50. Kellenberger, S, West, JW, Scheuer, T, et al. Molecular analysis of the putative inactivation particle in the inactivation gate of brain type IIA Na+ channels. J Gen Physiol. 1997; 109:589–605.
51. Kellenberger, S, Scheuer, T, Catterall, WA. Movement of the Na+ channel inactivation gate during inactivation. J Biol Chem. 1996; 271:30971–30979.
52. Kellenberger, S, West, JW, Catterall, WA, et al. Molecular analysis of potential hinge residues in the inactivation gate of brain type IIA Na+ channels. J Gen Physiol. 1997; 19:607–617.
53. Rohl, CA, Boeckman, FA, Baker, C, et al. Solution structure of the sodium channel inactivation gate. Biochemistry. 1999; 38:855–861.
54. McPhee, JC, Ragsdale, DS, Scheuer, T, et al. A critical role for transmembrane segment IVS6 of the sodium channel a subunit in fast inactivation. J Biol Chem. 1995; 270:12025–12034.
55. Smith, MR, Goldin, AL. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys J. 1997; 73:1885–1895.
56. Lerche, H, Peter, W, Fleischhauer, R, et al. Role in fast inactivation of the IV/S4-S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. J Physiol (Lond). 1997; 505:345–352.
57. McPhee, JC, Ragsdale, D, Scheuer, T, et al. A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel a subunit in fast inactivation. J Biol Chem. 1998; 273:1121–1129.
58. Tang, LH, Chehab, N, Wieland, SJ, et al. Glutamine substitution at Alanine1649 in the S4-S5 cytoplasmic loop of domain 4 removes the voltage sensitivity of fast inactivation in the human heart sodium channel. J Gen Physiol. 1998; 111:639–652.
59. Filatov, GN, Nguyen, TP, Kraner, SD, et al. Inactivation and secondary structure in the D4/S4-5 region of the SkM1 sodium channel. J Gen Physiol. 1998; 111:703–715.
60. Chen, LQ, Santarelli, V, Horn, R, et al. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol. 1996; 108:549–556.
61. Rogers, JC, Qu, Y, Tanada, TN, et al. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. J Biol Chem. 1996; 271:15950–15962.
62. Sheets, MF, Kyle, JW, Kallen, RG, et al. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys J. 1999; 77:747–757.
63. Cha, A, Ruben, PC, George, AL, et al. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999; 22:73–87.
64. Payandeh, J, Gamal El-Din, TM, Scheuer, T, et al. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012; 486:135–139.
65. Ragsdale, DS, McPhee, JC, Scheuer, T, et al. Molecular determinants of state-dependent block of sodium channels by local anesthetics. Science. 1994; 265:1724–1728.
66. Liu, G, Yarov-Yarovoy, V, Qu, Y, et al. Differential Interactions of lamotrigine and related drugs with transmembrane segment IVS6 of voltage-gated sodium channels. Neuropharm. 2003; 44:413–422.
67. Yarov-Yarovoy, V, Brown, J, Sharp, E, et al. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na+ channel a subunit. J Biol Chem. 2001; 276:20–27.
68. Yarov-Yarovoy, V, McPhee, JC, Idsvoog, D, et al. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the sodium channel alpha subunit in voltage-dependent gating and drug block. J Biol Chem. 2002; 277:35393–35401.
69. Hille, B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977; 69:497–515.
70. Hondeghem, LM, Katzung, BG. Antiarrhythmic agents: The modulated receptor mechanism of action of sodium and calcium channel blocking drugs. Annu Rev Pharmacol Toxicol. 1984; 24:387–423.
71. Catterall, WA. Common modes of drug action on Na+ channels: Local anesthetics, antiarrhythmics and anticonvulsants. Trends Pharmacol Sci. 1987; 8:57–65.
72. Ragsdale, DR, McPhee, JC, Scheuer, T, et al. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci USA. 1996; 93:9270–9275.
73. Wright, SN, Wang, SY, Wang, GK. Lysine point mutations in Na+ channel D4-S6 reduce inactivated channel block by local anesthetics. Mol Pharmacol. 1998; 54:733–739.
74. Nau, C, Wang, SY, Wang, GK. Point mutations at L1280 in Nav1. 4 channel D3-S6 modulate binding affinity and stereoselectivity of bupivacaine enantiomers. Mol Pharmacol. 2003; 63:1398–1406.
75. Weiser, T, Qu, Y, Catterall, WA, et al. Differential interaction of R-mexiletine with the local anesthetic receptor site on brain and heart sodium channel alpha-subunits. Mol Pharmacol. 1999; 56:1238–1244.
76. Lipkind, GM, Fozzard, HA. Molecular modeling of local anesthetic drug binding by voltage-gated sodium channels. Mol Pharmacol. 2005; 68:1611–1622.
77. Cestele, S, Catterall, WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000; 82:883–892.
78. Catterall, WA, Cestele, S, Yarov-Yarovoy, V, et al. Voltage-gated ion channels and gating modifier toxins. Toxicon. 2007; 49:124–141.
79. Yu, FH, Catterall, WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE. 2004; 2004:re15.
80. Goldin, AL, Barchi, RL, Caldwell, JH, et al. Nomenclature of voltage-gated sodium channels. Neuron. 2000; 28:365–368.
81. Rogart, RB, Cribbs, LL, Muglia, LK, et al. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc Natl Acad Sci USA. 1989; 86:8170–8174.
82. Satin, J, Kyle, JW, Chen, M, et al. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 1992; 256:1202–1205.
83. Sivilotti, L, Okuse, K, Akopian, AN, et al. A single serine residue confers tetrodotoxin insensitivity on the rat sensory-neuron-specific sodium channel SNS. FEBS Lett. 1997; 409:49–52.
84. Hiyama, TY, Watanabe, E, Okado, H, et al. The subfornical organ is the primary locus of sodium-level sensing by Na(x) sodium channels for the control of salt-intake behavior. J Neurosci. 2004; 24:9276–9281.
85. Westenbroek, R, Kaufmann, S, Lange, V, et al. Localization and function of sodium channel subtypes in human heart. Circulation. 2007; 116:11–12.
86. Maier, SK, Westenbroek, RE, Schenkman, KA, et al. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci USA. 2002; 99:4073–4078.
87. Maier, SK, Westenbroek, RE, Yamanushi, TT, et al. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci U S A. 2003; 100:3507–3512.
88. Maier, SK, Westenbroek, RE, McCormick, KA, et al. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation. 2004; 109:1421–1427.
89. Yarov-Yarovoy, V, DeCaen, PG, Westenbroek, RE, et al. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci U S A. 2012; 109:E93–E102.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]1
Voltage-Gated Sodium Channels and Electrical Excitability of the Heart
Voltage-gated sodium channels initiate action potentials in cardiac myocytes and other excitable cells, and they are responsible for propagation of action potentials through the atria, conduction system, and ventricles of the heart. As shown in Figure 1-1, action potentials in atrial and ventricular muscle fibers rise rapidly from a resting potential near −80 mV and reach their peak within 1 ms. During this brief interval, cardiac sodium channels respond to the change in pacemaker potential as it reaches threshold and open to allow rapid Na+ entry. Sodium channels begin to inactivate as soon as they open and inactivate to 98% or 99% completion within a few milliseconds. The plateau phase of the cardiac action potential is generated by opening of voltage-gated calcium channels (see Chapter 2), and the cell is finally repolarized by slower opening of voltage-gated potassium channels (see Chapter 3). The rate of conduction of the action potential through the cardiac tissue depends directly on the rate of rise of the cardiac action potential and therefore on the density of sodium channels and their rate of activation.
Figure 1-1 Cardiac action potential in sheep heart. Cardiac myocytes in left ventricle or left atrium were impaled with a microelectrode, and the cardiac action potential was recorded. (Courtesy J. Jalife.)
Subunit Structure of Sodium Channels
Sodium channel proteins purified from excitable cells are complexes composed of an approximately 260-kD α-subunit in association with one or two auxiliary β-subunits of approximately 33 to 39 kD.1 Purified sodium channel complexes of α- and β-subunits are sufficient for voltage-dependent gating and ion conduction in artificial lipid membranes and expression of the α-subunit alone is sufficient for physiologic function in recipient nonexcitable cells, indicating that this subunit has all the necessary structural elements for voltage-dependent gating and ion conduction.1–3 The primary sequence predicts that the sodium channel α-subunit folds into four internally repeated domains (I-IV), each of which contains six α-helical transmembrane segments (S1-S61,3–6; Figure 1-2). In each domain, the S1-S4 segments serve as the voltage-sensing module, and the S5 and S6 segments and the reentrant P loop between them serve as the pore-forming module. Extracellular loops connect the S5 and S6 transmembrane segments to the P loop in each domain, whereas the other extracellular loops are small. Large intracellular loops link the four homologous domains, and the large N-terminal and C-terminal domains also contribute substantially to the mass of the internal face of sodium channels. This view of sodium channel architecture, originally derived from hydrophobicity analysis of the amino acid sequence,4 has been largely confirmed by biochemical, electrophysiologic, and structural experiments.6
Figure 1-2 Transmembrane organization of sodium channel subunits. The primary structures of the subunits of the voltage-gated ion channels are illustrated as transmembrane folding diagrams. Cylinders represent probable alpha helical segments: blue, S1-S3; green, S4; yellow, S5; red, S6; shaded orange area, outer pore loop; purple, intracellular S4-S5 helix. Bold lines represent the polypeptide chains of each subunit with length approximately proportional to the number of amino acid residues in the brain sodium channel subtypes. The extracellular domains of the β1- and β2-subunits are shown as immunoglobulin-like folds Ψ, sites of probable N-linked glycosylation. P, sites of demonstrated protein phosphorylation by PKA (circles) and PKC (diamonds); white circles, the outer (EEDD) and inner (DEKA) rings of amino residues that form the ion selectivity filter and the tetrodotoxin binding site; ++, S4 voltage sensors; h in shaded circle, inactivation particle in the inactivation gate loop; open shaded circles, sites implicated in forming the inactivation gate receptor. The structure of the extracellular domain of the β-subunits is illustrated as an immunoglobulin-like fold based on amino acid sequence homology to the myelin P0 protein.8,22 Sites of binding of α- and β-scorpion toxins and a site of interaction between α- and β1-subunits are also shown. (Adapted from Catterall WA: From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26:13–25, 2000.)
The auxiliary β subunits were identified in the initial purification studies of sodium channels.1 These subunits have a single transmembrane segment, a large N-terminal extracellular domain that is homologous in structure to a variable chain (V-type) immunoglobulin-like fold, and a short C-terminal intracellular segment (see Figure 1-2).7–10 The β-subunits interact with α-subunits through their extracellular immunoglobulin (Ig)-fold domains, modulate α-subunit function, and enhance their cell surface expression.11 Like other proteins with an extracellular Ig-fold, they also serve as cell adhesion molecules by interacting with extracellular matrix proteins, cell adhesion molecules, and cytoskeletal linker proteins.12–17 These interactions are thought to localize and stabilize sodium channels in specific subcellular compartments and to bring crucial signaling molecules to the sodium channel to regulate it. Deletion of the genes encoding β-subunits causes alterations in sodium channel function; reduced action potential conduction and abnormal development of myelin folds in axons; hyperexcitability and epilepsy in the brain; and arrhythmias in the heart.18–20
Three-Dimensional Structure of Sodium Channels
Sodium channel architecture has been revealed in three dimensions by determination of the crystal structure of the bacterial sodium channel NavAb at high resolution (2.7 Å (0.27 nm); Figure 1-3). This ancient sodium channel has a simple structure—four identical subunits that are each similar to one homologous domain of a mammalian sodium channel without the large intracellular and extracellular loops of the mammalian protein.21 This structure has revealed a wealth of new information about the structural basis for sodium selectivity and conductance, the mechanism for block of the channel by therapeutically important drugs, and the mechanism of voltage-dependent gating. As viewed from the top, NavAb has a central pore surrounded by four pore-forming modules composed of S5 and S6 segments and the intervening pore loop (see Figure 1-3, A). Four voltage-sensing modules composed of S1-S4 segments are symmetrically positioned around the outer rim of the pore module (see Figure 1-3, A). The transmembrane architecture of NavAb shows that the adjacent subunits have swapped their functional domains such that each voltage-sensing module is most closely positional around with the pore-forming module of its neighbor (see Figure 1-3, B). It is likely that this domain-swapped arrangement enforces concerted gating of the four subunits or domains of sodium channels.
Figure 1-3 Three-dimensional structure of sodium channels. A, Top view of NavAb channels colored according to crystallographic temperature factors of the main-chain (blue < 50 Å2 to red > 150 Å2). B, Side view of NavAb. C, Structural elements in NavAb. The structural components of one subunit are highlighted. 1-6, Transmembrane segments S1-S6. (Adapted from Payandeh J, Scheuer T, Zheng N, et al: The crystal structure of a voltage-gated sodium channel. Nature 475:353–358, 2011.)
Comparison of the primary structures of the auxiliary β-subunits to those of other proteins revealed a close structural relationship to the family of proteins that contain Ig-like folds, which include many cell-adhesion molecules.8 The extracellular domains of these type-I single-membrane-spanning proteins are predicted to fold in a similar manner as myelin protein P0, whose Ig-like fold is known to be formed by a sandwich of two β-sheets held together by hydrophobic interactions (see Figure 1-222). Myelin P0 protein is a cell adhesion molecule involved in tight wrapping of myelin sheets, and many related cell adhesion molecules with extracellular Ig-folds and a single membrane-spanning segment are involved in cell-cell interactions among neurons and glia.22 As expected from their structure, NaVβ subunits interact with extracellular matrix molecules, other cell adhesion molecules, and intracellular cytoskeletal proteins and signaling proteins.12–16,23
Sodium Channel Structure and Function
Hodgkin and Huxley24 defined the three key functions of sodium channels: (1) voltage-dependent activation, (2) fast inactivation, and (3) selective ion conductance. Building on this foundation, detailed biophysical studies revealed the ion selectivity of the channel pore, detected the movement of the voltage sensors as gating current, and developed mechanistic models for these essential channel functions.25,26 Recent structure-function studies using molecular, biochemical, structural, and electrophysiologic techniques have provided clear understanding of the molecular and structural basis for these sodium channel functions.
Outer Pore and Selectivity Filter
Voltage clamp studies showed that sodium channels are highly selective for sodium versus potassium and other monovalent cations.26,27 Analysis of ion selectivity and block by tetrodotoxin and saxitoxin led to a model of tetrodotoxin and saxitoxin as plugs of the selectivity filter in the outer pore of sodium channels.26 Mutational analysis identified a key glutamate residue in the membrane-reentrant loop in domain I as a crucial residue for tetrodotoxin and saxitoxin binding.28 Additional studies revealed a pair of important amino acid residues, mostly negatively charged, in analogous positions in all four domains (see Figure 1-2, B, small white circles).29–31 Mutation of a set of four residues in analogous positions in each domain (aspartate in domain I, glutamate in domain II, lysine in domain III, and alanine in domain IV, DEKA) to glutamates confers calcium selectivity,32 indicating that the side chains of these amino acid residues are likely to interact with sodium ions as they are conducted through the ion selectivity filter of the pore. These results showed that the narrow selectivity filter in the outer pore is formed by the reentrant P loops between transmembrane segments S5 and S6 of each domain (see Figure 1-2). Mutations in this postulated ring of four amino acid residues have strong effects on selectivity for organic and inorganic monovalent cations, in agreement with the idea that they form the selectivity filter, and structure-function studies suggest specific structural interactions and functional roles for the P loops from the four domains.33–36
In the bacterial sodium channel NavAb, the overall pore architecture includes a large external vestibule, a narrow ion selectivity filter containing the amino acid residues shown to determine ion selectivity in vertebrate sodium and calcium channels, a large central cavity that is lined by the S6 segments and is filled with water, and an intracellular activation gate formed at the crossing of the S6 segments at the intracellular surface of the membrane (Figure 1-4, A). The activation gate is tightly closed in the NavAb structure (see Figure 1-4, B), and there is no space for ions or water to move through it. This general architecture resembles voltage-gated potassium channels (see Chapter 3). Although the overall pore architecture of sodium and potassium channels is similar, the structures of their ion selectivity filters and their mechanisms of ion selectivity and conductance are completely different. Potassium channels select K+ by direct interaction with a series of four ion coordination sites formed by the backbone carbonyls of the amino acid residues that comprise the ion selectivity filter (Chapter 3). No charged amino acid residues are involved, and no water molecules intervene between K+ and its interacting backbone carbonyls in the ion selectivity filter of potassium channels. In contrast, the NavAb ion selectivity filter has a high field strength site at its extracellular end (see Figure 1-4, C; Glu177 = ε177), which is formed by amino acid residues that are highly conserved and are key determinants of ion selectivity in vertebrate sodium and calcium channels (see Figure 1-2). Considering its dimensions of approximately 4.6 Å2, Na+ with two planar waters of hydration could fit in this high–field-strength site. This outer site is followed by two ion coordination sites formed by backbone carbonyls (see Figure 1-4, D). These two carbonyl sites are perfectly designed to bind Na+ with four planar waters of hydration, but would be much too large to bind Na+ directly. In fact, the NavAb selectivity filter is large enough to fit the entire K+ channel ion selectivity filter inside it. Thus, the chemistry of Na+ selectivity and conductance is opposite to that of K+: negatively charged residues interact with Na+ to remove most (but not all) of its waters of hydration, and Na+ is conducted as a hydrated ion interacting with the pore through its inner shell of bound waters. Theoretical considerations of sodium selectivity and conductance predicted an outer high–field-strength site that would only partially dehydrate the permeating ion and two inner sites that would conduct and rehydrate the permeant Na+ ion because of the high energy of hydration of Na+ (see Figure 1-4, C, D).37
Figure 1-4 NavAb pore and selectivity filter. A, Architecture of the NavAb pore. Purple, Glu177 side-chains; gray, pore volume. B, The closed activation gate at the intracellular end of the pore illustrating the close interaction of Met221 residues in closing the pore. C, Top view of the ion selectivity filter. Symmetry-related molecules are colored white and yellow; P-helix residues are colored green. Hydrogen bonds between Thr175 and Trp179 are indicated by gray dashes. Electron densities from Fo-Fc omit maps are contoured at 4.0 σ (blue and gray), and subtle differences can be appreciated (small arrows). D, Side view of the selectivity filter. Glu177 (purple) interactions with Gln172, Ser178, and the backbone of Ser180 are shown in the far subunit. Fo-Fc omit map, 4.75 σ (blue); putative cations or water molecules (red spheres, IonEX). Electron-density around Leu176 (gray; Fo-Fc omit map at 1.75 σ) and a putative water molecule are shown (grey sphere). Na+-coordination sites: SiteHFS, SiteCEN and SiteIN. (Adapted from Payandeh J, Scheuer T, Zheng N, et al: The crystal structure of a voltage-gated sodium channel. Nature 475:353–358, 2011.)
Voltage-Dependent Activation
The voltage dependence of the activation of sodium channels derives from outward movement of approximately 12 gating charges as a consequence of depolarization of the membrane and reduction of the membrane electric field.25,38 The S4 segments of each homologous domain serve as the primary voltage sensors for activation.4,5 They contain repeated motifs of a positively charged amino acid residue followed by two hydrophobic residues creating a transmembrane spiral of positive charges. Upon depolarization, outward movement and rotation of S4 is thought to initiate a conformational change that opens the sodium channel pore.4,5 This “sliding helix” or “helical screw” model is supported by strong evidence. For example, neutralization of the positively charged residues in S4 reduce the voltage-dependence of gating.39 The outward and rotational gating movement of S4 segment has been detected directly by reaction of substituted cysteine residues in S4 segments with extracellular sulfhydryl reagents following channel activation and by analysis of the movement of fluorescent probes incorporated into these substituted cysteine residues.40,41 Other support for this mechanism derives from a wide range of structure and function studies.42
In the structure of the bacterial sodium channel NavAb, the S4 segment is in a transmembrane position in its activated state and its positive charges are thought to be neutralized by negative charges in the nearby S1 and S2 segments (Figure 1-5, A).21 At the resting membrane potential, the force of the electric field, which is negative inside the cell, would pull the positive charges inward. Depolarization would abolish this force and allow an outward movement of the S4 helix and its gating charges, catalyzed by exchange of ion pair partners (see Figure 1-5, B; Movie 1-1). After conformational changes have occurred in all four domains, the transmembrane pore can open and conduct ions (see Movie 1-1). This structural model shows that the S4 segment and its gating charges move through a narrow gating pore that focuses the transmembrane electric field to a distance of approximately 5 Å normal to the membrane and allows the gating charges to move from an intracellular aqueous vestibule to an extracellular aqueous vestibule with a short transit through the channel protein (see Video 1-1).
Figure 1-5 The voltage-sensing domain (VSD). A, Side view of the VSD illustrating the extracellular negative charge-cluster (red; ENC), the intracellular negative charge-cluster (red; INC), hydrophobic constriction site (green; HCS), residues of the S1N helix (cyan), and phenylalanine of the S2-S3 loop (purple). S4 segment and gating charges (R1-R4) are in yellow. B, Transmembrane view of the lowest-energy Rosetta models of the VSD of NaChBac in Resting State 1 (left) and Activated State 3 (right). Side chains of the gating-charge-carrying arginines in S4 and key residues in S1, S2, and S3 segments are shown in stick representation and labeled. Gray, blue, and red atoms are C, N, and O, respectively. The HCS is highlighted by orange bars
[/not-level-membership-for-cardiovascular-category]
