Chapter 15 Viral, fungal, protozoal and helminthic infections
• Viruses present a more difficult problem of chemotherapy than do higher organisms, e.g. bacteria, for they are intracellular parasites that use the metabolism of host cells.1 Highly selective toxicity is, therefore, harder to achieve. In the past 15 years, identification of the molecular differences between viral and human metabolism has led to the development of many effective antiviral agents; four were available in 1990, now there are over 40.
• Fungal infections range from inconvenient skin conditions to life-threatening systemic diseases; the latter have become more frequent as opportunistic infections in patients immunocompromised by drugs or AIDS, or in those receiving intensive medical and surgical interventions in intensive care units.
• Protozoal infections. Malaria is the major transmissible parasitic disease in the world. Drug resistance is an increasing problem and differs with geographical location, and species of plasmodium.
• Helminthic infestations cause considerable morbidity. The drugs that are effective against these organisms are summarised.
Viral infections
Antiviral agents are most active when viruses are replicating. The earlier that treatment is given, therefore, the better the result. Apart from primary infection, viral illness is often the consequence of reactivation of latent virus in the body. Patients whose immune systems are compromised may suffer particularly severe illness. Viruses are capable of developing resistance to antimicrobial drugs, with similar implications for the individual patient, for the community and for drug development. An overview of drugs that have proved effective against virus diseases appears in Table 15.1.
| Organism | Drug of choice | Alternative |
|---|---|---|
| Varicella zoster | ||
| chickenpox | Aciclovir | Valaciclovir or famciclovir |
| zoster | Aciclovir or famciclovir | Valaciclovir |
| Herpes simplex | ||
| keratitis | Aciclovir (topical) | |
| labial | Aciclovir (topical and/or oral) | Valaciclovir or famciclovir |
| genital | Aciclovir (topical and/or oral) | Valaciclovir |
| Famciclovir (oral) | Penciclovir | |
| encephalitis | Aciclovir | |
| disseminated | Aciclovir | Foscarnet |
| Human immunodeficiency virus (HIV) | Lamivudine/emtricitabine | Abacavir |
| Tenofovir | Didanoside | |
| Zidovudine | Stavudine | |
| Lopinavir/ritonavir | Saquinavir | |
| Atazanavir | Darunavir | |
| Fosamprenavir | Tipranavir | |
| Efavirenz | Nevirapine | |
| Etravirine | ||
| Raltegravir | ||
| Enfuvirtide | ||
| Maraviroc | ||
| Hepatitis B | Pegylated interferon α-2a and interferon 2b, lamivudine | Adefovir, tenofovir, entecavir, telbivudine |
| Hepatitis C | Pegylated interferon α-2a or interferon 2b plus ribavirin | |
| Hepatitis D | Interferon-α | Pegylated interferon α-2a and interferon 2b |
| Influenza A | Zanamivir, oseltamivir | Amantadine |
| Cytomegalovirus (CMV) | Valganciclovir, ganciclovir | Foscarnet, cidofovir |
| Respiratory syncytial virus | Ribavirin | Palivizumab |
| Papillomavirus (genital warts) | Imiquimod | |
| Molluscum contagiosum | Imiquimod | Cidofovir |
Herpes simplex and varicella zoster
Aciclovir
• Skin infections, including initial and recurrent labial and genital herpes, most effective when new lesions are forming; skin and mucous membrane infections (as tablets or oral suspension).
• Ocular keratitis (topical treatment with ophthalmic ointment is standard, oral treatment is also effective).
• Prophylaxis and treatment in the immunocompromised (oral, as tablets or suspension).
Aciclovir-resistant herpes simplex virus has been reported in patients with AIDS but remains rare in immunocompetent patients. Foscarnet (p. 221) and cidofovir (p. 221) have been used in these cases.
• Chickenpox, particularly in the immunocompromised (i.v.) or in the immunocompetent with pneumonitis or hepatitis (i.v.).
• Shingles in immunocompetent persons (as tablets or suspension, and best started within 48 h of the appearance of the rash). Immunocompromised persons will often have more severe symptoms and require i.v. administration.
Human immunodeficiency virus (HIV)
General comments
• The aims of antiretroviral therapy are to delay disease progression and prolong survival by suppressing the replication of the virus. Optimal suppression also prevents the emergence of drug resistance and reduces the risks of onward transmission to sexual partners and the unborn children of HIV-infected mothers. Virological failure may be defined as primary where there is inability to reduce plasma HIV viral load to fewer than 50 copies per microlitre despite 6 months of antiretroviral therapy, or secondary if there is failure to maintain viral load suppression at less than 50 copies per microlitre.
• No current antiviral agents or combinations eliminate HIV infection, but the most effective combinations (so-called ‘highly active antiretroviral therapy’, HAART) produce profound suppression of viral replication in many patients and allow useful reconstitution of the immune system, measured by a fall in the plasma viral load and an increase in the numbers of cytotoxic T cells (CD4 count). Rates of opportunistic infections such as Pneumocystis carinii pneumonia and cytomegalovirus (CMV) retinitis are reduced when CD4 counts are restored, and life expectancy is markedly increased.
• Combination therapy reduces the risks of emergence of resistance to antiretroviral drugs, which is increasing in incidence even in patients newly diagnosed with HIV. Mutations in the viral genome either prevent binding of the drug to the active site of the protease or reverse transcriptase enzymes, or lead to removal of the drug from the reverse transcriptase active site. The potential for rapid development of resistance is immense because untreated HIV replicates rapidly (50% of circulating virus is replaced daily), the spontaneous mutation rate is high, the genome is small, the virus will develop single mutations at every codon every day, and for many antiretroviral agents a single mutation will render the virus fully resistant.
• The decision to begin antiretroviral therapy is based primarily on the CD4 cell count (most current recommendations are to start in patients with counts below 350 cells per microlitre). Early initiation of antiretroviral therapy should also be considered for patients with CD4 cell count above 350 cells per microlitre but a low CD4 percentage (e.g < 14%), those with an AIDS diagnosis (e.g. Kaposi sarcoma), hepatitis B and HIV co-infection where treatment is indicated, and in conditions where achieving a suppressed viral load is desired in order to prevent transmission (e.g. in pregnancy).
• There are currently more than 20 approved antiretroviral agents in four classes, plus various fixed drug combinations (Table 15.2).
• Current HAART regimens use a combination of drugs that act at different phases of the viral life cycle. The most frequently used combinations employ a backbone of two nucleoside analogue reverse transcriptase inhibitors (NRTIs) plus either a non-nucleoside reverse transcriptase inhibitor (NNRTI) or a ritonavir-boosted protease inhibitor (rPI). The choice for the individual patient is best made after reference to contemporary, expert advice (see the websites listed in the Guide to further reading).
• Alternative combinations are used if these variables deteriorate or unwanted drug effects occur. Antiretroviral resistance testing, both genetic (by searching viral RNA for sequences coding for resistance) and phenotypic (by testing antiretroviral agents against the patient’s virus in cell culture), also guide the choice of drug regimen, especially after virological failure.
• Pregnancy and breast feeding pose special problems. The objectives of therapy are to minimise drug toxicity to the fetus while reducing the maternal viral load and the catastrophic results of HIV transmission to the neonate. Prevention of maternal–fetal and maternal–infant spread is the most cost-effective way of using antiretroviral drugs in less developed countries. Maternal–fetal transmission rates are related to maternal viral load, with rates of 0.1% reported when maternal viral load is less than 50 copies per microlitre while on HAART. Where resources permit, access to safe alternatives to breast feeding should be provided to infected mothers.
• Combination antiretroviral therapy, especially the thymidine nucleoside analogue reverse transcriptase inhibitors zidovudine and stavudine, causes redistribution of body fat in some patients – the ‘lipodystrophy syndrome’. Protease inhibitors can disturb lipid and glucose metabolism to a degree that warrants a change to drugs with limited effects on lipid metabolism, e.g. ritonavir-boosted atazanavir, and the introduction of lipid-lowering agents.
• Impaired cell-mediated immunity leaves the host prey to opportunistic infections including: candidiasis, coccidioidomycosis, cryptosporidiosis, CMV disease, herpes simplex, histoplasmosis, Pneumocystis carinii pneumonia, toxoplasmosis and tuberculosis (often with multiply resistant organisms). Treatment of these conditions is referred to elsewhere in this text.2
• Improvement in immune function as a result of antiretroviral treatment may provoke an inflammatory reaction against residual opportunistic organisms (immune reconstitution inflammatory syndrome, IRIS). Although infrequent, this may present with development of new infections or worsening opportunistic infections, e.g. tuberculosis and cryptococcal disease.
• Antiretroviral drugs may also be used in combination to reduce the risks of infection with HIV from injuries, e.g. from HIV-contaminated needles and following sexual exposure to a high-risk partner. The decision to offer such post-exposure prophylaxis (PEP), and the optimal combination of drugs used, is a matter for experts; administration must begin within a few hours of exposure and continue for 28 days.
• Some drugs described here have found additional indications, or are used only, for therapy of non-HIV infections, e.g. adefovir for chronic hepatitis B infection.
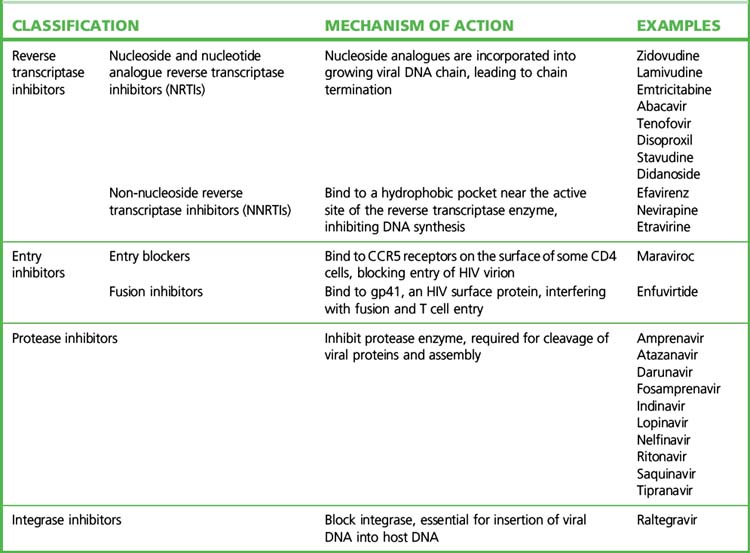
Nucleoside and nucleotide reverse transcriptase inhibitors
Protease inhibitors
• amprenavir, atazanavir, fosamprenavir (a prodrug of amprenavir), lopinavir, ritonavir, saquinavir, tipranavir, indinavir and darunavir.
Integrase inhibitors
Influenza A
Zanamivir (Relenza)
• At-risk patients (those with chronic respiratory or cardiovascular disease, immunosuppression or diabetes mellitus, or over the age of 65 years).
• When virological surveillance in the community indicates that influenza virus is circulating.
• Only those presenting within 48 h of the onset of influenza-like symptoms.
Zanamivir retains activity against amantadine-resistant and some oseltamivir-resistant strains.
Cytomegalovirus
Foscarnet
Foscarnet finds use i.v. for CMV retinitis in patients with HIV infection when ganciclovir is contraindicated, and for aciclovir-resistant herpes simplex virus infection (see p. 213). It is generally less well tolerated than ganciclovir; adverse effects include renal toxicity (usually reversible), nausea and vomiting, neurological reactions and marrow suppression. Hypocalcaemia is seen especially when foscarnet is given with pentamidine, e.g. during treatment of multiple infections in patients with AIDS. Renal toxicity can be minimised with good hydration and dose modification. Foscarnet causes a contact dermatitis which can lead to unpleasant genital ulcerations due to high urine drug concentrations; this is potentially preventable with good urinary hygiene.
Drugs that modulate the host immune system
Interferons
Virus infection stimulates the production of protective glycoproteins (interferons) which act:
• directly on uninfected cells to induce enzymes that degrade viral RNA
• indirectly by stimulating the immune system
• to modify cell regulatory mechanisms and inhibit neoplastic growth.
See also p. 553 for lamivudine and adefovir, use in chronic hepatitis B infection.
Fungal infections
Systemic mycoses
The principal treatment options are summarised in Table 15.3.
| Infection | Drug of first choice | Alternative |
|---|---|---|
| Aspergillosis | Amphotericin or voriconazole | Caspofungin, itraconazole, posaconazole |
| Blastomycosisa | Itraconazole or amphotericin | Fluconazole |
| Candidiasis | ||
| mucosal | Fluconazole or amphotericin | Caspofungin, voriconazole or fluconazole |
| systemic | Fluconazole or amphotericin ± flucytosine | Caspofungin, micafungin, anidulafungin, voriconazole |
| Coccidioidomycosisa | Fluconazole, amphotericin or itraconazole | |
| Cryptococcosis | Amphotericin + flucytosine (followed by fluconazole) | Fluconazole |
| Fusariosis | Voriconazole | Amphotericin |
| Histoplasmosis | Itraconazole or amphotericin | Fluconazole |
| chronic suppressionb | Itraconazole | Amphotericin |
| Mucormycosis | Amphotericin | Posaconazole |
| Paracoccidioidomycosis | Itraconazole or amphotericin | Ketoconazolec |
| Pseudallescheriasis | Voriconazole, ketoconazole or itraconazole | |
| Sporotrichosis | ||
| cutaneous | Itraconazole | Potassium iodide |
| deep | Amphotericin | Itraconazole or fluconazole |
| Tinea pedis | Terbinafine cream or topical azole (miconazole, clotrimazole, econazole) | Fluconazole |
This table was drawn substantially from The Medical Letter on Drugs and Therapeutics (2005, USA).
The authors are grateful to the Chairman of the Editorial Board for permission to publish the material.
a Patients with severe illness, meningitis, AIDS or some other causes of immunosuppression should receive amphotericin.
c Continue treatment for 6–12 months.




Drugs that disrupt the fungal cell membrane
Polyenes
Amphotericin (amphotericin B)
Amphotericin is at present the drug of choice for most systemic fungal infections (but see Table 15.3). The diagnosis of systemic infection should, whenever possible, be firmly established; tissue biopsy and culture may be necessary, and methods using the PCR to detect aspergillus DNA may revolutionise management of invasive infection.
Azoles
• Imidazoles (ketoconazole, miconazole, fenticonazole, clotrimazole, isoconazole, tioconazole) interfere with fungal oxidative enzymes to cause lethal accumulation of hydrogen peroxide; they also reduce the formation of ergosterol, an important constituent of the fungal cell wall which thus becomes permeable to intracellular constituents. Lack of selectivity in these actions results in important adverse effects.
• Triazoles (fluconazole, itraconazole, voriconazole, posaconazole) damage the fungal cell membrane by inhibiting lanosterol 14-α-demethylase, an enzyme crucial to ergosterol synthesis, resulting in accumulation of toxic sterol precursors. Triazoles have greater selectivity against fungi, better penetration of the CNS, resistance to degradation and cause less endocrine disturbance than do the imidazoles.
Ketoconazole
Ketoconazole is well absorbed from the gut (poorly where there is gastric hypoacidity; see below); it is widely distributed in tissues but concentrations in CSF and urine are low; its action is terminated by metabolism by cytochrome P450 3A (t½ 8 h). For systemic mycoses, ketoconazole (see Table 15.3) has been superseded by fluconazole and itraconazole on grounds of improved pharmacokinetics, tolerability and efficacy. Impairment of steroid synthesis by ketoconazole has been put to other uses, e.g. inhibition of testosterone synthesis lessens bone pain in patients with advanced androgen-dependent prostatic cancer.
Allylamine
Terbinafine
Terbinafine interferes with ergosterol biosynthesis, and thereby with the formation of the fungal cell membrane. It is absorbed from the gastrointestinal tract and undergoes extensive metabolism in the liver (t½ 14 h). Terbinafine is used topically for dermatophyte infections of the skin and orally for infections of hair and nails where the site, e.g. hair, severity or extent of the infection renders topical use inappropriate (see pp. 270–271). Treatment may need to continue for several weeks. Terbinafine may cause nausea, diarrhoea, dyspepsia, abdominal pain, headaches and cutaneous reactions.
Other antifungal drugs
Flucytosine
Flucytosine (5-fluorocytosine) is metabolised in the fungal cell to 5-fluorouracil, which inhibits nucleic acid synthesis. It is well absorbed from the gut, penetrates effectively into tissues, and almost all is excreted unchanged in the urine (t½ 4 h). The dose should be reduced for patients with impaired renal function, and the plasma concentration monitored. The drug is well tolerated when renal function is normal. Candida albicans rapidly becomes resistant to flucytosine, which ought not to be used alone; it may be combined with amphotericin (see Table 15.3), but this increases the risk of adverse effects (leucopenia, thrombocytopenia, enterocolitis) and it is reserved for serious infections where the risk:benefit balance is favourable, e.g. Cryptococcus neoformans meningitis.
Protozoal infections
Malaria
Life cycle of the malaria parasite and sites of drug action
The incubation period of malaria is 10–35 days. The principal features of the life cycle (Fig. 15.1) of the malaria parasite must be known in order to understand its therapy.
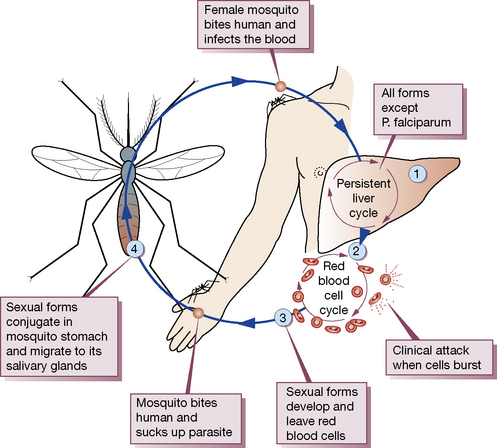
Hepatic cycle
Primaquine, proguanil and tetracyclines (tissue schizontocides) act at this site and are used for:
• Radical cure, i.e. an attack on persisting hepatic forms (hypnozoites, i.e. sleeping) once the parasite has been cleared from the blood; this is most effectively accomplished with primaquine; proguanil is only weakly effective.
• Preventing the initial hepatic cycle. This is also called causal prophylaxis. Primaquine was long regarded as too toxic for prolonged use but evidence now suggests it may be used safely, and it is inexpensive; proguanil is weakly effective. Doxycycline may be used short term.
Sexual forms
Some merozoites differentiate into male and female gametocytes in the erythrocytes and can develop further only if they are ingested by a mosquito, where they form sporozoites (site 4 in Fig. 15.1) and complete the transmission cycle.
Drugs used for malaria, and their principal actions, are classified in Table 15.4.
| Drug | Biological activity | |
|---|---|---|
| Blood schizontocide | Tissue schizontocide | |
| 4-Aminoquinolone | ||
| chloroquine | ++ | 0 |
| Arylaminoakohols | ||
| quinine | ++ | 0 |
| mefloquine | ++ | 0 |
| Phenanthrene methanol | ||
| halofantrine | ++ | 0 |
| lumefantrine | ++ | 0 |
| Antimetabolites | ||
| proguanil | + | + |
| pyrimethamine | + | 0 |
| sulfadoxine | + | 0 |
| dapsone | + | 0 |
| Antibiotics | ||
| tetracycline | + | + |
| doxycycline | + | + |
| minocycline | + | + |
| 8-Aminoquinolone | ||
| primaquine | 0 | + |
| Sesquiterpenes | ||
| artesunate | + | 0 |
| artemether | + | 0 |
Chemotherapy of an acute attack of malaria3
Successful management demands attention to the following points of principle:
• Whenever possible, the diagnosis should be confirmed before treatment by examination of blood smears; this is not often possible in the developing world, where clinically diagnosed illnesses may receive unnecessary courses of antimalarials, thus increasing the risks of plasmodial resistance.
• When the infecting organism is not known or infection is mixed, treatment should begin as for Plasmodium falciparum (below).
• Drugs used to treat Plasmodium falciparum malaria must always be selected with regard to the prevalence of local patterns of drug resistance.
• Patients not at risk of re-infection should be re-examined several weeks after treatment for signs of recrudescence, which may result from inadequate chemotherapy or survival of persistent hepatic forms.
Falciparum (‘malignant’) malaria
• A quinine salt:4 600 mg 8-hourly by mouth for 5–7 days, followed by doxycycline 200 mg daily for at least 7 days. This additional therapy is necessary as quinine alone tends to be associated with a higher rate of relapse. Clindamycin (450 mg four times daily for 7 days) may be given as an alternative follow-on therapy instead of doxycycline, and is particularly suitable for pregnant women. If the parasite is likely to be sensitive, Fansidar (pyrimethamine plus sulfadoxine) 3 tablets as a single dose is an alternative.
• Malarone (atovaquone and proguanil hydrochloride): 4 tablets once daily for 3 days.
• Riamet (artemether plus lumefantrine): if weight > 35 kg, 4 tablets initially, followed by five further doses of 4 tablets given at 8, 24, 36, 48 and 60 h.
• Mefloquine is also effective, but resistance has been reported in several regions, including South-East Asia. It is not necessary to use follow-on therapy after Riamet, mefloquine or Malarone.
• A quinine salt:4 20 mg/kg as a loading dose5 (maximum 1.4 g) infused i.v. over 4 h, followed 8 h later by a maintenance infusion of 10 mg/kg (maximum 700 mg) infused over 4 h, repeated every 8 h6 until the patient can swallow tablets to complete the 7-day course. Patients at increased risk of arrhythmias and the elderly should have ECG monitoring while on the infusion.
• Doxycycline or clindamycin should be given subsequently, as above (mefloquine is an alternative, but this must begin at least 12 h after parenteral quinine has ceased).
• Intravenous artesunate showed a clear benefit when compared to quinine in patients with severe falciparum malaria.7 A large randomised trial showed a 34% reduction in mortality with intravenous artesunate when compared to quinine; the number needed to treat to prevent one death was 13.8 Intravenous artesunate is not licensed in the European Union, but should be considered on a ‘named patient’ basis for severe cases not responsive to quinine or if quinine resistance is suspected, or there is a high parasite count (> 20%). Intravenous artesunate should be accompanied by a 7-day course of doxycycline.
Treatment in pregnancy should always be based on expert advice.
Non-falciparum (‘benign’) malarias
The drug of choice is chloroquine, which should be given by mouth as follows:
• Initial dose: 620 mg (base),9 then 310 mg as a single dose 6–8 h later.
• Primaquine, 15 mg/day for 14 days started after the chloroquine course has been completed. Plasmodium vivax infections require 30 mg/day for 14 days. Primaquine can lead to haemolysis in patients with G6PD deficiency (if mild G6PD deficiency, lower dosing of 45 mg once-weekly for 8 weeks may suffice without undue risk of haemolysis).
Chemoprophylaxis of malaria
General principles
• Chemoprophylaxis aims to prevent deaths from falciparum malaria, but only ever gives relative protection; travellers should guard against bites by using mosquito nets and repellents, and wearing well-covering clothing especially during high-risk times of day (after dusk).
• Mefloquine, doxycycline and atovaquone-proguanil (Malarone) are the most commonly advised prophylactic regimens, and are particularly recommended for areas of chloroquine-resistant falciparum malaria. Chloroquine, alone or in combination with proguanil, may be considered in areas of the world where the risk of acquiring chloroquine resistant falciparum malaria is low, although there is considerable concern regarding the protective efficacy of this regimen. Due to widespread P. falciparum resistance to proguanil, single-agent prophylaxis with this agent is rarely appropriate for most regions of the world.
• Effective chemoprophylaxis requires that there be a plasmodicidal concentration of drug in the blood when the first infected mosquito bites, and that it be sustained safely for long periods.
• The progressive rise in plasma concentration to steady state (after t½ × 5), sometimes attained only after weeks (consider mefloquine t½ 21 days, chloroquine t½ 50 days), allows unwanted effects (which can impair compliance or be unsafe) to be delayed, in some instances until after a subject has entered a malarial area. Thus, it is advised that prophylaxis begin long enough before travel to reveal acute intolerance and to impress on the subject the importance of compliance (to relate drug-taking to a specific daily or weekly event).
• Prompt achievement of efficacy and safety, i.e. plasmodicidal concentrations, by one (or two) doses is plainly important for travellers who cannot wait on dosage schedules to deliver both only when steady-state blood concentrations are attained; the schedules must reflect this need.
• Prophylaxis should continue for at least 4 weeks after leaving an endemic area to kill parasites that are acquired about the time of departure, are still incubating in the liver and will develop into the erythrocyte phase. Malarone, however, only needs to be taken for a week after return. The traveller should be aware that any illness occurring within a year, and especially within 3 months, of return, may be malaria.
• Chloroquine and proguanil may be used for periods of up to 5 years, and mefloquine for up to 1–2 years; expert advice should be taken by long-term travellers, especially those going to areas for which other prophylactic drugs are recommended.
• Naturally acquired immunity offers the most reliable protection for people living permanently in endemic areas (below). Repeated attacks of malaria confer partial immunity and the disease often becomes no more than an occasional inconvenience. Vaccines to confer active immunity are under development.
• A short course of prophylaxis (4–6 weeks) may be considered for pregnant women and young children returning to their permanent homes in malarious areas after a prolonged period of stay in a non-endemic area, pending suitable arrangements for health care.
• As a rule, the partially immune should not take a prophylactic. The reasoning is that immunity is sustained by the red cell cycle, loss of which through prophylaxis diminishes their resistance and leaves them highly vulnerable to the disease. There are, however, exceptions to this general advice, and the partially immune may or should use a prophylactic:



Examples of standard prophylactic regimens
• Chloroquine: 300 mg (base) once weekly (start 1 week before travel).
• Proguanil: 200 mg once daily (start 1 week before travel).
• Chloroquine plus proguanil in the above doses.
• Malarone: 1 tablet daily (start 1–2 days before travel).
• Mefloquine: 250 mg once weekly (start 1 week, preferably 2–3 weeks, before travel).
• Doxycycline: 100 mg once daily (start 1–2 days before travel).
Individual antimalarial drugs
Chloroquine
are infrequent at doses normally used for malaria prophylaxis and treatment, but are more common with the higher or prolonged doses given for resistant malaria or for rheumatoid arthritis or lupus erythematosus (see p. 251).
may be rapidly fatal without treatment, and indeed has even been described as a means of suicide.10 Pulmonary oedema is followed by convulsions, cardiac arrhythmias and coma; as little as 50 mg/kg can be fatal. These effects are principally due to the profound negative inotropic action of chloroquine. Diazepam was found fortuitously to protect the heart and adrenaline/epinephrine reduces intraventricular conduction time; this combination of drugs, given by separate i.v. infusions, improves survival.
Amoebiasis
• Bowel lumen amoebiasis is asymptomatic, and trophozoites (non-infective) and cysts (infective) are passed into the faeces. Treatment is directed at eradicating cysts with a luminal amoebicide; diloxanide furoate is the drug of choice; iodoquinol or paromomycin are alternatives.
• Tissue-invading amoebiasis gives rise to dysentery, hepatic amoebiasis and liver abscess. A systemically active drug (tissue amoebicide) effective against trophozoites must be used, e.g. metronidazole, tinidazole, ornidazole. Parenteral forms of these are available for patients too ill to take drugs by mouth. In severe cases of amoebic dysentery with suspected or confirmed peritonitis, broad-spectrum antibiotics should be administered and surgical intervention considered.
The drug treatment of other protozoal infections is summarised in Table 15.5.
| Infection | Drug and comment |
|---|---|
| Giardiasis | Metronidazole, tinidazole or mepacrine |
| Leishmaniasis | |
| visceral | Sodium stibogluconate or meglumine antimoniate; resistant cases may benefit from combining antimonials with paromomycin, miltefosine or amphotericin (including AmBisome). Pentamidine is rarely used now due to toxicity concerns |
| cutaneous | Mild lesions heal spontaneously, fluconazole; antimonials or paromomycin may be injected intralesionally |
| Toxoplasmosis | Most infections are self-limiting in the immunologically normal patient. Pyrimethamine with sulfadiazine for chorioretinitis, and active toxoplasmosis in immunodeficient patients; folinic acid is used to counteract the inevitable megaloblastic anaemia. Alternatives include pyrimethamine with clindamycin or azithromycin or atovaquone. Spiramycin for primary toxoplasmosis in pregnant women. Expert advice is essential |
| Trichomoniasis | Metronidazole or tinidazole is effective |
| Trypanosomiasis | |
| African (sleeping sickness) | Suramin or pentamidine is effective during the early stages but not for the later neurological manifestations for which melarsoprol should be used. Eflornithine is effective for both early and late stages. Expert advice is recommended |
| American (Chagas’ disease) | Prolonged (1–3 months) treatment with benznidazole or nifurtimox may be effective |
Notes on drugs for protozoal infections
Diloxanide furoate may cause troublesome flatulence, and pruritus and urticaria may occur.
Meglumine antimonate is a pentavalent antimony compound, similar to sodium stibogluconate (below).
Paromomycin, an aminoglycoside, is not absorbed from the gut; it is similar to neomycin.
Helminthic infections
Helminths have complex life cycles, special knowledge of which is required by those who treat infections. Table 15.6 will suffice here. Drug resistance has not so far proved to be a clinical problem, though it has occurred in animals on continuous chemoprophylaxis.
| Infection | Drug | Comment |
|---|---|---|
| Cestodes (tapeworms) | ||
| Beef tapeworm Taenia saginata | Niclosamide or praziquantel | Praziquantel cures with single dose |
| Pork tapeworm Taenia solium | Niclosamide or praziquantel | Praziquantel cures with single dose |
| Cysticercosis Taenia solium | Albendazole or praziquantel | Treat in hospital as dying and disintegrating cysts may cause cerebral oedema |
| Fish tapeworm Diphyllobothrium latum | Niclosamide or praziquantel | |
| Hydatid disease Echinococcus granulosus | Albendazole | Surgery for operable cyst disease |
| Nematodes (intestinal) | ||
| Ascariasis Ascaris lumbricoides | Levamisole, mebendazole, pyrantel pamoate, piperazine or albendazole | |
| Hookworm Ancylostoma duodenale | Mebendazole, pyrantel pamoate, or | Anaemic patients require iron or blood transfusion |
| Necator americanus | Albendazole | |
| Strongyloidiasis Strongyloides stercoralis | Thiabendazole or ivermectin | Alternatively, albendazole is better tolerated |
| Threadworm (pinworm) Enterobius vermicularis | Pyrantel pamoate, mebendazole, albendazole or piperazine salts | |
| Whipworm Trichuris trichiuria | Mebendazole or albendazole | |
| Nematodes (tissue) | ||
| Cutaneous larva migrans Ancylostoma braziliense; Ancylostoma caninum | Thiabendazole (topical for single tracks); ivermectin, albendazole or oral thiabendazole (for multiple tracks) | Calamine lotion for symptom relief |
| Guinea worm Dracunculus medinensis | Metronidazole, mebendazole | Rapid symptom relief |
| Trichinellosis Trichinella spiralis | Mebendazole | Prednisolone may be needed to suppress allergic and inflammatory symptoms |
| Visceral larva migrans Toxocara canis; Toxocara cati | Diethylcarbamazine, albendazole or mebendazole | Progressive escalation of dose lessens allergic reactions to dying larvae; prednisolone suppresses inflammatory response in ophthalmic disease |
| Lymphatic filariasis Wuchereria bancrofti; Brugia malayi; Brugia timori | Diethylcarbamazine | Destruction of microfilia may cause an immunological reaction (see below) |
| Onchocerciasis (river blindness) Onchocerca volvulus | Ivermectin | Cures with single dose. Suppressive treatment; a single annual dose prevents significant complications |
| Nematodes (tissue)—cont’d | ||
| Schistosomiasis (intestinal) | ||
| Schistosoma mansoni; Schistosoma japonicum | Praziquantel | Oxamniquine only for Schistosoma mansoni |
| Schistosomiasis (urinary) | ||
| Schistosoma haematobium | Praziquantel | Metriphonate only for Schistosoma haematobium |
| Flukes (intestinal, lung, liver) | Praziquantel | Alternatives: niclosamide for intestinal fluke, bithionol for lung fluke |
American Centers for Disease Control and Prevention (CDC-P). Their website includes a comprehensive travel section that contains high-quality and up-to-date information about prophylaxis, avoidance, diagnosis and treatment of infectious diseases of travel. Available online at http://www.cdc.gov/travel/ (accessed November 2011)
Baird J.K. Effectiveness of antimalarial drugs. N. Engl. J. Med.. 2005;352(15):1565–1577.
Beigel J.H., Farrar J., Han A.M., et alWriting Committee of WHO Consultation on Human Influenza A/H5, Avian influenza A (H5N1) infection in humans. of the. N. Engl. J. Med.. 2005;353:1374–1385.
Bethony J., Brooker S., Albonico M., et al. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet. 2006;367:1521–1532.
Bouchaud O., Imbert P., Touze J.E., et al. Fatal cardiotoxicity related to halofantrine: a review based on a worldwide safety data base. Malar. J.. 2009;8:289.
British HIV Association. The Association provides a wealth of information on best practice management of HIV infection and opportunistic infections. See their website, available online at http://www.bhiva.org/ClinicalGuidelines.aspx (accessed November 2011)
Bruce-Chwatt L.J. Three hundred and fifty years of the Peruvian fever bark. Br. Med. J.. 1988;296:1486–1487.
Chiodini P., Hill D., Lalloo D., et al. Guidelines for Malaria Prevention in Travellers to the United Kingdom. London.: Health Protection Agency; 2007. January 2007
Deeks S.G. Antiretroviral treatment of HIV infected adults. Br. Med. J.. 2006;332:1489–1493.
Edwards G., Biagini G.A. Resisting resistance: dealing with the irrepressible problem of malaria. Br. J. Clin. Pharmacol.. 2006;61(6):690–693.
Esté J.A., Telenti A. HIV entry inhibitors. Lancet. 2007;370:81–88.
European Group for Blood and Bone Marrow Transplantation. Their website has an Infectious Diseases Working Party section containing recent evidence-based recommendations for managing fungal and viral infections. See European Conference on Infection in Leukaemia (ECIL-3) Working Party Guidelines. Available online at: http://www.ebmt.org (accessed November 2011)
Fit for Travel. Another useful contemporary source is ‘Fit for Travel’, the NHS public access website providing travel health information for people travelling abroad from the UK. Available online at: http://www.fitfortravel.scot.nhs.uk/ (accessed November 2011)
Franco-Paredes C., Santos-Preciado J.I. Problem pathogens: prevention of malaria in travellers. Lancet Infect. Dis.. 2006;6(3):139–149.
Gazzard B.G., Anderson J., Babiker A., et al. British HIV Association Guidelines for the treatment of HIV-1-infected adults with antiretroviral therapy 2008. HIV Med.. 2008;9(8):563–608.
Geisbert T.W., Jahrling P.B., Exotic emerging viral diseases: progress and challenges. (supplement review). Nat Med., 2004;10(12):S110–S121.
Greenwood B.M., Bojang K., Whitty C.J., Targett G.A. Malaria. Lancet. 2005;365:1487–1498.
Gryseels B., Polman K., Clerinx J., Kestens L. Human schistosomiasis. Lancet. 2006;368:1106–1118.
Health Protection Agency. The Agency website has detailed information and links to common travel associated infections. Available online at: www.hpa.org.uk/ (accessed November 2011)
Jefferson T., Demicheli V., Rivetti D., et al. Antivirals for influenza in healthy adults: systematic review. Lancet. 2006;367:303–313.
Kremsner P.G., Krishna S. Antimalarial combinations. Lancet. 2004;364:285–294.
Lalloo D.G., Shingadia D., Pasvol G., et al. UK malaria treatment guidelines. J. Infect.. 2007;54:111–121.
HIV and AIDS. Lever A.M.L., Aliyu S.H., eds., Medicine Journal. 2009;37(7):313–390.
McManus D.P., Zhang W., Li J., Bartley P.B. Echinococcosis. Lancet. 2003;362:1295–1304.
Merson M.H. The HIV-AIDS pandemic at 25 – the global response. N. Engl. J. Med.. 2006;354(23):2414–2417.
Montoya J.G., Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976.
Moscona A. Neuraminidase inhibitors for influenza. N. Engl. J. Med.. 2005;353(13):1361–1373.
Murray H.W., Berman J.D., Davies C.R., Saravia N.G. Advances in leishmaniasis. Lancet. 2005;366:1561–1577.
Patterson T.F. Advances and challenges in management of invasive mycoses. Lancet. 2005;366:1013–1025.
Sepkowitz K.A. One disease, two epidemics – AIDS at 25. N. Engl. J. Med.. 2006;354(23):2411–2414.
Snow R.W., Marsh K. Malaria in Africa: progress and prospects in the decade since the Abuja Declaration. Lancet. 2010;376(9735):137–139.
University of Liverpool. interactive charts on antiretroviral drug interactions, information about advances in therapeutic drug monitoring and other resources. Available online at: http://www.hiv-druginteractions.org (accessed November 2011)
US Department of Health and Human Services. guidelines on use of antiretroviral agents in adults and adolescents, and children, which are updated several times per year. Available online at: http://www.aidsinfo.nih.gov (accessed November 2011)
World Health Organization. data on HIV infection. Available online at: http://www.who.int/topics/hiv_infections/en/ (accessed November 2011)
World Health Organization. guidelines for the treatment of malaria http://www.who.int/malaria/publications/atoz/9789241547925/en/index.html, 2010. (accessed November 2011)
1 The large-scale screening for natural compounds able to kill bacteria in vitro, which was the basis for the boom of antibiotics in the 1950s, was not successful for antivirals … The driving force for the boom of antivirals in this period has been the pressure to contain the HIV pandemic, combined with the increased understanding of the molecular mechanisms … which has allowed the identification of new targets for therapeutic intervention.’ (Rappuoli R 2004 From Pasteur to genomics: progress and challenges in infectious diseases. Nature Medicine 10:1177–1185.)
2 For a comprehensive review, see Kaplan J E, Benson C, Holmes K H et al 2009 Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recommendations and Reports 58(RR-4):1–207.
3 Treatment regimens vary in detail; those quoted here accord with the recommendations in the British National Formulary 2010 and national UK specialist guidelines; the BNF is a good source of contact numbers, addresses and websites to obtain expert advice on therapy and prophylaxis of malaria.
4 Acceptable as quinine hydrochloride, dihydrochloride or sulfate, but not quinine bisulfate, which contains less quinine.
5 The loading dose should not be given if the patient has received quinine, quinidine or mefloquine in the previous 24 h; see also warnings about halofantrine (below).
6 Reduced to 5–7 mg/kg of quinine salt if the infusion lasts for more than 48 h.
7 A Cochrane Review of six clinical trials showed a significantly reduced risk of death, reduced parasite clearance time and hypoglycaemia when artesunate was compared with quinine for the treatment of severe malaria. Jones K L, Donegan S, Lalloo D G 2007 Artesunate versus quinine for treating severe malaria. Cochrane Database Systematic Review Oct 17;(4):CD005967.
8 Dondorp A, Nosten F, Stepniewska K, et al 2005 Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet 366:717–725.
9 The active component of many drugs, whether acid or base, is relatively insoluble and may present a problem in formulation. This is overcome by adding an acid to a base or vice versa; the weight of the salt differs according to the acid or base component, i.e. chloroquine base 150 mg = chloroquine sulfate 200 mg = chloroquine phosphate 250 mg (approximately). Where there may be variation, therefore, the amount of drug prescribed is expressed as the weight of the active component, in the case of chloroquine, the base.