CHAPTER 27 VESTIBULAR SYSTEM DISORDERS
Humans have developed a sophisticated and complex mechanism for maintaining balance that relies on the integration and modulation of sensory inputs from vision, the vestibular receptors within the labyrinth, and proprioception. Within the central nervous system, the cerebellum, the extrapyramidal system, the limbic system, and the cerebral cortex facilitate processing to enable the perception of head and body position in space, eye movement control, and appropriate static and dynamic postural function (Fig. 27-1). An alteration in any one of the three sensory inputs, or within the central vestibular pathways and their connections, may give rise to disordered eye movements, disequilibrium or instability, and the perception of dizziness or vertigo. The complexity of this system is such that pathology in almost all body systems may be associated with dizziness/disequilibrium; thus, affected patients present to many different specialist departments but most commonly to otology or neurology outpatient offices (Table 27-1). Despite these ubiquitous presentations, most clinicians do not have a clear diagnostic strategy, including knowledge of detailed neuro-otological examination, to enable them to accurately diagnose and appropriately manage vestibular symptoms. This chapter provides a broad overview of peripheral and central vestibular syndromes, together with an outline of an appropriate clinical assessment based on an understanding of vestibular pathophysiology, a discussion of management strategies, and specific points with regard to common vestibular disorders.
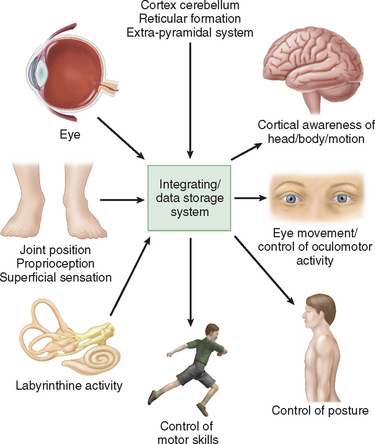
TABLE 27-1 Causes of Disequilibrium
EPIDEMIOLOGY
Dizziness is an extremely common symptom, both in primary care and at the tertiary level. One in four healthy subjects in the community reports symptoms of dizziness, with significant effects on their daily living.1 By the age of 70, 36% of women and 29% of men have balance problems, whereas by the ages of 88 to 90, 45% to 50% of the population suffer symptoms of balance dysfunction.2
In the community, many cases of vestibular dysfunction resolve spontaneously, without recourse to medical care, although each year 5 per 1000 patients consult their general practitioners because of symptoms that are classified as vertigo, and a further 10 per 1000 are seen for dizziness or giddiness.3 In a tertiary setting, dizziness is associated with significant morbidity, and in the older population, falls and mortality are common sequelae.4 Vestibular symptoms after head/whiplash injury are the commonest cause of failure to return to work, and two thirds of patients in a tertiary neuro-otological clinic suffer psychiatric symptoms in association with vestibular pathology.5
VESTIBULAR ANATOMY AND PHYSIOLOGY
The internal ear is a minute membranous structure within the bony labyrinth, buried in the temporal bone. Within the internal ear, the cochlea is the acoustic end-organ receptor, whereas, of the five vestibular end-organs, one lies within each of the three semicircular canals and one each in the utricle and saccule within the vestibule. The vestibular sensory epithelium is composed of type 1 and type 2 hair cells, covered with a gelatinous membrane that, in the saccule and utricle, contains calcium carbonate-rich crystals termed otoconia (Fig. 27-2).
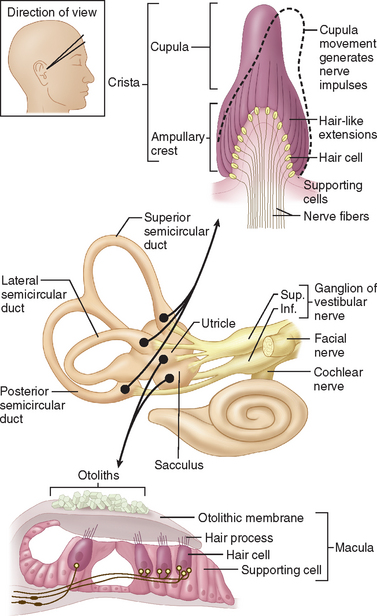
A force parallel to the surface of the sensory epithelium provides the maximal stimulus. Thus, the horizontal anterior and posterior semicircular canals are stimulated by angular acceleration in the three planes of space but are insensitive to gravity or head position.6 The saccule, which lies approximately vertically, senses vertical linear head acceleration and gravity; the utricle, which is oriented approximately horizontally, senses horizontal linear head motion and head position in space.
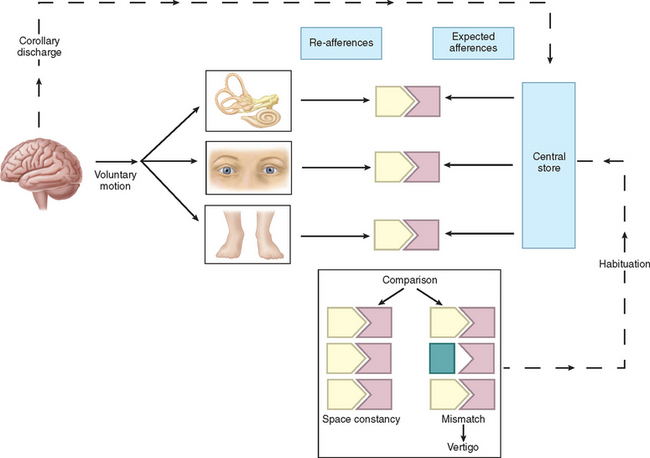
Physiologically, the vestibular apparatus can be considered in two halves, the right labyrinth and the left labyrinth, which are perfectly balanced and work in parallel. For example, when the head is turned to the right, the right horizontal semicircular canal increases its firing rate, whereas the left decreases its firing rate. This asymmetry in neural input is transmitted (1) to the vestibular nuclei and the cerebellum, which controls the amplitude and timing of movements, and (2) via the vestibular nuclei and the thalamus to the parietoinsular vestibular cortex. From birth, the vestibular, visual, and proprioceptive inputs associated with every type of movement are monitored, integrated, and stored in a “data bank,” which is considered to be the reticular formation of the brainstem.7 Subsequently, each movement generates signals that are then compared with the information in the “data bank.” Integration of movement-induced neural asymmetry with other sensory input and comparison with the “data bank” template allow for awareness of head and body position in space, together with the generation of compensatory oculomotor (vestibulo-ocular reflex) and motor (vestibulospinal) activity. In addition to the motor control, the extensive convergence of vestibular and autonomic afferent information in the brainstem and cerebellum allows for coordination of appropriate motor and autonomic responses during movement or changes in posture.
Thus, if there is any mismatch of the sensory input to the existing template, the patient senses disorientation, may develop an abnormal eye movement, frequently feels off balance, and may develop nausea lvomiting and other autonomic symptoms (Fig. 27-3). The classic physiological example of such a mismatch is motion sickness.8 However, any pathological lesion that results in a change in, for example, vestibular input to the central nervous system, as may occur in Meniere’s disease or vestibular neuritis, produces similar symptoms of disorientation, nausea, vomiting, and malaise as a consequence of the change in the vestibular signal, with no corresponding changes in visual and proprioceptive inputs. In addition, connections at various levels of the central vestibular system with the locus ceruleus, the limbic system, and other brain regions that control affective responses, mood, and arousal may underlie the observed overlap between psychiatric and vestibular disorders.9,10
AGING AND THE VESTIBULAR SYSTEM
Histopathological age-related changes in the human vestibular sensory organs include progressive hair cell degeneration, otoconial degeneration in the otolith organs, and decreasing numbers of vestibular nerve fibers,11,12 and age-dependent changes in both caloric and rotational test responses have been demonstrated.13,14 These changes alone are unlikely to generate vestibular symptoms, as they are symmetrical, and dizziness in elderly people is probably more multifactorial in origin.15 Thus, although older patients may be subject to the same common balance disorders as are younger patients, they have more problems with chronic disequilibrium and falls, and vertigo has been reported to rise with advancing age in parallel with the incidence of hearing loss.13 Correct diagnosis, prevention, and rehabilitation are particularly important in treating this group of patients.16
PERIPHERAL VESTIBULAR DISORDERS
Acute Unilateral Vestibular Deafferentation
Acute pathology of one labyrinth manifests as an acute clinical syndrome with profound motor and sensory abnormalities17 and with the same symptoms and signs irrespective of the cause. A patient with an acute total right vestibulopathy has the following signs (Table 27-2):
TABLE 27-2 Consequences of Unilateral Peripheral Vestibular Destruction
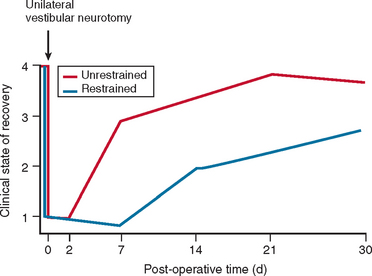
In most cases, the characteristic symptoms and signs of vestibular deafferentation abate, and the patient is rendered asymptomatic over a period of 2 weeks to several months. In this regard, the vestibular system has been shown to be extremely adaptable.18 The processes, which bring about the resolution of vestibular symptoms, are collectively known as cerebral compensation and are attributed to cerebral plasticity (Fig. 27-4; Table 27-3).
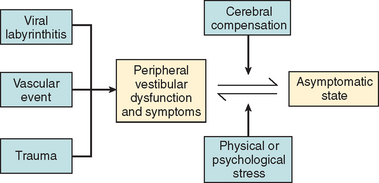
TABLE 27-3 Vestibular Compensation
The structures subserving compensation for vestibular dysfunction are unknown, but it has been shown that brainstem, cerebellar, and cortical structures are involved; the cerebellum is key to this recovery phenomenon,17,19,20 in addition to the requirement for all sensory inputs, including vision, somatosensory afferents, and remaining labyrinthine input.21–23 Furthermore, integrity of both the vestibular nerve24 and the central vestibular connections25 is required.
The physiological mechanisms on which compensation depends include physical activity26,27 and vision28 (Fig. 27-5). Moreover, Fetter and coworkers29 demonstrated that occipital lobectomy before labyrinthectomy impaired compensational recovery, and Schaefer and Meyer in 197330 also demonstrated that transsection of the cervical cord that led to loss of proprioception delayed vestibular compensation.
Bilateral Vestibular Hypofunction
 Sense of imbalance when standing or walking, especially on uneven surfaces (e.g., sand) or in the absence of vision (e.g., at night).
Sense of imbalance when standing or walking, especially on uneven surfaces (e.g., sand) or in the absence of vision (e.g., at night).

In these cases, the cervico-ocular reflex has been implicated in recovery of function31; other authorities have suggested that slippage of the retinal image in bilateral vestibular failure may be compensated for by central visual mechanisms, as occurs in congenital nystagmus32 and oculomotor palsies.33 In general, patients with bilateral vestibular function recover significantly, although a proportion remain handicapped by oscillopsia and instability.
Compensation/Decompensation
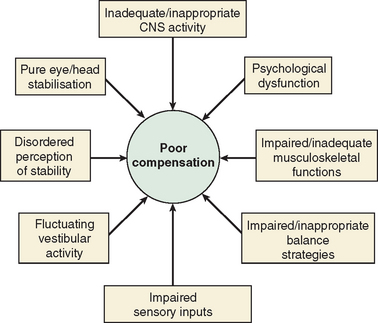
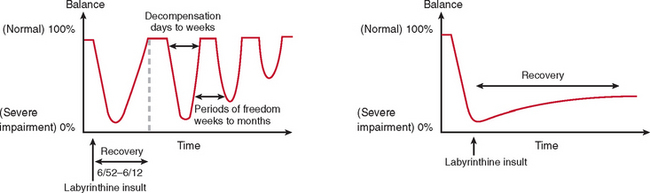
The majority of cases of a unilateral peripheral vestibular deficit recover by means of cerebral compensation. However, some patients do not recover spontaneously and require vestibular rehabilitation with physiotherapy. The basis of physical therapy intervention relies on a structured approach in promoting recovery with visual, proprioceptive, and vestibular stimulation by means of a standard or customized range of exercises. A number of factors that predispose to failure of compensation (Fig. 27-6) or decompensation from a previously recovered state (Fig. 27-7) have been identified. There is some evidence that there exists a critical period in which stimuli must be provided to the adaptive mechanisms and recalibration of the vestibular function must begin, or else the rate of recovery and perhaps the ultimate degree of recovery may decrease.34 However, Shepard and colleagues35 did not identify duration of symptoms, or age, as a negative prognostic factor of a vestibular rehabilitation program, although financial compensation, head injury, and severe postural control abnormalities have all been reported to indicate poor outcome.
DIAGNOSIS OF VESTIBULAR DISORDERS
The diagnosis of vertigo is critically dependent on a clear history that includes the following:



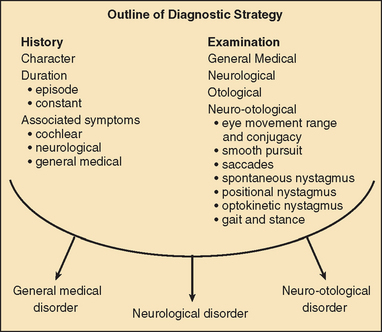
Vertigo of less than 1 minute’s duration is most commonly associated with benign paroxysmal positional vertigo (BPPV), whereas acute rotational vertigo of several hours’ duration is most commonly associated with migraine and Meniere’s disease. Vertigo lasting several days is common in viral vestibular neuritis and in ischemic and brainstem labyrinthitis. Pathology involving the labyrinth and cranial nerve VIII is commonly associated with hearing loss and/or tinnitus, whereas vertigo arising in the central vestibular pathways is most commonly associated with disordered eye movements. In order to make a correct neuro-otological diagnosis, a clinical examination of the vestibular and oculomotor systems is key and requires a clear understanding of vestibular and oculomotor pathology, together with regular clinical practice at examination.36
COMMON PERIPHERAL VESTIBULAR DISORDERS
Acute Vestibular Neuritis
Single episodes of acute rotational vertigo associated with nausea and vomiting, with or without cochlear symptoms, are a common occurrence in all age groups. The attacks are usually unprecipitated and are commonly ascribed to a viral infection, termed vestibular neuritis, vestibular neuronitis, labyrinthitis, or acute vestibulopathy.37,38
The signs and symptoms are as described earlier for an acute unilateral vestibular disorder, and the natural history is resolution of symptoms within a few days or weeks. The majority of patients recover spontaneously, but it appears that early mobilization and vestibular rehabilitation reduce the incidence of disability from chronic vestibular symptoms, which develops in about 20% of patients with acute vestibular neuritis.39
Most cases of vestibular neuritis affect the superior vestibular nerve, with a marked canal paresis on caloric testing, which shows progressive recovery in about 50% of patients on repeat testing.40,41 Frequently, it is possible to obtain a normal saccular response, as judged by the vestibular evoked myogenic potential, which depends on normal inferior vestibular nerve function. In 25% of the patients with vestibular neuritis,43 BPPV (described later) of the posterior canal variant may develop subsequently.
Ramsay-Hunt Syndrome
The Ramsay-Hunt syndrome is the clinical presentation of herpes zoster oticus with facial palsy, auricular rash, and hearing loss, which are often associated with acute vertigo. Abramovich and Prasher44 reported vertigo in 85% of their series; conversely, vestibular dysfunction has also been described with Bell’s palsy or idiopathic facial palsy,45,46 at an incidence of between 20% and 92%. A number of mechanisms of vestibular involvement in this latter condition have been postulated, including compression of cranial nerve VIII by the edematous cranial nerve VII and involvement of both cranial nerves VII and VIII in the same disease process.
Vertigo, imbalance, ataxia, and nausea have all been reported in human immunodeficiency virus infection, although it remains unclear whether the pathology is central or peripheral in type, and vestibular dysfunction is less common than auditory involvement.47
Meniere’s Disease
Meniere’s disease remains a clinical diagnosis characterized by fluctuating hearing loss, tinnitus, and vertigo, often associated with sensation of fullness or blockage in the ear. In 60% of patients affected, both vestibular and cochlear symptoms have developed within 6 months of the onset of the disease. The literature abounds with controversy on all aspects of this condition, and the diagnosis should be based on the strict American Academy of Otolaryngology—Head and Neck Surgery Committee on Hearing and Equilibrium Guidelines.48 The vertigo attacks usually last between 1 and 8 hours, but the tinnitus, hearing loss, and sensation of fullness in the ear may last for several days. Attacks tend to occur in clusters, with attack-free intervals. Initially, both vestibular function and cochlear function recover, so that the caloric test and audiometry may be normal between attacks. Later there is a progressive low-frequency hearing loss, which, in the older patient, may be superimposed on presbycusis to yield a tent-shaped audiogram, and with continuing progression, a plateau hearing loss emerges. Moreover, with progressive attacks, interval disorientation may accompany loss of vestibular function.
Clinical examination may show spontaneous nystagmus directed toward the affected ear (i.e., an irritative response), followed by an ablative phase, in which the nystagmus beats away from the affected ear, and a recovery phase, in which the nystagmus may again beat toward the affected side.49 Late in the disease, patients may develop drop attacks called Tumarkin or otolithic crises.50
The natural history of Meniere’s disease is variable, but in general there are clusters of episodes (relapses) with attack-free periods that may last several years (remission). Other patients, however, have a progressive course, with ultimate loss of auditory and vestibular function. Bilateral involvement is reported in 20% to 50% of cases.51
Electrocochleography with transtympanic recording at the promontory is the most sensitive and specific test for Meniere’s disease. Characteristically, there is broadening of the summating potential/action potential ratio; this ratio is often greater than 35%, in comparison with approximately 20% in normal subjects (Fig. 27-9).
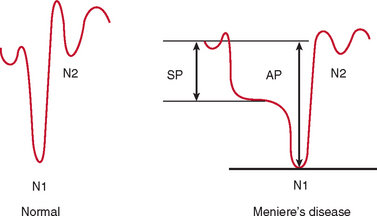
The underlying pathophysiology of Meniere’s disease is generally attributed to endolymphatic hydrops. In 75% of cases, the condition is considered idiopathic, whereas in 25%, a variety of other pathological conditions, including syphilis, trauma, infection, and otosclerosis, are reported to underlie the development of the condition. The disease may occur at any age, but the first attack most commonly occurs between the ages of 30 and 60. It is rare but not unknown in children and is uncommon as a presenting condition after the age of 60 years. About 10% of affected patients have a family history of this disease.52
A number of mechanisms have been hypothesized to predispose to a disorder of endolymph homeostasis, including a defect in normal endolymph absorption by the endolymphatic system. There may be hormonal factors53 or a viral etiology.54,55 Other hypotheses have been based on ischemia,56 and more complex disorders associated with autoimmune disease have been proposed.57
The differential diagnosis of Meniere’s disease includes perilymph fistula, vestibular neuritis with repeated decompensation, and vestibular migraine, which is a particularly difficult diagnosis in that there is a clear increased incidence of Meniere’s disease per se in migrainous subjects.58 Brief acute spells of dizziness may also occur in progressive bilateral vestibular failure of unknown etiology.59
Migrainous Vertigo
Migraine affects approximately 4% to 6% of men and 11% to 18% of women in both Europe and the United States. The incidence and frequency of disequilibrium in association with migraine are reported to range between 50% and 70%.60 Vertigo may occur independently of headache, particularly in children (benign paroxysmal vertigo of childhood).61 Normally, there is a personal or family history of migraine, with troublesome motion sickness in childhood.62 In association with the vertigo, there may be classic symptoms of sensory hyperexcitability, including photophobia, phonophobia, and osmophobia.
Overall, episodic vertigo occurs in about 25% of unselected patients with migraine.58 In one study, nonspecific dizziness occurred approximately equally in patients with migraine (N = 200) and patients with tension-type headaches (N = 166), but vertigo occurred in 27% of patients with migraine, as opposed to 8% of patients with tension-type headache (a significant difference at the level of P > 0.001).28
Young children exhibit multiple and diverse manifestations of migraine, with headache frequently being absent.61 Children may present with cyclical vomiting or attacks of abdominal pain. Basser63 described an episodic disorder that occurs in young children younger than 4 years, termed benign paroxysmal vertigo. The affected child suddenly becomes frightened, cries out, clings to the caregiver, staggers, becomes pale, and often vomits. Typically, the attack is brief, lasting only several minutes, the symptoms are exacerbated by head movement, and nystagmus and/or torticollis may be observed. The child’s condition rapidly returns to normal, and although the attacks may occur up to several times a month before the age of 4, they gradually decrease in number and disappear by the age of 7 or 8. Characteristically, these children develop migraine with aura in adult life.64
Benign recurrent vertigo in adults was described by both Slater in 197965 and Moretti and coworkers in 1980,66 but subsequent consideration of their patients, who complained of episodic vertigo, nausea, and vomiting, worse around menses in women, with no auditory symptoms nor interval symptoms and strong personal or family histories of migraine, suggests that this condition also represents migraine equivalents. Rassekh and Harker67 followed up 38 patients with the diagnosis of “vestibular Meniere’s syndrome.” Of these, 8 developed characteristic Meniere’s disease, 7 became asymptomatic, and the remaining 21 failed to develop the classic triad of Meniere’s disease. Of this latter group, 81% had migraine, which highlights the diagnostic difficulty between early Meniere’s disease and migraine.
It is currently believed that a specific pathophysiology underlines the association between vestibular and migrainous symptoms, termed migraine-related dizziness.68 Researchers have aimed to provide a diagnostic framework for this disorder on the basis of a combination of the International Headache Society criteria for migraine, the presence of specific other symptoms, and the exclusion of other pathology.69,70
Numerous studies have documented the familial pattern of migraine, although genetic studies to date have failed to define genetic abnormalities in the common forms of migraine. Familial hemiplegic migraine, a rare subtype of migraine with aura and autosomal dominant inheritance, is characterized by headache attacks that are preceded or accompanied by episodes of hemiplegia, usually lasting days. In about 50% of patients with familial hemiplegic migraine, mutations in a brain-specific P/Q-type calcium channel gene, CACNA1A,71 located on chromosome 19p, have been shown to produce this condition. Although the exact nature of the mutation has not been elucidated, certain features of migraine are compatible with ion channel dysfunction, including triggers, such as stress and menstruation. Research evaluating calcium channel genes in this population continues.
Benign Positional Paroxysmal Vertigo
BPPV was characterized by Dix and Hallpike72 in their seminal work on patients with vertigo in 1952. This condition is the most common cause of vertigo in adults in virtually all reported series. Schuknecht73 defined degenerative changes in the superior vestibular nerve, the utricle, and the horizontal and anterior semicircular canals in the temporal bones of patients with BPPV and postulated ischemia of the anterior vestibular artery. He further74 identified basophilic deposits on the cupulae of the posterior semicircular canals in two patients with BPPV before death. On the basis of these findings, he proposed the hypothesis of cupulolithiasis (i.e., a heavy cupula) as the mechanism giving rise to positional nystagmus of the paroxysmal type (Fig. 27-10A). However, later workers75 proposed the hypothesis of canalithiasis, with free-floating debris from the otolith organ moving within the posterior canal and acting as a plunger, thus dragging the cupula (see Fig. 27-10B).


Diagnosis is made by the Dix-Hallpike positioning test or roll tests. The typical features of BPPV (Table 27-4)76 are explained by free-floating otoconial debris, moving under the influence of gravity (so-called canalithiasis). In the upright position, a clot of calcium carbonate crystals is established around the most dependent portion of the posterior canal. As the patient moves backward and to the side, in the plane of the posterior canal (the movement induced by the Dix-Hallpike positioning test), the clot moves, producing an ampullar-fugal displacement of the cupula, as a result of a plunger effect within the narrow canal. The fatigability, which is characteristic of benign positional nystagmus of paroxysmal type, is explained by dispersion of the clot, making the plunger effect less marked on repeated maneuvers. The induced vertigo and nystagmus are brief, because once the clot settles in the lowest portion of the canal with regard to gravity, the cupula moves back to the primary position. Latency before the onset of nystagmus is best explained by a delay in the initial motion of the clot.
| BPPV | Central Positional Nystagmus | |
|---|---|---|
| Nystagmus direction | Rotational to undermost ear | Variable |
| Latent period | 2-20 seconds | None |
| Adaptation | Disappears in <50 seconds | Persists |
| Fatigability | Disappears on repetition | Persists |
| Vertigo | Strongly present | Typically absent |
| Incidence | Common | Uncommon |
BPPV, benign paroxysmal positional vertigo.
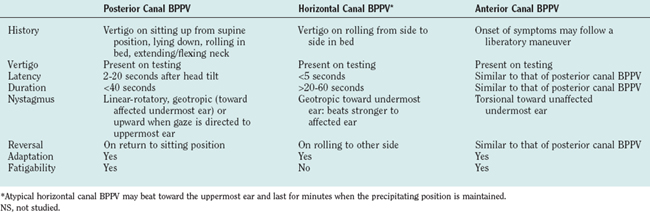
Thus, BPPV is not a disease but a clinical manifestation that may result from a variety of different inner ear pathological processes, most commonly labyrinthine concussion secondary to trauma and vestibular neuritis. The condition may affect any of the semicircular canals (Table 27-5), but the posterior canal variant is the most common.76 Characteristically, the patient complains of brief, severe spells of vertigo induced by turning over in bed, tipping the head backward to look at something in the sky, or reaching for something on a high shelf (i.e., “top shelf vertigo”).
Autoimmune Inner Ear Disease
Autoimmune inner ear disease is an uncommon but important cause of progressive bilateral loss of auditory and vestibular function.77 The condition commonly manifests with sequential stepwise loss of auditory and vestibular function. The disorder may be associated with systemic autoimmune phenomena, such as polyarthritis, Wegener’s granulomatosis, or otosclerosis; or it may be part of Cogan’s syndrome with associated interstitial keratitis or may occur in isolation as an inner ear organ-specific disorder.78 Initial symptoms may be suggestive of Meniere’s disease, with fluctuation of hearing and ear pressure, but unlike symptoms of Meniere’s disease, these symptoms progress rapidly over weeks or months to involve the opposite ear. Treatment requires high-dose steroids (60 to 80 mg of prednisolone for 10 days, tapering off, with or without the addition of cytotoxic drugs); plasmapheresis has also been advocated as treatment.
Labyrinthine Trauma
Damage to the vestibular apparatus can occur as a result of barotrauma, acoustic trauma, and physical trauma.79
Labyrinthine concussion with or without unilateral hearing loss is common after mild, moderate, and severe head injuries.80 Acute vertigo with evidence of a canal paresis on caloric testing and spontaneous nystagmus in the direction opposite the affected ear are common.79,80 The natural history of the condition is characteristic of that for any acute unilateral vestibular disorder, unless there is additional central vestibular dysfunction as a consequence of trauma, in which case recovery may be delayed. BPPV may also occur as an isolated late sequela of labyrinthine concussion—an important medicolegal consideration.
BILATERAL VESTIBULAR FAILURE
Bilateral vestibular failure may result from genetic derangements, meningitis, trauma, autoimmune disease, or, most commonly, ototoxicity.59 Aminoglycoside antibiotics are well recognized as giving rise to vestibular (and auditory) toxicity, and the effect of the drug on a bedridden patient may not manifest until the patient begins to ambulate, at which point he or she develops ataxia and bobbing oscillopsia. The different aminoglycoside antibiotics vary in their predilection for the auditory or vestibular system, but gentamicin is relatively specific for the vestibular apparatus. None of the aminoglycosides is metabolized; they are excreted by glomerular filtration. Thus, patients with renal impairment cannot excrete these drugs, which accumulate in the blood and inner ear fluids. The ototoxic effect is on the hair cells of the inner ear.
Clinically, there may be no nystagmus, as there is bilateral vestibular loss, but the patient has degradation of visual acuity with head motion, is unable to walk across a firm mat with the eyes closed, and has absent or severely reduced caloric and rotational responses. Intensive vestibular rehabilitation is of value in rehabilitation.
Neoplasia
In rare cases, both primary benign and malignant primary and secondary tumors involve the middle ear and temporal bone, but more rarely do they give rise to vertigo. Tumors of the cerebellopontine angle are also rare, but they are an important diagnostic entity in patients presenting with disequilibrium. A characteristic history is of a slowly progressive unilateral hearing loss, with tinnitus and instability, whereas only 10% to 20% of patients report vertigo. Together with the characteristic of the cranial nerve VIII, examination frequently reveals involvement of cranial nerve VII, followed by cranial nerve V, then the cerebellum, and, late in the disease process, the lower cranial nerves. Acoustic neuromata account for more than 90% of tumors in this region, but other tumors include meningiomas, epidermoid cysts, facial nerve schwannomas, cholesterol granulomas, and, in rare instances, secondary malignancies. Characteristically, there may be unidirectional horizontal nystagmus away from the affected ear, unless the tumor is compressing the brainstem, in which case Bruns bidirectional nystagmus is observed. The diagnosis should be suspected in all cases of progressive unilateral hearing loss and tinnitus and in unilateral hearing loss with an abnormal ipsilateral brainstem evoked response. The “gold standard” for diagnosis is magnetic resonance imaging (MRI) scan. Management may include watchful waiting, surgery, or radiotherapy.81
Acute Middle Ear Disorders
Serous otitis media in childhood is well recognized to give rise to symptoms of disequilibrium and vertigo, although the underlying mechanism is poorly understood.82 Infections of the ear and temporal bone may give rise to acute pain, hearing loss, and disequilibrium, and in all cases of vertigo, a detailed otological examination must be undertaken to rule out erosive middle ear disease with serious intracranial complications, including meningitis, brain abscess, subdural effusion, lateral sinus thrombosis, otitic hydrocephalus, and subdural empyema. Such disorders are more common in immunosuppressed or otherwise debilitated patients.
CENTRAL VESTIBULAR DISORDERS
Vertebrobasilar transient ischemic attacks are characterized by brief episodes of vertigo, usually of minutes’ duration, in association with one or more of the constellation of brainstem symptoms and signs characteristic of ischemia in the posterior circulation.83 Recurrent vertigo that is not associated with additional neurological symptoms should not be diagnosed as ischemic events; however, in posterior circulation ischemia, vertigo is usually associated with additional neurological symptoms, although, in general, there are no neurological signs, and MRI scans are often normal.
Nonetheless, vertigo in association with posterior circulation ischemia is common. The commonest cause is atherosclerosis of the subclavian, vertebral, and/or basilar arteries, but dissection, arteritis, emboli, polycythemia, and hyperviscosity syndromes may also manifest in this way. It should be emphasized that although cervical spondylosis is common in older patients, mechanical compression of the vertebral artery is extremely rare in this condition.84
Lateral medullary infarction (Wallenberg’s syndrome). This condition manifests with acute-onset vertigo, nausea, vomiting, imbalance, incoordination, facial numbness and weakness, diplopia, dysphagia, and dysphonia. This manifestation is commonly associated with ischemia of the posterior inferior cerebellar artery, although, in practice, it more commonly results from ipsilateral vertebral artery occlusion. Classic signs include ipsilateral Horner’s syndrome; ipsilateral paralysis of the palate, pharynx, and larynx; ipsilateral loss of pain and temperature sensation on the face with ipsilateral dysmetria, dysrhythmia, and dysdiadochokinesia; contralateral loss of pain and temperature sensation of the body; and spontaneous nystagmus. Classically, patients experience lateropulsion, a prominent, strong motor disturbance tending to push both the patient and the eyes to the side of the lesion. MRI reveals infarction in the dorsolateral medulla, with or without infarction of the posterior inferior cerebellum.85
Ischemia in the distribution of the anterior inferior cerebellar artery may result in infarction of the dorsolateral pontomedullary region and the anterior interior cerebellum.86 Because the labyrinthine artery arises from the anterior inferior cerebellar artery in about 85% of cases, there is commonly also infarction of the membranous labyrinth. This gives rise to a clinical syndrome characterized by both acute peripheral and central vestibular findings.
Diagnosis of cerebellar infarction is crucial, because surgical decompression may be required if massive edema results. There is acute-onset vertigo with nausea and vomiting, together with severe imbalance and incoordination. Clinically, spontaneous or gaze-evoked nystagmus with truncal ataxia and limb dysmetria is common. MRI shows infarction in the territory of one of the cerebellar arteries or in the watershed area between territories. The condition must be differentiated from acute unilateral peripheral vestibular loss87; the key lies in the finding of spontaneous nystagmus that changes direction with gaze and profound ataxia. After a latent period of 24 to 96 hours, some patients develop progressive brainstem dysfunction caused by compression from a swollen cerebellum and hydrocephalus. Death is inevitable unless the compression is surgically relieved.
Cerebellar hemorrhage is similar in presentation to cerebellar infarction, but frequently, in association with acute vertigo, nausea, and vomiting, there is headache and a complete inability to stand. In contrast to cerebellar infarction, in the early stages of examination there is frequently marked nuchal rigidity and prominent cerebellar signs, together with ipsilateral facial paralysis and ipsilateral gaze paralysis. Pupils are often small bilaterally but reactive. Approximately 50% of patients lose consciousness within 24 hours of the initial symptoms, and 75% become comatose within 1 week of onset. The condition is often fatal, unless surgical decompression is performed.88
In more than 50% of cases of brain tumors of the posterior fossa, vestibular and cochlear symptoms occur. In cerebellar tumors, positional vertigo, vomiting, headache, and gait imbalance are the norm. An MRI scan is diagnostic, and audiometry frequently shows normal pure-tone audiometry but abnormalities on brainstem evoked responses. Central gaze-evoked nystagmus with abnormalities of smooth pursuit, saccades, and visual-vestibular interaction are common.89 Such tumors are particularly common in children and adolescents when they are rapidly growing, whereas in adults, cerebellar secondary tumors are more common.
Multiple sclerosis is characterized by plaques of demyelination disseminated in time and space, and it is commonly associated with disorientation and/or imbalance. Symptoms commonly include monocular visual loss, vertigo, diplopia, weakness, numbness, and ataxia. Both the auditory and vestibular systems may be involved. Acute vertigo may manifest in 5% of patients with multiple sclerosis, frequently mimicking an acute peripheral vestibular episode. However, careful oculomotor examination90 usually reveals central vestibular findings of gaze-evoked nystagmus, disconjugate vertical nystagmus, central positional nystagmus, and/or disorders of smooth pursuit; saccades; and visual-vestibular interactions. Brainstem evoked responses and otoacoustic emission suppression evaluations of efferent auditory function frequently identify central auditory disorders. As in the majority of central vestibular syndromes, however, MRI is the definitive investigation.
Arnold-Chiari malformation is characterized by oscillopsia associated with downbeat nystagmus and gait unsteadiness. In addition to downbeat nystagmus, lower cranial nerve palsies and both gait and limb ataxia are typically noted on examination. Associated auditory disturbances caused by neural involvement may be present, and diagnosis is by means of MRI. Suboccipital decompression of the foramen magnum may alleviate progression of the neuro-otological abnormalities, but this procedure rarely brings about improvement.91
MANAGEMENT OF VESTIBULAR DISORDERS
Successful management of the patient with vertigo depends on accurate diagnosis, an understanding of vestibular physiology and compensation, appropriate intervention strategies, and the physician’s awareness of the overlap between vestibular, autonomic, and psychological aspects of vestibular pathology (Fig. 27-11).
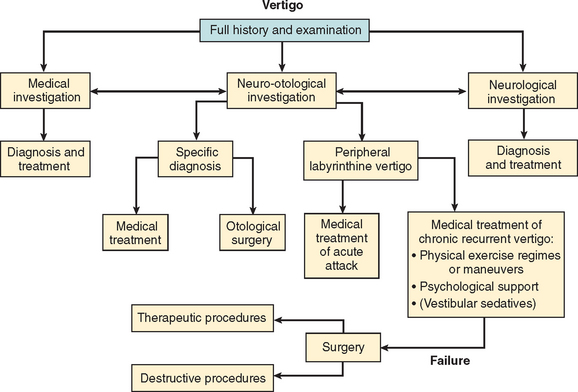
The five main branches of management intervention are as follows:92
On the basis of the diagnosis, an appropriate rehabilitation plan (Table 27-6) should be constructed for each patient, and it is important that the patient fully understands the aims and objectives of the rehabilitation package to ensure active compliance.
Pharmacological Treatment
Drug treatments of vestibular disorders include the following:
Specific treatment of vestibular disorders is appropriate for Meniere’s disease, migraine, and episodic ataxia. The treatment of Meniere’s disease remains controversial and empirical,93 with few double-blind randomized studies to assess treatment efficacy and an 80% placebo response in this condition. Medical treatment aims to influence the underlying pathology of endolymphatic hydrops or the postulated immunological pathogenesis of this disorder, whereas destructive procedures may be either medical (intratympanic injection of aminoglycosides) or surgical. A low-salt diet and diuretics (bendrofluazide, 10 mg/day), together with potassium supplements and regular serum potassium checks to avoid hypokalemia, appear to be highly effective. Betahistine, a histamine analog, is advocated for treatment of Meniere’s disease but is contraindicated in the presence of migraine. Steroids have been proposed both systematically and transtympanically, on the assumption of an autoimmune pathogenesis for Meniere’s disease.
The treatment of migraine-associated dizziness parallels the treatment of migrainous headache. General measures, including dietary restriction, lifestyle adaptations, stress reduction techniques, and vestibular rehabilitation are of value in the presence of a fixed vestibular deficit. Treatment may include over-the-counter analgesics, in addition to triptans, ergot derivatives, and acetazolamide. Prophylactic medication includes β blockers, calcium channel blockers, serotonin reuptake inhibitors, and amitriptyline. Episodic ataxia, which is rare in comparison with migraine, may manifest with acute vertigo and ataxia. Both acetazolamide and 4-aminopyridine are effective in episodic ataxia type 2.94,95 Standard antiemetics and vestibular sedative drugs may be tried in maximal dosages to alleviate acute vestibular symptoms.
On an empirical basis, nausea and disequilibrium associated with central eye movement disorders have reportedly been improved by a variety of drugs, including clonazepam, gabapentin, baclofen, flunarizine, and barbiturates. Each drug should be titrated against known side effects to obtain optimal suppression of symptoms.92 Ondansetron, a potent highly selective 5-hydroxytryptamine-3 receptor antagonist, has been effective in combating nausea, particularly in association with vertigo caused by brainstem stroke.96
Vestibular Rehabilitation
Management of peripheral vertigo hinges on the facilitation of compensation through physical exercise regimens, such as the Cawthorne-Cooksey exercises and customized regimens,97 together with appropriate psychological support for patients who develop the common sequelae of avoidance behavior, anxiety, and depression.5
Treatment of BPPV is particularly rewarding, because particle repositioning procedures have been shown to be highly effective.97 In 1980, Brandt and Daroff98 proposed specific repetitive positional exercises that were based on the hypothesis of cupulolithiasis, and they claimed a 98% success rate. More recent studies in which both the Epley and the Semont maneuvers, based on the hypothesis of canalithiasis, were used reported success rates of between 80% and 95% after the first maneuver. Repeated maneuvers improve the success rate so much that fewer than 5% of patients appear to continue suffering persistent symptoms despite these treatments. It may be appropriate to consider surgical intervention such as plugging the posterior canal to bring about resolution of positional vertigo in this small subset of patients in whom medical management fails.
Psychological Intervention
The interaction of psychological factors and vestibular symptoms in recovery from peripheral vestibular disorders cannot be overemphasized. Many studies have highlighted the association of agoraphobia, avoidance behavior, anxiety states, panic attacks, and depression with vestibular pathology.99 When a patient is not recovering with physiotherapy intervention, such factors should always be considered.
Surgical Intervention
It is now widely recognized that surgical intervention for vertigo is needed only in life-threatening complications of chronic middle ear disease, neoplasia involving otological structures (e.g., vestibular schwannoma), or trauma to the middle or inner ear, as may be seen in association with a perilymph fistula. Interventions such as endolymphatic sac decompression for Meniere’s disease have not been proved effective, and destructive surgical procedures such as labyrinthectomy or vestibular nerve section should be considered with caution in view of the reported incidence of bilateral pathology in this condition. Medical destruction of the inner ear with the aminoglycosides, through intratympanic instillation of gentamicin, remains an unproved therapeutic intervention in that although the episodes of vertigo may abate, there is a significant occurrence of sensorineural hearing loss, and the possible long-term consequence of bilateral disease must again be borne in mind.
CONCLUSION
Baloh RW, Halmagyi GM. Disorders of the Vestibular System. Oxford, UK: Oxford University Press, 1996.
Brandt T. Vertigo: Its Multisensory Syndromes, 2nd ed. London: Springer, 2003.
Furman JM, Cass SP. Balance Disorders: A Case-Study Approach, 2nd ed. Oxford, UK: Oxford University Press, 2003.
Herdman SJ. Vestibular Rehabilitation. Philadelphia: FA Davis, 2005.
Luxon LM, Furman JM, Martini A, et al, editors. Textbook of Audiological Medicine. London: Martin Duritz, 2003.
1 Yardley L, Owen O, Nazareth I, et al. Prevalence and presentation of dizziness in a general practice community sample of working age people. Br J Gen Pract. 1998;48:1131-1135.
2 Jonsson R, Sixt E, Landahl S, et al. Prevalence of dizziness and vertigo in an urban elderly population. J Vestib Res. 2004;14:47-52.
3 Royal College of General Practitioners and Office of Population Census and Surveys. Morbidity Statistics from General Practice. London: HMSO, 1986.
4 Downton JH. Falls in the Elderly. London: Edward Arnold, 1993.
5 Eagger S, Luxon LM, Davies RA, et al. Psychiatric morbidity in patients with peripheral vestibular disorder: a clinical and neuro-otological study. J Neurol Neurosurg Psychiatry. 1992;55:383-387.
6 Wuyts F: Physiology of Equilibrium. In Gleeson M, ed: Scott-Brown’s Otolaryngology: Head & Neck Surgery, 7th ed. London: Hodder Arnold. In press.
7 Roberts TDM. Neurophysiology of Postural Mechanisms, 2nd ed. London: Butterworths, 1978.
8 Watt DG. Sensory and motor conflict in motion sickness. Brain Behav Evol. 1983;23(1–2):32-35.
9 Balaban CD. Neural substrates linking balance control and anxiety. Physiol Behav. 2002;77:469-475.
10 Furman JM, Balaban CD, Jacob RG, et al. Migraine-anxiety related dizziness (MARD): a new disorder? J Neurol Neurosurg Psychiatry. 2005;76:1-8.
11 Rauch SD, Velazquez-Villasenor L, Dimitri PS, et al. Decreasing hair cell counts in aging humans. Ann N Y Acad Sci. 2001;942:220-227.
12 Nadol JBJr, Schuknecht HF. Pathology of peripheral vestibular disorders in the elderly. Am J Otolaryngol. 1990;11:213-227.
13 Enrietto JA, Jacobson KM, Baloh RW. Ageing effects of auditory and vestibular responses: a longitudinal study. Am J Otolaryngol. 1999;20:371-378.
14 Kazmierczak H, Pawlak-Osinski P. Visuoocular reflexes in presbyvertigo. Int Tinnitus J. 2001;7:112-114.
15 Drachman DA, Hart C. A new approach to the dizzy patient. Neurology. 1972;22:323-334.
16 Konrad H, Girardi M, Helfert R. Balance and aging. Laryngoscope. 1999;109:1454-1460.
17 Curthoys IS, Halmagyi GM. Vestibular compensation: a review of the oculomotor, neural and clinical consequences of unilateral vestibular loss. J Vestib Res. 1995;5:67-107.
18 Gonshor A, Melvill Jones G. Extreme vestibule-ocular adaptation induced by prolonged optical reversion of vision. J Physiol. 1976;256:381-414.
19 Ito M. The Cerebellum and Neural Control. New York: Raven Press, 1984.
20 Smith PF, Curthoys IS. Mechanisms of recovery following unilateral labyrinthectomy: a review. Brain Res Rev. 1989;14:155-180.
21 Lacour M, Xerri C. Lesion induced neuronal plasticity in sensorineural systems. In: Flohr H, Precht W, editors. Vestibular Compensation: New Perspectives. Berlin: Springer-Verlag; 1981:240-253.
22 Halmagyi GM, Cremer PD, Curthoys IS. Peripheral vestibular disorders and diseases in adults. In: Luxon LM, Furman JM, Martini A, et al, editors. Textbook of Audiological Medicine. London: Taylor & Francis; 2003:797-818.
23 Luxon LM. Vestibular compensation. In: Luxon LM, Davies RA, editors. Handbook of Vestibular Rehabilitation. London: Whurr; 1997:17-29.
24 Cass SP, Goshgarian HG. Vestibular compensation after labyrinthectomy and vestibular neurectomy in cats. Otolaryngol Head Neck Surg. 1991;104:14-19.
25 Petrone D, De Benedittis G, De Candia N. Experimental research on vestibular compensation using posturography. Boll Soc Ital Biol Sper. 1991;67:731-737.
26 Lacour M, Roll JP, Appaix M. Modifications and development of spinal reflexes in the alert baboon (Papio papio) following an unilateral vestibular neurotomy. Brain Res. 1976;113:255-269.
27 Igarashi M, Levy JK, O-Uchi T, et al. Further study of physical exercise and locomotor balance compensation after unilateral vestibular neurotomy. Acta Otolaryngol. 1981;92:101-105.
28 Courjon JH, Jeannerod M, Ossuzio I, et al. The role of vision in compensation of vestibulo-ocular reflex after hemilabyrinthectomy in the cat. Exper Brain Res. 1977;28:235-248.
29 Fetter M, Zee DS, Proctor LR. Effect of lack of vision and of occipital lobectomy upon recovery from unilateral labyrinthectomy in rhesus monkey. J Neurophysiol. 1988;59:394-407.
30 Schaefer KP, Meyer DL. Compensatory mechanisms following labyrinthine lesions in the guinea pig. A simple model of learning. In: Zippel HP, editor. Memory and Transfer of Information. New York: Plenum; 1973:203-232.
31 Bronstein AM, Hood JD. Oscillopsia of peripheral vestibular origin. Central and cervical compensatory mechanisms. Acta Otolaryngol. 1987;104:207-214.
32 Buchele W, Brandt T, Degner D. Ataxia and oscillopsia in downbeat-nystagmus vertigo syndrome. Adv Otorhinolaryngol. 1983;30:291-297.
33 Wist ER, Brandt T, Krafczyk S. Oscillopsia and retinal slip. Evidence supporting a clinical case. Brain. 1983;106(Part 1):153-168.
34 Lacour M. Relearning and critical postoperative period in the restoration of nerve function. Example of vestibular compensation and clinical implications. Ann Otolaryngol Chir Cervicofac. 1984;101:177-187.
35 Shepard NT, Telian SA, Smith-Wheelock M, et al. Vestibular and balance rehabilitation therapy. Ann Otol Rhinol Laryngol. 1993;102:198-205.
36 Luxon LM. Evaluation and management of the dizzy patient. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 4):iv45-iv52.
37 Strupp M, Brandt T. Vestibular neuritis. Adv Otorhinolaryngol. 1999;55:111-136.
38 Strupp M, Arbusow V. Acute vestibulopathy. Curr Opin Neurol. 1999;14:11-20.
39 Strupp M, Arbusow V, Maag KP, et al. Vestibular exercises improve central vestibulospinal compensation after vestibular neuritis. Neurology. 1998;51:838-844.
40 Herzog N, Allum JH, Probst R. [Follow-up of caloric test response after acute peripheral vestibular dysfunction]. HNO. 1997;45:123-127.
41 Schmid-Priscoveanu A, Bohmer A, Obzina H, et al. Caloric and search-coil head-impulse testing in patients after vestibular neuritis. J Assoc Res Otolaryngol. 2001;2:72-78.
42 Fetter M, Dichgans J. Vestibular neuritis spares the inferior division of the vestibular nerve. Brain. 1996;119:755-763.
43 Aw ST, Fetter M, Cremer PD, et al. Individual semicircular canal function in superior and inferior neuritis. Neurology. 2001;57:768-774.
44 Abramovich S, Prasher DK. Electrocochleography and brainstem potentials in Ramsay-Hunt syndrome. Arch Otolaryngol Head Neck Surg. 1986;112:925-928.
45 Koizuka I, Goto K, Okada M, et al. ENG findings in patients with Bell’s palsy. Relationship between visual suppression and clinical course. Acta Otolaryngol Suppl. 1988;446:85-92.
46 Yagi T, Yamaguchi J, Nonaka M. Neurotological findings in Bell’s palsy and Hunt’s syndrome. Acta Otolaryngol Suppl. 1988;446:97-100.
47 Pappas DGJr, Roland JTJr, Lim J, et al. Ultrastructural findings in the vestibular end-organs of AIDS cases. Am J Otol. 1995;16:140-145.
48 Committee on Hearing and Equilibrium, the American Academy of Ophthalmology and Otolaryngology. Committee on Hearing and Equilibrium guidelines for diagnosis and evaluation of therapy in Meniere’s disease. Otolaryngol Head Neck Surg. 1995;113:181-185.
49 Jacobson GP, Pearlstein R, Henderson J, et al. Recovery nystagmus revisited. J Am Acad Audiol. 1998;9:263-271.
50 Ishiyama G, Ishiyama A, Jacobson K, et al. Drop attacks in older patients secondary to an otologic cause. Neurology. 2001;57:1103-1106.
51 Conlon BJ, Gibson WP. Meniere’s disease: the incidence of hydrops in the contralateral asymptomatic ear. Laryngoscope. 1999;109:1800-1802.
52 Paparella MM, Djalilian HR. Etiology, pathophysiology of symptoms, and pathogenesis of Meniere’s disease. Otolaryngol Clin North Am. 2002;35:529-545. vi.
53 Brenner M, Hoistad DL, Hain TC. Prevalence of thyroid dysfunction in patients with Meniere’s disease. Arch Otolaryngol Head Neck Surg. 2004;130:226-228.
54 Pulec L, House WF. Meniere’s disease study: three-year progress report. Int J Equilib Res. 1973;3:156-165.
55 Selmani Z, Marttila T, Pyykko I. Incidence of virus infection as a cause of Meniere’s disease or endolymphatic hydrops assessed by electrocochleography. Eur Arch Otorhinolaryngol. 2005;262:331-334.
56 Friberg U, Rask-Andersen H. Vascular occlusion in the endolymphatic sac in Meniere’s disease. Ann Otorhinolaryngol. 2002;111:237-245.
57 Tomoda K, Suzuka Y, Iwai H, et al. Meniere’s disease and autoimmunity: clinical study and survey. Acta Otolaryngol Suppl. 1993;500:31-34.
58 Kayan A, Hood JD. Neuro-otological manifestations of migraine. Brain. 1983;107:1123-1142.
59 Bronstein A, Rinne T, Gresty M, et al. Bilateral loss of vestibular function. Acta Otolaryngol Suppl. 1994;520:247-250.
60 Harker LA. Migraine associated vertigo. In: Baloh RW, Halmagyi GM, editors. Disorders of the Vestibular System. Oxford, UK: Oxford University Press; 1996:407-417.
61 Al-Twaijri WA, Shevell MI. Pediatric migraine equivalents: occurrence and clinical features in practice. Pediatr Neurol. 2002;26:365-368.
62 Cutrer FM, Baloh RW. Migraine associated dizziness. Headache. 1992;32:300-304.
63 Basser LS. Benign paroxysmal vertigo of childhood (a variety of vestibular neuronitis). Brain. 1964;87:141-152.
64 Lanzi G, Balottin U, Fazzi E, et al. Benign paroxysmal vertigo of childhood: a long-term follow-up. Cephalalgia. 1994;14:458-460.
65 Slater R. Benign recurrent vertigo. J Neurol Neurosurg Psychiatry. 1979;42:363-367.
66 Moretti G, Manzoni GC, Caffarra P, et al. Benign recurrent vertigo and its connection with migraine. Headache. 1980;20:344-346.
67 Rassekh CH, Harker LA. The prevalence of migraine in Meniere’s disease. Laryngoscope. 1992;102:135-138.
68 Furman JM, Balaban CD, Jacob RG, et al. Migraine-anxiety related dizziness (MARD): a new disorder? J Neurol Neurosurg Psychiatry. 2005;76:1-8.
69 Furman JM, Marcus DA, Balaban CD. Migrainous vertigo: development of a pathogenetic model and structured diagnostic interview. Curr Opin Neurol. 2003;16:5-13.
70 Neuhauser H, Leopold M, von Brevern M, et al. The interrelations of migraine, vertigo and migrainous vertigo. Neurology. 2001;56:436-441.
71 Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543-552.
72 Dix MR, Hallpike CS. Pathology, symptomatology and diagnosis of certain common disorders of the vestibular system. Proc R Soc Med. 1952;45:341-354.
73 Schuknecht HF. Positional vertigo: clinical and experimental observations. Trans Am Acad Ophthalmol Otolaryngol. 1962;66:319-331.
74 Schuknecht HF. Cupulolithiasis. Arch Otolaryngol. 1969;90:765-778.
75 Hall SF, Ruby RRF, McLure JA. The mechanics of benign paroxysmal vertigo. J Otolaryngol. 1979;8:151-158.
76 Honrubia V, Baloh RW, Harris MR, et al. Paroxysmal positional vertigo syndrome. Am J Otol. 1999;20:465-470.
77 Yu TJ, Yazawa Y. Immunology of cochlear and vestibular disorders. In: Luxon LM, Furman JM, Martini A, et al, editors. Textbook of Audiological Medicine. London: Taylor & Francis; 2003:61-88.
78 Agrup C, Luxon LM. Autoimmune inner ear disease. Curr Opin Neurol. 2006;19(1):26-32.
79 Luxon LM. Post-traumatic vertigo. In: Baloh RW, Halmagyi M, editors. Disorders of the Vestibular System. New York: Oxford University Press; 1996:381-395.
80 Davies RA, Luxon LM. Dizziness following head injury: a neuro-otological study. J Neurol. 1995;242:222-230.
81 British Association of Otorhinolaryngologists—Head and Neck Surgeons. Clinical Effectiveness: Acoustic Neuroma (Vestibular Schwannoma) [Document 5]. London: ENT-UK, 2002.
82 Golz A, Netzer A, Angel-Yeger B, et al. Effects of middle ear effusion on the vestibular system in children. Otolaryngol Head Neck Surg. 1998;119:695-699.
83 Grad A, Baloh RW. Vertigo of vascular origin: clinical and electronystagmographic features in 84 cases. Arch Neurol. 1989;46:281-284.
84 Baloh RW, Honrubia V. Vascular disorders. In: Baloh RW, Honrubia V, editors. Clinical Neurophysiology of the Vestibular System. 3rd ed. New York: Oxford University Press; 2001:292.
85 Bjerner K, Silfverskold BJ. Lateropulsion and imbalance in Wallenberg’s syndrome. Acta Neurol Scand. 1968;44:91-98.
86 Osis JG, Baloh W. Vertigo and the anterior inferior cerebellar artery syndrome. Neurology. 1992;42:1274-1279.
87 Norrving B, Magnussen M, Holtas S. Isolated acute vertigo in the elderly: vestibular or vascular disease? Acta Neurol Scand. 1995;91:43-48.
88 Pollak L, Rabey JM, Gur R, et al. Indication to surgical management of cerebellar haemorrhage. Clin Neurol Neurosurg. 1998;100:99-103.
89 Hirose G, Halmagyi GM. Brain tumours and balance disorders. In: Baloh RW, Halmagyi GM, editors. Disorders of the vestibular system. Oxford, UK: Oxford University Press; 1996:446-460.
90 Williams NP, Roland PS, Yellin W. Vestibular evaluation in patients with early multiple sclerosis. Am J Otol. 1997;18:93-100.
91 Crtistante L, Westphal M, Herrmann HD. Cranio-cervical decompression for Chiari I malformation: a retrospective evaluation of functional outcome with particular attention to the motor deficits. Acta Neurochir (Wien). 1994;130(1–4):94-100.
92 Bamiou D-E, Luxon LM: Vertigo-clinical management and rehabilitation. In Gleeson M, Luxon LM, eds: Scott-Brown’s Otolaryngology: Head & Neck Surgery—Ear, Hearing and Balance, 7th ed, London: Hodder Arnold. In press.
93 Kim HH, Wiet RJ, Battista RA. Trends in the diagnosis and management of Meniere’s disease: results of a survey. Otolaryngol Head Neck Surg. 2005;132:722-726.
94 Jen J. Familial episodic ataxias and related ion channel disorders. Curr Treat Options Neurol. 2000;2:429-431.
95 Strupp M, Kalla R, Dichgans M, et al. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. 2004;62:1623-1625.
96 Rice GP, Ebers GC. Ondansetron for intractable vertigo complicating acute brainstem disorders. Lancet. 1995;345:1182-1189.
97 Luxon LM, Davies RA, editors. Handbook of Vestibular Medicine. London: Whurr, 1997.
98 Brandt T, Daroff RB. Physical therapy for paroxysmal positional vertigo. Arch Otolaryngol. 1980;106:484-485.
99 Jacobs RG, Furman JM, Cass SP. Psychiatric consequences of vestibular dysfunction. In: Luxon LM, Furman J, Martini A, et al, editors. Textbook of Audiological Medicine. London: Taylor & Francis; 2002:869-887.






