Chapter 16 Ventricular Arrhythmias
Please go to expertconsult.com for supplemental chapter material.
Ectopic (nonsinus) depolarizations frequently arise in the ventricles themselves, or the fascicular system, producing premature ventricular beats (or complexes), ventricular tachycardia (VT), discussed later, and in the most electrophysiologically unstable settings, sometimes ventricular fibrillation (VF) leading to immediate cardiac arrest (also see Chapter 19).
Ventricular Premature Beats
Ventricular premature beats (VPBs)∗ are premature depolarizations arising in the ventricles, analogous to atrial premature beats (APBs) and junctional premature beats (JPBs), which are supraventricular in origin. Recall that with APBs and JPBs, the QRS complex is usually of normal (“narrow”) width because the stimulus spreads synchronously through the bundle branches to the ventricles (unless a bundle branch block or some other cause of aberrancy is present).
With VPBs, however, the premature depolarizations typically arise in either the right or left ventricle. Therefore, the ventricles are not stimulated simultaneously, and the stimulus spreads through the ventricles in an aberrant direction and asynchronous way. Thus, the QRS complexes are wide with VPBs, just as they are with the bundle branch block patterns.† Examples of VPBs are shown in Figures 16-1 to 16-8.
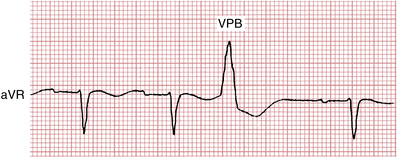
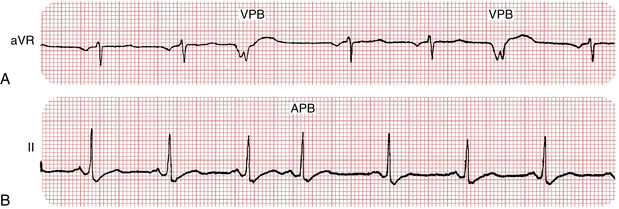
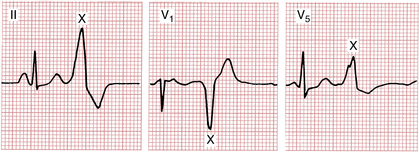
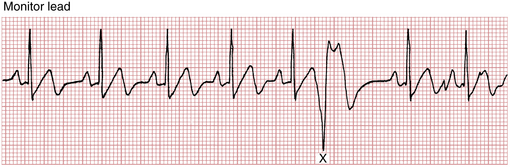
Figure 16-3 Notice that the same ventricular premature beat (marked X) recorded simultaneously in three different leads has different shapes. By comparison, notice that the multiform ventricular premature beats shown in Figure 16-11 have different shapes in the same lead.
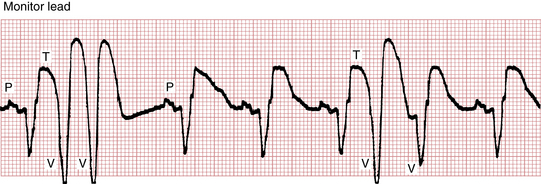
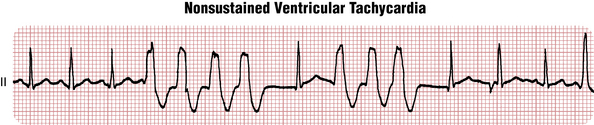
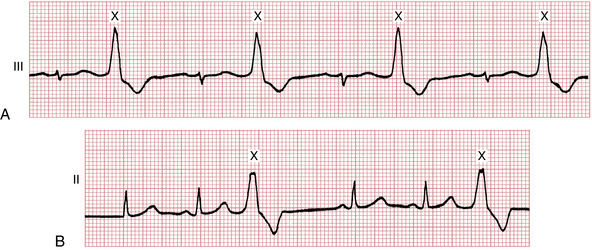
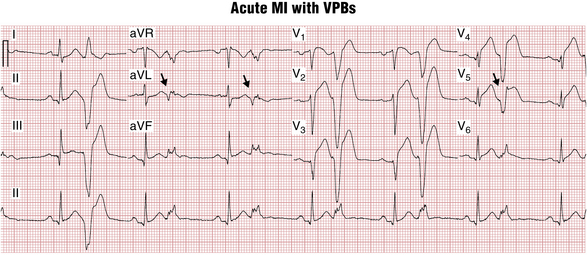
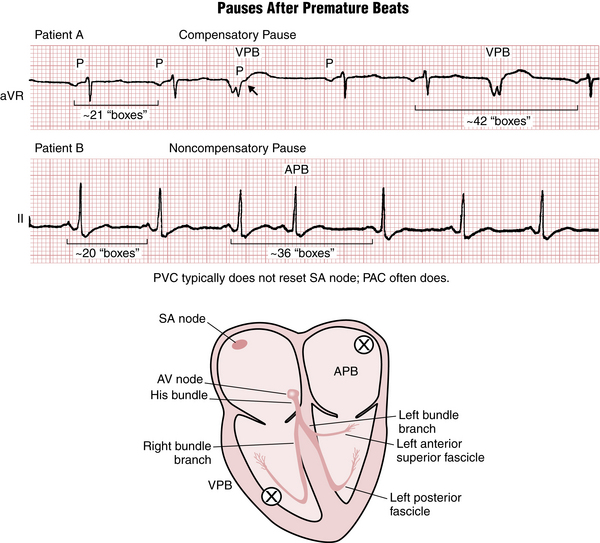
Figure 16-8 Difference between noncompensatory and compensatory pauses after atrial premature beats (APBs) and ventricular premature beats (VPBs), respectively. See text and Figure 16-9.
VPBs most often precede a sinus P wave. (Occasionally they appear just after a sinus P wave but before the normal QRS complex.) Sometimes, VPBs are followed by retrograde (nonsinus) P waves (negative in lead II) that arise because of reverse stimulation (from bottom to top) of the atria by each VPB. This sequence, called “VA” (ventricular-atrial) conduction, is identical to what may occur with ventricular pacing (see Chapter 21).
Features
Clinicians comment on a number of features of VPBs that may have clinical relevance.
Frequency
VPBs may occur in various combinations. Two in a row (see Fig. 16-4) are referred to as a pair or couplet. Three or more in a row are, by definition, VT (see Fig. 16-5). Sometimes, as shown in Figure 16-6A, isolated VPBs occur so frequently that each normal beat is followed by a VPB. This produces a distinctive repetitive grouping of one normal beat and one VPB, which is called ventricular bigeminy (see Figs. 16-6 and 16-7). The sequence of two normal beats with a VPB is ventricular trigeminy. Three normal beats followed by a VPB constitutes ventricular quadrigeminy.
Morphology and Origin
• If the ectopic beat originates in the left ventricle, then right ventricular activation will be delayed and the QRS of the VPB will resemble a right bundle branch block (RBBB).
• If the ectopic beat comes from the right ventricle, then left ventricular activation is delayed, and the QRS shape resembles a left bundle branch block (LBBB) pattern.
• VPBs from the interventricular septum often show an intermediate pattern between RBBB and LBBB. They are usually relatively narrow as both ventricles get activated simultaneously from the middle of the ventricles.
The further away from the middle of the heart VPB origin is, the wider is the QRS. VPBs coming from the top (base) of the heart have “inferior” QRS axis—pointing down and therefore positive in leads II, III, and aVF. They are often called “outflow tract” VPBs because their origin is located close to the pulmonary and aortic valves. Usually they have LBBB-like shape. Outflow tract VPBs (right ventricular outflow tract [RVOT] and left ventricular outflow tract [LVOT]) are the most common variety of “benign” VPBs occurring in a normal heart. VPBs originating closer to the apex and from inferior wall activate the heart from the bottom up and are said to have “superior” axis, with QRS negative in leads II, III, and aVF. VPBs originating in the area of postinfarct myocardial scar often have a qR configuration. Sometimes this allows diagnosing infarct even when no Q waves are seen during sinus rhythm (see Fig. 16-7).
The sequence of ventricular repolarization after VPBs is such that ST-T waves are directed in the opposite direction to the main QRS deflection (QRS-T “discordance”), often with prominent ST segment elevations/depressions as expected (see Fig. 16-2). Clinicians need to recognize that these secondary ST-T changes are not due to ischemia and are similar to the QRS-T discordance findings in wide complex beats due to bundle branch block and ventricular pacing. In fact, concordance between QRS and ST-T directions during VPBs may be a sign of myocardial injury.
Coupling Interval
The term coupling interval refers to the interval between the VPB and the QRS of the preceding normal beat. When multiple VPBs are present, fixed coupling often occurs, with the coupling interval approximately the same for each VPB (see Fig. 16-6). At other times, VPBs may show a variable coupling interval. Whether VPBs demonstrate fixed versus variable coupling does not have major clinical implications.
Compensatory Pauses
As you may have noticed, APBs and VPBs are usually followed by a pause before the next normal sinus beat. The pause after a VPB is usually but not always longer than the pause after an APB. A fully compensatory pause indicates that the interval between the normally sinus-generated QRS complexes immediately before and immediately after the VPB is twice the basic PP interval (Figs. 16-8 and 16-9). A fully compensatory pause is more characteristic of VPBs than APBs. The explanation for this difference (see Fig 16-9) relates to the fact that PVCs usually do not reset (delay) firing of the sinus node, although supraventricular beats often do.
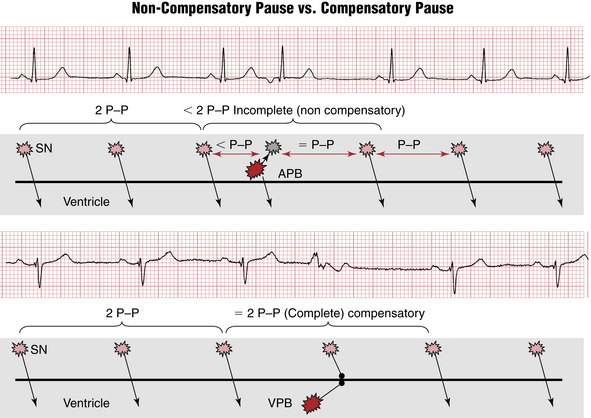
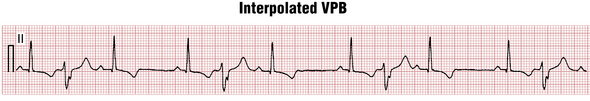
There are multiple exceptions to this “rule” that associates APBs with noncompensatory pause and VPBs with fully compensatory ones. For example, sometimes a VPB falls almost exactly between two normal beats; in such cases the VPB is said to be interpolated (Fig. 16-10).
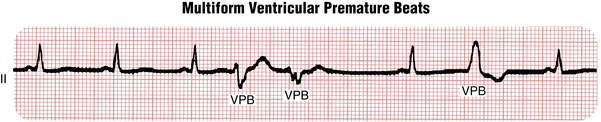
Uniform and Multiform VPBs
The terms uniform and multiform are used to describe the appearance of VPBs in any lead. Uniform VPBs have the same appearance in any lead and arise from the same anatomic site (focus) (see Fig. 16-6). (Of course, they have different shapes in different leads, just as normal beats do.) By contrast, multiform VPBs have different morphologies in the same lead (Fig. 16-11). Multiform VPBs often but not always arise from different foci. Uniform VPBs are unifocal, but multiform VPBs are not necessarily multifocal. Uniform VPBs may occur in normal hearts and hearts with underlying organic heart disease. Multiform VPBs usually indicate that organic heart disease is present, but are a nonspecific finding.
R on T Phenomenon
The R on T or VPB on T phenomenon refers to VPBs that are timed so that they fall near the peak or trough (most positive or negative point) of the T wave of the preceding normal beat (Fig. 16-12). This occurs in the setting of acute myocardial ischemia or infarction or with a long QT(U) interval. Those VPBs falling on the T wave are noteworthy in that they may precipitate VT or VF, particularly in the course of an acute myocardial infarction (MI) or myocardial ischemia or with very long QT intervals (discussed later in this chapter). Recall from the discussion of synchronized direct current (DC) cardioversion that the peak of the T wave is a time during which an external stimulus is especially likely to produce VF. However, VT and VF most commonly occur without a preceding “R on T” beat, and most “R on T” beats do not precipitate a sustained ventricular tachyarrhythmia.
Clinical Significance
VPBs are very common in healthy people of all ages as well as those with virtually any heart disease. They usually reflect increased automaticity of dormant ventricular pacemakers and may be provoked by adrenergic stimulation (stress, caffeine, sympathomimetic drugs, and by “recreational” drugs such as cocaine or other stimulants), electrolyte abnormalities (especially hypokalemia or hypomagnesemia), and certain drug toxicity (digoxin). One of the most common locations for benign VPBs is the outflow tract; however, VPBs can arise from any area of the normal heart.
Symptoms of VPBs can vary from none to severe palpitations (especially when electrical signal conducts retrogradely through the AV node to the atria causing almost simultaneous atrial contraction, see discussion of pacemaker syndrome, Chapter 21).
Ventricular Tachycardias
The usual definition of VT is three or more VPBs in a row at a rate of 100 or more beats/min (Figs. 16-13 and 16-14). As with NCTs, VTs can be due to focal (automatic or triggered) or reentrant mechanisms. They are usually initiated by VPBs.
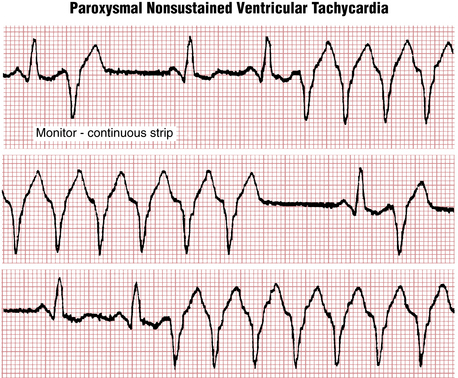
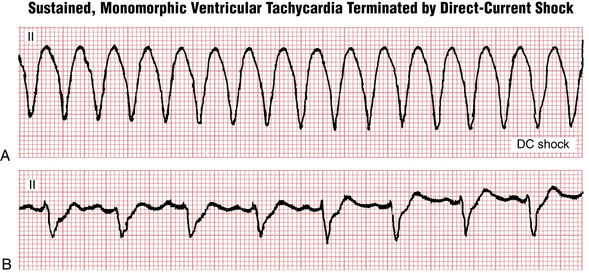
VT (Box 16-1) is most generally classified along two “axes”: one based on duration (Is it nonsustained or sustained?) and the other based on appearance in any given lead (Is it monomorphic or polymorphic?).
Monomorphic Ventricular Tachycardia
Similar to VPBs, the origin of monomorphic VT often can be determined by examining two features: the QRS morphology and the QRS axis. Arrhythmias coming from the left ventricle have RBBB-like QRS shape (Fig. 16-15); the ones coming from the right ventricle have an LBBB-like shape, which are best seen in lead V1 (Fig. 16-16). Those coming from the base (top) of the heart have QRS axes pointing in an inferior to rightward direction, toward leads II, III, and aVF; the ones coming from the inferior wall to the apex, in the opposite direction, have a superior axis (see Fig. 16-16).
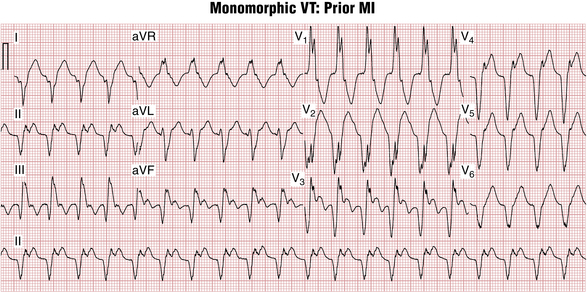
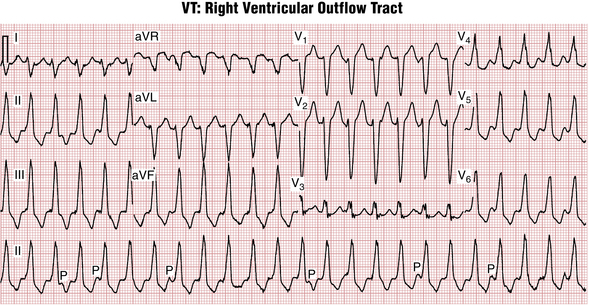
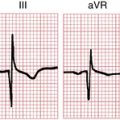
Figure 16-16 Monomorphic ventricular tachycardia (VT) originating in the right ventricular outflow tract (RVOT-VT). Note the QRS similarity to left bundle branch block (LBBB) in lead V1, with an inferior-rightward QRS axis. Key finding indicating VT, as opposed to a supraventricular tachycardia with aberration, is the presence of underlying sinus rhythm (P) at a slower, independent rate, a sign of atrioventricular (AV) dissociation. See Chapter 20.
VTs from the interventricular septum have a shape in between RBBB and LBBB and can have a relatively narrow QRS (120 ms) because both ventricles are activated in parallel fashion from the middle of the heart.
Finally, the presence of QR configuration in more than one lead, with an LBBB or RBBB configuration, suggests that VT originates in the area of a myocardial scar due to a prior MI (see Fig. 16-15).
Clinical Significance of Monomorphic Ventricular Tachycardia
Monomorphic VT can occur in a structurally normal heart (such as automatic “outflow tract” VT; see Fig. 16-16) or with virtually any structural heart disease. Symptoms of VT depend on its rate and the systolic function of the heart. Common symptoms include a sensation of palpitations, shortness of breath, and lightheadedness. Patients with structurally normal hearts and preserved left ventricular function can maintain their cardiac output and tolerate very fast VT rates (over 200 beats/min) with relatively few symptoms, whereas in patients with depressed left ventricular ejection fractions this rate would most likely result in syncope due to low cardiac output. Slow VTs, with rates between 100 and 130 beats/min or so are not unusual in patients receiving antiarrhythmic drugs and these episodes can be minimally symptomatic or completely asymptomatic.
Monomorphic VT in patients with a history of prior MI is usually caused by reentry around the areas of myocardial scar and not by acute ischemia. In the absence of other symptoms these patients do not need to be treated as having an “acute coronary syndrome.” However, decreased cardiac output and increased oxygen demands during sustained VT can cause demand ischemia and degeneration of VT into VF; therefore DC cardioversion should be considered even if the patient appears stable.
Key Point
Of note, sustained monomorphic VT in patients with structural heart disease is an indication for implantable cardioverter defibrillator (ICD) therapy (see Chapters 19 and 21).
Sustained monomorphic VT in patients without known heart disease should include appropriate imaging (usually echocardiogram and sometimes magnetic resonance imaging) to rule out areas of myocardial scar that can serve as the substrate for reentry (e.g., subclinical myocarditis or cardiomyopathy). VT with an LBBB morphology (originating in the right ventricle) can be the first sign of arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D), a potential substrate for sudden cardiac arrest (see Chapter 19).
Polymorphic Ventricular Tachycardias
Torsades de Pointes
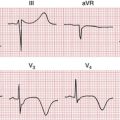
As noted, the distinct and clinical major type of polymorphic VT that occurs in the setting of QT interval prolongation is called torsades de pointes (TdP), from the French term meaning “twisting of the points” (Figs. 16-17 and 16-18). The hallmark of this tachycardia is the gradual variation in QRS direction (polarity) and amplitude in a given lead that resembles a spindle (see Fig. 16-18).
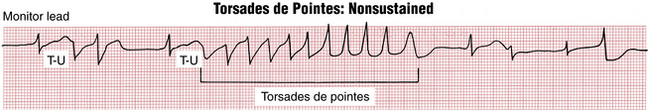
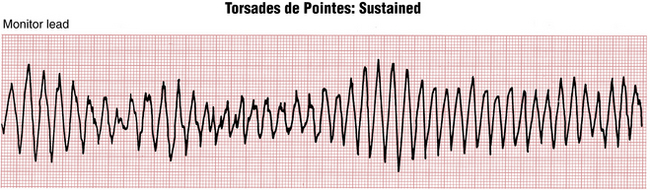
Figure 16-18 Classic pattern of the sustained torsades de pointes type of ventricular tachycardia. Notice the pattern of beats in which the QRS axis appears to rotate or turn in a systematic way. Figure 16-17 shows a short, nonsustained run of the same arrhythmia, which occurs in the setting of QT(U) prolongation.
TdP is usually initiated by a VPB starting at the peak of a prolonged T-U wave, as a type of “R on T’ beat (see Fig. 16-17). This sequence of events often starts with one or more VPBs followed by a postectopic pause and then a second VPB on the T-U wave of the next supraventricular beat, which develops even more prolonged repolarization due to the slight pause. TdP can occur with congenital (hereditary) or acquired QT interval prolongation, and can deteriorate into VF, causing sudden cardiac arrest (see Chapter 19).
QT prolongation syndromes that may give rise to TdP are usually classified as acquired or congenital (hereditary) (also see Chapter 24 for a more complete list).
Acquired Long QT Syndrome
Causes of acquired long QT syndrome include the following:
• Drugs, particularly quinidine (see Chapter 10 and Fig. 19-7) and related antiarrhythmic agents (disopyramide and procainamide), as well as ibutilide, dofetilide, sotalol, amiodarone, psychotropic agents (phenothiazines and tricyclic antidepressants), and many other noncardiac drugs (e.g., pentamadine, erythromycin, and certain other antibiotics)
• Electrolyte imbalances, especially hypokalemia and hypomagnesemia, and, less commonly, hypocalcemia, which prolong repolarization
• Severe bradyarrhythmias (especially high-grade AV heart block syndromes)
Hereditary Long QT Syndromes
General principles of management of TdP include review and discontinuation of any possible QT-prolonging medications and correction of relevant electrolyte abnormalities (especially hypokalemia or hypomagnesemia). Intravenous magnesium may be helpful to shorten the QT even with normal Mg2+ serum levels and to suppress the VPBs that trigger this arrhythmia. Increasing the heart rate by pacing, isoproterenol or dopamine infusion sometimes is necessary to shorten the QT interval and make repolarization more homogeneous.
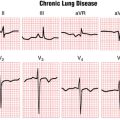
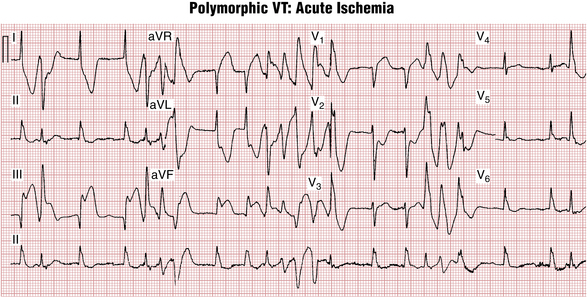
Polymorphic VT in the absence of QT interval prolongation most often indicates ischemia. It can be observed during acute MI (Fig. 16-19) but also with ischemia induced during exercise. The finding of polymorphic VT with no QT interval prolongation, especially during physical exertion, should prompt coronary artery evaluation. Much less frequent is so-called CPVT, which also presents during exercise and is caused by genetic defects in intracellular calcium handling. Most subjects are children or young adults. The Brugada syndrome (see Chapter 19) may also be associated with nonischemic polymorphic VT.
Accelerated Idioventricular Rhythm
Figures 16-20 and 16-21 present a distinctive arrhythmia that has been called accelerated idioventricular rhythm (AIVR) or slow VT. Recall that with typical VT the heart rate is more than 100 beats/min. With AIVR the heart rate is usually between 50 and 100 beats/min, and the ECG shows wide QRS complexes without associated sinus P waves.
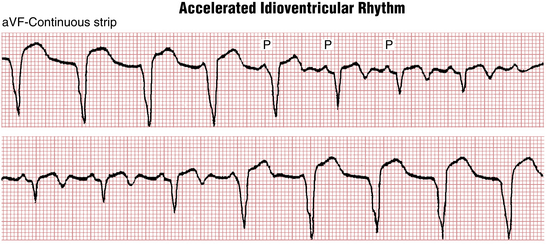
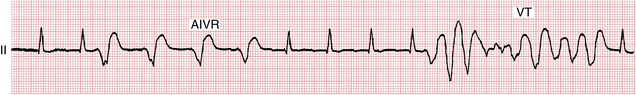
AIVR is particularly common with acute MI, and it may be a sign of reperfusion after the use of thrombolytic agents or after interventional coronary artery procedures, or occurring spontaneously. This arrhythmia is generally short-lived, lasting minutes or less, and usually requires no specific therapy. In these cases (see Fig. 16-15), AIVR appears to be a benign “escape” rhythm that competes with the underlying sinus mechanism. When the sinus rate slows, AIVR appears; when the sinus rate speeds up, the arrhythmia disappears.
More rarely (see Fig. 16-16), AIVR is initiated by premature beats rather than escape beats. This latter type is more likely to be associated with faster ventricular tachyarrhythmias.
Ventricular Fibrillation
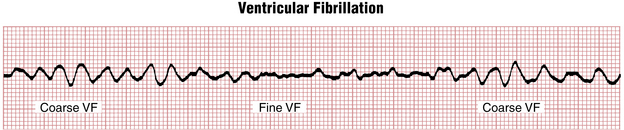
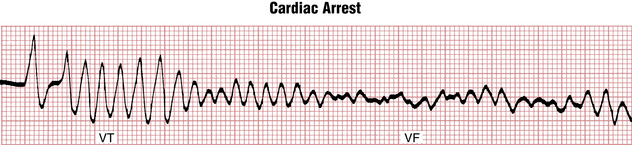
Ventricular fibrillation (VF, Figs. 16-22 and 16-23) is a completely disorganized ventricular rhythm resulting in immediate cessation of cardiac output and cardiac arrest unless electrical defibrillation is performed using unsynchronized DC shock. Based on the amplitude of fibrillatory waves VF is arbitrarily classified as coarse or fine. VF can appear suddenly as a primary arrhythmia (from the baseline of normal sinus or other supraventricular rhythm) or, more commonly, as a “degeneration” of monomorphic or polymorphic VT. If left untreated it progresses from coarse to fine VF, and then eventually to asystole (see Chapter 19).
Differential Diagnosis of Wide Complex Tachycardias
The differential diagnosis of wide complex tachycardias (WCTs), a major topic in clinical ECG interpretation, is discussed in Chapter 20. A boxed outline summary is presented in Chapter 24.
∗ In the ECG literature the terms ventricular premature beat, ventricular premature depolarization, ventricular extrasystole, and premature ventricular complex or contraction (PVC) are used interchangeably.
† The basic electrophysiologic mechanisms responsible for VPBs are a subject of active investigation. VPBs may arise by at least three mechanisms: reentrant waves, increased spontaneous depolarizations of ventricular cells (enhanced automaticity), and triggered activity or afterdepolarizations (i.e., premature firing of ventricular cells triggered by the previous depolarization).