79 Ventricular Arrhythmias
 Normal Electrophysiology
Normal Electrophysiology
Anatomic Synopsis
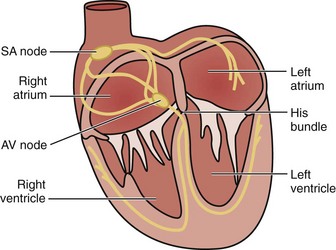
The cardiac electrical impulse originates in the sinoatrial (SA) node, located high on the right atrium near its junction with the superior vena cava (Figure 79-1). The impulse then propagates through muscle fibers and specialized internodal pathways (composed of Purkinje-type fibers) to converge on the AV node, located in the interatrial septum near the tricuspid valve and the opening of the coronary sinus. From the AV node, the impulse travels through the bundle of His, its left and right branches, and the Purkinje system to simultaneously activate the right and left ventricles. A ring of fibrous tissue interposed between the atria and the ventricles prevents spread of the electrical impulse through the muscle fibers, enabling the AV node to function as a relay and filter structure limiting the number of impulses that can be transmitted to the ventricles and thus maintaining the ventricular rate within a range that is physiologically permissible for stroke volume generation. Thus, the AV node prevents a 1 : 1 conduction under conditions of very rapid atrial activation such as atrial flutter (rate ≈ 300 s−1) and atrial fibrillation (rate ≈ 350 s−1 to 600 s−1), typically preventing increases in ventricular rate above 150 s−1 or 180 s−1 in instances of atrial flutter or atrial fibrillation, respectively.
Action Potential and Pacemaker Activity
Action potential results from rapid depolarization and repolarization of polarized cells driven by a coordinated sequence of opening and closing of channels that regulate ion currents across the cell membrane. The main ion currents are carried by channels that regulate influx of sodium ions (Na+) and calcium ions (Ca2+) (inward currents) and by efflux of potassium ions (K+) (outward currents).1–3
The action potential is essential to initiate and propagate the electrical impulse throughout the conduction system and ultimately reach cardiomyocytes where the action potential signals activating of contractile activity and therefore pump function through Ca2+-induced Ca2+-release from the sarcoplasmic reticulum. Accordingly, the electrical impulse typically precedes mechanical activity. However, cardiomyocytes can also react to mechanical forces through stretch-activated ion channels and other related mechanisms including mechanical modulation of Ca2+ handling and interaction with other mechanosensitive cells in the heart.4,5 This mechanism is in part responsible for commotion cordis,6,7 precordial thump,8 and fist pacing.9
The functional characteristics of the action potential differ contingent on the cell type. Cells from the Purkinje system and from atrial and ventricular muscle are primarily responsible for propagation of the action potential. These cells have a stable resting potential at approximately −90 mV (inside negative), which is largely the result of a K+ current known as the inward rectifier (IK1). IK1 “anchors” the membrane potential to a voltage close to the equilibrium potential of K+;2 it is turned off during depolarization (inward rectification). Initiation of an action potential requires depolarization of the membrane potential between −70 and −80 mV. This voltage is the threshold at which fast voltage-gated Na+ channels are activated, prompting Na+ influx driving an inward current (INa).3 Depolarization to the threshold potential typically occurs upon arrival of an action potential. The INa drives the membrane potential toward the equilibrium potential of Na+, causing further depolarization and reversal of the membrane potential to approximately +20 mV (overshoot). This phase is known as phase 0 of the action potential and ushers in a 4-phase repolarization (Figure 79-2). Phase 1 is the initial early repolarization (action potential notch) and results from rapid inactivation of Na+ channels (inner gate) and the opening of K+ channels carrying a rapidly activating and rapidly inactivating “transient” outward current (ITo). Based on recovery time, two distinct subpopulations of channels can be recognized carrying a rapidly “fast” recovering current (ITof) and a slowly recovering current (ITos). The difference in these currents rely on distinct pore-forming α-subunits with Kv4.2/Kv4.3 (KCND2/KCND3) genes encoding ITof and Kv1.4 encoding ITos.10 It appears that ITof is the predominant contributor to ITo in ventricular myocardium.11 Because the K+ channels carrying ITo are expressed in the subepicardial and mid-myocardial regions but not in the subendocardial region, they contribute to the inhomogeneity of repolarization.12
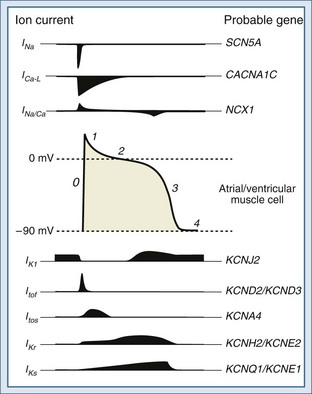
Phase 2 is the plateau phase of the action potential and results mainly from a Ca2+ current carried by the slow and prolonged opening of L-type voltage-gated Ca2+ channels (ICa-L).13,14 Opening of these channels begins during phase 0 at a membrane potential of −30 to −40 mV. These channels are inactivated in response to increases in cytosolic Ca2+ and are strongly regulated by neurotransmitters. Phase 3 corresponds to late repolarization and follows the closing of Ca2+ channels and opening of K+ channels with slow activation kinetics carrying currents known as delayed rectifiers (IK). The IK are the main repolarizing currents and have two components carried by distinct gene products: a rapid component (IKr) and a slow component (IKs).15,16 Both are implicated in the heritable forms of long QT syndrome (see later discussion).17 In addition, opening of IK1 (main contributor to the resting membrane potential as described earlier) contributes to repolarization. Phase 4 represents return to the resting membrane potential and the interval during which ionic balance is restituted, largely through the action of the Na+/K+ pump.
Cells of the SA and AV nodes lack voltage-gated Na+ channels, and phase 0 is carried by ICa-L.18 Because of their slower opening kinetics (relative to Na+ channels), they give rise to a slanted phase 0 and in part determine the lower conduction velocity of the SA and AV nodes (≈50 cm·s−1) compared with the His-Purkinje system (≈400 cm·s−1) and muscle cells (≈100 cm·s−1). SA and AV node cells also have pacemaker activity and slowly depolarize during phase 4 to a threshold potential of approximately −40 mV. The slow depolarization is called pre-potential or pacemaker potential and involves a background Na+ current (INa-B), a decay of K+ currents, and the opening of T-type voltage-gated Ca2+ channels (ICa-T) at a potential between the thresholds for INa and ICa-L, unleashing phase 0. Cells of the His-Purkinje system have latent pre-potential activity and can become active when SA or AV node activity is depressed or their impulse is blocked. Atrial and ventricular muscle cells exhibit pre-potential activity only under abnormal circumstances (see later discussion).
The preceding description is succinct and oversimplified. Various other ion channels, antiporters, pumps, and receptors play important roles in specific physiologic states and disease processes. For example, there is a nonselective cationic channel that is gated at resting potential by intracellular Ca2+ and produces an inward Na+ current (INS).19 This current may contribute to delayed afterdepolarizations following Ca2+ release by the sarcoplasmic reticulum. Ik(atp) is a K+ current carried through metabolically regulated channels that are inhibited by adenosine triphosphate (ATP) and opened under conditions of ischemia and hypoxia. Ik(atp) is the main contributor to the shortening of the action potential duration and the characteristic ST-segment elevation observed in the surface electrocardiogram during myocardial ischemia.20,21
The sarcolemmal Na+/Ca2+ exchanger is another important modulator of the action potential. Because it exchanges one Ca2+ for three Na+, it is electrogenic and generates a current (INa/Ca) whose direction is determined by the Na+ and Ca2+ gradients and the membrane potential.21,22 In settings in which there is cytosolic Ca2+ overload (e.g., ischemia and reperfusion, digitalis toxicity), Ca2+ may trigger Ca2+ release from the sarcoplasmic reticulum during phase 4, which in turn prompts reverse-mode operation of the Na+/Ca2+ exchanger, causing an inwardly directed INa/Ca (Na+ influx). This current contributes to the generation of delayed afterdepolarizations and triggered arrhythmias (see later discussion).
Adrenergic receptors also play important roles in modulating the action potential by modifying channel activity.23–25 For example, stimulation of β-adrenergic receptors increases the activity of ICa-L, leading to increased Ca2+ influx, signaling increased contractile activity. β-Adrenergic receptor stimulation can also activate K+ channels, shortening repolarization and the duration of the action potential.26 α1-Adrenergic receptors exert actions via G-protein on the Na+/K+ pump, K+ channels, and phospholipase C and can alter impulse initiation and repolarization. α1-Adrenergic stimulation has been linked to triggered rhythms via early and delayed afterdepolarizations and the development of abnormal automatic rhythms in the setting of ischemia and reperfusion.27,28
 Mechanisms of Ventricular Tachyarrhythmias
Mechanisms of Ventricular Tachyarrhythmias
Abnormalities in Impulse Generation
Abnormalities in impulse generation are generally the result of automaticity or triggered activity.
Automaticity
Abnormal automaticity refers to the generation of impulses in cells that normally do not exhibit pacemaker potential. This phenomenon can occur in cells that are partially depolarized as a result of a pathologic process (e.g., ischemia). Under these conditions, the reduction in the resting membrane potential (less negative; to −70 or even −50 mV) shifts the balance during phase 4 toward depolarizing currents.29 Through this mechanism, automaticity can developed in atrial and ventricular muscle cells and in specialized tissues other than the SA in which the firing conditions can be altered. Examples of abnormal automaticity include accelerated idioventricular rhythms and some VTs that develop 24 to 72 hours after an acute myocardial infarction.30,31
Triggered Activity
Triggered activity refers to arrhythmias that arise from afterdepolarizations. Afterdepolarizations are alterations in membrane potential that occur during the repolarization phase without intervening external triggers or cell-to-cell interactions.32 Afterdepolarizations can develop in different phases of the action potential. Those that develop during phase 2, phase 3, or early phase 4 and are called early afterdepolarizations and are characterized by transient retardations in repolarization with or without upturn of the membrane potential (Figure 79-3). An upturn of sufficient magnitude may trigger an “extra” action potential before the cycle is over. Early afterdepolarizations are typically associated with conditions that prolong the action potential duration, such as decreased inactivation of fast INa (e.g., long QT3 syndrome) or decreased outward K+ currents (e.g., IKs in long QT1 and IKr in long QT2 syndromes) prolonging Ca2+ entry through ICa-L.33 The development of early afterdepolarizations in this setting is thought to trigger torsades de pointes. Early afterdepolarizations are also associated with increased sympathetic tone, use of catecholamines, hypoxia, acidosis, and bradycardia.
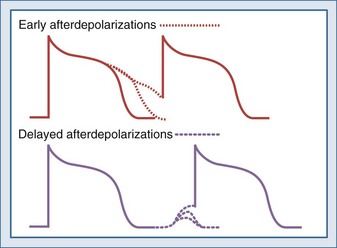
Afterdepolarizations that occur in late phase 4 are called delayed afterdepolarizations and are characterized by low-amplitude depolarizations that may reach threshold and trigger an action potential (see Figure 79-3). The main underlying abnormality in delayed afterdepolarizations is intracellular Ca2+ overload, promoting Ca2+ release from the sarcoplasmic reticulum33 and depolarizing currents (i.e., inward INa/Ca currents). Delayed afterdepolarizations are classically associated with digitalis toxicity; however, many other conditions favoring cytosolic Ca2+ overload can also produce delayed afterdepolarizations such as myocardial stretch, hypertrophy, catecholamines, ischemia, and reperfusion. In the setting of heart failure, increased expression of Na+-Ca2+ exchanger along with abnormalities in the ryanodine receptor has been shown to predispose to delayed afterdepolarizations.
Abnormalities in Impulse Conduction (Reentry)
Abnormalities in impulse conduction account for the vast majority of sustained ventricular tachyarrhythmias consequent to a phenomenon known as reentry. Reentry occurs when a normally propagating impulse reenters a region of previously excited tissue after its refractory period is over and excites it again. Reentry can continue to repeat, originating a tachyarrhythmia. Several forms of reentry have been described, including circus movement, phase 2, and reflection.34
Circus Movement
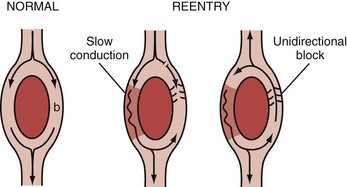
The ring model is the simplest of all35 and can be used to illustrate the basic mechanism of reentry (Figure 79-4). The ring model requires two anatomically contiguous paths in specialized tissue or in muscle fibers separated by a central area of unexcitable tissue. One of these paths (b in Figure 79-4) must exhibit a zone of unidirectional block allowing the impulse to propagate in only one direction. The alternative path (a in Figure 79-4) allows the impulse to circumvent the unidirectional block. Conduction through this alternative path should be slow or refractoriness proximal to the path should be brief to allow recovery of excitability. Once the impulse reaches the distal end of the alternative path, it propagates in a retrograde manner through the path of unidirectional block to reenter the proximal end of the alternative path. For the cycle to repeat (and establish a reentry circuit causing tachyarrhythmia), the wavelength of the circling impulse must be shorter than or at least equal to the length of the reentry circuit (or path length), thus enabling the leading edge of the circling impulse to find the tissue in an excitable state. The wavelength of the circling impulse is defined as the product of conduction velocity and duration of the refractory period.
The leading circle model is similar to the ring model but does not require anatomic obstacles and can develop in structurally uniform myocardium by a properly timed premature impulse.36,37 The figure-of-eight model was first described in experimental myocardial infarction. It refers to two reentry circuits moving alongside a functional conduction block (ischemia or infarct) in opposite directions, forming a pretzel-like configuration.38 The spiral wave model is considered a more complex version of the leading circle model. It involves a core and filaments and is usually described as reentry in two dimensions.39,40 The spiral wave model has been used to explain the electrocardiographic patterns associated with monomorphic and polymorphic VTs and also VF. In monomorphic VTs, the spiral wave is thought to be anchored and unable to drift within the myocardium, whereas in polymorphic VTs, such as torsades de pointes, the spiral is thought to drift. In the case of VF, the spiral wave is believed to break up into multiple rotating spiral waves that are continuously extinguishing and recreating. However, some authors have proposed a single rapidly shifting spiral, and others have postulated a stationary rotor whose frequency of excitation is exceedingly high, resulting in multiple areas of intermittent block.41
Phase 2
Phase 2 reentry refers to the generation of local reexcitation as a result of increased heterogeneity of repolarization. This phenomenon occurs when repolarization is markedly shortened in certain regions of the myocardium—essentially obliterating phase 2 of the action potential (plateau phase)—but is maintained in others. This creates conditions conducive to local reexcitation, which may precipitate ventricular tachyarrhythmias during myocardial ischemia.42 During ischemia, action potentials of normal duration alternate with ones of shorter duration, yielding beat-to-beat alternans (temporal dispersion) and site-to-site alternans (spatial dispersion) and promoting regions with conduction block and regions with injury current, leading to reentry and ventricular tachyarrhythmias. The degree of spatial and temporal dispersion progresses along with the duration of ischemia, suggesting that this mechanism may be an important trigger of VT and VF during acute myocardial ischemia.43,44 In the surface ECG, dispersion of the action potential duration manifests as T-wave alternans, which is a predictor of VF.45
Reflection
Reflection refers to a back-and-forth propagation of the impulse over the same functionally unexcitable tissue, with recurrent activation of the proximal region as a result of electrotonic currents.46,47 The area of unexcitable tissue could result from ischemia and lead to extrasystolic activity. Reflection differs from classic reentry in that the impulse travels along the same pathway in both directions.
 Conditions Predisposing to Ventricular Arrhythmias
Conditions Predisposing to Ventricular Arrhythmias
Channelopathies
The term “channelopathies” has been coined to identify a group of diseases characterized by abnormalities in the proteins that form ion channels.48,49 These abnormalities distort the normal action potential, primarily accentuating the inherent instability of repolarization and increasing the risk of polymorphic VT of the torsades de pointes type. Channelopathies may be hereditary or acquired.
Hereditary Channelopathies
Long QT Syndrome
The vast majority of hereditary channelopathies result from mutations in genes that encode for Na+ and K+ channels, with the most representative being the long QT syndrome.50–52 The long QT syndrome was first described in 1957 by Jervell and Lange-Nielsen in a group of patients with long QT intervals, episodes of torsades de pointes, and deafness.53 This syndrome is transmitted by autosomal recessive inheritance and is known as the Jervell and Lange-Nielsen syndrome. In 1963 and 1964, Romano and colleagues54 and Ward55 independently reported patients with an almost identical disorder but without deafness. This syndrome is transmitted by autosomal dominant inheritance and is known as the Romano-Ward syndrome.
It is now recognized that long QT syndrome results from mutations in at least 12 genes, leading to distinct types designated long QT1 through long QT12 (Table 79-1). Long QT1 is the principal genetic type responsible for both Jervell and Lange-Nielsen and Romano-Ward syndromes and accounts for nearly 50% of all genotyped families. Long QT2 accounts for nearly 40% and long QT3 for about 5%. The remaining types are much less frequent.56
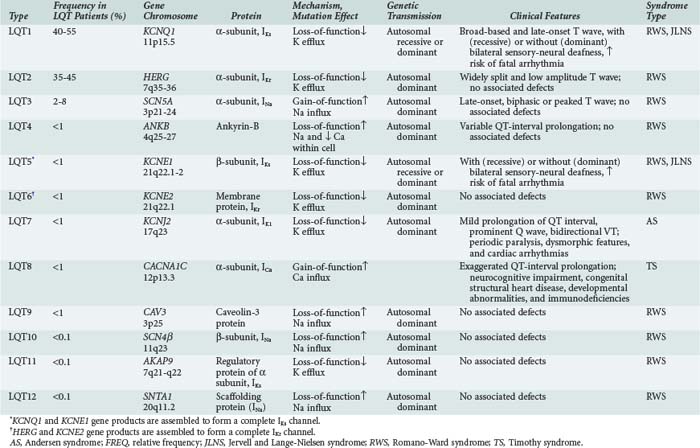
Long QT3 stems from a mutation in SCN5A, the gene that encodes the α-subunit of the fast cardiac Na+ channel. SCN5A mutation causes a gain of function leading to incomplete channel inactivation and persistence of INa during the plateau phase of the action potential.57,58
Long QT4 has been linked to a loss-of-function mutation in the ANKB gene.59 This gene encodes ankyrin-B, which is a member of a family of versatile membrane adapters. Ankyrin-B, among other functions, coordinates the opening and closing of calcium, potassium, sodium, and chloride channels. The failure to properly coordinate the opening and closing of ion channels leading to long QT syndrome and arrhythmias illustrates a novel mechanism of arrhythmias.
In long QT9, QT10, and QT12, the mutations affect the genes CAV3, SCN4β, and SNTA1, leading to a gain of function in INa, resulting in increased Na+ influx within the cardiac cell.60,61
Accordingly, the common mechanistic thread among long QT syndromes is perturbation of the balance between INa and IK during the plateau phase of the action potential, yielding prolongation of repolarization, a reduced rate of ICa-L inactivation, late Ca2+ influx, and early afterdepolarizations predisposing to torsades de pointes.62
The diagnosis is suspected in young individuals who present with syncope or episodes of sudden death typically during exercise, emotional distress, or exposure to factors that cause prolongation of the QT interval. A family history of unexplained syncope or sudden cardiac death, especially in young kindred, should raise suspicion. Sudden cardiac death occurs in approximately 4% of affected individuals. The diagnosis should be suspected when the corrected QT interval (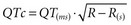 ) exceeds 470 milliseconds in males (normal <422 milliseconds) and 480 milliseconds in females (normal <432 milliseconds) in the absence of other conditions that may lengthen the QT interval. In addition, there may be sinus bradycardia with sinus pauses in about one-third of individuals (especially in long QT3), QT dispersion, and various T-wave abnormalities (e.g., notched, bifid, biphasic). Factors predisposing to sudden cardiac death include recurrent syncope, survival from cardiac arrest, congenital deafness, female sex, relative bradycardia, corrected QT interval greater than 600 milliseconds, and kinship with a symptomatic patient.50 Genetic testing for identifying the various long QT subtypes is becoming readily available, with its impact on risk stratification and guidance for placement of an implantable cardioverter-defibrillator (ICD) to be determined by future research.56,60
) exceeds 470 milliseconds in males (normal <422 milliseconds) and 480 milliseconds in females (normal <432 milliseconds) in the absence of other conditions that may lengthen the QT interval. In addition, there may be sinus bradycardia with sinus pauses in about one-third of individuals (especially in long QT3), QT dispersion, and various T-wave abnormalities (e.g., notched, bifid, biphasic). Factors predisposing to sudden cardiac death include recurrent syncope, survival from cardiac arrest, congenital deafness, female sex, relative bradycardia, corrected QT interval greater than 600 milliseconds, and kinship with a symptomatic patient.50 Genetic testing for identifying the various long QT subtypes is becoming readily available, with its impact on risk stratification and guidance for placement of an implantable cardioverter-defibrillator (ICD) to be determined by future research.56,60
In addition to long QT syndromes, recent population studies63 have shown that even milder prolongation of the QTc in adults (>450 milliseconds in men and > 470 milliseconds in women) increases the risk of sudden cardiac death by threefold after adjustment for age, gender, body mass index, hypertension, cholesterol/high-density lipoprotein ratio, diabetes mellitus, myocardial infarction, heart failure, and heart rate. In the same population, QTc prolongation was strongly associated with variant rs10494366 T>G and rs10918594 C>G of the nitric oxide synthase 1 adaptor protein (NOS1AP) gene.64 More recent studies show that use of the calcium channel blocker, verapamil, in patients with these variants prompts a greater QTc prolongation than in patients with the wild genotype.65
Short QT Syndrome
Short QT is a more recently described syndrome66 characterized by tall and peaked T waves with QT interval ≤ 300 milliseconds, insensitive to changes in heart rate, and a structurally normal heart. Individuals are at risk of developing VF and also atrial fibrillation and may complain of palpitations and episodes of syncope. They may also have family members with a similar history or a history of unexplained or sudden death at a young age.
Brugada Syndrome
Another important hereditary channelopathy is Brugada syndrome, described in 1992 by the Brugada brothers,67–70 who noticed an association between sudden cardiac death and ST-segment elevation in V1 to V3, with a pattern resembling right bundle branch block in individuals with structurally normal hearts.
Brugada syndrome results in part from mutations in the SCN5A gene (encoding for the INa α-subunit). In contrast to long QT3—which also affects the SCN5A gene—the mutations in Brugada syndrome lead to a loss of function resulting in accelerated inactivation of INa during phase 1, leaving the repolarizing ITo current unopposed and consequently prompting rapid repolarization and shortening of the action potential duration (refer to Figure 79-2 for the INa and ITo contributions to the action potential). Because ITo is expressed predominantly in the epicardium, the normally depolarized endocardium can re-excite the prematurely repolarized epicardium, leading to phase 2 reentry and generation of a phase 2 reentrant premature beat that could capture a vulnerable window and precipitate VT (which is typically polymorphic) and/or VF.
Brugada syndrome exhibits predominantly an autosomal dominant pattern of inheritance, with an average worldwide prevalence of 5 : 10,000. More than 100 mutations in seven genes have been identified. Loss-of-function mutations in the SCN5A gene cause approximately 20% of cases. A few mutations have been described in the GPD1L gene, which encodes the glycerol-3-phosphate dehydrogenase-1 like protein; the CACNA1C gene, which encodes the α-subunit of the Ca(v)1.2 ion channel conducting the inward current (ICa-L); the CACNB2 gene, which encodes the β2-subunit of the Ca(v)1.2 ion channel; the SCN1B and SCN3B genes, which in the heart encode the β-subunits of the Na(v)1.5 sodium ion channel; and the KCNE3 gene, which encodes the ancillary inhibitory β-subunit of several potassium channels including the Kv4.3 ion channel conducting the repolarizing potassium current ITos.71
The ST-segment elevation can adopt various shapes that have been related to the severity of the INa/ITo imbalance, including—in order of increasing severity—saddleback, coved, and triangular shapes.62 These changes are dynamic and can change in the same affected individual as shown in Figure 79-5.
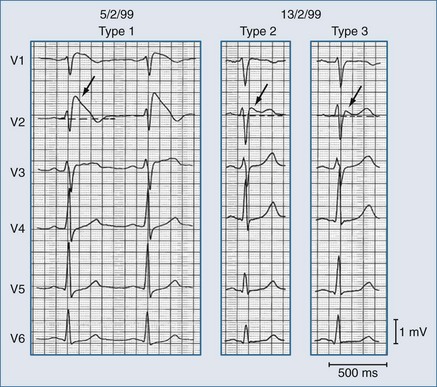
Brugada syndrome is the major but not the only cause of sudden unexpected death syndrome (SUDS)72,73 and is the most common cause of sudden death in young men without known underlying cardiac disease in Thailand and Laos.74 However, Brugada syndrome exhibits variable expressivity, reduced penetrance, and “mixed phenotypes” where families may contain members with Brugada syndrome as well as members with short QT syndrome, long QT syndrome, atrial fibrillation, disease of the conduction system, and even structural heart disease.71
Patient with Brugada syndrome may have concealed or intermittent forms that can be unmasked (or precipitated) by febrile states, vagotonic agents, α-adrenergic agonists, β-adrenergic blockers, tricyclic or tetracyclic antidepressants, a combination of glucose and insulin and hypokalemia, and alcohol and cocaine toxicity.75
A useful clinical test to identify concealed Brugada syndrome is the administration of class IC antiarrhythmic drugs: Na+ channel blockers such as ajmaline (1 mg/kg intravenous (IV) in 5 minutes), flecainide (2 mg/kg IV in 10 minutes), or procainamide (10 mg/kg IV in 10 minutes). The test should be performed in an environment with capability for immediate defibrillation, because there is a 0.5% risk of precipitating VF. Of the drugs listed above, ajmaline is the preferred drug because of its very short half-life.76 The test is considered positive if an additional 1-mm ST-segment elevation (measured 0.08 seconds after the J point) occurs in leads V1, V2, and V3. The test is highly specific and should be considered in patients who present with history of syncope of unknown origin or with idiopathic VF.
Patients with the Brugada syndrome must be treated with an ICD, because antiarrhythmic agents have not been found in general to be effective.69 However, recent studies have shown that quinidine and hydroquinidine can prevent spontaneous ECG changes and reduce the risk of VT and VF, presumably through inhibition of ITo.77,78 Although such a pharmacologic approach is currently regarded as an adjunct to ICD placement to reduce the number of shocks, it could be considered in high-risk patients when ICD placement is not possible.
Acquired Channelopathies
Acquired abnormalities in cardiac channels may result from a broad spectrum of conditions. One common mechanism is advanced heart failure affecting the expression of several ion channels regardless of the primary etiology.79,80 For example, there is down-regulation of ITo and IK1, leading to prolongation of the QT interval. Although such an effect could be considered adaptive, allowing more time for excitation-contraction coupling, it also predisposes to inhomogeneous repolarization and early afterdepolarizations. In addition, in heart failure there is up-regulation of the Na+/Ca2+ exchanger—consequent in part to down-regulation of the sarcoplasmic reticulum Ca2+-ATPase (SERCA2a)81—yielding larger INa/Ca, which predisposes to delayed afterdepolarizations and triggered arrhythmias, especially in the face of cytosolic Ca2+ overload.
Another increasingly important mechanism of acquired channelopathies is the use of medications that can interfere with channel function. By far the most common effect is prolongation of the QT interval leading to drug-induced long QT syndrome. Most of the drugs involved block the K+ repolarizing current carried by human ether-à-go-go (HERG) subunits corresponding to the IKr channel.82 IKr carries the same K+ current that is affected in congenital long QT2. The list of medications is long and includes antiarrhythmic agents, in which the primary target is ion channels, and many other drugs in which prolongation of the QT interval is an unintended effect.83 The intensivist should be familiar with this group of medications and capable of recognizing the arrhythmogenic risk of administering medications that could prolong the QT interval.
The University of Arizona, Health Sciences Center, maintains a list of drugs associated with risk of causing QT prolongation and promoting torsades de pointes (available at www.qtdrugs.org). Based on the risk level, drugs are classified by an advisory board as: (1) Torsades List, defined as drugs that are generally accepted to carry a risk of torsades de pointes; (2) Possible Torsades List, defined as drugs that prolong the QT interval and/or in some reports have been associated with torsades de pointes but lack substantial evidence for causing torsades de pointes; (3) Conditional Torsades List, defined as drugs that carry a risk of torsades de pointes and/or QT prolongation under certain conditions such as in patients with congenital long QT syndrome, drug overdose, or coadministration of interacting drugs; and (4) drugs to be avoided in patients with congenital long QT syndrome. Table 79-2 lists drugs from the Torsades List.
TABLE 79-2 Drugs Associated with QT Prolongation and Risk of Torsades de Pointes
| Antiarrhythmic Agents | Antipsychotic Agents |
| Amiodarone | Chlorpromazine |
| Disopyramide | Haloperidol |
| Dofetilide | Mesoridazine |
| Ibutilide | Pimozide |
| Procainamide | Thioridazine |
| Quinidine | Enterokinetic/Antinausea Agents |
| Sotalol | Cisapride |
| Antibiotics | Domperidone |
| Clarithromycin | Droperidol |
| Erythromycin | Opiate Agonists |
| Chloroquine | Levomethadyl |
| Halofantrine | Methadone |
| Pentamidine | Miscellaneous |
| Sparfloxacin | Arsenic trioxide |
| Antihistaminic Agents | Probucol |
| Astemizole | |
| Terfenadine |
Data from www.QTdrugs.org (Torsades list: Drugs with a risk of torsades de pointes); revised on 03/25/2008.
The importance of drug-induced long QT syndrome has mandated pharmaceutical companies to screen early in the process of drug selection to exclude compounds that can induce long QT syndrome, mostly screening for effects on the HERG gene products carrying the IKr current.82
Other Conditions
The QT interval may also be prolonged by cocaine abuse, organ phosphorus compound poisoning, subarachnoid hemorrhage, stroke, myocardial ischemia, fasting using liquid-protein-modified diets, autonomic neuropathy, and human immunodeficiency virus disease.84–88 Electrolyte abnormalities can not only prolong but also shorten the QT interval. Some of these conditions and others not associated with channelopathies are discussed next.
Electrolyte Abnormalities
Electrolyte abnormalities rarely precipitate but often contribute to the development of ventricular tachyarrhythmias, mostly in relation to abnormalities in serum K+, Mg2+, and Ca2+.89 Abnormalities in serum K+ are among the most common electrolyte abnormalities in critically ill patients.
Hypokalemia (serum K+ < 3.5 mM) decreases the resting membrane potential (making it more negative), rendering cells less excitable and lowering the firing rate of pacemaker cells. Hypokalemia also prolongs the QT interval and flattens the T wave.17 This effect is explained by the fact that conductivity of Ikr is proportional to the square root of external K+. Thus, at lower K+, IKr is reduced, prolonging repolarization. This effect is more pronounced in cells from the mid-myocardial region (which have a greater IKr/Iks ratio). Hypokalemia can develop in various settings, including the use of thiazide and loop diuretics, diabetic ketoacidosis, gastrointestinal fluid losses, alcohol abuse, hypomagnesemia, administration of insulin, and use of β-receptor agonists.
Hypothermia
Moderate hypothermia (32°C to 35°C) and severe hypothermia (<32°C) can also predispose to ventricular tachyarrhythmias by causing prolongation of the QT interval and QT dispersion.90 Typically, patients with hypothermia develop J waves (also known as Osborn waves) in the ECG, which reflects accentuation of the inhomogeneity of repolarization caused by the predominant distribution of ITo in subepicardial and mid-myocardial regions.91 Hypothermia may be complicated by the ingestion of drugs and the presence of electrolyte abnormalities that further increase the risk of ventricular tachyarrhythmias.
Hypoglycemia
Recent studies have shown that acute hypoglycemia can trigger VT and VF in patients with diabetes mellitus.92 The mechanism involves prolongation of the QT interval by direct suppression of repolarizing K+ currents. In addition, episodes of hypoglycemia trigger a neuroendocrine stress response with release of catecholamines which favors intracellular Ca2+ entry and reduces serum K+, further compounding the risk. Patient at particularly high risk are those with coronary artery disease or acute myocardial infarction, left ventricular hypertrophy, autonomic neuropathy, congestive heart failure, and on those taking medications that prolong the QT interval.
Arrhythmogenic Right Ventricular Cardiomyopathy
This disorder is characterized by progressive replacement of the normal right ventricular muscle cells by fibrous tissue and fat.93 The condition may be familial with autosomal dominant inheritance.94 Patients present with palpitations, syncope, and sometimes sudden death. It is considered an important cause of sudden death in subjects younger than 35 years, especially when related to exercise.95,96 The ECG is abnormal in 90% of cases, showing T-wave inversions beyond lead V1 and epsilon waves in leads V1 to V3. The QRS complex may be widened (>110 milliseconds), with complete or incomplete right bundle branch block morphology. There are ventricular premature beats with left bundle branch configuration.
 Clinical Diagnosis
Clinical Diagnosis
Ventricular Tachycardia
VT is defined as three or more consecutive ventricular ectopic beats with a rate that typically exceeds 100 beats per minute and often ranges between 130 and 170 beats per minute. VT usually have QRS complexes of 120 msec or longer and is therefore classified as wide-complex tachycardia. However, wide-complex tachycardia, as described earlier, can also be supraventricular when the impulse originates above the bifurcation of the bundle of His but is conducted with aberrancy (see later).97 VTs are classified as monomorphic if all QRS complexes have similar morphology and polymorphic if they have variable morphology. VTs are considered sustained if they last 30 seconds or longer and nonsustained if they last less than 30 seconds. Most sustained VTs present with palpitations, chest discomfort, and weakness or with more severe symptoms such as dizziness, angina, syncope, seizures, and even sudden cardiac death.98
Monomorphic Ventricular Tachycardia
Monomorphic VTs are the most common and are usually associated with structural heart disease, such as previous myocardial infarction and, less commonly, cardiomyopathy. The reentrant circuit can be small (microentry) or large (macroentry) and can be located in different regions of the myocardium. The mechanism is usually reentry operating within or around damaged myocardium. A representative tracing is shown in Figure 79-6.
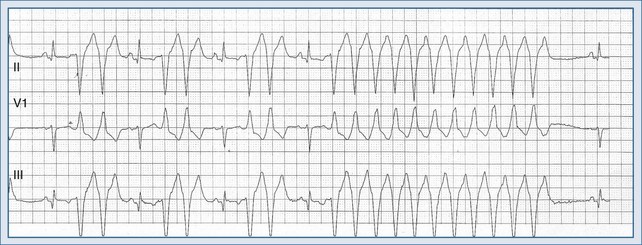
In nonemergency settings, a standard 12-lead ECG should be obtained to determine whether a wide-complex tachycardia is present and whether it is monomorphic or polymorphic. If monomorphic, the possibility of supraventricular tachycardia (SVT) with aberrancy should be considered, although most wide-complex tachycardias are ventricular. The presence of shock, heart failure, or cardiac arrest favors VT. SVT with aberrancy (in a stable patient) should be suspected whenever there is a history of previous aberrant rhythms, accessory pathways, and baseline or rate-induced bundle branch block. The ECG should be carefully examined for evidence of AV dissociation, which is specific for VT.99 If P waves are not visualized in V1 or in any of the other standard leads, a Lewis lead (arm electrode positioned on the parasternal area) or an esophageal lead can be used.100 AV dissociation is indicated by P waves and QRS complexes that present at different and uncoupled rates. Other manifestations of dissociation include captured beats (narrow QRS conducted beats) and fusion beats (merge of ectopic with conducted beats).
ECG criteria and algorithms are available to help differentiate ventricular from SVT.101–104 A widely accepted four-step algorithm developed by Brugada is shown in Figure 79-7.102 A similar algorithm that incorporates pertinent clinical information can be found at http://www.anaesthetist.com/icu/organs/heart/ecg/wct.htm#step0.
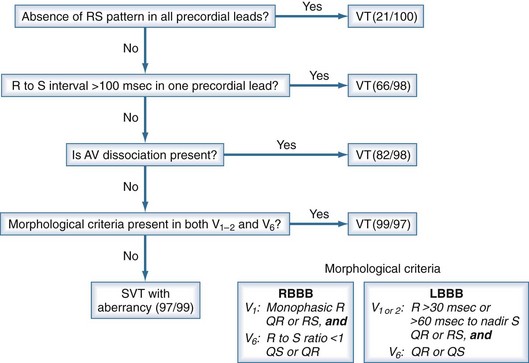
Figure 79-7 Four-step Brugada algorithm for diagnosis of wide-complex tachycardia. Ventricular tachycardia (VT) is diagnosed whenever an answer is positive within each successive step: in step 1, when an RS complex cannot be identified in any precordial lead; in step 2, when the longest RS complex (beginning of R to nadir of S) in a precordial lead exceeds 100 milliseconds; in step 3, when there is atrioventricular (AV) dissociation; and in step 4, when the morphologic criteria for tachycardia with right bundle branch block (RBBB) or left bundle branch block (LBBB) morphology are met. In parentheses are sensitivity and specificity reported in the original report based on 554 wide-complex tachycardias.102 SVT, supraventricular tachycardia.
Some special forms of VT tend to be mistaken for SVT with aberrancy.105 These include bundle branch reentrant tachycardia, in which the impulse travels down the right bundle branch, across the interventricular septum, and up the left bundle branch.106,107 The morphology resembles SVT with LBBB and is common among patients with nonischemic dilated cardiomyopathy.108 Right ventricular outflow tract tachycardia is another condition caused by triggered activity from delayed afterdepolarizations that most commonly originate in the right ventricular outflow tract.109 The tachycardia usually presents with LBBB morphology and right axis deviation. Right ventricular outflow tract tachycardias occur in structurally normal hearts, typically in young individuals, and are responsive to verapamil or adenosine.110 Finally, there are fascicular tachycardias that originate from either fascicle of the left bundle branch. They occur in structurally normal hearts, mimic SVT with aberrancy, and are responsive to beta-blockers and verapamil.111
Polymorphic Ventricular Tachycardia
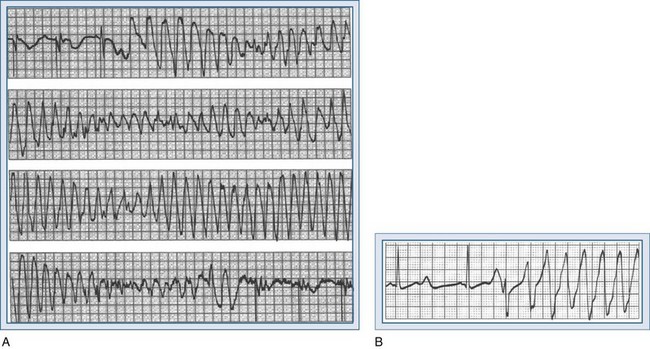
Polymorphic VTs have irregular rhythms, usually compromise hemodynamic function, and may quickly degenerate into VF. Variation in QRS morphology represents changes in the electrical axis. One special form of polymorphic VT is torsades de pointes. This is a descriptive term denoting a rotating electrical axis in 180 degrees along an imaginary axis (“twisting points”); it is typically associated with long QT syndrome. Representative tracings are shown in Figure 79-8.
Accelerated Ideoventricular Rhythm
Accelerated idioventricular rhythm (AIVR) is a form of automatic ventricular arrhythmia and is characterized by the presence of regularly wide QRS complexes with a rate between 50 and 120 beats per minute. It is often, but not always, slightly faster than the underlying sinus rhythm. Accelerated idioventricular rhythm is an electrocardiographic diagnosis and does not produce symptoms. Identifying this rhythm is important because it usually indicates underlying myocardial ischemia, and the treatments for VT may not apply.112
Incidence in the Critical Care Setting
The reported incidence of ventricular arrhythmias in critically ill patients is influenced by multiple factors including the underlying condition, predisposing factors, structural abnormalities, management in ICU, triggering events, and also the method for detection and the definition of the event.113,114
A recent multicenter study surveyed the incidence and prognostic implication of arrhythmias in critically ill patients in 26 ICUs over a period of 1 month.115 The study included 1341 patients, of whom 163 (12%) had episodes of sustained arrhythmias, encompassing 8% with supraventricular arrhythmias (atrial fibrillation the most common), 2% with ventricular arrhythmias, and 2% with conduction abnormalities. The in-hospital mortality was 29% in patients with arrhythmias and 17% in those without. However, adjusting for prognosis factors—including older age, past medical history of cardiovascular disease, admission for acute medical illness, sepsis, central nervous system or cardiovascular disease, higher SAPS II score, and ventilator management or vasopressor agents—and propensity scores, only ventricular arrhythmias were associated with increased mortality (OR = 3.53; 95% CI, 1.19-10.42). Supraventricular arrhythmias and conduction abnormalities were instead markers of severity of illness without independent contribution to the risk of death.
In another study, Reinelt and colleagues114 reported a higher incidence of ventricular tachyarrhythmias corresponding to 9% (65 of 756 patients), distributed among monomorphic VT in 83%, VF in 9%, and polymorphic VT in 8%. Factors present during the arrhythmic episodes included hypokalemia (10%), hypomagnesemia (12%), sedation (60%), mechanical ventilation (77%), and administration of catecholamines such as norepinephrine, epinephrine, or dobutamine (75%). In 23% of the episodes there was a history of previous myocardial infarction, and in 40% a history of recent myocardial infarction. Fifty-two percent of the episodes occurred during a postoperative period and 35% while a pulmonary artery catheter was in place. The presence of arrhythmias was associated with an increased length of stay and lower survival.
Special consideration should be given to patients admitted for evaluation of an acute coronary syndrome. Before the advent of thrombolysis in the 1980s, the incidence of VT ranged between 3% and 39%.116 With the widespread use of thrombolysis, the incidence has decreased.117 This is thought to reflect less ventricular dysfunction and dilatation as a result of successful reperfusion. For similar reasons, VT is less frequent in patients with non–ST-segment elevation (<1%) compared with those with ST-segment elevation (≈4%) myocardial infarction.117
Episodes of ventricular arrhythmias associated with acute myocardial infarction have different mechanisms contingent on whether they occur early or late and whether they are sustained or nonsustained. Early usually refers to the initial 48 hours after the onset of symptoms.118 Early nonsustained VTs are relatively common, with an incidence between 9% and 12%,119,120 and reflect electrical instability during the acute ischemic event but have little prognostic implication.121 Early sustained VTs or VF occur less frequently122 but identify a population at higher risk of death.117
In a recent study,123 investigators analyzed the incidence and long-term prognosis of early VF or sustained VT (VF/VT) in 16,588 patients with acute myocardial infarction from the GUSTO V trial, and also examined the impact of baseline use of angiotensin-converting enzyme inhibitors (ACEI) and angiotensin receptor blockers (ARB). Early VF/VT occurred in 732 patients (4.4%). Compared to patients without VF/VT, the development of VF or VT was associated with significantly higher 30-day mortality (22% versus 5%, P < 0.001). Baseline use of an ACEI or ARB was associated with decreased incidence of VF/VT (OR 0.65, 0.47-0.89, P = 0.008). Moreover, of patients who developed VF/VT, those on baseline ACEI or ARB had a lower 30-day mortality compared with those not on an ACEI or ARB (17.7% versus 24.2%, P = 0.04). The association between baseline ACEI or ARB persisted after adjustment for multiple confounders.
Late VTs coincide with the phase of myocardial healing and may signal the presence of persistent ischemia, left ventricle dysfunction, or electrophysiologic instability.118 Nonsustained VTs occur in approximately 6% of patients.52,124 Sustained VTs occur in approximately 1% and convey a worse prognosis than do nonsustained episodes.117,125
One particularly arrhythmogenic period in patients with acute myocardial infarction is during reperfusion after thrombolysis.52,119 There are frequent PVCs and episodes of nonsustained VT but rarely episodes of sustained VT or VF.126 AIVR is also common, with an incidence as high as 50% to 75%. It occurs within 24 hours after the start of thrombolysis and then subsides.112,126
Efforts to predict life-threatening arrhythmias in the ICU have not been successful. In one study, 127 ECG markers of autonomic tone, ventricular irritability, and repolarization lability measured within 12 hours before an episode of monomorphic VT, polymorphic VT, or VF failed to predict the event.
 Acute Management
Acute Management
Ventricular Arrhythmias with Preserved Blood Flow
Monomorphic Ventricular Tachycardia
The American College of Cardiology/American Heart Association/European Society of Cardiology (ACC/AHA/ESC) 2006 Guidelines for management of patients with ventricular arrhythmias128 recognize various drugs available in IV formulation that could be used for treating VT, including flecainide, propafenone, sotalol, procainamide, lidocaine, and amiodarone, with availability contingent on the specific country. The same 2006 Guidelines recommend IV procainamide (or ajmaline in some European countries) as a reasonable initial choice for patients with stable sustained monomorphic VT.129,130 Close monitoring is recommended, as IV procainamide can cause transient hypotension,131 especially in patients with severe left ventricular dysfunction. For patients with sustained monomorphic VT but who are hemodynamically unstable, are refractory to conversion after electrical shocks, or have recurrent episodes despite procainamide or other agents, IV amiodarone is considered a reasonable choice.132–135 The initial effect of amiodarone is to slow down AV nodal conduction and block adrenergic stimulation. However, effects on ventricular conduction and refractoriness develop more gradually, achieving the maximal effect only after weeks or months of treatment.136–138
In patients in whom stable sustained monomorphic VT is specifically associated with an acute ischemic substrate (i.e., unstable angina or myocardial infarction), lidocaine is considered a reasonable initial choice.139,140
Ventricular Arrhythmias with Cessation of Effective Blood Flow
Ventricular Fibrillation and Pulseless VT
Consistent with the AHA/ERC 2005 guidelines,141 the energy levels of the initial electric shock depends on the waveform and specific device. For biphasic waveform defibrillators, the initial device-specific energy level typically ranges from 150 to 200 J. In the absence of a recommended dose, 200 J should be used. Equal or higher energy level dose is recommended for the second and subsequent shocks. If the available defibrillator uses monophasic waveforms, the energy level should be 360 J for all shocks. Following shock delivery, providers should be prepared to provide advanced cardiac life support according to the most recent guidelines for cardiopulmonary resuscitation and emergency cardiovascular care (i.e., 2010 Guidelines). A 1-shock strategy followed by immediate chest compression is now recommended to minimize interruptions on chest compression. The general goals of advanced cardiac life support are to reestablish and maintain a hemodynamically effective cardiac rhythm.
The time for appropriate intervention is critically important. The probability of survival after VF and pulseless VT is inversely related to the time elapsed between the onset of the arrhythmia and the delivery of electric shocks.142,143 Recent studies have shown that immediate defibrillation is highly effective and is associated with high survival rates when the duration of untreated VF is short (<4 minutes).144,145 With more protracted untreated VF, mounting evidence from animal and human studies indicates that a period of closed-chest resuscitation before attempting defibrillation could improve outcome.144,146–148 For patients with shock-refractory VF or pulseless VT, use of amiodarone has been shown to facilitate the restoration of cardiac activity.149,150
Electrical storm is a rather uncommon but highly lethal phenomenon defined as recurrent episodes of VF, occurring mainly in the course of an acute myocardial infarction. Conventional antiarrhythmic drug therapy—including lidocaine and procainamide—often fails to secure a stable sinus rhythm. The underlying mechanism seems to be excessive (and probably unbalanced) sympathetic activity. Recent studies have shown that outcome can be dramatically improved by sympathetic blockade using IV beta-blockers or stellate ganglionic blockade.151
Polymorphic Ventricular Tachycardia
Polymorphic VT with a normal QT interval is most frequently seen when acute myocardial ischemia is present but is also associated with cardiomyopathies, idiopathic polymorphic VT, and catecholaminergic VT.152,153 In this setting, use of IV beta-blockers153 or IV amiodarone154 has been shown to be effective. Coronary angiography should be considered in the setting of recurrent polymorphic VT when ischemia is suspected.155
 Conclusion
ConclusionKey Points
1 Catterall WA. Structure and function of voltage-sensitive ion channels. Science. 1988;242:50-61.
2 Jan LY, Jan YN. Voltage-gated and inwardly rectifying potassium channels. J Physiol. 1997;505:267-282.
3 Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. J Physiol. 1998;508:647-657.
4 Garny A, Kohl P. Mechanical induction of arrhythmias during ventricular repolarization: modeling cellular mechanisms and their interaction in two dimensions. Ann N Y Acad Sci. 2004;1015:133-143.
5 Chorro FJ, Trapero I, Such-Miquel L, Pelechano F, Mainar L, Canoves J, et al. Pharmacological modifications of the stretch-induced effects on ventricular fibrillation in perfused rabbit hearts. Am J Physiol Heart Circ Physiol. 2009;297:H1860-H1869.
6 Madias C, Maron BJ, sheikh-Ali AA, Estes NAIII, Link MS. Commotio cordis. Indian Pacing Electrophysiol J. 2007;7:235-245.
7 Madias C, Maron BJ, Supron S, Estes NA, Link MS. Cell membrane stretch and chest blow-induced ventricular fibrillation: commotio cordis. J Cardiovasc Electrophysiol. 2008;19:1304-1309.
8 Madias C, Maron BJ, sheikh-Ali AA, Rajab M, Estes NAIII, Link MS. Precordial thump for cardiac arrest is effective for asystole but not for ventricular fibrillation. Heart Rhythm. 2009;6:1495-1500.
9 Eich C, Bleckmann A, Schwarz SK. Percussion pacing—an almost forgotten procedure for haemodynamically unstable bradycardias? A report of three case studies and review of the literature. Br J Anaesth. 2007;98:429-433.
10 Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol. 2010;48:12-25.
11 McKinnon D. Molecular identity of Ito: Kv1.4 redux. Circ Res. 1999;84:620-622.
12 Nabauer M, Beuckelmann DJ, Uberfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 1996;93:168-177.
13 Pelzer D, Pelzer S, McDonald TF. Properties and regulation of calcium channels in muscle cells. Rev Physiol Biochem Pharmacol. 1990;114:107-207.
14 Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J. 1995;68:949-964.
15 Giles WR, Imaizumi Y. Comparison of potassium currents in rabbit atrial and ventricular cells. J Physiol. 1988;405:123-145.
16 Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol. 1990;96:195-215.
17 Yan GX, Antzelevitch C. Cellular basis for the normal T wave and the electrocardiographic manifestations of the long-QT syndrome. Circulation. 1998;98:1928-1936.
18 Brown HF. Electrophysiology of the sinoatrial node. Physiol Rev. 1982;62:505-530.
19 Ehara T, Noma A, Ono K. Calcium-activated non-selective cation channel in ventricular cells isolated from adult guinea-pig hearts. J Physiol. 1988;403:117-133.
20 Hamada K, Yamazaki J, Nagao T. Shortening of monophasic action potential duration during hyperkalemia and myocardial ischemia in anesthetized dogs. Jpn J Pharmacol. 1998;76:149-154.
21 Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular perspective. Annu Rev Physiol. 2000;62:111-133.
22 Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198-205.
23 Apkon M, Nerbonne JM. Alpha 1-adrenergic agonists selectively suppress voltage-dependent K+ current in rat ventricular myocytes. Proc Natl Acad Sci U S A. 1988;85:8756-8760.
24 Hamra M, Rosen MR. Alpha-adrenergic receptor stimulation during simulated ischemia and reperfusion in canine cardiac Purkinje fibers. Circulation. 1988;78:1495-1502.
25 del Balzo U, Rosen MR, Malfatto G, Kaplan LM, Steinberg SF. Specific alpha 1-adrenergic receptor subtypes modulate catecholamine-induced increases and decreases in ventricular automaticity. Circ Res. 1990;67:1535-1551.
26 Yazawa K, Kameyama M. Mechanism of receptor-mediated modulation of the delayed outward potassium current in guinea-pig ventricular myocytes. J Physiol. 1990;421:135-150. 135-50
27 Sheridan DJ, Penkoske PA, Sobel BE, Corr PB. Alpha adrenergic contributions to dysrhythmia during myocardial ischemia and reperfusion in cats. J Clin Invest. 1980;65:161-171.
28 Kimura S, Cameron JS, Kozlovskis PL, Bassett AL, Myerburg RJ. Delayed afterdepolarizations and triggered activity induced in feline Purkinje fibers by alpha-adrenergic stimulation in the presence of elevated calcium levels. Circulation. 1984;70:1074-1082.
29 Dangman KH, Hoffman BF. Studies on overdrive stimulation of canine cardiac Purkinje fibers: maximal diastolic potential as a determinant of the response. J Am Coll Cardiol. 1983;2:1183-1190.
30 Norris RM, Mercer CJ. Significance of idioventricular rhythms in acute myocardial infarction. Prog Cardiovasc Dis. 1974;16:455-468.
31 Sclarovsky S, Strasberg B, Fuchs J, Lewin RF, Arditi A, Klainman E, et al. Multiform accelerated idioventricular rhythm in acute myocardial infarction: electrocardiographic characteristics and response to verapamil. Am J Cardiol. 1983;52:43-47.
32 Fozzard HA. Afterdepolarizations and triggered activity. Basic Res Cardiol. 1992;87(Suppl 2):105-113.
33 Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185-1192.
34 Antzelevitch C. Basic mechanisms of reentrant arrhythmias. Curr Opin Cardiol. 2001;16:1-7.
35 Wit AL, Cranefield PF. Reentrant excitation as a cause of cardiac arrhythmias. Am J Physiol. 1978;235:H1-17.
36 Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. Circ Res. 1973;33:54-62.
37 Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res. 1977;41:9-18.
38 El Sherif N, Smith RA, Evans K. Canine ventricular arrhythmias in the late myocardial infarction period. 8. Epicardial mapping of reentrant circuits. Circ Res. 1981;49:255-265.
39 Davidenko JM, Pertsov AV, Salomonsz R, Baxter W, Jalife J. Stationary and drifting spiral waves of excitation in isolated cardiac muscle. Nature. 1992;355:349-351.
40 Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res. 1993;72:631-650.
41 Samie FH, Jalife J. Mechanisms underlying ventricular tachycardia and its transition to ventricular fibrillation in the structurally normal heart. Cardiovasc Res. 2001;50:242-250.
42 Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res. 1996;32:593-603.
43 Kurz RW, Mohabir R, Ren XL, Franz MR. Ischaemia induced alternans of action potential duration in the intact-heart: dependence on coronary flow, preload and cycle length. Eur Heart J. 1993;14:1410-1420.
44 Kurz RW, Xiao-Lin R, Franz MR. Increased dispersion of ventricular repolarization and ventricular tachyarrhythmias in the globally ischaemic rabbit heart. Eur Heart J. 1993;14:1561-1571.
45 Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385-1394.
46 Rozanski GJ, Jalife J, Moe GK. Reflected reentry in nonhomogeneous ventricular muscle as a mechanism of cardiac arrhythmias. Circulation. 1984;69:163-173.
47 Lukas A, Antzelevitch C. Reflected reentry, delayed conduction, and electrotonic inhibition in segmentally depressed atrial tissues. Can J Physiol Pharmacol. 1989;67:757-764.
48 Clancy CE, Kass RS. Defective cardiac ion channels: from mutations to clinical syndromes. J Clin Invest. 2002;110:1075-1077.
49 Marban E. Cardiac channelopathies. Nature. 2002;415:213-218.
50 Khan IA. Long QT syndrome: diagnosis and management. Am Heart J. 2002;143:7-14.
51 Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866-1874.
52 Vincent GM. The long-QT syndrome—bedside to bench to bedside. N Engl J Med. 2003;348:1837-1838.
53 Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59-68.
54 Romano C, Gemme G, Pongiglione R. [Rare cardiac arrhythmias of the pediatric age. II. syncopal attacks due to paroxysmal ventricular fibrillation (presentation of 1st case in Italian pediatric literature)]. Clin Pediatr (Bologna). 1963;45:656-683.
55 Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103-106.
56 Lu JT, Kass RS. Recent progress in congenital long QT syndrome. Curr Opin Cardiol. 2010.
57 Bendahhou S, Fournier E, Gallet S, Menard D, Larroque MM, Barhanin J. Corticosteroid-exacerbated symptoms in an Andersen’s syndrome kindred. Hum Mol Genet. 2007;16:900-906.
58 Jamshidi Y, Nolte IM, Spector TD, Snieder H. Novel genes for QTc interval. How much heritability is explained, and how much is left to find? Genome Med. 2010;2:35.
59 Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634-639.
60 Delaney J, Mittal S, Sherrid MV. Current perspectives on congenital long QT syndrome. Anadolu Kardiyol Derg. 2009;9(Suppl 2):3-11.
61 Hedley PL, Jorgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, et al. The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat. 2009;30:1486-1511.
62 Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res. 2002;90:889-896.
63 Straus SM, Kors JA, De Bruin ML, van der Hooft CS, Hofman A, Heeringa J, et al. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J Am Coll Cardiol. 2006;47:362-367.
64 Aarnoudse AJ, Newton-Cheh C, de Bakker PI, Straus SM, Kors JA, Hofman A, et al. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation. 2007;116:10-16.
65 van NC, Aarnoudse AJ, Eijgelsheim M, Sturkenboom MC, Straus SM, Hofman A, et al. Calcium channel blockers, NOS1AP, and heart-rate-corrected QT prolongation. Pharmacogenet Genomics. 2009;19:260-266.
66 Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calo L, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494-1499.
67 Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391-1396.
68 Rook MB, Bezzina AC, Groenewegen WA, van Gelder IC, van Ginneken AC, Jongsma HJ, et al. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res. 1999;44:507-517.
69 Grant AO, Carboni MP, Neplioueva V, Starmer CF, Memmi M, Napolitano C, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest. 2002;110:1201-1209.
70 Mattu A, Rogers RL, Kim H, Perron AD, Brady WJ. The Brugada syndrome. Am J Emerg Med. 2003;21:146-151.
71 Hedley PL, Jorgensen P, Schlamowitz S, Moolman-Smook J, Kanters JK, Corfield VA, et al. The genetic basis of Brugada syndrome: a mutation update. Hum Mutat. 2009;30:1256-1266.
72 Brugada J, Brugada P, Brugada R. The syndrome of right bundle branch block ST segment elevation in V1 to V3 and sudden death—the Brugada syndrome. Europace. 1999;1:156-166.
73 Vatta M, Dumaine R, Varghese G, Richard TA, Shimizu W, Aihara N, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337-345.
74 Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96:2595-2600.
75 Riera AR, Zhang L, Uchida AH, Schapachnik E, Dubner S, Ferreira C. The management of Brugada syndrome patients. Cardiol J. 2007;14:97-106.
76 Hong K, Brugada J, Oliva A, Berruezo-Sanchez A, Potenza D, Pollevick GD, et al. Value of electrocardiographic parameters and ajmaline test in the diagnosis of Brugada syndrome caused by SCN5A mutations. Circulation. 2004;110:3023-3027.
77 Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110:1731-1737.
78 Hermida JS, Denjoy I, Clerc J, Extramiana F, Jarry G, Milliez P, et al. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol. 2004;43:1853-1860.
79 Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379-385.
80 Borlak J, Thum T. Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J. 2003;17:1592-1608.
81 Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, et al. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007;42:852-861.
82 Farkas AS, Nattel S. Minimizing repolarization-related proarrhythmic risk in drug development and clinical practice. Drugs. 2010;70:573-603.
83 Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, et al. The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Cardiovasc Res. 2000;47:219-233.
84 Isner JM, Sours HE, Paris AL, Ferrans VJ, Roberts WC. Sudden, unexpected death in avid dieters using the liquid-protein-modified-fast diet. Observations in 17 patients and the role of the prolonged QT interval. Circulation. 1979;60:1401-1412.
85 Bellavere F, Ferri M, Guarini L, Bax G, Piccoli A, Cardone C, et al. Prolonged QT period in diabetic autonomic neuropathy: a possible role in sudden cardiac death? Br Heart J. 1988;59:379-383.
86 Kocheril AG, Bokhari SA, Batsford WP, Sinusas AJ. Long QTc and torsades de pointes in human immunodeficiency virus disease. Pacing Clin Electrophysiol. 1997;20:2810-2816.
87 Machado C, Baga JJ, Kawasaki R, Reinoehl J, Steinman RT, Lehmann MH. Torsades de pointes as a complication of subarachnoid hemorrhage: a critical reappraisal. J Electrocardiol. 1997;30:31-37.
88 Bauman JL, DiDomenico RJ. Cocaine-induced channelopathies: emerging evidence on the multiple mechanisms of sudden death. J Cardiovasc Pharmacol Ther. 2002;7:195-202.
89 Gettes LS. Electrolyte abnormalities underlying lethal and ventricular arrhythmias. Circulation. 1992;85:I70-I76.
90 Durakovic Z, Misigoj-Durakovic M, Corovic N. Q-T and JT dispersion in the elderly with urban hypothermia. Int J Cardiol. 2001;80:221-226.
91 Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372-379.
92 Nordin C. The case for hypoglycaemia as a proarrhythmic event: basic and clinical evidence. Diabetologia. 2010;53:1552-1561.
93 Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983-991.
94 Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G, et al. Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol. 1988;12:1222-1228.
95 Maron BJ. Right ventricular cardiomyopathy: another cause of sudden death in the young. N Engl J Med. 1988;318:178-180.
96 Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 1990;89:588-596.
97 Janeira LF. Wide-complex tachycardias. The importance of identifying the mechanism. Postgrad Med. 100, 1996. 259–256, 269
98 Wyse DG, Friedman PL, Brodsky MA, Beckman KJ, Carlson MD, Curtis AB, et al. Life-threatening ventricular arrhythmias due to transient or correctable causes: high risk for death in follow-up. J Am Coll Cardiol. 2001;38:1718-1724.
99 Shah CP, Thakur RK, Xie B, Hoon VK. Clinical approach to wide QRS complex tachycardias. Emerg Med Clin North Am. 1998;16:331-360.
100 Shaw M, Niemann JT, Haskell RJ, Rothstein RJ, Laks MM. Esophageal electrocardiography in acute cardiac care. Efficacy and diagnostic value of a new technique. Am J Med. 1987;82:689-696.
101 Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med. 1978;64:27-33.
102 Brugada P, Brugada J, Mont L, Smeets J, Andries EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation. 1991;83:1649-1659.
103 Antunes E, Brugada J, Steurer G, Andries E, Brugada P. The differential diagnosis of a regular tachycardia with a wide QRS complex on the 12-lead ECG: ventricular tachycardia, supraventricular tachycardia with aberrant intraventricular conduction, and supraventricular tachycardia with anterograde conduction over an accessory pathway. Pacing Clin Electrophysiol. 1994;17:1515-1524.
104 Steurer G, Gursoy S, Frey B, Simonis F, Andries E, Kuck K, Brugada P. The differential diagnosis on the electrocardiogram between ventricular tachycardia and preexcited tachycardia. Clin Cardiol. 1994;17:306-308.
105 Lau EW, Ng GA. The reliable electrocardiographic diagnosis of regular broad complex tachycardia: a holy grail that will forever elude the clinician’s grasp? Pacing Clin Electrophysiol. 2002;25:1756-1761.
106 Lloyd EA, Zipes DP, Heger JJ, Prystowsky EN. Sustained ventricular tachycardia due to bundle branch reentry. Am Heart J. 1982;104:1095-1097.
107 Caceres J, Jazayeri M, McKinnie J, Avitall B, Denker ST, Tchou P, et al. Sustained bundle branch reentry as a mechanism of clinical tachycardia. Circulation. 1989;79:256-270.
108 Tchou P, Mehdirad AA. Bundle branch reentry ventricular tachycardia. Pacing Clin Electrophysiol. 1995;18:1427-1437.
109 Buxton AE, Waxman HL, Marchlinski FE, Simson MB, Cassidy D, Josephson ME. Right ventricular tachycardia: clinical and electrophysiologic characteristics. Circulation. 1983;68:917-927.
110 Griffith MJ, Garratt CJ, Rowland E, Ward DE, Camm AJ. Effects of intravenous adenosine on verapamil-sensitive “idiopathic” ventricular tachycardia. Am J Cardiol. 1994;73:759-764.
111 Ward DE, Nathan AW, Camm AJ. Fascicular tachycardia sensitive to calcium antagonists. Eur Heart J. 1984;5:896-905.
112 Gorgels AP, Vos MA, Letsch IS, Verschuuren EA, Bar FW, Janssen JH, et al. Usefulness of the accelerated idioventricular rhythm as a marker for myocardial necrosis and reperfusion during thrombolytic therapy in acute myocardial infarction. Am J Cardiol. 1988;61:231-235.
113 Artucio H, Pereira M. Cardiac arrhythmias in critically ill patients: epidemiologic study. Crit Care Med. 1990;18:1383-1388.
114 Reinelt P, Karth GD, Geppert A, Heinz G. Incidence and type of cardiac arrhythmias in critically ill patients: a single center experience in a medical-cardiological ICU. Intensive Care Med. 2001;27:1466-1473.
115 Annane D, Sebille V, Duboc D, Le Heuzey JY, Sadoul N, Bouvier E, et al. Incidence and prognosis of sustained arrhythmias in critically ill patients. Am J Respir Crit Care Med. 2008;178:20-25.
116 Bigger JTJr, Dresdale FJ, Heissenbuttel RH, Weld FM, Wit AL. Ventricular arrhythmias in ischemic heart disease: mechanism, prevalence, significance, and management. Prog Cardiovasc Dis. 1977;19:255-300.
117 Newby KH, Thompson T, Stebbins A, Topol EJ, Califf RM, Natale A. Sustained ventricular arrhythmias in patients receiving thrombolytic therapy: incidence and outcomes. The GUSTO Investigators. Circulation. 1998;98:2567-2573.
118 Cheema AN, Sheu K, Parker M, Kadish AH, Goldberger JJ. Nonsustained ventricular tachycardia in the setting of acute myocardial infarction: tachycardia characteristics and their prognostic implications. Circulation. 1998;98:2030-2036.
119 Hohnloser SH, Klingenheben T, Zabel M, Schopperl M, Mauss O. Prevalence, characteristics and prognostic value during long-term follow-up of nonsustained ventricular tachycardia after myocardial infarction in the thrombolytic era. J Am Coll Cardiol. 1999;33:1895-1902.
120 Keelan PC, Bunch TJ, White RD, Packer DL, Holmes DRJr. Early direct coronary angioplasty in survivors of out-of-hospital cardiac arrest. Am J Cardiol. 2003;91:1461-1463. A6
121 Eldar M, Sievner Z, Goldbourt U, Reicher-Reiss H, Kaplinsky E, Behar S. Primary ventricular tachycardia in acute myocardial infarction: clinical characteristics and mortality. The SPRINT Study Group. Ann Intern Med. 1992;117:31-36.
122 Berger PB, Ruocco NA, Ryan TJ, Frederick MM, Podrid PJ. Incidence and significance of ventricular tachycardia and fibrillation in the absence of hypotension or heart failure in acute myocardial infarction treated with recombinant tissue-type plasminogen activator: results from the Thrombolysis in Myocardial Infarction (TIMI) Phase II trial. J Am Coll Cardiol. 1993;22:1773-1779.
123 Askari AT, Shishehbor MH, Kaminski MA, Riley MJ, Hsu A, Lincoff AM. The association between early ventricular arrhythmias, renin-angiotensin-aldosterone system antagonism, and mortality in patients with ST-segment-elevation myocardial infarction: Insights from Global Use of Strategies to Open coronary arteries (GUSTO) V. Am Heart J. 2009;158:238-243.
124 Michaud GF, Sticherling C, Tada H, Oral H, Pelosi FJr, Knight BP, et al. Relationship between serum potassium concentration and risk of recurrent ventricular tachycardia or ventricular fibrillation. J Cardiovasc Electrophysiol. 2001;12:1109-1112.
125 Volpi A, Cavalli A, Turato R, Barlera S, Santoro E, Negri E. Incidence and short-term prognosis of late sustained ventricular tachycardia after myocardial infarction: results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI-3) Data Base. Am Heart J. 2001;142:87-92.
126 Heidbuchel H, Tack J, Vanneste L, Ballet A, Ector H, Van de WF. Significance of arrhythmias during the first 24 hours of acute myocardial infarction treated with alteplase and effect of early administration of a beta-blocker or a bradycardiac agent on their incidence. Circulation. 1994;89:1051-1059.
127 Sachdev M, Fetics BJ, Lai S, Dalal D, Insel J, Berger RD. Failure in short-term prediction of ventricular tachycardia and ventricular fibrillation from continuous electrocardiogram in intensive care unit patients. J Electrocardiol. 2010;43:400-407.
128 Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e385-e484.
129 Callans DJ, Marchlinski FE. Dissociation of termination and prevention of inducibility of sustained ventricular tachycardia with infusion of procainamide: evidence for distinct mechanisms. J Am Coll Cardiol. 1992;19:111-117.
130 Gorgels AP, van den DA, Hofs A, Mulleneers R, Smeets JL, Vos MA, et al. Comparison of procainamide and lidocaine in terminating sustained monomorphic ventricular tachycardia. Am J Cardiol. 1996;78:43-46.
131 Sharma AD, Purves P, Yee R, Klein G, Jablonsky G, Kostuk WJ. Hemodynamic effects of intravenous procainamide during ventricular tachycardia. Am Heart J. 1990;119:1034-1041.
132 Mooss AN, Mohiuddin SM, Hee TT, Esterbrooks DJ, Hilleman DE, Rovang KS, et al. Efficacy and tolerance of high-dose intravenous amiodarone for recurrent, refractory ventricular tachycardia. Am J Cardiol. 1990;65:609-614.
133 Hohnloser SH, Meinertz T, Dammbacher T, Steiert K, Jahnchen E, Zehender M, et al. Electrocardiographic and antiarrhythmic effects of intravenous amiodarone: results of a prospective, placebo-controlled study. Am Heart J. 1991;121:89-95.
134 Levine JH, Massumi A, Scheinman MM, Winkle RA, Platia EV, Chilson DA, et al. Intravenous amiodarone for recurrent sustained hypotensive ventricular tachyarrhythmias. Intravenous Amiodarone Multicenter Trial Group. J Am Coll Cardiol. 1996;27:67-75.
135 Hamaad A, Lip GY. Intravenous water-soluble amiodarone improved 24-hour survival in incessant ventricular tachycardia. ACP J Club. 2003;138:62.
136 Mitchell LB, Wyse DG, Gillis AM, Duff HJ. Electropharmacology of amiodarone therapy initiation. Time courses of onset of electrophysiologic and antiarrhythmic effects. Circulation. 1989;80:34-42.
137 Kowey PR, Marinchak RA, Rials SJ, Filart RA. Intravenous amiodarone. J Am Coll Cardiol. 1997;29:1190-1198.
138 Kulakowski P, Karczmarewicz S, Karpinski G, Soszynska M, Ceremuzynski L. Effects of intravenous amiodarone on ventricular refractoriness, intraventricular conduction, and ventricular tachycardia induction. Europace. 2000;2:207-215.
139 Lie KI, Wellens HJ, van Capelle FJ, Durrer D. Lidocaine in the prevention of primary ventricular fibrillation. A double-blind, randomized study of 212 consecutive patients. N Engl J Med. 1974;291:1324-1326.
140 Nasir NJr, Taylor A, Doyle TK, Pacifico A. Evaluation of intravenous lidocaine for the termination of sustained monomorphic ventricular tachycardia in patients with coronary artery disease with or without healed myocardial infarction. Am J Cardiol. 1994;74:1183-1186.
141 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care, Part 3: Overview of CPR. Circulation. 2005;112(suppl IV):IV12-IV18.
142 Larsen MP, Eisenberg MS, Cummins RO, Hallstrom AP. Predicting survival from out-of-hospital cardiac arrest: a graphic model. Ann Emerg Med. 1993;22:1652-1658.
143 Ekstrom L, Herlitz J, Wennerblom B, Axelsson A, Bang A, Holmberg S. Survival after cardiac arrest outside hospital over a 12-year period in Gothenburg. Resuscitation. 1994;27:181-187.
144 Cobb LA, Fahrenbruch CE, Walsh TR, Copass MK, Olsufka M, Breskin M, et al. Influence of cardiopulmonary resuscitation prior to defibrillation in patients with out-of-hospital ventricular fibrillation. JAMA. 1999;281:1182-1188.
145 Valenzuela TD, Roe DJ, Nichol G, Clark LL, Spaite DW, Hardman RG. Outcomes of rapid defibrillation by security officers after cardiac arrest in casinos. N Engl J Med. 2000;343:1206-1209.
146 Niemann JT, Cairns CB, Sharma J, Lewis RJ. Treatment of prolonged ventricular fibrillation. Immediate countershock versus high-dose epinephrine and CPR preceding countershock. Circulation. 1992;85:281-287.
147 Kolarova J, Ayoub IM, Yi Z, Gazmuri RJ. Optimal timing for electrical defibrillation after prolonged untreated ventricular fibrillation. Crit Care Med. 2003;31:2022-2028.
148 Wik L, Hansen TB, Fylling F, Steen T, Vaagenes P, Auestad BH, Steen PA. Delaying defibrillation to give basic cardiopulmonary resuscitation to patients with out-of-hospital ventricular fibrillation: a randomized trial. JAMA. 2003;289:1389-1395.
149 Kudenchuk PJ, Cobb LA, Copass MK, Cummins RO, Doherty AM, Fahrenbruch CE, et al. Amiodarone for resuscitation after out-of-hospital cardiac arrest due to ventricular fibrillation. N Engl J Med. 1999;341:871-878.
150 Dorian P, Cass D, Schwartz B, Cooper R, Gelaznikas R, Barr A. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002;346:884-890.
151 Nademanee K, Taylor R, Bailey WE, Rieders DE, Kosar EM. Treating electrical storm : sympathetic blockade versus advanced cardiac life support-guided therapy. Circulation. 2000;102:742-747.
152 Viskin S, Belhassen B. Polymorphic ventricular tachyarrhythmias in the absence of organic heart disease: classification, differential diagnosis, and implications for therapy. Prog Cardiovasc Dis. 1998;41:17-34.
153 Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, et al. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89:66-70.
154 Kowey PR, Marinchak RA, Rials SJ, Bharucha DB. Intravenous antiarrhythmic therapy in the acute control of in-hospital destabilizing ventricular tachycardia and fibrillation. Am J Cardiol. 1999;84:46R-51R.
155 Dorian P, Cass D. An overview of the management of electrical storm. Can J Cardiol. 1997;13(Suppl A):13A-17A.
