Chapter 15 Vasculitis
| VESSEL SIZE | VASCULITIC SYNDROME |
|---|---|
| Large vessel vasculitis | Giant cell (temporal) arteritis Takayasu arteritis |
| Medium vessel vasculitis | Polyarteritis nodosa (classic PAN) Kawasaki disease |
| Small vessel vasculitis | Wegener’s granulomatosis Churg-Strauss syndrome Microscopic polyangiitis (polyarteritis) Henoch-Schönlein purpura Essential cryoglobulinemic vasculitis Cutaneous leukocytoclastic vasculitis |
Adapted from Jennette JC, Falk RJ, Andrassy K, et al: Nomenclature of systemic vasculitides: proposal of an International Consensus Conference, Arthritis Rheum 37:187–192, 1994. Adopted by the Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis, 1994.
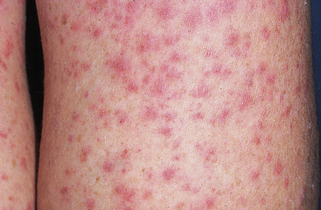
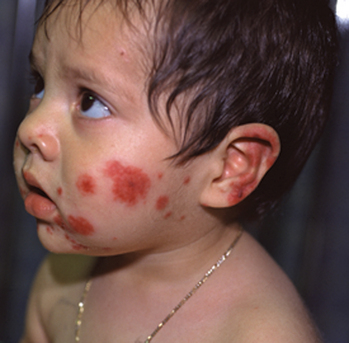
| FINDING | SENSITIVITY |
|---|---|
| Asthma | 100% |
| Blood eosinophilia >10% | 95% |
| Paranasal sinus abnormalities | 86% |
| Mononeuropathy or polyneuropathy | 75% |
| Pulmonary infiltrates | 40% |
| Extravascular (perivascular) eosinophils | 14% |
| History of seasonal allergies | − |
de Groot K, Gross WL: Wegener’s granulomatosis, Lupus 7:285–291, 1998.
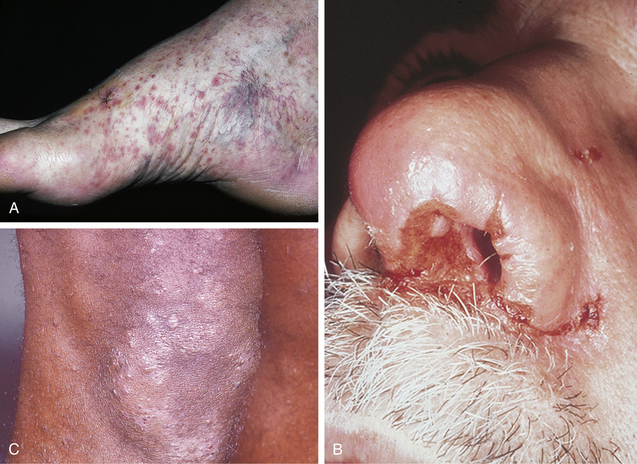
The use of cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) for differentiating between these two disorders is helpful (c-ANCA is usually present in high titers in Wegener’s but not in Churg-Strauss), but is not included in the current diagnostic criteria for either syndrome. The presence or absence of other features should confirm the diagnosis in most cases (Table 15-3).
Table 15-3 Wegener’s Granulomatosis versus Churg-Strauss Syndrome
| WEGENER’S | CHURG-STRAUSS | |
|---|---|---|
| Asthma | − | + |
| Blood eosinophilia | − | + |
| Perivascular eosinophils on biopsy | − | + |
| Hemoptysis | + | − |
| Microhematuria | + | − |
Key Points: Antineutrophil Cytoplasmic Antibodies
Stein SL, Miller LC, Konnikov N: Wegener’s granulomatosis, Pediatr Dermatol 15:352–356, 1998.
Ishiguro N, Kawashima M: Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work, J Dermatol 37:85–93, 2010.
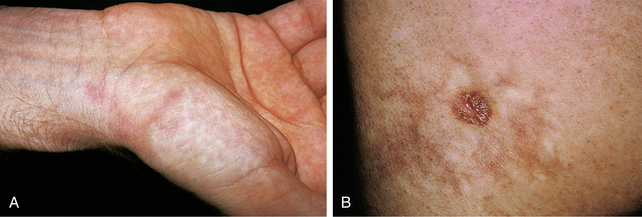
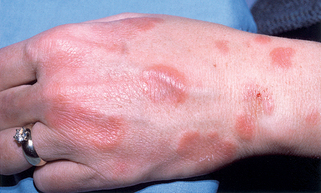
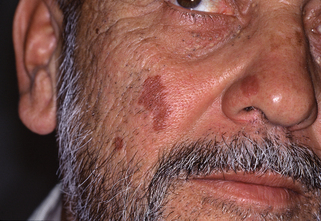
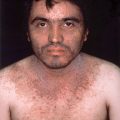
Figure 15-6. Granuloma faciale demonstrating purplish indurated plaques of the nose and cheeks.
(Courtesy of the Joanna Burch Collection.)
Thiyanaratnam J, Doherty SD, Krishnan B, Hsu S: Granuloma faciale: case report and review, Dermatol Online J 15(12):3, 2009.