[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 36 Vascular Lesions of the Gastrointestinal Tract
Through the widespread use of endoscopy and angiography, as well as advances in imaging techniques such as computed tomographic angiography (CTA) and magnetic resonance angiography (MRA), vascular lesions of the gastrointestinal (GI) tract are being increasingly characterized. Vascular lesions are a common cause of GI hemorrhage and may be solitary or multiple, benign or malignant, isolated or part of a syndrome or systemic disorder (Table 36-1). It is important at the outset to understand the nomenclature for the commonest lesions. Vas and its derivative vascular are Latin words meaning vessel; the Greek equivalent is angeion. Ectasia is a word of Greek derivation that refers to the process whereby a blood vessel becomes dilated or lengthened; the resulting lesion also can be referred to as an ectasia. Telangiectasia is the lesion resulting from dilation of the terminal aspect (tele) of a vessel. Angiodysplasia is used as a general term to describe the lesion or process whereby a badly formed (dys, “bad”; plasis, “molded”) vessel develops. An arteriovenous malformation is a congenital lesion, whereas an angioma is a neoplasm. This chapter discusses the more important vascular lesions that cause GI bleeding and that are representative of the spectrum of vascular lesions of the GI tract.
Table 36-1 Vascular Lesions of the Gastrointestinal Tract
| Primary Vascular Lesions |
| Diseases and Syndromes with Vascular Lesions |
| Systemic Disorders Associated with Vascular Lesions |
CREST, calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia; GAVE, gastric antral vascular ectasia.
VASCULAR LESIONS
ANGIOECTASIA
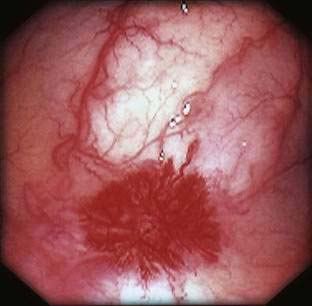
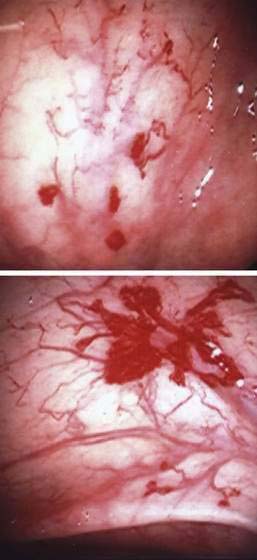
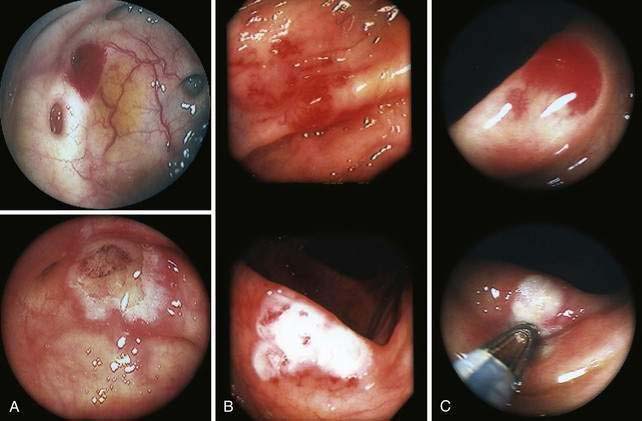
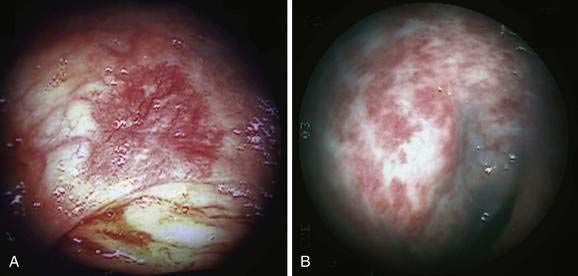
Angioectasia (AE) of the colon is a distinct clinical and pathologic entity.1–3 It is the most common vascular abnormality of the GI tract and probably the most frequent cause of recurrent or chronic lower intestinal bleeding in persons older than 60 years of age.4 AEs are probably acquired with aging, and there does not appear to be a gender predominance. In contrast to congenital or neoplastic vascular lesions of the GI tract, AEs are not associated with lesions of the skin or other viscera. However, when patients with vascular lesions of the colon are aggressively studied with angiography or enteroscopy, concomitant lesions may be seen in the small intestine in approximately 10% of patients.3,5,6 AEs almost always are confined to the cecum or ascending colon, usually are multiple rather than single, and usually are smaller than 10 mm in diameter. They are seldom identified by the surgeon at operation or by the pathologist using standard histologic techniques, but usually they can be diagnosed by angiography; colonoscopy (Figs. 36-1 and 36-2); or, as recently shown, helical CTA.7
The roles of computed tomography (CT) and magnetic resonance imaging (MRI) for vascular lesions of all types are evolving but are certain to increase as these sophisticated modes of diagnosis become more widely available; it is also clear that conventional angiography at present is more important for therapy than for diagnosis. To determine the precise nature of a vascular lesion, histologic examination, with or without injection studies of the vasculature, is necessary. In one report in which histologic confirmation of vascular lesions was not performed, AEs reportedly occurred distal to the hepatic flexure in 46% of patients8; review of tissue sections from supposed AEs in the small bowel or left colon revealed histologic changes different from those of AEs in the right colon (personal review by S. J. Boley and L. J. Brandt).
Bleeding from cecal AEs was first shown in 1961 by intraoperative angiography and has since become well recognized, especially after the introduction of selective angiography and colonoscopy for identifying the source of intestinal bleeding (see Chapter 19).8 In older literature, AEs and diverticulosis were considered the two most common causes of severe lower GI hemorrhage in older adults; however, more recent publications have cited AEs and diverticulosis to be responsible for 3% to 37% (mean: 10%) and 15% to 55% (mean: 30%) of major lower intestinal bleeding episodes, respectively (see Chapters 19 and 117).9 The problem of attributing bleeding to one or the other cause, when bleeding from the lesion is not demonstrated by endoscopy or by extravasation of contrast material on radiologic imaging studies, is compounded by the frequency and coexistence of these disorders without bleeding in people older than 60 years of age. The prevalence of diverticulosis is estimated to be as high as 50% in the population older than age 60; mucosal and submucosal AEs of the right colon can be found by injection studies of colons removed at surgery in more than 25% and 50%, respectively, of patients in this age range without evidence of bleeding.1,10 In large series of colonoscopic examinations, AEs have been seen in 0.2% to 2.9% of nonbleeding persons and 2.6% to 6.2% of patients evaluated specifically for occult blood in the stool, anemia, or hemorrhage.3,11–12 In a patient being studied for GI bleeding, in whom the site of active bleeding is unproven, the only basis for determining that an identified ectasia or diverticulosis is responsible for bleeding is the indirect evidence provided by the patient’s course after ablation or resection of the suspected lesion. It is unusual for AEs found incidentally to bleed, and an AE, even in a patient with a history of bleeding, cannot be assumed to be the cause.13
Bleeding from AEs typically is recurrent and low grade, although approximately 15% of patients present with massive hemorrhage. The nature and degree of bleeding frequently vary in the same patient with different episodes: Patients may have bright red blood, maroon stools, or melena on separate occasions. In 20% to 25% of episodes, only tarry stools are passed, and in 10% to 15% of patients, bleeding is evidenced solely by iron deficiency anemia, with stools that are intermittently positive for occult blood.4 This spectrum reflects the varied rate of bleeding from the ectatic capillaries, venules, and arteriovenous communications, depending on the developmental stage of the lesions (see later). In more than 90% of instances, bleeding stops spontaneously.
In 1958 E. C. Heyde described what is still a controversial association of AEs, GI bleeding, and aortic stenosis; aortic valve replacement had even been recommended for “Heyde’s syndrome” when bleeding could not be managed adequately. Numerous reports of Heyde’s syndrome appeared in the literature; subsequent analysis14 and many studies,15 however, failed to support the association. This association recently has been suggested again16 in a retrospective study in which the frequency of aortic stenosis was 31.7% in patients with “AVMs” compared with 14% in the general population; severe aortic stenosis was also more likely in the group with intestinal vascular lesions. The additional postulate has been offered that deficiencies of the largest forms of von Willebrand factor multimers (von Willebrand syndrome, type 2A) result in hemostatic abnormalities that may predispose preexisting AEs to bleed.17 Preoperative deficiency of these multimers reverses after aortic valve replacement,18 but the general recommendation to replace the aortic valve to control bleeding from AEs seems premature now that a variety of transendoscopic and angiographic means are available to ablate AEs.
Pathology
Histologic identification of AEs is difficult unless special techniques are used.1 Although usually less than one third of lesions are found by routine pathologic examination, almost all can be identified by injecting the colonic vasculature with silicone rubber, dehydrating the cells with increasing concentrations of ethyl alcohol, clearing the specimen by immersing it for 24 hours in a bath of methyl salicylate, and then viewing the specimen by dissecting stereomicroscopy (Fig. 36-3).1 In a study using these methods, surgically resected colons were analyzed and found to have one or more mucosal AEs measuring 1 mm to 1 cm in diameter. AEs were usually multiple, and in this study, all were located within the cecum and ascending colon; the most distal one was 23 cm beyond the ileocecal valve.1
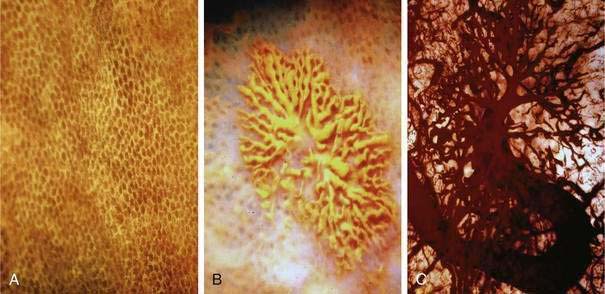
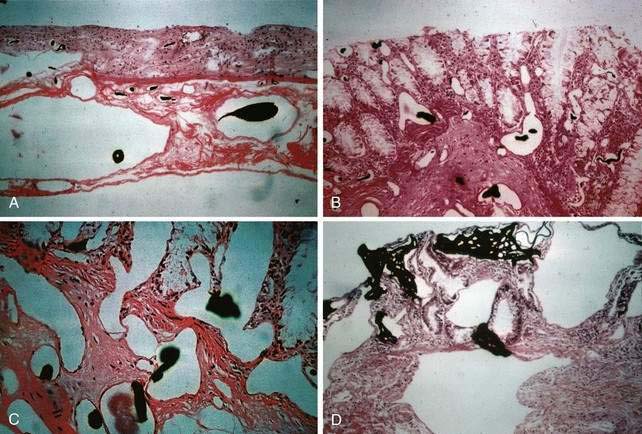
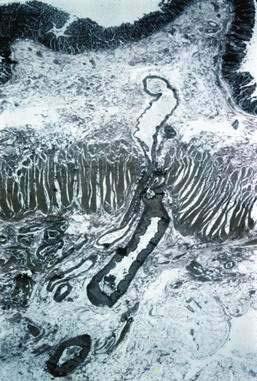
Microscopically, mucosal AEs consist of ectatic, distorted, thin-walled venules, capillaries, and arterioles, vessels that are lined by endothelium, and, infrequently, a small amount of smooth muscle. The earliest abnormality is the presence of dilated, tortuous, submucosal veins (Fig. 36-4A), often in areas where mucosal vessels appear normal. More extensive lesions show increasing numbers of dilated and deformed vessels traversing the muscularis mucosa and involving the mucosa (see Fig. 36-4B and C) until, in the most severe lesions, the mucosa is replaced by a maze of distorted, dilated vascular channels (see Fig. 36-4D). Enlarged arteries and thick-walled veins occasionally are seen in advanced lesions, in which the dilated arteriolar-capillary-venular unit has become a small arteriovenous fistula because of loss of prearteriolar sphincter function. Large thick-walled arteries are more typical of congenital arteriovenous malformations.
Pathogenesis
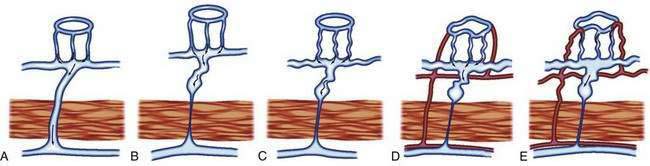
The previously described studies using injection and clearing techniques indicated that AEs are acquired lesions associated with aging and that they represent a unique clinical and pathologic entity.1 That AEs are common lesions associated with aging is supported by their frequent identification at colonoscopy in older adults and in injected colons resected from older patients with no history of bleeding.1,11 Boley postulated that the likely cause of AEs is partial, intermittent, low-grade obstruction of submucosal veins at the site where these vessels pierce the muscular layers of the colon1 (Figs. 36-5 and 36-6). He further suggested that repeated episodes of transiently elevated pressure during muscular contraction and distention of the cecum over many years conceivably result in dilation and tortuosity of the submucosal vein and, later, of the venules and capillaries of the mucosal units that drain into it. Finally, he postulated that the capillary rings dilate, the precapillary sphincters lose their competency, and a small arteriovenous fistula is produced. The latter is responsible for the “early-filling vein,” which was the original angiographic hallmark of this lesion (Fig. 36-7). Prolonged increased flow through the arteriovenous fistula can then produce alterations in the arteries supplying the area and in the extramural veins that drain it. This developmental concept of the cause of AEs was based on the finding of (1) a prominent submucosal vein, either in the absence of any mucosal lesion, or underlying only a minute mucosal AE supplied by a normal artery; (2) dilation of the veins, starting where they traverse the muscularis propria (see Fig. 36-5); and (3) previous studies showing that venous flow in the bowel may be diminished by increases in colon motility, intramural tension, and intraluminal pressure.19 Following this logic, the prevalence of AEs in the right colon can be attributed to the greater tension in the cecal wall compared with that in other parts of the colon, according to LaPlace’s principle: T ∝ pDP (where T is tension, D is diameter, and P is intraluminal pressure).
An alternative concept for the development of AEs is based on the demonstration that AEs have been shown to express vascular endothelial growth factor (VEGF) and its receptors along the endothelial lining in surgical specimens from patients who have undergone colectomy for recurrent bleeding20; this indicates a proliferative phase of angiogenesis. VEGF and VEGF receptor 1 have been shown to be up-regulated by hypoxia21 and therefore a role has been suggested for hypoxia in the pathogenesis of AEs. Further research still is needed to clarify the pathophysiology of AEs.
Diagnosis and Management
Management of bleeding AEs consists of three phases: (1) diagnosis; (2) conversion of an emergency situation to an elective one by control of acute bleeding; and (3) definitive treatment of the AE by colonoscopic ablation or surgical removal. The diagnostic approach to colonic AEs is essentially the same as that for lower intestinal bleeding in general and includes radionuclide bleeding scans; colonoscopy; angiography; and, to exclude the small intestine as a site of bleeding, push enteroscopy and wireless capsule endoscopy (WCE). WCE is a relatively new diagnostic technique that enables visualization of the entire small intestine, and rarely the very proximal colon where colonic AEs are found. WCE is particularly useful for evaluating patients with obscure and occult GI bleeding.22 WCE has been shown to be superior to push enteroscopy in the evaluation of patients with small intestinal bleeding, yielding a diagnosis in 55% to 75% of patients with obscure bleeding that required transfusions, most common of which was angiodysplasia.23,24 Radionuclide scans are used to determine whether a patient is actively bleeding and, if so, to localize the site (see Chapter 19). Although angiography previously had been the principal means of identifying AE as the source of bleeding, colonoscopy currently is the preferred method. Helical CTA is a relatively new, sensitive, specific, and well-tolerated technique to diagnose colonic AEs, although prospective studies comparing CTA with other imaging techniques are necessary.7
The endoscopist’s ability to diagnose the specific nature of a vascular lesion is limited by the similar appearance of different types of lesions. AEs, spider angiomas, hereditary hemorrhagic telangiectasia, angiomas, the focal hypervascularity of radiation colitis, ulcerative colitis, Crohn’s disease, ischemic colitis, certain infections (e.g., syphilis, Pneumocystis), hyperplastic and adenomatous polyps, and malignancies, including lymphoma and leukemic infiltrations, can all, on occasion, resemble each other (Table 36-2). Because traumatic and endoscopic suction artifacts may resemble vascular lesions, all lesions must be evaluated on insertion of the colonoscope, rather than during withdrawal. Pinch biopsy samples of vascular lesions obtained during endoscopy usually are nonspecific; therefore, the risk of performing biopsies of these abnormalities is not justified.
Table 36-2 Lesions That May Be Confused with Angioectasias on Endoscopy
| Vascular Lesions |
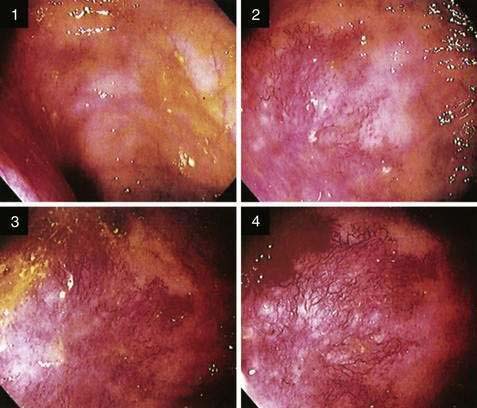
Because the appearance of vascular lesions is influenced by a patient’s blood pressure, blood volume, and state of hydration, such lesions may not be evident in those with severely reduced blood volumes or shock; thus accurate evaluation may not be possible until red cell and volume deficits are corrected. Meperidine also may diminish the prominence of some vascular abnormalities (e.g., AEs and the telangiectasias of hereditary hemorrhagic telangiectasia); use of meperidine, therefore, should be minimized and its effects reversed by naloxone so that vascular lesions can be detected accurately. Such a masking effect does not seem to occur with fentanyl. Naloxone has been shown to enhance the appearance of normal colonic vasculature in approximately 10% of patients and cause existing AEs to appear (2.7%) or increase in size (5.4%) (Fig. 36-8).25 For these reasons, naloxone is an important adjunctive medication for patients undergoing endoscopic evaluation for lower intestinal bleeding. Cold water lavage of the colon, as is sometimes done to cleanse the luminal surface of debris during colonoscopy, also may cause underlying AEs to disappear transiently.26
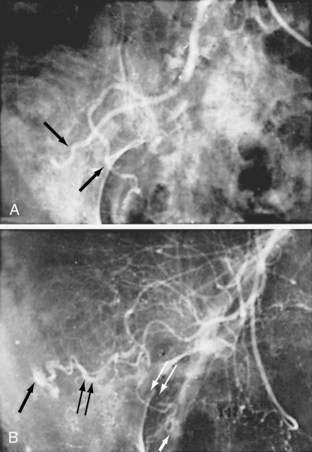
Angiography is used to determine the site and nature of lesions during active bleeding and can identify some vascular lesions even after bleeding has ceased. The three reliable angiographic signs of AEs are a densely opacified, slowly emptying, dilated, tortuous vein; a vascular tuft; and an early-filling vein (see Fig. 36-7).27 A fourth sign, extravasation of contrast material, identifies the site of bleeding when bleeding volume is at least 0.5 mL/minute but is not specific for AE. The slowly emptying vein (see Fig. 36-7A) persists late into the venous phase, after the other mesenteric veins have emptied. Vascular tufts (see Fig. 36-7B) are created by the ectatic venules that join the mucosal AE and the submucosal vein. They are seen best in the arterial phase; are usually located at the termination of a branch of the ileocolic artery; appear as small candelabra-like or oval clusters of vessels; and still are seen in the venous phase communicating with a dilated, tortuous, intramural vein. The early-filling vein is seen in the arterial phase within four or five seconds of injection (see Fig. 36-7B); it is not a valid sign of AE if vasodilators such as papaverine or tolazoline (Priscoline) have been used to enhance the study. When the lesion is bleeding, intraluminal extravasation of contrast material usually appears during the arterial phase of angiography and persists throughout the study. Extravasation identifies the site of active bleeding, but in the absence of other signs of AEs, it suggests another cause for the bleeding.
Management of incidental (nonbleeding) AEs detected by colonoscopy is expectant. The natural history of colonic AE is benign in healthy, asymptomatic people, and the risk of bleeding is small.13,28,29 In such cases, endoscopic therapy is not warranted.30
Bleeding can be controlled endoscopically or angiographically in most patients, thereby avoiding the morbidity and mortality of emergency operation. In decades past, intra-arterial embolization and vasopressin were used to control upper and lower GI bleeding, respectively. Vasopressin, given via an angiographic catheter placed into the feeding splanchnic vessel, arrested hemorrhage successfully from AE in more than 80% of patients in whom extravasation was demonstrated. Now, superselective microcoil embolization has largely replaced intra-arterial vasopressin infusion for the treatment of lower intestinal hemorrhage.31 Such embolization is highly effective and safe but complicated by ischemic events in approximately 5% of cases.32 Vasopressin still is recommended, however, when intestinal lesions are diffuse throughout the bowel or when superselective catheterization is not possible.32
Hormonal therapy, using estrogens in combination with progestins, has been used to treat patients with a variety of vascular lesions of the GI tract, in an attempt to reduce or terminate bleeding. The mechanisms by which such agents work are not known, although procoagulant effects and endothelial injury are popular theories. Although one long-term observational study33 showed that combination hormonal therapy stopped bleeding in patients with occult GI bleeding of obscure origin (likely to have resulted from small bowel angiodysplasia), current studies do not support the use of these agents to prevent rebleeding from GI angiodysplasia.34 It is likely that hormonal therapy affects different vascular lesions differently and that vascular lesions in the small intestine may respond differently to such treatment than the same lesions in the colon; no study of hormonal therapy has been done for known colonic AEs.
A novel therapy for AEs, and perhaps other vascular lesions in the gastrointestinal tract, is the use of antiangiogenic factors. Thalidomide was developed in the 1950s as a sedative, sleeping pill, and antiemetic for pregnant women, but it soon became notorious for causing phocomelia and other malformations in the newborn.34 In 1994, D’Amato and colleagues reported that thalidomide inhibited VEGF and basic fibroblast growth factor-mediated angiogenesis, which led to further characterization and subsequent clinical applications of its antiangiogenic activity.35,36 Recent data suggest the mechanism for its antiangiogenic effect is related to reduced expression of integrin genes and resulting decreased cell-cell surface interactions and response to angiogenic cytokines.37 Several case reports and case series have described the successful use of thalidomide to treat life-threatening or refractory bleeding from intestinal AEs and Crohn’s disease with refractory bleeding.38–42 After treatment with thalidomide for three months, substantial reductions in the number, size, and color intensity of AEs were observed by WCE.39
Of the available antiangiogenic biologic therapies, most information regarding clinical efficacy and toxicity is available for bevacizumab (Avastatin), a humanized monoclonal antibody against VEGF that is effective against colon and renal cancers and that also has a strong antiangiogenic activity.43 Curiously, dose-dependent nasal and GI bleeding is observed in up to 59% of patients during treatment, possibly caused by a loss of vascular integrity as a result of bevacizumab-induced endothelial-cell shedding in highly regenerative mucosal tissues with active angiogenesis. It is unclear why some antiangiogenic substances like bevacizumab cause mucosal bleeding and others like thalidomide do not; this disparity effect may be related to the phase of angiogenesis that is antagonized, or might reflect a particular strong antiangiogenic activity. Although VEGF-based antiangiogenic therapy is a promising therapy, the issue of aggravation of bleeding from vascular lesions needs further study. A more detailed understanding of the angiogenic cascade and how antiangiogenic substances act within it will be needed to resolve this issue.
Neodymium:yttrium-aluminum-garnet (Nd:YAG) laser3,6,44,45; endoscopic sclerosis11; monopolar46 and bipolar47 electrocoagulation; heater probe47; and, recently, hemoclips in combination with cautery,48 endoscopic band ligation,49 and argon plasma coagulation (APC)50 have been used to ablate vascular lesions throughout the GI tract and can be used to control active bleeding (Fig. 36-9). Control of bleeding has been obtained with a variety of endoscopic thermal means in 47% to 88% of cases,3 and no technique has been established as superior to the next.11 Severe delayed bleeding occurs in 5% of patients with colonic AEs after thermal therapy.46 Recurrent bleeding from colonic AEs appears to be reduced after these therapies, but more than one treatment session is usually necessary.47 Rebleeding can be expected to increase with time after the procedure and has been seen in 28% to 52% of patients over a follow-up period ranging from 15 to 36 months.3
Right hemicolectomy is indicated when AE has been identified by either colonoscopy or angiography and when therapy by either or both of these two modalities is unsuccessful, cannot be performed, or is unavailable. The presence or absence of diverticulosis in the left colon does not alter the extent of colonic resection in this circumstance; only the right half of the colon is removed, but it is important that the entire right half of the colon be removed to ensure that no AEs are left behind. If the site of bleeding (and its cause) is not identified, and bleeding recurs or is continuous, recent experience suggests that a subtotal colectomy is appropriate surgical therapy. Older literature emphasized that the morbidity and mortality rates of a right hemicolectomy (which would remove all bleeding AEs and 50% to 70% of bleeding diverticula) followed by a left hemicolectomy (if bleeding recurred postoperatively) were less than the morbidity and mortality rates of a subtotal colectomy. More recent literature suggests that morbidity and mortality rates of a subtotal colectomy are not statistically different from those accompanying a “blind” hemicolectomy, that is, when the bleeding site is not identified.51,52 In one surgical series and review, mortality for subtotal colectomy was 0% to 40% with a rebleeding rate of 0% to 8%, and mortality and rebleeding rates for a directed limited colectomy were 2% to 22% and 0% to 15% respectively51; in contrast, the mortality and rebleeding rates for a blind limited colectomy were 20% to 57% and 35% to 75%, respectively. In another surgical series, frequency of bowel movements after limited colectomy was 2.4 per day, a number not substantially different from the 3.5 bowel movements per day documented after subtotal colectomy.52
HEREDITARY HEMORRHAGIC TELANGIECTASIA (OSLER-WEBER-RENDU DISEASE)
This autosomal dominant familial disorder is characterized by telangiectasia of the skin and mucous membranes, as well as recurrent GI bleeding.53–55 The pathogenesis may relate to mutations of the endoglin (ENG) and activin receptor–like kinase 1 (ALK-1) genes, which have an important role in determining the properties of endothelial cells during angiogenesis (see later).56 Lesions typically are noticed in the first few years of life, and recurrent epistaxis in childhood is characteristic of the disease. By age 10, about half of patients have had some GI bleeding. Severe hemorrhage is unusual before the fourth decade and has a peak incidence in the sixth decade. In most patients, bleeding presents as melena; bright red blood per rectum and hematemesis are less frequent. Hematochezia in a patient with hereditary hemorrhagic telangiectasia (HHT) suggests bleeding from a source other than telangiectasia. Bleeding is chronic and may be severe; patients may receive more than 60 transfusions in a lifetime. A family history of the disease has been reported in 80% of patients with HHT but is less common in those who bleed later in life. Telangiectasias usually are present on the lips, oral and nasopharyngeal membranes, tongue, and periungual areas; lack of involvement of these sites casts suspicion on the diagnosis (Fig. 36-10).
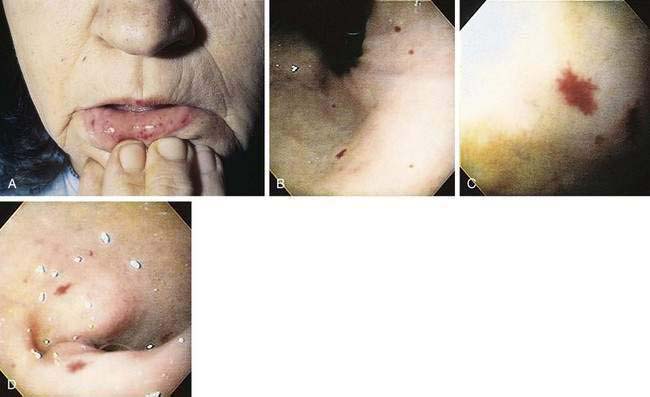
The clinical diagnosis of HHT currently requires the presence of at least three of four relevant clinical criteria. These so-called Curaçao criteria include epistaxis (spontaneous and recurrent nosebleeds); telangiectases (multiple at characteristic sites, e.g., lips, oral cavity, fingers or nose); visceral lesions (e.g., pulmonary, hepatic, cerebral, spinal, or GI vascular malformations); and family history (a first-degree relative with HHT).57 The clinical diagnosis of HHT can be confirmed by molecular genetic analysis. In most cases, HHT is caused by mutations in one of the two known HHT genes. Mutations of the ENG lead to type 1 HHT.58 The ENG gene is located on chromosome 9q34.1 and encodes for endoglin, a type III transforming growth factor-β (TGF-β) receptor. Type 2 HHT is attributed to mutations of the activin A receptor type II-like 1 gene (also termed activin receptor-like kinase-1 (ALK1),59 which codes for the ACVRL1 protein, a type I TGF-β receptor. Both receptors are members of the TGF-β receptor family, are expressed predominantly on vascular endothelium, and play essential roles in maintaining vascular integrity. Evidence for the existence of two other as yet unidentified HHT genes has been reported.60,61 Despite genotypic heterogeneity in HHT, the clinical expression of the different HHT genotypes appears to be the same. HHT and nonhereditary intestinal AE (see earlier) are characterized by increased production of VEGF. High serum levels of VEGF, which also correlate with severity of bleeding, are found in patients with HHT.62,63 Vascular involvement of the liver is common in HHT and frequently is asymptomatic; hepatic manifestations during the course of the disease are seen in 8% to 31% of patients. Typical clinical presentations of liver involvement are high-output heart failure resulting from arteriovenous shunting, portal hypertension, and biliary tract disease.64,65 Serious complications, including liver failure necessitating liver transplantation, have been reported. Telangiectasias occur in the colon but are more common in the stomach and small bowel, where they also are more apt to cause major bleeding.
Telangiectasias are seen easily on endoscopy, although in the presence of severe anemia, blood loss, or hypotension, they transiently may become less obvious or even invisible; after correction of blood volume and blood pressure, they become prominent again. Evaluation by conventional angiography or newer techniques such as helical CTA66 and MRA may be unrevealing or demonstrate arteriovenous communications, conglomerate masses of abnormal vessels, phlebectasia, and aneurysms.67 Angiography may be misleading when it demonstrates multiple vascular abnormalities because some of these lesions may be in the mesentery rather than in the bowel and are not potential sites of GI blood loss.
Grossly, the telangiectasias are the size of millet seeds and typically appear as cherry red, smooth hillocks. Pathologically the major changes involve the capillaries and venules, but arterioles also may be affected. Lesions consist of irregular, ectatic, tortuous blood spaces lined by a delicate single layer of endothelial cells and supported by a fine layer of fibrous connective tissue. No elastic lamina or muscular tissue is present in these vessels, so they cannot contract; this property may explain why the lesions tend to bleed. Arterioles show intimal proliferation and commonly have thrombi in them, suggesting vascular stasis. In contrast to the thinned venules of AEs, venules are abnormally thick in HHT; have prominent, well-developed longitudinal muscles; and apparently play a major role in regulating blood flow in telangiectasias.66
Many forms of treatment have been recommended for telangiectasias, including estrogens,68 aminocaproic acid,69 endoscopic thermal ablation,6,45 and resection of involved bowel. Endoscopic ablation, including the use of the APC and thermal contact devices, is most promising when lesions are within reach of the endoscope and not too diffuse. Endoscopic therapy may be performed during active bleeding or between bleeding episodes and has reduced the need for emergency bowel resection. Long-term follow-up studies are necessary to evaluate the ultimate efficacy of the various forms of therapy.
Bevacizumab, the humanized monoclonal antibody against VEGF discussed earlier, was used to treat a 47-year-old woman with HHT and severe liver involvement. Significant clinical improvement was observed three months after initiating treatment with reversal of cholestasis, resolution of cardiac failure and ascites, and improvement in nutritional status. A marked reduction in liver vascularity and liver volume also were seen over a six-month interval.70
PROGRESSIVE SYSTEMIC SCLEROSIS (see also Chapter 35)
Vascular lesions are a prominent feature of progressive systemic sclerosis, especially in the calcinosis, Raynaud’s phenomenon, esophageal dysmotility, scleroderma, and telangiectasia (CREST) variant.71 Sites most frequently involved by these telangiectasias are the hands, lips, tongue, and face, but gastric, intestinal, and colorectal lesions have been reported. These tiny lesions may be the source of occult or clinically significant bleeding and are best treated, if possible, by endoscopic thermal ablation.72
GASTRIC ANTRAL VASCULAR ECTASIA (WATERMELON STOMACH) AND PORTAL HYPERTENSIVE GASTROPATHY
Gastric antral vascular ectasia (GAVE), or watermelon stomach, describes a vascular lesion of the gastric antrum that consists of tortuous, dilated vessels radiating outward from the pylorus-like spokes of a wheel and resembling the dark stripes on the surface of a watermelon.73 This lesion may cause acute hemorrhage and chronic occult bleeding. Its cause is unknown, although it has been proposed that gastric peristalsis causes prolapse of the loose antral mucosa with consequent elongation and ectasia of the mucosal vessels (Fig. 36-11).73 GAVE also has been thought to result from delayed gastric emptying, as well as from humoral factors such as hypergastrinemia, prostaglandin E2, 5-hydroxytryptamine (serotonin) produced by neuroendocrine cells, and vasoactive intestinal polypeptide (VIP).
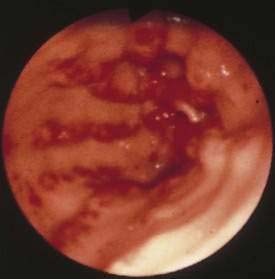
Figure 36-11. Endoscopic appearance of watermelon stomach, also referred to as gastric antral vascular ectasia (GAVE).
GAVE is seen particularly in middle-aged or older women and in association with achlorhydria, atrophic gastritis, cirrhosis, and the CREST syndrome, as well as after bone marrow transplantation.71,74 The association with cirrhosis and portal hypertension in approximately 40% of reported cases of GAVE suggests that this lesion may be caused by portal hypertension or hepatic veno-occlusive disease.75 Microscopic features of GAVE include dilated capillaries with focal thrombosis, dilated and tortuous submucosal venous channels, and fibromuscular hyperplasia of the muscularis mucosa.
Some researchers believe that GAVE and portal hypertensive gastropathy (PHG) are different manifestations of the same pathogenetic process, whereas others view them as separate entities with distinct clinical and histologic features. Recent evidence suggests that these are distinct entities.76 The pathophysiology of GAVE does not appear to involve portal hypertension because it does not respond to therapies directed at reducing portal pressure. Given case reports of GAVE resolution after liver transplantation, however, it is possible that liver insufficiency may play a role in its pathophysiology.77,78
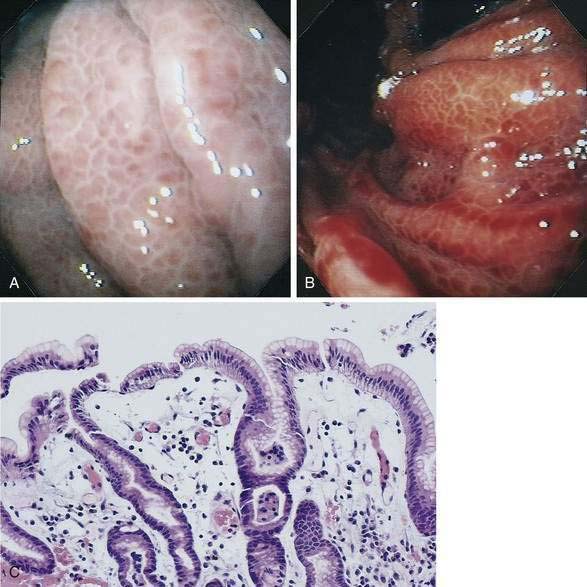
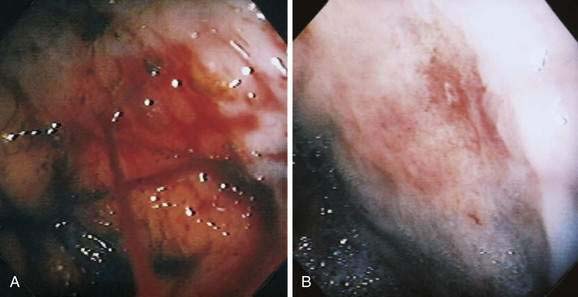
PHG is characterized endoscopically by three patterns: (1) fine red speckling of the mucosa; (2) superficial reddening, especially on the tips of the gastric rugae; and, most commonly, (3) the presence of a mosaic pattern with red spots (snakeskin appearance) in the gastric fundus or body (Fig. 36-12). Histologically, the stomach in PHG contains dilated, tortuous, irregular veins in the mucosa and submucosa, sometimes with intimal thickening, usually in the absence of significant inflammation.76
Limited therapeutic options are available for treating GAVE. Estrogen-progesterone has been tried79 and appears to have some efficacy. Successful use of tranexamic acid, an antifibrinolytic agent,80,81 and thalidomide82 also have been reported. Transjugular intrahepatic portosystemic shunting (TIPS) does not appear to be effective for GAVE.83 This is not surprising given that the pathophysiology of GAVE may not be related to portal hypertension.78,84 Antrectomy has been used for patients in whom pharmacologic and endoscopic therapies have failed.85 Portal hypertension in a patient with GAVE and GI bleeding makes the bleeding more difficult to manage; bleeding is usually greater and more resistant to treatment in the presence of portal hypertension.86 Several case reports have detailed reversal of GAVE after liver transplantation,77 however, the data are insufficient to recommend this therapy unless the patient is otherwise a liver transplant candidate.
Iron therapy and blood transfusions were the mainstays of medical treatment for GAVE, and antrectomy often was required in severe cases before development of transendoscopic thermal ablation techniques. All of the available endoscopic therapies have been used successfully to ablate GAVE, and now antrectomy is rarely required.87 TIPS offers another modality when GAVE is associated with portal hypertension or when bleeding resulting from PHG is not controlled by transendoscopic coagulation therapy (see also Chapters 19 and 90).
The initial management of PHG is with iron supplementation and nonselective beta blockers. Propranolol is the nonselective beta blocker that was investigated in the classic randomized controlled trial evaluating the role of beta blockade in preventing recurrent bleeding in severe PHG.88 In this study, patients who received propranolol had a significantly lower rebleeding rate at 12 months (35% versus 62%) and at 30 months (48% versus 93%) compared with patients taking a placebo. If a patient is refractory to beta blocker therapy, however, shunt therapy (TIPS or shunt surgery) is indicated. TIPS placement or shunt surgery has been effective in almost all cases.83,89–92 The choice between TIPS or surgery should be made based on local expertise.
Somatostain analogs such as octreotide, which are established as effective treatment for acute variceal bleeding, also have been shown to be effective for bleeding from PHG. In two studies evaluating the effect of these vasoactive drugs in acute hemorrhage from PHG,93,94 bleeding was successfully treated in all patients who received somatostatin or octreotide. Vasopressin and its analog, terlipressin, also have been tried, but results have been mixed.93,95
PORTAL COLOPATHY AND ENTEROPATHY
Portal colopathy is the term used to describe vascular manifestations of portal hypertension in the colon. Manifestations include hemorrhoids, varices, and spider-like telangiectasias (Fig. 36-13A and B). Mucosal lesions of portal colopathy typically resemble those seen in PHG and may have a diffuse, colitis-like appearance, including granularity, erythema, telangiectases, and friability. Varices and spider-like telangiectases also may be seen in the small intestine, warranting the term portal enteropathy. Histologic changes of portal colopathy and enteropathy are similar to those of portal gastropathy.96 The lesions of portal colopathy and enteropathy are amenable to the same thermal therapies used for GAVE and PHG.97
DIEULAFOY’S LESION
This vascular lesion is an unusual cause of massive GI hemorrhage, usually from the stomach, but sometimes from the small or large bowel (Fig. 36-14).98 It is twice as common in men as in women and presents at a mean age of 52 years. The vascular abnormality is the presence of arteries of persistently large caliber in the submucosa and, in some instances, the mucosa, typically with a small, overlying mucosal defect. Dieulafoy called the lesion “exulceratio simplex” because he thought it was the initial stage of a gastric ulcer. This lesion also has been called an atherosclerotic aneurysm, an inaccurate term because the caliber of the artery’s walls is uniform throughout and shows no unusual degree of arteriosclerosis. It is believed that focal pressure from these large “caliber-persistent” vessels thins the overlying mucosa, leading to erosion of the exposed vascular wall and resulting hemorrhage. Massive hematemesis or melena typically is not preceded by any GI tract symptoms and usually is followed by intermittent and severe bleeding over several days. The most common site of bleeding is 6 cm distal to the cardioesophageal junction, where the arteries are largest, but many lesions have been reported in extragastric locations, including the esophagus, small bowel, rectum,98 and even outside the GI tract in the bronchus, presenting as hemoptysis. It may be difficult to find a Dieulafoy lesion in a patient with upper GI bleeding because the overlying mucosal defect may be small and hidden between the gastric rugae, and the caliber-persistent vessel may constrict and retract after the bleeding episode.
The mortality rate for older adult patients with this lesion has been high, nearly 80% before diagnostic endoscopy was available and more than 20% in cases reported between 1970 and 1986. The high mortality rate resulted from the inability to localize the bleeding site and the frequent need for emergency gastric surgery. Current angiographic and endoscopic techniques used to localize and treat bleeding lesions have led to an improvement in 30-day mortality rates, now reported to be 13%. Therapeutic approaches to bleeding Dieulafoy’s lesions include injection therapy, heater probe, laser, APC, band ligation, and hemoclip placement.99
HEMANGIOMAS
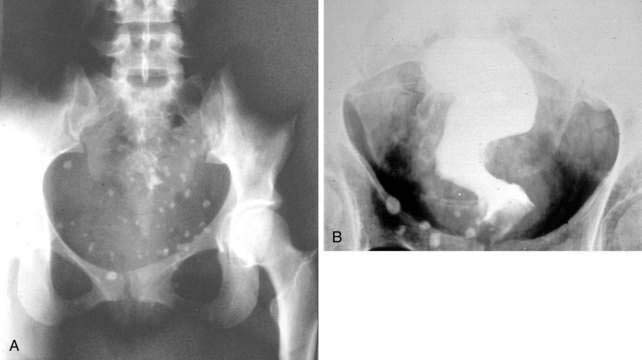
Bleeding from colonic hemangiomas usually is slow, producing occult blood loss with anemia or melena. Hematochezia is less common, except with large cavernous hemangiomas of the rectum, which may cause massive hemorrhage. The diagnosis is best established by endoscopy, including enteroscopy because roentgen studies, including angiography, frequently are normal. The diagnosis of cavernous hemangioma of the rectum often can be suggested on plain films of the abdomen by the presence of phleboliths and displacement or distortion of the rectal air column (Fig. 36-15). On barium enema, the affected rectal lumen typically shows narrowing and rigidity, scalloping of the rectal wall, and widening of the presacral space (see Fig. 36-15). Endoscopically, one sees elevated plum-red nodules or vascular congestion; ulcers and proctitis also may be present. Angiography can demonstrate these lesions but seldom is necessary to establish the diagnosis.
Small hemangiomas that are solitary or few in number and can be approached endoscopically are locally ablated. Most large or multiple lesions require resection of either the hemangioma alone or the involved segment of colon. Local measures to control massive bleeding from cavernous hemangioma of the rectum usually are effective only temporarily. Embolization and surgical ligation of major feeding vessels also have been used, but ultimately excision of the rectum often is required.100
In a particular entity known as diffuse intestinal hemangiomatosis, numerous lesions, usually of the cavernous type, involve the stomach, small bowel, and colon; hemangiomas of the skin or soft tissues of the head and neck frequently are present. The occurrence of bleeding or anemia in childhood typically leads to the diagnosis, which usually is made after various studies including endoscopy, barium series, scintigraphy with 99mTc-labeled red blood cells,101 and contrast-enhanced CT. Angiographic findings can be normal despite the number of lesions. Surgical intervention may be required for continuous, slow bleeding or for intussusception. At operation, all identifiable lesions should be excised either through enterotomies or by limited bowel resections. Intraoperative endoscopy may be helpful in finding small lesions. Repeated operations may be necessary to control blood loss.102 Semaxanib, a small-molecule inhibitor of VEGF receptor 2, recently has been used for treatment of hemangioblastoma in patients with von Hippel-Lindau disease (vHLD). In vHLD, a loss of von Hippel-Lindau protein results in an accumulation of hypoxia-inducible factor and subsequently, excessive production of VEGF.103 Recent studies have reported regression or stabilization, with improvement of macular edema, in patients with hemangioblastoma treated with semaxanib, which suggests inhibition of VEGF103,104 and a role for VEGF inhibitors in the management of hemangiomas.
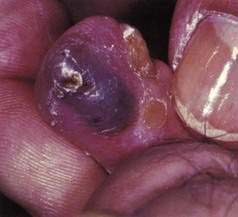
BLUE RUBBER BLEB NEVUS SYNDROME
In 1860, an association among cutaneous vascular nevi, intestinal lesions, and GI bleeding was described, and almost a century later this constellation of findings was named blue rubber bleb syndrome by Bean to distinguish it from other cutaneous vascular lesions (Fig. 36-16). Although the GI tract is most frequently involved, other sites may be affected, including the eyes, nasopharynx, parotid glands, lungs, liver, spleen, heart, brain, skeletal muscles, urinary bladder, and penis. Orthopedic abnormalities may be present, and calcification, thrombosis, and consumptive coagulopathy (with thrombocytopenia) may occur within the lesions.105 A familial history is infrequent, although a few cases of autosomal dominant transmission have been reported106 and one analysis has identified a responsible locus on chromosome 9.
CONGENITAL ARTERIOVENOUS MALFORMATIONS
Arteriovenous malformations (AVMs) are embryonic growth defects and are considered to be developmental anomalies. Although AVMs are found mainly in the extremities, they may occur anywhere in the vascular tree. In the colon they may be small and resemble AEs or they may involve a long segment of bowel. The most extensive lesions typically are in the rectum and sigmoid. Histologically, AVMs are persistent congenital communications between arteries and veins located primarily in the submucosa. Characteristically, there is “arterialization” of the veins (i.e., tortuosity, dilatation, and thick walls with smooth muscle hypertrophy and intimal thickening or sclerosis). In long-standing AVMs, the arteries are dilated with atrophic and sclerotic degeneration. Angiography is the primary means of diagnosis (Fig. 36-17). Early-filling veins in small lesions and extensive dilatation of arteries or veins in large lesions are typical. Patients with significant bleeding from large AVMs should undergo resection of the involved segment; transendoscopic therapy may be beneficial for smaller lesions.
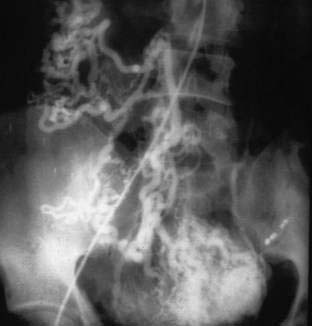
KLIPPEL-TRENAUNAY AND PARKES WEBER SYNDROMES
In its initial description, the Klippel-Trenaunay syndrome consisted of (1) a vascular nevus involving the lower limb; (2) varicose veins limited to the affected side and appearing at birth or in childhood; and (3) hypertrophy of all tissues of the involved limb, especially the bones.107 Subsequently, a variety of vascular lesions associated with the hypertrophic limb were described and some authors now divide the syndrome into two: Klippel-Trenaunay and Parkes Weber; the former is a pure low-flow condition, whereas the latter is characterized by arteriovenous fistulas. Several genetic defects in the regulation of the angiogenic factor VG5Q have been shown in patients with this syndrome.108 The cause of bony elongation is controversial, but one theory invokes in utero venous hypertension and stasis.107 Edema of the involved leg is common, and if the thigh is involved, a variety of lymphatic abnormalities are usually present (e.g., chylous mesenteric cysts, chyloperitoneum, protein-losing enteropathy; see Chapters 28 and 37).
Symptomatic GI involvement is rare. In the largest series, the most common GI symptom was hematochezia, reported by only 6 of 588 patients.107 GI bleeding may be recurrent and mild or severe and usually is caused by a rectal hemangioma, localized rectovaginal varices resulting from obstruction of the internal iliac system, or portal hypertension with varices. Bleeding may be intensified by consumption coagulopathy, which may occur within the smaller sinusoids of the vascular lesion. Physical examination is diagnostic, and various imaging techniques are used to define the anatomy and plan surgical repair.109 Most recently, MRA has been used for diagnosis and to detect arteriovenous shunting.110 Endoscopic thermal ablation therapy is useful in controlling hemorrhage and preventing or minimizing recurrent GI bleeding, especially when the lesions are relatively well localized (L. Brandt, personal experience).
ABDOMINAL AORTIC ANEURYSM
Approximately 95% of abdominal aortic aneurysms (AAAs) are atherosclerotic in origin, but other factors such as genetic predisposition are also important; less common causes include trauma, vasculitis, infection, and congenital abnormalities. Eighty-five percent of affected people are men. Familial clustering of AAAs has been noted in 15% to 20% of cases, and in some families an abnormality has been identified on chromosome 16111; defects in procollagen III in patients with Ehlers-Danlos syndrome type IV and altered gene expression causing abnormalities of the elastin and collagen content of aneurysms have been shown in other families.111
Abdominal plain films may show a soft tissue mass with peripheral calcification in the region of the abdominal aorta. With large aneurysms, erosion of the lumbar vertebrae or displacement of surrounding viscera, including bowel, kidneys, and ureters, may be seen. Because plain film studies are not sufficiently sensitive to establish the presence or size of an aneurysm, ultrasonography, CT, and MRI have become the standard means of evaluation. These procedures are simple, safe, and accurate in the diagnosis and sizing of aneurysms. Ultrasonography is less sensitive than CT in determining the extent of the aneurysmal process, but it is useful for following changes in the size of the aneurysm.112 CT and MRI are used preoperatively to demonstrate aortic and vascular anatomy. Preoperative angiography is not used as frequently as in the past and is most appropriate in patients with evidence of peripheral vascular disease, severe hypertension, symptoms of chronic mesenteric ischemia (see Chapter 114), if thoracic or iliac artery involvement is suspected and, in cases of horseshoe or pelvic kidneys, to demonstrate renal artery anatomy. Angiography is not used to estimate the size of the aneurysm because intraluminal laminated thrombus limits delineation of the entire lumen.
The major complication of AAAs is rupture, which is heralded by the sudden onset or worsening of pain in the abdomen, flank, or back; pain may be present for several weeks and is attributable to “leakage” that precedes overt rupture. Pain may be exacerbated by lying recumbent and relieved by sitting or leaning forward. In one series, only 14% of patients referred for treatment of rupture had been known to have an aneurysm previously.113 Severe abdominal pain also may be seen with aortic dissection as the splanchnic vessels become compromised and acute intestinal ischemia develops. The consensus among vascular surgeons is that the most important predictor of rupture is the size of the aneurysm. The risk of rupture for small aneurysms is negligible and in one series was reported to be 0% at five years114; the five-year risk for aneurysms that are 5.5 to 5.9 cm in size is about 20% to 25%; for a 6-cm aneurysm, the risk is 35% to 40%; and for those larger than 7 cm, the risk is 75%.111 Other risk factors for rupture include hypertension and the presence of chronic obstructive pulmonary disease. AAAs most commonly rupture into the retroperitoneal tissues that surround the aorta. Less commonly, the aneurysm may communicate with the peritoneal cavity, in which case shock develops rapidly. Patients whose aneurysm ruptures into the small intestine, usually the third or fourth portions of the duodenum, typically present with massive GI bleeding; bleeding may be intermittent because a clot alternately forms and is dislodged from the eroded bowel or fistulous opening. Indeed, many of these patients will have a “herald bleed” followed by massive hemorrhage several hours or days later.115 Endoscopy is the most sensitive method for diagnosing this complication. Rarely, abdominal aneurysms rupture into the inferior vena cava; if so, a loud bruit can be heard.
Operative management of an AAA usually consists of replacement of the aneurysm with a prosthetic graft, which may be done via laparotomy through a retroperitoneal or groin incision and use of an endovascular graft. In elective cases, preoperative angiography is useful for demonstrating additional vascular disease (e.g., stenosis or occlusion of the splanchnic arteries) and, by allowing planned vascular reconstruction, may help avoid postoperative bowel ischemia. The mortality of aneurysm repair in good-risk patients is 1% to 4%115; mortality increases sharply to 34% to 85% when surgery is done as an emergency for rupture or impending rupture.113,116,117 Increasingly, endovascular aneurysm repair (EVAR) is being used as an alternative to open repair of an AAA. Recent studies have shown that both can be performed safely in patients treated for elective infrarenal AAAs. EVAR has the perioperative advantages of reduced blood loss, and reduced length of intensive care unit and hospital stay118,119; however, concerns have been raised about endovascular leaks and late rupture.115,120
Aneurysms larger than 5 cm, symptomatic aneurysms, or enlarging aneurysms of any size should be treated electively in good-risk patients. Patients who cannot tolerate an open operation may sometimes still be treated with an endovascular graft.120–122 Patients with asymptomatic and nonexpanding aneurysms that are 4 to 5 cm in diameter are best treated conservatively because rupture of such small AAAs is rare. Aneurysms that are not treated surgically should be followed by ultrasound every three to six months. The growth rate of AAAs is variable and has been less in recent studies than in older ones. Study of the growth rate of small aneurysms (average initial size of 4 cm) in a large population of well-studied patients revealed that over an average of 3.3 years, 58.4% of patients had no change or a decrease in aneurysm size, 25.3% had an expansion between 0.1 and 0.25 cm, 12.6 had an increase of greater than 0.25 cm, and only 3.7% had an enlargement of more than 0.5 cm.111 On average, the growth rate of an AAA is 0.35 cm per year.
MYCOTIC ANEURYSMS
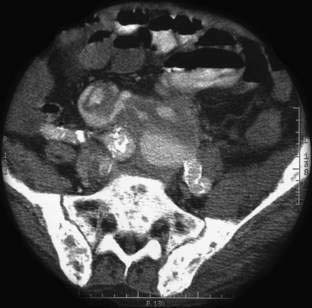
Mycotic aneurysms of the aorta and splanchnic vessels are rare. They were so-named by Sir William Osler because their appearance reminded him of fungi (mykes, fungus). In the past, mycotic aneurysms were most commonly caused by septic emboli from bacterial endocarditis. Today the main risk factor is intravenous drug use. Other important risk factors include contiguous spread from adjacent infectious processes, arterial manipulation, and immunocompromise (e.g., alcoholism, diabetes mellitus, chemotherapy, and treatment with glucocorticoids). Salmonella (especially Salmonella choleraesuis) and Staphylococcus are the most common infecting organisms. The celiac artery (CA) is most often affected, followed by the superior mesenteric artery (SMA) and inferior mesenteric artery (IMA). Early in the course, symptoms of mycotic aneurysms are nonspecific. Later, fever, chills, and abdominal pain are typical. Diagnosis is by imaging the vasculature: mycotic aneurysms typically are lobulated and saccular and affect the upper abdominal aorta (Fig. 36-18). The destructive process can develop quickly, leading to rapid expansion and rupture. Treatment is surgical, usually with resection of the aneurysm and vascular reconstruction.123
PARAPROSTHETIC ENTERIC AND AORTOENTERIC FISTULAS
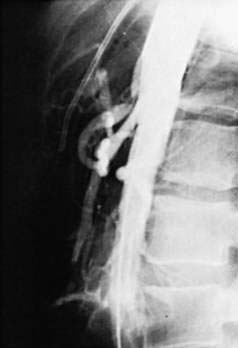
An uncommon but potentially catastrophic complication of aortic aneurysmectomy and other procedures in which vascular prostheses are placed in the retroperitoneum or abdomen is the formation of a fistula between the graft and the adjacent bowel, usually the third or fourth portion of the duodenum (Fig. 36-19).124,125 The frequency of this complication is between 0.6% and 2.35%. Such fistulas develop as early as 21 days postoperatively, but in most cases, they are delayed beyond two years; in one case, an interval of 14 years was documented. This complication is thought to result from local conditions at the time of, or subsequent to, graft placement, including infection, damage to the duodenum or its blood supply during the dissection, and subsequent erosion of the duodenal wall by the graft. Newer surgical techniques, including the use of nonabsorbable sutures and antibiotics, strict hemostasis, and covering of suture lines with retroperitoneal tissue and peritoneum, as well as the increasing use of endovascular grafts, may reduce the frequency of fistula formation.
Patients with aortoenteric fistulas present with upper or lower GI bleeding that if untreated may be massive and rapidly fatal. Upper GI endoscopy is the procedure of choice for aiding diagnosis by excluding other obvious lesions, but CT imaging is helpful.126 The most important clue to the diagnosis is awareness of the possibility in a patient with GI bleeding who has had an aortoiliac artery graft. Prompt diagnosis and expedient surgical repair are essential for survival.
SUPERIOR MESENTERIC ARTERY SYNDROME
The third portion of the duodenum is cradled in an angle of about 45 degrees formed by the root of the SMA and the wall of the aorta. When this angle is narrowed to less than 25 degrees, the SMA impinges on the duodenum, thereby leading to gastric and intestinal obstruction, a condition referred to as Wilkie’s syndrome or the superior mesenteric artery syndrome (Fig. 36-20).127,128 The latter term may be confusing because the condition is not one of vascular insufficiency. Symptoms may be acute or chronic and typically include epigastric pain, vomiting, and early satiety. The syndrome has been associated with immobilization in a body cast; rapid growth in children; and marked, rapid weight loss in adults, particularly young women with an eating disorder (see Chapter 8). Rarely, anatomic anomalies predispose to the condition, including a high ligament of Treitz or low origin of the SMA.
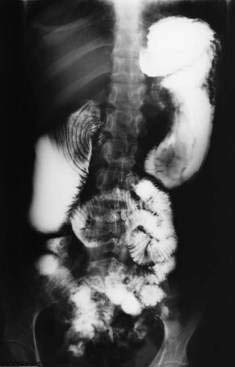
Barium studies may show an abrupt cutoff in the third portion of the duodenum with dilatation proximally, particularly when the patient is supine. Treatment approaches have included small feedings or a liquid diet. Modern imaging techniques such as CTA and MRA can provide noninvasive and detailed anatomic information that can be used in diagnosing the condition and planning surgical approaches.129,130 Symptoms typically improve after restoration of lost weight or removal of a body cast. Surgery is necessary only rarely. Duodenojejunostomy may relieve the symptoms and has been performed for this condition laparoscopically.131
CELIAC AXIS COMPRESSION SYNDROME
Whether celiac axis compression syndrome (CACS) is a cause of GI ischemia has been a subject of controversy ever since the description of postprandial pain and an epigastric bruit in a patient in whom angiography showed narrowing of the CA caused by compression of a fibrotic celiac ganglion.132 After release of the artery, the murmur and postprandial pain disappeared. Since that description, compression of the CA by the median arcuate ligament of the diaphragm and the celiac ganglion has been identified but is not well understood.
A major difficulty in determining the validity of CACS as an entity, also sometimes referred to as Dunbar syndrome, arises from the different criteria used by various investigators to define it.133,134 At the least, clinical features that should be present to diagnose CACS include postprandial epigastric pain, diarrhea, weight loss, and an abdominal bruit that intensifies with expiration.
Compression of the CA is demonstrated by lateral aortography or selective studies of the CA. Endoscopic ultrasound, CTA, and MRA are noninvasive means of demonstrating the anatomy and compression of the CA.135 Compression by the crural fibers of the diaphragm or the celiac ganglion produces a smooth, asymmetrical narrowing of the superior aspect of the celiac axis and displaces it toward the SMA (Fig. 36-21). These findings are shown best during expiration.
Operative approaches to CACS include division of the median arcuate ligament, with or without gangliectomy, or arterial reconstruction or bypass. Laparoscopy has been successful in releasing the compression.135 Results of operations for CACS have varied as much as have the criteria used to diagnose them. In the largest study of the long-term results of patients treated for CACS, Evans found that 83% of patients were asymptomatic 6 months after a decompression procedure, but only 41% remained asymptomatic 3 to 11 years later.136 Furthermore, no correlation existed between the presenting symptoms and the results of surgery and no clinical patterns emerged to identify those patients who might benefit from surgery. Additionally, of 12 patients treated nonoperatively, 9 remained free of pain at the time of Evans’s report.
The controversy concerning CACS continues. A small number of patients who have otherwise unexplained abdominal pain not helped by standard regimens are relieved by some aspect of the operations performed for celiac axis compression.137 If surgery is performed only in patients who fulfill the criteria previously described, unnecessary procedures should be kept to a minimum.
Azuma H. Genetic and molecular pathogenesis of hereditary hemorrhagic telangiectasia. J Med Invest. 2000;47:81-90. (Ref 56.)
Bauditz J, Lochs H, Voderholzer W. Macroscopic appearance of intestinal angiodysplasias under antiangiogenic treatment with thalidomide. Endoscopy. 2006;38:1036-9. (Ref 41.)
Boley SJ, Brandt LJ. Vascular ectasias of the colon. Dig Dis Sci. 1986;31:26S-42S. (Ref 4.)
Boley SJ, Sammartano RJ, Adams A, et al. On the nature and etiology of vascular ectasias of the colon. Degenerative lesions of aging. Gastroenterology. 1977;72:650-60. (Ref 1.)
Brandt LJ, Spinell MK. Ability of naloxone to enhance the colonoscopic appearance of normal colon vasculature and colon vascular ectasias. Gastrointest Endosc. 1999;49:79-83. (Ref 25.)
Carey EJ, Leighton JA, Heigh RI, et al. A single-center experience of 260 consecutive patients undergoing capsule endoscopy for obscure gastrointestinal bleeding. Am J Gastroenterol. 2007;102:89-95. (Ref 23.)
Chahwan S, Comerota AJ, Pigott JP, et al. Elective treatment of abdominal aortic aneurysm with endovascular or open repair: The first decade. J Vasc Surg. 2007;45:258-62. (Ref 118.)
Foutch PG, Rex DK, Lieberman DA. Prevalence and natural history of colonic angiodysplasia among healthy asymptomatic people. Am J Gastroenterol. 1995;90:564-7. (Ref 29.)
Junquera F, Feu F, Papo M, et al. A multicenter, randomized, clinical trial of hormonal therapy in the prevention of rebleeding from gastrointestinal angiodysplasia. Gastroenterology. 2001;121:1073-9. (Ref 34.)
Junquera F, Quirga S, Saperas E, et al. Accuracy of helical computed tomographic angiography for the diagnosis of colonic angiodysplasia. Gastroenterology. 2000;119:293-9. (Ref 7.)
Kuo WT, Lee DE, Saad WE, et al. Superselective microcoil embolization for the treatment of lower gastrointestinal hemorrhage. J Vasc Interv Radiol. 2003;14:1503-9. (Ref 32.)
Ripoll C, Garcia-Tsao G. Treatment of gastropathy and gastric antral vascular ectasia in patients with portal hypertension. Curr Treat Options Gastroenterol. 2007;10:483-94. (Ref 88.)
Savastano S, Teso S, Corra S, et al. Multislice CT angiography of the celiac and superior mesenteric arteries: Comparison with arteriographic findings. Radiol Med (Torino). 2002;103:456-63. (Ref 130.)
Shurafa M, Kamboj G. Thalidomide for the treatment of bleeding angiodysplasias. Am J Gastroenterol. 2003;98:221. (Ref 38.)
Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349:343-9. (Ref 18.)
1. Boley SJ, Sammartano RJ, Adams A, et al. On the nature and etiology of vascular ectasias of the colon. Degenerative lesions of aging. Gastroenterology. 1977;72:650-60.
2. Naveau S, Leger-Ravet MB, Houdayer C, et al. Nonhereditary colonic angiodysplasias: Histomorphometric approach to their pathogenesis. Dig Dis Sci. 1995;40:839.
3. Foutch PG. Angiodysplasia of the gastrointestinal tract. Am J Gastroenterol. 1993;88:807.
4. Boley SJ, Brandt LJ. Vascular ectasias of the colon. Dig Dis Sci. 1986;31:26S-42S.
5. Trudel JL, Fazio VW, Sivak MV. Colonoscopic diagnosis and treatment of arteriovenous malformations in chronic lower gastrointestinal bleeding. Clinical accuracy and efficacy. Dis Colon Rect. 1988;31:107.
6. Gostout CJ, Bowyer BA, Ahlquist DA, et al. Mucosal vascular malformations of the gastrointestinal tract. Clinical observations and results of endoscopic neodymium: yttrium-aluminum-garnet laser therapy. Mayo Clin Proc. 1988;63:993.
7. Junquera F, Quirga S, Saperas E, et al. Accuracy of helical computed tomographic angiography for the diagnosis of colonic angiodysplasia. Gastroenterology. 2000;119:293-9.
8. Hochter WJ, Weingart W, Kunner E, et al. Angiodysplasia in the colon and rectum: Endoscopic morphology, localization and frequency. Endoscopy. 1985;17:182.
9. Elta GH. Urgent colonoscopy for acute lower-GI bleeding. Gastrointest Endosc. 2004;59:402.
10. Reinus JF, Brandt LJ. Vascular ectasias and diverticulosis: Common causes of lower GI bleeding. Gastroenterol Clin North Am. 1994;23:1.
11. Danesh BJ, Spiliadis C, Williams CB, et al. Angiodysplasia, an uncommon cause of colonic bleeding: Colonic evaluation of 1,050 patients with rectal bleeding and anemia. Int J Colon Dis. 1987;2:218.
12. Zuckerman G, Benitez J. A prospective study of bidirectional endoscopy (colonoscopy and upper endoscopy) in the evaluation of patients with occult gastrointestinal bleeding. Am J Gastroenterol. 1992;87:62.
13. Richter JM, Christensen MR, Colditz GA, et al. Angiodysplasia: Natural history and efficacy of therapeutic interventions. Dig Dis Sci. 1989;34:1542.
14. Imperiale TF, Ransohoff DF. Aortic stenosis, idiopathic gastrointestinal bleeding, and angiodysplasia: Is there an association? A methodologic critique of the literature. Gastroenterology. 1998;95:1670.
15. Bhutani MS, Gupta SC, Markert RJ, et al. A prospective controlled evaluation of endoscopic detection of angiodysplasia and its association with aortic valve disease. Gastrointest Endosc. 1995;42:398.
16. Batur P, Stewart WJ, Isaacson JH. Increased prevalence of aortic stenosis in patients with arteriovenous malformations of the gastrointestinal tract in Heyde syndrome. Arch Intern Med. 2003;163:1821.
17. Veyradier A, Balian A, Wolf M, et al. Abnormal von Willebrand factor in bleeding angiodysplasias of the digestive tract. Gastroenterology. 2001;120:346.
18. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349:343-9.
19. Semba T, Fujii Y. Relationship between venous flow and colonic peristalsis. Jpn J Physiol. 1970;20:408.
20. Junquera F, Saperas E, de Torres I, et al. Increased expression of angiogenic factors in human colonic angiodysplasia. Am J Gastroenterol. 1999;94:1070.
21. Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci U S A. 1998;95:15809.
22. Pennazio M, Santucci R, Rondonotti E, et al. Outcome of patients with obscure gastrointestinal bleeding after capsule endoscopy: Report of 100 consecutive cases. Gastroenterology. 2004;126:643.
23. Carey EJ, Leighton JA, Heigh RI, et al. A single-center experience of 260 consecutive patients undergoing capsule endoscopy for obscure gastrointestinal bleeding. Am J Gastroenterology. 2007;102:89-95.
24. Lewis B, Swain P. Capsule endoscopy in the evaluation of patients with suspected small intestinal bleeding: Results of a pilot study. Gastrointest Endosc. 2003;56:349-53.
25. Brandt LJ, Spinell MK. Ability of naloxone to enhance the colonoscopic appearance of normal colon vasculature and colon vascular ectasias. Gastrointest Endosc. 1999;49:79-83.
26. Brandt LJ, Mukhopadhyay D. Masking of colon vascular ectasias by cold water lavage. Gastrointest Endosc. 1999;49:141.
27. Boley SJ, Sprayregen S, Sammartano RJ, et al. The pathophysiologic basis for the angiographic signs of vascular ectasias of the colon. Radiology. 1977;125:615.
28. Wilcox CM, Alexander LN, Clark WS. Prospective evaluation of the gastrointestinal tract in patients with iron deficiency anemia and no systemic or gastrointestinal signs or symptoms. Am J Med. 1997;103:405.
29. Foutch PG, Rex DK, Lieberman DA. Prevalence and natural history of colonic angiodysplasia among healthy asymptomatic people. Am J Gastroenterol. 1995;90:564-7.
30. Brandt LJ. A cecal angiodysplastic lesion is discovered during diagnostic colonoscopy performed for iron-deficiency anemia associated with stool positive for occult blood. What therapy would you recommend? Am J Gastroenterol. 1988;83:710.
31. Darcy M. Treatment of lower gastrointestinal bleeding: Vasopressin infusion versus embolization. J Vasc Interv Radiol. 2003;14:535.
32. Kuo WT, Lee DE, Saad WE, et al. Superselective microcoil embolization for the treatment of lower gastrointestinal hemorrhage. J Vasc Interv Radiol. 2003;14:1503-9.
33. Barkin JS, Ross BS. Medical therapy for chronic gastrointestinal bleeding of obscure origin. Am J Gastroenterol. 1998;93:1250.
34. Junquera F, Feu F, Papo M, et al. A multicenter, randomized, clinical trial of hormonal therapy in the prevention of rebleeding from gastrointestinal angiodysplasia. Gastroenterology. 2001;121:1073-9.
35. D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082-5.
36. Franks ME, Macpherson GR, Figg WD. Thalidomide. Lancet. 2004;363:1802-11.
37. Barlogie B, Tricot G, Anaissie E, et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N Engl J Med. 2006;354:1021.
38. Shurafa M, Kamboj G. Thalidomide for the treatment of bleeding angiodysplasias. Am J Gastroenterol. 2003;98:221.
39. Kirkham SE, Lindley KJ, Elawad MA, et al. Treatment of multiple small bowel angiodysplasias causing severe life-threatening bleeding with thalidomide. J Pediatr Gastroenterol Nutr. 2006;42:585-7.
40. Bauditz J, Schachschal G, Wedel S, Lochs H. Thalidomide for treatment of severe intestinal bleeding. Gut. 2004;53:609.
41. Bauditz J, Lochs H, Voderholzer W. Macroscopic appearance of intestinal angiodysplasias under antiangiogenic treatment with thalidomide. Endoscopy. 2006;38:1036-9.
42. Shurafa M, Kamboj G. Thalidomide for the treatment of bleeding angiodysplasias. Am J Gastroenterol. 2003;98:221.
43. Kabbinavar F, Hurwitz HI, Fehrenbacher L, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21:60.
44. Cello JP, Grendell JH. Endoscopic laser treatment for gastrointestinal vascular ectasias. Ann Intern Med. 1986;104:352.
45. Naveau S, Aubert A, Poynard AT, et al. Long-term results of treatment of vascular malformations of the gastrointestinal tract by neodymium YAG laser photocoagulation. Dig Dis Sci. 1990;35:821.
46. Rogers BH. Endoscopic diagnosis and therapy of mucosal vascular abnormalities of the gastrointestinal tract occurring in elderly patients and associated with cardiac, vascular, and pulmonary disease. Gastrointest Endosc. 1980;26:134.
47. Jensen DM, Machicado GA. Colonoscopy for diagnosis and treatment of severe lower gastrointestinal bleeding: Routine outcomes and cost analysis. Gastroenterol Clin North Am. 1997;7:477.
48. Moparty B, Raju GS. Role of hemoclips in a patient with cecal angiodysplasia at high risk of recurrent bleeding from antithrombotic therapy to maintain coronary stent patency: A case report. Gastrointest Endosc. 2005;62:41.
49. Junquera F, Brullet E, Campo R, et al. Usefulness of endoscopic band ligation for bleeding small bowel vascular lesions. Gastrointest Endosc. 2003;58:274.
50. Vargo JJ. Clinical applications of the argon plasma coagulator. Gastrointest Endosc. 2004;59:81.
51. Farmer R, Lichliter W, Kuhn J, et al. Total colectomy versus limited colonic resection for acute lower intestinal bleeding. Am J Surg. 1999;178:587.
52. Renzulli P, Maurer CA, Netzer P, et al. Subtotal colectomy with primary ileorectoscopy is effective for unlocalized, diverticular hemorrhage. Lagenbeck’s Arch Surg. 2002;387:67.
53. Kjeldsen AD, Kjeldsen J. Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol. 2000;95:415.
54. Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918.
55. Sharma VK, Howden CW. Gastrointestinal and hepatic manifestations of hereditary hemorrhagic telangiectasia. Dig Dis. 1998;16:169.
56. Azuma H. Genetic and molecular pathogenesis of hereditary hemorrhagic telangiectasia. J Med Invest. 2000;47:81-90.
57. Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91:66.
58. McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345.
59. Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189.
60. Cole SG, Begbie ME, Wallace GM, et al. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42:577.
61. Bayrak-Toydemir P, McDonald J, Akarsu N, et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140:2155.
62. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669.
63. Cirulli A, Liso A, D’Ovidio F, et al. Vascular endothelial growth factor serum levels are elevated in patients with hereditary hemorrhagic telangiectasia. Acta Haematol. 2003;110:29.
64. Garcia-Tsao G, Korzenik JR, Young L. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2000;343:931.
65. Martini GA. The liver in hereditary hemorrhagic telangiectasia: An inborn error of vascular structure with multiple manifestations. A reappraisal. Gut. 1978;19:531.
66. Ravard G, Soyer P, Boudiaf M, et al. Hepatic involvement in hereditary hemorrhagic telangiectasia: helical computed tomography features in 24 consecutive patients. J Comput Assist Tomogr. 2004;28:488.
67. Halpern M, Turner AF, Citron BP. Hereditary hemorrhagic telangiectasia. An angiographic study of abdominal visceral angiodysplasias associated with gastrointestinal hemorrhage. Radiology. 1968;90:1143.
68. Van Cutsam E, Rutgeerts P, Geboes K, et al. Estrogen-progesterone treatment of Osler-Weber-Rendu disease. J Clin Gastroenterol. 1988;10:676.
69. Saba HI, Morelli GA, Logrono LA. Brief report: Treatment of bleeding in hereditary hemorrhagic telangiectasia with aminocaproic acid. N Engl J Med. 1995;330:1789.
70. Mitchell A, Adams LA, MacQuillan G, et al. Bevacizumab reverses need for liver transplantation in hereditary hemorrhagic telangiectasia. Liver Transpl. 2008;14:210.
71. Sjogren RW. Gastrointestinal features of scleroderma. Curr Opin Rheumatol. 1996;8:569.
72. Duchini A, Sessoms SL. Gastrointestinal hemorrhage in patients with systemic sclerosis and CREST syndrome. Am J Gastroenterol. 1998;93:1453.
73. Jabbari M, Cherry R, Lough JO, et al. Gastric antral vascular ectasia: The watermelon stomach. Gastroenterology. 1984;87:1165.
74. Toyota M, Hinoda Y, Nakagawa N, et al. Gastric antral vascular ectasia causing severe anemia. J Gastroenterol. 1996;31:710.
75. Fisher NC. Gastric antral vascular ectasia and its relation to portal hypertension. Gut. 2000;46:441.
76. Payen JL, Cales P, Voigt JJ. Severe portal hypertensive gastropathy and antral vascular ectasia are distinct entities in patients with cirrhosis. Gastroenterology. 1995;108:138.
77. Vincent C, Pomier-Layrargues G, Dagenais M, et al. Cure of gastric antral vascular ectasia by liver transplantation despite persistent portal hypertension: a clue for pathogenesis. Liver Transpl. 2002;8:717-20.
78. Spahr L, Villeneuve JP, Dufresne MP, et al. Gastric antral vascular ectasia in cirrhotic patients: absence of relation with portal hypertension. Gut. 1999;44:739-42.
79. Tran A, Villeneuve JP, Bilodeau M, et al. Treatment of chronic bleeding from gastric antral vascular ectasia (GAVE) with estrogen-progesterone in cirrhotic patients: an open pilot study. Am J Gastroenterol. 1999;94:2909-11.
80. McCormick PA, Ooi H, Crosbie O. Tranexamic acid for severe bleeding gastric antral vascular ectasia in cirrhosis. Gut. 1998;42:750-2.
81. Park RH, Danesh BJ, Upadhyay R, et al. Gastric antral vascular ectasia (watermelon stomach)—therapeutic options. Postgrad Med J. 1990;66:720.
82. Dunne KA, Hill J, Dillon JF. Treatment of chronic transfusion- dependent gastric antral vascular ectasia (watermelon stomach) with thalidomide. Eur J Gastroenterol Hepatol. 2006;18:455.
83. Kamath PS, Lacerda M, Ahlquist DA, et al. Gastric mucosal responses to intrahepatic portosystemic shunting in patients with cirrhosis. Gastroenterology. 2000;118:905.
84. Spahr L, Villeneuve JP, Dufresne MP, et al. Gastric antral vascular ectasia in cirrhotic patients: absence of relation with portal hypertension. Gut. 1999;44:739.
85. Mann NS, Rachut E. Gastric antral vascular ectasia causing severe hypoalbuminemia and anemia cured by antrectomy. J Clin Gastroenterol. 2002;34:284-6.
86. Brandt LJ. Gastric antral vascular ectasia: Is there to be a consensus? Gastrointest Endosc. 1996;44:355.
87. Sebastian S, O’Morain CA, Buckley MJ. Current therapeutic options for gastric antral vascular ectasia. Aliment Pharmacol Ther. 2003;18:157.
88. Ripoll C, Garcia-Tsao G. Treatment of gastropathy and gastric antral vascular ectasia in patients with portal hypertension. Curr Treat Options Gastroenterol. 2007;10:483-94.
89. Orloff MJ, Orloff MS, Orloff SL, Haynes KS. Treatment of bleeding from portal hypertensive gastropathy by portacaval shunt. Hepatology. 1995;21:1011-17.
90. Urata J, Yamashita Y, Tsuchigame T, et al. The effects of transjugular intrahepatic portosystemic shunt on portal hypertensive gastropathy. J Gastroenterol Hepatol. 1998;13:1061.
91. Soin AS, Acharya SK, Mathur M, et al. Portal hypertensive gastropathy in noncirrhotic patients. The effect of lienorenal shunts. J Clin Gastroenterol. 1998;26:64.
92. Mezawa S, Homma H, Ohta H, et al. Effect of transjugular intrahepatic portosystemic shunt formation on portal hypertensive gastropathy and gastric circulation. Am J Gastroenterol. 2001;96:1155.
93. Zhou Y, Qiao L, Wu J, et al. Comparison of the efficacy of octreotide, vasopressin, and omeprazole in the control of acute bleeding in patients with portal hypertensive gastropathy: A controlled study. J Gastroenterol Hepatol. 2002;17:973.
94. Kouroumalis EA, Koutroubakis IE, Manousos ON. Somatostatin for acute severe bleeding from portal hypertensive gastropathy. Eur J Gastroenterol Hepatol. 1998;10:509.
95. Bruha R, Marecek Z, Spicak J, et al. Double-blind randomized, comparative multicenter study of the effect of terlipressin in the treatment of acute esophageal variceal and/or hypertensive gastropathy bleeding. Hepatogastroenterology. 2002;49:1161-6.
96. Misra V, Misra SP, Dwivedi M, et al. Colonic mucosa in patients with portal hypertension. J Gastroenterol Hepatol. 2003;18:302.
97. Kozarek RA, Botoman VA, Bredfeldt JE, et al. Portal colopathy: Prospective study of colonoscopy in patients with portal hypertension. Gastroenterology. 1991;101:1192.
98. Schmulewitz N, Baillie J. Dieulafoy lesions: A review of 6 years of experience at a tertiary referral center. Am J Gastroenterol. 2001;96:1688.
99. Park CH, Sohn YH, Lee WS, et al. The usefulness of endoscopic hemoclipping for bleeding Dieulafoy lesions. Endoscopy. 2003;35:388.
100. Tanaka N, Onda M, Seya T, et al. Diffuse cavernous hemangioma of the rectum. Eur J Surg. 1999;165:280.
101. Iwata Y, Shiomi S, Otso R, et al. A case of cavernous hemangioma of the small intestine diagnosed by scintigraphy with Tc-99m-labeled red blood cells. Ann Nucl Med. 2000;14:373-376.
102. Fremond B, Yazbeck S, Dubois J, et al. Intestinal vascular anomalies in children. J Pediatr Surg. 1997;6:873.
103. Maxwell PH, Wiesener MS, Chang GW, et al. The tumor suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271.
104. Madhusudan S, Deplanque G, Braybrooke JP, et al. Antiangiogenic therapy for von Hippel-Lindau disease. JAMA. 2004;291:943.
105. Rodrigues D, Bourrol ML, Ferrer AP, et al. Blue rubber bleb nevus syndrome. Rev Hosp Clin Fac Med Sao Paulo. 2000;55:29.
106. Oksuzoglu BC, Oksuzoglu G, Cakir U, et al. Blue rubber bleb nevus syndrome. Am J Gastroenterol. 1996;91:780.
107. Servelle M, Bastin R, Loygue J, et al. Hematuria and rectal bleeding in the child with Klippel and Trenaunay syndrome. Ann Surg. 1976;183:418.
108. Tian XL, Kadaba R, You SA, et al. Identification of an angiographic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature. 2004;427:592.
109. Wilson CL, Song LM, Chua H, et al. Bleeding from cavernous angiomatosis of the rectum in Klippel-Trenaunay syndrome: Report of three cases and review of the literature. Am J Gastroenterol. 2001;96:2783.
110. Ziyeh S, Spreer J, Rossler J, et al. Parkes Weber or Klippel-Trenaunay syndrome? Non-invasive diagnosis with MR projection angiography. Eur Radiol. 2004;14:2025.
111. Kalman PG, Johnston KW. Abdominal aortic aneurysms. In: Hobson RW, Wilson SE, Veith FJ, editors. Vascular surgery: Principles and practice. New York: Marcel Dekker, 2004.
112. Delin A, Ohlson H, Swendenborg J. Growth rates of abdominal aortic aneurysms as measured by computed tomography. Br J Surg. 1985;7:530.
113. Ottinger LW. Ruptured arteriosclerotic aneurysms of the abdominal aorta. JAMA. 1975;233:147.
114. Nevitt MP, Ballard DJ, Hallet JW. Prognosis of abdominal aortic aneurysms: A population-based study. N Engl J Med. 1989;15:1009.
115. Champion MC, Sullivan SN, Coles JC, et al. Aortoenteric fistula: Incidence, presentation, recognition, and management. Ann Surg. 1982;195:314.
116. Ohki T, Veith FJ. Abdominal aortic aneurysms. Curr Treat Options Cardiovasc Med. 1999;1:19.
117. Collen J, Murie J, Morris PJ. 2 year prospective analysis of the Oxford experience with surgical treatment of abdominal aortic aneurysm. Surg Gynecol Obstet. 1989;169:527.
118. Chahwan S, Comerota AJ, Pigott JP, et al. Elective treatment of abdominal aortic aneurysm with endovascular or open repair: the first decade. J Vasc Surg. 2007;45:258-62.
119. Chaikof EL, Lin PH, Brinkman WT, et al. Endovascular repair of abdominal aortic aneurysms: Risk stratified outcomes. Ann Surg. 2002;235:833.
120. Hollier LA, Taylor LM, Ochner J. Recommended indications for operative treatment of abdominal aortic aneurysms. J Vasc Surg. 1992;15:1046.
121. Zarins CK, White RA, Schwarten D, et al. Aneurysm stent graft versus open surgical repair of abdominal aortic aneurysms: Multicenter prospective clinical trial. J Vasc Surg. 1999;29:292.
122. Seelig MH, Oldenberg WA, Hakaim AG, et al. Endovascular repair of abdominal aortic aneurysms: Where do we stand? Mayo Clin Proc. 1999;74:999.
123. Gomes MN, Chokye PL, Wallace RB. Infected aortic aneurysms. A changing entity. Ann Surg. 1992;215:435.
124. Bashir RM, al-Kiwas FH. Rare causes of occult small intestinal bleeding including aortoenteric fistulas, small bowel tumors and small bowel ulcers. Gastrointest Endosc Clin North Am. 1996;6:709.
125. Gozzetti G, Poggioli G, Spolaore R, et al. Aortoenteric fistulae: Spontaneous and after aorto-iliac operations. J Cardiovasc Surg. 1984;25:420.
126. Perks FJ, Gillespie I, Patel D. Multidetector computed tomography imaging of aortoenteric fistula. J Comput Assist Tomogr. 2004;28:343.
127. Baltazar U, Dunn J, Floresguerra C, et al. Superior mesenteric artery syndrome: An unusual cause of intestinal obstruction. South Med J. 2000;93:606.
128. Lee CS, Mangla JC. Superior mesenteric artery compression syndrome. Am J Gastroenterol. 1978;70:141.
129. Konen E, Amitai M, Apter S, et al. CT angiography of superior mesenteric artery syndrome. Am J Roentgenol. 1998;171:1279.
130. Savastano S, Teso S, Corra S, et al. Multislice CT angiography of the celiac and superior mesenteric arteries: Comparison with arteriographic findings. Radiol Med (Torino). 2002;103:456-63.
131. Richardson WS, Surowiec WJ. Laparoscopic repair of superior mesenteric artery syndrome. Am J Surg. 2001;181:377.
132. Harjola PT. A rare obstruction of the celiac artery. Ann Chir Gynaecol Fenn. 1963;52:547.
133. Brandt LJ, Boley S. Celiac axis compression syndrome: A critical review. Am J Gastroenterol. 1978;23:633.
134. Szilagyi DE, Rian RL, Elliot JP, et al. The celiac artery compression syndrome: Does it exist? Surgery. 1972;6:849.
135. Roayaie S, Jossart G, Gitlitz D, et al. Laparoscopic release of celiac artery compression syndrome facilitated by laparoscopic ultrasound scanning to confirm restoration of flow. J Vasc Surg. 2000;32:814.
136. Evans WE. Long-term evaluation of the celiac band syndrome. Surgery. 1974;76:867.
137. Bech FR. Celiac artery compression syndromes. Surg Clin North Am. 1997;77:409.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 36 Vascular Lesions of the Gastrointestinal Tract
Through the widespread use of endoscopy and angiography, as well as advances in imaging techniques such as computed tomographic angiography (CTA) and magnetic resonance angiography (MRA), vascular lesions of the gastrointestinal (GI) tract are being increasingly characterized. Vascular lesions are a common cause of GI hemorrhage and may be solitary or multiple, benign or malignant, isolated or part of a syndrome or systemic disorder (Table 36-1). It is important at the outset to understand the nomenclature for the commonest lesions. Vas and its derivative vascular are Latin words meaning vessel; the Greek equivalent is angeion. Ectasia is a word of Greek derivation that refers to the process whereby a blood vessel becomes dilated or lengthened; the resulting lesion also can be referred to as an ectasia. Telangiectasia is the lesion resulting from dilation of the terminal aspect (tele) of a vessel. Angiodysplasia is used as a general term to describe the lesion or process whereby a badly formed (dys, “bad”; plasis, “molded”) vessel develops. An arteriovenous malformation is a congenital lesion, whereas an angioma is a neoplasm. This chapter discusses the more important vascular lesions that cause GI bleeding and that are representative of the spectrum of vascular lesions of the GI tract.
Table 36-1 Vascular Lesions of the Gastrointestinal Tract
| Primary Vascular Lesions |
| Diseases and Syndromes with Vascular Lesions |
| Systemic Disorders Associated with Vascular Lesions |
CREST, calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia; GAVE, gastric antral vascular ectasia.
VASCULAR LESIONS
ANGIOECTASIA
Angioectasia (AE) of the colon is a distinct clinical and pathologic entity.1–3 It is the most common vascular abnormality of the GI tract and probably the most frequent cause of recurrent or chronic lower intestinal bleeding in persons older than 60 years of age.4 AEs are probably acquired with aging, and there does not appear to be a gender predominance. In contrast to congenital or neoplastic vascular lesions of the GI tract, AEs are not associated with lesions of the skin or other viscera. However, when patients with vascular lesions of the colon are aggressively studied with angiography or enteroscopy, concomitant lesions may be seen in the small intestine in approximately 10% of patients.3,5,6 AEs almost always are confined to the cecum or ascending colon, usually are multiple rather than single, and usually are smaller than 10 mm in diameter. They are seldom identified by the surgeon at operation or by the pathologist using standard histologic techniques, but usually they can be diagnosed by angiography; colonoscopy (Figs. 36-1 and 36-2); or, as recently shown, helical CTA.7
The roles of computed tomography (CT) and magnetic resonance imaging (MRI) for vascular lesions of all types are evolving but are certain to increase as these sophisticated modes of diagnosis become more widely available; it is also clear that conventional angiography at present is more important for therapy than for diagnosis. To determine the precise nature of a vascular lesion, histologic examination, with or without injection studies of the vasculature, is necessary. In one report in which histologic confirmation of vascular lesions was not performed, AEs reportedly occurred distal to the hepatic flexure in 46% of patients8; review of tissue sections from supposed AEs in the small bowel or left colon revealed histologic changes different from those of AEs in the right colon (personal review by S. J. Boley and L. J. Brandt).
Bleeding from cecal AEs was first shown in 1961 by intraoperative angiography and has since become well recognized, especially after the introduction of selective angiography and colonoscopy for identifying the source of intestinal bleeding (see Chapter 19).8 In older literature, AEs and diverticulosis were considered the two most common causes of severe lower GI hemorrhage in older adults; however, more recent publications have cited AEs and diverticulosis to be responsible for 3% to 37% (mean: 10%) and 15% to 55% (mean: 30%) of major lower intestinal bleeding episodes, respectively (see Chapters 19 and 117).9 The problem of attributing bleeding to one or the other cause, when bleeding from the lesion is not demonstrated by endoscopy or by extravasation of contrast material on radiologic imaging studies, is compounded by the frequency and coexistence of these disorders without bleeding in people older than 60 years of age. The prevalence of diverticulosis is estimated to be as high as 50% in the population older than age 60; mucosal and submucosal AEs of the right colon can be found by injection studies of colons removed at surgery in more than 25% and 50%, respectively, of patients in this age range without evidence of bleeding.1,10 In large series of colonoscopic examinations, AEs have been seen in 0.2% to 2.9% of nonbleeding persons and 2.6% to 6.2% of patients evaluated specifically for occult blood in the stool, anemia, or hemorrhage.3,11–12 In a patient being studied for GI bleeding, in whom the site of active bleeding is unproven, the only basis for determining that an identified ectasia or diverticulosis is responsible for bleeding is the indirect evidence provided by the patient’s course after ablation or resection of the suspected lesion. It is unusual for AEs found incidentally to bleed, and an AE, even in a patient with a history of bleeding, cannot be assumed to be the cause.13
Bleeding from AEs typically is recurrent and low grade, although approximately 15% of patients present with massive hemorrhage. The nature and degree of bleeding frequently vary in the same patient with different episodes: Patients may have bright red blood, maroon stools, or melena on separate occasions. In 20% to 25% of episodes, only tarry stools are passed, and in 10% to 15% of patients, bleeding is evidenced solely by iron deficiency anemia, with stools that are intermittently positive for occult blood.4 This spectrum reflects the varied rate of bleeding from the ectatic capillaries, venules, and arteriovenous communications, depending on the developmental stage of the lesions (see later). In more than 90% of instances, bleeding stops spontaneously.
In 1958 E. C. Heyde described what is still a controversial association of AEs, GI bleeding, and aortic stenosis; aortic valve replacement had even been recommended for “Heyde’s syndrome” when bleeding could not be managed adequately. Numerous reports of Heyde’s syndrome appeared in the literature; subsequent analysis14 and many studies,15 however, failed to support the association. This association recently has been suggested again16 in a retrospective study in which the frequency of aortic stenosis was 31.7% in patients with “AVMs” compared with 14% in the general population; severe aortic stenosis was also more likely in the group with intestinal vascular lesions. The additional postulate has been offered that deficiencies of the largest forms of von Willebrand factor multimers (von Willebrand syndrome, type 2A) result in hemostatic abnormalities that may predispose preexisting AEs to bleed.17 Preoperative deficiency of these multimers reverses after aortic valve replacement,18 but the general recommendation to replace the aortic valve to control bleeding from AEs seems premature now that a variety of transendoscopic and angiographic means are available to ablate AEs.
Pathology
Histologic identification of AEs is difficult unless special techniques are used.1 Although usually less than one third of lesions are found by routine pathologic examination, almost all can be identified by injecting the colonic vasculature with silicone rubber, dehydrating the cells with increasing concentrations of ethyl alcohol, clearing the specimen by immersing it for 24 hours in a bath of methyl salicylate, and then viewing the specimen by dissecting stereomicroscopy (Fig. 36-3).1 In a study using these methods, surgically resected colons were analyzed and found to have one or more mucosal AEs measuring 1 mm to 1 cm in diameter. AEs were usually multiple, and in this study, all were located within the cecum and ascending colon; the most distal one was 23 cm beyond the ileocecal valve.1
Microscopically, mucosal AEs consist of ectatic, distorted, thin-walled venules, capillaries, and arterioles, vessels that are lined by endothelium, and, infrequently, a small amount of smooth muscle. The earliest abnormality is the presence of dilated, tortuous, submucosal veins (Fig. 36-4A), often in areas where mucosal vessels appear normal. More extensive lesions show increasing numbers of dilated and deformed vessels traversing the muscularis mucosa and involving the mucosa (see Fig. 36-4B and C) until, in the most severe lesions, the mucosa is replaced by a maze of distorted, dilated vascular channels (see Fig. 36-4D). Enlarged arteries and thick-walled veins occasionally are seen in advanced lesions, in which the dilated arteriolar-capillary-venular unit has become a small arteriovenous fistula because of loss of prearteriolar sphincter function. Large thick-walled arteries are more typical of congenital arteriovenous malformations.
Pathogenesis
The previously described studies using injection and clearing techniques indicated that AEs are acquired lesions associated with aging and that they represent a unique clinical and pathologic entity.1 That AEs are common lesions associated with aging is supported by their frequent identification at colonoscopy in older adults and in injected colons resected from older patients with no history of bleeding.1,11 Boley postulated that the likely cause of AEs is partial, intermittent, low-grade obstruction of submucosal veins at the site where these vessels pierce the muscular layers of the colon1 (Figs. 36-5 and 36-6). He further suggested that repeated episodes of transiently elevated pressure during muscular contraction and distention of the cecum over many years conceivably result in dilation and tortuosity of the submucosal vein and, later, of the venules and capillaries of the mucosal units that drain into it. Finally, he postulated that the capillary rings dilate, the precapillary sphincters lose their competency, and a small arteriovenous fistula is produced. The latter is responsible for the “early-filling vein,” which was the original angiographic hallmark of this lesion (Fig. 36-7). Prolonged increased flow through the arteriovenous fistula can then produce alterations in the arteries supplying the area and in the extramural veins that drain it. This developmental concept of the cause of AEs was based on the finding of (1) a prominent submucosal vein, either in the absence of any mucosal lesion, or underlying only a minute mucosal AE supplied by a normal artery; (2) dilation of the veins, starting where they traverse the muscularis propria (see Fig. 36-5); and (3) previous studies showing that venous flow in the bowel may be diminished by increases in colon motility, intramural tension, and intraluminal pressure.19 Following this logic, the prevalence of AEs in the right colon can be attributed to the greater tension in the cecal wall compared with that in other parts of the colon, according to LaPlace’s principle: T ∝ pDP (where T is tension, D is diameter, and P is intraluminal pressure).
An alternative concept for the development of AEs is based on the demonstration that AEs have been shown to express vascular endothelial growth factor (VEGF) and its receptors along the endothelial lining in surgical specimens from patients who have undergone colectomy for recurrent bleeding20; this indicates a proliferative phase of angiogenesis. VEGF and VEGF receptor 1 have been shown to be up-regulated by hypoxia21 and therefore a role has been suggested for hypoxia in the pathogenesis of AEs. Further research still is needed to clarify the pathophysiology of AEs.
Diagnosis and Management
Management of bleeding AEs consists of three phases: (1) diagnosis; (2) conversion of an emergency situation to an elective one by control of acute bleeding; and (3) definitive treatment of the AE by colonoscopic ablation or surgical removal. The diagnostic approach to colonic AEs is essentially the same as that for lower intestinal bleeding in general and includes radionuclide bleeding scans; colonoscopy; angiography; and, to exclude the small intestine as a site of bleeding, push enteroscopy and wireless capsule endoscopy (WCE). WCE is a relatively new diagnostic technique that enables visualization of the entire small intestine, and rarely the very proximal colon where colonic AEs are found. WCE is particularly useful for evaluating patients with obscure and occult GI bleeding.22 WCE has been shown to be superior to push enteroscopy in the evaluation of patients with small intestinal bleeding, yielding a diagnosis in 55% to 75% of patients with obscure bleeding that required transfusions, most common of which was angiodysplasia.23,24 Radionuclide scans are used to determine whether a patient is actively bleeding and, if so, to localize the site (see Chapter 19). Although angiography previously had been the principal means of identifying AE as the source of bleeding, colonoscopy currently is the preferred method. Helical CTA is a relatively new, sensitive, specific, and well-tolerated technique to diagnose colonic AEs, although prospective studies comparing CTA with other imaging techniques are necessary.7
The endoscopist’s ability to diagnose the specific nature of a vascular lesion is limited by the similar appearance of different types of lesions. AEs, spider angiomas, hereditary hemorrhagic telangiectasia, angiomas, the focal hypervascularity of radiation colitis, ulcerative colitis, Crohn’s disease, ischemic colitis, certain infections (e.g., syphilis, Pneumocystis), hyperplastic and adenomatous polyps, and malignancies, including lymphoma and leukemic infiltrations, can all, on occasion, resemble each other (Table 36-2). Because traumatic and endoscopic suction artifacts may resemble vascular lesions, all lesions must be evaluated on insertion of the colonoscope, rather than during withdrawal. Pinch biopsy samples of vascular lesions obtained during endoscopy usually are nonspecific; therefore, the risk of performing biopsies of these abnormalities is not justified.
Table 36-2 Lesions That May Be Confused with Angioectasias on Endoscopy
| Vascular Lesions |
Because the appearance of vascular lesions is influenced by a patient’s blood pressure, blood volume, and state of hydration, such lesions may not be evident in those with severely reduced blood volumes or shock; thus accurate evaluation may not be possible until red cell and volume deficits are corrected. Meperidine also may diminish the prominence of some vascular abnormalities (e.g., AEs and the telangiectasias of hereditary hemorrhagic telangiectasia); use of meperidine, therefore, should be minimized and its effects reversed by naloxone so that vascular lesions can be detected accurately. Such a masking effect does not seem to occur with fentanyl. Naloxone has been shown to enhance the appearance of normal colonic vasculature in approximately 10% of patients and cause existing AEs to appear (2.7%) or increase in size (5.4%) (Fig. 36-8).25 For these reasons, naloxone is an important adjunctive medication for patients undergoing endoscopic evaluation for lower intestinal bleeding. Cold water lavage of the colon, as is sometimes done to cleanse the luminal surface of debris during colonoscopy, also may cause underlying AEs to disappear transiently.26
Angiography is used to determine the site and nature of lesions during active bleeding and can identify some vascular lesions even after bleeding has ceased. The three reliable angiographic signs of AEs are a densely opacified, slowly emptying, dilated, tortuous vein; a vascular tuft; and an early-filling vein (see Fig. 36-7).27 A fourth sign, extravasation of contrast material, identifies the site of bleeding when bleeding volume is at least 0.5 mL/minute but is not specific for AE. The slowly emptying vein (see Fig. 36-7A) persists late into the venous phase, after the other mesenteric veins have emptied. Vascular tufts (see Fig. 36-7B) are created by the ectatic venules that join the mucosal AE and the submucosal vein. They are seen best in the arterial phase; are usually located at the termination of a branch of the ileocolic artery; appear as small candelabra-like or oval clusters of vessels; and still are seen in the venous phase communicating with a dilated, tortuous, intramural vein. The early-filling vein is seen in the arterial phase within four or five seconds of injection (see Fig. 36-7B); it is not a valid sign of AE if vasodilators such as papaverine or tolazoline (Priscoline) have been used to enhance the study. When the lesion is bleeding, intraluminal extravasation of contrast material usually appears during the arterial phase of angiography and persists throughout the study. Extravasation identifies the site of active bleeding, but in the absence of other signs of AEs, it suggests another cause for the bleeding.
Management of incidental (nonbleeding) AEs detected by colonoscopy is expectant. The natural history of colonic AE is benign in healthy, asymptomatic people, and the risk of bleeding is small.13,28,29 In such cases, endoscopic therapy is not warranted.30
Bleeding can be controlled endoscopically or angiographically in most patients, thereby avoiding the morbidity and mortality of emergency operation. In decades past, intra-arterial embolization and vasopressin were used to control upper and lower GI bleeding, respectively. Vasopressin, given via an angiographic catheter placed into the feeding splanchnic vessel, arrested hemorrhage successfully from AE in more than 80% of patients in whom extravasation was demonstrated. Now, superselective microcoil embolization has largely replaced intra-arterial vasopressin infusion for the treatment of lower intestinal hemorrhage.31 Such embolization is highly effective and safe but complicated by ischemic events in approximately 5% of cases.32 Vasopressin still is recommended, however, when intestinal lesions are diffuse throughout the bowel or when superselective catheterization is not possible.32
Hormonal therapy, using estrogens in combination with progestins, has been used to treat patients with a variety of vascular lesions of the GI tract, in an attempt to reduce or terminate bleeding. The mechanisms by which such agents work are not known, although procoagulant effects and endothelial injury are popular theories. Although one long-term observational study33 showed that combination hormonal therapy stopped bleeding in patients with occult GI bleeding of obscure origin (likely to have resulted from small bowel angiodysplasia), current studies do not support the use of these agents to prevent rebleeding from GI angiodysplasia.34 It is likely that hormonal therapy affects different vascular lesions differently and that vascular lesions in the small intestine may respond differently to such treatment than the same lesions in the colon; no study of hormonal therapy has been done for known colonic AEs.
A novel therapy for AEs, and perhaps other vascular lesions in the gastrointestinal tract, is the use of antiangiogenic factors. Thalidomide was developed in the 1950s as a sedative, sleeping pill, and antiemetic for pregnant women, but it soon became notorious for causing phocomelia and other malformations in the newborn.34 In 1994, D’Amato and colleagues reported that thalidomide inhibited VEGF and basic fibroblast growth factor-mediated angiogenesis, which led to further characterization and subsequent clinical applications of its antiangiogenic activity.35,36 Recent data suggest the mechanism for its antiangiogenic effect is related to reduced expression of integrin genes and resulting decreased cell-cell surface interactions and response to angiogenic cytokines.37 Several case reports and case series have described the successful use of thalidomide to treat life-threatening or refractory bleeding from intestinal AEs and Crohn’s disease with refractory bleeding.38–42 After treatment with thalidomide for three months, substantial reductions in the number, size, and color intensity of AEs were observed by WCE.39
Of the available antiangiogenic biologic therapies, most information regarding clinical efficacy and toxicity is available for bevacizumab (Avastatin), a humanized monoclonal antibody against VEGF that is effective against colon and renal cancers and that also has a strong antiangiogenic activity.43 Curiously, dose-dependent nasal and GI bleeding is observed in up to 59% of patients during treatment, possibly caused by a loss of vascular integrity as a result of bevacizumab-induced endothelial-cell shedding in highly regenerative mucosal tissues with active angiogenesis. It is unclear why some antiangiogenic substances like bevacizumab cause mucosal bleeding and others like thalidomide do not; this disparity effect may be related to the phase of angiogenesis that is antagonized, or might reflect a particular strong antiangiogenic activity. Although VEGF-based antiangiogenic therapy is a promising therapy, the issue of aggravation of bleeding from vascular lesions needs further study. A more detailed understanding of the angiogenic cascade and how antiangiogenic substances act within it will be needed to resolve this issue.
Neodymium:yttrium-aluminum-garnet (Nd:YAG) laser3,6,44,45; endoscopic sclerosis11; monopolar46 and bipolar47 electrocoagulation; heater probe47; and, recently, hemoclips in combination with cautery,48 endoscopic band ligation,49 and argon plasma coagulation (APC)50 have been used to ablate vascular lesions throughout the GI tract and can be used to control active bleeding (Fig. 36-9). Control of bleeding has been obtained with a variety of endoscopic thermal means in 47% to 88% of cases,3 and no technique has been established as superior to the next.11 Severe delayed bleeding occurs in 5% of patients with colonic AEs after thermal therapy.46 Recurrent bleeding from colonic AEs appears to be reduced after these therapies, but more than one treatment session is usually necessary.47 Rebleeding can be expected to increase with time after the procedure and has been seen in 28% to 52% of patients over a follow-up period ranging from 15 to 36 months.3
[/not-level-membership-for-gastroenterology-and-hepatology-category]
