339 |
Polycystic Kidney Disease and Other Inherited Disorders of Tubule Growth and Development |
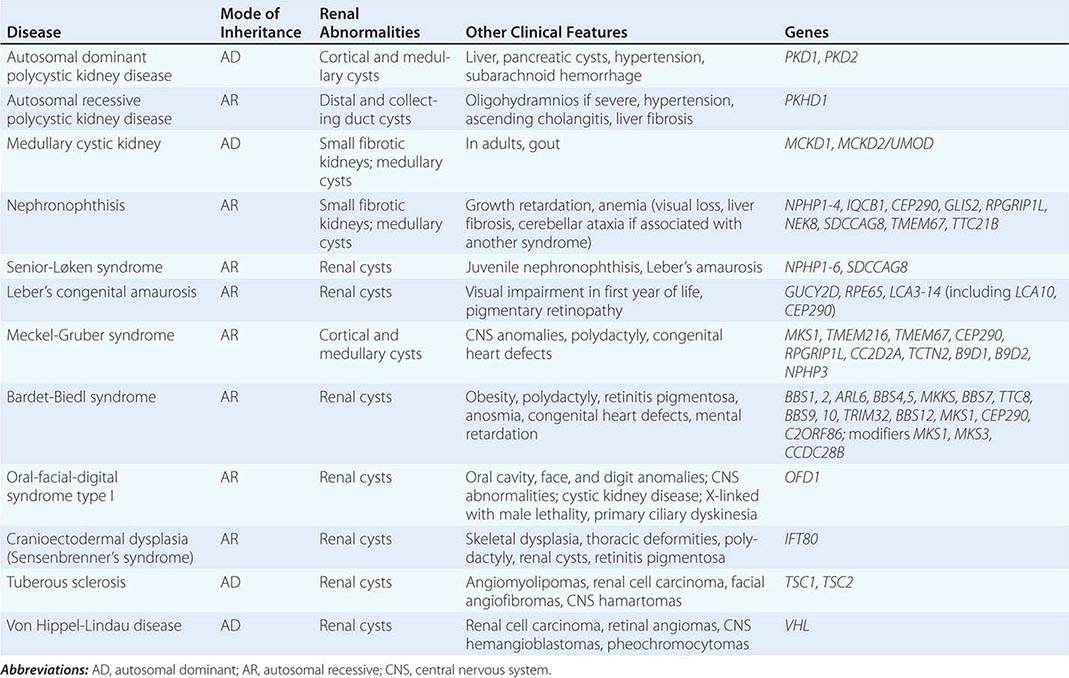
The polycystic kidney diseases are a group of genetically heterogeneous disorders and a leading cause of kidney failure. The autosomal dominant form of polycystic kidney disease (ADPKD) is the most common life-threatening monogenic disease, affecting 12 million people worldwide. The autosomal recessive form of polycystic kidney disease (ARPKD) is rarer but affects the pediatric population. Kidney cysts are often seen in a wide range of syndromic diseases. Recent studies have shown that defects in the structure or function of the primary cilia may underlie this group of genetic diseases collectively termed ciliopathies (Table 339-1).
|
INHERITED DISEASES COMMONLY ASSOCIATED WITH A CYSTIC PHENOTYPE |
AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE
Etiology and Pathogenesis (Fig. 339-1) ADPKD is characterized by progressive formation of epithelial-lined cysts in the kidney. Although cysts only occur in 5% of the tubules in the kidney, the enormous growth of these cysts ultimately leads to the loss of normal surrounding tissues and loss of renal function. The cellular defects in ADPKD that have been known for a long time are increased cell proliferation and fluid secretion, decreased cell differentiation, and abnormal extracellular matrix. ADPKD is caused by mutations in PKD1 and PKD2, which, respectively, code for polycystin-1 (PC1) and polycystin-2 (PC2). PC1 is a large 11-transmembrane protein that functions like a G protein–coupled receptor. PC2 is a calcium-permeable six-transmembrane protein that structurally belongs to the transient receptor potential (TRP) cation channel family. PC1 and PC2 are widely expressed in almost all tissues and organs. PC1 expression is high in development and low in the adult, whereas PC2 expression is relatively constant. PC1 and PC2 are found on the primary cilium, a hair-like structure present on the apical membrane of a cell, in addition to the cell membranes and cell-cell junctions of tubular epithelial cells. Defects in the primary cilia are linked to a wide spectrum of human diseases, collectively termed ciliopathies. The most common phenotype shared by many ciliopathies is kidney cysts. PC1 and PC2 bind to each other via their respective C-terminal tails to form a receptor-channel complex and regulate each other’s function. The PC1/2 protein complex serves as a mechanosensor or chemical sensor and regulates calcium and G-protein signaling. The PC1/2 protein complex may also directly regulate a number of cellular functions including the cell cycle, the actin cytoskeleton, planar cell polarity (PCP), and cell migration. This protein complex has also been implicated in regulating a number of signaling pathways, including Wnt, mammalian target of rapamycin (mTOR), STAT3, cMET, phosphoinositide 3-kinase (PI3K)/AKT, G protein–coupled receptor (GPCR), and epidermal growth factor receptor (EGFR), as well as in the localization and activity of cystic fibrosis transmembrane conductance (CFTR). One hypothesis is that loss of ciliary function of PC1 and PC2 leads to reduced calcium signaling and a subsequent increase of adenylyl cyclase activity and decrease of phosphodiesterase activity, which, in turn, causes increased cellular cyclic AMP (cAMP). Increased cAMP promotes protein kinase A activity, among other effectors, and, in turn, leads to cyst growth by promoting proliferation and fluid secretion of cyst-lining cells through chloride and aquaporin channels in ADPKD kidneys.
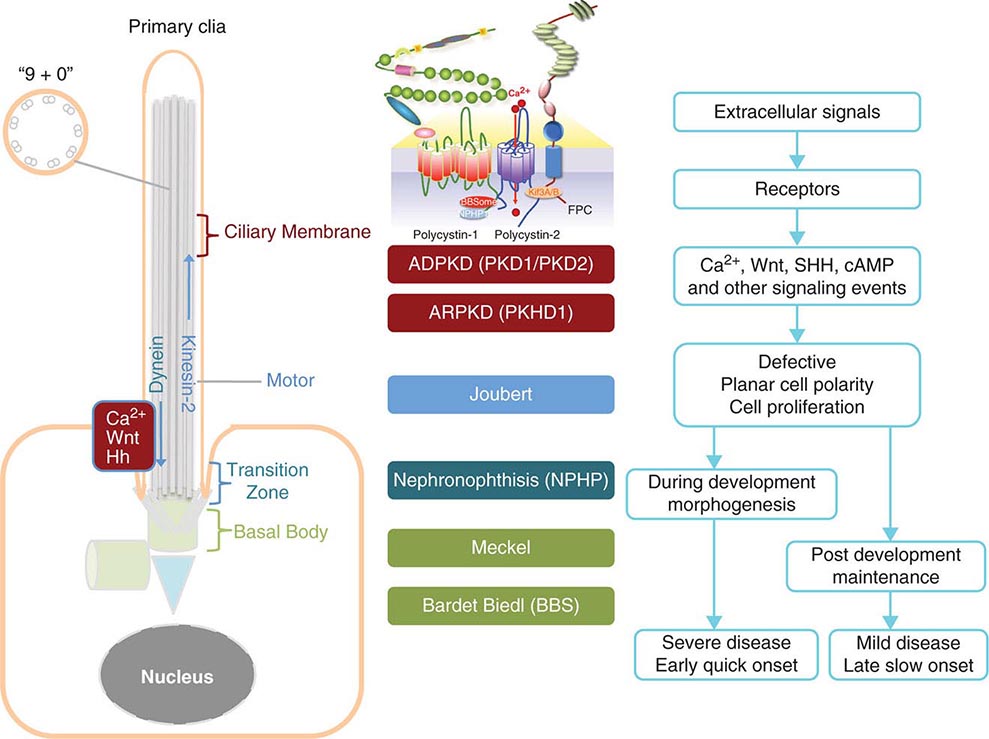
FIGURE 339-1 Scheme of the primary cilium and cystic kidney disease proteins. Left. A scheme of the primary cilium. Primary cilia share a “9+0” organization of microtubule doublets. Proteins are transported into the cilium by motor protein kinesin 2 and transported out of the cilium by dynein. The cilium is connected to the basal body through the transition zone. Middle. Topology of autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD) proteins polycystin-1, polycystin-2, and fibrocystin/polyductin (FPC) are shown. PC1 also interacts with other proteins such as components of the BBSome and NPHP1. PC2 and FPC both interact with kinesin 2 (KIF 3A/B). Localization of disease proteins in the cilium, the transition zone, and the basal body is color coded. Right. Potential disease mechanisms due to cilium-mediated signaling events.
![]() Genetic Considerations ADPKD is inherited as an autosomal dominant trait with complete penetrance but variable expressivity. The disease affects all ethnic groups worldwide with an estimated prevalence of 1:1000 to 1:400. Only half of the patients with ADPKD are clinically diagnosed during their lifetime. ADPKD is genetically heterogeneous. The first disease gene (PKD1) was localized to the region of the α-globin gene on chromosome 16p13 in 1985, and a second disease gene (PKD2) locus was mapped to chromosome 4q21-q23 in 1993. Mutations of PKD1 and PKD2 are responsible for ~85% and ~15% of ADPKD cases, respectively. However, patients with PKD2 mutations may be higher than 15% because they tend to have milder clinical disease and, as a result, may be underdiagnosed. Embryonic lethality of Pkd1 and Pkd2 knockout mice suggests that human homozygotes may be lethal and thus not clinically recognized.
Genetic Considerations ADPKD is inherited as an autosomal dominant trait with complete penetrance but variable expressivity. The disease affects all ethnic groups worldwide with an estimated prevalence of 1:1000 to 1:400. Only half of the patients with ADPKD are clinically diagnosed during their lifetime. ADPKD is genetically heterogeneous. The first disease gene (PKD1) was localized to the region of the α-globin gene on chromosome 16p13 in 1985, and a second disease gene (PKD2) locus was mapped to chromosome 4q21-q23 in 1993. Mutations of PKD1 and PKD2 are responsible for ~85% and ~15% of ADPKD cases, respectively. However, patients with PKD2 mutations may be higher than 15% because they tend to have milder clinical disease and, as a result, may be underdiagnosed. Embryonic lethality of Pkd1 and Pkd2 knockout mice suggests that human homozygotes may be lethal and thus not clinically recognized.
PKD1 is comprised of 46 exons occupying ~52 kb of genomic DNA. It produces an ~14-kb transcript that encodes PC1, a protein of ~4300 amino acids. A feature of the PKD1 gene is that the 5´ three-quarters of PKD1 have been duplicated at six other sites on chromosome 16p, and many of them produce mRNA transcripts, which provides a major challenge for genetic analysis of the duplicated region. PKD2 is a single-copy gene with 15 exons producing an ~5.3-kb mRNA transcript that encodes PC2, a protein of 968 amino acids. The presence of additional genes for ADPKD was suggested based on several families linked to neither PKD1 nor PKD2 genes. However, careful analyses have excluded the existence of a third ADPKD gene.
In ADPKD patients, every cell carries a germline mutant allele of either PKD1 or PKD2. However, cysts develop in only a small fraction of the nephrons. Cysts are thought to originate from clonal growth of single cells that have received a somatic “second hit” mutation in the “normal” allele of the PKD1 or PKD2 gene. Accumulating evidence in mouse models now shows that partial loss of function of the second allele of Pkd1 in a proliferative environment is sufficient for cystogenesis, suggesting that a critical amount of PKD1 is needed in a cell. Somatic inactivation of the second allele of Pkd1 in adult mice results in very slow onset of cyst development in the kidney, but a “third hit,” such as an additional genetic or epigenetic event, the inactivation of a growth-suppressor gene, the activation of a growth-promoting gene(s), or an event like renal injury that activates the developmental program, may promote rapid cyst formation.
Clinical Manifestations ADPKD is characterized by the progressive bilateral formation of renal cysts. Focal renal cysts are typically detected in affected subjects before 30 years of age. Hundreds to thousands of cysts are usually present in the kidneys of most patients in the fifth decade (Fig. 339-2). Enlarged kidneys can each reach a fourfold increase in length and weigh up to 20 times the normal weight. The clinical presentations of ADPKD are highly variable. Although many patients are asymptomatic until the fourth to fifth decade of life and are diagnosed by incidental discoveries of hypertension or abdominal masses, back or flank pain is a frequent symptom in ~60% of patients with ADPKD. The pain may result from renal cyst infection, hemorrhage, or nephrolithiasis. Gross hematuria resulting from cyst rupture occurs in ~40% of patients during the course of their disease, and many of them will have recurrent episodes. Flank pain and hematuria may coexist if the cyst that ruptures is connected with the collecting system. Proteinuria is usually a minor feature of ADPKD. Infection is the second most common cause of death for patients with ADPKD. Up to half of patients with ADPKD will have one or more episodes of renal infection during their lifetime. An infected cyst and acute pyelonephritis are the most common renal infections often due to gram-negative bacteria, which are associated with fever and flank pain, with or without bacteremia. These complications and renal insufficiency often correlate with structural abnormality of the renal parenchyma. Kidney stones occur in ~20% of patients with ADPKD. Different from the general population, more than half of the stones in patients with ADPKD are composed of uric acid, with the remainder due to calcium oxalate. Distal acidification defects, abnormal ammonium transport, low urine pH, and hypocitraturia may be important in the pathogenesis of renal stones in ADPKD. Renal cell carcinoma is a rare complication of ADPKD with no apparent increased frequency compared to the general population. However, in ADPKD, these tumors are more often bilateral at presentation, multicentric, and sarcomatoid in type. Radiologic imaging is often not helpful in distinguishing cyst infection and cyst hemorrhage because of their complexity. Computed tomography (CT) scan and magnetic resonance imaging (MRI) are often useful in distinguishing a malignancy from a complex cyst. Cardiovascular complications are the major cause of mortality in patients with ADPKD. Hypertension is common and typically occurs before any reduction in glomerular filtration rate (GFR). Hypertension is a risk factor for both cardiovascular and kidney disease progression in ADPKD. Notably, some normotensive patients with ADPKD may also have left ventricular hypertrophy. Hypertension in ADPKD may result from the increased activation of the renin-angiotensin-aldosterone system, increased sympathetic nerve activity, and impaired endothelial cilium function-dependent relaxation of small resistant blood vessels.
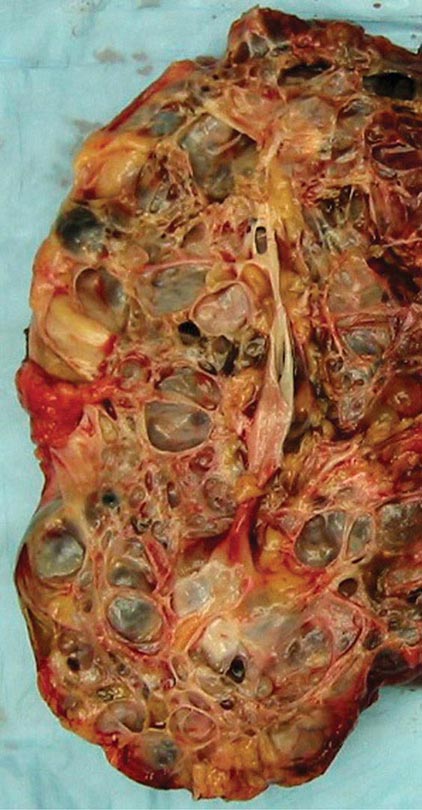
FIGURE 339-2 Photograph showing a kidney from a patient with autosomal dominant polycystic kidney disease. The kidney has been cut open to expose the parenchyma and internal aspects of cysts.
The progression of ADPKD has striking inter- and intrafamilial variability. The disease can present as early as in utero, but end-stage renal disease typically occurs in late middle age. Risk factors include early diagnosis of ADPKD, hypertension, gross hematuria, multiple pregnancies, and large kidney size. Liver cysts derived from the biliary epithelia are the most common extrarenal complication. Polycystic liver disease associated with ADPKD is different from autosomal dominant polycystic liver disease (ADPLD), which is caused by mutations in at least two distinct genes (PRKCSH and SEC63) and does not progress to renal failure. Massive polycystic liver disease occurs almost exclusively in women with ADPKD, particularly those with multiple pregnancies.
Intracranial aneurysm (ICA) occurs four to five times more frequently in ADPKD patients than in the general population and causes high mortality. The disease gene products PC1 and PC2 may be directly responsible for defects in arterial smooth muscle cells and myofibroblasts. The focal nature and the natural history of ICA in ADPKD remain unclear. A family history of ICA is a risk factor of aneurysm rupture in ADPKD, but whether hypertension and cigarette smoking are independent risk factors is not clear. About 20–50% of patients may experience “warning headaches” preceding the index episode of subarachnoid hemorrhage due to ruptured ICA. A CT scan is generally used as the first diagnostic test. A lumbar puncture may be used to confirm the diagnosis. The role of radiologic screening for ICA in asymptomatic patients with ADPKD remains unclear. ADPKD patients with a positive family history of ICAs may undergo presymptomatic screening of ICAs by magnetic resonance angiography. Other vascular abnormalities in ADPKD patients include diffuse arterial dolichoectasias of the anterior and posterior cerebral circulation, which can predispose to arterial dissection and stroke. Mitral valve prolapse occurs in up to 30% of patients with ADPKD, and tricuspid valve prolapse is less common. Other valvular abnormalities occurring with increased frequency in ADPKD patients include insufficiency of the mitral, aortic, and tricuspid valves. Most patients are asymptomatic, but some may progress and require valve replacement. The prevalence of colonic diverticulae and abdominal wall hernias is also increased in ADPKD patients.
Diagnosis Diagnosis is typically made from a positive family history consistent with autosomal dominant inheritance and multiple kidney cysts bilaterally. Renal ultrasonography is often used for presymptomatic screening of at-risk subjects and for evaluation of potential living-related kidney donors from ADPKD families. The presence of at least two renal cysts (unilateral or bilateral) is sufficient for diagnosis among at-risk subjects between 15 and 29 years of age with a sensitivity of 96% and specificity of 100%. The presence of at least two cysts in each kidney and the presence at least four cysts in each kidney are required for the diagnosis of at-risk subjects age 30 to 59 years and age 60 years or older, respectively, with a sensitivity of 100% and specificity of 100%. This is because there is an increased frequency of developing simple renal cysts with age. Conversely, in subjects between age 30 and 59 years, the absence of at least two cysts in each kidney, which is associated with a false-negative rate of 0%, can be used for disease exclusion. These criteria have a lower sensitivity for patients with a PKD2 mutation because of a late onset of ADPKD2. CT scan and T2-weighted MRI, with and without contrast enhancement, are more sensitive than ultrasonography and can detect cysts of smaller size. However, a CT scan exposes the patient to radiation and radiocontrast, which may cause serious allergic reactions and nephrotoxicity in patients with renal insufficiency. T2-weighted MRI, with gadolinium as a contrast agent, has minimal renal toxicity and can detect cysts of only 2–3 mm in diameter. However, a large majority of cysts may still be below the detection level. Genetic testing by linkage analyses and mutational analyses is available for ambiguous cases. Because of the large size of the PKD1 gene and the presence of multiple highly homologous pseudogenes, mutational analysis of the PKD1 gene is difficult and costly. Application of new technologies, such as paired-end next-generation sequencing with multiplexing individually bar-coded long-range polymerase chain reaction libraries, may reduce the costs and improve the sensitivity for clinical genetic testing.
AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE
![]() Genetic Considerations ARPKD is a significant hereditary renal disease in childhood, with an estimated prevalence of 1 in 20,000 live births. A carrier frequency of up to 1:70 has been reported. Mutations in a single gene, PKHD1, are responsible for all the clinical presentations of ARPKD. PKHD1, localized on human chromosome region 6p21.1-6p12.2, is one of the largest genes in the genome, occupies ~450 kb of DNA, and contains at least 86 exons. It produces multiple alternatively spliced transcripts. The largest transcript encodes fibrocystin/polyductin (FPC), which is a large receptor-like integral membrane protein of 4074 amino acids. FPC has a single transmembrane, a large N-terminal extracellular region, and a short intracellular cytoplasmic domain. FPC is localized on the primary cilia of epithelia cells of cortical and medullary collecting ducts and cholangiocytes of bile ducts, similar to polycystins and several other ciliopathy proteins. FPC is also expressed on the basal body and plasma membrane. The large extracellular domain of FPC is presumed to bind to an as yet unknown ligand(s) and is involved in cell-cell and cell-matrix interactions. FPC interacts with ADPKD protein PC2 and may also participate in regulation of the mechanosensory function of the primary cilia, calcium signaling, and PCP, suggesting a common mechanism underlying cystogenesis between ADPKD and ARPKD. FPC is also found on the centrosomes and mitotic spindle and may regulate centrosome duplication and mitotic spindle assembly during cell division. A large number of various mutations have been found throughout PKHD1 and are unique to individual families. Most patients are compound heterozygotes for PKHD1 mutations. Patients with two truncation mutations appear to have an earlier onset of the disease.
Genetic Considerations ARPKD is a significant hereditary renal disease in childhood, with an estimated prevalence of 1 in 20,000 live births. A carrier frequency of up to 1:70 has been reported. Mutations in a single gene, PKHD1, are responsible for all the clinical presentations of ARPKD. PKHD1, localized on human chromosome region 6p21.1-6p12.2, is one of the largest genes in the genome, occupies ~450 kb of DNA, and contains at least 86 exons. It produces multiple alternatively spliced transcripts. The largest transcript encodes fibrocystin/polyductin (FPC), which is a large receptor-like integral membrane protein of 4074 amino acids. FPC has a single transmembrane, a large N-terminal extracellular region, and a short intracellular cytoplasmic domain. FPC is localized on the primary cilia of epithelia cells of cortical and medullary collecting ducts and cholangiocytes of bile ducts, similar to polycystins and several other ciliopathy proteins. FPC is also expressed on the basal body and plasma membrane. The large extracellular domain of FPC is presumed to bind to an as yet unknown ligand(s) and is involved in cell-cell and cell-matrix interactions. FPC interacts with ADPKD protein PC2 and may also participate in regulation of the mechanosensory function of the primary cilia, calcium signaling, and PCP, suggesting a common mechanism underlying cystogenesis between ADPKD and ARPKD. FPC is also found on the centrosomes and mitotic spindle and may regulate centrosome duplication and mitotic spindle assembly during cell division. A large number of various mutations have been found throughout PKHD1 and are unique to individual families. Most patients are compound heterozygotes for PKHD1 mutations. Patients with two truncation mutations appear to have an earlier onset of the disease.
Clinical Features Classic ARPKD is generally diagnosed in utero or within the neonatal period and characterized by greatly enlarged echogenic kidneys in diseased fetuses. Reduced fetal urine production may contribute to oligohydramnios and pulmonary hypoplasia. About 30% of affected neonates die shortly after birth due to respiratory insufficiency. Close to 60% of mortality occurs within the first month of life. In the classic group, most patients are born with renal insufficiency and ESRD. However, infants often have a transient improvement in their GFR; death from renal insufficiency at this stage is rare. Some patients are diagnosed after the neonatal stage and form the older group. Morbidity and mortality in this group often involve systemic hypertension, progressive renal insufficiency, and liver manifestations. The hallmarks of ARPKD liver disease are biliary dysgenesis due to a primary ductal plate malformation with associated periportal fibrosis, namely congenital hepatic fibrosis (CHF) and dilatation of intrahepatic bile ducts (Caroli’s disease). CHF and Caroli’s disease can then lead to portal hypertension exhibiting hepatosplenomegaly, variceal bleeding, and cholangitis. Some patients with the diagnosis of ARPKD at 1 year of age with nephromegaly exhibit slowly declining renal function over 20 years with only minimally enlarged kidneys at ESRD and markedly atrophic kidneys following renal transplantation. The slow progression of renal disease is likely due to increasing fibrosis rather than the development of cysts. Systemic hypertension is common in all ARPKD patients, even those with normal renal function.
Diagnosis Ultrasonography, CT, and MRI all can be used for diagnosis. Ultrasonography reveals large, echogenic kidneys with poor corticomedullary differentiation. The diagnosis can be made in utero after 24 weeks of gestation in severe cases. Macrocysts generally are not common at birth in ARPKD patients. The absence of renal cysts in either parent, particularly if they are more than 40 years of age on ultrasonography, helps distinguish ARPKD from ADPKD in older patients. Clinical, laboratory, or radiographic evidence of hepatic fibrosis, hepatic pathology demonstrating characteristic ductal plate abnormalities, family history of affected siblings, or parental consanguinity suggestive of autosomal recessive inheritance is helpful. The lack of mutational hotspots and the large and complex genomic structure of PKHD1 make molecular diagnosis difficult; however, presymptomatic screening of other at-risk members in a family with already identified ARPKD mutations is straightforward and inexpensive.
OTHER DISEASES CHARACTERIZED BY LARGE KIDNEY CYSTS
TUBEROUS SCLEROSIS
Tuberous sclerosis (TS) is a rare autosomal dominant syndrome caused by mutations in one of two genes, TSC1, encoding hamartin, or, TSC2, encoding tuberin. Published estimates of prevalence vary widely, but it certainly occurs in less than 1:5000 births. Kidney cysts are a frequent feature of this condition, as are two other abnormalities of kidney growth, renal cell carcinoma and renal angiomyolipomas. TS is a syndrome affecting multiple organ systems. Other features of TS include benign growths in the nervous system, eyes, heart, lung, liver, and skin. Essentially all TS patients have associated skin lesions, and a large proportion of patients have neurologic and cognitive manifestations. The TSC2 gene is adjacent to PKD1 in the human genome. Some patients have deletions in their genomic DNA that inactivate these two genes. Such individuals may have manifestations of both ADPKD and TS.
The most common kidney finding in TS is the presence of angiomyolipomas. These growths tend to be multiple and bilateral. Although they are usually benign, they may bleed. Surgical removal is often recommended as a prophylactic measure in people with angiomyolipomas larger than 4 cm in diameter. The cysts in TS are radiographically similar to those seen in ADPKD. In contrast to ADPKD, there is a clearly increased risk of renal cell carcinoma in TS patients. Regular periodic imaging is recommended in TS patients with kidney involvement to screen for the development of renal cell carcinoma.
Although not common, TS may lead to significant chronic kidney disease (CKD) and progress to end-stage kidney failure. Patients with TS and CKD typically have an unremarkable urine sediment and only minimal to mild amounts of proteinuria.
Mechanistically, the TSC1 and TSC2 gene products tuberin and hamartin interact physically. This protein complex is localized to the base of the cilia and inhibits intracellular signaling processes mediated by mTOR, leading to abnormal growth in a number of tissues. Investigation of mTOR inhibitors as therapy for TS is ongoing.
VON HIPPEL-LINDAU DISEASE
Von Hippel-Lindau disease (VHL) is an inherited cancer syndrome with renal manifestations. VHL is an autosomal dominant condition caused by mutations in the VHL tumor-suppressor gene. VHL is localized to the primary cilia and is necessary for the formation of primary cilia. Like many autosomal dominant cancer syndromes, VHL is recessive at the cellular level: a somatic mutation in the second VHL allele leads to loss of VHL in the cell and abnormal growth. Kidney manifestations of VHL include multiple bilateral kidney cysts and renal cell carcinomas. Kidney cysts and carcinoma affect the majority of VHL patients. Nonrenal features of VHL include pheochromocytomas, cerebellar hemangioblastomas, and retinal hemangiomas.
Annual screening of the kidneys by imaging with CT or MRI is recommended for early detection of renal cell carcinomas. Increasingly, nephron-sparing surgical approaches are being used for removal of cancerous lesions in order to preserve kidney function.
OTHER INHERITED DISEASES OF TUBULE GROWTH AND DEVELOPMENT
ADPKD is by far the most common adult-onset, single-gene form of kidney disease. The large cysts that are sometimes seen in VHL and TS are similar in appearance to the cysts seen in ADPKD. A variety of other inherited disorders affecting primarily tubule and renal interstitial function can lead to CKD and eventual end-stage kidney disease in the absence of large tubule-derived cysts.
Inherited diseases affecting the tubulointerstitial compartment of the kidney can lead to secondary glomerular stress and glomerulosclerosis with some degree of concomitant proteinuria. Similarly, disorders of glomerular function will typically lead to secondary interstitial fibrosis and tubule atrophy. From a clinical perspective, therefore, distinguishing between a genetic disease of the renal tubules and a disease of the glomerulus may not be easy, particularly in the absence of a gross phenotype such as large kidney cysts.
MEDULLARY CYSTIC KIDNEY DISEASE (AUTOSOMAL DOMINANT INTERSTITIAL KIDNEY DISEASE)
The medullary cystic kidney diseases (MCKD) are autosomal dominant disorders. Despite the nosology, kidney cysts are not invariably present. Older literature often grouped MCKD together with the childhood-onset disorders known as the nephronophthises, but these are distinct clinical and genetic entities.
Medullary Cystic Kidney Disease Type I Patients with MCKD type I (MCKD I) have mutations in the mucin 1 gene MUC1. In contrast to MCKD type II (MCKD II) patients, individuals with MCKD I do not have elevated uric acid levels. The disease-causing MUC1 mutations that have been reported all alter a repeat region within the MUC1 gene, leading to a large “neoprotein” fragment that may lead to toxic effects on the kidney tubule.
Clinically, patients with MCKD I exhibit slowly progressive CKD in adulthood, with only minimal amounts of increased urine protein and occasional renal cysts seen on ultrasound examination. Kidney histology shows tubulointerstitial fibrosis and tubular atrophy. The mechanisms by which MUC1 mutations cause human kidney disease are not known.
Medullary Cystic Kidney Disease Type II MCKD II is caused by mutations in the UMOD gene, which encodes the protein uromodulin, also known as Tamm-Horsfall protein. Uromodulin is also found on the centrosome, the mitotic spindle, and the primary cilia; it colocalizes with nephrocystin-1 and KIF3A on the cilia. UMOD mutations also cause the conditions that have been referred to as familial juvenile hyperuricemic nephropathy (HNFJ1) and glomerulocystic kidney disease (GCKD), although it is not clear that these different names represent clearly distinct disorders. The term uromodulin-associated kidney disease (UAKD) has been suggested as a better name for MCKD II and the various other related UMOD-associated diseases. Despite the name, kidney cysts are not a common feature of MCKD II. MCKD II should be suspected clinically in patients with a family history of late-onset kidney disease, benign urine sediments, absence of significant proteinuria, and hyperuricemia. Large genome-wide association studies have suggested that certain common noncoding sequence variants in UMOD are associated with a moderately increased risk of CKD in the general population.
Other Forms of Familial Tubulointerstitial Kidney Disease A small number of families have been identified with autosomal dominant tubulointerstitial kidney disease and hyperuricemia who lack UMOD mutations. Some of these families carry disease-segregating mutations in the renin gene REN. There are other families who lack mutations in UMOD, MUC1, or REN. Thus, mutations in other yet-to-be identified genes are able to produce similar interstitial kidney disease, both with and without hyperuricemia.
Kidney biopsies in patients with any of the various forms of MCKD typically show interstitial fibrosis. These histologic features are not diagnostic of any particular genetic entity, and the specific diagnosis must be made by other means. Genetic tests for alterations in specific genes are increasingly available in the clinical setting.
Patients with autosomal dominant interstitial kidney disease, UMOD or REN mutations, or hyperuricemia and gout should be treated similarly to others with these findings, with uric acid–lowering agents, such allopurinol or febuxostat.
NEPHRONOPHTHISIS
A large and growing number of genetically distinct but related autosomal recessive disorders are referred to as nephronophthises. These should not be confused with the adult-onset autosomal dominant medullary cystic kidney diseases discussed above, despite the often confusing nomenclature seen in older medical literature. Nephronophthisis is quite rare but is nevertheless the most common inherited childhood form of kidney failure requiring kidney replacement therapy.
Like ADPKD and ARPKD, the various genetically heterogeneous entities that fall under the category of nephronophthisis (NPHP) are disorders of ciliary function. Mutations in a very large number of genes have been identified that lead to NPHP under an autosomal recessive pattern of inheritance. The various forms of NPHP share common features, including tubulointerstitial fibrosis, corticomedullary cysts, and progressive CKD, leading to renal failure. Proteinuria is absent or mild, and the urine sediment is not active.
NPHP is often divided into infantile, juvenile, and adolescent forms. The juvenile form is the most frequent and usually caused by mutations in the NPHP2 gene. The infantile form, usually caused by NPHP2 mutations, is associated with end-stage kidney failure in early childhood. Patients with the adolescent form of NPHP typically develop end-stage kidney failure in early adulthood. The products of the NPHP genes are referred to as nephrocystins. NPHP1 through NPHP16 have been reported; some are referred to by other names as well.
NPHP can present as an isolated finding or be part of several multiorgan syndromes. Neurologic abnormalities are present in a significant number of patients. Bone and liver abnormities are seen in some NPHP patients. Senior-Løken syndrome is defined by the presence of NPHP with retinitis pigmentosa. Joubert’s syndrome is defined by multiple neurologic findings, including hypoplasia of the cerebellar vermis. Some forms of this genetically heterogeneous syndrome include NPHP as a component.
The multisystem disease Bardet-Biedl syndrome (BBS) is defined clinically by a spectrum of features, including truncal obesity, cognitive impairment, retinal dystrophy, polydactyly, developmental urogenital abnormalities, and kidney cysts. The kidney phenotype is NPHP-like, with small cysts deriving from the tubules, tubulointerstitial and often secondary glomerular disease, and urine concentrating defects. There are 18 BBS genes cloned. BBS follows autosomal recessive inheritance. Like ADPKD, ARPKD, and NPHP, BBS is a disease of abnormal ciliary function.
The multiple genes and gene products (nephrocystins) that are responsible for NPHP are expressed in cilia, basal bodies, and the centrosomes of kidney tubule cells. It has been hypothesized that all of the NPHP gene defects lead to a clinical phenotype by interfering with the regulation of PCP.
There are no specific clinical tests that define NPHP. Genetic diagnosis is possible but cumbersome because of the large number of genes that can be responsible. There are no specific therapies for NPHP. Rather, therapy is aimed at treating signs of these diseases as well as the systemic abnormalities seen with all CKDs. Chronic dialysis or kidney transplantation is eventually required for NPHP-affected individuals.
KARYOMEGALIC TUBULOINTERSTITIAL NEPHRITIS
Karyomegalic tubulointerstitial nephritis is an exceptionally rare form of kidney disease with adult-onset progressive kidney failure. Kidney biopsy shows chronic tubulointerstitial nephritis, as well as interstitial fibrosis. This is a recessive disorder caused by inheritance of two mutant copies of the FAN1 gene. FAN1 encodes a component of a DNA repair machinery complex. Individuals with two mutant FAN1 genes are genetically sensitized to the effect of DNA damage. Kidney histology shows karyomegaly in addition to the nonspecific findings of interstitial fibrosis and tubular atrophy.
MEDULLARY SPONGE KIDNEY
Medullary sponge kidney (MSK) is often grouped together with inherited disorders of the kidney affecting tubule growth and development, although it is usually a sporadic finding rather than an inherited phenotype. MSK is caused by developmental malformation and cystic dilatation of the renal collecting ducts. The medullary cysts seen in this entity can be quite variable in size.
MSK is usually a benign entity. The diagnosis of MSK is often made incidentally. In the past, the diagnosis of MSK was often made by intravenous pyelography (IVP). CT scans, which have replaced IVPs for much routine kidney imaging, are not as sensitive in detecting MSK.
MSK is associated with an increased frequency of calcium phosphate and calcium oxalate kidney stones. Altered flow characteristics in the kidney tubules may lead to the development of formation of a nidus for stone formation. Kidney stones in this group are treated the same as are kidney stones in the general population. MSK patients also often exhibit reduced kidney concentrating ability and an increased frequency of urinary tract infections.
CONGENITAL ABNORMALITIES OF THE KIDNEY AND URINARY TRACT
The structural abnormalities known as the congenital abnormalities of the kidney and urinary tract (CAKUTs) are a group of etiologically and phenotypically heterogeneous disorders. Some form of CAKUT is estimated to occur in up to 1 in 500 live births. Specific abnormalities classified as part of the CAKUT spectrum include kidney hypoplasia, kidney agenesis, ureteropelvic junction obstruction, and vesicoureteral reflux.
CAKUT can be the cause of clinically significant problems in both adults and children. However, it is a major contributor to kidney failure in children, accounting for more than one-third of end-stage kidney disease in this group.
CAKUT is typically a sporadic finding but can also cluster in families. Familial forms can be observed as parts of multisystem developmental syndromes. A growing list of specific genes have been identified, which when mutated lead to syndromic forms of CAKUT. For example, the branchio-oto-renal syndrome, characterized by developmental abnormalities in the neck, ears, and kidney, can be caused by mutations in the EYA1 and SIX1 genes. Mutations in the PAX2 transcription factor gene can cause the autosomal dominant renal coloboma syndrome, characterized by optic nerve malformations and hypoplastic kidneys.
In many instances, CAKUT is caused by environmental influences rather than genetic alterations. For example, renal tubular dysgenesis, defined by altered tubule development, can be caused by prenatal exposure to angiotensin-converting enzyme inhibitors or angiotensin receptor blockers.
MITOCHONDRIAL DISEASE
Inherited disorders of the mitochondrial genome (discussed elsewhere in this text [Chap. 85e]) commonly affect kidney function. Thirteen of the genes involved in encoding components of the mitochondrial respiratory chain are located on the mitochondrial genome that is inherited maternally. The remainder of these components is encoded by the nuclear genome. These defects of oxidative phosphorylation may affect multiple organs and tissues.
Neuromuscular disease is the best recognized part of this complex phenotype. Kidney disease is now recognized as a common component as well. Tubulointerstitial disease may be seen on kidney biopsy, and progression to kidney failure may occur. Glomerular involvement, manifest as proteinuria and glomerulosclerosis, can also develop. Changes in proximal tubule activity are the most common renal phenotype. Patients may have several defects in proximal tubule transport, including the Fanconi syndrome. Some patients may also have acidosis, hypophosphatemic rickets, hypercalciuria, glycosuria, and tubular proteinuria. Decreased urine concentrating ability is common.
GLOBAL CONSIDERATIONS
![]() The disorders discussed above are all seen worldwide. In addition, a previously unrecognized epidemic of kidney disease is leading to very high rates of kidney failure in and near the western coast of Central America. This Mesoamerican nephropathy is particularly common in Nicaragua and El Salvador. Mesoamerican nephropathy patients do not have significant proteinuria, suggesting that this is a disease of the kidney tubules and interstitium. The cause is unknown, but some have suggested that a combination of toxic environmental factors and heat stress underlies the development of this kidney disease, which has a striking male predominance. However, the fact that, in many families, a large fraction of the men are affected with kidney disease has suggested that a strong genetic component may be involved as well.
The disorders discussed above are all seen worldwide. In addition, a previously unrecognized epidemic of kidney disease is leading to very high rates of kidney failure in and near the western coast of Central America. This Mesoamerican nephropathy is particularly common in Nicaragua and El Salvador. Mesoamerican nephropathy patients do not have significant proteinuria, suggesting that this is a disease of the kidney tubules and interstitium. The cause is unknown, but some have suggested that a combination of toxic environmental factors and heat stress underlies the development of this kidney disease, which has a striking male predominance. However, the fact that, in many families, a large fraction of the men are affected with kidney disease has suggested that a strong genetic component may be involved as well.
340 |
Tubulointerstitial Diseases of the Kidney |
Inflammation or fibrosis of the renal interstitium and atrophy of the tubular compartment are common consequences of diseases that target the glomeruli or vasculature. Distinct from these secondary phenomena, however, are a group of disorders that primarily affect the tubules and interstitium, with relative sparing of the glomeruli and renal vessels. Such disorders are conveniently divided into acute and chronic tubulointerstitial nephritis (TIN) (Table 340-1).
|
CLASSIFICATION OF THE CAUSES OF TUBULOINTERSTITIAL DISEASES OF THE KIDNEY |
Abbreviations: CMV, cytomegalovirus; COX, cyclooxygenase; EBV, Epstein-Barr virus.
Acute TIN most often presents with acute renal failure (Chap. 334). The acute nature of this group of disorders may be caused by aggressive inflammatory infiltrates that lead to tissue edema, tubular cell injury, and compromised tubular flow, or by frank obstruction of the tubules with casts, cellular debris, or crystals. There is sometimes flank pain due to distention of the renal capsule. Urinary sediment is often active with leukocytes and cellular casts, but depends on the exact nature of the disorder in question.
The clinical features of chronic TIN are more indolent and may manifest with disorders of tubular function, including polyuria from impaired concentrating ability (nephrogenic diabetes insipidus), defective proximal tubular reabsorption leading to features of Fanconi’s syndrome (glycosuria, phosphaturia, aminoaciduria, hypokalemia, and type II renal tubular acidosis [RTA] from bicarbonaturia), or non-anion-gap metabolic acidosis and hyperkalemia (type IV RTA) due to impaired ammoniagenesis, as well as progressive azotemia (rising creatinine and blood urea nitrogen [BUN]). There is often modest proteinuria (rarely >2 g/d) attributable to decreased tubular reabsorption of filtered proteins; however, nephrotic-range albuminuria may occur in some conditions due to the development of secondary focal segmental glomerulosclerosis (FSGS). Renal ultrasonography may reveal changes of “medical renal disease,” such as increased echogenicity of the renal parenchyma with loss of corticomedullary differentiation, prominence of the renal pyramids, and cortical scarring in some conditions. The predominant pathology in chronic TIN is interstitial fibrosis with patchy mononuclear cell infiltration and widespread tubular atrophy, luminal dilation, and thickening of tubular basement membranes. Because of the nonspecific nature of the histopathology, biopsy specimens rarely provide a specific diagnosis. Thus, diagnosis relies on careful analysis of history, drug or toxin exposure, associated symptoms, and imaging studies.
ACUTE INTERSTITIAL NEPHRITIS
In 1897, Councilman reported on eight cases of acute interstitial nephritis (AIN) in the Medical and Surgical Reports of the Boston City Hospital; three as a postinfectious complication of scarlet fever and two from diphtheria. Later, he described the lesion as “an acute inflammation of the kidney characterized by cellular and fluid exudation in the interstitial tissue, accompanied by, but not dependant on, degeneration of the epithelium; the exudation is not purulent in character, and the lesions may be both diffuse and focal.” Today AIN is far more often encountered as an allergic reaction to a drug (Table 340-1). Immune-mediated AIN may also occur as part of a known autoimmune syndrome, but in some cases there is no identifiable cause despite features suggestive of an immunologic etiology (Table 340-1).
ALLERGIC INTERSTITIAL NEPHRITIS
Although biopsy-proven AIN accounts for no more than ~15% of cases of unexplained acute renal failure, this is likely a substantial underestimate of the true incidence. This is because potentially offending medications are more often identified and empirically discontinued in a patient noted to have a rising serum creatinine, without the benefit of a renal biopsy to establish the diagnosis of AIN.
Clinical Features The classic presentation of AIN, namely, fever, rash, peripheral eosinophilia, and oliguric renal failure occurring after 7–10 days of treatment with methicillin or another β-lactam antibiotic, is the exception rather than the rule. More often, patients are found incidentally to have a rising serum creatinine or present with symptoms attributable to acute renal failure (Chap. 334). Atypical reactions can occur, most notably nonsteroidal anti-inflammatory drug (NSAID)-induced AIN, in which fever, rash, and eosinophilia are rare, but acute renal failure with heavy proteinuria is common. A particularly severe and rapid-onset AIN may occur upon reintroduction of rifampin after a drug-free period. More insidious reactions to the agents listed in Table 340-1 may lead to progressive tubulointerstitial damage. Examples include proton pump inhibitors and, rarely, sulfonamide and 5-aminosalicylate (mesalazine and sulfasalazine) derivatives and antiretrovirals.
Diagnosis Finding otherwise unexplained renal failure with or without oliguria and exposure to a potentially offending agent usually points to the diagnosis. Peripheral blood eosinophilia adds supporting evidence but is present in only a minority of patients. Urinalysis reveals pyuria with white blood cell casts and hematuria. Urinary eosinophils are neither sensitive nor specific for AIN; therefore, testing is not recommended. Renal biopsy is generally not required for diagnosis but reveals extensive interstitial and tubular infiltration of leukocytes, including eosinophils.
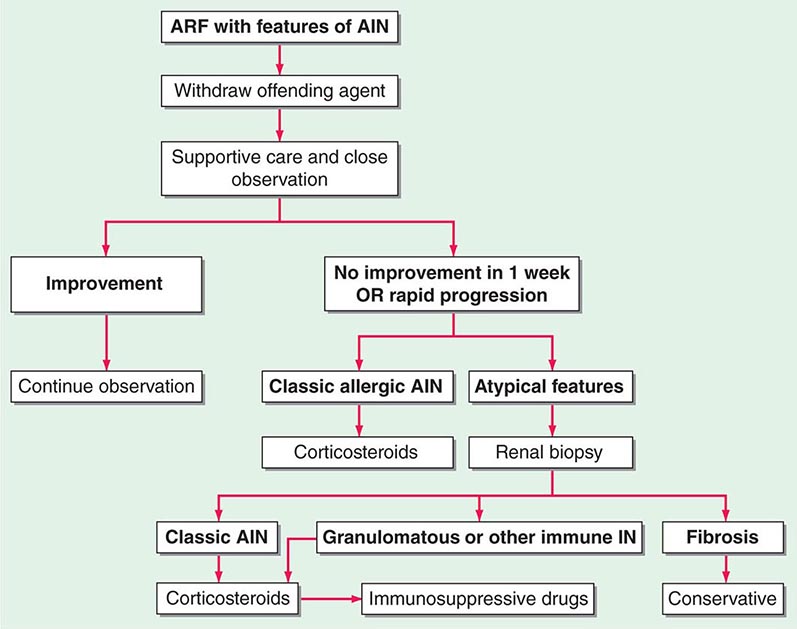
FIGURE 340-1 Algorithm for the treatment of allergic and other immune-mediated acute interstitial nephritis (AIN). ARF, acute renal failure; IN, interstitial nephritis. See text for immunosuppressive drugs used for refractory or relapsing AIN. (Modified from S Reddy, DJ Salant: Ren Fail 20:829, 1998.)
|
INDICATIONS FOR CORTICOSTEROIDS AND IMMUNOSUPPRESSIVES IN INTERSTITIAL NEPHRITIS |
Abbreviations: AIN, acute interstitial nephritis; SLE, systemic lupus erythematosus; TINU, tubulointerstitial nephritis with uveitis.
Source: Modified from S Reddy, DJ Salant: Ren Fail 20:829, 1998.
SJÖGREN’S SYNDROME
Sjögren’s syndrome is a systemic autoimmune disorder that primarily targets the exocrine glands, especially the lacrimal and salivary glands, and thus results in symptoms, such as dry eyes and mouth, that constitute the “sicca syndrome” (Chap. 383). Tubulointerstitial nephritis with a predominant lymphocytic infiltrate is the most common renal manifestation of Sjögren’s syndrome and can be associated with distal RTA, nephrogenic diabetes insipidus, and moderate renal failure. Diagnosis is strongly supported by positive serologic testing for anti-Ro (SS-A) and anti-La (SS-B) antibodies. A large proportion of patients with Sjögren’s syndrome also have polyclonal hypergammaglobulinemia. Treatment is initially with glucocorticoids, although patients may require maintenance therapy with azathioprine or mycophenolate mofetil to prevent relapse (Fig. 340-1 and Table 340-2).
TUBULOINTERSTITIAL NEPHRITIS WITH UVEITIS (TINU)
TINU is a systemic autoimmune disease of unknown etiology. It accounts for fewer than 5% of all cases of AIN, affects females three times more often than males, and has a median age of onset of 15 years. Its hallmark feature, in addition to a lymphocyte-predominant interstitial nephritis (Fig. 340-2), is a painful anterior uveitis, often bilateral and accompanied by blurred vision and photophobia. Diagnosis is often confounded by the fact that the ocular symptoms precede or accompany the renal disease in only one-third of cases. Additional extrarenal features include fever, anorexia, weight loss, abdominal pain, and arthralgia. The presence of such symptoms as well as elevated creatinine, sterile pyuria, mild proteinuria, features of Fanconi’s syndrome, and elevated erythrocyte sedimentation rate should raise suspicion for this disorder. Serologies suggestive of the more common autoimmune diseases are usually negative, and TINU is often a diagnosis of exclusion after other causes of uveitis and renal disease, such as Sjögren’s syndrome, Behçet’s disease, sarcoidosis, and systemic lupus erythematosus, have been considered. Clinical symptoms are typically self-limited in children, but are more apt to follow a relapsing course in adults. The renal and ocular manifestations generally respond well to oral glucocorticoids, although maintenance therapy with agents such as methotrexate, azathioprine, or mycophenolate may be necessary to prevent relapses (Fig. 340-1 and Table 340-2).
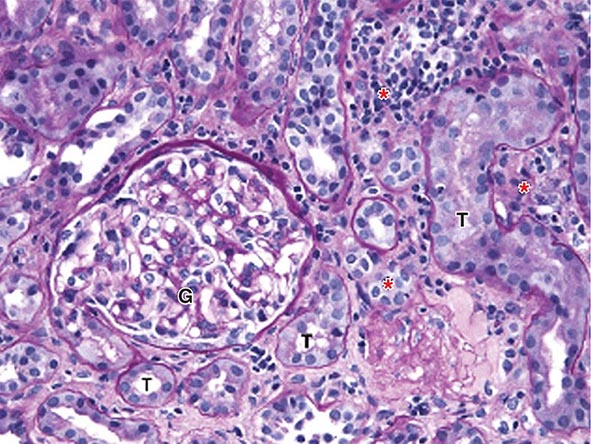
FIGURE 340-2 Acute interstitial nephritis (AIN) in a patient who presented with acute iritis, low-grade fever, erythrocyte sedimentation rate of 103, pyuria and cellular casts on urinalysis, and a newly elevated serum creatinine of 2.4 mg/dL. Both the iritis and AIN improved after intravenous methylprednisolone. This PAS-stained renal biopsy shows a mononuclear cell interstitial infiltrate (asterisks) and edema separating the tubules (T) and a normal glomerulus (G). Some of the tubules contain cellular debris and infiltrating inflammatory cells. The findings in this biopsy are indistinguishable from those that would be seen in a case of drug-induced AIN. PAS, Periodic acid–Schiff.
SYSTEMIC LUPUS ERYTHEMATOSUS
An interstitial mononuclear cell inflammatory reaction often accompanies the glomerular lesion in most cases of class III or IV lupus nephritis (Chap. 338), and deposits of immune complexes can be identified in tubule basement membranes in about 50% of cases. Occasionally, however, the tubulointerstitial inflammation predominates and may manifest with azotemia and type IV RTA rather than features of glomerulonephritis.
GRANULOMATOUS INTERSTITIAL NEPHRITIS
Some patients may present with features of AIN but follow a protracted and relapsing course. Renal biopsy in such patients reveals a more chronic inflammatory infiltrate with granulomas and multinucleated giant cells. Most often, no associated disease or cause is found; however, some of these cases may have or subsequently develop the pulmonary, cutaneous, or other systemic manifestations of sarcoidosis such as hypercalcemia. Most patients experience some improvement in renal function if treated early with glucocorticoids before the development of significant interstitial fibrosis and tubular atrophy (Table 340-2). Other immunosuppressive agents may be required for those who relapse frequently upon steroid withdrawal. Other immunosuppressive agents may be required for those who relapse frequently upon steroid withdrawal (Fig. 340-1). Tuberculosis should be ruled out before starting treatment because this too is a rare cause of granulomatous interstitial nephritis.
IgG4-RELATED SYSTEMIC DISEASE
A form of AIN characterized by a dense inflammatory infiltrate containing IgG4-expressing plasma cells can occur as a part of a syndrome known as IgG4-related systemic disease. Autoimmune pancreatitis, sclerosing cholangitis, retroperitoneal fibrosis, and a chronic sclerosing sialadenitis (mimicking Sjögren’s syndrome) may variably be present as well. Fibrotic lesions that form pseudotumors in the affected organs soon replace the initial inflammatory infiltrates and often lead to biopsy or excision for fear of true malignancy. Although the involvement of IgG4 in the pathogenesis is not understood, glucocorticoids have been successfully used as first-line treatment in this group of disorders, once they are correctly diagnosed.
IDIOPATHIC AIN
Some patients present with typical clinical and histologic features of AIN but have no evidence of drug exposure or clinical or serologic features of an autoimmune disease. The presence in some cases of autoantibodies to a tubular antigen, similar to that identified in rats with an induced form of interstitial nephritis, suggests that an autoimmune response may be involved. Like TINU and granulomatous interstitial nephritis, idiopathic AIN is responsive to glucocorticoid therapy but may follow a relapsing course requiring maintenance treatment with another immunosuppressive agent (Fig. 340-1 and Table 340-2).
INFECTION-ASSOCIATED AIN
AIN may also occur as a local inflammatory reaction to microbial infection (Table 340-1) and should be distinguished from acute bacterial pyelonephritis (Chap. 162). Acute bacterial pyelonephritis does not generally cause acute renal failure unless it affects both kidneys or causes septic shock. Presently, infection-associated AIN is most often seen in immunocompromised patients, particularly renal transplant recipients with reactivation of polyomavirus BK (Chaps. 169 and 337).
CRYSTAL DEPOSITION DISORDERS AND OBSTRUCTIVE TUBULOPATHIES
Acute renal failure may occur when crystals of various types are deposited in tubular cells and interstitium or when they obstruct tubules. Oliguric acute renal failure, often accompanied by flank pain from tubular obstruction, may occur in patients treated with sulfadiazine for toxoplasmosis, indinavir and atazanavir for HIV, and intravenous acyclovir for severe herpesvirus infections. Urinalysis reveals “sheaf of wheat” sulfonamide crystals, individual or parallel clusters of needle-shaped indinavir crystals, or red-green birefringement needle-shaped crystals of acyclovir. This adverse effect is generally precipitated by hypovolemia and is reversible with saline volume repletion and drug withdrawal. Distinct from the obstructive disease, a frank AIN from indinavir crystal deposition has also been reported.
Acute tubular obstruction is also the cause of oliguric renal failure in patients with acute urate nephropathy. It typically results from severe hyperuricemia from tumor lysis syndrome in patients with lympho- or myeloproliferative disorders treated with cytotoxic agents, but also may occur spontaneously before the treatment has been initiated (Chap. 331). Uric acid crystallization in the tubules and collecting system leads to partial or complete obstruction of the collecting ducts, renal pelvis, or ureter. A dense precipitate of birefringent uric acid crystals is found in the urine, usually in association with microscopic or gross hematuria. Prophylactic allopurinol reduces the risk of uric acid nephropathy but is of no benefit once tumor lysis has occurred. Once oliguria has developed, attempts to increase tubular flow and solubility of uric acid with alkaline diuresis may be of some benefit; however, emergent treatment with hemodialysis or rasburicase, a recombinant urate oxidase, is usually required to rapidly lower uric acid levels and restore renal function.
Calcium oxalate crystal deposition in tubular cells and interstitium may lead to permanent renal dysfunction in patients who survive ethylene glycol intoxication, in patients with enteric hyperoxaluria from ileal resection or small-bowel bypass surgery, and in patients with hereditary hyperoxaluria (Chap. 342). Acute phosphate nephropathy is an uncommon but serious complication of oral Phospho-soda used as a laxative or for bowel preparation for colonoscopy. It results from calcium phosphate crystal deposition in tubules and interstitium and occurs especially in subjects with underlying renal impairment and hypovolemia. Consequently, Phospho-soda should be avoided in patients with chronic kidney disease.
LIGHT CHAIN CAST NEPHROPATHY
Patients with multiple myeloma may develop acute renal failure in the setting of hypovolemia, infection, or hypercalcemia or after exposure to NSAIDs or radiographic contrast media. The diagnosis of light chain cast nephropathy (LCCN)—commonly known as myeloma kidney—should be considered in patients who fail to recover when the precipitating factor is corrected or in any elderly patient with otherwise unexplained acute renal failure.
In this disorder, filtered monoclonal immunoglobulin light chains (Bence-Jones proteins) form intratubular aggregates with secreted Tamm-Horsfall protein in the distal tubule. Casts, in addition to obstructing the tubular flow in affected nephrons, incite a giant cell or foreign body reaction and can lead to tubular rupture, resulting in interstitial fibrosis (Fig. 340-3). Although LCCN generally occurs in patients with known multiple myeloma and a large plasma cell burden, the disorder should also be considered as a possible diagnosis in patients who have known monoclonal gammopathy even in the absence of frank myeloma. Filtered monoclonal light chains may also cause less pronounced renal manifestations in the absence of obstruction, due to direct toxicity to proximal tubular cells and intracellular crystal formation. This may result in isolated tubular disorders such as RTA or full Fanconi’s syndrome.
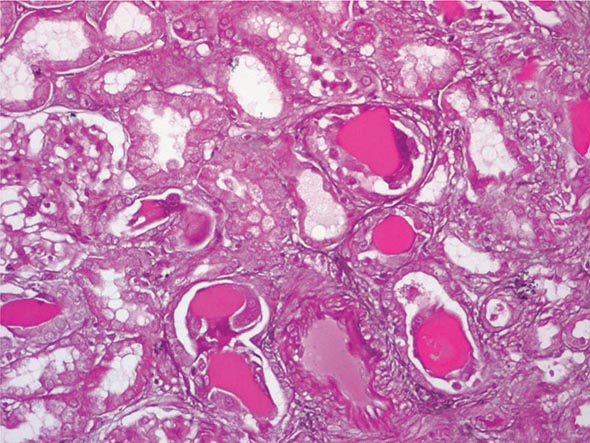
FIGURE 340-3 Histologic appearance of myeloma cast nephropathy. A hematoxylin-eosin–stained kidney biopsy shows many atrophic tubules filled with eosinophilic casts (consisting of Bence-Jones protein), which are surrounded by giant cell reactions. (Courtesy of Dr. Michael N. Koss, University of Southern California Keck School of Medicine; with permission.)
Diagnosis Clinical clues to the diagnosis include anemia, bone pain, hypercalcemia, and an abnormally narrow anion gap due to hypoalbuminemia and hypergammaglobulinemia. Urinary dipsticks detect albumin but not immunoglobulin light chains; however, laboratory detection of increased amounts of protein in a spot urine specimen and a negative dipstick result are highly suggestive that the urine contains Bence-Jones protein. Serum and urine should both be sent for protein electrophoresis and for immunofixation for the detection and identification of a potential monoclonal band. A sensitive method is available to detect urine and serum free light chains.
LYMPHOMATOUS INFILTRATION OF THE KIDNEY
Interstitial infiltration by malignant B lymphocytes is a common autopsy finding in patients dying of chronic lymphocytic leukemia and non-Hodgkin’s lymphoma; however, this is usually an incidental finding. Rarely, such infiltrates may cause massive enlargement of the kidneys and oliguric acute renal failure. Although high-dose glucocorticoids and subsequent chemotherapy often result in recovery of renal function, the prognosis in such cases is generally poor.
CHRONIC TUBULOINTERSTITIAL DISEASES
Improved occupational and public health measures, together with the banning of over-the-counter phenacetin-containing analgesics, has led to a dramatic decline in the incidence of chronic interstitial nephritis (CIN) from heavy metal—particularly lead and cadmium—exposure and analgesic nephropathy in North America. Today, CIN is most often the result of renal ischemia or secondary to a primary glomerular disease (Chap. 338). Other important forms of CIN are the result of developmental anomalies or inherited diseases such as reflux nephropathy or sickle cell nephropathy and may not be recognized until adolescence or adulthood. Although it is impossible to reverse damage that has already occurred, further deterioration may be prevented or at least slowed in such cases by treating glomerular hypertension, a common denominator in the development of secondary FSGS and progressive loss of functioning nephrons. Therefore, awareness and early detection of patients at risk may prevent them from developing end-stage renal disease (ESRD).
VESICOURETERAL REFLUX AND REFLUX NEPHROPATHY
Reflux nephropathy is the consequence of vesicoureteral reflux (VUR) or other urologic anomalies in early childhood. It was previously called chronic pyelonephritis because it was believed to result from recurrent urinary tract infections (UTIs) in childhood. VUR stems from abnormal retrograde urine flow from the bladder into one or both ureters and kidneys because of mislocated and incompetent ureterovesical valves (Fig. 340-4). Although high-pressure sterile reflux may impair normal growth of the kidneys, when coupled with recurrent UTIs in early childhood, the result is patchy interstitial scarring and tubular atrophy. Loss of functioning nephrons leads to hypertrophy of the remnant glomeruli and eventual secondary FSGS. Reflux nephropathy often goes unnoticed until early adulthood when chronic kidney disease is detected during routine evaluation or during pregnancy. Affected adults are frequently asymptomatic, but may give a history of prolonged bed-wetting or recurrent UTIs during childhood, and exhibit variable renal insufficiency, hypertension, mild to moderate proteinuria, and unremarkable urine sediment. When both kidneys are affected, the disease often progresses inexorably over several years to ESRD, despite the absence of ongoing urinary infections or reflux. A single affected kidney may go undetected, except for the presence of hypertension. Renal ultrasound in adults characteristically shows asymmetric small kidneys with irregular outlines, thinned cortices, and regions of compensatory hypertrophy (Fig. 340-4).
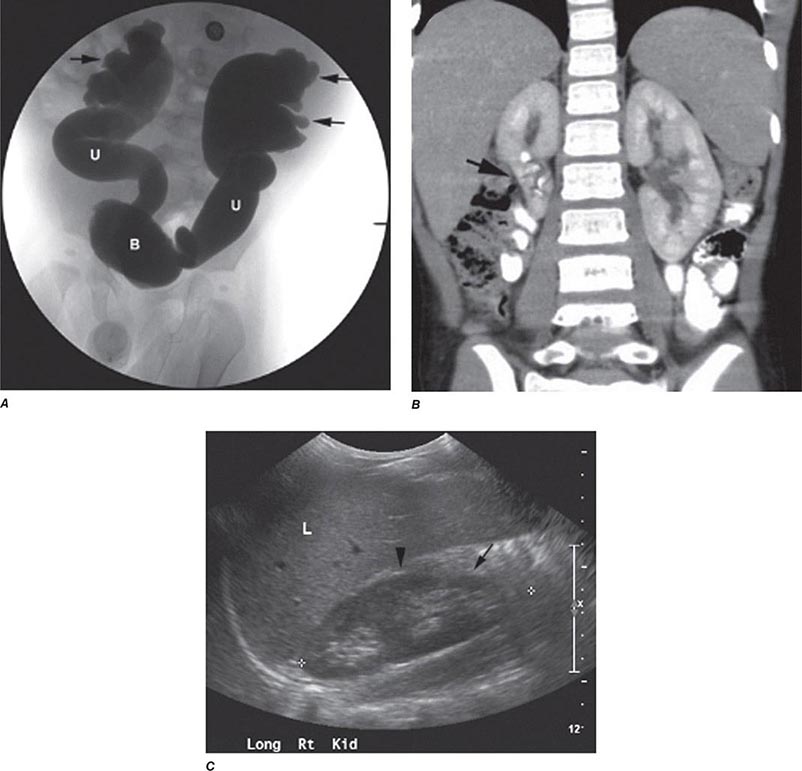
FIGURE 340-4 Radiographs of vesicoureteral reflux (VUR) and reflux nephropathy. A. Voiding cystourethrogram in a 7-month-old baby with bilateral high-grade VUR evidenced by clubbed calyces (arrows) and dilated tortuous ureters (U) entering the bladder (B). B. Abdominal computed tomography scan (coronal plane reconstruction) in a child showing severe scarring of the lower portion of the right kidney (arrow). C. Sonogram of the right kidney showing loss of parenchyma at the lower pole due to scarring (arrow) and hypertrophy of the mid-region (arrowhead). (Courtesy of Dr. George Gross, University of Maryland Medical Center; with permission.)
SICKLE CELL NEPHROPATHY
The pathogenesis and clinical manifestations of sickle cell nephropathy are described in Chap. 341. Evidence of tubular injury may be evident in childhood and early adolescence in the form of polyuria due to decreased concentrating ability or type IV renal tubular acidosis years before there is significant nephron loss and proteinuria from secondary FSGS. Early recognition of these subtle renal abnormalities or development of microalbuminuria in a child with sickle cell disease may warrant consultation with a nephrologist and/or therapy with low-dose ACEIs. Papillary necrosis may result from ischemia due to sickling of red cells in the relatively hypoxemic and hypertonic medullary vasculature and present with gross hematuria and ureteric obstruction by sloughed ischemic papillae (Table 340-3).
|
MAJOR CAUSES OF PAPILLARY NECROSIS |
Abbreviation: NSAID, nonsteroidal anti-inflammatory drug.
TUBULOINTERSTITIAL ABNORMALITIES ASSOCIATED WITH GLOMERULONEPHRITIS
Primary glomerulopathies are often associated with damage to tubules and interstitium. This may occasionally be due to the same pathologic process affecting the glomerulus and tubulointerstitium, as is the case with immune-complex deposition in lupus nephritis. More often, however, chronic tubulointerstitial changes occur as a secondary consequence of prolonged glomerular dysfunction. Potential mechanisms by which glomerular disease might cause tubulointerstitial injury include proteinuria-mediated damage to the epithelial cells, activation of tubular cells by cytokines and complement, or reduced peritubular blood flow leading to downstream tubulointerstitial ischemia, especially in the case of glomeruli that are globally obsolescent due to severe glomerulonephritis. It is often difficult to discern the initial cause of injury by renal biopsy in a patient who presents with advanced renal disease in this setting.
ANALGESIC NEPHROPATHY
Analgesic nephropathy results from the long-term use of compound analgesic preparations containing phenacetin (banned in the United States since 1983), aspirin, and caffeine. In its classic form, analgesic nephropathy is characterized by renal insufficiency, papillary necrosis (Table 340-3) attributable to the presumed concentration of the drug to toxic levels in the inner medulla, and a radiographic constellation of small, scarred kidneys with papillary calcifications best appreciated by computed tomography (Fig. 340-5). Patients may also have polyuria due to impaired concentrating ability and non-anion-gap metabolic acidosis from tubular damage. Shedding of a sloughed necrotic papilla can cause gross hematuria and ureteric colic due to ureteral obstruction. Individuals with ESRD as a result of analgesic nephropathy are at increased risk of a urothelial malignancy compared to patients with other causes of renal failure. Recent cohort studies in individuals with normal baseline renal function suggest that the moderate chronic use of current analgesic preparations available in the United States, including acetaminophen and NSAIDs, does not seem to cause the constellation of findings known as analgesic nephropathy, although volume-depleted individuals and those with chronic kidney disease are at higher risk of NSAID-related renal toxicity. Nonetheless, it is recommended that heavy users of acetaminophen and NSAIDs be screened for evidence of renal disease.
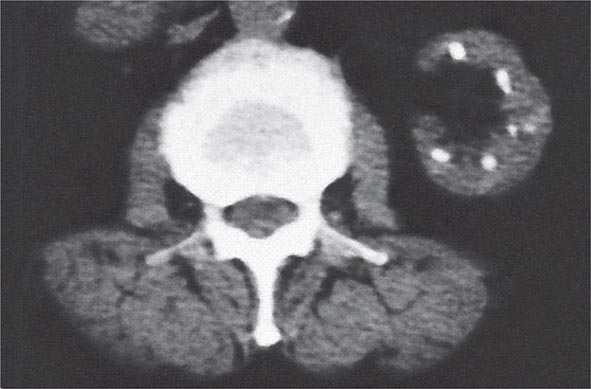
FIGURE 340-5 Radiologic appearance of analgesic nephropathy. A noncontrast computed tomography scan shows an atrophic left kidney with papillary calcifications in a garland pattern. (Reprinted by permission from Macmillan Publishers, Ltd., MM Elseviers et al: Kidney International 48:1316, 1995.)
ARISTOLOCHIC ACID NEPHROPATHY
Two seemingly unrelated forms of CIN, Chinese herbal nephropathy and Balkan endemic nephropathy, have recently been linked by the underlying etiologic agent aristolochic acid and are now collectively termed aristolochic acid nephropathy (AAN). In Chinese herbal nephropathy, first described in the early 1990s in young women taking traditional Chinese herbal preparations as part of a weight-loss regimen, one of the offending agents has been identified as aristolochic acid, a known carcinogen from the plant Aristolochia. Multiple Aristolochia species have been used in traditional herbal remedies for centuries and continue to be available despite official bans on their use in many countries. Molecular evidence has also implicated aristolochic acid in Balkan endemic nephropathy, a chronic tubulointerstitial nephritis found primarily in towns along the tributaries of the Danube River and first described in the 1950s. Although the exact route of exposure is not known with certainty, contamination of local grain preparations with the seeds of Aristolochia species seems most likely. Aristolochic acid, after prolonged exposure, produces renal interstitial fibrosis with a relative paucity of cellular infiltrates. The urine sediment is bland, with rare leukocytes and only mild proteinuria. Anemia may be disproportionately severe relative to the level of renal dysfunction. Definitive diagnosis of AAN requires two of the following three features: characteristic histology on kidney biopsy; confirmation of aristolochic acid ingestion; and detection of aristolactam-DNA adducts in kidney or urinary tract tissue. These latter lesions represent a molecular signature of aristolochic acid–derived DNA damage and often consist of characteristic A:T-to-T:A transversions. Due to this mutagenic activity, AAN is associated with a very high incidence of upper urinary tract urothelial cancers, with risk related to cumulative dose. Surveillance with computed tomography, ureteroscopy, and urine cytology is warranted, and consideration should be given to bilateral nephroureterectomy once a patient has reached ESRD.
KARYOMEGALIC INTERSTITIAL NEPHRITIS
Karyomegalic interstitial nephritis is an unusual form of slowly progressive chronic kidney disease with mild proteinuria, interstitial fibrosis, tubular atrophy, and oddly enlarged nuclei of proximal tubular epithelial cells. It has been linked to mutations in FAN1, a nuclease involved in DNA repair, which may render carriers of the mutation susceptible to environmental DNA-damaging agents.
LITHIUM-ASSOCIATED NEPHROPATHY
The use of lithium salts for the treatment of manic-depressive illness may have several renal sequelae, the most common of which is nephrogenic diabetes insipidus manifesting as polyuria and polydipsia. Lithium accumulates in principal cells of the collecting duct by entering through the epithelial sodium channel (ENaC), where it inhibits glycogen synthase kinase 3β and downregulates vasopressin-regulated aquaporin water channels. Less frequently, chronic tubulointerstitial nephritis develops after prolonged (>10–20 years) lithium use and is most likely to occur in patients who have experienced repeated episodes of toxic lithium levels. Findings on renal biopsy include interstitial fibrosis and tubular atrophy that are out of proportion to the degree of glomerulosclerosis or vascular disease, a sparse lymphocytic infiltrate, and small cysts or dilation of the distal tubule and collecting duct that are highly characteristic of this disorder. The degree of interstitial fibrosis correlates with both duration and cumulative dose of lithium. Individuals with lithium-associated nephropathy are typically asymptomatic, with minimal proteinuria, few urinary leukocytes, and normal blood pressure. Some patients develop more severe proteinuria due to secondary FSGS, which may contribute to further loss of renal function.
CALCINEURIN-INHIBITOR NEPHROTOXICITY
The calcineurin inhibitor (CNI) immunosuppressive agents cyclosporine and tacrolimus can cause both acute and chronic renal injury. Acute forms can result from vascular causes such as vasoconstriction or the development of thrombotic microangiopathy, or can be due to a toxic tubulopathy. Chronic CNI-induced renal injury is typically seen in solid organ (including heart-lung and liver) transplant recipients and manifests with a slow but irreversible reduction of glomerular filtration rate, with mild proteinuria and arterial hypertension. Hyperkalemia is a relatively common complication and is caused, in part, by tubular resistance to aldosterone. The histologic changes in renal tissue include patchy interstitial fibrosis and tubular atrophy, often in a “striped” pattern. In addition, the intrarenal vasculature often demonstrates hyalinosis, and focal glomerulosclerosis can be present as well. Similar changes may occur in patients receiving CNIs for autoimmune diseases, although the doses are generally lower than those used for organ transplantation. Dose reduction or CNI avoidance appears to mitigate the chronic tubulointerstitial changes, but may increase the risk of rejection and graft loss.
HEAVY METAL (LEAD) NEPHROPATHY
Heavy metals, such as lead or cadmium, can lead to a chronic tubulointerstitial process after prolonged exposure. The disease entity is no longer commonly diagnosed, because such heavy metal exposure has been greatly reduced due to the known health risks from lead and the consequent removal of lead from most commercial products and fuels. Nonetheless, occupational exposure is possible in workers involved in the manufacture or destruction of batteries, removal of lead paint, or manufacture of alloys and electrical equipment (cadmium) in countries where industrial regulation is less stringent. In addition, ingestion of moonshine whiskey distilled in lead-tainted containers has been one of the more frequent sources of lead exposure.
Early signs of chronic lead intoxication are attributable to proximal tubule dysfunction, particularly hyperuricemia as a result of diminished urate secretion. The triad of “saturnine gout,” hypertension, and renal insufficiency should prompt a practitioner to ask specifically about lead exposure. Unfortunately, evaluating lead burden is not as straightforward as ordering a blood test; the preferred methods involve measuring urinary lead after infusion of a chelating agent or by radiographic fluoroscopy of bone. Several recent studies have shown an association between chronic low-level lead exposure and decreased renal function, although either of these two factors may have been the primary event. In those patients who have CIN of unclear origin and an elevated total body lead burden, repeated treatments of lead chelation therapy have been shown to slow the decline in renal function.
METABOLIC DISORDERS
Disorders leading to excessively high or low levels of certain electrolytes and products of metabolism can also lead to chronic kidney disease if untreated.
CHRONIC URIC ACID NEPHROPATHY
The constellation of pathologic findings that represent gouty nephropathy are very uncommon nowadays and are more of historical interest than clinical importance, as gout is typically well managed with allopurinol and other agents. However, there is emerging evidence that hyperuricemia is an independent risk factor for the development of chronic kidney disease, perhaps through endothelial damage. The complex interactions of hyperuricemia, hypertension, and renal failure are still incompletely understood.
Presently, gouty nephropathy is most likely to be encountered in patients with severe tophaceous gout and prolonged hyperuricemia from a hereditary disorder of purine metabolism (Chap. 431e). This should be distinguished from juvenile hyperuricemic nephropathy, a form of medullary cystic kidney disease caused by mutations in uromodulin (UMOD) (Chap. 339). Histologically, the distinctive feature of gouty nephropathy is the presence of crystalline deposits of uric acid and monosodium urate salts in the kidney parenchyma. These deposits not only cause intrarenal obstruction but also incite an inflammatory response, leading to lymphocytic infiltration, foreign-body giant cell reaction, and eventual fibrosis, especially in the medullary and papillary regions of the kidney. Since patients with gout frequently suffer from hypertension and hyperlipidemia, degenerative changes of the renal arterioles may constitute a striking feature of the histologic abnormality, out of proportion to the other morphologic defects. Clinically, gouty nephropathy is an insidious cause of chronic kidney disease. Early in its course, glomerular filtration rate may be near normal, often despite morphologic changes in medullary and cortical interstitium, proteinuria, and diminished urinary concentrating ability. Treatment with allopurinol and urine alkalinization is generally effective in preventing uric acid nephrolithiasis and the consequences of recurrent kidney stones; however, gouty nephropathy may be intractable to such measures. Furthermore, the use of allopurinol in asymptomatic hyperuricemia has not been consistently shown to improve renal function.
HYPERCALCEMIC NEPHROPATHY
(See also Chap. 424) Chronic hypercalcemia, as occurs in primary hyperparathyroidism, sarcoidosis, multiple myeloma, vitamin D intoxication, or metastatic bone disease, can cause tubulointerstitial disease and progressive renal failure. The earliest lesion is a focal degenerative change in renal epithelia, primarily in collecting ducts, distal tubules, and loops of Henle. Tubular cell necrosis leads to nephron obstruction and stasis of intrarenal urine, favoring local precipitation of calcium salts and infection. Dilation and atrophy of tubules eventually occur, as do interstitial fibrosis, mononuclear leukocyte infiltration, and interstitial calcium deposition (nephrocalcinosis). Calcium deposition may also occur in glomeruli and the walls of renal arterioles.
Clinically, the most striking defect is an inability to maximally concentrate the urine, due to reduced collecting duct responsiveness to arginine vasopressin and defective transport of sodium and chloride in the loop of Henle. Reductions in both glomerular filtration rate and renal blood flow can occur, both in acute and in prolonged hypercalcemia. Eventually, uncontrolled hypercalcemia leads to severe tubulointerstitial damage and overt renal failure. Abdominal x-rays may demonstrate nephrocalcinosis as well as nephrolithiasis, the latter due to the hypercalciuria that often accompanies hypercalcemia.
Treatment consists of reducing the serum calcium concentration toward normal and correcting the primary abnormality of calcium metabolism (Chap. 424). Renal dysfunction of acute hypercalcemia may be completely reversible. Gradual progressive renal insufficiency related to chronic hypercalcemia, however, may not improve even with correction of the calcium disorder.
HYPOKALEMIC NEPHROPATHY
Patients with prolonged and severe hypokalemia from chronic laxative or diuretic abuse, surreptitious vomiting, or primary aldosteronism may develop a reversible tubular lesion characterized by vacuolar degeneration of proximal and distal tubular cells. Eventually, tubular atrophy and cystic dilation accompanied by interstitial fibrosis may ensue, leading to irreversible chronic kidney disease. Timely correction of the hypokalemia will prevent further progression, but persistent hypokalemia can cause ESRD.
GLOBAL PERSPECTIVE
![]() The causes of acute and chronic interstitial nephritis vary widely across the globe. Analgesic nephropathy continues to be seen in countries where phenacetin-containing compound analgesic preparations are readily available. Adulterants in unregulated herbal and traditional medicaments pose a threat of toxic interstitial nephritis, as exemplified by aristolochic acid contamination of herbal slimming preparations. Contamination of food sources with toxins, such as the recent outbreak of nephrolithiasis and acute renal failure from melamine contamination of infant milk formula, poses a continuing risk. Large-scale exposure to aristolochic acid remains prevalent in many Asian countries where traditional herbal medicine use is common. Although industrial exposure to lead and cadmium has largely disappeared as a cause of chronic interstitial nephritis in developed nations, it remains a risk for nephrotoxicity in countries where such exposure is less well controlled. New endemic forms of chronic kidney disease continue to be described, such as the nephropathy found among Pacific coastal plantation workers in Central America, which may be related to repetitive heat exposure and fluid losses.
The causes of acute and chronic interstitial nephritis vary widely across the globe. Analgesic nephropathy continues to be seen in countries where phenacetin-containing compound analgesic preparations are readily available. Adulterants in unregulated herbal and traditional medicaments pose a threat of toxic interstitial nephritis, as exemplified by aristolochic acid contamination of herbal slimming preparations. Contamination of food sources with toxins, such as the recent outbreak of nephrolithiasis and acute renal failure from melamine contamination of infant milk formula, poses a continuing risk. Large-scale exposure to aristolochic acid remains prevalent in many Asian countries where traditional herbal medicine use is common. Although industrial exposure to lead and cadmium has largely disappeared as a cause of chronic interstitial nephritis in developed nations, it remains a risk for nephrotoxicity in countries where such exposure is less well controlled. New endemic forms of chronic kidney disease continue to be described, such as the nephropathy found among Pacific coastal plantation workers in Central America, which may be related to repetitive heat exposure and fluid losses.
341 |
Vascular Injury to the Kidney |
The renal circulation is complex and is characterized by a highly perfused arteriolar network, reaching cortical glomerular structures adjacent to lower-flow vasa recta that descend into medullary segments. Disorders of the larger vessels, including renal artery stenosis and atheroembolic disease, are discussed elsewhere (Chap. 354). This chapter examines primary disorders of the renal microvessels, many of which are associated with thrombosis and hemolysis.
THROMBOTIC MICROANGIOPATHY
Thrombotic microangiopathy (TMA) is characterized by injured endothelial cells that are thickened, swollen, or detached mainly from arterioles and capillaries. Platelet and hyaline thrombi causing partial or complete occlusion are integral to the histopathology of TMA. TMA is usually the result of microangiopathic hemolytic anemia (MAHA), with its typical features of thrombocytopenia and schistocytes. In the kidney, TMA is characterized by swollen endocapillary cells (endotheliosis), fibrin thrombi, platelet plugs, arterial intimal fibrosis, and a membranoproliferative pattern. Fibrin thrombi may extend into the arteriolar vascular pole, producing glomerular collapse and at times cortical necrosis. In kidneys that recover from acute TMA, secondary focal segmental glomerulosclerosis may be seen. Diseases associated with this lesion include thrombotic thrombocytopenic purpura (TTP), hemolytic-uremic syndrome (HUS), malignant hypertension, scleroderma renal crisis, antiphospholipid syndrome, preeclampsia/HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome, HIV infection, and radiation nephropathy.
HEMOLYTIC-UREMIC SYNDROME/THROMBOTIC THROMBOCYTOPENIC PURPURA
HUS and TTP are the prototypes for MAHA. Historically, HUS and TTP were distinguished mainly by their clinical and epidemiologic differences. TTP develops more commonly in adults and was thought to include neurologic involvement more often. HUS occurs more commonly in children, particularly when associated with hemorrhagic diarrhea. However, atypical HUS (aHUS) can first appear in adulthood, and better testing has revealed that neurologic involvement is as common in HUS as in TTP. Accordingly, HUS and TTP now should be differentiated and treated according to their specific pathophysiologic features.
Hemolytic-Uremic Syndrome HUS is loosely defined by the presence of MAHA and renal impairment. At least four variants are recognized. The most common is Shiga toxin–producing Escherichia coli (STEC) HUS, which is also known as D+HUS or enterohemorrhagic E. coli (EHEC) HUS. Most cases involve children <5 years of age, but adults also are susceptible, as evidenced by a 2011 outbreak in northern Europe. Diarrhea, often bloody, precedes MAHA within 1 week in >80% of cases. Abdominal pain, cramping, and vomiting are frequent, whereas fever is typically absent. Neurologic symptoms, including dysphasia, hyperreflexia, blurred vision, memory deficits, encephalopathy, perseveration, and agraphia, often develop, especially in adults. Seizures and cerebral infarction can occur in severe cases. STEC HUS is caused by the Shiga toxins (Stx1 and Stx2), which are also referred to as verotoxins. These toxins are produced by certain strains of E. coli and Shigella dysenteriae. In the United States and Europe, the most common STEC strain is O157:H7, but HUS due to other strains (O157/H–, O111:H–, O26:H11/H–, O145:H28, and O104:H4) has occurred. After entry into the circulation, Shiga toxin binds to the glycolipid surface receptor globotriaosylceramide (Gb3), which is richly expressed on cells of the renal microvasculature. Upon binding, the toxin enters the cells, inducing inflammatory cytokines (interleukin 8 [IL-8], monocyte chemotactic protein 1 [MCP-1], and stromal cell–derived factor 1 [SDF-1]) and chemokine receptors (CXCR4 and CXCR7); this action results in platelet aggregation and the microangiopathic process. Streptococcus pneumoniae can also cause HUS. Certain strains produce a neuraminidase that cleaves the N-acetylneuraminic acid moieties covering the Thomsen-Friedenreich antigen on platelets and endothelial cells. Exposure of this normally cryptic antigen to preformed IgM results in severe MAHA.
Atypical HUS is the result of congenital complement dysregulation. The affected patients have the low C3 and normal C4 levels characteristic of alternative pathway activation. Factor H deficiency, the most common defect, has been linked to families with aHUS. Factor H competes with factor B to prevent the formation of C3bBb and acts as a cofactor for factor I, which proteolytically degrades C3b. More than 70 mutations of the factor H gene have been identified. Most are missense mutations that produce abnormalities in the C-terminus region, affecting its binding to C3b but not its concentration. Other mutations result in low levels or the complete absence of the protein. Deficiencies in other complement-regulatory proteins, such as factor I, factor B, membrane cofactor protein (CD46), C3, complement factor H–related protein 1 (CFHR1), CFHR3, CFHR5, and thrombomodulin, have also been reported. Finally, an autoimmune variant of aHUS has been discovered. DEAP (deficient for CFHR protein and positive for factor H autoantibody) HUS occurs when an autoantibody to factor H is formed. DEAP HUS is often associated with a deletion of an 84-kb fragment of the chromosome that encodes for CFHR1 and CFHR3. The autoantibody blocks the binding of factor H to C3b and surface-bound C3 convertase.
Thrombotic Thrombocytopenic Purpura Traditionally, TTP is characterized by the pentad: MAHA, thrombocytopenia, neurologic symptoms, fever, and renal failure. The pathophysiology of TTP involves the accumulation of ultra-large multimers of von Willebrand factor as a result of the absence or markedly decreased activity (<5–10%) of the plasma protease ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13. These ultra-large multimers form clots and shear erythrocytes, resulting in MAHA; however, the absence of ADAMTS13 alone may not itself produce TTP. Often, an additional trigger (such as infection, surgery, pancreatitis, or pregnancy) is required to initiate clinical TTP.
Data from the Oklahoma TTP/HUS Registry suggest an incidence rate of 11.3 cases/106 patients in the United States. The median age of onset is 40 years. The incidence is more than nine times higher among blacks than among non-blacks. Like that of systemic lupus erythematosus, the incidence of TTP is nearly three times higher among women than among men. If untreated, TTP has a mortality rate exceeding 90%. Even with modern therapy, 20% of patients die within the first month from complications of microvascular thrombosis.
The classic form of TTP is idiopathic TTP, which is usually the result of a deficiency in ADAMTS13. While TTP had traditionally been associated with infection, malignancy, and intense inflammation (e.g., pancreatitis), ADAMTS13 activity usually is not decreased in these conditions. In idiopathic TTP, the formation of an autoantibody to ADAMTS13 (IgG or IgM) either increases its clearance or inhibits its activity. Upshaw-Schülman syndrome is a hereditary condition characterized by congenital deficiency of ADAMTS13. TTP in these patients can start within the first weeks of life but in some instances may not present until the patient is several years of age. Both environmental and genetic factors are thought to influence the development of TTP. Plasma transfusion is an effective strategy for prevention and treatment.
Drug-induced TMA is a recognized complication of treatment with some chemotherapeutic agents, immunosuppressive agents, antiplatelet agents, and quinine. Two different mechanisms have been described. Endothelial damage (pathologically similar to that in HUS) is the main cause of the TMA that develops in association with chemotherapeutic agents (e.g., mitomycin C, gemcitabine) and immunosuppressive agents (cyclosporine, tacrolimus, and sirolimus). This process is usually dose-dependent. Alternatively, TMA may develop as a result of drug-induced autoantibodies. This form is less likely to be dose-dependent and can, in fact, occur after a single dose in patients with previous exposure. Ticlopidine produces TTP by inducing an autoantibody to ADAMTS13, but ADAMTS13 deficiency is found in fewer than half of patients with clopidogrel-associated TTP. Quinine appears to induce autoantibodies to granulocytes, lymphocytes, endothelial cells, and platelet glycoprotein IbB/IX or IIb/IIIa complexes, but not to ADAMTS13. Quinine-associated TTP is more common among women. TMA has been reported with drugs that inhibit vascular endothelial growth factor, such as bevacizumab; the mechanism is not completely understood.
HEMATOPOIETIC STEM CELL TRANSPLANTATION–ASSOCIATED THROMBOTIC MICROANGIOPATHY (HSCT-TMA)
HSCT-TMA develops after HSCT, with an incidence of 8.2%. Etiologic factors include conditioning regimens, immunosuppression, infections, and graft-versus-host disease. Other risk factors include female sex and human leukocyte antigen (HLA)–mismatched donor grafts. HSCT-TMA usually occurs within the first 100 days of HSCT. Table 341-1 lists definitions of HSCT-TMA currently used for clinical trials. Diagnosis may be difficult since thrombocytopenia, anemia, and renal insufficiency are common after HSCT. HSCT-TMA carries a high mortality rate (75% within 3 months). The majority of patients have >5% ADAMTS13 activity, and plasma exchange is beneficial in <50% of patients. Discontinuation of calcineurin inhibitors and substitution with daclizumab (antibody to the IL-2 receptor) are recommended. Treatment with rituximab and defibrotide may also be helpful.
|
CRITERIA FOR ESTABLISHING MICROANGIOPATHIC KIDNEY INJURY ASSOCIATED WITH HEMATOPOIETIC STEM CELL TRANSPLANTATION |
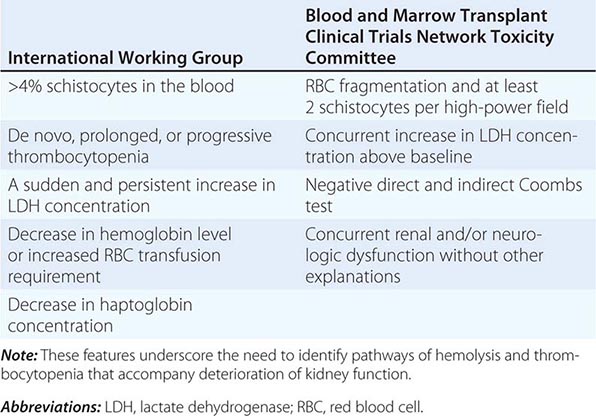
HIV-RELATED TMA
HIV-related TMA is a complication encountered mainly before widespread use of highly active antiretroviral therapy. It is seen in patients with advanced AIDS and low CD4+ T cell counts although it can be the first manifestation of HIV infection. The presence of MAHA, thrombocytopenia, and renal failure are suggestive, but renal biopsy is required for diagnosis since other renal diseases are also associated with HIV infection. Thrombocytopenia may prohibit renal biopsy in some patients. The mechanism of injury is unclear, although HIV can induce apoptosis in endothelial cells. ADAMTS13 activity is not reduced in these patients. Cytomegalovirus co-infection may also be a risk factor. Effective antiviral therapy is key, while plasma exchange should be limited to patients who have evidence of TTP.
RADIATION NEPHROPATHY
Either local or total body irradiation can produce microangiopathic injury. The kidney is one of the most radiosensitive organs, and injury can result with as little as 4–5 Gy. Such injury is characterized by renal insufficiency, proteinuria, and hypertension usually developing ≥6 months after radiation exposure. Renal biopsy reveals classic TMA with damage to glomerular, tubular, and vascular cells, but systemic evidence of MAHA is uncommon. Because of its high incidence after allogeneic HSCT, radiation nephropathy is often referred to as bone marrow transplant nephropathy. No specific therapy is available, although observational evidence supports renin-angiotensin system blockade.
SCLERODERMA (PROGRESSIVE SYSTEMIC SCLEROSIS)
Kidney involvement is common (up to 52%) in patients with widespread scleroderma, with 20% of cases resulting directly from scleroderma renal crisis. Other renal manifestations in scleroderma include transient (prerenal) or medication-related forms of acute kidney injury (e.g., associated with D-penicillamine, nonsteroidal anti-inflammatory drugs, or cyclosporine). Scleroderma renal crisis occurs in 12% of patients with diffuse systemic sclerosis but in only 2% of those with limited systemic sclerosis. Scleroderma renal crisis is the most severe manifestation of renal involvement, and is characterized by accelerated hypertension, a rapid decline in renal function, nephrotic proteinuria, and hematuria. Retinopathy and encephalopathy may accompany the hypertension. Salt and water retention with microvascular injury can lead to pulmonary edema. Cardiac manifestations, including myocarditis, pericarditis, and arrhythmias, denote an especially poor prognosis. Although MAHA is present in more half of patients, coagulopathy is rare.
The renal lesion in scleroderma renal crisis is characterized by arcuate artery intimal and medial proliferation with luminal narrowing. this lesion is described as “onion-skinning” and can be accompanied by glomerular collapse due to reduced blood flow. Histologically, scleroderma renal crisis is indistinguishable from malignant hypertension, with which it can coexist. Fibrinoid necrosis and thrombosis are common. Before the availability of angiotensin-converting enzyme (ACE) inhibitors, the mortality rate for scleroderma renal crisis was >90% at 1 month. Introduction of renin-angiotensin system blockade has lowered the mortality rate to 30% at 3 years. Nearly two-thirds of patients with scleroderma renal crisis may require dialysis support, with recovery of renal function in 50% (median time, 1 year). Glomerulonephritis and vasculitis associated with antineutrophil cytoplasmic antibodies and systemic lupus erythematosus have been described in patients with scleroderma. An association has been found with a speckled pattern of antinuclear antibodies and with antibodies to RNA polymerases I and III. Anti-U3-RNP may identify young patients at risk for scleroderma renal crisis. Anticentromere antibody, in contrast, is a negative predictor of this disorder. Because of the overlap between scleroderma renal crisis and other autoimmune disorders, a renal biopsy is recommended for patients with atypical renal involvement, especially if hypertension is absent.
Treatment with ACE inhibition is the first-line therapy unless contraindicated. The goal of therapy is to reduce systolic and diastolic blood pressure by 20 mmHg and 10 mmHg, respectively, every 24 h until blood pressure is normal. Additional antihypertensive therapy may be given once the dose of drug for ACE inhibition is maximized. Both ACE inhibitors and angiotensin II receptor antagonists are effective, although data suggest that treatment with ACE inhibitors is superior. ACE inhibition alone does not prevent scleroderma renal crisis, but it does reduce the impact of hypertension. Intravenous iloprost has been used in Europe for blood pressure management and improvement of renal perfusion. Kidney transplantation is not recommended for 2 years after the start of dialysis since delayed recovery may occur.
ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome (Chap. 379) can be either primary or secondary to systemic lupus erythematosus. It is characterized by a predisposition to systemic thrombosis (arterial and venous) and fetal morbidity mediated by antiphospholipid antibodies—mainly anticardiolipin antibodies (IgG, IgM, or IgA), lupus anticoagulant, or anti-β-2 glycoprotein I antibodies (antiβ2GPI). Patients with both anticardiolipin antibodies and antiβ2GPI appear to have the highest risk of thrombosis. The vascular compartment within the kidney is the main site of renal involvement. Arteriosclerosis is commonly present in the arcuate and intralobular arteries. In the intralobular arteries, fibrous intimal hyperplasia characterized by intimal thickening secondary to intense myofibroblastic intimal cellular proliferation with extracellular matrix deposition is frequently seen along with onion-skinning. Arterial and arteriolar fibrous and fibrocellular occlusions are present in more than two-thirds of biopsy samples. Cortical necrosis and focal cortical atrophy may result from vascular occlusion. TMA is commonly present in renal biopsies, although signs of MAHA and platelet consumption are usually absent. TMA is especially common in the catastrophic variant of antiphospholipid syndrome. In patients with secondary antiphospholipid syndrome, other glomerulopathies may be present, including membranous nephropathy, minimal change disease, focal segmental glomerulosclerosis, and pauci-immune crescentic glomerulonephritis.
Large vessels can be involved in antiphospholipid syndrome and may form the proximal nidus near the ostium for thrombosis of the renal artery. Renal vein thrombosis can occur and should be suspected in patients with lupus anticoagulant who develop nephrotic-range proteinuria. Progression to end-stage renal disease can occur, and a thrombosis may form in the vascular access and the renal allografts. Hypertension is common. Treatment entails lifelong anticoagulation. Glucocorticoids may be beneficial in accelerated hypertension. Immunosuppression and plasma exchange may be helpful for catastrophic episodes of antiphospholipid syndrome but by themselves do not reduce recurrent thrombosis.
HELLP SYNDROME
HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome is a dangerous complication of pregnancy associated with microvascular injury. Occurring in 0.2–0.9% of all pregnancies and in 10–20% of women with severe preeclampsia, this syndrome carries a mortality rate of 7.4–34%. Most commonly developing in the third trimester, 10% of cases occur before week 27 and 30% post-partum. Although a strong association exists between HELLP syndrome and preeclampsia, nearly 20% of cases are not preceded by recognized preeclampsia. Risk factors include abnormal placentation, family history, and elevated levels of fetal mRNA for FLT1 (vascular endothelial growth factor receptor 1) and endoglin. Patients with HELLP syndrome have higher levels of inflammatory markers (C-reactive protein, IL-1Ra, and IL-6) and soluble HLA-DR than do those with preeclampsia alone.
Renal failure occurs in half of patients with HELLP syndrome, although the etiology is not well understood. Limited data suggest that renal failure is the result of both preeclampsia and acute tubular necrosis. Renal histologic findings are those of TMA with endothelial cell swelling and occlusion of the capillary lumens, but luminal thrombi are typically absent. However, thrombi become more common in severe eclampsia and HELLP syndrome. Although renal failure is common, the organ that defines this syndrome is the liver. Subcapsular hepatic hematomas sometimes produce spontaneous rupture of the liver and can be life-threatening. Neurologic complications such as cerebral infarction, cerebral and brainstem hemorrhage, and cerebral edema are other potentially life-threatening complications. Nonfatal complications include placental abruption, permanent vision loss due to Purtscher-like (hemorrhagic and vaso-occlusive vasculopathy) retinopathy, pulmonary edema, bleeding, and fetal demise.
Many features are shared by HELLP syndrome and MAHA. Diagnosis of HELLP syndrome is complicated by the fact that aHUS and TTP also can be triggered by pregnancy. Patients with antiphospholipid syndrome also have an elevated risk of HELLP syndrome. A history of MAHA before pregnancy is of diagnostic value. Serum levels of ADAMTS13 activity are reduced (by 30–60%) in HELLP syndrome but not to the levels seen in TTP (<5%). Determination of the ratio of lactate dehydrogenase to aspartate aminotransferase may be helpful; this ratio is 13:1 in patients with HELLP syndrome and preeclampsia as opposed to 29:1 in patients without preeclampsia. Other markers, such as antithrombin III (decreased in HELLP syndrome but not in TTP) and D-dimer (elevated in HELLP syndrome but not in TTP), may also be useful. HELLP syndrome usually resolves spontaneously after delivery, although a small percentage of HELLP cases occur post-partum. Glucocorticoids may decrease inflammatory markers, although two randomized controlled trials failed to show much benefit. Plasma exchange should be considered if hemolysis is refractory to glucocorticoids and/or delivery, especially if TTP has not been ruled out.
SICKLE CELL NEPHROPATHY
Renal complications in sickle cell disease result from occlusion of the vasa recta in the renal medulla. The low partial pressure of oxygen and high osmolarity predispose to hemoglobin S polymerization and erythrocyte sickling. Sequelae include hyposthenuria, hematuria, and papillary necrosis (which can also occur in sickle trait). The kidney responds by increases in blood flow and glomerular filtration rate mediated by prostaglandins. This dependence on prostaglandins may explain the greater reduction of glomerular filtration rate by nonsteroidal anti-inflammatory drugs in these patients than in others. The glomeruli are typically enlarged. Intracapillary fragmentation and phagocytosis of sickled erythrocytes are thought to be responsible for the membranoproliferative glomerulonephritis–like lesion, and focal segmental glomerulosclerosis is seen in more advanced cases. Proteinuria is present in 20–30%, and nephrotic-range proteinuria is associated with progression to renal failure. ACE inhibitors reduce proteinuria, although data are lacking on prevention of renal failure. Patients with sickle cell disease are also more prone to acute renal failure. The cause is thought to reflect microvascular occlusion associated with nontraumatic rhabdomyolysis, high fever, infection, and generalized sickling. Chronic kidney disease is present in 12–20% of patients. Despite the frequency of renal disease, hypertension is uncommon in patients with sickle cell disease.
RENAL VEIN THROMBOSIS
Renal vein thrombosis either can present with flank pain, tenderness, hematuria, rapid decline in renal function, and proteinuria or can be silent. Occasionally, renal vein thrombosis is identified during a workup for pulmonary embolism. The left renal vein is more commonly involved, and two-thirds of cases are bilateral. Etiologies can be divided into three broad categories: endothelial damage, venous stasis, and hypercoagulability. Homocystinuria, endovascular intervention, and surgery can produce vascular endothelial damage. Dehydration, which is more common among male patients, is a common cause of stasis in the pediatric population. Stasis also can result from compression and kinking of the renal veins from retroperitoneal processes such as retroperitoneal fibrosis and abdominal neoplasms. Thrombosis can occur throughout the renal circulation, including the renal veins, with antiphospholipid antibody syndrome. Renal vein thrombosis can also be secondary to nephrotic syndrome, particularly membranous nephropathy. Other hypercoagulable states less commonly associated with renal vein thrombosis include proteins C and S, antithrombin deficiency, factor V Leiden, disseminated malignancy, and oral contraceptives. Severe nephrotic syndrome may also predispose patients to renal vein thrombosis.
Diagnostic screening can be performed with Doppler ultrasonography, which is more sensitive than ultrasonography alone. CT angiography is nearly 100% sensitive. Magnetic resonance angiography is another option but is more expensive. Treatment for renal vein thrombosis consists of anticoagulation and therapy for the underlying cause. Endovascular thrombolysis may be considered in severe cases. Occasionally, nephrectomy may be undertaken for life-threatening complications. Vena caval filters are often used to prevent migration of thrombi.
342 |
Nephrolithiasis |
Nephrolithiasis, or kidney stone disease, is a common, painful, and costly condition. Each year, billions of dollars are spent on nephrolithiasis-related activity, with the majority of expenditures on surgical treatment of existing stones. While a stone may form due to crystallization of lithogenic factors in the upper urinary tract, it can subsequently move into the ureter and cause renal colic. Although nephrolithiasis is rarely fatal, patients who have had renal colic report that it is the worst pain they have ever experienced. The evidence on which to base clinical recommendations is not as strong as desired; nonetheless, most experts agree that the recurrence of most, if not all, types of stones can be prevented with careful evaluation and targeted recommendations. Preventive treatment may be lifelong; therefore, an in-depth understanding of this condition must inform the implementation of tailored interventions that are most appropriate for and acceptable to the patient.
There are various types of kidney stones. It is clinically important to identify the stone type, which informs prognosis and selection of the optimal preventive regimen. Calcium oxalate stones are most common (~75%); next, in order, are calcium phosphate (~15%), uric acid (~8%), struvite (~1%), and cystine (<1%) stones. Many stones are a mixture of crystal types (e.g., calcium oxalate and calcium phosphate) and also contain protein in the stone matrix. Rarely, stones are composed of medications, such as acyclovir, indinavir, and triamterene.
Infectious stones, if not appropriately treated, can have devastating consequences and lead to end-stage renal disease. Consideration should be given to teaching practitioners strategies to prevent stone recurrence and its related morbidity.
EPIDEMIOLOGY
![]() Nephrolithiasis is a global disease. Data suggest an increasing prevalence, likely due to Westernization of lifestyle habits (e.g., dietary changes, increasing body mass index). National Health and Nutrition Examination Survey data for 2007–2010 indicate that up to 19% of men and 9% of women will develop at least one stone during their lifetime. The prevalence is ~50% lower among black individuals than among whites. The incidence of nephrolithiasis (i.e., the rate at which previously unaffected individuals develop their first stone) also varies by age, sex, and race. Among white men, the peak annual incidence is ~3.5 cases/1000 at age 40 and declines to ~2 cases/1000 by age 70. Among white women in their thirties, the annual incidence is ~2.5 cases/1000; the figure decreases to ~1.5/1000 at age 50 and beyond. In addition to the medical costs associated with nephrolithiasis, this condition also has a substantial economic impact, as those affected are often of working age. Once an individual has had a stone, the prevention of a recurrence is essential. Published recurrence rates vary by the definitions and diagnostic methods used. Some reports have relied on symptomatic events, while others have been based on imaging. Most experts agree that radiographic evidence of a second stone should be considered to represent a recurrence, even if the stone has not yet caused symptoms.
Nephrolithiasis is a global disease. Data suggest an increasing prevalence, likely due to Westernization of lifestyle habits (e.g., dietary changes, increasing body mass index). National Health and Nutrition Examination Survey data for 2007–2010 indicate that up to 19% of men and 9% of women will develop at least one stone during their lifetime. The prevalence is ~50% lower among black individuals than among whites. The incidence of nephrolithiasis (i.e., the rate at which previously unaffected individuals develop their first stone) also varies by age, sex, and race. Among white men, the peak annual incidence is ~3.5 cases/1000 at age 40 and declines to ~2 cases/1000 by age 70. Among white women in their thirties, the annual incidence is ~2.5 cases/1000; the figure decreases to ~1.5/1000 at age 50 and beyond. In addition to the medical costs associated with nephrolithiasis, this condition also has a substantial economic impact, as those affected are often of working age. Once an individual has had a stone, the prevention of a recurrence is essential. Published recurrence rates vary by the definitions and diagnostic methods used. Some reports have relied on symptomatic events, while others have been based on imaging. Most experts agree that radiographic evidence of a second stone should be considered to represent a recurrence, even if the stone has not yet caused symptoms.
ASSOCIATED MEDICAL CONDITIONS
Nephrolithiasis is a systemic disorder. Several conditions predispose to stone formation, including gastrointestinal malabsorption (e.g., Crohn’s disease, gastric bypass surgery), primary hyperparathyroidism, obesity, type 2 diabetes mellitus, and distal renal tubular acidosis. A number of other medical conditions are more likely to be present in individuals with a history of nephrolithiasis, including hypertension, gout, cholelithiasis, reduced bone mineral density, and chronic kidney disease.
Individuals with medullary sponge kidney (MSK), a condition designated by an anatomic description, often have metabolic abnormalities, such as higher levels of urine calcium and lower levels of urine citrate, and are more likely to form calcium phosphate stones. As intravenous urography is now rarely used, the diagnosis of MSK has become less frequent. Fortunately, the diagnosis of MSK does not change either the evaluation or the treatment recommendations; thus, it is not essential in pursuing the diagnosis of nephrolithiasis.
Although nephrolithiasis does not directly cause upper urinary tract infections (UTIs), a UTI in the setting of an obstructing stone is a urologic emergency (“pus under pressure”) and requires urgent intervention to reestablish drainage.
PATHOGENESIS
In the consideration of the processes involved in crystal formation, it is helpful to view urine as a complex solution. A clinically useful concept is supersaturation (the point at which the concentration product exceeds the solubility product). However, even though the urine in most individuals is supersaturated with respect to one or more types of crystals, the presence of inhibitors of crystallization prevents the majority of the population from continuously forming stones. The most clinically important inhibitor of calcium-containing stones is urine citrate. While supersaturation is a calculated value (rather than being directly measured) and does not perfectly predict stone formation, it is a useful guide as it integrates the multiple factors that are measured in a 24-h urine collection.
Recent studies have changed the paradigm for the site of initiation of stone formation. Renal biopsies of stone formers have revealed calcium phosphate in the renal interstitium. It is hypothesized that this calcium phosphate extends down to the papilla and erodes through the papillary epithelium, where it provides a site for deposition of calcium oxalate and calcium phosphate crystals. The majority of calcium oxalate stones grow on calcium phosphate at the tip of the renal papilla (Randall’s plaque). Thus, the process of stone formation may begin years before a clinically detectable stone is identified. The processes involved in interstitial deposition are under active investigation.
RISK FACTORS
Risk factors for nephrolithiasis can be categorized as dietary, nondietary, or urinary. These risk factors vary by stone type and by clinical characteristics.
Dietary Risk Factors Patients who develop stones often change their diet; therefore, studies that retrospectively assess diet may be hampered by recall bias. Some studies have examined the relation between diet and changes in the lithogenic composition of the urine, often using calculated supersaturation. However, the composition of the urine does not perfectly predict risk, and not all components that modify risk are included in the calculation of supersaturation. Thus, dietary associations are best investigated by prospective studies that examine actual stone formation as the outcome. Dietary factors that are associated with an increased risk of nephrolithiasis include animal protein, oxalate, sodium, sucrose, and fructose. Dietary factors associated with a lower risk include calcium, potassium, and phytate.
CALCIUM The role of dietary calcium deserves special attention. Although in the past dietary calcium had been suspected of increasing the risk of stone disease, several prospective observational studies and a randomized controlled trial have demonstrated that higher dietary calcium intake is related to a lower risk of stone formation. The reduction in risk associated with higher calcium intake may be due to a reduction in intestinal absorption of dietary oxalate that results in lower urine oxalate. Low calcium intake is contraindicated as it increases the risk of stone formation and may contribute to lower bone density in stone formers.
Despite similar bioavailability, supplemental calcium may increase the risk of stone formation. The discrepancy between the risks from dietary calcium and calcium supplements may be due to the timing of supplemental calcium intake or to higher total calcium consumption leading to higher urinary calcium excretion.
OXALATE Urinary oxalate is derived from both endogenous production and absorption of dietary oxalate. Owing to its low and often variable bioavailability, much of the oxalate in food may not be readily absorbed. However, absorption may be higher in stone formers. Although observational studies demonstrate that dietary oxalate is only a weak risk factor for stone formation, urinary oxalate is a strong risk factor for calcium oxalate stone formation, and efforts to avoid high oxalate intake should thus be beneficial.
OTHER NUTRIENTS Several other nutrients have been studied and implicated in stone formation. Higher intake of animal protein may lead to increased excretion of calcium and uric acid as well as to decreased urinary excretion of citrate, all of which increase the risk of stone formation. Higher sodium and sucrose intake increases calcium excretion independent of calcium intake. Higher potassium intake decreases calcium excretion, and many potassium-rich foods increase urinary citrate excretion due to their alkali content. Other dietary factors that have been inconsistently associated with lower stone risk include magnesium and phytate.
Vitamin C supplements are associated with an increased risk of calcium oxalate stone formation, possibly because of raised levels of oxalate in urine. Thus, calcium oxalate stone formers should be advised to avoid vitamin C supplements. Although high doses of supplemental vitamin B6 may be beneficial in selected patients with type 1 primary hyperoxaluria, the benefit of supplemental vitamin B6 in other patients is uncertain.
FLUIDS AND BEVERAGES The risk of stone formation increases as urine volume decreases. When the urine output is less than 1 L/d, the risk of stone formation more than doubles. Fluid intake is the main determinant of urine volume, and the importance of fluid intake in preventing stone formation has been demonstrated in observational studies and in a randomized controlled trial. Observational studies have found that coffee, tea, beer, and wine are associated with a reduced risk of stone formation. Sugar-sweetened carbonated beverage consumption may increase risk.
Nondietary Risk Factors Age, race, body size, and environment are important risk factors for nephrolithiasis. The incidence of stone disease is highest in middle-aged white men, but stones can form in infants as well as in the elderly. There is geographic variability, with the highest prevalence in the southeastern United States. Weight gain increases the risk of stone formation, and the increasing prevalence of nephrolithiasis in the United States may be due in part to the increasing prevalence of obesity. Environmental and occupational influences that may lead to lower urine volume, such as working in a hot environment or lack of ready access to water or a bathroom, are important considerations.
Urinary Risk Factors • URINE VOLUME As mentioned above, lower urine volume results in increased concentrations of lithogenic factors and is a common and readily modifiable risk factor. A randomized trial has demonstrated the effectiveness of elevated fluid intake in increasing urine volume and reducing the risk of stone recurrence.
URINE CALCIUM Higher urine calcium excretion increases the likelihood of formation of calcium oxalate and calcium phosphate stones. While the term hypercalciuria is often used, there is no widely accepted cutoff that distinguishes between normal and abnormal urine calcium excretion. In fact, the relation between urine calcium and stone risk appears to be continuous; thus the use of an arbitrary threshold should be avoided. Levels of urine calcium excretion are higher in individuals with a history of nephrolithiasis; however, the mechanisms remain poorly understood. Greater gastrointestinal calcium absorption is one important contributor, and greater bone turnover (with a resultant reduction in bone mineral density) may be another. Primary renal calcium loss, with lower serum calcium concentrations and elevated serum levels of parathyroid hormone (PTH) (and a normal 25-hydroxy vitamin D level), is rare.
URINE OXALATE Higher urine oxalate excretion increases the likelihood of calcium oxalate stone formation. As for urine calcium, no definition for “abnormal” urine oxalate excretion is widely accepted. Given that the relation between urine oxalate and stone risk is continuous, simple dichotomization of urine oxalate excretion is not helpful in assessing risk. The two sources of urine oxalate are endogenous generation and dietary intake. Dietary oxalate is the major contributor and also the source that can be modified. Notably, higher dietary calcium intake reduces gastrointestinal oxalate absorption and thereby reduces urine oxalate.
URINE CITRATE Urine citrate is a natural inhibitor of calcium-containing stones; thus, lower urine citrate excretion increases the risk of stone formation. Citrate reabsorption is influenced by the intracellular pH of proximal tubular cells. Metabolic acidosis will lead to a reduction in citrate excretion by increasing reabsorption of filtered citrate. However, a notable proportion of patients have lower urine citrate for reasons that remain unclear.
URINE URIC ACID Higher urine levels of uric acid—a risk factor for uric acid stone formation—are found in individuals with excess purine consumption and rare genetic conditions that lead to overproduction of uric acid. This characteristic does not appear to be associated with the risk of calcium oxalate stone formation.
URINE pH Urine pH influences the solubility of some crystal types. Uric acid stones form only when the urine pH is consistently ≤5.5 or lower, whereas calcium phosphate stones are more likely to form when the urine pH is ≥6.5 or higher. Cystine is more soluble at higher urine pH. Calcium oxalate stones are not influenced by urine pH.
![]() Genetic Risk Factors The risk of nephrolithiasis is more than twofold greater in individuals with a family history of stone disease. This association is likely due to a combination of genetic predisposition and similar environmental exposures. While a number of monogenic disorders cause nephrolithiasis, the genetic contributors to common forms of stone disease remain to be determined.
Genetic Risk Factors The risk of nephrolithiasis is more than twofold greater in individuals with a family history of stone disease. This association is likely due to a combination of genetic predisposition and similar environmental exposures. While a number of monogenic disorders cause nephrolithiasis, the genetic contributors to common forms of stone disease remain to be determined.
The two most common and well-characterized rare monogenic disorders that lead to stone formation are primary hyperoxaluria and cystinuria. Primary hyperoxaluria is an autosomal recessive disorder that causes excessive endogenous oxalate generation by the liver, with consequent calcium oxalate stone formation and crystal deposition in organs. Intraparenchymal calcium oxalate deposition in the kidney can eventually lead to renal failure. Cystinuria is an autosomal recessive disorder that causes abnormal reabsorption of filtered dibasic amino acids. The excessive urinary excretion of cystine, which is poorly soluble, leads to cystine stone formation. Cystine stones are visible on plain radiographs and often manifest as staghorn calculi or multiple bilateral stones. Repeat episodes of obstruction and instrumentation can cause chronic renal impairment.
APPROACH TO THE PATIENT:
Nephrolithiasis
At present, there are no widely accepted, evidence-based guidelines for the evaluation and treatment of nephrolithiasis. However, there are standard approaches to patients with acute and chronic presentations that can reasonably guide the clinical evaluation.
It typically requires weeks to months (and often much longer) for a kidney stone to grow to a clinically detectable size. Although the passage of a stone is a dramatic event, stone formation and growth are characteristically clinically silent. A stone can remain asymptomatic in the kidney for years or even decades before signs (e.g., hematuria) or symptoms (e.g., pain) become apparent. Thus, it is important to remember that the onset of symptoms, typically attributable to a stone moving into the ureter, does not provide insight into when the stone actually formed. The factors that induce stone movement are unknown.
Clinical Presentation and Differential Diagnosis There are two common presentations for individuals with an acute stone event: renal colic and painless gross hematuria. Renal colic is a misnomer because pain typically does not subside completely; rather, it varies in intensity. When a stone moves into the ureter, the discomfort often begins with a sudden onset of unilateral flank pain. The intensity of the pain can increase rapidly, and there are no alleviating factors. This pain, which is accompanied often by nausea and occasionally by vomiting, may radiate, depending on the location of the stone. If the stone lodges in the upper part of the ureter, pain may radiate anteriorly; if the stone is in the lower part of the ureter, pain can radiate to the ipsilateral testicle in men or the ipsilateral labium in women. Occasionally, a patient has gross hematuria without pain.
Other diagnoses may be confused with acute renal colic. If the stone is lodged at the right ureteral pelvic junction, symptoms may mimic those of acute cholecystitis. If the stone blocks the ureter as it crosses over the right pelvic brim, symptoms may mimic acute appendicitis, whereas blockage at the left pelvic brim may be confused with acute diverticulitis. If the stone lodges in the ureter at the ureterovesical junction, the patient may experience urinary urgency and frequency. In female patients, the latter symptoms may lead to an incorrect diagnosis of bacterial cystitis; the urine will contain red and white blood cells, but the urine culture will be negative. An obstructing stone with proximal infection may present as acute pyelonephritis. A UTI in the setting of ureteral obstruction is a medical emergency that requires immediate restoration of drainage by placement of either a ureteral stent or a percutaneous nephrostomy tube. Other conditions to consider in the differential diagnosis include muscular or skeletal pain, herpes zoster, duodenal ulcer, abdominal aortic aneurysm, gynecologic conditions, ureteral stricture, and ureteral obstruction by materials other than a stone, such as a blood clot or sloughed papilla. Extraluminal processes can lead to ureteral compression and obstruction; however, because of the gradual onset, these conditions do not typically present with renal colic.
Diagnosis and Intervention Serum chemistry findings are typically normal, but the white blood cell count may be elevated. Examination of the urine sediment will usually reveal red and white blood cells and occasionally crystals (Fig. 342-1). The absence of hematuria does not exclude a stone, particularly when urine flow is completely obstructed by a stone.
FIGURE 342-1 Urine sediment from a patient with calcium oxalate stones (left) and a patient with cystine stones (right). Calcium oxalate dihydrate crystals are bipyramidally shaped, and cystine crystals are hexagonal. (Left panel image courtesy of Dr. John Lieske, Mayo Clinic.)
The diagnosis is often made on the basis of the history, physical examination, and urinalysis. Thus, it may not be necessary to wait for radiographic confirmation before treating the symptoms. The diagnosis is confirmed by an appropriate imaging study—preferably helical CT, which is highly sensitive, allows visualization of uric acid stones (traditionally considered “radiolucent”), and is able to avoid radiocontrast (Fig. 342-2). Helical CT detects stones as small as 1 mm that may be missed by other imaging modalities. Typically, helical CT reveals a ureteral stone or evidence of recent passage (e.g., perinephric stranding or hydronephrosis), whereas a plain abdominal radiograph (kidney/ureter/bladder, or KUB) can miss a stone in the ureter or kidney, even if it is radiopaque, and does not provide information on obstruction. Abdominal ultrasound offers the advantage of avoiding radiation and provides information on hydronephrosis, but it is not as sensitive as CT and images only the kidney and possibly the proximal segment of the ureter; thus most ureteral stones are not detectable by ultrasound.
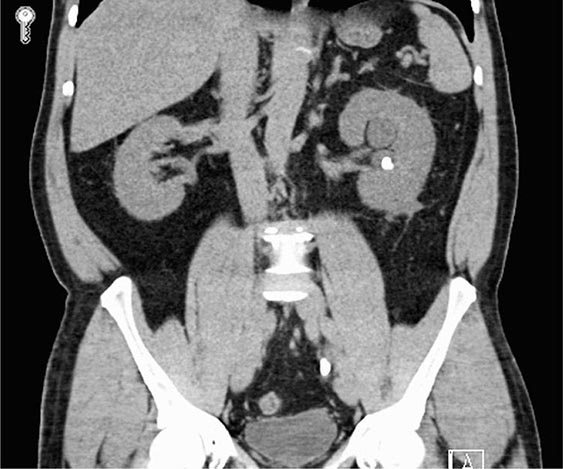
FIGURE 342-2 Coronal noncontrast CT image from a patient who presented with left-sided renal colic. An obstructing calculus, present in the distal left ureter at the level of S1, measures 10 mm in maximal dimension. There is severe left hydroureteronephrosis and associated left perinephric fat stranding. In addition, there is a nonobstructing 6-mm left renal calculus in the interpolar region. (Image courtesy of Dr. Stuart Silverman, Brigham and Women’s Hospital.)
Many patients who experience their first episode of colic seek emergent medical care. Randomized trials have demonstrated that parenterally administered nonsteroidal anti-inflammatory drugs (such as ketorolac) are just as effective as opioids in relieving symptoms and have fewer side effects. Excessive fluid administration has not been shown to be beneficial; therefore, the goal should be to maintain euvolemia. If the pain can be adequately controlled and the patient is able to take fluids orally, hospitalization can be avoided. Use of an alpha-blocker may increase the rate of spontaneous stone passage.
Urologic intervention should be postponed unless there is evidence of UTI, a low probability of spontaneous stone passage (e.g., a stone measuring ≥6 mm or an anatomic abnormality), or intractable pain. A ureteral stent may be placed cystoscopically, but this procedure typically requires general anesthesia, and the stent can be quite uncomfortable, may cause gross hematuria, and may increase the risk of UTI.
If an intervention is indicated, the selection of the most appropriate intervention is determined by the size, location, and composition of the stone; the urinary tract anatomy; and the experience of the urologist. Extracorporeal shockwave lithotripsy, the least invasive option, uses shock waves generated outside the body to fragment the stone. An endourologic approach can remove a stone by basket extraction or laser fragmentation. For large upper-tract stones, percutaneous nephrostolithotomy has the highest likelihood of rendering the patient stone-free. Advances in urologic approaches and instruments have nearly eliminated the need for open surgical procedures such as ureterolithotomy or pyelolithotomy.
Evaluation for Stone Prevention More than half of first-time stone formers will have a recurrence within 10 years. A careful evaluation is indicated to identify predisposing factors, which can then be modified to reduce the risk of new stone formation. It is appropriate to proceed with an evaluation even after the first stone because recurrences are common and are usually preventable with inexpensive lifestyle modifications or other treatments.
History A detailed history, obtained from the patient and from a thorough review of medical records, should include the number and frequency of episodes (distinguishing stone passage from stone formation) and previous imaging studies, interventions, evaluations, and treatments. Inquiries about the patient’s medical history should cover UTIs, bariatric surgery, gout, hypertension, and diabetes mellitus. A family history of stone disease may reveal a genetic predisposition. A complete list of current prescription and over-the-counter medications as well as vitamin and mineral supplements is essential. The review of systems should focus on identifying possible etiologic factors related to low urine volume (e.g., high insensible losses; low fluid intake) and gastrointestinal malabsorption as well as on ascertaining how frequently the patient voids during the day and overnight.
A large body of compelling evidence has demonstrated the important role of diet in stone disease. Thus, the dietary history should encompass information on usual dietary habits (meals and snacks), calcium intake, consumption of high-oxalate foods (spinach, rhubarb, potatoes), and fluid intake (including specific beverages typically consumed).
Physical Examination The physical examination should assess weight, blood pressure, costovertebral angle tenderness, and lower-extremity edema as well as signs of other systemic conditions such as primary hyperparathyroidism and gout.
Laboratory Evaluation If not recently measured, the following serum levels should be determined: electrolytes (to uncover hypokalemia or renal tubular acidosis), creatinine, calcium, and uric acid. The PTH level should be measured if indicated by high-normal or elevated serum and urine calcium concentrations. Often, 25-hydroxy vitamin D is measured in concert with PTH to investigate the possible role of secondarily elevated PTH levels in the setting of vitamin D insufficiency.
The urinalysis, including examination of the sediment, can provide useful information. In individuals with asymptomatic residual renal stones, red and white blood cells are frequently present in urine. If there is concern about the possibility of an infection, a urine culture should be performed. The sediment may also reveal crystals (Fig. 342-1), which may help identify the stone type and also provide prognostic information, as crystalluria is a strong risk factor for new stone formation.
The results from 24-h urine collections serve as the cornerstone on which therapeutic recommendations are based. Recommendations on lifestyle modification should be deferred until urine collection is complete. As a baseline assessment, patients should collect at least two 24-h urine samples while consuming their usual diet and usual volume of fluid. The following factors should be measured: total volume, calcium, oxalate, citrate, uric acid, sodium, potassium, phosphorus, pH, and creatinine. When available, the calculated supersaturation is also informative. There is substantial day-to-day variability in the 24-h excretion of many relevant factors; therefore, obtaining values from two collections is important before committing a patient to long-term lifestyle changes or medication. The interpretation of the 24-h urine results should take into account that the collections are usually performed on a weekend day when the patient is staying at home; an individual’s habits may differ dramatically (beneficially or detrimentally) at work or outside the home. Specialized testing, such as calcium loading or restriction, is not recommended as it does not influence clinical recommendations.
Stone composition analysis is essential if a stone or fragment is available; patients should be encouraged to retrieve passed stones. The stone type cannot be determined with certainty from 24-h urine results.
Imaging The “gold standard” diagnostic test is helical CT without contrast. If not already performed during an acute episode, a CT should be considered to definitively establish the baseline stone burden. A suboptimal imaging study may not detect a residual stone that, if subsequently passed, would be mistaken for a new stone. In this instance, the preventive medical regimen might be unnecessarily changed as the result of a preexisting stone.
Recommendations for follow-up imaging should be tailored to the individual patient. While CT provides the best information, the radiation dose is substantially higher than from other modalities; therefore, CT should be performed only if the results will lead to a change in clinical recommendations. Although they are less sensitive, renal ultrasound or a KUB exam are typically used to minimize radiation exposure, with recognition of the limitations.
Prevention of New Stone Formation Recommendations for preventing stone formation depend on the stone type and the results of metabolic evaluation. After remediable secondary causes of stone formation (e.g., primary hyperparathyroidism) are excluded, the focus should turn to modification of the urine composition to reduce the risk of new stone formation. The urinary constituents are continuous variables, and the associated risk is continuous; thus, there is no definitive threshold. Dichotomization into “normal” and “abnormal” can be misleading and should be avoided.
For all stone types, consistently diluted urine reduces the likelihood of crystal formation. The urine volume should be at least 2 L/d. Because of differences in insensible fluid losses and fluid intake from food sources, the required total fluid intake will vary from person to person. Rather than specify how much to drink, it is more helpful to educate patients about how much more they need to drink in light of their 24-h urine volume. For example, if the daily urine volume is 1.5 L, then the patient should be advised to drink at least 0.5 L more per day in order to increase the urine volume to the goal of 2 L/day.
RECOMMENDATIONS FOR SPECIFIC STONE TYPES
Calcium Oxalate Risk factors for calcium oxalate stones include higher urine calcium, higher urine oxalate, and lower urine citrate. This stone type is insensitive to pH in the physiologic range.
Individuals with higher urine calcium excretion tend to absorb a higher percentage of ingested calcium. Nevertheless, dietary calcium restriction is not beneficial and, in fact, is likely to be harmful (see “Dietary Risk Factors,” above). In a randomized trial in men with high urine calcium and recurrent calcium oxalate stones, a diet containing 1200 mg of calcium and a low intake of sodium and animal protein significantly reduced subsequent stone formation from that with a low-calcium diet (400 mg/d). Excessive calcium intake (>1200 mg/d) should be avoided.
A thiazide diuretic, in doses higher than those used to treat hypertension, can substantially lower urine calcium excretion. Several randomized controlled trials have demonstrated that thiazide diuretics can reduce calcium oxalate stone recurrence by ~50%. When a thiazide is prescribed, dietary sodium restriction is essential to obtain the desired reduction in urinary calcium excretion. While bisphosphonates may reduce urine calcium excretion in some individuals, there are no data on whether this class of medication can reduce stone formation; therefore, bisphosphonates cannot be recommended solely for stone prevention at present.
A reduction in urine oxalate will in turn reduce the supersaturation of calcium oxalate. In patients with the common form of nephrolithiasis, avoiding high-dose vitamin C supplements is the only known strategy that reduces endogenous oxalate production.
Oxalate is a metabolic end product; therefore, any dietary oxalate that is absorbed will be excreted in the urine. Reducing absorption of exogenous oxalate involves two approaches. First, the avoidance of foods that contain high amounts of oxalate, such as spinach, rhubarb, and potatoes, is prudent. However, extreme oxalate restriction has not been demonstrated to reduce stone recurrence and could be harmful to overall health, given other health benefits of many foods that are erroneously considered to be high in oxalate. Controversy exists regarding the most clinically relevant measure of the oxalate content of foods (e.g., bioavailability). Notably, the absorption of oxalate is reduced by higher calcium intake; therefore, individuals with higher-than-desired urinary oxalate should be counseled to consume adequate calcium. Oxalate absorption can be influenced by the intestinal microbiota, depending on the presence of oxalate-degrading bacteria. Currently, however, there are no available therapies to alter the microbiota that beneficially affect urinary oxalate excretion over the long term.
Citrate is a natural inhibitor of calcium oxalate and calcium phosphate stones. Higher-level consumption of foods rich in alkali (i.e., fruits and vegetables) can increase urine citrate. For patients with lower urine citrate in whom dietary modification does not adequately increase urine citrate, the addition of supplemental alkali (typically potassium citrate) will lead to an increase in urinary citrate excretion. Sodium salts, such as sodium bicarbonate, while successful in raising urine citrate, are typically avoided due to the adverse effects of sodium on urine calcium excretion.
Past reports suggested that higher levels of urine uric acid may increase the risk of calcium oxalate stones, but more recent studies do not support this association. However, allopurinol reduced stone recurrence in one randomized controlled trial in patients with calcium oxalate stones and high urine uric acid levels. The lack of association between urine uric acid level and calcium oxalate stones suggests that a different mechanism underlies the observed beneficial effect of allopurinol.
Additional dietary modifications may be beneficial in reducing stone recurrence. Restriction of nondairy animal protein (e.g., meat, chicken, seafood) is a reasonable approach and may result in higher excretion of citrate and lower excretion of calcium. In addition, reducing sodium intake to <2.5 g/d may decrease urinary excretion of calcium. Sucrose and fructose intake should be minimized.
For adherence to a dietary pattern that is more manageable for patients than manipulating individual nutrients, the DASH (Dietary Approaches to Stop Hypertension) diet provides an appropriate and readily available option. Randomized trials have conclusively shown the DASH diet to reduce blood pressure. At present, only data from observational studies are available, but these demonstrate a strong and consistent inverse association between the DASH diet and risk of stone formation.
Calcium Phosphate Calcium phosphate stones share risk factors with calcium oxalate stones, including higher concentrations of urine calcium and lower concentrations of urine citrate, but additional factors deserve attention. Higher urine phosphate levels and higher urine pH (typically ≥6.5) are associated with an increased likelihood of calcium phosphate stone formation. Calcium phosphate stones are more common in patients with distal renal tubular acidosis and primary hyperparathyroidism.
There are no randomized trials on which to base preventive recommendations for calcium phosphate stone formers, so the interventions are focused on modification of the recognized risk factors. Thiazide diuretics (with sodium restriction) may be used to reduce urine calcium, as described above for calcium oxalate stones. In patients with low urine citrate levels, alkali supplements (e.g., potassium citrate) may be used to increase these concentrations. However, the urine pH of these patients should be monitored carefully because supplemental alkali can raise urine pH, thereby potentially increasing the risk of stone formation. Reduction of dietary phosphate may be beneficial by reducing urine phosphate excretion.
Uric Acid The two main risk factors for uric acid stones are persistently low urine pH and higher uric acid excretion. Urine pH is the predominant influence on uric acid solubility; therefore, the mainstay of prevention of uric acid stone formation entails increasing urine pH. While acidifying the urine is not easily done, alkalinizing the urine can be readily achieved by increasing the intake of foods rich in alkali (e.g., fruits and vegetables) and reducing the intake of foods that produce acid (e.g., animal flesh). If necessary, supplementation with bicarbonate or citrate salts (preferably potassium citrate) can be used to reach the recommended pH goal of 6 to 7 throughout the day and night.
Urine uric acid excretion is determined by uric acid generation. Uric acid is the end product of purine metabolism; thus reduced consumption of purine-containing foods can lower urine uric acid excretion. It is noteworthy that the serum uric acid level is dependent on the fractional excretion of uric acid and therefore does not provide information on urine uric acid excretion. For example, an individual with high uric acid generation and concurrent high fractional excretion of uric acid will have high urine uric acid excretion with a normal (or even low) serum uric acid level. If alkalinization of the urine alone is not successful and if dietary modifications do not reduce urine uric acid sufficiently, then the use of a xanthine oxidase inhibitor, such as allopurinol or febuxostat, can reduce urine uric acid excretion by 40–50%.
Cystine Cystine excretion is not easily modified. Long-term dietary cystine restriction is not feasible and is unlikely to be successful; thus the focus for cystine stone prevention is on increasing cystine solubility. This goal may be achieved by treatment with medication that covalently binds to cystine (tiopronin and penicillamine) and a medication that raises urine pH. Tiopronin is the preferred choice due to its better adverse event profile. The preferred alkalinizing agent is potassium citrate as sodium salts may increase cystine excretion. As with all stone types, and especially in patients with cystinuria, maintaining a high urine volume is an essential component of the preventive regimen.
Struvite Struvite stones, also known as infection stones or triple-phosphate stones, form only when the upper urinary tract is infected with urease-producing bacteria such as Proteus mirabilis, Klebsiella pneumoniae, or Providencia species. Urease produced by these bacteria hydrolyzes urea and may elevate the urine pH to a supraphysiologic level (>8.0). Struvite stones may grow quickly and fill the renal pelvis (staghorn calculi).
Struvite stones require complete removal by a urologist. New stone formation can be avoided by the prevention of UTIs. In patients with recurrent upper UTIs (e.g., some individuals with surgically altered urinary drainage or spinal cord injury), the urease inhibitor acetohydroxamic acid can be considered; however, this agent should be used with caution because of potential side effects.
LONG-TERM FOLLOW-UP
In general, the preventive regimens described above do not cure the underlying pathophysiologic process. Thus these recommendations typically need to be followed for the patient’s lifetime, and it is essential to tailor recommendations in a way that is acceptable to the patient. Because the memory of the acute stone event fades and patients often return to old habits (e.g., insufficient fluid intake), long-term follow-up is important to ensure that the preventive regimen has been implemented and has resulted in the desired reduction in the risk of new stone formation.
Follow-up imaging should be planned thoughtfully. Many patients with recurrent episodes of renal colic that lead to emergency room visits often undergo repeat CT studies. While CT does provide the best information, the radiation dose is substantially higher than that with plain abdominal radiography (KUB). Small stones may be missed by KUB, and ultrasound has a limited ability to determine size and number of stones. Minimizing radiation exposure should be a goal of the long-term follow-up plan and must be balanced against the gain in diagnostic information.
343 |
Urinary Tract Obstruction |
Obstruction to the flow of urine, with attendant stasis and elevation in urinary tract pressure, impairs renal and urinary conduit functions and is a common cause of acute and chronic kidney disease (obstructive nephropathy). With early relief of obstruction, the defects in function usually disappear completely. However, chronic obstruction may produce permanent loss of renal mass (renal atrophy) and excretory capability, as well as enhanced susceptibility to local infection and stone formation. Early diagnosis and prompt therapy are, therefore, essential to minimize the otherwise devastating effects of obstruction on kidney structure and function.
ETIOLOGY
Obstruction to urine flow can result from intrinsic or extrinsic mechanical blockade as well as from functional defects not associated with fixed occlusion of the urinary drainage system. Mechanical obstruction can occur at any level of the urinary tract, from the renal calyces to the external urethral meatus. Normal points of narrowing, such as the ureteropelvic and ureterovesical junctions, bladder neck, and urethral meatus, are common sites of obstruction. When obstruction is above the level of the bladder, unilateral dilatation of the ureter (hydroureter) and renal pyelocalyceal system (hydronephrosis) occurs; lesions at or below the level of the bladder cause bilateral involvement.
Common forms of obstruction are listed in Table 343-1. Childhood causes include congenital malformations, such as narrowing of the ureteropelvic junction and abnormal insertion of the ureter into the bladder, the most common cause. Vesicoureteral reflux in the absence of urinary tract infection or bladder neck obstruction often resolves with age. Reinsertion of the ureter into the bladder is indicated if reflux is severe and unlikely to improve spontaneously, if renal function deteriorates, or if urinary tract infections recur despite chronic antimicrobial therapy. Vesicoureteral reflux may cause prenatal hydronephrosis and, if severe, can lead to recurrent urinary infections and renal scarring in childhood. Posterior urethral valves are the most common cause of bilateral hydronephrosis in boys. In adults, urinary tract obstruction (UTO) is due mainly to acquired defects. Pelvic tumors, calculi, and urethral stricture predominate. Ligation of, or injury to, the ureter during pelvic or colonic surgery can lead to hydronephrosis which, if unilateral, may remain undetected. Obstructive uropathy may also result from extrinsic neoplastic (carcinoma of cervix or colon) or inflammatory disorders. Lymphomas and pelvic or colonic neoplasms with retroperitoneal involvement are causes of ureteral obstruction. As many as 50% of men over 40 years old may have lower urinary tract symptoms associated with benign prostatic hypertrophy, but these symptoms may occur without bladder outlet obstruction.
|
COMMON MECHANICAL CAUSES OF URINARY TRACT OBSTRUCTION |
Functional impairment of urine flow occurs when voiding is altered by abnormal pontine or sacral centers of micturition control. It may be asymptomatic or associated with lower urinary tract symptoms such as frequency, urgency, urge and postmicturition incontinence, nocturia, straining to void, slow stream, hesitancy, or a feeling of incomplete emptying. A history should be sought for trauma, back injury, surgery, diabetes, neurologic or psychiatric conditions, and medications. Causes include neurogenic bladder, often with adynamic ureter, and vesicoureteral reflux. Reflux in children may result in severe unilateral or bilateral hydroureter and hydronephrosis. Urinary retention may be the consequence of α-adrenergic and anticholinergic agents, as well as opiates. Hydronephrosis in pregnancy is due to relaxational effects of progesterone on smooth muscle of the renal pelvis, as well as ureteral compression by the enlarged uterus.
Diagnostic tools to identify anatomic obstruction include urinary flow measurements and a postvoid residual. Cystourethroscopy and urodynamic studies may be reserved for the symptomatic patient to assess the filling phase (cystometry), pressure-volume relationship of the bladder, bladder compliance, and capacity. Pressure-flow analysis evaluates bladder contractility and bladder outlet resistance during voiding. Bladder obstruction is characterized by high pressures in women, whereas in men, a diagnosis of bladder outlet obstruction is based on flow rate and voiding pressures. A voiding cystourethrogram may be useful in evaluating incomplete emptying and bladder neck and urethral pathology.
CLINICAL FEATURES AND PATHOPHYSIOLOGY
The pathophysiology and clinical features of UTO are summarized in Table 343-2. Pain, the symptom that most commonly leads to medical attention, is due to distention of the collecting system or renal capsule. Pain severity is influenced more by the rate at which distention develops than by the degree of distention. Acute supravesical obstruction, as from a stone lodged in a ureter (Chap. 342), is associated with excruciating pain, known as renal colic. This pain often radiates to the lower abdomen, testes, or labia. By contrast, more insidious causes of obstruction, such as chronic narrowing of the ureteropelvic junction, may produce little or no pain and yet result in total destruction of the affected kidney. Flank pain that occurs only with micturition is pathognomonic of vesicoureteral reflux.
|
PATHOPHYSIOLOGY OF BILATERAL URETERAL OBSTRUCTION |
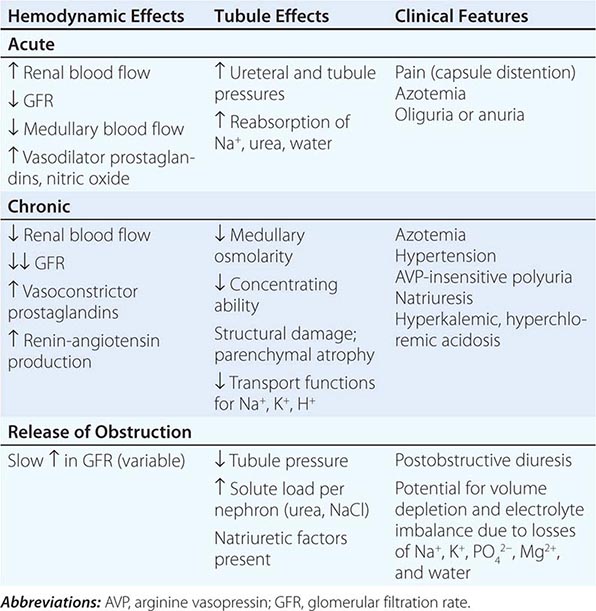
Obstruction of urine flow results in an increase in hydrostatic pressures proximal to the site of obstruction. It is this buildup of pressure that leads to the accompanying pain, the distention of the collecting system in the kidney, and elevated intratubular pressures that initiate tubular dysfunction. As the increased hydrostatic pressure is expressed in the urinary space of the glomeruli, further filtration decreases or stops completely.
Azotemia develops when overall excretory function is impaired, often in the setting of bladder outlet obstruction, bilateral renal pelvic or ureteric obstruction, or unilateral disease in a patient with a solitary functioning kidney. Complete bilateral obstruction should be suspected when acute renal failure is accompanied by anuria. Any patient with renal failure otherwise unexplained, or with a history of nephrolithiasis, hematuria, diabetes mellitus, prostatic enlargement, pelvic surgery, trauma, or tumor should be evaluated for UTO.
In the acute setting, partial, bilateral obstruction may mimic prerenal azotemia with concentrated urine and sodium retention. However, with more prolonged obstruction, symptoms of polyuria and nocturia commonly accompany partial UTO and result from diminished renal concentrating ability. Impairment of transcellular salt reabsorption in the proximal tubule, medullary thick ascending limb of Henle, and collecting duct cells is due to downregulation of transport proteins including the Na+, K+ adenosine triphosphatase (ATPase), NaK2Cl cotransporter (NKCC) in the thick ascending limb, and the epithelial Na+ channel (ENaC) in collecting duct cells. Consequences include failure to produce urine free of salt (natriuresis) and loss of medullary hypertonicity producing a urinary concentrating defect. In addition to direct effects on renal transport mechanisms, increased prostaglandin E2 (PGE2) (due to induction of cyclooxygenase-2 [COX-2]), angiotensin II (with its downregulation of Na+ transporters), and atrial or B-type natriuretic peptides (ANP or BNP) (due to volume expansion in the azotemic patient) contribute to the decreased salt reabsorption along the nephron.
Dysregulation of aquaporin-2 water channels in the collecting duct contributes to the polyuria. The defect usually does not improve with administration of vasopressin and is therefore a form of acquired nephrogenic diabetes insipidus.
Wide fluctuations in urine output in a patient with azotemia should always raise the possibility of intermittent or partial UTO. If fluid intake is inadequate, severe dehydration and hypernatremia may develop. However, as with other causes of poor renal function, excesses of salt and water intake may result in edema and hyponatremia.
Partial bilateral UTO often results in acquired distal renal tubular acidosis, hyperkalemia, and renal salt wasting. The H+-ATPase, situated on the apical membrane of the intercalated cells of the collecting duct, is critical for distal H+ secretion. The trafficking of intracellular H+ pumps from the cytoplasm to the cell membrane is disrupted in UTO. The decreased function of the ENaC, in the apical membrane of neighboring collecting duct principal cells, contributes to decreased Na+ reabsorption (salt-wasting), decreased electronegativity of the tubule lumen, and therefore decreased K+ secretion via K+ channels (hyperkalemia) and H+ secretion via the H+-ATPases (distal renal tubular acidosis [RTA]). Proximal tubule ammoniagenesis, important to the elimination of H+ as NH4+, is impaired. These defects in tubule function are often accompanied by renal tubulointerstitial damage. Azotemia with hyperkalemia and metabolic acidosis should prompt consideration of UTO.
The renal interstitium becomes edematous and infiltrated with mononuclear inflammatory cells early in UTO. Later, interstitial fibrosis and atrophy of the papillae and medulla occur and precede these processes in the cortex. The increase in angiotensin II noted in UTO contributes to the inflammatory response and fibroblast accumulation through mechanisms involving profibrotic cytokines. With time, this process leads to chronic kidney damage.
UTO must always be considered in patients with urinary tract infections or urolithiasis. Urinary stasis encourages the growth of organisms. Urea-splitting bacteria are associated with magnesium ammonium phosphate (struvite) calculi. Hypertension is frequent in acute and subacute unilateral obstruction and is usually a consequence of increased release of renin by the involved kidney. Chronic kidney disease from bilateral UTO, often associated with extracellular volume expansion, may result in significant hypertension. Erythrocytosis, an infrequent complication of obstructive uropathy, is secondary to increased erythropoietin production.
DIAGNOSIS
A history of difficulty in voiding, pain, infection, or change in urinary volume is common. Evidence for distention of the kidney or urinary bladder can often be obtained by palpation and percussion of the abdomen. A careful rectal and genital examination may reveal enlargement or nodularity of the prostate, abnormal rectal sphincter tone, or a rectal or pelvic mass.
Urinalysis may reveal hematuria, pyuria, and bacteriuria. The urine sediment is often normal, even when obstruction leads to marked azotemia and extensive structural damage. An abdominal scout film may detect nephrocalcinosis or a radiopaque stone. As indicated in Fig. 343-1, if UTO is suspected, a bladder catheter should be inserted. Abdominal ultrasonography should be performed to evaluate renal and bladder size, as well as pyelocalyceal contour. Ultrasonography is approximately 90% specific and sensitive for detection of hydronephrosis. False-positive results are associated with diuresis, renal cysts, or the presence of an extrarenal pelvis, a normal congenital variant. Congenital ureteropelvic junction (UPJ) obstruction may be mistaken for renal cystic disease. Hydronephrosis may be absent on ultrasound when obstruction is less than 48 h in duration or associated with volume contraction, staghorn calculi, retroperitoneal fibrosis, or infiltrative renal disease. Duplex Doppler ultrasonography may detect an increased resistive index in urinary obstruction.
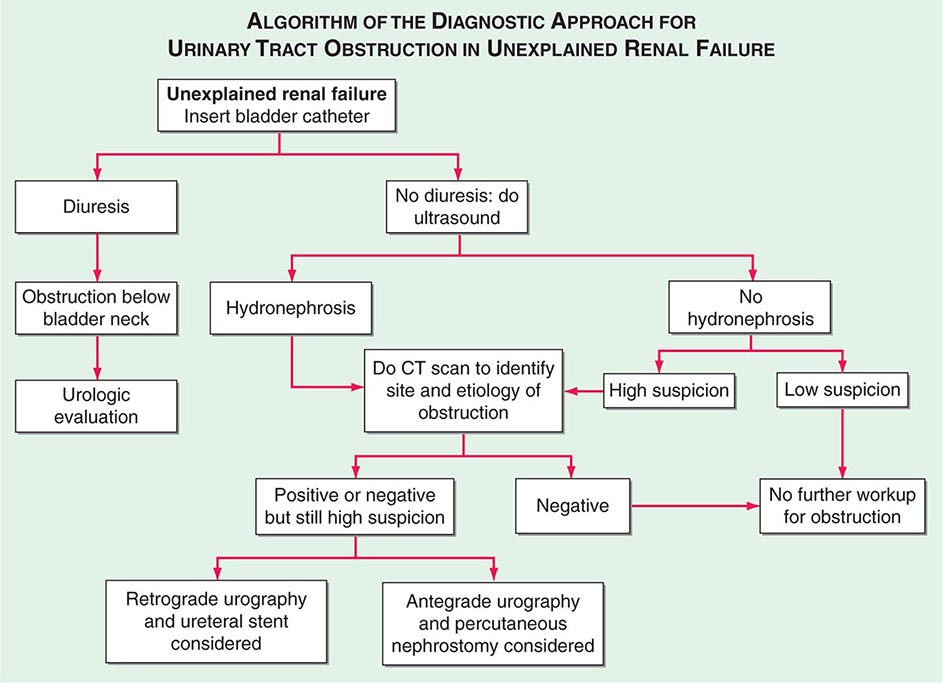
FIGURE 343-1 Diagnostic approach for urinary tract obstruction in unexplained renal failure. CT, computed tomography.
Recent advances in technology have led to alternatives and have largely replaced the once standard intravenous urogram in the further evaluation of UTO. The high-resolution multidetector row computed tomography (CT) scan in particular has advantages of visualizing the retroperitoneum, as well as identifying both intrinsic and extrinsic sites of obstruction. Noncontrast CT scans improve visualization of the urinary tract in the patient with renal impairment and are safer for patients at risk for contrast nephropathy. Magnetic resonance urography is a promising technique but, at this time, not superior to the CT scan and carries the risk of certain gadolinium agents in patients with renal insufficiency, i.e., nephrogenic systemic fibrosis. The intravenous urogram may define the site of obstruction and demonstrate dilatation of the calyces, renal pelvis, and ureter above the obstruction. The ureter may be tortuous in chronic obstruction. Radionuclide scans are able to give differential renal function but give less anatomic detail than CT or intravenous urography (IVU).
To facilitate visualization of a suspected lesion in a ureter or renal pelvis, retrograde or antegrade urography should be attempted. These procedures do not carry risk of contrast-induced acute renal failure in patients with renal insufficiency. The retrograde approach involves catheterization of the involved ureter under cystoscopic control, whereas the antegrade technique necessitates percutaneous placement of a catheter into the renal pelvis. Although the antegrade approach may provide immediate decompression of a unilateral obstructing lesion, many urologists initially attempt the retrograde approach unless the catheterization is unsuccessful.
Voiding cystourethrography is of value in the diagnosis of vesicoureteral reflux and bladder neck and urethral obstructions. Postvoiding films reveal residual urine. Endoscopic visualization by the urologist often permits precise identification of lesions involving the urethra, prostate, bladder, and ureteral orifices.
PROGNOSIS
With relief of obstruction, the prognosis regarding return of renal function depends largely on whether irreversible renal damage has occurred. When obstruction is not relieved, the course will depend mainly on whether the obstruction is complete or incomplete and bilateral or unilateral, as well as whether or not urinary tract infection is also present. Complete obstruction with infection can lead to total destruction of the kidney within days. Partial return of glomerular filtration rate may follow relief of complete obstruction of 1 and 2 weeks’ duration, but after 8 weeks of obstruction, recovery is unlikely. In the absence of definitive evidence of irreversibility, every effort should be made to decompress the obstruction in the hope of restoring renal function at least partially. A renal radionuclide scan, performed after a prolonged period of decompression, may be used to predict the reversibility of renal dysfunction.
POSTOBSTRUCTIVE DIURESIS
Relief of bilateral, but not unilateral, complete obstruction commonly results in polyuria, which may be massive. The urine is usually hypotonic and may contain large amounts of sodium chloride, potassium, phosphate, and magnesium. The natriuresis is due in part to the normal correction of extracellular volume expansion, the increase in natriuretic factors accumulated during the period of renal failure, and depressed salt and water reabsorption when urine flow is reestablished. The retained urea is excreted with improved GFR, resulting in an osmotic diuresis which increases the urine volume of electrolyte-free water. In the majority of patients, this diuresis results in the appropriate excretion of the excesses of retained salt and water. When extracellular volume and composition return to normal, the diuresis usually abates spontaneously. Occasionally, iatrogenic expansion of extracellular volume is responsible for, or sustains, the diuresis observed in the postobstructive period. Replacement with intravenous fluids in amounts less than urinary losses usually prevents this complication. More aggressive fluid management is required in the setting of hypovolemia, hypotension, or disturbances in serum electrolyte concentrations.
The loss of electrolyte-free water with urea may result in hypernatremia. Serum and urine sodium and osmolal concentrations should guide the use of appropriate intravenous replacement. Often replacement with 0.45% saline is required. Relief of obstruction may be followed by urinary salt and water losses severe enough to provoke profound dehydration and vascular collapse. In these patients, decreased tubule reabsorptive capacity is probably responsible for the marked diuresis. Appropriate therapy in such patients includes intravenous administration of salt-containing solutions to replace sodium and volume deficits.




