CHAPTER 83 Vascular Diseases of the Liver
Vascular disorders of the liver are relatively uncommon but frequently result in serious liver disease and portal hypertension.1,2 The continuously high metabolic activity of the liver makes it particularly susceptible to vascular compromise; however, its complex dual blood supply offers unique protection against ischemic injury. Hypercoagulable states play an important role in the pathogenesis of many of these disorders, and knowledge of them is essential for understanding and treating the associated hepatic vascular conditions. This chapter reviews a heterogeneous group of disorders resulting from hepatic vascular and cardiovascular diseases. Vasculitis involving the liver is discussed in Chapter 35.
BUDD-CHIARI SYNDROME
Hepatic venous outflow obstruction is the hallmark of the Budd-Chiari syndrome. Reductions in hepatic venous outflow can occur anywhere from the right atrium to the small hepatic venules and result in dramatic anatomic and physiologic changes. Classic Budd-Chiari syndrome results from thrombosis of one or more hepatic veins at their openings into the inferior vena cava. The deleterious physiologic changes of hepatic venous obstruction are transmitted directly to the hepatic sinusoids, resulting in sinusoidal congestion, portal vein hypertension, and reduced portal vein blood flow. The result is hepatomegaly, pain, ascites, and impaired hepatic function. The ascitic fluid typically has a high serum-ascites albumin gradient and a high protein content as a result of the increased filtration of serum proteins through the highly permeable sinusoidal spaces (see Chapter 91). The progression of disease is rarely fulminant; in most patients, the clinical course is subacute and less than six months in duration.1 In older series, the mortality rate in untreated cases was as high as 90% at three and one half years.3 With advances in diagnostic imaging and improved medical, surgical, and radiologic treatments for Budd-Chiari syndrome, however, survival has improved significantly since 1980.
The literature on Budd-Chiari syndrome is extensive. Two major reviews of the world literature collected data on cases reported before 1980.4,5 Reviews of more than 100 cases have appeared from Japan, India, China, and South Africa.6–9 The review from India is especially helpful in demonstrating the geographic diversity of this syndrome.7
ETIOLOGY
Anatomically, Budd-Chiari syndrome results from hepatic vein obstruction, inferior vena cava obstruction (above or at the level of the hepatic veins), or both. The main causes of the syndrome are listed in Table 83-1. In Western countries, thrombosis of the hepatic veins is the most common presentation, whereas in Asia and Africa, membranous obstruction of the inferior vena cava (MOVC) accounts for more than 40% of cases.10 Thrombogenic states can be identified in at least 75% of cases not caused by MOVC. Increasingly sophisticated testing for hypercoagulable states has reduced the frequency of idiopathic cases to less than 10%.1,2,11 Hematologic disorders are the most common causes of Budd-Chiari syndrome. Primary myeloproliferative diseases, particularly polycythemia vera, may account for 50% of cases.1,2 In addition, latent myeloproliferative disorders may be detected using cell culture techniques12 or by testing for mutations (specifically the V617F mutation) in the gene coding for the tyrosine kinase Janus kinase 2 (JAK2).13 Testing for JAK2 mutations is likely to play an increasing role in the evaluation of patients with Budd-Chiari syndrome and a suspected myeloproliferative disorder. Tumors, infections, and pregnancy each account for about 10% of cases. Other hypercoagulable states associated with Budd-Chiari syndrome include paroxysmal nocturnal hemoglobinuria, antiphospholipid syndrome,14 and deficiencies of antithrombin, protein C, and protein S.15 More recently recognized causes include factor V Leiden mutation16 and mutations of the prothrombin gene and the methylenetetrahydrofolate reductase gene.17 Oral contraceptive use increases the risk of Budd-Chiari syndrome by more than two-fold, especially in the presence of other hypercoagulable states. More than 25% of patients are found to have multiple thrombogenic risk factors.17
| Hypercoagulable States |
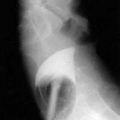
The pathophysiologic characteristics of MOVC are poorly understood. The disorder is much more common in developing countries, especially Asia and Africa, than in Western countries. More than 70% of cases of Budd-Chiari syndrome in China are due to MOVC. In India the incidence of MOVC, in contrast to classic Budd-Chiari syndrome, seems to be decreasing.18 The clinical presentation usually is subacute or chronic. The membranous webs may be thick or thin and typically occur in the intrahepatic inferior vena cava, often with occlusion of the ostia of the hepatic veins (Fig. 83-1). A congenital origin for the lesion has been proposed on the basis of the complex embryologic development of the inferior vena cava and reported presentations in childhood. An acquired origin, however, is supported by the peak occurrence in the fourth decade of life and histologic studies suggesting that the membrane develops from an organizing thrombus. Hypercoagulability is relatively less common in MOVC than in hepatic vein thrombosis. Therefore, other explanations for thrombosis in MOVC have been proposed, such as chronic infection, endothelial trauma caused by movement of the diaphragm with respiration and coughing, and venous turbulence resulting from the right-angle flow of blood from the hepatic veins into the inferior vena cava. Another feature of MOVC is the propensity of affected persons to develop hepatocellular carcinoma, which is less common in classic Budd-Chiari syndrome.19,20 The distinctive features of MOVC have led some investigators to consider MOVC a separate clinical entity termed obliterative hepatocavopathy.21
CLINICAL FEATURES
The epidemiologic characteristics of Budd-Chiari syndrome (except those cases associated with MOVC) parallel those of its underlying conditions. The syndrome is rare in infants and young children; the largest pediatric series describes South African children with MOVC. More than one half of the cases of classic Budd-Chiari syndrome occur between the ages of 20 and 39 years.5 Budd-Chiari syndrome occasionally may be identified in asymptomatic persons undergoing evaluation for mildly elevated liver biochemical test levels.22 In these patients, the lack of symptoms probably is the result of thrombosis of only one hepatic vein or the development of large venous collaterals.
PATHOLOGY
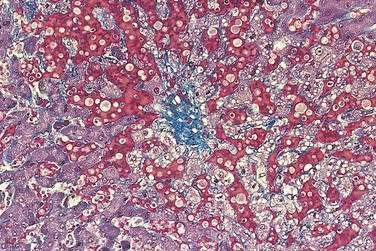
Acutely, the hepatic histologic features of centrilobular congestion, hemorrhage, sinusoidal dilatation, and noninflammatory cell necrosis predominate (Fig. 83-2). Within weeks, fibrosis develops in the centrilobular areas, more so than in periportal areas. Over time, these lesions evolve into cirrhosis. Large regenerative nodules are common, especially in areas of decreased portal venous perfusion.23 Indeed, portal vein thrombosis can be seen in 10% of cases and in 50% of hepatic explants at the time of liver transplantation for Budd-Chiari syndrome.24 Because hepatic vein occlusion is asymmetrical, the pathologic effects may vary in different regions of the liver. Massive caudate lobe hypertrophy is a common feature, probably because of preservation of venous drainage directly into the inferior vena cava, and may contribute to compression of the inferior vena cava. Pathologic changes (except for cirrhosis) can be reversed with adequate decompression of the hepatic sinusoids (see later).
DIAGNOSIS
Doppler ultrasonography, with sensitivity and specificity rates greater than 80%, is the diagnostic procedure of first choice.25 It is relatively inexpensive, safe, and available in most hospitals. Typical Doppler ultrasonographic features of Budd-Chiari syndrome include lack of visualization of normal hepatic venous connections to the inferior vena cava, comma-shaped intrahepatic or subcapsular collateral vessels, and absence of flow signal in the hepatic veins. The diagnostic accuracy of ultrasonography is decreased by a large body habitus and is operator dependent.
Magnetic resonance imaging (MRI) and computed tomography (CT)26 may also demonstrate characteristic features of Budd-Chiari syndrome but do not add much to the findings of an adequate ultrasonographic examination.27 MRI may be a better second-line test than CT because of the ability to provide accurate angiographic detail of the hepatic vein and inferior vena cava anatomy with minimal risk of nephrotoxicity. The combination of Doppler ultrasonography and either MRI or CT imaging should be sufficient to diagnose most cases of Budd-Chiari syndrome.
For years, venography was the standard for diagnosis of Budd-Chiari syndrome; however, with improvements in less invasive radiologic imaging, venography is often unnecessary for diagnostic purposes alone. Venography should be performed in cases of suspected Budd-Chiari syndrome when first- and second-line imaging tests are nondiagnostic and when surgery and other therapeutic interventions are planned. Measurement of the hepatic venous pressure gradient is required when vena cava stenosis is present, to plan for portosystemic shunt surgery (see Chapter 90). Venography also allows access for transjugular biopsy of both the right and left hepatic lobes, to confirm the diagnosis of Budd-Chiari syndrome and guide therapy. Liver biopsy is not essential for making the diagnosis of Budd-Chiari syndrome, however, and clinical staging of hepatic synthetic function (e.g., Child-Turcotte-Pugh score) may be better for planning treatment (see later).28
TREATMENT
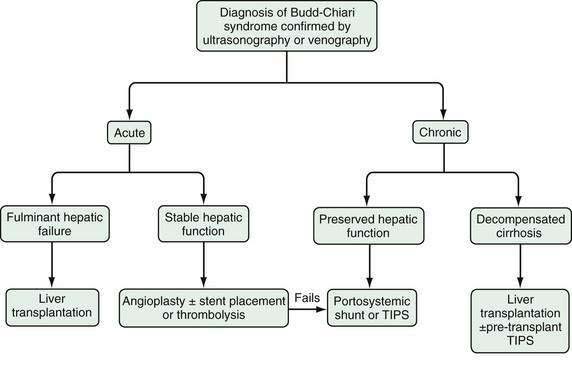
The therapy of Budd-Chiari syndrome depends on the etiology, anatomic characteristics, and pace of the disease (Fig. 83-3). The precipitating causes of Budd-Chiari syndrome must be evaluated and treated. Most patients need some form of intervention, and collaboration among a hepatologist, interventional radiologist, and hepatobiliary surgeon is optimal. Treatment options consist primarily of combinations of medical therapy with diuretics and anticoagulants, interventional therapy to decompress the hepatic sinusoids and prevent further hepatic necrosis, and liver transplantation for hepatic failure.
Medical management alone may be appropriate for the few patients with milder forms of Budd-Chiari syndrome. Treatment typically consists of diuretic therapy with spironolactone and furosemide and dietary sodium restriction to achieve a negative sodium balance (see Chapter 91). Large-volume paracentesis may be needed to relieve tense ascites. Anticoagulation is recommended and consists of intravenous heparin followed by warfarin to achieve an international normalized ratio (INR) for prothrombin of 2.0 to 2.5. Medical therapy is considered successful if ascites is controlled, liver biochemical test results improve or normalize, and symptoms resolve. Most patients with Budd-Chiari syndrome do not respond adequately to medical therapy alone, however, and require some form of intervention to decompress the hepatic sinusoids.
Many case reports and small series have described the use of thrombolytic therapy for acute Budd-Chiari syndrome. Thrombolytic agents are most effective if administered within three weeks of the onset of symptoms, if flow is demonstrated in the thrombosed vein, and if the agent is infused directly into the occluded hepatic vein. Systemic and hepatic arterial infusions are less effective.29 Angioplasty is often performed in conjunction with thrombolysis to improve vein patency and to reopen an acutely thrombosed stent or transjugular intrahepatic portosystemic shunt (TIPS) (see later).
The role of interventional radiology in the treatment of Budd-Chiari syndrome has expanded greatly. Angioplasty, with or without placement of a stent through short and localized stenoses of the hepatic veins or the inferior vena cava, can relieve symptoms in more than 80% of patients with MOVC and is the primary treatment for this disorder in many parts of the world.30–33 The rate of restenosis is high, and regular follow-up with Doppler ultrasonography is required.34 Angioplasty can also be combined successfully with surgical creation of a portacaval shunt in patients with both inferior vena cava and hepatic vein obstruction (see later).35
Use of TIPS has gained popularity as a treatment for refractory complications of portal hypertension (see Chapter 90). Procedural mortality is low, and shunt placement is usually successful,36 even in complete hepatic vein occlusion, in which a shunt can be placed between the intrahepatic vena cava and portal vein.37 TIPS is also useful for treating combined hepatic vein and inferior vena cava obstruction and can be effective in patients with fulminant Budd-Chiari syndrome awaiting liver transplantation. Short-term mortality rates in the latter setting may be as high as 50%38; however, long-term clinical stabilization after TIPS placement without liver transplantation also has been reported.39 TIPS has been used successfully in patients with acute Budd-Chiari syndrome in whom thrombolytic therapy and angioplasty have failed. Finally, TIPS is an effective “bridge” to liver transplantation in patients with chronic Budd-Chiari syndrome who have liver failure and refractory ascites or variceal bleeding. Unfortunately, TIPS dysfunction requiring revision occurs in as many as 70% of patients40 and seems to be more common in patients with Budd-Chiari syndrome than in those with other indications for TIPS placement. Polytetrafluoroethylene-covered stents significantly reduce rates of TIPS dysfunction and the need for reintervention and improve primary patency rates at one year from 19% to 67%.41 TIPS placement that extends too far into the portal vein or into the suprahepatic vena cava makes liver transplantation more complicated.
In Western countries, surgical therapy for Budd-Chiari syndrome consists mainly of portosystemic shunting and liver transplantation. Surgical therapy of MOVC that is unsuitable for or refractory to angioplasty and stent placement may entail resection or transatrial “finger fracture” of vena cava webs42 or dorsocranial liver resection with a hepaticoatrial anastomosis (the Senning procedure).43 For classic Budd-Chiari syndrome, portosystemic shunting relieves portal hypertension effectively, thereby alleviating hepatic ischemic necrosis, refractory ascites, and variceal bleeding. When shunt surgery is successful, the portal vein becomes the hepatic outflow tract, hepatomegaly resolves, hepatic histologic findings improve and may even normalize, and survival is prolonged in more than 90% of cases.44
The choice of a portosystemic shunt depends on the degree of hepatomegaly and caudate lobe hypertrophy, presence or absence of inferior vena cava stenosis, and surgical expertise. Portacaval and mesocaval shunts are associated with the best results. With mesocaval shunts, the rate of shunt thrombosis is higher—33% at five years45—but placement is technically simpler when portal dissection is impeded because of caudate lobe hypertrophy. Thrombosis of portacaval shunts is uncommon, with a rate of only 3% over 13 years,44 but portacaval shunts increase the technical difficulty of subsequent liver transplantation more than do mesocaval shunts. Dysfunction of a surgical portosystemic shunt significantly reduces long-term survival and is more likely with prosthetic grafts or with high portal venous pressures.46 Placement of a portacaval or mesocaval shunt is contraindicated if inferior vena cava stenosis is present and the vena cava pressure is greater than 20 mm Hg or if the portacaval pressure gradient is less than 10 mm Hg. Surgical options in these patients include placement of a mesoatrial shunt,47,48 a combined portacaval and cavoatrial shunt,44 and combinations of surgical shunt creation with vena cava angioplasty and stent placement.35 Because of high rates of shunt thrombosis, however, TIPS may be the best option in these patients.
Liver transplantation is appropriate in patients with liver failure resulting from fulminant or chronic Budd-Chiari syndrome and in patients with a failed surgical portosystemic shunt (see Chapter 95).49 In patients with protein C, protein S, or antithrombin deficiency, liver transplantation also cures the underlying hypercoagulable state, although most patients will require lifelong anticoagulation. Underlying myeloproliferative disorders can be managed with hydroxyurea and aspirin after liver transplantation,50 but there is a risk of progression and leukemic transformation.51 Recurrent Budd-Chiari syndrome after liver transplantation occurs in 4% to 10% of patients, and the risk of thrombosis of the hepatic artery and portal vein is increased as well.52,53 In addition, bleeding complications are more common because of anticoagulant therapy.54 Despite these drawbacks, five-year survival rates for patients with Budd-Chiari syndrome who undergo liver transplantation exceed 85%.53
The choices of therapy in patients with Budd-Chiari syndrome are complicated, in great part because of the lack of large, controlled studies comparing various treatments and the lack of standardization in classifying Budd-Chiari syndrome into acute, subacute, and chronic forms.55 One multivariate analysis failed to show a survival advantage in surgically shunted patients compared with those receiving medical treatment alone.56 Although medium-term studies of TIPS as definitive therapy in Budd-Chiari syndrome have been reported, there are no long-term controlled studies comparing TIPS with surgical therapy. TIPS is clearly an effective and widely available therapy; however, shunt dysfunction is common and leads to frequent reinterventions. On the other hand, surgical shunts are associated with significant perioperative morbidity and mortality, but when performed successfully, they offer good long-term outcomes. Surgical expertise in complex shunt surgery, however, is declining with the increasing use of TIPS. Surgical shunt surgery seems to be most efficacious in patients with acute or chronic Budd-Chiari syndrome with refractory symptoms and preserved hepatic synthetic function (Child classes A and B [see Chapter 90]).57 Patients with fulminant or chronic Budd-Chiari syndrome with impaired hepatic synthetic function (Child classes B and C) should be considered for liver transplantation, with or without prior TIPS placement. (As compared with the Child class, the Model for End-stage Liver Disease (MELD) score does not incorporate an assessment of refractory ascites and is less useful for estimating prognosis in patients with Budd-Chiari syndrome than in those with other causes of advanced chronic liver disease [see Chapter 95]).
In patients with acute Budd-Chiari syndrome, attempts should be made to decompress the hepatic sinusoids with combinations of angioplasty, stent placement, and thrombolytic therapy, followed by TIPS placement or creation of a surgical portosystemic shunt in those in whom these treatment approaches fail. A proposed stepwise (“minimally invasive”) approach beginning with anticoagulation therapy, then attempted hepatic vein recanalization, followed by TIPS, and then liver transplantation in treatment failures has been reported to achieve an overall five-year survival rate of nearly 90%.58
SINUSOIDAL OBSTRUCTION SYNDROME (VENO-OCCLUSIVE DISEASE)
Occlusion of the terminal hepatic venules and hepatic sinusoids resembles the Budd-Chiari syndrome clinically; however, the causes, epidemiologic and pathophysiologic characteristics, and prognosis of this entity are sufficiently distinct to justify a separate designation. In the past, the entity was known as veno-occlusive disease. Because the occlusion consistently involves the hepatic sinusoids, the term sinusoidal obstruction syndrome has been proposed as a more appropriate name for this disorder.59
ETIOLOGY
Liver disease caused by Senecio poisoning was originally reported from South Africa in 1920 (see Chapter 87),60 but the term veno-occlusive disease was not used until 1954, when it was related to the ingestion of pyrrolizidine alkaloids contained in plants of the genera Senecio, Crotalaria, and Heliotropium in Jamaica.61 Ingestion of alkaloids in inadequately winnowed wheat or in “bush tea,” especially in malnourished persons, is the main cause of veno-occlusive disease (sinusoidal obstruction syndrome) worldwide. Epidemics have been reported in India, Afghanistan, South Africa, the Middle East, and the United States. More recently, the herbal remedy comfrey (genus Symphytum) has been associated with sinusoidal obstruction syndrome. Rare familial clusters have been reported in association with immunodeficiency states.62
Since the advent of cancer chemotherapy in the 1950s, sinusoidal obstruction syndrome in Western countries has occurred most commonly after bone marrow transplantation (see Chapter 34).63 A variety of antineoplastic drugs have been implicated as causes of sinusoidal obstruction syndrome including gemtuzumab ozogamicin, actinomycin D, dacarbazine, cytosine arabinoside, mithramycin, and 6-thioguanine. Hepatic irradiation and therapy with busulfan plus cyclophosphamide also are established risk factors (see Chapters 34 and 39). In addition, long-term immunosuppression with azathioprine and 6-thioguanine used in patients with inflammatory bowel disease and in kidney and liver transplant recipients has been reported to cause sinusoidal obstruction syndrome.63 The frequency of sinusoidal obstruction syndrome after bone marrow transplantation varies, ranging from 0% to 70%, depending on patient and treatment-related variables.
CLINICAL FEATURES AND DIAGNOSIS
In the early stages of the disorder, features of portal hypertension predominate. Classically, sinusoidal obstruction syndrome manifests with mild hyperbilirubinemia (bilirubin levels greater than 2 mg/dL), painful hepatomegaly, weight gain of more than 2%, and development of ascites. Weight gain and painful hepatomegaly usually precede the onset of jaundice, which can be followed by the development of ascites, encephalopathy, and multiorgan failure. Sinusoidal obstruction syndrome occurs most commonly within 10 to 20 days after bone marrow transplantation (see Chapter 34). Serum bilirubin levels typically peak at day 17 after transplantation. Predisposing factors include pretreatment with norethisterone to suppress menses, preexisting elevated serum aminotransferase levels and chronic liver diseases such as hepatitis C, the HFE C282Y mutation for genetic hemochromatosis,64 advanced age, recent systemic bacterial or viral infections, previous bone marrow transplantation, and allogeneic (as opposed to autologous) bone marrow transplantation.63
The diagnosis of sinusoidal obstruction syndrome is often based on characteristic clinical features and exclusion of other conditions. Distinguishing this disease from the many other disorders that may complicate bone marrow transplantation is challenging. The most frequent conditions that can mimic sinusoidal obstruction syndrome include graft-versus-host disease, hepatic dysfunction caused by sepsis and drug toxicity, and cholestasis resulting from hemolysis and congestive heart failure. Graft-versus-host disease is rare before day 15 after transplantation, and sepsis and drug toxicity rarely cause painful hepatomegaly and ascites. Routine liver biochemical test results are not specific for sinusoidal obstruction syndrome. Serum alkaline phosphatase and aminotransferase elevations can accompany the hyperbilirubinemia and probably indicate coexisting hepatic ischemic necrosis. Thrombocytopenia is common and may be aggravated by portal hypertension and splenic sequestration. Findings on ultrasound examination, CT, and MRI are nonspecific in early sinusoidal obstruction syndrome but are helpful in excluding biliary obstruction, infiltrative liver lesions, and hepatic vein occlusion. Common findings in sinusoidal obstruction syndrome include gallbladder wall thickening, hepatosplenomegaly, ascites, portal vein enlargement with sluggish or reversed flow, and umbilical vein recanalization. Ultrasound and Doppler findings can predict disease severity noninvasively.65 If the diagnosis is uncertain, a transjugular liver biopsy and measurement of the hepatic venous pressure gradient can be performed (see Chapter 90). In this setting, a gradient of greater than 10 mm Hg is highly suggestive of sinusoidal obstruction syndrome and predictive of increased disease severity.66
Serum levels of a variety of mediators of coagulation, inflammation, and fibrosis have been found to be predictive of sinusoidal obstruction syndrome before it becomes clinically apparent. Levels of protein C, factor VII, and antithrombin are decreased in plasma before the onset of sinusoidal obstruction syndrome, whereas levels of plasminogen activator inhibitor type 1, tumor necrosis factor-α, and procollagen type III are increased.67 In patients who undergo hematopoietic stem cell transplantation, an elevated procollagen type III level—even before chemotherapy—is a risk factor for the later development of the disorder.68 It is unclear if these abnormalities cause the occlusion or result from the occlusion or hepatic injury.
In most patients with sinusoidal obstruction syndrome, the disorder resolves gradually over two to three weeks. The overall mortality rate is 20% to 50%, with most deaths resulting from multiorgan failure rather than hepatic failure.63 Severe sinusoidal obstruction syndrome, which carries a mortality rate of almost 100%, can be predicted by more rapid increases in the serum bilirubin level and weight. Other predictors of severe disease include the presence of ascites, a hepatic venous pressure gradient greater than 20 mm Hg, and the onset of multiorgan failure.69
PATHOLOGY
The principal histologic features of sinusoidal obstruction syndrome result from toxic injury to the centrilobular (zone 3) endothelial cells in the hepatic sinusoids and terminal hepatic venules (see Fig. 34-5A and B).70 The resulting cellular debris, exfoliated hepatocytes, activated coagulation factors, and extravasated red blood cells produce progressive occlusion of the sinusoids and venules, causing sinusoidal dilatation and severe hepatic congestion. Inflammation is notably absent. Progressive venular sclerosis ensues, with deposition of collagen in the sinusoids and venules eventually leading to venular obliteration, hepatocellular ischemic necrosis, and widespread fibrosis. Animal studies have shown that toxin-mediated depolymerization of F-actin in sinusoidal epithelial cells leads to rounding and swelling of the cells with dissection of the sinusoidal lining off the space of Disse. This leads to embolization of cellular debris and obstruction of the hepatic sinusoids. F-actin depolymerization seems to be mediated by metalloproteinases, which in turn are affected by nitric oxide production; sinusoidal obstruction syndrome can be prevented in animal models by inhibition of metalloprotenase-971 and stimulation of nitric oxide production.72 In addition, sinusoidal obstruction syndrome is less likely to occur in humans who have enhanced urea cycle activity because of the carbamyl-phosphate synthetase (CPS) A allele, which increases hepatic nitric oxide production.64
TREATMENT
The lack of effective and safe treatment for sinusoidal obstruction syndrome has increased interest in preventive strategies. Recognizing risk factors for the development of the disorder and altering chemotherapeutic regimens and doses can decrease associated morbidity and mortality. Studies of ursodeoxycholic acid, heparin, and low-molecular-weight heparin (LMWH) for the prevention of sinusoidal obstruction syndrome have yielded inconclusive results. Ursodeoxycholic acid has minimal toxicity, and in two studies it reduced the frequency of the disease by more than 50% compared with placebo or no treatment but had no effect on overall survival.73,74 One large study failed to show any preventive benefit of ursodeoxycholic acid. Similarly, heparin reduced the frequency of sinusoidal obstruction syndrome in a large, prospective, randomized trial from 13.7% in control subjects who received no treatment to 2.5% in patients in the group treated with heparin,75 but two other studies showed no preventive benefit from heparin and a 7% risk of hemorrhagic complications.74,76 The combination of ursodeoxycholic acid and heparin offers no advantage over heparin alone.77 Use of LMWH is associated with a lower rate of bleeding complications than that observed with heparin and may be more effective. In a retrospective study in which LMWH was compared with heparin and no treatment, the frequency of sinusoidal obstruction syndrome was 4% with LMWH, 11% with heparin, and 22% with no treatment.78 Studies of prostaglandin E1 to prevent sinusoidal obstruction syndrome have also yielded inconclusive results, and use of this agent is limited by its significant toxicity.
Treatment of sinusoidal obstruction syndrome primarily entails supportive care, including diuresis, analgesia for pain, paracentesis for tense ascites, and avoidance of nephrotoxins and other hepatotoxins. Survival when multiorgan failure has occurred is dismal. Treatment with tissue plasminogen activator, as described in many case reports and small series, has shown response rates of 30% and a rate of life-threatening hemorrhage of 20% to 30%.74 Treatment is ineffective in patients in whom multiorgan failure has developed. Other treatments reported in small studies or in trials with inconclusive results include the use of high-dose glucocorticoids, N-acetylcysteine, antithrombin,74 and protein C.
Defibrotide is a novel oligodeoxyribonucleotide with anti-ischemic, antithrombotic, and thrombolytic activity but minimal systemic anticoagulant effect. Several studies have shown efficacy of defibrotide in the prevention and treatment of sinusoidal obstruction syndrome, with no major toxicity. Prophylactic defibrotide plus heparin prevented the development of sinusoidal obstruction syndrome in 52 successive bone marrow or stem cell transplant patients, compared with a 19% disease incidence in a historical control group.79 A large multi-institutional study of defibrotide in the treatment of severe sinusoidal obstruction syndrome showed a 36% rate of response (improvement in serum bilirubin levels and multiorgan failure) in a group of patients with an otherwise dismal prognosis.80 Defibrotide has gained orphan drug status in the United States, and a large prospective phase III study is in progress.
TIPS has been performed successfully in patients with sinusoidal obstruction syndrome, but with generally poor results. Clinical improvement after TIPS may be seen in 50% of patients, but only 10% survive long term.81 Until more studies are available, TIPS should be reserved for patients with severe sinusoidal obstruction syndrome who have refractory ascites or who are candidates for liver transplantation. Liver transplantation has been performed successfully for liver failure caused by sinusoidal obstruction syndrome. Most patients with sinusoidal obstruction syndrome, however, are unsuitable for liver transplantation because of underlying malignancy and the severity of the associated multiorgan failure.
PORTAL VEIN THROMBOSIS
Portal vein obstruction results from thrombosis, constriction, or invasion of the portal vein. The resulting portal hypertension leads to splenomegaly and formation of portosystemic collaterals and esophageal, gastric, duodenal, and jejunal varices. Varices proliferate in the porta hepatis and involve the gallbladder and bile duct. As portal vein thrombosis evolves, fibroblasts transform the clot into a firm, collagenous plug in which tortuous venous channels develop. This cavernous transformation (resulting in a portal cavernoma) begins within days after acute thrombosis and continues to evolve over weeks to months. Upstream from the obstruction, the small intestine and colon become congested, and the stomach exhibits changes of portal hypertensive gastropathy (see Chapter 90). Mesenteric ischemia can occur if the thrombus extends into the mesenteric veins (see Chapter 114). Downstream from the clot, the liver usually maintains normal function and appears unaffected. Ascites, abdominal pain, and fever may develop with acute thrombosis, but usually recede subsequently. As more venous collaterals form, a state of equilibrium is reached in which some portal perfusion is maintained and some portal hypertension persists. Clinically, chronic portal vein thrombosis usually is asymptomatic until variceal bleeding occurs.
ETIOLOGY
Most cases of portal vein thrombosis have an identifiable cause related to hypercoagulability or to local factors such as inflammation, trauma, or malignancy (Table 83-2). Fewer than 20% of cases are considered idiopathic. Better understanding of the multiple causes of hypercoagulability has led to the recognition that multiple coexisting risk factors are present in as many as 40% of affected patients.82 Infection, most often from umbilical vein sepsis, is the main cause of portal vein thrombosis in children. Portal vein thrombosis is well documented after neonatal umbilical vein catheterization but resolves in more than 50% of cases.83 In adults, cirrhosis or abdominal malignancies are responsible for more than 50% of the cases of portal vein thrombosis.84 The disorder occurs in at least 10% of patients with cirrhosis, presumably as a result of sluggish portal vein blood flow, but acquired and inherited hypercoagulable states can be identified in many patients with cirrhosis and portal vein thrombosis.85 Prior variceal sclerotherapy, abdominal surgeries,86 and hepatocellular carcinoma are major risk factors for portal vein thrombosis in patients with cirrhosis. Hepatocellular and pancreatic carcinomas are the most common malignant causes of portal vein thrombosis,84 usually because of a combination of hypercoagulability and invasion or constriction of the portal vein. Local inflammatory reactions resulting from acute or chronic pancreatitis are a common cause of portal vein thrombosis. Pylephlebitis, or septic portal vein thrombosis, can complicate intra-abdominal infections such as appendicitis, diverticulitis, and cholangitis. Splenic vein trauma during splenectomy results in portal vein thrombosis in 8% of cases; the risk increases to 40% if a myeloproliferative disorder is present.87 In addition, portal vein thrombosis acutely complicates 5% of pancreaticoduodenectomies with portal vein reconstruction and is chronic in up to 17% of such cases.88 In this setting, portal vein thrombosis seems to have a low morbidity but is commonly associated with recurrent malignancy.
| Hypercoagulable StatesAntiphospholipid syndrome | Complications of Therapeutic Interventions |
|---|---|
| Antithrombin deficiency | Alcohol injection |
| Factor V Leiden mutation | Colectomy |
| Methylenetetrahydrofolate reductase mutation TT677 | Endoscopic sclerotherapyFundoplication |
| Myeloproliferative disorder (including polycythemia vera) | Gastric bandingHepatic chemoembolizationHepatobiliary surgery |
| Nephrotic syndrome | Islet cell injection |
| Oral contraceptives | Liver transplantation |
| Paroxysmal nocturnal hemoglobinuriaPregnancy | Peritoneal dialysisRadiofrequency ablation of hepatic tumor(s) |
| Prothrombin mutation G20210A | SplenectomyTIPS procedure |
| Protein C deficiency | Umbilical vein catheterization |
| Protein S deficiency | Impaired Portal Vein Flow |
| Sickle cell disease | Budd-Chiari syndrome |
| Inflammatory Diseases | Cirrhosis |
| Behçet’s syndrome | Cholangiocarcinoma |
| Inflammatory bowel disease | Hepatocellular carcinoma |
| Pancreatitis | Nodular regenerative hyperplasia |
| Infections | Pancreatic carcinoma |
| Appendicitis | Sinusoidal obstruction syndrome |
| Cholangitis | Miscellaneous |
| Cholecystitis | Bladder cancer |
| Diverticulitis | Choledochal cyst |
| Liver abscess (amebic or pyogenic) | Living at high altitude |
| Schistosomiasis | |
| Umbilical vein infection |
TIPS, transjugular intrahepatic portosystemic shunt.
CLINICAL FEATURES AND DIAGNOSIS
Portal vein thrombosis is found with equal frequency in adults (mean age, 40 years) and children (mean age, 6 years). The initial manifestation is almost always hematemesis from variceal bleeding. Abdominal pain is unusual unless the thrombosis is acute or involves the mesenteric veins and causes intestinal ischemia. Splenomegaly is usually present, but ascites is uncommon, except in acute portal vein thrombosis or when the thrombosis complicates cirrhosis. Liver biochemical test results are usually normal. Occasionally, bile duct varices can cause biliary obstruction89 and even mimic cholangiocarcinoma on cholangiography.90 Other unusual locations for ectopic varices in portal vein thrombosis include the gallbladder, duodenum, and rectum.
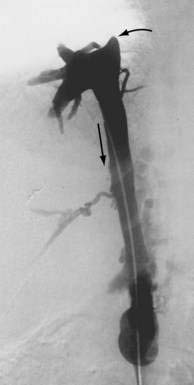
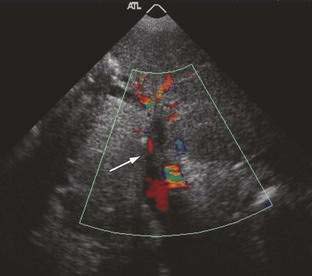
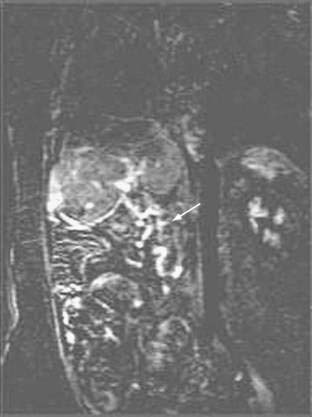
Doppler ultrasonography is highly sensitive for detection of portal vein thrombosis and reveals an echogenic thrombus in the portal vein (Fig. 83-4), extensive collateral vessels in the porta hepatis, an enlarged spleen, and occasionally nonvisualization of the portal vein. When the diagnosis of portal vein thrombosis is still uncertain, magnetic resonance angiography is better than CT in demonstrating the typical changes of portal vein thrombosis (portal cavernoma) (Fig. 83-5). Portal venography usually is unnecessary unless a surgical portosystemic shunt is being considered. Evaluating the patient for precipitating hypercoagulable risk factors may require a consultation with a hematologist.
The natural history of portal vein thrombosis is related primarily to the underlying disorder. In the absence of cirrhosis, cancer, and mesenteric vein thrombosis, the 10-year survival rate for patients with portal vein thrombosis is greater than 80%; only 2% experience fatal variceal hemorrhage.84 Variceal bleeding caused by portal vein thrombosis has a much better outcome than that observed with variceal bleeding caused by cirrhosis because of preserved hepatic function and lack of coagulopathy in patients with thrombosis alone. In addition, development of spontaneous portosystemic collaterals can lead to a reduced frequency of recurrent variceal bleeding in patients with portal vein thrombosis.
TREATMENT
Endoscopic band ligation or sclerotherapy is first-line therapy for variceal bleeding in patients with portal vein thrombosis (see Chapter 90). Sessions should be repeated until the varices are obliterated. Therapy with a beta blocker is beneficial in preventing initial91 and, in combination with endoscopic therapy, recurrent variceal bleeding. Recurrent or refractory variceal bleeding or bleeding from varices distal to the esophagus is an indication for placement of a portosystemic shunt. TIPS is an option if the technical challenge of gaining access to the portal vein can be overcome. Focal malignant portal vein obstruction can be stented via a percutaneous approach to control refractory variceal bleeding and ascites.92 Elective mesocaval and splenorenal shunts93 and the extended Sugiura procedure (esophagogastric devascularization and transection)94 have also been performed successfully in patients with portal vein thrombosis, with low mortality and long survival. In children with refractory complications of portal vein thrombosis, a Rex shunt (mesenteric-to-left-portal-vein bypass) is also an effective option.95
Anticoagulation is recommended in patients with acute portal vein thrombosis, to prevent cavernous transformation and complications of portal hypertension. Spontaneous recanalization with acute thrombosis is rare, but therapeutic recanalization can be achieved in more than 80% of the cases with anticoagulants (intravenous heparin or subcutaneous LMWH, followed by warfarin to achieve an INR of 2.0 to 2.5 for at least six months).96 Prompt use of broad-spectrum antibiotics in cases of septic pylephlebitis also promotes thrombolysis. Selective transcatheter infusions of thrombolytic agents have been used successfully in acute portal vein thrombosis97 when associated with mesenteric vein thrombosis and intestinal ischemia but are associated with a high complication rate. Long-term anticoagulation should be considered in patients with portal vein thrombosis and a recognized hypercoagulable state, surgical portosystemic shunt, or concomitant mesenteric vein thrombosis. The safety of chronic anticoagulation in patients with portal vein thrombosis and varices has been a major concern, but one study has shown that long-term anticoagulation does not increase the risk or severity of variceal bleeding and prevents further portal and mesenteric venous thrombotic complications.98 In general, however, anticoagulants are not recommended for chronic portal vein thrombosis, especially when associated with cavernous transformation. Liver transplantation for liver failure complicated by portal vein thrombosis is associated with increased technical complexity and a high rethrombosis rate, but overall survival rates are not reduced when compared with rates for other indications.99
ISCHEMIC HEPATITIS
Because hepatitis refers to inflammation of the liver, the term ischemic hepatitis is somewhat of a misnomer because inflammation is typically not present. A more physiologic term would be hypoxic hepatitis because the primary cause of this syndrome is tissue hypoxia, which may be the result of hypoperfusion from cardiac failure, systemic hypoxemia from respiratory failure, or increased oxygen requirements from sepsis.100 The name ischemic hepatitis is used, however, because of clinical similarities to other forms of acute hepatitis and the characteristic pathologic feature of acute centrilobular necrosis. Ischemic hepatitis is probably the most commonly encountered form of vascular liver disease.
ETIOLOGY
Of all cases of extreme serum aspartate aminotransferase (AST) elevations (to more than 3000 U/L), ischemic hepatitis accounts for about half.101 The most common cause of ischemic hepatitis is cardiovascular disease, which accounts for more than 70% of cases, followed in frequency by respiratory failure and sepsis, each of which accounts for less than 15% of cases.100 Hypotension is documented as a precipitating factor in more than 50% of patients with ischemic hepatitis but does not need to be evident for ischemic hepatitis to occur. Hypotension often is clinically apparent as a result of acute myocardial infarction, severe congestive heart failure, or sepsis but may be less obvious following a transient arrhythmia or silent coronary ischemic event. The presence of congestive heart failure significantly increases the likelihood that a drop in the cardiac output from any cause will result in ischemic hepatitis. Indeed, more than 80% of cases of ischemic hepatitis occur in the setting of congestive heart failure.102 Acute trauma, hemorrhage, burns, and heat stroke can also cause ischemic hepatitis, but the likelihood is substantially less in the absence of underlying heart disease.
CLINICAL FEATURES AND DIAGNOSIS
Ischemic hepatitis often is first considered when extreme serum aminotransferase elevations are detected in a patient hospitalized for problems not primarily associated with the liver. Findings on physical examination are usually dominated by the underlying precipitating medical condition. The patient’s mental status is often altered because of diminished cerebral perfusion. Laboratory studies show extreme elevations of the aminotransferase levels (more than 3000 U/L). The serum lactate dehydrogenase (LDH) level is profoundly elevated, often more so than the alanine aminotransferase (ALT), and an ALT-to-LDH ratio of less than 1.5 is more typical of ischemic hepatitis than of viral hepatitis.103 The prothrombin time may be prolonged by two or three seconds, and the serum bilirubin level is often mildly increased, with peak levels seen after the aminotransferase levels peak. Serum creatinine and blood urea nitrogen levels are often elevated because of acute tubular necrosis. Characteristically, serum aminotransferase levels peak 1 to 3 days after the hemodynamic insult and return to normal within 7 to 10 days.
The differential diagnosis for this type of severe acute injury includes acute hepatitis caused by viral infections, autoimmunity, toxins, and medications (see also Chapter 73). Liver biopsy specimens, although usually unnecessary, reveal bland, centrilobular necrosis with preservation of the hepatic architecture (Fig. 83-6). Occasionally a definitive diagnosis of ischemic hepatitis can be difficult to make, but the typical prompt rise in serum aminotransferase and LDH levels followed by a rapid fall within a few days is more characteristic of ischemic hepatitis than of other causes of severe acute liver injury (Fig. 83-7).104
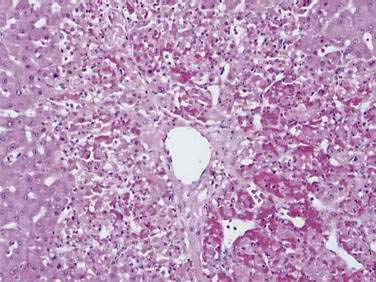
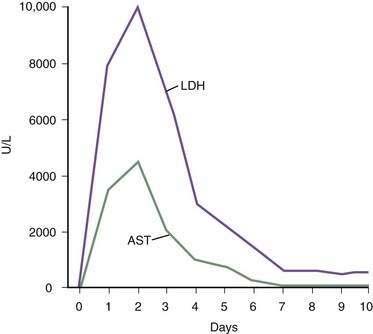
CONGESTIVE HEPATOPATHY
The effects of congestive heart failure on the liver predictably include decreased hepatic blood flow, increased hepatic vein pressure, and decreased arterial oxygen saturation.105 Right-sided heart failure results in transmission of increased central venous pressure from the heart directly to the hepatic sinusoids. The result is centrilobular congestion and sinusoidal edema that further decrease oxygen delivery. The injurious effects of superimposed ischemic hepatitis are common in these patients. The acute and chronic damage results in progressive centrilobular fibrosis. Sinusoidal hypertension and congestion can lead to the development of ascites, with a characteristically high serum-ascites albumin gradient and a high protein concentration (see Chapter 91).
Clinically, the symptoms and signs of congestive heart failure are the predominant features. Dull right upper quadrant pain in association with hepatomegaly is common. The liver may be pulsatile if tricuspid regurgitation is present, and hepatojugular reflux is often apparent on compression over the liver. Spider angiomata and varices are usually not present, and variceal bleeding caused by congestive hepatopathy alone does not occur. Mild elevation of the serum bilirubin level (to less than 3 mg/dL) is common, and jaundice is seen in fewer than 10% of patients, occurring in those with severe or acute congestive heart failure.106 The prothrombin time is prolonged in more than 75% of cases and usually is resistant to therapy with vitamin K. Other liver biochemical test levels are often normal or only mildly elevated. Liver test results improve slowly or normalize with effective therapy of the underlying congestive heart failure. Ultrasound is useful in excluding other hepatobiliary diseases. Typical CT findings in congestive hepatopathy include hepatomegaly, ascites, dilatation of the inferior vena cava and hepatic veins, and inhomogeneous hepatic enhancement during the portal phase of contrast administration.
The histologic features of congestive hepatopathy include atrophy of hepatocytes, sinusoidal distention, and centrilobular fibrosis (Fig. 83-8). Centrilobular necrosis, consistent with ischemic hepatitis, is frequent in liver biopsy specimens that show congestive hepatopathy and usually correlates with recent hypotension.107 Bridging fibrosis typically extends between central veins (rather than between portal tracts) to produce a pattern of “reverse lobulation” characteristic of cardiac cirrhosis. The distribution of fibrosis throughout the liver is highly variable and correlates with focal sinusoidal thrombosis, with obliteration of central and portal veins that leads in turn to localized ischemia, parenchymal extinction, and fibrosis.108
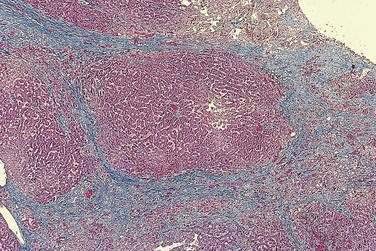
PELIOSIS HEPATIS
Peliosis hepatis is characterized by the presence of multiple blood-filled cavities distributed randomly throughout the liver. The cavities range in size from a few millimeters to 3 cm across and usually are seen in association with dilated hepatic sinusoids (Fig. 83-9). Two histologic types of peliosis hepatis occur. In the parenchymal type, blood-filled cavities are lined by hepatocytes, and hemorrhagic parenchymal necrosis and congestion usually are present. In the phlebectatic type, the cavities are lined by endothelial cells associated with aneurysmal dilation of the central vein.109 Fibrosis, cirrhosis, regenerative nodules, and tumors also may be seen with peliosis. In the past, peliosis hepatis was largely a histologic curiosity, but its association with renal transplantation and the acquired immunodeficiency syndrome (AIDS) have increased clinical awareness of this syndrome.
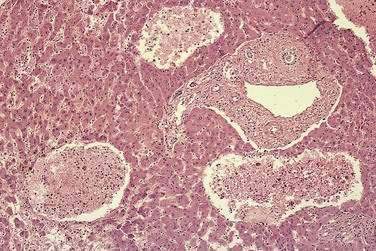
Although jaundice, painful hepatomegaly, liver failure, and fatal hemorrhage may be manifestations of peliosis, more often the disorder is detected during evaluation of abnormal liver biochemical test results in an asymptomatic patient. If the hemorrhagic cavities are large enough, they can be detected by ultrasonography, CT, and magnetic resonance imaging.110
The pathogenesis of peliosis hepatis is unknown, but the leading theories include damage to sinusoidal endothelial cells, outflow obstruction of blood flow at the sinusoidal level, and hepatocellular necrosis. Before the 1970s, peliosis was most often identified in patients dying of wasting diseases, particularly tuberculosis and carcinomatosis.109 Since then, peliosis has been associated with the use of anabolic steroids, oral contraceptives, tamoxifen, danazol, vitamin A, glucocorticoids, 6-thioguanine and azathioprine, and exposure to urethane, vinyl chloride, and thorium dioxide (see Chapter 87). The syndrome can regress with discontinuation of the causative agent. Myeloproliferative diseases such as agnogenic myeloid metaplasia111 and malignant histiocytosis,112 infections such as E. coli pyelonephritis,113 and Castleman disease (giant lymph node hyperplasia)114 have also been associated with peliosis.
Bacillary peliosis is caused by the bacterium responsible for cat-scratch disease, in the genus Bartonella. Traumatic exposure to cats is a recognized risk factor. The syndrome has been reported in immunocompetent persons but is associated primarily with AIDS (see Chapters 33 and 82).115 In affected patients, vascular proliferative lesions are found commonly in the skin (where they are termed bacillary angiomatosis) or in the liver and spleen (where they are termed bacillary peliosis and where bacilli can be identified histologically adjacent to the peliotic lesions). Symptoms and signs include anorexia, abdominal pain, fever, lymphadenopathy, hepatosplenomegaly, and cutaneous vascular lesions or nodules.116 Anemia, an elevated serum alkaline phosphatase level, and a CD4+ lymphocyte count of less than 200/mm3 are typical laboratory findings. Bacillary peliosis responds to antibiotic therapy (e.g., erythromycin, doxycycline) (see Chapter 82).
Peliosis hepatis occurs in 20% of patients after kidney transplantation and is associated mainly with the prolonged use of azathioprine and possibly cyclosporine.117 The lesions often are asymptomatic or associated with abnormal liver biochemical test levels, but progressive fibrosis, cirrhosis, and portal hypertension may be additional findings. Hepatic lesions may regress on withdrawal of azathioprine, but the overall course is not clearly modified and the risk of transplant rejection is increased substantially.118
HEPATIC ARTERY ANEURYSM
Hepatic artery aneurysms (HAAs) are uncommon, but they are the second leading category of visceral artery aneurysms (after splenic artery aneurysms) and account for more than 20% of cases. A majority of true HAAs are isolated, saccular, extrahepatic lesions involving the full arterial wall. In the past, HAAs were mainly mycotic (infectious) in etiology, but today they typically result from atherosclerosis, mediointimal degeneration, trauma, and, less commonly, infection. Other rare causes of true HAA are vasculitides, such as polyarteritis nodosa, systemic lupus erythematosus, Takayasu arteritis, and Kawasaki disease, and connective tissue disorders, such as Marfan syndrome, Ehlers-Danlos syndrome, and Osler-Weber-Rendu disease.119 Approximately one half of HAAs are pseudoaneurysms (aneurysms resulting from injury). Procedures commonly associated with hepatic artery pseudoaneurysms include liver biopsy, transhepatic biliary drainage, cholecystectomy, hepatectomy, and liver transplantation.120
Symptoms of HAA include epigastric or right subchondral pain, but most affected persons are asymptomatic until the aneurysm ruptures. Rarely, a pulsatile right upper quadrant mass or thrill may be detected. Patients may present with rupture into the biliary tree, with hemobilia, epigastric pain, and icterus; rupture into the portal vein, with portal hypertension and variceal bleeding; or rupture into the peritoneal cavity, with abdominal pain and shock. The mortality rate from rupture of a HAA is more than 30%. Nonatherosclerotic aneurysms and multiple HAAs carry an increased risk of rupture and should be treated. Although the risk of rupture of an aneurysm is independent of its size, atherosclerotic aneurysms greater than 2 cm in diameter also should be treated.119
Doppler ultrasound studies and CT readily demonstrate HAAs, but angiography is especially useful for defining these lesions, accessing the collateral circulation, and planning treatment. Hepatic artery pseudoaneurysms are treated effectively by angiographic embolization.120 True extrahepatic aneurysms may be treated with embolization, provided that presence of collateral circulation, distance from the gastroduodenal artery, absence of cirrhosis, and patency of the portal vein can be confirmed, but surgical resection of the aneurysm may be preferable, to minimize the risk of hepatic infarction.121
HEPATIC ARTERY ATHEROSCLEROSIS
Despite its frequency in the general population, atherosclerosis is rarely a cause of liver disease. Intimal thickening and atherosclerosis in hepatic arteries are less common and occur later in life than is typical for coronary arteries.122 Hepatic infarction resulting from atherosclerosis alone is rare. The dual blood supply to the liver undoubtedly confers protection from ischemia. Nevertheless, atherosclerosis is the primary cause of approximately one third of HAAs (see earlier).121 In addition, because the common bile duct derives all of its blood supply from the hepatic artery, atherosclerosis can result in ischemic cholangiopathy with biliary strictures and obstruction.123 The presence of atherosclerosis occasionally prevents the use of some donor livers for transplantation. Atherosclerosis makes arterial anastomoses technically more difficult to secure and may predispose the liver to ischemic injury during transport and reperfusion.
Abbas MA, Fowl RJ, Stone WM, et al. Hepatic artery aneurysm: Factors that predict complications. J Vasc Surg. 2003;38:41-5. (Ref 119.)
Bearman SI. Avoiding hepatic veno-occlusive disease: What do we know and where are we going? Bone Marrow Transplant. 2001;27:1113-20. (Ref 74.)
Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: Risk and benefit of anticoagulant therapy. Gastroenterology. 2001;120:490-7. (Ref 98.)
DeLeve LD, Valla DC, Garcia Tsao G, American Association for the Study of Liver Disease. Vascular disorders of the liver. Hepatology. 2009;49:1729-64. (Ref 2.)
Henrion J, Schapira M, Luwaert R, et al. Hypoxic hepatitis: Clinical and hemodynamic study in 142 consecutive cases. Medicine. 2003;82:392-406. (Ref 100.)
Janssen HL, Wijnhoud A, Haagsma EB, et al. Extrahepatic portal vein thrombosis: Aetiology and determinants of survival. Gut. 2001;49:720-4. (Ref 84.)
Mitchell MC, Boitnott JK, Kaufman S, et al. Budd-Chiari syndrome: Etiology, diagnosis and management. Medicine. 1982;61:199-218. (Ref 4.)
Mohle-Boetani JC, Koehler JE, Berger TG, et al. Bacillary angiomatosis and bacillary peliosis in patients infected with human immunodeficiency virus: Clinical characteristics in a case-control study. Clin Infect Dis. 1996;22:794-800. (Ref 116.)
Okuda K. Inferior vena cava thrombosis at its hepatic portion (obliterative hepatocavopathy). Semin Liver Dis. 2002;22:15-26. (Ref 21.)
Orloff MJ, Daily PO, Orloff SL, et al. A 27 year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg. 2000;232:340-52. (Ref 44.)
Plessier A, Sibert A, Consigny Y, et al. Aiming at minimal invasiveness as a therapeutic strategy for Budd-Chiari syndrome. Hepatology. 2006;44:1308-16. (Ref 58.)
Richman SM, Delman AJ, Grob D. Alterations in indices of liver function in congestive heart failure with particular reference to serum enzymes. Am J Med. 1961;30:211-25. (Ref 106.)
Rossle M, Olschewski M, Siegerstetter V, et al. The Budd-Chiari syndrome: Outcome after treatment with the transjugular intrahepatic portosystemic shunt. Surgery. 2004;135:394-403. (Ref 36.)
Valla DC. Primary Budd-Chiari syndrome. J Hepatol. 2009;50:195-203. (Ref 1.)
Yoshimoto K, Ono N, Okamura T, et al. Recent progress in the diagnosis and therapy for veno-occlusive disease of the liver. Leukemia Lymphoma. 2003;44:229-34. (Ref 67.)
1. Valla DC. Primary Budd-Chiari syndrome. J Hepatol. 2009;50:195-203.
2. DeLeve LD, Valla DC, Garcia Tsao G, American Association for the Study of Liver Disease. Vascular disorders of the liver. Hepatology. 2009;49:1729-64.
3. Tavill AS, Wood EJ, Kreel L, et al. The Budd-Chiari syndrome: Correlation between hepatic scintigraphy and the clinical, radiological, and pathological findings in nineteen cases of hepatic venous outflow obstruction. Gastroenterology. 1975;68:509-18.
4. Mitchell MC, Boitnott JK, Kaufman S, et al. Budd-Chiari syndrome: Etiology, diagnosis and management. Medicine. 1982;61:199-218.
5. Parker RGF. Occlusion of the hepatic veins in man. Medicine. 1959;38:369-402.
6. Okuda H, Yamagata H, Obata H, et al. Epidemiological and clinical features of Budd-Chiari syndrome in Japan. J Hepatol. 1995;22:1-9.
7. Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Medicine. 1994;73:21-36.
8. Wang Z. Recognition and management of Budd-Chiari syndrome: Experience with 143 patients. Chinese Med J. 1989;102:338-46.
9. Simson IW. Membranous obstruction of the inferior vena cava and hepatocellular carcinoma in South Africa. Gastroenterology. 1982;82:171-8.
10. Okuda K, Kage M, Shrestha SM. Proposal of a new nomenclature for Budd-Chiari syndrome: Hepatic vein thrombosis versus thrombosis of the inferior vena cava at its hepatic portion. Hepatology. 1998;28:1191-8.
11. Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med. 2004;350(6):578-85.
12. Valla D, Casadevall N, Lacombe C, et al. Primary myeloproliferative disorder and hepatic vein thrombosis. Ann Intern Med. 1985;103:329-34.
13. Patel RK, Lea NC, Heneghan MA, et al. Prevalence of the activating JAK2 tyrosine kinase mutation V617F in the Budd-Chiari syndrome. Gastroenterology. 2006;130:2031-8.
14. Espinosa G, Font J, Garcia-Pagan JC, et al. Budd-Chiari syndrome secondary to antiphospholipid syndrome. Medicine. 2001;80:345-54.
15. Mohanty D, Shetty S, Ghosh K, et al. Hereditary thrombophilia as a cause of Budd-Chiari syndrome: A study from Western India. Hepatology. 2001;34(4 Pt 1):666-70.
16. Deltenre P, Denninger MH, Hillaire S, et al. Factor V Leiden related Budd-Chiari syndrome. Gut. 2001;48:264-8.
17. Denninger MH, Chait Y, Casadevall N, et al. Cause of portal or hepatic venous thrombosis in adults: The role of multiple concurrent factors. Hepatology. 2000;31:587-91.
18. Eapen CE, Mammen T, Moses V, Shyamkumar NK. Changing Profile of Budd Chiari syndrome in India. Indian J Gastroenterol. 2007;26:77-81.
19. Matsui S, Ichida T, Watanabe M, et al. Clinical features and etiology of hepatocellular carcinoma arising in patients with membranous obstruction of the inferior vena cava: In reference to hepatitis viral infection. J Gastroenterol Hepatol. 2000;15:1205-11.
20. Moucari R, Rautou P, Cazals-Hatem D, et al. Hepatocellular carcinoma in Budd-Chiari syndrome: Characteristics and risk factors. Gut. 2008;57:828-35.
21. Okuda K. Inferior vena cava thrombosis at its hepatic portion (obliterative hepatocavopathy). Semin Liver Dis. 2002;22:15-26.
22. Hadengue A, Poliquin M, Vilgrain V, et al. The changing scene of hepatic vein thrombosis: Recognition of asymptomatic cases. Gastroenterology. 1994;106:1042-7.
23. Ibarrola C, Castellano VM, Colina F. Focal hyperplastic hepatocellular nodules in hepatic venous outflow obstruction: A clinicopathological study of four patients and 24 nodules. Histopathology. 2004;44:172-9.
24. Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: A study in 17 explanted livers. Hepatology. 2003;37:510-19.
25. Bolondi L, Gaiani S, Li Bassi S, et al. Diagnosis of Budd-Chiari syndrome by pulsed Doppler ultrasound. Gastroenterology. 1991;100(5 Pt 1):1324-31.
26. Camera L, Mainenti PP, Di Giacomo A, et al. Triphasic helical CT in Budd-Chiari syndrome: Patterns of enhancement in acute, subacute and chronic disease. Clinical Radiology. 2006;61:331-7.
27. Kane R, Eustace S. Diagnosis of Budd-Chiari syndrome: Comparison between sonography and MR angiography. Radiology. 1995;195:117-21.
28. Tang TJ, Batts KP, de Groen PC, et al. The prognostic value of histology in the assessment of patients with Budd-Chiari syndrome. J Hepatol. 2001;35:338-43.
29. Sharma S, Texeira A, Texeira P, et al. Pharmacological thrombolysis in Budd Chiari syndrome: A single centre experience and review of the literature. J Hepatol. 2004;40:172-80.
30. Xu K, Feng B, Zhong H, et al. Clinical application of interventional techniques in the treatment of Budd-Chiari syndrome. Chin Med J. 2003;116:609-15.
31. Zhang CQ, Fu LN, Xu L, et al. Long term effect of stent placement in 115 patients with Budd-Chiari syndrome. World J Gastroenterol. 2003;9:2587-91.
32. De BK, Biswas PK, Sen S, et al. Management of the Budd-Chiari syndrome by balloon cavoplasty. Indian J Gastroenterol. 2001;20:151-4.
33. Wu T, Wang L, Xiao Q, et al. Percutaneous balloon angioplasty of inferior vena cava in Budd-Chiari syndrome-R1. Int J Cardiol. 2002;83:175-8.
34. Pelage JP, Denys A, Valla D, et al. Budd-Chiari syndrome due to prothrombotic disorder: Mid-term patency and efficacy of endovascular stents. Eur Radiol. 2003;13:286-93.
35. Mourad FH, Khalifeh M, Al-Kutoubi A, et al. Inferior vena cava obstruction in Budd-Chiari syndrome: Successful treatment by radiological stenting followed by a portosystemic shunt. Eur J Gastroenterol Hepatol. 2001;13:275-7.
36. Rossle M, Olschewski M, Siegerstetter V, et al. The Budd-Chiari syndrome: Outcome after treatment with the transjugular intrahepatic portosystemic shunt. Surgery. 2004;135:394-403.
37. Gasparini D, Del Forno M, Sponza M, et al. Transjugular intrahepatic portosystemic shunt by direct transcaval approach in patients with acute and hyperacute Budd-Chiari syndrome. Eur J Gastroenterol Hepatol. 2002;14:567-71.
38. Mancuso A, Fung K, Mela M, et al. TIPS for acute and chronic Budd-Chiari syndrome: A single-centre experience. J Hepatol. 2003;38:751-4.
39. Kavanagh P, Roberts J, Gibney R, et al. Acute Budd-Chiari syndrome with liver failure: The experience of a policy of initial interventional radiological treatment using transjugular intrahepatic portosystemic shunt. J Gastroenterol Hepatol. 2004;19:1135-9.
40. Perello A, Garcia-Pagan JC, Gilabert R, et al. TIPS is a useful long-term derivative therapy for patients with Budd-Chiari syndrome uncontrolled by medical therapy. Hepatology. 2002;35:132-9.
41. Hernandez-Guerra M, Turnes J, Rubinstein P, et al. PTFE-covered stents improve TIPS patency in Budd-Chiari syndrome. Hepatology. 2004;40:1197-202.
42. Xu PQ, Dang XW. Treatment of membranous Budd-Chiari syndrome: Analysis of 480 cases. Hepatobiliary Pacreat Dis Int. 2004;3:73-6.
43. Pasic M, Senning A, von Segesser L, et al. Transcaval liver resection with hepatoatrial anastomosis for treatment of patients with the Budd-Chiari syndrome. J Thorac Cardiovasc Surg. 1993;106:275-82.
44. Orloff MJ, Daily PO, Orloff SL, et al. A 27 year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg. 2000;232:340-52.
45. Terpstra OT, Ausema B, Bruining HA, et al. Late results of mesocaval interposition shunting for bleeding oesophageal varices. Br J Surg. 1987;74:787-90.
46. Bachet JB, Condat B, Hagege H, et al. Long-term portosystemic shunt patency as a determinant of outcome in Budd-Chiari syndrome. J Hepatol. 2007;46:60-8.
47. Behera A, Menakuru SR, Thingnam S, et al. Treatment of Budd-Chiari syndrome with inferior vena caval occlusion by mesoatrial shunt. Eur J Surg. 2002;168:355-9.
48. Slakey DP, Klein AS, Venbrux AC, et al. Budd-Chiari syndrome: Current management options. Ann Surg. 2001;233:522-7.
49. Ringe B, Lang H, Oldhafer KJ, et al. Which is the best surgery for Budd-Chiari syndrome: Venous decompression or liver transplantation? A single-center experience with 50 patients. Hepatology. 1995;21:1337-44.
50. Melear JM, Goldstein RM, Levy MF, et al. Hematologic aspects of liver transplantation for Budd-Chiari syndrome with special reference to myeloproliferative disorders. Transplantation. 2002;74:1090-5.
51. Briere JB. Budd-Chiari syndrome and portal vein thrombosis associated with myelopoliferative disorders: Diagnosis and management. Semin Thromb Hemost. 2006;32:208-18.
52. Bahr MJ, Schubert J, Bleck JS, et al. Recurrence of Budd-Chiari syndrome after liver transplantation in paroxysmal nocturnal hemoglobinuria. Transpl Int. 2003;16:890-4.
53. Srinivasan P, Rela M, Prachalias A, et al. Liver transplantation for Budd-Chiari syndrome. Transplantation. 2002;73:973-7.
54. Cruz E, Ascher NL, Roberts JP, et al. High incidence of recurrence and hematologic events following liver transplantation for Budd-Chiari syndrome. Clin Transplant. 2005;19:501-6.
55. Janssen HL, Garcia-Pagan JC, Elias E, et al. Budd-Chiari syndrome: A review by an expert panel. J Hepatol. 2003;38:364-71.
56. Langlet P, Escolano S, Valla D, et al. Clinicopathological forms and prognostic index in Budd-Chiari syndrome. J Hepatol. 2003;39:496-501.
57. Murad SD, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology. 2004;39:500-8.
58. Plessier A, Sibert A, Consigny Y, et al. Aiming at minimal invasiveness as a therapeutic strategy for Budd-Chiari syndrome. Hepatology. 2006;44:1308-16.
59. DeLeve LD, Shulman HM, McDonald GB. Toxic injury to hepatic sinusoids: Sinusoidal obstruction syndrome (veno-occlusive disease). Semin Liver Dis. 2002;22:27-42.
60. Willmot FC, Robertson GW. Senecio disease, or cirrhosis of the liver due to Senecio poisoning. Lancet. 1920;2:848.
61. Bras G, Jelliffe DB, Stuart KL. Veno-occlusive disease of liver with nonportal type of cirrhosis, occurring in Jamaica. Arch Pathol. 1954;57:285-300.
62. Mellis C, Bale PM. Familial hepatic veno-occlusive disease with probable immune deficiency. J Pediatr. 1976;88:236-42.
63. Kumar S, DeLeve LD, Kamath PS, et al. Hepatic veno-occlusive disease (sinusoidal obstruction syndrome) after hematopoietic stem cell transplantation. Mayo Clin Proc. 2003;78:589-98.
64. Kallianpur AR, Hall LD, Yadav M, et al. The hemochromatosis C282Y allele: A risk factor for hepatic veno-occlusive disease after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2005;35:1155-64.
65. Lassau N, Auperin A, Leclere J, et al. Prognostic value of Doppler-ultrasonography in hepatic veno-occlusive disease. Transplantation. 2002;74:60-6.
66. Shulman HM, Gooley T, Dudley MD, et al. Utility of transvenous liver biopsies and wedged hepatic venous pressure measurements in sixty marrow transplant recipients. Transplantation. 1995;59:1015-22.
67. Yoshimoto K, Ono N, Okamura T, et al. Recent progress in the diagnosis and therapy for veno-occlusive disease of the liver. Leukemia Lymphoma. 2003;44:229-34.
68. Tanikawa S, Mori S, Ohhashi K, et al. Predictive markers for hepatic veno-occlusive disease after hematopoietic stem cell transplantation in adults: A prospective single center study. Bone Marrow Transplant. 2000;26:881-6.
69. Richardson P, Guinan E. Hepatic veno-occlusive disease following hematopoietic stem cell transplantation. Acta Haematol. 2001;106(1-2):57-68.
70. Shulman HM, Fisher LB, Schoch HG, et al. Veno-occlusive disease of the liver after marrow transplantation: Histological correlates of clinical signs and symptoms. Hepatology. 1994;19:1171-81.
71. DeLeve LD, Wang X, Tsai J. Sinusoidal obstruction syndrome (veno-occlusive disease) in the rat is prevented by matrix metalloproteinase inhibition. Gastroenterology. 2003;125:882-90.
72. DeLeve LD, Wang X, Kanel GC. Decreased hepatic nitric oxide production contributes to the development of rat sinusoidal obstruction syndrome. Hepatology. 2003;38:900-8.
73. Essell JH, Schroeder MT, Harman GS, et al. Ursodiol prophylaxis against hepatic complications of allogeneic bone marrow transplantation: A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1998;128(12 Pt 1):975-81.
74. Bearman SI. Avoiding hepatic veno-occlusive disease: What do we know and where are we going? Bone Marrow Transplant. 2001;27:1113-20.
75. Attal M, Huguet F, Rubie H, et al. Prevention of hepatic veno-occlusive disease after bone marrow transplantation by continuous infusion of low-dose heparin: A prospective, randomized trial. Blood. 1992;79:2834-40.
76. Reiss U, Cowan M, McMillan A, et al. Hepatic veno-occlusive disease in blood and bone marrow transplantation in children and young adults: Incidence, risk factors, and outcome in a cohort of 241 patients. J Pediatr Hematol Oncol. 2002;24:746-50.
77. Park SH, Lee MH, Lee H, et al. A randomized trial of heparin plus ursodiol vs. heparin alone to prevent hepatic veno-occlusive disease after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2002;29:137-43.
78. Simon M, Hahn T, Ford LA, et al. Retrospective multivariate analysis of hepatic veno-occlusive disease after blood or marrow transplantation: Possible beneficial use of low molecular weight heparin. Bone Marrow Transplant. 2001;27:627-33.
79. Chalandon Y, Roosnek E, Mermillod B, et al. Prevention of veno-occlusive disease with defibrotide after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2004;10:347-54.
80. Richardson PG, Murakami C, Jin Z, et al. Multi-institutional use of defibrotide in 88 patients after stem cell transplantation with severe veno-occlusive disease and multisystem organ failure: Response without significant toxicity in a high-risk population and factors predictive of outcome. Blood. 2002;100:4337-43.
81. Azoulay D, Castaing D, Lemoine A, et al. Transjugular intrahepatic portosystemic shunt (TIPS) for severe veno-occlusive disease of the liver following bone marrow transplantation. Bone Marrow Transplant. 2000;25:987-92.
82. Janssen HL. Changing perspectives in portal vein thrombosis. Scand J Gastroenterol. 2000;232:69-73.
83. Kim JH, Lee YS, Kim SH, et al. Does umbilical vein catheterization lead to portal venous thrombosis? Prospective US evaluation in 100 neonates. Radiology. 2001;219:645-50.
84. Janssen HL, Wijnhoud A, Haagsma EB, et al. Extrahepatic portal vein thrombosis: Aetiology and determinants of survival. Gut. 2001;49:720-4.
85. Amitrano L, Guardascione MA, Brancaccio V, et al. Risk factors and clinical presentation of portal vein thrombosis in patients with liver cirrhosis. J Hepatol. 2004;40:736-41.
86. Mangia A, Villani MR, Cappucci G, et al. Causes of portal venous thrombosis in cirrhotic patients: The role of genetic and acquired factors. Eur J Gastroenterol Hepatol. 2005;17:745-51.
87. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg. 2002;184:631-5.
88. Smoot RL, Christein JD, Farnell MB. Durability of portal venous reconstruction following resection during pancreaticoduodenectomy. J Gastrointest Surg. 2006;10:1371-5.
89. Lohr JM, Kuchenreuter S, Grebmeier H, et al. Compression of the common bile duct due to portal-vein thrombosis in polycythemia vera. Hepatology. 1993;17:586-92.
90. Bayraktar Y, Balkanci F, Kayhan B, et al. Bile duct varices or “pseudo-cholangiocarcinoma sign” in portal hypertension due to cavernous transformation of the portal vein. Am J Gastroenterol. 1992;87:1801-6.
91. Gurakan F, Eren M, Kocak N, et al. Extrahepatic portal vein thrombosis in children: Etiology and long-term follow-up. J Clin Gastroenterol. 2004;38:368-72.
92. Yamakado K, Nakatsuka A, Tanaka N, et al. Portal venous stent placement in patients with pancreatic and biliary neoplasms invading portal veins and causing portal hypertension: Initial experience. Radiology. 2001;220:150-6.
93. Boles ET, Wise WE, Birken G. Extrahepatic portal hypertension in children: Long-term evaluation. Am J Surg. 1986;151:734-9.
94. Dagenais M, Langer B, Taylor BR, et al. Experience with radical esophagogastric devascularization procedures (Sugiura) for variceal bleeding outside Japan. World J Surg. 1994;18:222-8.
95. Superina R, Bambini DA, Lokar J, et al. Correction of extrahepatic portal vein thrombosis by the mesenteric to left portal vein bypass. Ann Surg. 2006;243:515-21.
96. Valla DC, Condat B, Lebrec D. Spectrum of portal vein thrombosis in the West. J Gastroenterol Hepatol. 2002;17(Suppl 3):S224-7.
97. Hollingshead M, Burke CT, Mauro MA, et al. Transcatheter thrombolytic therapy for acute mesenteric and portal vein thrombosis. J Vasc Interv Radiol. 2005;16:651-61.
98. Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: Risk and benefit of anticoagulant therapy. Gastroenterology. 2001;120:490-7.
99. Llado L, Fabregat J, Castellote J, et al. Management of portal vein thrombosis in liver transplantation: influence on morbidity and mortality. Clin Transplant. 2007;21:716-21.
100. Henrion J, Schapira M, Luwaert R, et al. Hypoxic hepatitis: Clinical and hemodynamic study in 142 consecutive cases. Medicine. 2003;82:392-406.
101. Johnson RD, O’Connor ML, Kerr RM. Extreme serum elevations of aspartate aminotransferase. Am J Gastroenterol. 1995;90:1244-5.
102. Seeto RK, Fenn B, Rockey DC. Ischemic hepatitis: Clinical presentation and pathogenesis. Am J Med. 2000;109:109-13.
103. Cassidy WM, Reynolds TB. Serum lactate dehydrogenase in the differential diagnosis of acute hepatocellular injury. J Clin Gastroenterol. 1994;19:118-21.
104. Gitlin N, Serio KM. Ischemic hepatitis: Widening horizons. Am J Gastroenterol. 1992;87:831-6.
105. Giallourakis CC, Rosenberg PM, Friedman LS. The liver in heart failure. Clin Liver Disease. 2002;6:947-67. viii-ix
106. Richman SM, Delman AJ, Grob D. Alterations in indices of liver function in congestive heart failure with particular reference to serum enzymes. Am J Med. 1961;30:211-25.
107. Arcidi JM, Moore GW, Hutchins GM. Hepatic morphology in cardiac dysfunction: A clinicopathologic study of 1000 subjects at autopsy. Am J Pathol. 1981;104:159-66.
108. Wanless IR, Liu JJ, Butany J. Role of thrombosis in the pathogenesis of congestive hepatic fibrosis (cardiac cirrhosis). Hepatology. 1995;21:1232-7.
109. Yanoff M, Rawson AJ. Peliosis hepatis: An anatomical study with demonstration of two varieties. Arch Pathol Lab Med. 1964;77:159-65.
110. Iannaccone R, Federle MP, Brancatelli G, et al. Peliosis hepatis: Spectrum of imaging findings. Am J Roentgenol. 2006;187:W43-52.
111. Makdisi WJ, Cherian R, Vanveldhuizen PJ, et al. Fatal peliosis of the liver and spleen in a patient with agnogenic myeloid metaplasia treated with danazol. Am J Gastroenterol. 1995;90:317-18.
112. Fine KD, Solano M, Polter DE, et al. Malignant histiocytosis in a patient presenting with hepatic dysfunction and peliosis hepatis. Am J Gastroenterol. 1995;90:485-8.
113. Jacquemin E, Pariente D, Fabre M, et al. Peliosis hepatis with initial presentation as acute hepatic failure and intraperitoneal hemorrhage in children. J Hepatol. 1999;30:1146-50.
114. Saritas U, Ustundag Y, Isitan G, et al. Abdominal Castleman disease with mixed histopathology in a patient with iron deficiency anemia, growth retardation and peliosis hepatis. Am J Med Sci. 2006;331:51-4.
115. Tappero JW, Mohle-Boetani J, Koehler JE, et al. The epidemiology of bacillary angiomatosis and bacillary peliosis. JAMA. 1993;269:770-5.
116. Mohle-Boetani JC, Koehler JE, Berger TG, et al. Bacillary angiomatosis and bacillary peliosis in patients infected with human immunodeficiency virus: Clinical characteristics in a case-control study. Clin Infect Dis. 1996;22:794-800.
117. Izumi S, Nishiuchi M, Kameda Y, et al. Laparoscopic study of peliosis hepatis and nodular transformation of the liver before and after renal transplantation: Natural history and aetiology in follow-up cases. J Hepatol. 1994;20:129-37.
118. Cavalcanti R, Pol S, Carnot F, et al. Impact and evolution of peliosis hepatis in renal transplant recipients. Transplantation. 1994;58:315-16.
119. Abbas MA, Fowl RJ, Stone WM, et al. Hepatic artery aneurysm: Factors that predict complications. J Vasc Surg. 2003;38:41-5.
120. Tessier DJ, Fowl RJ, Stone WM, et al. Iatrogenic hepatic artery pseudoaneurysms: An uncommon complication after hepatic, biliary, and pancreatic procedures. Ann Vasc Surg. 2003;17:663-9.
121. Baggio E, Migliara B, Lipari G, et al. Treatment of six hepatic artery aneurysms. Ann Vasc Surg. 2004;18:93-9.
122. Krus S, Turjman MW, Fiejka E. Comparative morphology of the hepatic and coronary artery walls. Part 1. Differences in the distribution and intensity of atherosclerotic intimal thickening and atherosclerosis. Med Sci Monit. 2000;6:19-23.
123. Saiura A, Umekita N, Inoue S, et al. Benign biliary stricture associated with atherosclerosis. Hepatogastroenterology. 2001;48:81-2.