[level-membership-for-neurology-category]
CHAPTER 47 VASCULAR DEMENTIA
Cerebrovascular disease constitutes a well-defined cause of dementia first recognized by Sir Thomas Willis in 1672 under the name postapoplectic dementia. Vascular dementia is considered the second major cause of dementia in elderly persons, after Alzheimer’s disease, representing 15% to 20% of all cases of dementia worldwide.1,2 Furthermore, it has been recognized that cerebrovascular disease may serve as a catalyst for converting low-grade Alzheimer’s disease to clinical dementia. The diagnosis of vascular dementia is relatively straightforward, particularly in cases with abrupt loss of cognitive functions after stroke or in individuals with a genetic history of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL).3 The diagnosis may be less obvious in very old patients, in whom cerebrovascular disease and degenerative pathology (neurofibrillary tangles, amyloid deposits, Lewy bodies) may coexist. However, even in these cases of so-called mixed dementia, the contribution of the vascular pathology seems to outweigh the age-associated degenerative pathology. Moreover, vascular dementia is usually unrecognized in patients in whom cerebral hypoperfusion complicates cardiac failure and circulatory diseases. Judicious treatment of vascular risk factors at population level may lessen the predicted epidemic increase of dementia among elderly persons.
DEFINITIONS
Vascular Dementia
Vascular dementia is an etiological category of dementia characterized by severe cognitive impairment resulting from ischemic or hemorrhagic stroke or from hypoperfusion affecting brain regions important for memory, cognition, and behavior.
Vascular dementia is most frequently caused by the following lesions:
Diagnostic Criteria
In most controlled trials in vascular dementia, investigators have used the clinical criteria proposed by Román and colleagues4 on behalf of the National Institute of Neurological Disorders and Stroke–Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINDS-AIREN criteria). Most cases of acute poststroke vascular dementia fulfill criteria for probable vascular dementia, modified as follows:5
Vascular Cognitive Disorder
Numerous diseases and syndromes can produce a vascular cognitive disorder.5 Table 47-1 provides a comprehensive list of potential causes of vascular cognitive disorder.
TABLE 47-1 Potential Etiologies of the Vascular Cognitive Disorder
| Primary vascular causes or hypoperfusion | Amyloidosis angiopathy |
| Arterial dissection | |
| Atherosclerosis of large or mid-sized arteries | |
| Cardiopulmonary arrest | |
| Diabetes mellitus | |
| Dural arteriovenous malformation | |
| Fibromuscular dysplasia | |
| Giant cerebral aneurysm with compression | |
| Hyperlipidemia | |
| Hypertensive arteriolosclerosis | |
| Moyamoya disease | |
| Recurrent hypotension with syncope | |
| Vasospasm/migraine | |
| Hemorrhage | Amyloid angiopathy |
| Aneurysms | |
| Arteriovenous malformations | |
| Cavernous angioma | |
| Charcot-Bouchard aneurysm | |
| CNS hemosiderosis with recurrent bleeding | |
| Hematologic bleeding diathesis | |
| Hemorrhagic transformation of infarction | |
| Hypertension | |
| Neoplasm (glioblastoma or metastatic) | |
| Pharmacologic (e.g., anticoagulants) | |
| Posthemorrhagic normal-pressure hydrocephalus | |
| Telangiectasis | |
| Vasculitis | |
| Venous angioma | |
| Venous thrombosis | |
| Embolic causes | Atherosclerotic stenosis with embolization |
| Atrial fibrillation | |
| Atrial myxoma | |
| Cardiac surgery, including CAGB | |
| Cardiomyopathy | |
| Congenital heart disease | |
| Libman-Sacks endocarditis | |
| Marantic endocarditis | |
| Mitral valve prolapse syndrome | |
| Myocardial infarction with mural thrombus | |
| Nitrogen bubble emboli | |
| Prosthetic valves | |
| Rheumatic endocarditis and valvulopathies | |
| Septal defect with paradoxical emboli | |
| Septic, air, or fat emboli | |
| Subacute bacterial endocarditis | |
| Hematologic disorders | Antiphospholipid/anticardiolipin antibodies |
| Antithrombin III deficiency | |
| Arylsulfatase A pseudodeficiency | |
| Cryoglobulinemia | |
| Disseminated intravascular coagulation | |
| Dysfibrinogenemias | |
| Dysproteinemias | |
| Factor V Leyden mutation | |
| Factors V, VII, XII, and XIII deficiencies | |
| Hemoglobinopathies (e.g., sickle cell disease) | |
| Heparin cofactor II deficiency | |
| Hyperviscosity syndromes | |
| Idiopathic thrombocytosis | |
| Leukemias | |
| Nephrotic syndrome | |
| Polycythemia vera | |
| Pregnancy and oral contraceptives | |
| Protein C or S deficiency | |
| Thrombotic thrombocytopenic purpura | |
| Waldenström’s macroglobulinemia | |
| Toxic causes | Amphetamines |
| Arsenic | |
| Carbon monoxide | |
| Cocaine or crack | |
| Ergot alkaloids | |
| Oral contraceptives | |
| Radiation-induced vasculopathy | |
| Noninfectious inflammatory or autoimmune causes | Allergic or hypersensitivity angiitis |
| Anticardiolipin or antiphospholipid antibodies | |
| Behçet’s syndrome | |
| Burgher’s disease | |
| Cogan’s syndrome | |
| Dermatomyositis-polymyositis | |
| Disseminated neocortical and subcortical encephalopathies | |
| Drug-induced vasculitis | |
| Endocardial fibroelastosis | |
| Henoch-Schönlein purpura (radiation, tumor) | |
| Kawasaki syndrome | |
| Köhlmeier-Degos disease | |
| Lupus anticoagulant | |
| Polyarteritis nodosa | |
| Primary CNS vasculitis | |
| Relapsing polychondritis | |
| Rheumatoid arthritis with arteritis | |
| Sarcoidosis | |
| Scleroderma | |
| Sjögren’s syndrome | |
| Systemic lupus erythematosus | |
| Takayasu’s arteritis | |
| Temporal (giant cell) arteritis | |
| Thromboangiitis obliterans | |
| Thrombotic microangiopathy | |
| Ulcerative colitis | |
| Vogt-Koyanagi-Harada syndrome | |
| Wegener’s granulomatosis | |
| Infectious causes of stroke | AIDS |
| Bacterial meningitis with arteritis | |
| Cat-scratch disease | |
| Cysticercosis | |
| Herpes zoster ophthalmicus | |
| Lyme disease | |
| Meningovascular infectious arteritis | |
| Parasites and ova | |
| Rickettsial arteritis | |
| Syphilis | |
| Tuberculosis | |
| Viral arteritis | |
| Yeast and fungal arteritis | |
| Genetic causes | 11β-hydroxylase deficiency |
| 11β-ketoreductase deficiency | |
| 17α-hydroxylase deficiency | |
| Antithrombin III deficiency | |
| β-Thalassemia major | |
| Bannayan-Zonana syndrome | |
| Bloom’s syndrome | |
| CADASIL | |
| Chronic familial cerebral vasculopathy | |
| Cockayne’s syndrome | |
| Dysfibrinogenemia | |
| Ehlers-Danlos syndrome, especially type IV | |
| Fabry’s disease | |
| Factors VII to XIII deficiencies | |
| Familial atrial myxoma | |
| Familial cavernous angioma | |
| Familial hemiplegic migraine | |
| Familial hypercholesterolemia | |
| Familial hypoalphalipoproteinemia | |
| Familial intracranial aneurysm | |
| Familial oculoleptomeningeal amyloidosis | |
| Familial porencephaly | |
| Familial triglyceridemia | |
| Fibromuscular dysplasia | |
| Hemoglobin SC | |
| Heparin cofactor II deficiency | |
| Hereditary arteriovenous malformations | |
| Hereditary cardiac conduction disorder | |
| Hereditary cardiomyopathies | |
| Hereditary cerebral amyloidosis | |
| Hereditary platelet defect (Wiskott-Aldrich syndrome) | |
| Hereditary polycythemia | |
| Homocystinuria | |
| Hyperlipoproteinemia (types III and IV) | |
| Klippel-Trénaunay-Weber syndrome | |
| Leigh’s disease | |
| Marfan syndrome | |
| MELAS syndrome | |
| Menkes’ syndrome | |
| Methylmalonic, propionic, and isovaleric acidemia; glutamic aciduria type I | |
| Mitral valve prolapse | |
| Moyamoya disease | |
| Neurofibromatosis | |
| Plasminogen deficiency | |
| Progeria | |
| Prekallikrein deficiency | |
| Protein C or protein S deficiency | |
| Pseudoxanthoma elasticum | |
| Rendu-Osler-Weber syndrome | |
| Rhabdomyomas | |
| Sickle-cell disease | |
| Sturge-Weber syndrome | |
| Sulfite oxidase deficiency | |
| Tangier disease | |
| Tuberous sclerosis | |
| Von Hippel–Lindau syndrome | |
| Miscellaneous causes | Metastatic deposits |
| Neoplastic anagioendotheliosis | |
| Sneddon’s syndrome | |
| Spatz-Lindenberg disease | |
| Susac’s disease |
AIDS, acquired immunodeficiency syndrome; CABG, coronary artery bypass graft; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CNS, central nervous system; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and strokelike symptoms (syndrome).
Modified From Mendez MF, Cummings JL: Dementia: A Clinical Approach, 3rd ed. Philadelphia: Butterworth-Heinemann (Elsevier Science), 2003.
Vascular Cognitive Impairment
This term designates a wide range of severity of cognitive alterations of vascular nature. However, the term is preferably restricted to cases of vascular cognitive impairment without dementia (vascular mild cognitive impairment).5
EPIDEMIOLOGY
In some of the most heavily populated regions of the world—including China, Japan, India, and Mexico—a vascular etiology is the most frequent cause of dementia. As the population in most of the world ages, likely increases in stroke and heart disease associated with old age will render vascular dementia the most frequent cause of dementia worldwide.
Prevalence
Jorm and Jolley6 performed a meta-analysis of 47 international studies on the prevalence of vascular dementia and found the following consistent trends:
Incidence
Incidence data for vascular dementia are quite limited. Dubois and Herbert2 used age-standardized incidence ratios to analyze data from 10 incidence studies of vascular dementia. These ratios ranged from 0.42 to 2.68, confirming the geographic and racial variations of vascular dementia.
CLINICAL FEATURES
Despite the apparent complexity of vascular dementia, the clinical diagnosis may be simplified into two major groups, acute and subacute, according to the temporal profile of presentation (Table 47-2).
TABLE 47-2 Clinical Forms of Vascular Dementia According to Temporal Pattern of Presentation
| Acute Forms | |
Clinical featuresAmnesia, visual disturbances (homonymous hemianopsia, color agnosia, visual agnosia)Left-sided lesions: transcortical sensory aphasia, alexia without agraphiaRight-sided lesions: spatial disorientationBilateral occipital lesions: Anton’s syndromeBilateral parieto-occipital lesions: Balint’s syndrome
LesionsRuptured AComA aneurysm or proximal ACAClinical featuresSevere anterograde amnesia for verbal orvisuospatial materialSevere apathyLack of initiative and spontaneityExecutive dysfunction
a. Thalamic dementiaLesionsAnterior (polar) thalamic territories (polar thalamic artery, from PComA)DMn or mammillothalamic tractClinical featuresImpairments of attention, motivation, initiative, executive functions, and memoryVerbal and motor slowness and apathyVertical gaze paresis, medial rectus paresis, and absent convergence; dysarthria and mild hemiparesisb. Inferior genu strokeLesionsLacune in inferior genu of internal capsuleInferior capsular genu stroke, producing diaschisis of frontal lobes and cerebellumClinical featuresSudden change in cognitionFluctuating attentionConfusionAbuliaStriking psychomotor retardationInattentionExecutive dysfunctionMild to absent focal findings such as hemiparesis or dysarthria
Subacute Forms
LesionsSmall-vessel disease with presence of white matter ischemic lesions and lacunae in basal ganglia and white matterClinical featuresSubacute onset, chronic course, fluctuations, and progressive worseningSubcortical-type dementia, frontal lobe deficits, depressive mood, psychomotor slowing, parkinsonian features, pseudobulbar palsy, urinary urgencyMain forms of SIVD:Binswanger’s disease, CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), and some types of cerebral amyloid angiopathy
ACA, anterior cerebral artery; AComA, anterior communicating artery; DMn, dorsomedial nucleus of the thalamus; PCA, posterior cerebral artery; PComA, posterior communicating artery.
Acute Forms of Vascular Dementia
In acute forms, patients exhibit new-onset dementia after a vascular event (cerebral hemorrhage, ischemic “strategic” stroke). For instance, occlusion of the left posterior parietal branch of the middle cerebral artery that damages the supramarginal gyrus usually produces cortical sensory loss, Wernicke’s aphasia, and Gerstmann’s syndrome with right-left disorientation, finger agnosia, acalculia, and agraphia.
Multi-infarct Dementia
MID results from multiple cortical strokes; about one third (range, 25% to 41%) of all ischemic stroke survivors aged 65 years and older may develop vascular dementia.7,8 This represents about 125,000 new cases per year of poststroke vascular dementia in the United States. Table 47-3 lists the most important risk factors for poststroke vascular dementia.
TABLE 47-3 Main Risk Factors for Poststroke Vascular Dementia
| Age | Older age |
| Education | Lower educational level |
| Personal factors | Lower income, current smokers |
| Genetic factors | Family history of dementia |
| Stroke type | Recurrent strokes |
| Stroke location | Left-sided lesions, “strategic strokes” |
| Stroke volume | Lesions larger than 50-100 mL of tissue destruction, large perilesional incomplete ischemic areas involving white matter, larger periventricular white matter ischemic lesions |
| Stroke complications | Hypoxic and ischemic complications of acute stroke (i.e., seizures, cardiac arrhythmias, aspiration pneumonia, hypotension) |
| Stroke manifestations | Dysphagia, gait limitations, urinary impairment |
Strategic Strokes
Single ischemic strokes in three possible vascular territories—the posterior cerebral artery, anterior cerebral artery, and thalamus-basal ganglia—may produce vascular dementia.
Posterior cerebral artery infarctions involving the ventral-medial temporal lobe, occipital structures, and thalamus
About 25% of affected patients develop amnesia from damage of the hippocampus-mammillothalamic-cingulum circuit. Left-sided lesions cause verbal amnesia, whereas right-sided lesions alter visuospatial (location) memory; bilateral damage causes global amnesia. Visual signs (homonymous hemianopsia, color agnosia, visual agnosia) occur in 80% or more of patients with posterior cerebral artery strokes. Left-sided lesions may produce transcortical sensory aphasia, or alexia without agraphia. Spatial disorientation occurs with right-sided lesions and Anton’s syndrome (cortical blindness with anosognosia) with bilateral occipital lesions. Bilateral parieto-occipital infarctions above the calcarine fissure may result in Balint’s syndrome (inability to direct the eyes to a certain point in the visual field) with simultagnosia, optic ataxia, and ocular apraxia.
Anterior cerebral artery territory and medial frontal lobe lesions
Ischemic injury of cholinergic nuclei in the basal forebrain may occur with subarachnoid hemorrhage from ruptured aneurysms of the anterior communicating artery or proximal anterior cerebral artery, usually after surgical repair. Such hemorrhage leads to severe anterograde amnesia, apathy, and executive dysfunction.
Lesions of the thalamus and basal ganglia
Thalamic dementia occurs after paramedian thalamic ischemic strokes. The lesions involve the anterior thalamus, which is irrigated by the polar thalamic artery, a branch of the posterior communicating artery, or the medial and central thalamus, involving the dorsomedial nucleus and the mammillothalamic tract. Damage of the mammillothalamic tract is critical in thalamic amnesia. Patients are initially unresponsive but improve over days to weeks, revealing impairments in attention, motivation, initiative, executive functions, and memory; verbal and motor slowness; and apathy. Gaze abnormalities include vertical gaze, medial rectus paresis, and absence of convergence. Dysarthria and mild hemiparesis occur with larger lesions. Left-sided thalamic lesions cause memory deficits more often than do right-sided ones. Global amnesia occurs with bilateral lesions or with simultaneous damage to the mammillothalamic tract and inferior thalamic peduncle. Severe attentional and motivational deficits play roles in the amnesia.
Inferior genu stroke is a lacunar infarction of the inferior genu of the internal capsule, manifested by sudden change in cognitive function, often in association with fluctuating attention, confusion, abulia, striking psychomotor retardation, inattention, executive dysfunction, and other features of frontal lobe dysfunction but with mild focal findings such as hemiparesis or dysarthria. These deficits are caused by ipsilateral blood flow reduction to the inferomedial frontal cortex, ipsilateral temporal lobe, and contralateral cerebellar hemisphere by a mechanism of diaschisis. Severed pathways that contain cholinergic fibers from the nucleus basalis of Meyner may have an effect in reducing blood flow.
Subacute Forms of Vascular Dementia
Subcortical ischemic vascular dementia (SIVD) refers to forms of vascular dementia characterized by subacute onset, caused by small-vessel disease that produces either arteriolar occlusion and lacunes or widespread incomplete infarction of white matter as a result of critical stenosis of medullary arterioles and hypoperfusion (Binswanger’s disease).7 The term subcortical refers both to the location of lesions that involve predominantly basal ganglia, cerebral white matter, and brainstem and to their manifestations with preponderant executive dysfunction (subcortical dementia as opposed to cortical dementia). The main mechanism of dementia causation in SIVD is ischemic brain injury; this label includes both complete infarction (mainly lacunar strokes and microinfarcts) as well as incomplete infarction of deep cerebral white matter.
The temporal profile is typically subacute, with a chronic course marked by fluctuations and slowly progressive worsening. This subcortical type of dementia is manifested by motor and cognitive executive function slowing, forgetfulness, dysarthria, mood changes, pseudobulbar palsy, urinary symptoms, parkinsonian features, and short-stepped gait. Manifestations probably result from ischemic interruption of parallel circuits from prefrontal cortex to basal ganglia and corresponding thalamocortical connections. Many strokes in this common type of vascular dementia are clinically silent; that is, they do not appear with the usual sensorimotor manifestations associated with stroke but with changes in personality, mood, behavior, or cognition. Therefore, brain imaging (computed tomography/magnetic resonance imaging) is crucial for correct diagnosis.
The main forms of SIVD are Binswanger’s disease, CADASIL, lacunar dementia (lacunar state, or état lacunaire) with multiple lacunes and extensive perifocal incomplete infarctions or with abundant microinfarctions, and some types of cerebral amyloid angiopathy.
Binswanger’s Disease
The hallmark of this condition is an ischemic periventricular leukoencephalopathy that typically spares the arcuate subcortical U fibers. These periventricular, distal-territory, white matter lesions in the elderly brain are caused by chronic ischemia with incomplete infarction. Small-vessel disease and multiple lacunes often coexist in Binswanger’s disease.
Clinical manifestations include a subcortical dementia with cognitive and motor executive dysfunction, loss of verbal fluency, perseveration, impersistence, inattention, difficulties with set shifting, and abnormal performance on Luria’s kinetic melody tests. Memory loss is characterized by poor retrieval and intact recognition. Apathy, depression, and behavioral problems are common. Mild residual hemiparesis, discrete focal findings, and short-stepped gait (marche à petits pas), dysarthria, and pseudobulbar palsy are common. Extrapyramidal features, such as inexpressive facies, slowness of movement, axial rigidity, loss of postural reflexes, frequent falls, increased urinary frequency, and nocturia are also common findings. Brain T2-weighted magnetic resonance imaging findings include extensive hyperintense lesions in the periventricular regions with frontal and parietal preponderance.
CADASIL
This genetic form of vascular dementia was first identified in France in 1991.3 CADASIL is a systemic autosomal dominant arteriopathy caused by mutations of the Notch3 gene in chromosome 19.8 The vascular lesion shows fragmentation of the internal elastica and eosinophilic deposits in media. Electron microscopy reveals typical granular osmiophilic material in the basement membranes of vascular smooth muscle cells of arterioles (100 to 400 μm in diameter) and in capillaries, primarily in the brain but also in other organs. The diagnosis may be established by genetic studies and by skin biopsy with confirmation by immunostaining with a Notch3 monoclonal antibody. Neuropathology examination demonstrates lacunar strokes associated with extensive, confluent areas of frontal ischemic leukoencephalopathy.
Clinically, CADASIL is a subcortical vascular dementia identical to Binswanger’s disease. Onset is early for stroke (mean age, 46 years) with recurrent transient ischemic attacks or lacunar strokes, in the absence of vascular risk factors, culminating in vascular dementia and death, usually about 20 years after onset. Migraine with aura and depression are common. The dementia is subcortical, frontal, and accompanied by gait disturbances, urinary incontinence, and pseudobulbar palsy. Magnetic resonance imaging shows striking white matter changes with a typical predilection for the temporal lobes, along with lacunar strokes, in both symptomatic and asymptomatic members of families affected by CADASIL.
PATHOLOGY AND PATHOPHYSIOLOGY
The main mechanism of production of vascular dementia is ischemic brain injury, occurring as the result of either vascular occlusion or hypoperfusion. From the neuropathological viewpoint, ischemia includes both complete and incomplete infarctions.
Complete Infarctions
These are typical embolic or atherothrombotic large-vessel ischemic infarcts involving well-defined arterial territories, cortical or cortical-subcortical in location, usually with perifocal incomplete white matter infarction. Small-vessel occlusion causes lacunes and microinfarcts.
Lacunar Infarcts
Lacunes (Latin lacuna, plural lacunae: a minute cavity, hole, or pit) are small cavitating ischemic infarcts, less than 15 mm in diameter, typically located in the basal ganglia, internal capsule, thalamus, pons, corona radiata, and centrum semiovale. White matter lacunes may overlap with nonconfluent areas of ischemic white matter changes.
Incomplete Infarctions
These lesions are caused by hypoperfusion9 and typically affect watershed territories such as the CA1 sector of the hippocampus; the cortex posterior to the interparietal sulcus or the central white matter between deep territories at the boundaries between the anterior cerebral artery and middle cerebral artery in cases of extracranial internal carotid artery occlusion; and the periventricular white matter fed by long-penetrating end-arterioles, as in Binswanger’s disease.
White matter lesions include diffuse myelin pallor that spares the U fibers, astrocytic gliosis, loss of oligodendrocytes progressing to rarefaction, spongiosis (vacuolization), widening of perivascular spaces (état criblé), with myelin and axonal loss but without definite necrosis (incomplete white matter infarction) that culminates in necrosis and cavitations (lacunes). Pure incomplete white matter infarction, similar to that observed in the penumbra of large infarcts, is typically seen in hypoperfusive disease.
Figures 47-1 and 47-2 illustrate the two main pathophysiological pathways leading to the aforementioned ischemic lesions:
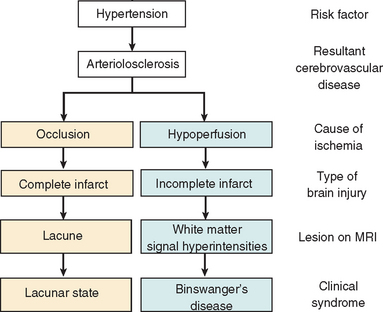
Figure 47-1 Two pathophysiological pathways of ischemic brain injury. The pathway on the left is initiated by occlusion of an arterial lumen. This leads to discrete areas of complete infarction (i.e., lacunar infarcts) and functional disruption within a distributed network (e.g., dementia). The pathway on the right is defined by critical stenosis and hypoperfusion involving multiple small arterioles mainly in deep white matter. These two pathways often coexist in the same patient. MRI, magnetic resonance imaging.
(From Román GC, Erkinjuntti T, Wallin A, et al: Subcortical ischaemic vascular dementia. Lancet Neurology 2002; 1:426-436.)
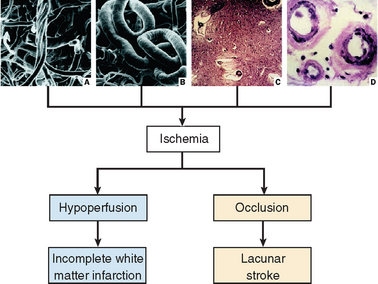
Figure 47-2 Factors leading to brain ischemia and hypoperfusion in elderly people. Striking age-dependent morphological changes in brain vessels include tortuosity with formation of skeins in cortical arterioles (A) and elongation, which results in loops and tangles (B), particularly of long penetrating arterioles supplying deep white matter. Increase in blood vessel length raises the blood pressure threshold for perfusion of periventricular white matter at the distal ends of the these vessels. Furthermore, the lumen is stenosed by senile concentric arteriolosclerosis (D), often with calcification of vessels (C, top right). Under normal conditions, autoregulatory mechanisms induce vasodilation in response to decreases in mean arterial perfusion pressure; these mechanisms become inoperative in patients with arteriolosclerosis and calcified vessels (C). The elderly brain is therefore more susceptible to hypotension and pump failure (cardiac arrhythmias and congestive heart failure). Delivery of oxygen to tissues and other metabolic exchanges are impeded by increased thickness of vessel walls (D) and widespread widening of perivascular spaces (état criblé) (C) with enlargement of perivascular spaces of Virchow-Robin, which results from tortuosity of elongated arterioles.
(A from Duvernoy HM, Delon S, Vanson JL: Cortical blood vessels of the human brain. Brain Res Bull 1981; 7:519-579. B to D from GC Román: Vascular dementia. In Fisher M, ed: Clinical Atlas of Cerebrovascular Disorders. London: Wolfe/Mosby/Year Book Europe, 1994, pp 13.1-13.23.)
Table 47-4 summarizes important physical principles and pathophysiological mechanisms relevant to the microcirculation in SIVD; these include hemorheological factors, increased resistance to flow, decreased autoregulation, endothelial changes, blood-brain barrier dysfunction, and dilatation of perivascular spaces. Their combined effects result in hypoperfusion and incomplete infarction of deep white matter.
TABLE 47-4 Relevant Hemorheological and Autoregulatory Circulatory Factors in White Matter Hypoperfusion
Blood flow = perfusion pressure · π · radius4/8 · η · length
rCMRO2 = OEF * arterial oxygen content * rCBFWhen failing perfusion pressure exceeds compensatory mechanisms, oxygen metabolism is compromised. Neuronal-glial function is lost, followed by irreversible necrosis. In infarcted tissue, both CMRO2 and OEF values are minimal. Thus, increased OEF is a transitional phenomenon, signaling a critical period of incipient ischemia.
From Román GC, Erkinjuntti T, Wallin A, et al: Subcortical ischaemic vascular dementia. Lancet Neurology 2002;1:426-436.
Earlier concepts of MID required the loss of more than 50 to 100 mL of brain tissue from ischemia. Stroke location appears to be equally important. In fact, small lesions in strategic areas are capable of causing vascular dementia by interrupting memory and behavioral circuits, such as the prefrontal–basal ganglia–thalamocortical loops that are crucial for executive function and independent living.
DIAGNOSIS
In practical terms, the diagnosis of vascular dementia should be suspected in patients with significant vascular risk factors, stroke, or heart disease who exhibit abrupt changes in cognition. Brain imaging demonstrating the presence of relevant lesions of cerebrovascular disease is crucial for the diagnosis of vascular dementia. Most of the diagnostic difficulties in vascular dementia occur with the subacute, slowly progressive forms of vascular dementia that, in the opinion of some clinicians, could be confused with Alzheimer’s disease. A closer look at these subcortical forms of vascular dementia reveals significant differences, as listed in Table 47-5.
TABLE 47-5 Important Feature Differences between Subcortical Vascular Dementia and Alzheimer’s Disease
| Clinical Feature | Alzheimer’s Disease | Vascular Dementia |
|---|---|---|
| Memory loss | Early, severe amnesia | Forgetfulness, poor recall |
| Language disorder | Early aphasia | Loss of verbal fluency, dysarthria |
| Gait | Normal | Abnormal, marche à petits pas |
| Falls | Rare | Frequent |
| Executive dysfunction | Late | Early and severe |
| Mood and personality | Intact | Changes: apathy, depression, crying spells |
| Urinary problems | Late incontinence | Urgency, nocturia |
| Onset | Slow | Abrupt |
| Progression | Rapid | Slow, stepwise, fluctuating |
TREATMENT
Recently, a number of treatment trials in patients with vascular dementia were successfully completed. Despite progress, there are no approved drugs for symptomatic treatment of vascular dementia.
The antiplatelet agents aspirin, triflusal, and Ginkgo biloba have been used in patients with MID or with Alzheimer’s disease plus cerebrovascular disease, with modest results, mainly by decreasing vascular dementia progression by lowering the risk of recurrent stroke. Vasodilators have no proved effects on vascular dementia, except for niacin, which has lipid-lowering effects. The calcium-channel blockers nimodipine and nicardipine have moderate efficacy in tests of attention and psychomotor performance in SIVD. Of the neuroprotective or nootropic agents, piracetam, oxiracetam, and nicergoline appear to be modestly effective for the MID form of vascular dementia. Citicoline showed positive short-term effects on memory and behavior in patients with vascular dementia.
In the multicenter European Pentoxifylline Multi-Infarct Dementia Study,10 pentoxifylline, a xanthine derivative with hemorheological and immunomodulatory properties, produced significant cognitive improvement in MID, in comparison with placebo. Pentoxifylline is useful in Binswanger’s disease, improving hyperviscosity, sedimentation rate, hyperfibrinogenemia, and increased acute-phase reactants (tumor necrosis factor α).
Currently, the most promising therapies for vascular dementia and vascular cognitive impairment include the cholinesterase inhibitors donepezil (Aricept), galantamine (Reminyl), and rivastigmine (Exelon), as well as the N-methyl-D-aspartate (NMDA)–glutamate receptor antagonist memantine. Cholinesterase inhibitors, approved for the treatment of Alzheimer’s disease, appear to be an effective and safe alternative for patients with vascular dementia.11
PREVENTION
Age, gender, ethnicity, and genetic makeup are nonmodifiable risk factors for stroke. However, a number of risk factors for vascular dementia (Table 47-6) can be prevented. Primary prevention is directed to the control of vascular risk factors in an effort to prevent stroke, ischemic heart disease, and vascular dementia. Secondary prevention addresses prevention of further ischemic episodes.
TABLE 47-6 Risk Factors for Vascular Dementia
Primary Prevention
Treatment of hypertension appears to decrease the incidence of dementia. Other treatable risk factors include diabetes mellitus, the metabolic syndrome, raised homocysteine level, and smoking. Homocysteine is lowered with oral folate and parenteral vitamin B12 supplementation. The beneficial cardiovascular effects of a Mediterranean diet rich in fruits and antioxidants have been well documented. Statins (3-hydroxy-3-methylglutaryl–coenzyme A [HMG-CoA] reductase inhibitors) may reduce dementia risk, but its use in nondemented, nonhyperlipidemic patients cannot currently be recommended.
Secondary Prevention
Optimal management of acute stroke and swift implementation of secondary prevention are mandatory to prevent vascular dementia. In the Perindopril Protection Against Recurrent Stroke Study (PROGRESS),12 blood pressure lowering in patients with previous stroke or transient ischemic attack reduced the incidence of secondary stroke by 28%, that of major vascular events by 26%, and that of major coronary events by 26%. These reductions were all magnified by approximately 50% in a subgroup of patients in whom the angiotensin-converting enzyme inhibitor perindopril was routinely combined with the diuretic indapamide. This combination alone could reduce the burden of stroke and could avert between 0.5 million and 1 million strokes each year worldwide.
In brief, control of blood pressure in the general population could significantly decrease the social and economic burdens of dementia in years to come.
Bowler JV, Hachinski V, editors. Vascular Cognitive Impairment: preventable Dementia. Oxford, UK: Oxford University Press, 2003.
Erkinjuntti T, Román G, Gauthier S, et al. Emerging therapies for vascular dementia and vascular cognitive impairment. Stroke. 2004;35:1010-1017.
Paul RH, Cohen R, Ott BR, et al, editors. Vascular Dementia: Cerebrovascular Mechanisms and Clinical Management. Totowa, NJ: Humana Press, 2005.
Román GC, editor. Advances in vascular dementia [whole issue]. Semin Cerebrovasc Dis Stroke. 2004;4(2).
Román GC, Erkinjuntti T, Wallin A, et al. Subcortical ischaemic vascular dementia. Lancet Neurology. 2002;1:426-436.
1 Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurology. 2000;54(Suppl 5):S4-S9.
2 Dubois MF, Herbert R. The incidence of vascular dementia in Canada: a comparison with Europe and East Asia. Neuroepidemiology. 2001;20:179-187.
3 Bousser MG, Tournier-Lasserve E. Summary of the Proceedings of the First International Workshop on CADASIL. Paris, May 19–21, 1993. Stroke. 1994;25:704-707.
4 Román GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: diagnostic criteria for research studies: report of the NINDS-AIREN International Workshop. Neurology. 1993;43:250-260.
5 Román GC, Sachdev P, Royall DR, et al. Vascular cognitive disorder: a new diagnostic category updating vascular cognitive impairment and vascular dementia. J Neurol Sci. 2004;226:81-87.
6 Jorm AF, Jolley D. The incidence of dementia: a meta-analysis. Neurology. 1998;51:728-733.
7 Román GC, Erkinjuntti T, Wallin A, et al. Subcortical ischaemic vascular dementia. Lancet Neurology. 2002;1:426-436.
8 Tournier-Lasserve E, Joutel A, Melki J, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat Genet. 1993;3:256-259.
9 Román GC. Brain hypoperfusion: a critical factor in vascular dementia. Neurol Res. 2004;26:454-458.
10 European Pentoxifylline Multi-Infarct Dementia Study. Eur Neurol. 1996;36:315-321.
11 Erkinjuntti T, Román G, Gauthier S, et al. Emerging therapies for vascular dementia and vascular cognitive impairment. Stroke. 2004;35:1010-1017.
12 Tzourio C, Anderson C, Chapman N, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069-1075.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]<
[/not-level-membership-for-neurology-category]
