[level-membership-for-pediatrics-category]
Vascular and Lymphatic Anomalies
Vascular anomalies are broadly divided into two groups based on biologic and clinical behavior: vascular tumors and vascular malformations.1 Vascular tumors are true neoplasms that arise from cellular hyperplasia. In contrast, vascular malformations are congenital lesions originating from errors of embryonic development and exhibit normal endothelial cell turnover.1 Historically, the field of vascular anomalies has been hindered by a myriad of confusing and misused terminology and nomenclature. This, along with the rarity and often complex nature of some of these disorders, has combined to make diagnosis and treatment of vascular anomalies difficult. However, the last several decades have brought better insight and understanding into the field of vascular anomalies, with improved knowledge of blood vessel angiogenesis and the development of a more logical classification system.
Classification
In 1996, the International Society of the Study of Vascular Anomalies formally accepted the biological classification system in use today (Table 72-1).2,3 This system divides these anomalies into vascular tumors and vascular malformations based on physical characteristics, natural history, and cellular features. Examples of vascular tumors are infantile hemangioma (IH), kaposiform hemangioendothelioma (KHE), and tufted angioma (TA). Vascular malformations can be divided based on vascular channel type (capillary, lymphatic, venous, arterial, or combined) or by flow (slow or fast). Examples of slow-flow lesions are capillary malformations (CM), lymphatic malformations (LM), and venous malformations (VM). Fast-flow lesions include arteriovenous fistulas (AVF) and arteriovenous malformations (AVM).

Nomenclature
In the 19th century, Virchow first described the histologic features of vascular nevi.4 He initiated the term ‘angioma’, which became the default term to describe all such nevi regardless of natural history or other clinical features. He also labeled the IH ‘angioma simplex’, a lesion that has been historically referred to as ‘capillary hemangioma’ and ‘strawberry hemangioma.’ Virchow’s ‘angioma cavernosum’ was used to label two distinct lesions, IH (when located deep to the skin) and VM, because both have similar appearance on physical examination. ‘Angioma racemosum’ was Virchow’s designation for what today is termed an arteriovenous malformation and which has previously been called an ‘arteriovenous hemangioma.’
Wegener, a student of Virchow, described the histology of LMs, which he called ‘lymphangiomas.’5 The classic term ‘cystic hygroma’, referring to LM, unfortunately also continues to have common usage. Thus, both the terms cystic hygroma and lymphangioma should be abandoned in favor of LM (macrocystic and microcystic, respectively). The problems with this jumble of descriptive and histologic terms are obvious. The same lesion can often have several different names, and simultaneously, the same name can refer to several different lesions. For example, the term ‘hemangioma’, combined with descriptive modifiers such as ‘strawberry’, ‘cavernous’, and ‘lympho-’, is used to describe tumors, birthmarks, and vascular malformations alike. Vascular anomalies with quite distinct features, whether congenital or acquired, or whether they spontaneously regress or progress over time, become lumped under the umbrella term ‘hemangioma.’ These faulty designations lead to improper diagnosis and treatment for patients as well as leading to misguided interdisciplinary communication and research efforts.
Vascular Tumors
Infantile Hemangioma
IH are the most common tumor of infancy. They occur in about 4% of infants, though early studies were as high as 10%, probably due to the inclusion of other vascular lesions.6 The incidence is higher in premature infants, Caucasians, and females (by a 3 to 5 : 1 ratio). 6,7 Advanced maternal age, multiple gestations, and placental abnormalities are also risk factors.8 IH have a unique and characteristic life cycle consisting of three phases: proliferative, involuting, and involuted.
Pathophysiology
The precise etiology of IH remains unknown. Viral causes have been speculated, but none elucidated. Some studies suggest that they arise from the clonal expansion of endothelial stem/progenitor cells, the source of which is unclear.9–11 One report concluded these cells arise from a population of resident angioblasts, arrested in an early stage of vascular development.12 As hemangiomas are more common in females, estrogen, which has a stimulatory effect on endothelial cells, may factor in the development of these lesions. Its receptors are present on endothelial cells and elevated levels of estradiol have been found in infants with hemangiomas.13,14
The expression of placental markers (including CD 32, Fcγ-RIIb, glucose transporter 1 [GLUT-1], indoleamine deoxygenase [IDO], insulin growth factor 2 [IGF-2] Lewis Y antigen, merosin, type II 17-hydroxysteroid dehydrogenase [17HSDβ2], tissue factor pathway inhibitor 2 [TFPI-2], and type III iodothyronine deoidinase) by hemangioma endothelial cells suggests a placental origin.15–21 GLUT-1, an erythrocyte type glucose transporter, is a specific marker for endothelial cells of hemangiomas, but is not found in other vascular anomalies.15 The placental cells may arrive at fetal tissue following local placental disruption as an embolic nidus though the permissive right to left shunt of fetal circulation. This may occur during chorionic villus sampling or placental complications such as pre-eclampsia and placenta previa, which have shown to be predisposing factors for hemangiomas.8,22,23
The dysregulation of angiogenesis can be seen during the proliferation and involution of hemangiomas, and is suspected to be a cause of the disease. IH in the proliferative phase express high levels of fibroblast growth factor (FGF), TIE-2, angiopoietins, matrix metalloproteinases (MMPs) and vascular endothelial growth factor A (VEGF-A) and its receptor (VEGFR), all of which play critical roles in the formation of blood vessels during and after embryogenesis.24–30 The tumor during this phase is composed of plump, rapidly dividing endothelial cells forming a mass of sinusoidal vascular channels. Enlarged feeding arteries and draining veins often vascularize the tumor. Markers for mature endothelium, CD-31 and von Willebrand factor, are present on these neoplastic endothelial cells. Involuted hemangiomas express normal levels of these factors, but elevated levels of tissue inhibitor of TIMP1, a metalloproteinase that inhibits new blood vessel formation, and interferon-β.28,29 The endothelial cells of the tumor flatten as apoptosis progresses, the vascular channels dilate, and the tumor assumes a lobular architecture with replacement by fibrofatty stroma.31 All that remains in the involuted phase is a residuum of fibrofatty tissue with tiny capillaries and mildly dilated draining vessels.
Clinical Features
IH are not fully developed at birth and first appear in the neonatal period with a median age of onset of 2 weeks. A premonitory cutaneous sign may be present at birth in 30–50% of cases.1 They are most often cutaneous (80%) and have an anatomic predilection for the head and neck region (60%). They occur in the trunk and extremities 25% and 15% of the time, respectively.2 Internal and visceral lesions are uncommon. Up to 20% of patients can have multiple lesions, and these cases are most likely to have internal involvement affecting organs such as the liver and gastrointestinal (GI) tract.1
The proliferative phase of IH is marked by rapid growth for the first six to eight months that typically plateaus by age 10–12 months. Tumors that involve the superficial dermis present as a red, raised lesion (Fig. 72-1A). Superficial tumors that are larger or that exhibit more rapid growth can occasionally cause ulceration of the skin with bleeding. Tumors in the lower dermis, subcutaneous tissue, or muscle appear bluish in color with slightly raised overlying skin (previously incorrectly termed ‘cavernous’ hemangiomas). With experience, history and physical examination can establish an accurate diagnosis for most of these tumors. The involuting phase of hemangiomas occurs from age 1 to 7 years during which time the tumor slowly regresses, although it may grow in proportion with the child. This phase is notable for fading color of the tumor from crimson to a dull purple, accompanied by a deflation of the tumor mass (Fig. 72-1B).1 The skin may become pale, usually in the center of the tumor first, spreading outwards. Fifty per cent of tumors have completed involution by 5 years of age, and 70% by age 7 years. There is continued gradual improvement in these aspects until the regression is entirely complete by age 10–12 years.32 In the final involuted phase of the tumor, 50% of patients have nearly normal skin in the area of the prior lesion. Patients with larger tumors can have lax or redundant skin and yellowish discoloration. Scars will persist if parts of the tumor were previously ulcerated.1
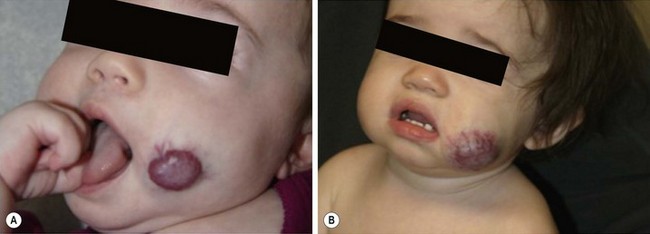
FIGURE 72-1 (A) This infant has an infantile hemangioma that is in the proliferative phase. This hemangioma was not present at birth but was noted at several weeks of age. (B) This hemangioma is in its involuting phase.
The differential diagnosis of cutaneous hemangiomas consists primarily of other vascular anomalies. CM that involves the skin can be mistaken for superficial hemangiomas, or vice versa. Deeper hemangiomas can be confused for VM or LM as all can appear as bluish masses through the skin. Hemangiomas with fast-flow vascularity of the parenchyma could be confused for AVM, but the age of onset and history generally distinguishes the two. Congenital hemangiomas, discussed in a later section, may be misdiagnosed as vascular malformations, which are congenital by definition. Pyogenic granulomas may be differentiated from hemangiomas by their rare appearance before six months (mean age is 6 to 7 years), and their frequent association with minor trauma.33,34 Other tumors such as TA, hemangiopericytoma, and fibrosarcoma can also be confused.35
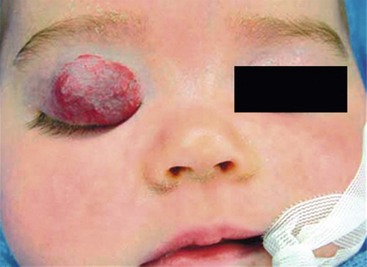
The primary local complications that can occur with cutaneous hemangiomas are ulceration, bleeding, and pain. Hemangiomas are rarely life-threatening, but complications can be anticipated by recognition of the anatomic distribution of the lesion.36 Lesions in the cervicofacial region can lead to airway obstruction as they grow during the proliferative phase. Very large hemangiomas, notably in the liver, can lead to high-output congestive heart failure (secondary to fast-flow and vascular shunting within the tumor), hypothyroidism, and abdominal compartment syndrome. Facial lesions involving the eyelid, nose, lip, or ear can result in tissue destruction with cosmetic consequences. Periorbital and eyelid hemangiomas can cause visual axis obstruction, leading to deprivation amblyopia (Fig. 72-2). Alternatively, distortion of the cornea can cause astigmatic amblyopia. GI hemangiomas are very rare, but may manifest with GI bleeding.
Associated Congenital Anomalies
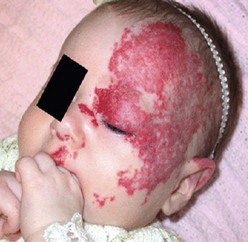
IH are rarely associated with other anomalies. However, a few such anomalies have been described, more commonly with larger and midline hemangiomas. Cervicothoracic hemangiomas can be seen in conjunction with sternal nonunion.37 Tumors of the lumbosacral area have been noted to occur along with spinal dysraphism abnormalities such as meningocele and tethered spinal cord.38,39 Hemangiomas of the pelvis and perineum have been found in association with urogenital and anorectal anomalies. PHACES association describes hemangiomas associated with congenital ocular abnormalities such as microophthalmia, cataracts, optic nerve hypoplasia, posterior fossa cystic malformations, hypoplasia or absence of carotid and vertebral vessels, as well as malformation of the aortic arch (Fig 72-3).40,41 Females are affected in 90% of cases. These patients are at risk for stroke in the neonatal period and migraines in older ages.
Other Manifestations
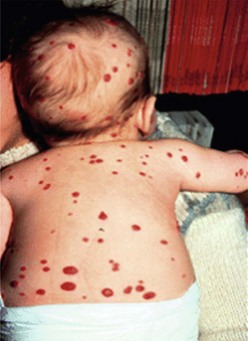
The presence of multiple disseminated hemangiomas is termed hemangiomatosis. The cutaneous tumors are usually tiny (<5 mm) and dome-like. When five or more lesions are present, occult visceral lesions, most commonly in the liver, may also be present (Fig. 72-4). Screening patients with ultrasound (US) and/or magnetic resonance imaging (MRI) may be indicated for these patients.
Imaging
Proper radiologic diagnosis of vascular anomalies is dependent on specific expertise and clinical experience with the radiologic features of these lesions.42 Ultrasound and MRI are the most useful imaging modalities. Ultrasound of proliferative phase hemangiomas demonstrates a mass with dense parenchyma exhibiting fast-flow vascularity.43,44 This distinguishes deep IH from VM, which exhibit slow-flow vascularity and larger blood-filled spaces. MRI of proliferating hemangiomas shows a lobulated solid mass of intermediate intensity with T1 spin-echo sequences, and moderate hyperintensity on T2 spin-echo. Flow voids that represent fast flow and shunting are seen in and around the tumor. During the involuting phase, MRI demonstrates decreased flow voids and vascularity, with the mass taking on a more lobular and fatty appearance.45
Treatment
The majority of IH do not require any specific treatment other than observation and reassurance of the parents.46 Even tumors that exhibit rapid growth or fiery red skin will spontaneously regress and leave behind little to no evidence of their presence. However, regular follow-up is important as the potential complications have few prognostic indicators. Serial photographs are very helpful in documenting progression and subsequent improvement. Reasons for treatment or referral to a vascular anomalies specialist or center include dangerous locations (impinging on a vital structure such as the airway or eye), unusually large size or rapid growth, and local or endangering complications (skin ulceration or high-output heart failure).
Hemangiomas exhibiting the above risk factors or complications should be considered for treatment. Since hemangiomas are tumors of pure angiogenesis, pharmacologic therapy involves angiogenesis inhibition. Systemic corticosteroids, which inhibit the expression of VEGF-A by hemangioma-derived stem cells and thus angiogenesis, were first line therapy for decades.47–49 Oral prednisone is given at a dose of 2–3 mg/kg/day. Doses up to 5 mg/kg/day have been used for life-threatening complications of large hemangiomas causing airway obstruction or heart failure. The overall response rate is 80–90% with initial improvement in the color and tension of the mass usually noted within one week. The steroids are maintained with a very gradual taper every two to four weeks with the goal of discontinuation around age 10–11 months. Live vaccines such as polio, measles, mumps, rubella, and varicella should be withheld while children are taking prednisone. Hemangiomas will have rebound growth if steroids are tapered or stopped too quickly. Return to the initial dosage and slower tapering will usually treat this problem. Potential complications of steroid use in infants and children include impaired growth and weight gain in about one-third of cases. Almost all children will have ‘catch up’ growth and return to pretreatment growth curves by age 14–24 months.50 Cushingoid facies occur in almost all patients and normalizes upon tapering. In rare circumstances, steroids may induce hypertension or hypertrophic cardiomyopathy, both of which are indications to wean or change therapy.50
Intralesional corticosteroids are used for small cutaneous hemangiomas that cause local deformity or ulceration, especially for lesions of the eyelid, nose, cheek, or lip.51 A total of three to five injections are typically given at intervals of six to eight weeks at a dose of 3–5 mg/kg/injection.52 The response rate approaches that of systemic steroids. Subcutaneous atrophy is a potential complication of steroid injection, but is usually temporary. There have been reported cases of blindness following intralesional steroid injection for periorbital hemangiomas.53 This is presumed to be secondary to particle embolization into the retinal artery through feeding vessels. Manual compression around the periphery of the tumor is recommended during injection to minimize embolization through draining veins.
Propranolol, a nonselective beta blocker, has recently been recognized as an important treatment option for hemangiomas and in most centers has become first line pharmacotherapy. A child with a nasal capillary hemangioma treated with propranolol for steroid-induced cardiomyopathy had regression of his lesion.54 This revelation led to the publication of several more studies supporting this finding.55–58 Propranolol is given orally at 2–3 mg/kg/day, in two or three divided doses, and discontinued following regression of the lesion.55,59 Treatment often leads to a consistent, rapid, therapeutic effect with softening of the lesion on palpation and color shift from intense red to purple.55 Propranolol is well tolerated but can cause rare side effects such as bradycardia, gastroesophageal reflux, hypoglycemia, hypotension, rash, somnolence, and wheezing.55,56,58–61 Several mechanisms of action have been proposed.62 Propranolol inhibits β-adrenoreceptors, which are activated by adrenaline, causing vasoconstriction of capillaries supplying the hemangioma.63,64 This likely leads to the visible changes in color and palpable softening. Blockage of β-adrenoreceptors also results in decreased expression of VEGF and MMPs, thereby inhibiting angiogenesis, and induces apoptosis in endothelial cells.65,66
Recombinant interferon was once considered as a second line agent, but has fallen out of favor except in very limited circumstances.67–71 A small subset of patients (5–12%) may develop a severe complication known as spastic diplegia.72,73 Spasticity usually resolves if the drug is terminated quickly. Children receiving IFN should be followed carefully by a neurologist. Though experience is limited, low-dose, high frequency anti-angiogenic regimens using vincristine can be effective.74 The use of interferon and vincristine has waned since the introduction of propranolol therapy.
Although attractive in concept, laser therapy is not often beneficial for IHs, except for a few specific indications.75 The flash lamp pulse-dye laser penetrates the dermis to a depth of only 0.75–1.2 mm. Most cutaneous hemangiomas are deeper than this, and therefore not affected by laser treatment. Additionally, laser therapy carries risks of scarring, skin hypopigmentation, and ulceration, which may lead to a poor result compared to observation alone. One instance in which the laser is advantageous is the treatment of telangiectasias that often remain in the involuted phase of hemangioma. The use of endoscopic continuous wave carbon dioxide laser has been shown to be a good strategy for controlling proliferative phase hemangiomas in the unilateral subglottic location.76 Lastly, intralesional photocoagulation with bare fiber Nd:YAG laser can be useful for hemangiomas in certain locations, such as the upper eyelid when visual obstruction is a concern.
Indications for resection of IH vary with patient age. During the proliferative phase in infancy, well-localized or pedunculated tumors can be expeditiously resected with linear closure, especially for tumors complicated by bleeding and ulceration. Sites that are most amenable to resection are the scalp, trunk, and extremities. Other modalities to treat ulceration include wound care with dressing changes, topical antibiotics, and topical steroids, which can accelerate healing.77 Tumors of the upper eyelid that obstruct vision and that do not respond to pharmacologic therapy may also require excision or debulking. Focal lesions of the GI tract with bleeding that fail medical management may require enterotomy and resection, or endoscopic band ligation. Diffuse, patchy involvement is the more common presentation of GI hemangiomas. Management is difficult but most lesions eventually involute and stop bleeding.78 Preoperative localization with capsule endoscopy and/or intraoperative endoscopy may be necessary to identify lesions in the small bowel.79
During the involuting phase, resection may be needed for hemangiomas that are large and protuberant, and therefore likely to create excess and lax overlying skin.80 Indications for resection include: (1) it is obvious that resection will be necessary sooner or later; (2) the surgical scar will be identical regardless of timing of operation; and (3) the surgical scar can easily be hidden. Lesions of the nose, eyelids, lips, and ears require special expertise. It is often preferable to perform the operation for the above indications during the preschool years before children become aware of and focus on body image differences that may lead to low self-esteem.
Finally, for the difficult to treat and life-threatening large hemangiomas, especially in the liver, angiographic embolization may be required to manage high-output cardiac failure. Arterial catheterization in infants carries significant risks and should generally be limited to those situations with cardiac compromise in which there is the capacity and intent to perform simultaneous embolization. In these rare cases, anti-angiogenic pharmacotherapy remains the first line of therapy and should continue along with angiographic procedures. Repeat embolization procedures may be required. Success with embolization is dependent on occlusion of macrovascular shunts within the tumor rather than occlusion of feeding vessels.81
Congenital Hemangioma
Two types of rare congenital hemangiomas exist that are fully developed at birth and that do not usually exhibit postnatal tumor growth.82 These have been termed rapidly involuting congenital hemangioma (RICH) and the noninvoluting congenital hemangioma (NICH) (Fig. 72-5). Lesions are solitary and affect both genders equally. Unlike IHs, they do not stain positive for GLUT-1.83 The diagnosis is typically made on physical exam at birth, although some can be diagnosed prenatally as early as 12 weeks of gestation.82 Most do not require therapy.
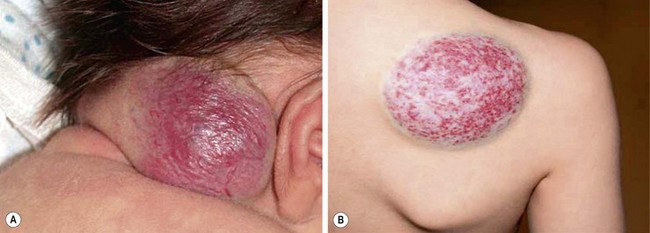
FIGURE 72-5 These two patients are examples of rapid involuting congenital hemangiomas (RICH) and noninvoluting congenital hemangiomas (NICH). (A) The newborn baby has a RICH. This lesion was present at birth and will spontaneously regress, much more quickly than the typical infantile hemangioma. (B) This 9-year-old child has a NICH. This hemangioma has not resolved. If treatment is needed, arterial embolization may be beneficial as these lesions have significant flow to them.
As opposed to IH, RICH is more common on the extremities. Also, RICH will spontaneously regress, much more quickly than IH, by 6 to 14 months; NICH do not involute and grow with the child.82 RICH appear raised and violaceous, often with a central depression or scar, ulceration, telangiectasia, or surrounding pale rim.82,83 MRI will show enhancing hyperintense masses, high-flow vessels within and adjacent to the mass, and the presence of vascular flow voids on T2-weighted imaging.83 NICH are well-circumscribed, plaque-like lesions, often with coarse telangiectasias, areas of pallor, or a white to bluish rim.84 They appear on MRI as homogeneous lesions with isointense signal on T1-weighted imaging and hyperintense on T2-weighted sequences. Both lesions are fast flow on Doppler ultrasound. If treatment is needed for NICH, arterial embolization may be beneficial as these lesions have significant flow to them. Operative excision is reserved for cases with equivocal diagnosis, poor cosmetic appearance, or for complications such as ulceration, bleeding, obstruction, or congestive heart failure.83,85
Hepatic Hemangioma
Hepatic hemangiomas (HH) in infants should be differentiated from ‘hepatic hemangiomas’ that present in adulthood.85 Adult ‘hepatic hemangiomas’, which are sometimes called ‘cavernous hemangiomas’, are in fact VMs. In contrast, HH of infancy are true tumors and have a similar pattern of involution to cutaneous IH. Contrary to popular belief, not all liver hemangiomas are life-threatening. The classic triad of heart failure, anemia, and hepatomegaly is unusual, and most involute without long-term sequelae.
The majority of HH can be divided into three categories: focal, multifocal, and diffuse, each with distinct features (Fig. 72-6).86 Focal HH are the hepatic equivalent of the cutaneous RICH, and also do not stain positive for GLUT-1. They are histologically distinct from the typical IH.15,87 They are fully grown at birth and regress much faster than IH. Many focal lesions are detected antenatally on prenatal ultrasound, or are discovered as an abdominal mass in otherwise healthy infants.88 They are usually asymptomatic, found in equal numbers in both genders, and rarely associated with cutaneous hemangiomas. Transient anemia and moderate thrombocytopenia, due to intralesional thrombosis, are observed in some infants and generally resolve spontaneously. This is in contradistinction to the profound thrombocytopenia seen with Kasabach–Merritt phenomenon (KMP). However, a subset of focal hepatic RICH-type hemangiomas will have macrovascular shunts from the hepatic arteries and/or portal veins to the hepatic veins. These shunts can cause a large steal, accounting for blood-flow demands above and beyond the hypervascular tumor parenchyma, and can result in high-output cardiac failure. These shunts may close as the tumors involute. However, cardiac strain can often mandate interruption of the shunts via embolization. Steroids have been used for solitary focal hepatic lesions with in-utero cardiac failure/cardiomegaly.88,89 However, these lesions may have undergone rapid spontaneous involution and the benefit of steroids remains unproven, though still should be attempted before employing more invasive therapies. Resection is rarely indicated.
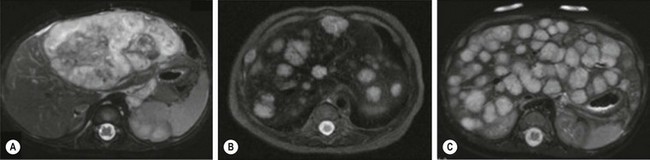
FIGURE 72-6 Most hepatic hemangiomas can be divided into three categories: focal, multifocal, and diffuse. (A) A large focal hepatic hemangioma. (B) The scan depicts multifocal hepatic hemangiomas. (C) Diffuse hepatic hemangioma.
Multifocal HH are true IH and biologically distinct from focal HH. They undergo involution similar to cutaneous IH and stain positive for GLUT-1.87,90,91 They are more common in females and Caucasians.92 Infants with multifocal lesions are typically born earlier and are asymptomatic, but are diagnosed later than focal lesions. These patients often have cutaneous IH and are identified following screening for visceral hemangiomas. Some of these patients will have hypothyroidism. Thus, a serum TSH should be checked in patients with multifocal disease. Infants who are asymptomatic should be observed; however, some can have high-output cardiac failure from macrovascular shunting. Treatment with steroids or propranolol can often close these shunts.54,55 Embolization can be employed in those who fail medical therapy. Serial abdominal ultrasound should be performed in infants with multifocal HH until the lesions involute.
Diffuse HH are also true IH, but are far more serious lesions with a more difficult clinical course. Like multifocal lesions, diagnosis occurs in the weeks following birth. They are more common in Caucasians and females, are associated with cutaneous IH and follow a similar course of involution, and express GLUT-1.92 These patients all develop severe hypothyroidism from high levels of type 3 iodothyronine deiodinase, which inactivates circulating thyroid hormones.20 Aggressive exogenous thyroid hormone replacement is essential to prevent hypothyroid complications such as mental retardation and cardiac failure. Involution of the lesions will usually result in amelioration of the hypothyroidism.93 These innumerable lesions often almost completely replace the normal hepatic parenchyma. Those with considerable disease may have respiratory compromise from compression of the diaphragm and thoracic cavity, but can also develop abdominal compartment syndrome and multisystem organ failure. Aggressive pharmacotherapy with propranolol, steroids, and occasionally low-dose vincristine is warranted in those with cardiac failure, hemodynamically significant shunting, or hypothyroidism to hasten involution.55,86,94–96 Hepatic transplantation is the last resort for critically ill infants.86
Tufted Angioma and Kaposiform Hemangioendothelioma
These vascular tumors of childhood are more aggressive and invasive than IH. TA and KHE probably exist within the same spectrum as they share many overlapping clinical and histologic features.21,97 Both tumors typically present at birth, although they occur postnatally as well. Males and females are affected equally. The tumors are unifocal and are most often located on the trunk, shoulder, thigh, or retroperitoneum. TA present as erythematous macules or plaques and histology reveals small tufts of capillaries.97 KHE are more extensive tumors that present with deep, red-purple skin discoloration and overlying and surrounding ecchymosis (Fig. 72-7). The natural history of these tumors is one of continued proliferation into early childhood followed by subsequent, but incomplete regression. These tumors usually persist, albeit in a smaller form.98 Fortunately, they are usually asymptomatic in later stages, though may cause musculoskeletal pain. Histology of these lesions reveals infiltrating sheets of spindle-shaped endothelial cells in the form of irregular lobules, sheets, and lacy network.99 Imaging of KHE depicts an enhancing lesion on MRI with poorly defined margins that extend across tissue planes. This is in contrast to IH, which are well-circumscribed and respective of tissue planes.
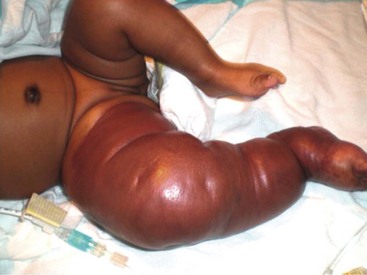
FIGURE 72-7 This infant has a kaposiform hemangioendothelioma (KHE). KHE are more extensive tumors that present with deep, red-purple skin discoloration and overlying ecchymosis. These lesions usually regress later in childhood, although usually not completely.
Kasabach–Merritt Phenomenon
KMP was first reported in 1940 in a case of profound thrombocytopenia, petechiae, and bleeding in conjunction with a ‘giant hemangioma.’100 As with many terms in the field of vascular anomalies, this term has been often misused in connection with coagulopathy and other vascular lesions, most prominently VM. However, the profound and persistent thrombocytopenia that occurs with KMP does not occur with either VM or IH. The only known true associations are with TA and KHE.99,101,102 The platelet count with KMP is typically under 10,000/μL, and may be associated with decreased fibrinogen levels, increased D-dimer, and mildly elevated partial prothrombin time (PT) and partial thromboplastin time (PTT). Bleeding can result from this platelet trapping coagulopathy at many sites, including intracranial, GI, peritoneal, pleural, and pulmonary. A microangiopathic hemolytic anemia is also present. Treatment for KHE with KMP is primarily medical as the tumor is usually too large and extensive to be resected. Corticosteroids and interferon-alfa have been effective in about 50% of cases; actinomycin, anti-platelet therapy, cyclophosphamide, doxorubicin, gemcitabine, propranolol, sirolimus, and vincristine have also been found to be beneficial in several case series, as single drugs or in combination, but none of these agents have been shown to be consistently successful.103–111 Platelet transfusions are ineffective and should be avoided unless there is active bleeding. Additionally, heparin may stimulate tumor growth and worsens the thrombocytopenia of KMP and should likewise be avoided. Mortality rates with KHE and TA remain high at 20–30%. KHE not associated with KMP can be followed without treatment as long as the size and location of tumor are limited.
Vascular Malformations
Vascular malformations are congenital lesions of vascular dysmorphogenesis that can be local or diffuse. The majority of vascular malformations are sporadic, though some rare varieties are familial.112–114 They occur in 1.5% of the population.115 Vascular malformations probably result from genetic mutations that lead to dysfunction in the regulation of endothelial proliferation and apoptosis, cellular differentiation, maturation, and cell-to-cell adhesions.116
Embryology and Development of the Vascular and Lymphatic Systems
The vascular system develops during embryogenesis through the processes of vasculogenesis, the formation of new vascular channels from mesodermally derived endothelial precursor cells (hemangioblasts), and angiogenesis, the formation of new blood vessels from preexisting blood vessels. The destiny of endothelial precursors to create different types of blood vessels appears to be imprinted early in embryogenesis by unique cell surface markers.117 The differentiation of angioblasts into an early vascular plexus leads to the creation of primitive blood vessels.118 Following formation of the primary vascular plexus, endothelial cells proliferate and sprout or split from their vessel of origin to form new capillaries. A process called ‘pruning’ then occurs, during which the vascular plexus is remodeled into a system with larger and smaller vessels. The signals for microvascular endothelial cells to proliferate and differentiate for vessel development are controlled by a number of growth factors and their receptors, including VEGF, basic fibroblast growth factor 2 (FGF-2), and angiopoietin 1 (Ang-1).119
The lymphatic system develops around midgestation after the blood vasculature forms, and is thought to derive from either venous endothelial cells or mesenchymal progenitor cells.120–123 It is a one-way valve system that collects fluid and macromolecules from tissue. In 1902, Sabin described the prevailing model of lymphangiogenesis in which venous endothelial cells commit to become lymphatic endothelium and then migrate and proliferate to form lymph sacs.122 These sacs then form a lymphatic plexus, which remodel and mature into the lymphatic vasculature. 120,123 Venous endothelial cells along the anterior cardinal vein capable of differentiating into lymphatic endothelial cells (LECs) begin to express lymphatic endothelial hyaluronan receptor-1 (LYVE-1) on embryonic day (E) 9, a marker specific for lymphatic endothelium.120,123,124 A subgroup of endothelial cells on one side of the vein then begin to express prospero-related homeobox 1 (Prox-1). 125 This transcription factor is required for the maturation and differentiation of LECs.125,126 VEGFR3, also known as Flt4, plays an important role in lymphatic development as well. VEGFR is expressed in both blood and lymphatic vasculature in early embryogenesis, but is restricted to mostly lymphatic vessels later in development127–129 VEGFR3 knockout mice die on E9 with major venous anomalies before any lymphatic sprouting has occurred.116 In contrast, transgenic mice that overexpress the ligand for VEGFR3 (VEGF-C) will develop distended lymphatic channels.130
Capillary Malformations
CM, previously referred to as ‘port-wine stains’, are present at birth as permanent flat, pink-red cutaneous lesions (Fig. 72-8). In the newborn nursery, CM can be confused with nevus flammeus neonatorum, commonly called ‘angel’s kiss’ when located on the face, or ‘stork bite’ when in the posterior cervical location. However, these discolorations are due to transient dilations of dermal vessels and fade with time, whereas CM does not. CM occur with equal gender distribution in 0.3% of infants.131 Multiple CM are rare. The majority of CM appear sporadically, but some familial forms exist that are inherited in an autosomally dominant fashion.132 Capillary malformation–arteriovenous malformations (CM-AVM) are associated with mutations in RASA1, a gene coding for p120-RasGTPase.133 Histologically, cutaneous CM consist of dilated capillary- to venule-sized vessels located in the superficial dermis, with a paucity of surrounding normal nerve fibers.134 These abnormal vessels gradually dilate over time leading to darkening color and occasionally nodular ectasias. CM can also be found in complex-combined vascular malformations.
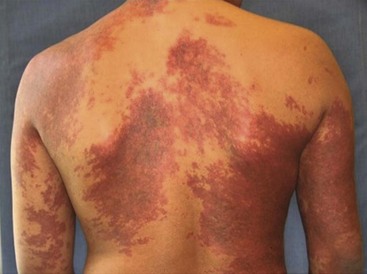
FIGURE 72-8 This child has a capillary malformation. Such lesions were previously referred to as a port-wine stain.
CM can be associated with underlying soft tissue and skeletal overgrowth, as well as other internal abnormalities. CM of the occiput can signal an underlying encephalocele or ectopic meninges. When located over the spine, underlying spinal dysraphism is a concern. Facial CM affecting the trigeminal dermatomes can be associated with ipsilateral ocular and leptomeningeal vascular anomalies in Sturge–Weber syndrome. Ocular lesions lead to increased risk for retinal detachment, glaucoma, and blindness. Leptomeningeal involvement can manifest with seizures, hemiplegia, and impaired motor and cognitive function. MRI reveals the CNS abnormalities showing pial vascular enhancement and gyriform calcifications.135
Treatment for CM is primarily related to cosmesis. Flash lamp pulse-dye laser therapy will cause photothermolysis and improve the appearance by lightening the color of the lesions in most (70%) patients.136 Repeated treatments are usually needed and the timing of therapy remains controversial.137 Treatment in infants less than six months of age has been shown to improve facial CM.138 Ablative and orthopedic surgical procedures can be tailored to treat cosmetic and functional problems related to soft tissue and bony hypertrophy.
Cutis Marmorata Telangiectatica Congenita
Cutis marmorata telangiectatica congenita (CMTC) is an uncommon congenital vascular anomaly, first described in 1922, and characterized by a deep purple reticular vascular pattern.139 Lesions are noted at birth or shortly after, and both genders are equally affected. CMTC can have a localized or generalized distribution; localized lesions are more common on the extremities.140,141 Histopathology demonstrates dilated capillaries in the papillary dermis and proliferation of blood vessels in the reticular dermis.142 Approximately 50% of cases are associated with other congenital anomalies. The most common is hypertrophy or atrophy of an involved extremity, but cardiovascular, craniofacial, cutaneous, neurologic, and skeletal abnormalities have also been described.142–148 Partial regression of the capillary stains begins in the first year of life, but prominent dilated veins and discoloration often remain in adults.
Telangiectasias
Telangiectasias are tiny acquired vascular marks that can appear on children in the preschool and school-aged years, and are commonly known as ‘spider nevus’ or ‘spider telangiectasias.’ They may be present in nearly half of all children, with no preference for gender.115 Some may spontaneously disappear, but can be removed with pulsed-dye laser.
Hereditary hemorrhagic telangiectasia (HHT or Rendu–Osler–Weber disease) was the first vascular anomaly to be elucidated at a genetic level.149 This autosomal dominant disease has five genetic types linked to mutations in genes coding for endoglin (an endothelial glycoprotein), activin receptor-like kinase 1, and Smad4, an intracellular signaling protein, all of which are involved in the binding and signaling of transforming growth factor-β.150–153 Definite diagnosis of HHT is based on the presence of three of the following criteria: epistaxis, multiple telangiectasias (lips, oral cavity, fingers, nose), visceral lesions (GI, pulmonary, hepatic cerebral, or spinal AVMs), and family history (first degree relative with HHT).154 Children often present with recurrent, spontaneous nosebleeds before school age. Telangiectasias of the skin and buccal mucosa present in the third decade of life. Chronic anemia from lower GI bleeds occurs in about one-third of patients.155
Lymphatic Malformations
Clinical Features
LM are usually noted at birth, but can manifest at any age or even be identified prenatally on fetal ultrasound.156,157 The etiology of this anomaly is unknown. Lymphedema, a type of LM, does have heritable forms.113 LM are classified as microcytic (diameter <1 cm), macrocystic (diameter >1 cm), or a combination thereof. These descriptions are also useful therapeutically as size determines whether or not the cystic cavity can be aspirated or compressed. Clinical presentation varies across a wide spectrum, from localized masses, to areas of diffuse infiltration, to chylous fluid accumulations in various body cavities.
The skin and soft tissues are most commonly affected, but LM can also involve the subcutaneous tissues, muscle, bone, and more rarely, internal organs such as the GI tract and lungs. As with CM, underlying localized soft tissue and skeletal hypertrophy is often associated with LM. LM appear as soft compressible masses, similarly to VM, and may have a bluish hue, although not to the same extent as VM (Fig. 72-9). LM appear histologically as thin-walled vascular channels lined by LECs, whose lumen can be empty or filled with a proteinaceous fluid containing macrophages and lymphocytes.158 These cells stain positive for podoplanin (D2-40) and LYVE-1.159 Involvement of the dermis may produce puckering of the skin or vesicles that weep clear yellowish fluid. Diffuse infiltration of the subcutaneous tissue can produce extensive lymphedema that also falls within the spectrum of LM. One unique factor among the vascular anomalies is that LMs are at risk for infection that can lead to cellulitis or even systemic illness. Similarly, infections located elsewhere in the body, or viral illnesses, can cause increased size and tension in the LM. The cystic components of LM are also subject to intralesional bleeding secondary to trauma or abnormal venous connections. The vesicles from cutaneous involvement can also leak thin sanguineous fluid or appear as red, purple, or black nodules.
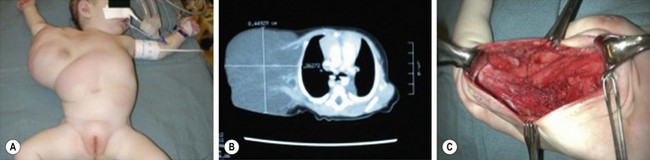
FIGURE 72-9 This baby has a (A) large right axillary lymphatic malformation which is seen on the CT scan (B). (C) This operative photograph shows the residual cavity after resection of the mass.
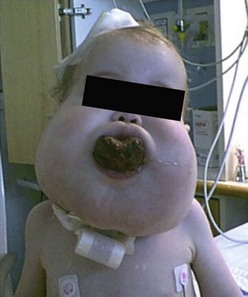
LM at various anatomic locations are prone to unique associated anomalies. Periorbital LM can lead to proptosis. Facial LM can cause the associated deformities of macrocheilia, macroglossia, and macromala. Overgrowth of the mandible, sometimes massive, can be seen with cervicofacial LM.160 Congenital airway obstruction is rare but also possible (Fig. 72-10). Lesions of the tongue and floor of the mouth, on the other hand, may more commonly produce obstruction of the oropharynx. LM in the cervical and axillary regions can signal associated LM in the mediastinum. Anomalies of the central conducting lymphatic channels, the thoracic duct and cisterna chyli, can lead to very problematic and recurrent chylous effusions that affect the pleural, pericardial, and/or peritoneal cavities. In addition, LM of the GI tract can lead to loss of chyle and subsequent protein losing enteropathy. In the pelvis, associated problems include recurrent infection and bladder outlet obstruction. LM of the extremities are seen in conjunction with overgrowth and limb length discrepancy.
Gorham Syndrome
A rare but very difficult problem arises with Gorham syndrome, in which soft tissue and skeletal LM lead to progressive osteolysis and ‘disappearing bone disease’ (Fig. 72-11).161 It presents most frequently in the second and third decades of life, and is seen slightly more often in males.162,163 Presenting symptoms include pain, limping, extremity weakness, and spontaneous fractures, most commonly involving the shoulder, facial, spine, and pelvic bones.164,165 The clinical course is variable, ranging from mild disability to paraplegia. On imaging, well-circumscribed intramedullary and subcortical lucencies resembling osteoporosis are seen early.166 Biopsy typically demonstrates a matrix of thin-walled vessels lined with a single layer of endothelium surrounded by extensive fibrovascular connective tissue, but without signs of inflammation or malignancy.164 A variety of treatments have been reported. Interferon alpha-2b is believed to have anti-angiogenic activity and has been shown to induce remission.167,168 Bisphophonates can also stabilize disease, presumably by inhibition of osteoclasts.169 The mTOR inhibitor rapamycin is currently under investigation and has been effective for treatment in children with refractory lymphatic anomalies.170 Surgery, reserved for symptomatic lesions, involves resection of affected areas and reconstruction.
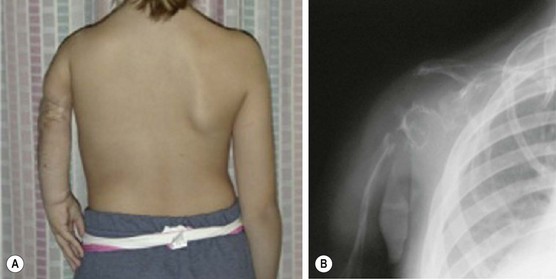
FIGURE 72-11 This child has Gorham disease in which soft tissue and skeletal lymphatic malformations leads to progressive osteolysis. (A) Note the foreshortening of the left arm due to osteolysis of the left shoulder. (B) On the left upper extremity radiograph, note the loss of the humerus, clavicle, and shoulder joint.
Lymphedema
Lymphedema, which occurs when protein-rich fluid leaks into the subcutaneous tissue, should be considered a type of LM. It can be inherited or acquired.171 Milroy disease is an autosomal dominant, congenital lymphedema caused by a mutation in the VEGR3 gene.172,173 Bilateral, below-the-knee swelling is a consistent phenotypic feature of the disorder, and is usually present at birth. Cellulitis, dilated lower extremity veins, upslanting toenails, and papillomatosis are also present.174 Males may often develop a hydrocele. In contrast, Meige disease and lymphedema-distichiasis syndrome (LD) present in puberty or later in adulthood with lymphedema. Both are autosomal dominant disorders. Meige disease is more common in females. No genetic cause has been found. LD patients can also present with distichiasis (double rows of eyelashes), varicose veins, ptosis, cleft palate, and cardiac abnormalities.175 A mutation in the FOXC2 forkhead/winged-helix transcription factor, which plays a role in somite formation, is responsible for the disease.176
Imaging
Well-localized and cystic LM are easily characterized by ultrasound and CT (see Fig. 72-9B). MRI, however, provides the most reliable diagnosis and is best for documenting the full extent of more complex LM as well as their macrocystic and microcystic components. LM are hyperintense on T2 sequences because of their high water content. Within the cysts, fluid-fluid levels denote layering of protein and/or blood. Cystic rims and intralesional septae are highlighted by contrast enhancement. Adjacent enlarged or anomalous venous channels may be apparent as well. The differential diagnosis of these cystic lesions in the infant includes teratoma and infantile fibrosarcoma. For lymphatic anomalies of the thoracic duct and chylous effusions, contrast lymphangiography, although technically difficult to perform, can be helpful to identify the abnormal lymphatic channels or site of leakage.177
Treatment
The indications for LM treatment vary with the extent and location of the lesions.178 Focal and macrocystic LM are amenable to ablation by both sclerotherapy and resection. In contrast, more diffuse and predominantly microcystic LM are difficult to eradicate by any method. For local intralesional bleeding that causes sudden enlargement of LM and pain, conservative management with rest and pain medications is sufficient. Similarly, the enlargement of LM that coincides with systemic viral or bacterial infections can be managed expectantly as it is usually harmless. On the other hand, bacterial infections presenting with cellulitis require treatment. Infected LM become tense and swollen producing erythema, pain, and toxicity; the incidence of this complication is around 15–20%. Treatment consists of systemic antibiotics; hospitalization for intravenous antibiotics is often necessary.
Resection for complex LM can also be of significant benefit (see Fig. 72-9C), but staging is often needed.179 The operations may be long and tedious, and often require meticulous dissection to preserve vital structures. General guidelines for resection are: (1) each operation should focus on a defined anatomic region, removing as much of the lesion as possible including neurovascular dissection, but without injuring vital structures; (2) limit blood loss to less than the patient’s blood volume; and (3) prolonged closed-suction drainage of the resulting cavity is required. The recurrence rate following ‘macroscopically complete excision’ ranges from 15–40%. This recurrence is thought to be secondary to regrowth and re-expansion of unexcised lymphatic channels. Sclerotherapy of the residual cavity following excision may be helpful in this regard. Following resection, it is common for cutaneous vesicles to occur within the scar. These can be controlled to some extent by local intravesicular sclerotherapy or laser. Alternatively, additional staged excision, pulling uninvolved dermis over the resection bed, may prevent this annoying result.
Some other caveats about operation for LM are worthy of mention. Cervicofacial LM will often require staged orthognathic procedures to improve bite and speech impediments related to maxillary and mandibular overgrowth. Tracheostomy may be needed in cases of oropharyngeal and airway obstruction, and should precede attempts at sclerotherapy for cervicofacial LM. Reactive inflammatory swelling can be dramatic in the initial period following sclerotherapy, and can exacerbate partial oropharyngeal obstruction. Lesions of the cervical and axillary regions often involve the brachial plexus. The use of nerve stimulators is a useful adjunct to prevent injury in these cases. Resection of thoracic and mediastinal LM to treat recurrent pleural and pericardial effusions involves dissection and skeletonization of the great vessels, and vagus and phrenic nerves. For pelvic and anorectal LM, detailed knowledge of the anatomy of the ischiorectal fossa and sciatic nerve are important. Preoperative sclerotherapy is often useful as well to shrink lesions, but discernment is necessary as scarring can impede the preservation of important nerves. Lastly, for the specific type of cutaneous LM, ‘lymphangioma circumscriptum’, wide resection and closure, if necessary with split thickness skin grafts, can be curative. However, serial resections, allowing adjacent skin to grow, is generally preferable to grafting.
Venous Malformations
Clinical Features
VM, often mistermed ‘cavernous hemangiomas’, are slow-flow lesions consisting of venous channels that can develop anywhere in the body, most commonly in the skin and soft tissues. VM may be seen at birth or become apparent later, depending on the anatomic location. A wide spectrum of presentations is possible, including simple varicosities and ectasias, discreet spongy masses, and complex channels that can permeate any tissue or organ system. VMs are probably the most common of the vascular malformations, and are more likely to be multiple as well. They tend to slowly enlarge with normal growth of the patient, but can dilate and become symptomatic at any time. As with other VMs, the proportional growth that occurs may become exaggerated during puberty. On examination, these soft, bluish, compressible lesions can expand with dependent position and Valsalva maneuver (Fig. 72-12). Episodes of phlebothrombosis secondary to stasis may lead to acute pain and swelling. Phleboliths can be palpated in many VM. Associated local overgrowth and limb length discrepancy are not uncommon. Involvement of bones and joints creates risk for pathologic fractures and hemarthroses, with subsequent arthritis.
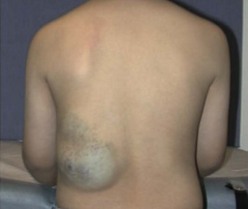
FIGURE 72-12 This adolescent has a venous malformation in the subcutaneous tissues of his back. These soft, bluish, compressible lesions can expand with dependent position and during Valsalva maneuver.
Approximately 90% of VM are sporadic; half of those result from a mutation in the vascular endothelial cell-specific receptor tyrosine kinase TIE-2 and its associated TEK gene.180,181 The TIE-2 signaling pathways play an important role in angiogenic remodeling and vessel stabilization during development.182 Cutaneomucosal venous malformations (VMCM), inherited through autosomal dominant transmission, are caused by a gain-of-function mutation in TIE-2 and represent 1–2% of VM.181,183 GVM, also autosomal dominant, represents 5% of VM and results from loss-of-function mutations in glomulin, which affects vascular smooth muscle differentiation.184
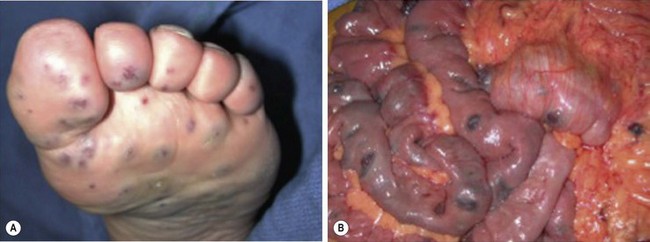
VM of the GI tract are often multiple as well, and can affect every part from mouth to anus. They are more common in the left colon and rectum when associated with VM of the pelvis and perineum. GI bleeding, typically chronic in nature, can result. Blue rubber bleb nevus syndrome (or ‘Bean syndrome’) represents a specific rare disorder consisting of multifocal VM that affect the skin and GI tract primarily.185 The skin lesions are unique in that they are often quite numerous and resemble tiny ‘blue rubber nipples.’ These skin lesions present diffusely and are classically seen on the palms and soles of the feet (Fig. 72-13). As with other GI VM, chronic bleeding, and intussusception can result. Diagnosis of GI VM is generally based on endoscopy. Patients with rectal VM can have associated ectatic mesenteric veins and are at risk for developing portomesenteric venous thrombosis.186
Imaging
Radiologic modalities useful for the diagnosis of VM include ultrasound, MRI, and venography. MRI is most informative and demonstrates hyperintense lesions with T2 sequences. Contrast enhancement of the vascular spaces distinguishes VM from LM, as does the presence of pathognomonic phleboliths. Intralesional bleeding within LM can represent an exception to this rule. In contrast to AVM, VM do not demonstrate evidence of arterial flow on MRI.
Treatment
Intralesional sclerotherapy is the mainstay of treatment for most VM.187 Sclerosing agents, most commonly ethanol and sodium tetradecyl sulfate, cause direct endothelial damage, thrombosis, and scarring. For small VM, the injection process is similar to that for simple varicosities. Larger lesions are accessed by direct puncture and the therapeutic agents are injected under fluoroscopy, with the use of tourniquets and compression of venous drainage to prevent systemic administration of the sclerosants. General anesthesia is required in most instances. Staged therapy and occasional embolization of large venous channels are useful for more complex VM. The more complex lesions are best treated by a skilled interventional radiologist who has experience with vascular anomalies.
Unifocal GI lesions can be excised. Diffuse colorectal malformations causing significant bleeding may be treated by colectomy, anorectal mucosectomy, and coloanal pull-through.188 For multifocal VM in the blue rubber bleb nevus syndrome, complete resection of all lesions, combined with endoscopy of the entire GI tract at the time of operation, provides the only chance for possible cure. Bowel resection for these lesions is rarely indicated. Rather, wedge excision and polypectomy by intussusception of successive lengths of intestine are the preferred methods of resection.189
Arteriovenous Malformations
Clinical Features
AVM are fast-flow vascular malformations characterized by abnormal connections or shunts between feeding arteries and draining veins, without an intervening capillary bed. These shunts define the nidus of the malformation. Lesions tend to be localized, but can be extensive as well. AVM are one of the most common vascular anomalies that occur in the central nervous system and are more frequent than extracranial AVM. A clinical staging system was developed to describe the natural history of progression (Table 72-2).190 At birth, they appear as a pink cutaneous blemish that can be confused with both CM and the premonitory sign of an IH. The fast flow through the shunt becomes more evident in childhood and adolescence as the lesion expands and develops into a mass.191 Lesions will feel warm to the touch, often with a bruit or thrill. With continued expansion, they become more red and prominent. Puberty, pregnancy, or local trauma tends to trigger more rapid expansion. Skin ischemia can develop from expansion or local steal phenomenon, leading to pain, ulceration, and bleeding (Fig. 72-14). Large AVM can cause high-output cardiac failure.
TABLE 72-2
Schobinger Clinical Staging System for Arteriovenous Malformations
| Stage | Clinical Findings |
| I (quiescent) | Pink to bluish stain, cutaneous warmth, and arteriovenous shunting by Doppler ultrasound imaging |
| II (expanding) | Same as stage I, plus enlargement, pulsation, thrill, bruit, and tortuous and tense veins |
| III (destructive) | Same as stage II, plus skin ulceration, bleeding, persistent pain, or tissue necrosis |
| IV (decompensating) | Same as stage III, plus cardiac failure |
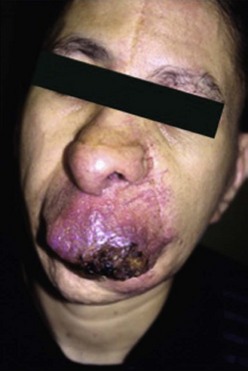
FIGURE 72-14 A facial arteriovenous malformation (stage III) with ulceration of the skin is seen in this patient.
Most AVM are sporadic, but heritable forms exist. Mutations in RASA1 cause the autosomal dominant disorder CM-AVM.133 The CM are multifocal, small, round-to-oval, pinkish-to-red lesions, and are usually randomly distributed. They can be associated with an AVM or arteriovenous fistula.192
Treatment
The majority of AVM require treatment because of continued expansion, which can lead to local tissue ischemia and pain.190,193 The mainstays of treatment are angiographic embolization alone or in combination with excision. Local recurrence following early intervention can complicate future procedures. Very well-localized stage I AVM may be amenable to excision. Intervention is often delayed until symptoms (focal pain, ulceration, and bleeding) develop which is indicative of stage III. Treatment during infancy is indicated in cases of heart failure. The proximal feeding arteries should not be embolized or ligated during treatment as they provide the only avenue by which to reach the nidus of the AVM for subsequent embolization. The nidus will recruit other nearby arteries after the primary feeding vessels are occluded, and the AVM will recur and continue to enlarge.
Combined Vascular Malformations
Klippel–Trenaunay Syndrome
Klippel–Trenaunay syndrome, or KT, is a slow-flow combined vascular malformation involving abnormal capillaries, lymphatics, and veins.194–196 This capillary-lymphatico-venous malformation (CLVM) usually involves one or more extremities, most often a lower limb, and is associated with prominent soft tissue and bony hypertrophy (Fig. 72-15). This syndrome is sporadic and obvious at birth, with a wide range in severity. The CM component can be multiple, and typically presents as a large geographic pattern affecting the extremity, buttock, and trunk. It is macular at birth and develops lymphatic vesicles over time. The lymphatic anomalies have variable presentation including hypoplasia, lymphedema, and macrocystic LM, and are common in the buttock, pelvis, and perineum. Anomalous lateral superficial veins in the extremity, usually dilated with incompetent valves, are persistent embryonic vessels, the most common being the marginal vein of Servelle. There are anomalous deep system veins as well that may be hypoplastic or even absent. Thrombophlebitis of the anomalous veins occurs with a frequency of 20–45%, and pulmonary emboli are found in up to one-quarter of the patients.
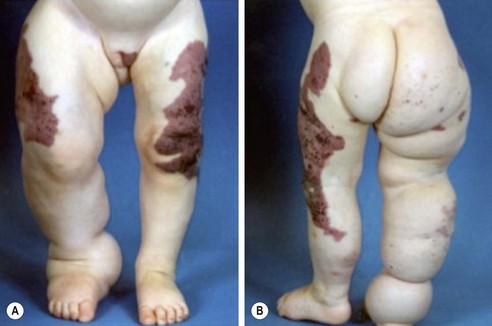
FIGURE 72-15 This patient has Klippel-Trenaunay syndrome. This capillary-lymphatico-venous malformation usually involves one or more extremities, most often a lower limb, and is associated with prominent soft tissue and bony hypertrophy. The capillary malformation component typically presents as a large geographic pattern affecting the extremity, buttocks, and trunk.
Operative therapies, mainly debulking procedures, can manage some of the specific problems encountered, primarily for overgrowth.197 Staged contour resection can be used to treat areas of soft tissue overgrowth and lymphedema (Fig. 72-16).198 It is critical to determine the location of the overgrowth, which can be either extrafascial or intrafascial. Intrafascial overgrowth should not be debulked secondary to risk of injury to major neurovascular structures and immobility. Debulking of the trunk and thoracic wall is feasible, but excision of intrathoracic and mediastinal malformations should generally not be undertaken, especially if asymptomatic. Tissue with poor lymphatic drainage and altered circulation makes for poor tissue flaps, leading to delayed postoperative healing and protracted use of closed-suction drains. Perioperative management during significant resections should include consideration for anticoagulation and temporary inferior vena cava filter placement to help prevent deep venous thrombosis and PE.
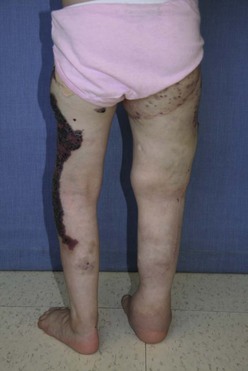
FIGURE 72-16 This is a photograph of the same patient depicted in Figure 72-15. The patient is now older and has undergone staged debulking procedures for management of the soft tissue overgrowth and lymphedema in the right leg. Most of the capillary hemangioma component has also been excised.
CLOVES Syndrome
This syndrome is named for its phenotypic features: congenital lipomatous overgrowth, vascular malformations, epidermal nevi, seizures, scoliosis, and skeletal/spinal anomalies (Fig. 72-17).199,200 It may be misdiagnosed as Proteus syndrome. Truncal lipomatous masses are present at birth, typically with an overlying capillary malformation, and may be identified on prenatal imaging.201 The masses can extend into the mediastinum, pleural cavity, retroperitoneum, and paraspinal-intraspinal space. Both low-flow and high-flow vascular malformation are seen in CLOVES. Musculoskeletal abnormalities usually involve the extremities and include macrodactyly, large hands, wide triangular feet, or widened sandal gap.199 Recently, mutations in PIK3CA, which encodes for a subunit of PI3K, have been identified as the cause of CLOVES.202 Activation of PI3K eventually leads to phosphorylation of AKT, levels of which are increased by the PI3K mutations seen in CLOVES. Mutations in this gene have been shown to induce angiogenesis and malignant cell growth.203
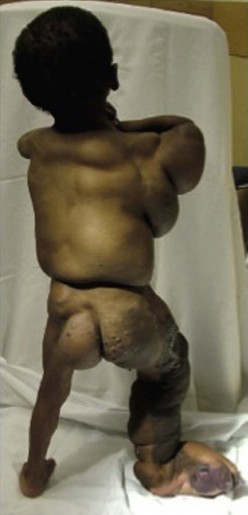
FIGURE 72-17 This patient has CLOVES syndrome. Truncal lipomatous masses are present at birth often with an overlying capillary malformation. Musculoskeletal abnormalities usually involve extremities and include macrodactyly, wide triangular feet, or widened sandal gap. Both high-flow and low-flow venous malformations can also be found in this syndrome.
Maffucci Syndrome
This syndrome consists of exophytic VM of the soft tissue and bones, bony exostoses, and endochondromas. This sporadic disorder is not usually evident at birth and can be uni- or bilateral. The bony lesions and endochondromas manifest first in childhood, while the venous anomalies appear later. Spindle cell hemangioendotheliomas commonly occur and denote reactive vascular proliferation within the preexisting VM, rather than true tumors.204 The endochondromas can undergo malignant transformation in 20% to 30% of cases, leading to chondrosarcomas.205 Two studies have recently identified somatic mosaic mutations in IDH-1 and IDH-2, which code for different forms of the enzyme isocitrate dehydrogenase, as the cause of Maffucci syndrome.206,207
PTEN Hamartoma Tumor Syndrome
Bannayan–Riley–Ruvalcaba (BRRS) and Cowden syndromes (CS) are autosomal dominant disorders caused by mutations of the tumor-suppressor gene PTEN (phosphatase tensin homolog on chromosome 10), collectively termed PTEN hamartoma tumor syndrome.208 BRRS is primarily an overgrowth syndrome that has vascular malformations as a minor component. The more prominent clinical features are macrocephaly, developmental delay, multiple lipomas, hamartomatous polyps of the ileum and colon, genital lentiginosis, and Hashimoto thyroiditis.209 CS is characterized by mucocutaoneous and papillomatous lesions, lipomas, hamartomatous intestinal lesions, and an increased risk for thyroid, breast, and endometrial cancer. Multifocal fast-flow vascular anomalies associated with ectopic fat are seen in about half of patients with PTEN mutations.210 Macrocephalic patients with fast-flow vascular anomalies or multiple intracranial developmental venous anomalies should be tested for PTEN mutations.
Proteus Syndrome
Proteus syndrome is probably diagnosed more often than it actually occurs.211 This overgrowth disorder is associated with activating mutations in the oncogene AKT-1 and is progressive over time.212 Vascular, skeletal, and soft tissue anomalies tend to be asymmetrical and variably expressed. Common features include lipomas or lipomatosis, macrocephaly, and gigantism of the hands or feet (or both).
Parkes Weber Syndrome
Parkes Weber syndrome is a sporadic, combined fast-flow vascular malformation affecting the limb and trunk, with the lower extremity being the most common site.213,214 Capillary arteriovenous fistulae (CAVF) and CM-AVM are combined with hypertrophy of the bone and muscle of the affected limb. CM-AVM is obvious at birth, appearing as overgrowth with a large geographic macular pink stain. In contrast to CLVM seen with KT syndrome, the limb hypertrophy is symmetrical along the length and substance of the extremity. The macular stain associated with CM-AVM has much greater cutaneous warmth than does typical CM. The finding of bruits or thrills on examination confirms the diagnosis. MRI demonstrates symmetric muscular and bony overgrowth, with generalized enlargement of the normal named arteries and veins within the affected limb. Angiography depicts the discrete arteriovenous fistulae. In rare cases, superselective embolization is used to occlude the arteriovenous shunts if symptoms of ischemia, pain, or high-output congestive heart failure occur.
Interdisciplinary Vascular Anomalies Center
The clarification of nomenclature and biologic classification of vascular anomalies has provided a useful clinical framework for the diagnosis and treatment of these lesions. Nevertheless, patients with vascular anomalies often provide complex exceptions to these designations. Lesions that are congenital malformations may not become apparent until adulthood, either due to anatomic location or progressive expansion over time. Neoplastic lesions, such as IH, often have a premonitory cutaneous sign at birth. Additionally, hemangiomas, when they have a significant fast-flow component, can be difficult to distinguish from AVM. VM can also at times exhibit enlargement and even endothelial hyperplasia triggered by clotting, ischemia, or partial resection, which leads to their propensity for recurrence after treatment. For these reasons, several regional and international centers have developed interdisciplinary vascular anomalies teams that serve as referral centers and which combine the medical, surgical, and radiologic expertise required to effectively diagnose and treat these often complex disorders. Guidelines have been established for referral to vascular anomalies centers.215 Patients with unclear diagnoses, vascular malformation (except for CM), and rare vascular tumors should be referred to these centers for management and treatment.
References
1. Mulliken, JB, Fishman, SJ, Burrows, PE. Vascular anomalies. Curr Probl Surg. 2000; 37:517–584.
2. Finn, MC, Glowacki, J, Mulliken, JB. Congenital vascular lesions: Clinical application of a new classification. J Pediatr Surg. 1983; 18:894–900.
3. Mulliken, JB, Glowacki, J. Hemangiomas and vascular malformations in infants and children: A classification based on endothelial characteristics. Plast Reconstr Surg. 1982; 69:412–422.
4. Virchow, R. Angioma in die krankhaften Geschwüstele; vol. 3. Hirshwald, Berlin, 1863.
5. Wegener, G. Ueber lymphangiome. Arch Klin Chir. 1877; 20:641–707.
6. Holmdahl, K. Cutaneous hemangiomas in premature and mature infants. Acta Paediatri Scand. 1955; 44:370–379.
7. Amir, J, Metzker, A, Krikler, R, et al. Strawberry hemangioma in preterm infants. Pediatr Dermatol. 1986; 3:331–332.
8. Haggstrom, AN, Drolet, BA, Baselga, E, et al. Prospective study of infantile hemangiomas: Demographic, prenatal, and perinatal characteristics. J Pediatr. 2007; 150:291–294.
9. Smoller, BR, Apfelberg, DB. Infantile (juvenile) capillary hemangioma: A tumor of heterogeneous cellular elements. J Cutan Pathol. 1993; 20:330–336.
10. Khan, ZA, Melero-Martin, JM, Wu, X, et al. Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood. 2006; 108:915–921.
11. Boye, E, Yu, Y, Paranya, G, et al. Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest. 2001; 107:745–752.
12. Dadras, SS, North, PE, Bertoncini, J, et al. Infantile hemangiomas are arrested in an early developmental vascular differentiation state. Mod Pathol. 2004; 17:1068–1079.
13. Lui, W, Zhang, S, Hu, T, et al. Sex hormone receptors of hemangiomas in children. Chin Med J (Engl). 1997; 110:349–351.
14. Sasaki, GH, Pang, CY, Wittliff, JL. Pathogenesis and treatment of infant skin strawberry hemangiomas: Clinical and in vitro studies of hormonal effects. Plast Reconstr Surg. 1984; 73:359–370.
15. North, PE, Waner, M, Mizeracki, A, et al. GLUT1: A newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000; 31:11–22.
16. Barnes, CM, Huang, S, Kaipainen, A, et al. Evidence by molecular profiling for a placental origin of infantile hemangioma. Proc Natl Acad Sci U S A. 2005; 102:19097–19102.
17. Ritter, MR, Dorrell, MI, Edmonds, J, et al. Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc Natl Acad Sci U S A. 2002; 99:7455–7460.
18. Yu, Y, Wylie-Sears, J, Boscolo, E, et al. Genomic imprinting of IGF2 is maintained in infantile hemangioma despite its high level of expression. Mol Med. 2004; 10:117–123.
19. Huang, SA, Dorfman, DM, Genest, DR, et al. Type 3 iodothyronine deiodinase is highly expressed in the human uteroplacental unit and in fetal epithelium. J Clin Endocrinol Metab. 2003; 88:1384–1388.
20. Huang, SA, Tu, HM, Harney, JW, et al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med. 2000; 343:185–189.
21. Ritter, MR, Moreno, SK, Dorrell, MI, et al. Identifying potential regulators of infantile hemangioma progression through large-scale expression analysis: A possible role for the immune system and indoleamine 2,3 dioxygenase (IDO) during involution. Lymph Res Biol. 2003; 1:291–299.
22. Burton, SK, Schulz, CJ, Angle, B, et al. An increased incidence of haemangiomas in infants born following chorionic villus sampling (CVS). Prenat Diagn. 1995; 15:209–214.
23. Kaplan, P, Normandin, JJ, Wilson, GN, et al. Malformations and minor anomalies in children whose mothers had prenatal diagnosis: Comparison between CVS and amniocentesis. Am J Med Genet. 1990; 37:366–370.
24. Sato, TN, Tozawa, Y, Deutsch, U, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995; 376:70–74.
25. Suri, C, Jones, PF, Patan, S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996; 87:1171–1180.
26. Maisonpierre, PC, Suri, C, Jones, PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997; 277:55–60.
27. Roberts, DM, Kearney, JB, Johnson, JH, et al. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am J Pathol. 2004; 164:1531–1535.
28. Bielenberg, DR, Bucana, CD, Sanchez, R, et al. Progressive growth of infantile cutaneous hemangiomas is directly correlated with hyperplasia and angiogensis of adjacent epidermis and inversely correlated with expression of the endogenous angiogenesis inhibitor, INF-beta. Int J Oncol. 1999; 14:401–408.
29. Takahashi, K, Mulliken, JB, Kozakewich, HP, et al. Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest. 1994; 93:2357–2364.
30. Yu, Y, Varughese, J, Brown, LF, et al. Increased Tie2 expression, enhanced response to angiopoietin-1, and dysregulated angiopoietin-2 expression in hemangioma-derived endothelial cells. Am J Pathol. 2001; 159:2271–2280.
31. Razon, MJ, Kraling, BM, Mulliken, JB, et al. Increased apoptosis coincides with onset of involution in infantile hemangioma. Microcirculation. 1998; 5:189–195.
32. Bowers, RE, Graham, EA, Tomlinson, KM. The natural history of the strawberry nevus. Arch Dermatol. 1960; 82:667–680.
33. Patrice, SJ, Wiss, K, Mulliken, JB. Pyogenic granuloma (lobular capillary hemangioma): A clinicopathologic study of 178 cases. Pediatr Dermatol. 1991; 8:267–276.
34. Kirschner, RE, Low, DW. Treatment of pyogenic granuloma by shave excision and laser photocoagulation. Plast Reconstr Surg. 1999; 104:1346–1349.
35. Boon, LM, Fishman, SJ, Lund, DP, et al. Congenital fibrosarcoma masquerading as congenital hemangioma: Report of two cases. J Pediatr Surg. 1995; 30:1378–1381.
36. Enjolras, O, Gelbert, F. Superficial hemangiomas: Association and management. Pediatr Dermatol. 1997; 14:173–179.
37. Hersh, JH, Waterfill, D, Rutledge, J, et al. Sternal malformatio/vascular dysplasia association. Am J Med Genet. 1985; 21:177–186.
38. Goldberg, NS, Hebert, AA, Esterly, NB. Sacral hemangiomas and multiple congenital anomalies. Arch Dermatol. 1986; 122:684–687.
39. Albright, AL, Gartner, JC, Wiener, ES. Lumbar cutaneous hemangiomas as indicators of tethered spinal cords. Pediatrics. 1989; 83:977–980.
40. Gorlin, RJ, Kantaputra, P, Aughton, DJ, et al. Marked female predilection in some syndromes associated with facial hemangiomas. Am J Med Genet. 1994; 52:130–135.
41. Frieden, IJ, Reese, V, Cohen, D. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996; 132:307–311.
42. Burrows, PE, Laor, T, Paltiel, H, et al. Diagnostic imaging in the evaluation of vascular birthmarks. Dermatol Clin. 1998; 16:455–488.
43. Dubois, J, Patriquin, HB, Garel, L, et al. Soft-tissue hemangiomas in infants and children: Diagnosis using Doppler sonography. AJR Am J Roentgenol. 1998; 171:247–252.
44. Paltiel, HJ, Burrows, PE, Kozakewich, HP, et al. Soft-tissue vascular anomalies: Utility of ultrasound for diagnosis. Radiology. 2000; 214:747–754.
45. Meyer, JS, Hoffer, FA, Barnes, PD, et al. Biological classification of soft-tissue vascular anomalies: MR correlation. AJR Am J Roentgenol. 1991; 157:559–564.
46. Margileth, AM, Museles, M. Cutaneous hemangiomas in children. Diagnosis and conservative management. JAMA. 1965; 194:523–526.
47. Bennett, ML, Fleischer, ABJ, Chamlin, SL, et al. Oral corticosteroid use is effective for cutaneous hemangiomas: An evidence-based evaluation. Arch Dermatol. 2001; 137:1208–1213.
48. Greenberger, S, Boscolo, E, Adini, I, et al. Corticosteroid suppression of VEGF-A in infantile hemangioma-derived stem cells. N Engl J Med. 2010; 362:1005–1013.
49. Crum, R, Szabo, S, Folkman, J. A new class of steroids inhibits angiogenesis in the presence of heparin or a heparin fragmnet. Science. 1985; 230:1375–1378.
50. Boon, LM, MacDonald, DM, Mulliken, JB. Complications of systemic corticosteroid therapy for problematic hemangiomas. Plast Reconstr Surg. 1999; 104:1616–1623.
51. Sloan, GM, Reinisch, JF, Nichter, LS, et al. Intralesional corticosteroid therapy for infantil hemangiomas. Plast Reconstr Surg. 1989; 83:459–467.
52. Marler, JJ, Mulliken, JB. Vascular Anomalies, 2nd ed. Philadephia: Elsevier; 2006.
53. Ruttum, MS, Abrams, GW, Harris, GJ, et al. Bilateral retinal embolization associated with intralesional corticosteroid injection for capillary hemangioma of infancy. J Pediatr Ophthalmol Strabismus. 1993; 30:4–7.
54. Leute-Labreze, C, Dumas de la Roque, E, Hubiche, T, et al. Propanolol for severe hemangiomas of infancy. N Engl J Med. 2008; 358:2649–2651.
55. Sans, V, de la Roque, ED, Berge, J, et al. Propanolol for severe infantile hemangiomas: Follow-up report. Pediatrics. 2009; 124:e423–e431.
56. Buckmiller, LM, Munson, PD, Dyamenahalli, U, et al. Propranolol for infantile hemangiomas: Early experience at a tertiary vascular anomalies center. Laryngoscope. 2010; 120:676–681.
57. Bigorre, M, Van Kien, AK, Valette, H. Beta-blocking agent for treatment of infantile hemangioma. Plast Reconstr Surg. 2009; 123:195e–196e.
58. Bagazgoitia, L, Torrelo, A, Gutierrez, JC, et al. Propranolol for infantile hemangiomas. Pediatr Dermatol. 2011; 28:108–114.
59. Cushing, SL, Boucek, RJ, Manning, SC, et al. Initial experience with a multidisciplinary strategy for initiation of propranolol therapy for infantile hemangiomas. Otolaryngol Head Neck Surg. 2011; 144:78–84.
60. Holland, KE, Frieden, IJ, Frommelt, PC, et al. Hypoglycemia in children taking propranolol for the treatment of infantile hemangioma. Arch Dermatol. 2010; 146:775–778.
61. Harrison, DC, Meffin, PJ, Winkle, RA. Clinical pharmacokinetics of antiarrhythmic drugs. Prog Cardiovasc Dis. 1977; 20:217–242.
62. Storch, CH, Hoeger, PH. Propranolol for infantile haemangiomas: Insights into the molecular mechanisms of action. Br J Dermatol. 2010; 163:269–274.
63. Westfall, TC, Westfall, DP. Neurotransmission: The autonomic and somatic motor nervous system, 11th ed. New York: McGraw-Hill; 2006.
64. Guimaraes, S, Moura, D. Vascular adrenoceptors: An update. Pharmacol Rev. 2001; 53:319–356.
65. Sommers Smith, SK, Smith, DM. Beta blockade induces apoptosis in cultured capillary endothelial cells. In Vitro Cell Dev Biol Anim. 2002; 38:298–304.
66. Yang, EV, Sood, AK, Chen, M, et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006; 66:10357–10364.
67. White, CW, Wolf, SJ, Korones, DN, et al. Treatment of childhood angiomatous diseases with recombinant interferon alfa-2a. J Pediatr. 1991; 118:59–66.
68. Tamayo, L, Ortiz, DM, Orozco-Covarrubias, L, et al. Therapeutic efficacy of interferon alfa-2b in infants with life-threatening giant hemangiomas. Arch Dermatol. 1997; 133:1567–1571.
69. Ricketts, RR, Hatley, RM, Corden, BJ, et al. Interferon-alpha-2a for the treatment of complex hemangiomas of infancy and childhood. Ann Surg. 1994; 219:605–612.
70. Greinwald, JH, Jr., Burke, DK, Bonthius, DJ, et al. An update on the treatment of hemangiomas in children with interferon alfa-2a. Arch Otolaryngol Head Neck Surg. 1999; 125:21–27.
71. Ezekowitz, RA, Mulliken, JB, Folkman, J. Interferon alfa-2a therapy for life-threatening hemangiomas of infancy. N Engl J Med. 1992; 326:1456–1463.
72. Barlow, CF, Priebe, CJ, Mulliken, JB, et al. Spastic diplegia as a complication of interferon Alfa-2a treatment of hemangiomas of infancy. J Pediatr. 1998; 132:527–530.
73. Deb, G, Jenkner, A, Donfrancesco, A. Spastic diplegia and interferon. J Pediatr. 1999; 134:382.
74. Perez, J, Pardo, J, Gomez, C. Vincristine–an effective treatment of corticoid-resistant life-threatening infantile hemangiomas. Acta Oncol. 2002; 41:197–199.
75. Scheepers, JH, Quaba, AA. Does the pulsed tunable dye laser have a role in the management of infantile hemangiomas: Observations based on 3 years experience. Plast Reconstr Surg. 1995; 95:305–312.
76. Sie, KC, McGill, T, Healy, GB. Subglottic hemangioma: Ten years’ experience with the carbon dioxide laser. Ann Otol Rhinol Laryngol. 1994; 103:167–172.
77. Morelli, JG, Tan, OT, Yohn, JJ, et al. Treatment of ulcerated hemangiomas infancy. Arch Pediatr Adolesc Med. 1994; 148:1104–1105.
78. Fishman, SJ, Fox, VL. Visceral vascular anomalies. Gastrointest Endosc Clin N Am. 2001; 11:813–834.
79. Fishman, SJ, Burrows, PE, Leichtner, AM, et al. Gastrointestinal manifestations of vascular anomlies in childhood: Varied etiologies require multiple therapeutic modalities. J Pediatr Surg. 1998; 33:1163–1167.
80. Mulliken, JB, Rogers, GF, Marler, JJ. Circular excision of hemangioma and purse-string closure: The smallest possible scar. Plast Reconstr Surg. 2002; 109:1544–1554.
81. Enjolras, O, Riche, MC, Merland, JJ, et al. Management of alarming hemangiomas in infancy: A review of 25 cases. Pediatrics. 1990; 85:491–498.
82. Boon, LM, Enjolras, O, Mulliken, JB. Congenital hemangioma: Evidence of accelerated involution. J Pediatr. 1996; 128:329–335.
83. Berenguer, B, Mulliken, JB, Enjolras, O, et al. Rapidly involuting congenital hemangioma: Clinical and histopathologic features. Pediatr Dev Pathol. 2003; 6:495–510.
84. Enjolras, O, Mulliken, JB, Boon, LM, et al. Noninvoluting congenital hemangioma: A rare cutaneous vascular anomaly. Plast Reconstr Surg. 2001; 107:1647–1654.
85. Boon, LM, Burrows, PE, Paltiel, HJ, et al. Hepatic vascular anomalies in infancy: A twenty-seven-year experience. J Pediatr. 1996; 129:346–354.
86. Christison-Lagay, ER, Burrows, PE, Alomari, A, et al. Hepatic hemangiomas: Subtype classification and development of a clinical practice algorithm and registry. J Pediatr Surg. 2007; 42:62–67.
87. Mo, JQ, Dimashkieh, HH, Bove, KE. GLUT1 endothelial reactivity distinguishes hepatic infantile hemangioma from congenital hepatic vascular malformation with associated capillary proliferation. Hum Pathol. 2004; 35:200–209.
88. Morris, J, Abbott, J, Burrows, P, et al. Antenatal diagnosis of fetal hepatic hemangioma treated with maternal corticosteroids. Obstet Gynecol. 1999; 94:813–815.
89. Mejides, AA, Adra, AM, O’Sullivan, MJ, et al. Prenatal diagnosis and therapy for a fetal hepatic vascular malformation. Obstet Gynecol. 1995; 85:850–853.
90. Hernandez, F, Navarro, M, Encinas, JL, et al. The role of GLUT1 immunostaining in the diagnosis and classification of liver vascular tumors in children. J Pediatr Surg. 2005; 40:801–804.
91. Drut, RM, Drut, R. Extracutaneous infantile haemangioma is also Glut1 positive. J Clin Pathol. 2004; 57:1197–1200.
92. Kulungowski, AM, Alomari, AI, Chawla, A, et al. Lessons from a liver hemangioma registry: Subtype classification. J Pediatr Surg. 2012; 47:165–170.
93. Konrad, D, Ellis, G, Perlman, K. Spontaneous regression of severe acquired infantile hypothyroidism associated with multiple liver hemangiomas. Pediatrics. 2003; 112:1424–1426.
94. Draper, H, Diamond, IR, Temple, M, et al. Multimodal management of endangering hepatic hemangioma: Impact on tranplast avoidance: A descriptive case series. J Pediatr Surg. 2008; 43:120–125.
95. Dickie, B, Dasgupta, R, Nair, R, et al. Spectrum of hepatic hemangiomas: Management and outcome. J Pediatr Surg. 2009; 44:125–133.
96. Perez-Valle, S, Peinador, M, Herraiz, P, et al. Vincristine, an efficacious alternative for diffuse neonatal haemangiomatosis. Acta Paediatr. 2010; 99:311–315.
97. Jones, EW, Orkin, M. Tufted angioma (angioblastoma): A benign progressive angioma, not to be confused with Kaposi’s sarcoma or low grade angiosarcoma. J Am Acad Dermatol. 1989; 20:214–225.
98. Enjolras, O, Mulliken, JB, Wassef, M, et al. Residual lesions after Kasabach-Merritt phenomenon in 41 patients. J Am Acad Dermatol. 2000; 42:225–235.
99. Sarkar, M, Mulliken, JB, Kozakewich, HP, et al. Thrombocytopenic coagulopathy (Kasabach-Merritt phenomenon) is associated with Kaposiform hemangioendothelioma and not with common infantile hemangioma. Plast Reconstr Surg. 1997; 100:1377–1386.
100. Kasabach, HH, Merritt, KK. Capillary hemangioma with extensive purpura: A report of a case. Am J Dis Child. 1940; 59:1063–1070.
101. Enjolras, O, Wassef, M, Mazoyer, E, et al. Infants with Kasabach-Merritt syndrome do not have ‘true’ hemangiomas. J Pediatr. 1997; 130:631–640.
102. Zuckerberg, LR, Nikoloff, BJ, Weiss, SW. Kaposiform hemangioendothelioma of infancy and childhood: An aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol. 1993; 17:321–328.
103. Vin-Christian, K, McCalmont, TH, Frieden, IJ. Kaposiform hemangioendothelioma: An aggressive, locally invasive vascular tumor that can mimic hemangioma of infancy. Arch Dermatol. 1997; 133:1573–1578.
104. Hu, B, Lachman, R, Phillips, J, et al. Kasabach-Merritt syndrome-associated kaposiform hemangioendothelioma successfully treated with cyclophosphagmide, vincristine and actionmycin D. J Pediatr Hematol Oncol. 1998; 20:567–569.
105. Haisley-Royster, C, Enjolras, O, Frieden, IJ, et al. Kasabach-merritt phenomenon: A retrospective study of treatment with vincristine. J Pediatr Hematol Oncol. 2002; 24:459–462.
106. Enjolras, O, Wassef, M, Dosquet, C, et al. [Kasabach-Merritt syndrome on a congenital tufted angioma]. Ann Dermatol Venereol. 1998; 125:257–260.
107. Blatt, J, Stavas, J, Moats-Staats, B, et al. Treatment of childhood kaposiform hemangioendothelioma with sirolimus. Pediatr Blood Cancer. 2010; 55:1396–1398. [15].
108. Chiu, YE, Drolet, BA, Blei, F, et al. Variable response to propranolol treatment of kaposiform hemangioendothelioma, tufted angioma, and Kasabach-Merritt phenomenon. Pediatr Blood Cancer. Pediatr Blood Cancer. 2012; 59:934–938.
109. Adams, DM. The nonsurgical management of vascular lesions. Facial Plast Surg Clin North Am. 2001; 9(4):601–608.
110. Hermans, DJ, van Beynum, IM, van der Vijver, RJ, et al. Kaposiform hemangioendothelioma with Kasabach-Merritt syndrome: A new indication for propranolol treatment. J Pediatr Hematol Oncol. 2011; 33:e171–e173.
111. Lopez, V, Marti, N, Pereda, C, et al. Successful management of Kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon using vincristine and ticlopidine. Pediatr Dermatol. 2009; 26:365–366.
112. Vikkula, M, Boon, LM, Mulliken, JB, et al. Molecular basis of vascular anomalies. Trends Cardiovasc Med. 1998; 8:281–292.
113. Brouillard, P, Vikkula, M. Genetic causes of vascular malformations. Hum Mol Genet. 2007; 16(Spec No. 2):R140–R149.
114. Limaye, N, Boon, LM, Vikkula, M. From germline towards somatic mutations in the pathophysiology of vascular anomalies. Hum Mol Genet. 2009; 18:R65–R74.
115. Christison-Lagay, ER, Fishman, SJ. Vascular anomalies. Surg Clin North Am. 2006; 86:393–425.
116. Dumont, DJ, Fong, GH, Puri, MC, et al. Vascularization of the mouse embryo: A study of flk-1, tek, tie, and vascular endothelial growth factor expression during development. Dev Dyn. 1995; 203:80–92.
117. Folkman, J, D’Amore, PA. Blood vessel formation: What is its molecular basis? Cell. 1996; 87:1153–1155.
118. Risau, W. Mechanisms of angiogenesis. Nature. 1997; 386:671–674.
119. Ramsauer, M, D’Amore, PA. Contextual role for angiopoietins and TGFbeta1 in blood vessel stabilization. J Cell Sci. 2007; 120:1810–1817.
120. Karpanen, T, Alitalo, K. Molecular biology and pathology of lymphangiogenesis. Annu Rev Pathol. 2008; 3:367–397.
121. Huntington, GS, McClure, CFW. The anatomy and development of the jugular lymph sac in the domestic cat (Felis domestica). Anat Rec. 1908; 2:1–19.
122. Sabin, FR. The lymphatic system in human embryos, with a consideration of the morphology of the system as a whole. Am J Anat. 1909; 9:43–91.
123. Oliver, G. Lymphatic vasculature development. Nat Rev Immunol. 2004; 4:35–45.
124. Banerji, S, Ni, J, Wang, SX, et al. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J Cell Biol. 1999; 144:789–801.
125. Wigle, JT, Oliver, G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999; 98:769–778.
126. Wigle, JT, Harvey, N, Detmar, M, et al. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J. 2002; 21:1505–1513.
127. Karkkainen, MJ, Alitalo, K. Lymphatic endothelial regulation, lymphoedema, and lymph node metastasis. Semin Cell Dev Biol. 2002; 13:9–18.
128. Kaipainen, A, Korhonen, J, Mustonen, T, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A. 1995; 92:3566–3570.
129. Dumont, DJ, Jussila, L, Taipale, J, et al. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998; 282:946–949.
130. Jeltsch, M, Kaipainen, A, Joukov, V, et al. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science. 1997; 276:1423–1425.
131. Jacobs, AH, Walton, RG. The incidence of birthmarks in the neonate. Pediatrics. 1976; 58:218–222.
132. Eerola, I, Boon, LM, Watanabe, S, et al. Locus for susceptibility for familial capillary malformation (‘port-wine stain’) maps to 5q. Eur J Hum Genet. 2002; 10:375–380.
133. Eerola, I, Boon, LM, Mulliken, JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003; 73:1240–1249.
134. Smoller, BR, Rosen, S. Port-wine stains. A disease of altered neural modulation of blood vessels? Arch Dermatol. 1986; 122:177–179.
135. Enjolras, O, Riche, MC, Merland, JJ. Facial port-wine stains and Sturge-Weber syndrome. Pediatrics. 1985; 76:48–51.
136. Tan, OT, Sherwood, K, Gilchrest, BA. Treatment of children with port-wine stains using the flashlamp pumped tunable dye laser. N Engl J Med. 1989; 320:416–421.
137. van der Horst, CM, Koster, PH, de Borgie, CA, et al. Effect of the timing of treatment of port-wine stains with the flash-lamp-pumped pulsed-dye laser. N Engl J Med. 1998; 338:1028–1033.
138. Chapas, AM, Eickhorst, K, Geronemus, RG. Efficacy of early treatment of facial port wine stains in newborns: A review of 49 cases. Lasers Surg Med. 2007; 39:563–568.
139. Van Lohuizen, CHJ. Uber eine seltene angeborene Hautanomalie (cutis marmorata telangiectatica congenita). Acta Derm Venereol. 1922; 3:202–211.
140. Kienast, AK, Hoeger, PH. Cutis marmorata telangiectatica congenita: A prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clin Exp Dermatol. 2009; 34:319–323.
141. Amitai, DB, Fichman, S, Merlob, P, et al. Cutis marmorata telangiectatica congenita: Clinical findings in 85 patients. Pediatr Dermatol. 2000; 17:100–104.
142. Fujita, M, Darmstadt, GL, Dinulos, JG. Cutis marmorata telangiectatica congenita with hemangiomatous histopathologic features. J Am Acad Dermatol. 2003; 48:950–954.
143. South, DA, Jacobs, AH. Cutis marmorata telangiectatica congenita (congenital generalized phlebectasia). J Pediatr. 1978; 93:944–949.
144. Requena, L, Sangueza, OP. Cutaneous vascular proliferation. Part II. Hyperplasias and benign neoplasms. J Am Acad Dermatol. 1997; 37:887–919.
145. Picascia, DD, Esterly, NB. Cutis marmorata telangiectatica congenita: Report of 22 cases. J Am Acad Dermatol. 1989; 20:1098–1104.
146. O’Toole, EA, Deasy, P, Watson, R. Cutis marmorata telangiectatica congenita associated with a double aortic arch. Pediatr Dermatol. 1995; 12:348–350.
147. Devillers, AC, de Waard-van der Spek, FB, Oranje, AP. Cutis marmorata telangiectatica congenita: Clinical features in 35 cases. Arch Dermatol. 1999; 135:34–38.
148. Vogel, AM, Paltiel, HJ, Kozakewich, HP, et al. Iliac artery stenosis in a child with cutis marmorata telangiectatica congenita. J Pediatr Surg. 2005; 40:e9–12.
149. Guttmacher, AE, Marchuk, DA, White, RI, Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995; 333:918–924.
150. McDonald, J, Damjanovich, K, Millson, A, et al. Molecular diagnosis in hereditary hemorrhagic telangiectasia: Findings in a series tested simultaneously by sequencing and deletion/duplication analysis. Clin Genet. 2011; 79:335–344.
151. McAllister, KA, Grogg, KM, Johnson, DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994; 8:345–351.
152. Johnson, DW, Berg, JN, Baldwin, MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996; 13:189–195.
153. Gallione, CJ, Repetto, GM, Legius, E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004; 363:852–859.
154. Shovlin, CL, Guttmacher, AE, Buscarini, E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000; 91(1):66–67.
155. Govani, FS, Shovlin, CL. Hereditary haemorrhagic telangiectasia: A clinical and scientific review. Eur J Hum Genet. 2009; 17:860–871.
156. Marler, JJ, Fishman, SJ, Upton, J, et al. Prenatal diagnosis of vascular anomalies. J Pediatr Surg. 2002; 37:318–326.
157. Gallagher, PG, Mahoney, MJ, Gosche, JR. Cystic hygroma in the fetus and newborn. Semin Perinatol. 1999; 23:341–356.
158. Bruder, E, Perez-Atayde, AR, Jundt, G, et al. Vascular lesions of bone in children, adolescents, and young adults. A clinicopathologic reappraisal and application of the ISSVA classification. Virchows Arch. 2009; 454:161–179.
159. Florez-Vargas, A, Vargas, SO, Debelenko, LV, et al. Comparative analysis of D2-40 and LYVE-1 immunostaining in lymphatic malformations. Lymphology. 2008; 41:103–110.
160. Padwa, BL, Hayward, PG, Ferraro, NF, et al. Cervicofacial lymphatic malformation: Clinical course, surgical intervention, and pathogenesis of skeletal hypertrophy. Plast Reconstr Surg. 1995; 95:951–960.
161. Gorham, LW, Stout, AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone); Its relation to hemangiomatosis. J Bone Joint Surg Am. 1955; 37-A:985–1004.
162. Pedicelli, G, Mattia, P, Zorzoli, AA, et al. Gorham syndrome. JAMA. 1984; 252:1449–1451. [21].
163. Kulenkampff, HA, Richter, GM, Hasse, WE, et al. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990; 14:361–366.
164. Venkatramani, R, Ma, NS, Pitukcheewanont, P, et al. Gorham’s disease and diffuse lymphangiomatosis in children and adolescents. Pediatr Blood Cancer. 2011; 56:667–670.
165. Papadakis, SA, Khaldi, L, Babourda, EC, et al. Vanishing bone disease: Review and case reports. Orthopedics. 2008; 31:278.
166. Vinee, P, Tanyu, MO, Hauenstein, KH, et al. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994; 18:985–989.
167. Takahashi, A, Ogawa, C, Kanazawa, T, et al. Remission induced by interferon alfa in a patient with massive osteolysis and extension of lymph-hemangiomatosis: A severe case of Gorham-Stout syndrome. J Pediatr Surg. 2005; 40:E47–E50.
168. Hagberg, H, Lamberg, K, Astrom, G. Alpha-2b interferon and oral clodronate for Gorham’s disease. Lancet. 1997; 350:1822–1823.
169. Hammer, F, Kenn, W, Wesselmann, U, et al. Gorham-Stout disease–stabilization during bisphosphonate treatment. J Bone Miner Res. 2005; 20:350–353.
170. Hammill, AM, Wentzel, M, Gupta, A, et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer. 2011; 57:1018–1024.
171. Smeltzer, DM, Stickler, GB, Schirger, A. Primary lymphedema in children and adolescents: A follow-up study and review. Pediatrics. 1985; 76:206–218.
172. Karkkainen, MJ, Ferrell, RE, Lawrence, EC, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000; 25:153–159.
173. Irrthum, A, Karkkainen, MJ, Devriendt, K, et al. Congenital hereditary lymphedema caused by a mutation that inactivated VEGR3 tyrosine kinase. Am J Hum Genet. 2000; 67:295–301.
174. Brice, G, Child, AH, Evans, A, et al. Milroy disease and the VEGFR-3 mutation phenotype. J Med Genet. 2005; 42:98–102.
175. Brice, G, Mansour, S, Bell, R, et al. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet. 2002; 39:478–483.
176. Fang, J, Dagenais, SL, Erickson, RP, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000; 67:1382–1388.
177. Fishman, SJ, Burrows, PE, Upton, J, et al. Life-threatening anomalies of the thoracic duct: Anatomic delineation dictates management. J Pediatr Surg. 2001; 36:1269–1272.
178. Alqahtani, A, Nguyen, LT, Flageole, H, et al. 25 years’ experience with lymphangiomas in children. J Pediatr Surg. 1999; 34:1164–1168.
179. Upton, J, Coombs, CJ, Mulliken, JB, et al. Vascular malformations of the upper limb: A review of 270 patients. J Hand Surg Am. 1999; 24:1019–1035.
180. Limaye, N, Wouters, V, Uebelhoer, M, et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet. 2009; 41:118–124.
181. Vikkula, M, Boon, LM, Carraway, KL, 3rd., et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996; 87:1181–1190.
182. Jones, N, Iljin, K, Dumont, DJ, et al. Tie receptors: New modulators of angiogenic and lymphangiogenic responses. Nat Rev Mol Cell Biol. 2001; 2:257–267.
183. Calvert, JT, Riney, TJ, Kontos, CD, et al. Allelic and locus heterogeneity in inherited venous malformations. Hum Mol Genet. 1999; 8:1279–1289.
184. Brouillard, P, Boon, LM, Mulliken, JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (‘glomangiomas’). Am J Hum Genet. 2002; 70:866–874.
185. AP, O. Blue rubber bleb nevus syndrome. Pediatr Dermatol. 1986; 3:304–310.
186. Kulungowski, AM, Fox, VL, Burrows, PE, et al. Portomesenteric venous thrombosis associated with rectal venous malformations. J Pediatr Surg. 2010; 45:1221–1227.
187. Berenguer, B, Burrows, PE, Zurakowski, D, et al. Sclerotherapy of craniofacial venous malformations: Complications and results. Plast Reconstr Surg. 1999; 104:1–11.
188. Fishman, SJ, Shamberger, RC, Fox, VL, et al. Endorectal pull-through abates gastrointestinal hemorrhage from colorectal venous malformations. J Pediatr Surg. 2000; 35:982–984.
189. Fishman, SJ, Smithers, CJ, Folkman, J, et al. Blue rubber bleb nevus syndrome: Surgical eradication of gastrointestinal bleeding. Ann Surg. 2005; 241:523–528.
190. Kohout, MP, Hansen, M, Pribaz, JJ, et al. Arteriovenous malformations of the head and neck: Natural history and management. Plast Reconstr Surg. 1998; 102:643–654.
191. Liu, AS, Mulliken, JB, Zurakowski, D, et al. Extracranial arteriovenous malformations: Natural progression and recurrence after treatment. Plast Reconstr Surg. 2010; 125:1185–1194.
192. Revencu, N, Boon, LM, Mulliken, JB, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008; 29:959–965.
193. Yakes, WF, Rossi, P, Odink, H. How I do it. Arteriovenous malformation management. Cardiovasc Intervent Radiol. 1996; 19:65–71.
194. Cohen, MM, Jr. Klippel-Trenaunay syndrome. Am J Med Genet. 2000; 93:171–175.
195. Jacob, AG, Driscoll, DJ, Shaughnessy, WJ, et al. Klippel-Trenaunay syndrome: Spectrum and management. Mayo Clin Proc. 1998; 73:28–36.
196. Klippel, M, Trenaunay, P. Du naevus variqueux osteohypertrophique. Arch Gen Med (Paris). 1900; 185:641–672.
197. Servelle, M. Klippel and Trenaunay’s syndrome. 768 operated cases. Ann Surg. 1985; 201:365–373.
198. Kulungowski, AM, Fishman, SJ. Management of combined vascular malformations. Clin Plast Surg. 2011; 38:107–120.
199. Alomari, AI. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: A descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol. 2009; 18:1–7.
200. Sapp, JC, Turner, JT, van de Kamp, JM, et al. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am J Med Genet A. 2007; 143A:2944–2958.
201. Fernandez-Pineda, I, Fajardo, M, Chaudry, G, et al. Perinatal clinical and imaging features of CLOVES syndrome. Pediatr Radiol. 2010; 40:1436–1439.
202. Kurek, KC, Luks, VL, Ayturk, UM, et al. Somatic Mosaic Activating Mutations in PIK3CA Cause CLOVES Syndrome. Am J Hum Genet. 2012; 90:1108–1115.
203. Bader, AG, Kang, S, Vogt, PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006; 103:1475–1479.
204. Perkins, P, Weiss, SW. Spindle cell hemangioendothelioma. An analysis of 78 cases with reassessment of its pathogenesis and biologic behavior. Am J Surg Pathol. 1996; 20:1196–1204.
205. Sun, TC, Swee, RG, Shives, TC, et al. Chondrosarcoma in Maffucci’s syndrome. J Bone Joint Surg Am. 1985; 67:1214–1219.
206. Amary, MF, Damato, S, Halai, D, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011; 43:1262–1265.
207. Pansuriya, TC, van Eijk, R, d’Adamo, P, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011; 43:1256–1261.
208. Cohen, MM, Jr. Bannayan-Riley-Ruvalcaba syndrome: Renaming three formerly recognized syndromes as one etiologic entity. Am J Med Genet. 1990; 35:291–292.
209. Parisi, MA, Dinulos, MB, Leppig, KA, et al. The spectrum and evolution of phenotypic findings in PTEN mutation positive cases of Bannayan-Riley-Ruvalcaba syndrome. J Med Genet. 2001; 38:52–58.
210. Tan, WH, Baris, HN, Burrows, PE, et al. The spectrum of vascular anomalies in patients with PTEN mutations: Implications for diagnosis and management. J Med Genet. 2007; 44:594–602.
211. Biesecker, LG, Happle, R, Mulliken, JB, et al. Proteus syndrome: Diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999; 84:389–395.
212. Lindhurst, MJ, Sapp, JC, Teer, JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011; 365:611–619.
213. Weber, FP. Angioma formation in connection with hypertrophy of limbs and hemi-hypertrophy. Br J Dermatol Syph. 1907; 19:231–235.
214. Weber, FP. Haemangiectatic hypertrophy of limbs: Congenital phlebarteriectasis and so-called congenital varicose veins. Br J Dis Child. 1918; 15:13–17.
215. Greene, AK, Liu, AS, Mulliken, JB, et al. Vascular anomalies in 5621 patients: guidelines for referral. J Pediatr Surg. 2011; 46:1784–1789.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Vascular and Lymphatic Anomalies
Vascular anomalies are broadly divided into two groups based on biologic and clinical behavior: vascular tumors and vascular malformations.1 Vascular tumors are true neoplasms that arise from cellular hyperplasia. In contrast, vascular malformations are congenital lesions originating from errors of embryonic development and exhibit normal endothelial cell turnover.1 Historically, the field of vascular anomalies has been hindered by a myriad of confusing and misused terminology and nomenclature. This, along with the rarity and often complex nature of some of these disorders, has combined to make diagnosis and treatment of vascular anomalies difficult. However, the last several decades have brought better insight and understanding into the field of vascular anomalies, with improved knowledge of blood vessel angiogenesis and the development of a more logical classification system.
Classification
In 1996, the International Society of the Study of Vascular Anomalies formally accepted the biological classification system in use today (Table 72-1).2,3 This system divides these anomalies into vascular tumors and vascular malformations based on physical characteristics, natural history, and cellular features. Examples of vascular tumors are infantile hemangioma (IH), kaposiform hemangioendothelioma (KHE), and tufted angioma (TA). Vascular malformations can be divided based on vascular channel type (capillary, lymphatic, venous, arterial, or combined) or by flow (slow or fast). Examples of slow-flow lesions are capillary malformations (CM), lymphatic malformations (LM), and venous malformations (VM). Fast-flow lesions include arteriovenous fistulas (AVF) and arteriovenous malformations (AVM).
Nomenclature
In the 19th century, Virchow first described the histologic features of vascular nevi.4 He initiated the term ‘angioma’, which became the default term to describe all such nevi regardless of natural history or other clinical features. He also labeled the IH ‘angioma simplex’, a lesion that has been historically referred to as ‘capillary hemangioma’ and ‘strawberry hemangioma.’ Virchow’s ‘angioma cavernosum’ was used to label two distinct lesions, IH (when located deep to the skin) and VM, because both have similar appearance on physical examination. ‘Angioma racemosum’ was Virchow’s designation for what today is termed an arteriovenous malformation and which has previously been called an ‘arteriovenous hemangioma.’
Wegener, a student of Virchow, described the histology of LMs, which he called ‘lymphangiomas.’5 The classic term ‘cystic hygroma’, referring to LM, unfortunately also continues to have common usage. Thus, both the terms cystic hygroma and lymphangioma should be abandoned in favor of LM (macrocystic and microcystic, respectively). The problems with this jumble of descriptive and histologic terms are obvious. The same lesion can often have several different names, and simultaneously, the same name can refer to several different lesions. For example, the term ‘hemangioma’, combined with descriptive modifiers such as ‘strawberry’, ‘cavernous’, and ‘lympho-’, is used to describe tumors, birthmarks, and vascular malformations alike. Vascular anomalies with quite distinct features, whether congenital or acquired, or whether they spontaneously regress or progress over time, become lumped under the umbrella term ‘hemangioma.’ These faulty designations lead to improper diagnosis and treatment for patients as well as leading to misguided interdisciplinary communication and research efforts.
Vascular Tumors
Infantile Hemangioma
IH are the most common tumor of infancy. They occur in about 4% of infants, though early studies were as high as 10%, probably due to the inclusion of other vascular lesions.6 The incidence is higher in premature infants, Caucasians, and females (by a 3 to 5 : 1 ratio). 6,7 Advanced maternal age, multiple gestations, and placental abnormalities are also risk factors.8 IH have a unique and characteristic life cycle consisting of three phases: proliferative, involuting, and involuted.
Pathophysiology
The precise etiology of IH remains unknown. Viral causes have been speculated, but none elucidated. Some studies suggest that they arise from the clonal expansion of endothelial stem/progenitor cells, the source of which is unclear.9–11 One report concluded these cells arise from a population of resident angioblasts, arrested in an early stage of vascular development.12 As hemangiomas are more common in females, estrogen, which has a stimulatory effect on endothelial cells, may factor in the development of these lesions. Its receptors are present on endothelial cells and elevated levels of estradiol have been found in infants with hemangiomas.13,14
The expression of placental markers (including CD 32, Fcγ-RIIb, glucose transporter 1 [GLUT-1], indoleamine deoxygenase [IDO], insulin growth factor 2 [IGF-2] Lewis Y antigen, merosin, type II 17-hydroxysteroid dehydrogenase [17HSDβ2], tissue factor pathway inhibitor 2 [TFPI-2], and type III iodothyronine deoidinase) by hemangioma endothelial cells suggests a placental origin.15–21 GLUT-1, an erythrocyte type glucose transporter, is a specific marker for endothelial cells of hemangiomas, but is not found in other vascular anomalies.15 The placental cells may arrive at fetal tissue following local placental disruption as an embolic nidus though the permissive right to left shunt of fetal circulation. This may occur during chorionic villus sampling or placental complications such as pre-eclampsia and placenta previa, which have shown to be predisposing factors for hemangiomas.8,22,23
The dysregulation of angiogenesis can be seen during the proliferation and involution of hemangiomas, and is suspected to be a cause of the disease. IH in the proliferative phase express high levels of fibroblast growth factor (FGF), TIE-2, angiopoietins, matrix metalloproteinases (MMPs) and vascular endothelial growth factor A (VEGF-A) and its receptor (VEGFR), all of which play critical roles in the formation of blood vessels during and after embryogenesis.24–30 The tumor during this phase is composed of plump, rapidly dividing endothelial cells forming a mass of sinusoidal vascular channels. Enlarged feeding arteries and draining veins often vascularize the tumor. Markers for mature endothelium, CD-31 and von Willebrand factor, are present on these neoplastic endothelial cells. Involuted hemangiomas express normal levels of these factors, but elevated levels of tissue inhibitor of TIMP1, a metalloproteinase that inhibits new blood vessel formation, and interferon-β.28,29 The endothelial cells of the tumor flatten as apoptosis progresses, the vascular channels dilate, and the tumor assumes a lobular architecture with replacement by fibrofatty stroma.31 All that remains in the involuted phase is a residuum of fibrofatty tissue with tiny capillaries and mildly dilated draining vessels.
Clinical Features
IH are not fully developed at birth and first appear in the neonatal period with a median age of onset of 2 weeks. A premonitory cutaneous sign may be present at birth in 30–50% of cases.1 They are most often cutaneous (80%) and have an anatomic predilection for the head and neck region (60%). They occur in the trunk and extremities 25% and 15% of the time, respectively.2 Internal and visceral lesions are uncommon. Up to 20% of patients can have multiple lesions, and these cases are most likely to have internal involvement affecting organs such as the liver and gastrointestinal (GI) tract.1
The proliferative phase of IH is marked by rapid growth for the first six to eight months that typically plateaus by age 10–12 months. Tumors that involve the superficial dermis present as a red, raised lesion (Fig. 72-1A). Superficial tumors that are larger or that exhibit more rapid growth can occasionally cause ulceration of the skin with bleeding. Tumors in the lower dermis, subcutaneous tissue, or muscle appear bluish in color with slightly raised overlying skin (previously incorrectly termed ‘cavernous’ hemangiomas). With experience, history and physical examination can establish an accurate diagnosis for most of these tumors. The involuting phase of hemangiomas occurs from age 1 to 7 years during which time the tumor slowly regresses, although it may grow in proportion with the child. This phase is notable for fading color of the tumor from crimson to a dull purple, accompanied by a deflation of the tumor mass (Fig. 72-1B).1 The skin may become pale, usually in the center of the tumor first, spreading outwards. Fifty per cent of tumors have completed involution by 5 years of age, and 70% by age 7 years. There is continued gradual improvement in these aspects until the regression is entirely complete by age 10–12 years.32 In the final involuted phase of the tumor, 50% of patients have nearly normal skin in the area of the prior lesion. Patients with larger tumors can have lax or redundant skin and yellowish discoloration. Scars will persist if parts of the tumor were previously ulcerated.1
FIGURE 72-1 (A) This infant has an infantile hemangioma that is in the proliferative phase. This hemangioma was not present at birth but was noted at several weeks of age. (B) This hemangioma is in its involuting phase.
The differential diagnosis of cutaneous hemangiomas consists primarily of other vascular anomalies. CM that involves the skin can be mistaken for superficial hemangiomas, or vice versa. Deeper hemangiomas can be confused for VM or LM as all can appear as bluish masses through the skin. Hemangiomas with fast-flow vascularity of the parenchyma could be confused for AVM, but the age of onset and history generally distinguishes the two. Congenital hemangiomas, discussed in a later section, may be misdiagnosed as vascular malformations, which are congenital by definition. Pyogenic granulomas may be differentiated from hemangiomas by their rare appearance before six months (mean age is 6 to 7 years), and their frequent association with minor trauma.33,34 Other tumors such as TA, hemangiopericytoma, and fibrosarcoma can also be confused.35
The primary local complications that can occur with cutaneous hemangiomas are ulceration, bleeding, and pain. Hemangiomas are rarely life-threatening, but complications can be anticipated by recognition of the anatomic distribution of the lesion.36 Lesions in the cervicofacial region can lead to airway obstruction as they grow during the proliferative phase. Very large hemangiomas, notably in the liver, can lead to high-output congestive heart failure (secondary to fast-flow and vascular shunting within the tumor), hypothyroidism, and abdominal compartment syndrome. Facial lesions involving the eyelid, nose, lip, or ear can result in tissue destruction with cosmetic consequences. Periorbital and eyelid hemangiomas can cause visual axis obstruction, leading to deprivation amblyopia (Fig. 72-2). Alternatively, distortion of the cornea can cause astigmatic amblyopia. GI hemangiomas are very rare, but may manifest with GI bleeding.
Associated Congenital Anomalies
IH are rarely associated with other anomalies. However, a few such anomalies have been described, more commonly with larger and midline hemangiomas. Cervicothoracic hemangiomas can be seen in conjunction with sternal nonunion.37 Tumors of the lumbosacral area have been noted to occur along with spinal dysraphism abnormalities such as meningocele and tethered spinal cord.38,39 Hemangiomas of the pelvis and perineum have been found in association with urogenital and anorectal anomalies. PHACES association describes hemangiomas associated with congenital ocular abnormalities such as microophthalmia, cataracts, optic nerve hypoplasia, posterior fossa cystic malformations, hypoplasia or absence of carotid and vertebral vessels, as well as malformation of the aortic arch (Fig 72-3).40,41 Females are affected in 90% of cases. These patients are at risk for stroke in the neonatal period and migraines in older ages.
Other Manifestations
The presence of multiple disseminated hemangiomas is termed hemangiomatosis. The cutaneous tumors are usually tiny (<5 mm) and dome-like. When five or more lesions are present, occult visceral lesions, most commonly in the liver, may also be present (Fig. 72-4). Screening patients with ultrasound (US) and/or magnetic resonance imaging (MRI) may be indicated for these patients.
Imaging
Proper radiologic diagnosis of vascular anomalies is dependent on specific expertise and clinical experience with the radiologic features of these lesions.42 Ultrasound and MRI are the most useful imaging modalities. Ultrasound of proliferative phase hemangiomas demonstrates a mass with dense parenchyma exhibiting fast-flow vascularity.43,44 This distinguishes deep IH from VM, which exhibit slow-flow vascularity and larger blood-filled spaces. MRI of proliferating hemangiomas shows a lobulated solid mass of intermediate intensity with T1 spin-echo sequences, and moderate hyperintensity on T2 spin-echo. Flow voids that represent fast flow and shunting are seen in and around the tumor. During the involuting phase, MRI demonstrates decreased flow voids and vascularity, with the mass taking on a more lobular and fatty appearance.45
Treatment
The majority of IH do not require any specific treatment other than observation and reassurance of the parents.46 Even tumors that exhibit rapid growth or fiery red skin will spontaneously regress and leave behind little to no evidence of their presence. However, regular follow-up is important as the potential complications have few prognostic indicators. Serial photographs are very helpful in documenting progression and subsequent improvement. Reasons for treatment or referral to a vascular anomalies specialist or center include dangerous locations (impinging on a vital structure such as the airway or eye), unusually large size or rapid growth, and local or endangering complications (skin ulceration or high-output heart failure).
Hemangiomas exhibiting the above risk factors or complications should be considered for treatment. Since hemangiomas are tumors of pure angiogenesis, pharmacologic therapy involves angiogenesis inhibition. Systemic corticosteroids, which inhibit the expression of VEGF-A by hemangioma-derived stem cells and thus angiogenesis, were first line therapy for decades.47–49 Oral prednisone is given at a dose of 2–3 mg/kg/day. Doses up to 5 mg/kg/day have been used for life-threatening complications of large hemangiomas causing airway obstruction or heart failure. The overall response rate is 80–90% with initial improvement in the color and tension of the mass usually noted within one week. The steroids are maintained with a very gradual taper every two to four weeks with the goal of discontinuation around age 10–11 months. Live vaccines such as polio, measles, mumps, rubella, and varicella should be withheld while children are taking prednisone. Hemangiomas will have rebound growth if steroids are tapered or stopped too quickly. Return to the initial dosage and slower tapering will usually treat this problem. Potential complications of steroid use in infants and children include impaired growth and weight gain in about one-third of cases. Almost all children will have ‘catch up’ growth and return to pretreatment growth curves by age 14–24 months.50 Cushingoid facies occur in almost all patients and normalizes upon tapering. In rare circumstances, steroids may induce hypertension or hypertrophic cardiomyopathy, both of which are indications to wean or change therapy.50
Intralesional corticosteroids are used for small cutaneous hemangiomas that cause local deformity or ulceration, especially for lesions of the eyelid, nose, cheek, or lip.51 A total of three to five injections are typically given at intervals of six to eight weeks at a dose of 3–5 mg/kg/injection.52 The response rate approaches that of systemic steroids. Subcutaneous atrophy is a potential complication of steroid injection, but is usually temporary. There have been reported cases of blindness following intralesional steroid injection for periorbital hemangiomas.53 This is presumed to be secondary to particle embolization into the retinal artery through feeding vessels. Manual compression around the periphery of the tumor is recommended during injection to minimize embolization through draining veins.
Propranolol, a nonselective beta blocker, has recently been recognized as an important treatment option for hemangiomas and in most centers has become first line pharmacotherapy. A child with a nasal capillary hemangioma treated with propranolol for steroid-induced cardiomyopathy had regression of his lesion.54 This revelation led to the publication of several more studies supporting this finding.55–58 Propranolol is given orally at 2–3 mg/kg/day, in two or three divided doses, and discontinued following regression of the lesion.55,59 Treatment often leads to a consistent, rapid, therapeutic effect with softening of the lesion on palpation and color shift from intense red to purple.55 Propranolol is well tolerated but can cause rare side effects such as bradycardia, gastroesophageal reflux, hypoglycemia, hypotension, rash, somnolence, and wheezing.55,56,58–61 Several mechanisms of action have been proposed.62 Propranolol inhibits β-adrenoreceptors, which are activated by adrenaline, causing vasoconstriction of capillaries supplying the hemangioma.63,64 This likely leads to the visible changes in color and palpable softening. Blockage of β-adrenoreceptors also results in decreased expression of VEGF and MMPs, thereby inhibiting angiogenesis, and induces apoptosis in endothelial cells.65,66
Recombinant interferon was once considered as a second line agent, but has fallen out of favor except in very limited circumstances.67–71 A small subset of patients (5–12%) may develop a severe complication known as spastic diplegia.72,73 Spasticity usually resolves if the drug is terminated quickly. Children receiving IFN should be followed carefully by a neurologist. Though experience is limited, low-dose, high frequency anti-angiogenic regimens using vincristine can be effective.74 The use of interferon and vincristine has waned since the introduction of propranolol therapy.
Although attractive in concept, laser therapy is not often beneficial for IHs, except for a few specific indications.75 The flash lamp pulse-dye laser penetrates the dermis to a depth of only 0.75–1.2 mm. Most cutaneous hemangiomas are deeper than this, and therefore not affected by laser treatment. Additionally, laser therapy carries risks of scarring, skin hypopigmentation, and ulceration, which may lead to a poor result compared to observation alone. One instance in which the laser is advantageous is the treatment of telangiectasias that often remain in the involuted phase of hemangioma. The use of endoscopic continuous wave carbon dioxide laser has been shown to be a good strategy for controlling proliferative phase hemangiomas in the unilateral subglottic location.76 Lastly, intralesional photocoagulation with bare fiber Nd:YAG laser can be useful for hemangiomas in certain locations, such as the upper eyelid when visual obstruction is a concern.
Indications for resection of IH vary with patient age. During the proliferative phase in infancy, well-localized or pedunculated tumors can be expeditiously resected with linear closure, especially for tumors complicated by bleeding and ulceration. Sites that are most amenable to resection are the scalp, trunk, and extremities. Other modalities to treat ulceration include wound care with dressing changes, topical antibiotics, and topical steroids, which can accelerate healing.77 Tumors of the upper eyelid that obstruct vision and that do not respond to pharmacologic therapy may also require excision or debulking. Focal lesions of the GI tract with bleeding that fail medical management may require enterotomy and resection, or endoscopic band ligation. Diffuse, patchy involvement is the more common presentation of GI hemangiomas. Management is difficult but most lesions eventually involute and stop bleeding.78 Preoperative localization with capsule endoscopy and/or intraoperative endoscopy may be necessary to identify lesions in the small bowel.79
During the involuting phase, resection may be needed for hemangiomas that are large and protuberant, and therefore likely to create excess and lax overlying skin.80 Indications for resection include: (1) it is obvious that resection will be necessary sooner or later; (2) the surgical scar will be identical regardless of timing of operation; and (3) the surgical scar can easily be hidden. Lesions of the nose, eyelids, lips, and ears require special expertise. It is often preferable to perform the operation for the above indications during the preschool years before children become aware of and focus on body image differences that may lead to low self-esteem.
Finally, for the difficult to treat and life-threatening large hemangiomas, especially in the liver, angiographic embolization may be required to manage high-output cardiac failure. Arterial catheterization in infants carries significant risks and should generally be limited to those situations with cardiac compromise in which there is the capacity and intent to perform simultaneous embolization. In these rare cases, anti-angiogenic pharmacotherapy remains the first line of therapy and should continue along with angiographic procedures. Repeat embolization procedures may be required. Success with embolization is dependent on occlusion of macrovascular shunts within the tumor rather than occlusion of feeding vessels.81
Congenital Hemangioma
Two types of rare congenital hemangiomas exist that are fully developed at birth and that do not usually exhibit postnatal tumor growth.82 These have been termed rapidly involuting congenital hemangioma (RICH) and the noninvoluting congenital hemangioma (NICH) (Fig. 72-5). Lesions are solitary and affect both genders equally. Unlike IHs, they do not stain positive for GLUT-1.83 The diagnosis is typically made on physical exam at birth, although some can be diagnosed prenatally as early as 12 weeks of gestation.82 Most do not require therapy.
FIGURE 72-5 These two patients are examples of rapid involuting congenital hemangiomas (RICH) and noninvoluting congenital hemangiomas (NICH). (A) The newborn baby has a RICH. This lesion was present at birth and will spontaneously regress, much more quickly than the typical infantile hemangioma. (B) This 9-year-old child has a NICH. This hemangioma has not resolved. If treatment is needed, arterial embolization may be beneficial as these lesions have significant flow to them.
As opposed to IH, RICH is more common on the extremities. Also, RICH will spontaneously regress, much more quickly than IH, by 6 to 14 months; NICH do not involute and grow with the child.82 RICH appear raised and violaceous, often with a central depression or scar, ulceration, telangiectasia, or surrounding pale rim.82,83 MRI will show enhancing hyperintense masses, high-flow vessels within and adjacent to the mass, and the presence of vascular flow voids on T2-weighted imaging.83 NICH are well-circumscribed, plaque-like lesions, often with coarse telangiectasias, areas of pallor, or a white to bluish rim.84 They appear on MRI as homogeneous lesions with isointense signal on T1-weighted imaging and hyperintense on T2-weighted sequences. Both lesions are fast flow on Doppler ultrasound. If treatment is needed for NICH, arterial embolization may be beneficial as these lesions have significant flow to them. Operative excision is reserved for cases with equivocal diagnosis, poor cosmetic appearance, or for complications such as ulceration, bleeding, obstruction, or congestive heart failure.83,85
Hepatic Hemangioma
Hepatic hemangiomas (HH) in infants should be differentiated from ‘hepatic hemangiomas’ that present in adulthood.85 Adult ‘hepatic hemangiomas’, which are sometimes called ‘cavernous hemangiomas’, are in fact VMs. In contrast, HH of infancy are true tumors and have a similar pattern of involution to cutaneous IH. Contrary to popular belief, not all liver hemangiomas are life-threatening. The classic triad of heart failure, anemia, and hepatomegaly is unusual, and most involute without long-term sequelae.
The majority of HH can be divided into three categories: focal, multifocal, and diffuse, each with distinct features (Fig. 72-6).86 Focal HH are the hepatic equivalent of the cutaneous RICH, and also do not stain positive for GLUT-1. They are histologically distinct from the typical IH.15,87 They are fully grown at birth and regress much faster than IH. Many focal lesions are detected antenatally on prenatal ultrasound, or are discovered as an abdominal mass in otherwise healthy infants.88 They are usually asymptomatic, found in equal numbers in both genders, and rarely associated with cutaneous hemangiomas. Transient anemia and moderate thrombocytopenia, due to intralesional thrombosis, are observed in some infants and generally resolve spontaneously. This is in contradistinction to the profound thrombocytopenia seen with Kasabach–Merritt phenomenon (KMP). However, a subset of focal hepatic RICH-type hemangiomas will have macrovascular shunts from the hepatic arteries and/or portal veins to the hepatic veins. These shunts can cause a large steal, accounting for blood-flow demands above and beyond the hypervascular tumor parenchyma, and can result in high-output cardiac failure. These shunts may close as the tumors involute. However, cardiac strain can often mandate interruption of the shunts via embolization. Steroids have been used for solitary focal hepatic lesions with in-utero cardiac failure/cardiomegaly.88,89 However, these lesions may have undergone rapid spontaneous involution and the benefit of steroids remains unproven, though still should be attempted before employing more invasive therapies. Resection is rarely indicated.
FIGURE 72-6 Most hepatic hemangiomas can be divided into three categories: focal, multifocal, and diffuse. (A) A large focal hepatic hemangioma. (B) The scan depicts multifocal hepatic hemangiomas. (C) Diffuse hepatic hemangioma.
Multifocal HH are true IH and biologically distinct from focal HH. They undergo involution similar to cutaneous IH and stain positive for GLUT-1.87,90,91 They are more common in females and Caucasians.92 Infants with multifocal lesions are typically born earlier and are asymptomatic, but are diagnosed later than focal lesions. These patients often have cutaneous IH and are identified following screening for visceral hemangiomas. Some of these patients will have hypothyroidism. Thus, a serum TSH should be checked in patients with multifocal disease. Infants who are asymptomatic should be observed; however, some can have high-output cardiac failure from macrovascular shunting. Treatment with steroids or propranolol can often close these shunts.54,55 Embolization can be employed in those who fail medical therapy. Serial abdominal ultrasound should be performed in infants with multifocal HH until the lesions involute.
Diffuse HH are also true IH, but are far more serious lesions with a more difficult clinical course. Like multifocal lesions, diagnosis occurs in the weeks following birth. They are more common in Caucasians and females, are associated with cutaneous IH and follow a similar course of involution, and express GLUT-1.92 These patients all develop severe hypothyroidism from high levels of type 3 iodothyronine deiodinase, which inactivates circulating thyroid hormones.20 Aggressive exogenous thyroid hormone replacement is essential to prevent hypothyroid complications such as mental retardation and cardiac failure. Involution of the lesions will usually result in amelioration of the hypothyroidism.93 These innumerable lesions often almost completely replace the normal hepatic parenchyma. Those with considerable disease may have respiratory compromise from compression of the diaphragm and thoracic cavity, but can also develop abdominal compartment syndrome and multisystem organ failure. Aggressive pharmacotherapy with propranolol, steroids, and occasionally low-dose vincristine is warranted in those with cardiac failure, hemodynamically significant shunting, or hypothyroidism to hasten involution.55,86,94–96 Hepatic transplantation is the last resort for critically ill infants.86
Tufted Angioma and Kaposiform Hemangioendothelioma
These vascular tumors of childhood are more aggressive and invasive than IH. TA and KHE probably exist within the same spectrum as they share many overlapping clinical and histologic features.21,97 Both tumors typically present at birth, although they occur postnatally as well. Males and females are affected equally. The tumors are unifocal and are most often located on the trunk, shoulder, thigh, or retroperitoneum. TA present as erythematous macules or plaques and histology reveals small tufts of capillaries.97 KHE are more extensive tumors that present with deep, red-purple skin discoloration and overlying and surrounding ecchymosis (Fig. 72-7). The natural history of these tumors is one of continued proliferation into early childhood followed by subsequent, but incomplete regression. These tumors usually persist, albeit in a smaller form.98 Fortunately, they are usually asymptomatic in later stages, though may cause musculoskeletal pain. Histology of these lesions reveals infiltrating sheets of spindle-shaped endothelial cells in the form of irregular lobules, sheets, and lacy network.99 Imaging of KHE depicts an enhancing lesion on MRI with poorly defined margins that extend across tissue planes. This is in contrast to IH, which are well-circumscribed and respective of tissue planes.
FIGURE 72-7 This infant has a kaposiform hemangioendothelioma (KHE). KHE are more extensive tumors that present with deep, red-purple skin discoloration and overlying ecchymosis. These lesions usually regress later in childhood, although usually not completely.
Kasabach–Merritt Phenomenon
KMP was first reported in 1940 in a case of profound thrombocytopenia, petechiae, and bleeding in conjunction with a ‘giant hemangioma.’100 As with many terms in the field of vascular anomalies, this term has been often misused in connection with coagulopathy and other vascular lesions, most prominently VM. However, the profound and persistent thrombocytopenia that occurs with KMP does not occur with either VM or IH. The only known true associations are with TA and KHE.99,101,102 The platelet count with KMP is typically under 10,000/μL, and may be associated with decreased fibrinogen levels, increased D-dimer, and mildly elevated partial prothrombin time (PT) and partial thromboplastin time (PTT). Bleeding can result from this platelet trapping coagulopathy at many sites, including intracranial, GI, peritoneal, pleural, and pulmonary. A microangiopathic hemolytic anemia is also present. Treatment for KHE with KMP is primarily medical as the tumor is usually too large and extensive to be resected. Corticosteroids and interferon-alfa have been effective in about 50% of cases; actinomycin, anti-platelet therapy, cyclophosphamide, doxorubicin, gemcitabine, propranolol, sirolimus, and vincristine have also been found to be beneficial in several case series, as single drugs or in combination, but none of these agents have been shown to be consistently successful.103–111 Platelet transfusions are ineffective and should be avoided unless there is active bleeding. Additionally, heparin may stimulate tumor growth and worsens the thrombocytopenia of KMP and should likewise be avoided. Mortality rates with KHE and TA remain high at 20–30%. KHE not associated with KMP can be followed without treatment as long as the size and location of tumor are limited.
Vascular Malformations
Vascular malformations are congenital lesions of vascular dysmorphogenesis that can be local or diffuse. The majority of vascular malformations are sporadic, though some rare varieties are familial.112–114 They occur in 1.5% of the population.115 Vascular malformations probably result from genetic mutations that lead to dysfunction in the regulation of endothelial proliferation and apoptosis, cellular differentiation, maturation, and cell-to-cell adhesions.116
Embryology and Development of the Vascular and Lymphatic Systems
The vascular system develops during embryogenesis through the processes of vasculogenesis, the formation of new vascular channels from mesodermally derived endothelial precursor cells (hemangioblasts), and angiogenesis, the formation of new blood vessels from preexisting blood vessels. The destiny of endothelial precursors to create different types of blood vessels appears to be imprinted early in embryogenesis by unique cell surface markers.117 The differentiation of angioblasts into an early vascular plexus leads to the creation of primitive blood vessels.118 Following formation of the primary vascular plexus, endothelial cells proliferate and sprout or split from their vessel of origin to form new capillaries. A process called ‘pruning’ then occurs, during which the vascular plexus is remodeled into a system with larger and smaller vessels. The signals for microvascular endothelial cells to proliferate and differentiate for vessel development are controlled by a number of growth factors and their receptors, including VEGF, basic fibroblast growth factor 2 (FGF-2), and angiopoietin 1 (Ang-1).119
The lymphatic system develops around midgestation after the blood vasculature forms, and is thought to derive from either venous endothelial cells or mesenchymal progenitor cells.120–123 It is a one-way valve system that collects fluid and macromolecules from tissue. In 1902, Sabin described the prevailing model of lymphangiogenesis in which venous endothelial cells commit to become lymphatic endothelium and then migrate and proliferate to form lymph sacs.122 These sacs then form a lymphatic plexus, which remodel and mature into the lymphatic vasculature. 120,123 Venous endothelial cells along the anterior cardinal vein capable of differentiating into lymphatic endothelial cells (LECs) begin to express lymphatic endothelial hyaluronan receptor-1 (LYVE-1) on embryonic day (E) 9, a marker specific for lymphatic endothelium.120,123,124 A subgroup of endothelial cells on one side of the vein then begin to express prospero-related homeobox 1 (Prox-1). 125 This transcription factor is required for the maturation and differentiation of LECs.125,126 VEGFR3, also known as Flt4, plays an important role in lymphatic development as well. VEGFR is expressed in both blood and lymphatic vasculature in early embryogenesis, but is restricted to mostly lymphatic vessels later in development127–129 VEGFR3 knockout mice die on E9 with major venous anomalies before any lymphatic sprouting has occurred.116 In contrast, transgenic mice that overexpress the ligand for VEGFR3 (VEGF-C) will develop distended lymphatic channels.130
Capillary Malformations
CM, previously referred to as ‘port-wine stains’, are present at birth as permanent flat, pink-red cutaneous lesions (Fig. 72-8). In the newborn nursery, CM can be confused with nevus flammeus neonatorum, commonly called ‘angel’s kiss’ when located on the face, or ‘stork bite’ when in the posterior cervical location. However, these discolorations are due to transient dilations of dermal vessels and fade with time, whereas CM does not. CM occur with equal gender distribution in 0.3% of infants.131 Multiple CM are rare. The majority of CM appear sporadically, but some familial forms exist that are inherited in an autosomally dominant fashion.132 Capillary malformation–arteriovenous malformations (CM-AVM) are associated with mutations in RASA1, a gene coding for p120-RasGTPase.133 Histologically, cutaneous CM consist of dilated capillary- to venule-sized vessels located in the superficial dermis, with a paucity of surrounding normal nerve fibers.134 These abnormal vessels gradually dilate over time leading to darkening color and occasionally nodular ectasias. CM can also be found in complex-combined vascular malformations.
FIGURE 72-8 This child has a capillary malformation. Such lesions were previously referred to as a port-wine stain.
[/not-level-membership-for-pediatrics-category]
