[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 115 Ulcers of the Small and Large Intestine
ISOLATED ULCERS
NONSPECIFIC OR IDIOPATHIC SMALL INTESTINAL ULCERATION
Solitary ulcers of the small intestine result from a wide variety of causes (Table 115-1). Radiation injury is a known cause of small intestinal ulceration and is discussed in Chapter 39. Solitary ulcers beyond the duodenum that cannot be explained on the basis of any known etiology are referred to as nonspecific or idiopathic intestinal ulcers. Such solitary nonspecific ulcers are rare, with an incidence of 4 per 100,000.1
Table 115-1 Causes of Small Intestinal Ulceration
Clinical Features
In a review of the Mayo Clinic’s experience with 59 cases of small intestinal ulcers over a 22-year period ending in 1979, Boydstun and associates1 showed that 53 (89.8%) patients had no identifiable cause of ulceration. Patients ranged in age from 17 to 77 years, with most presenting in the fifth and sixth decades of life; no gender predominance was found. The most common presenting symptom was intermittent small bowel obstruction (63%). Physical findings ranged from nonspecific abdominal tenderness and distention to an acute abdomen resulting from intestinal perforation. Laboratory evaluation was notable only for anemia in one half of the patients. Radiologic studies localized the ulcer in a minority of patients.
Pathology
In the Mayo Clinic series,1 the ileum was the most common location of nonspecific ulceration (78%), and perforation (13 cases, 22%) occurred most commonly in the jejunum (78%). At surgery, 41 patients were found to have solitary ulcers, five patients had two ulcers, and six patients had more than three ulcers. Ulcer size varied between 0.3 and 5 cm. On pathologic examination, the ulcers were predominantly on the antimesenteric border of the small intestine and, in some cases, were associated with a fibrous scar that narrowed the lumen. Microscopy revealed nonspecific chronic inflammation that ended abruptly at the ulcer edge. The intervening bowel and vasculature were normal.1
SOLITARY RECTAL ULCER SYNDROME
Solitary rectal ulcer syndrome (SRUS) is an uncommon or under-reported disorder of defecation that affects patients of all ages. The term is a misnomer: Patients can present with hyperemic mucosa only, a solitary ulcer, multiple ulcers, or even a polypoid lesion resembling carcinoma.2 Regardless, the histology of SRUS is typical, showing fibromuscular obliteration of the lamina propria and smooth muscle fibers extending from a hypertrophied muscularis mucosa to the lumen.2 The diagnosis of SRUS often is delayed because of its varied endoscopic appearance and a lack of awareness of the disorder.
Pathogenesis
SRUS is a disorder of defecation, but its pathogenesis is uncertain, and it has a spectrum of disease presentations. A large subgroup of patients with SRUS strain excessively during defecation, and some have a behavioral disorder. Occult or overt rectal prolapse with paradoxical contraction of the pelvic floor during defecation appears to be involved in most patients3; evidence of inappropriate pelvic floor contraction has been shown in electromyographic and video-proctographic studies.3 It has been suggested that the rectal mucosa can be traumatized from the pressure of being prolapsed against a closed anal canal4 and that straining during defecation results in prolapse and high fecal voiding pressures that reduce local blood flow, causing ischemia and ulceration.4 The mucosa of the anterior rectal wall, 7 to 10 cm above the anal verge, is the most common area of such prolapse into the anal canal, and this corresponds to the usual location of ulceration in SRUS.
SRUS also has been associated with the use of ergotamine suppositories and is well known after radiotherapy, further supporting a pathogenic role for ischemia.5,6 Successful treatment of SRUS using biofeedback has been associated with an increase in local blood flow, additionally suggesting that SRUS may be associated with reduced rectal blood flow from impaired extrinsic autonomic cholinergic nerve activity.7
The association of SRUS and rectal prolapse, however, is neither pathogenically clear nor universal; the incidence of associated rectal prolapse varies from 13% to 94%.2 It is assumed that the ulcer develops as a result of local trauma to the apex of the prolapse, either because of manual attempts to reduce the prolapse digitally or because of contractions of the external anal sphincter when the mucosa prolapses through the anal canal.4
Du Boulay and colleagues have shown that the histology of the rectal mucosa in patients with SRUS is similar to that seen at other sites of mucosal prolapse, suggesting that prolapse of the mucosa alone rather than the entire rectal wall is important in SRUS pathogenesis.8 Ischemia results in fibromuscular obliteration of the lamina propria and the formation of an ulcer. Once the ulcer is formed, it can further intensify the urge to defecate; this urge combined with straining and changes in local blood flow causes persistent symptoms and chronic ulceration.
Clinical Features
Patients with SRUS present with varied symptoms, but most patients typically complain of passage of mucus and blood per rectum on defecation.9,10 Some patients also complain of tenesmus, straining, altered bowel habits, and the sensation of incomplete evacuation. Men and women are affected equally, and they usually present in the third and fourth decades, respectively.11 The mean duration of symptoms is long, ranging from 3.5 to 5 years, possibly reflecting a delay in diagnosis.
Diagnosis and Pathology
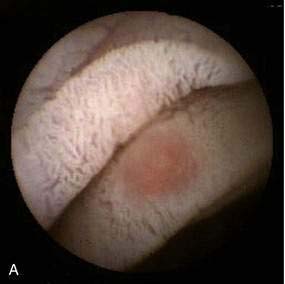
Diagnosis of SRUS is based on clinical symptoms, physical examination, endoscopic findings, and typical histology. Physical examination can demonstrate tenderness in the left lower quadrant. On digital rectal examination, there may be reduced anal sphincter tone and an indurated area, or thickened folds may be palpated.12 Overt rectal prolapse may be demonstrated by having the patient assume a squatting position and straining as if to have a bowel movement. Sigmoidoscopy may demonstrate single or multiple ulcers or a patch of erythematous mucosa on the anterior rectal wall within 10 cm of the anal verge. The lesion has a polypoid appearance (Fig. 115-1) in 25% to 44% of patients.2,12 Differential diagnosis includes inflammatory bowel disease, malignancy, ischemic colitis, stercoral ulcer, medication-induced ulceration, trauma, and infections, including amebiasis and secondary syphilis.11
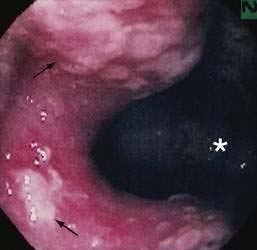
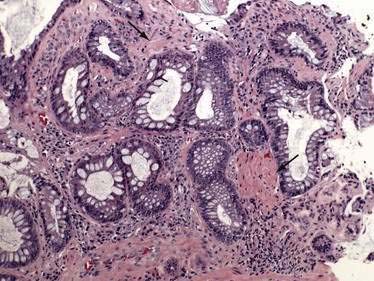
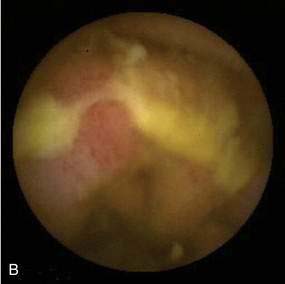
Biopsy specimens always should be taken from the ulcer margin and from any abnormal-appearing mucosa. In 1969, Madigan and Morson9 first described the histologic features of SRUS. There is fibromuscular obliteration of the lamina propria by collagen from fibroblasts and smooth muscle fibers derived from the muscularis mucosae. The muscularis mucosae is often hypertrophied, and its fibers are in continuity with those in the lamina propria. There is no significant increase in the number of inflammatory cells. The polypoid variant is similar to the ulcerative variant except for regenerative hyperplastic changes, such as cystic dilatation and mucus cell production.11 Epithelial elements and lamina propria can be displaced into the submucosa (Fig. 115-2). This displaced tissue can then undergo cystic dilatation because of mucus retention. The misplaced and dysplastic-appearing glands may be misdiagnosed as adenocarcinoma, especially when the histologic and macroscopic features of SRUS are not recognized; at times SRUS is present in association with an carcinoma, further confusing the issue.
Histology typically provides a definitive diagnosis; however, defecography may be useful to shed light on the pathophysiology of SRUS, especially if surgery is being considered. Defecography may be used to demonstrate mucosal prolapse, intussusception, rectal prolapse, a nonrelaxing puborectalis muscle, and incomplete or delayed evacuation.11 Endorectal ultrasound can demonstrate the presence and components of rectal wall thickening, particularly the muscularis propria, and may be useful to distinguish SRUS from other conditions such as invasive cancer.11
Treatment
Asymptomatic patients might not require any treatment, and in some patients, SRUS resolves spontaneously. Treatment includes improving bowel habits; consuming a high-fiber diet; using bulk laxatives, local agents, and biofeedback; and undergoing surgery. The addition of fiber as a bulking agent along with bowel habit training to reduce straining can improve symptoms in patients with mild disease. Local agents such as topical glucocorticoids and aminosalicylates are not effective. Sucralfate enemas and human fibrin sealant have been effective in small studies.13 Argon plasma coagulation (APC) has been used to treat hemorrhage from SRUS; continued treatment with APC has been associated with symptomatic and endoscopic improvement.14
Behavioral therapy or biofeedback is the first line of therapy for those with more-severe disease, and it improves symptoms in more than 50% of patients; ulcer healing, however, is seen in a minority of patients. Behavioral therapy aims at bowel habit training with normalization of pelvic floor coordination. Jarrett and associates demonstrated that biofeedback resulted in improved rectal blood flow, which was associated with a successful clinical outcome.7
Surgery is indicated in symptomatic patients who have severe disease and who do not respond to medical or biofeedback therapy, but the best surgical option depends upon the underlying anatomic pathology, and every patient must be assessed individually.15 Surgical procedures include excision of the ulcer, low anterior resection, colostomy, or anterior resection with rectopexy.10 It is difficult to compare surgical treatments for SRUS because of the small number of patients in surgical series and the variety of anatomic pathology that underlies SRUS.10
STERCORAL ULCERS OF THE COLON
Stercoral ulcers result from pressure necrosis of the mucosa caused by the direct effect of a hard fecal mass (scybalum). Over time, the pressure of the scybalum results in local ischemic necrosis and ulceration and can eventuate in perforation. These ulcers are rare and usually asymptomatic until they manifest with lower gastrointestinal bleeding or colonic perforation. Fecal disimpaction occasionally precipitates rectal hemorrhage when the scybalum is removed, along with an adherent blood vessel in the subjacent ulcer crater. Maurer and coworkers observed that 3.2% of colonic perforations in their series were caused by stercoral ulcers.16
Chronic constipation is the major risk factor for stercoral ulceration and, although described in patients of all ages, it is more common among elderly patients with clinical features that may be associated with constipation.17 Although constipation and fecal impaction are observed commonly, complications of stercoral ulceration are relatively uncommon. Serpell and Nicholls reviewed 64 cases of stercoral perforation of the colon. The median age of these patients was 60 years, and 23% of them were nursing home residents. Factors that increase constipation and formation of a scybalum, such as antacids containing aluminum hydroxide, use of narcotic analgesics, constipating sedatives and psychiatric medications, and chronic renal failure, were observed in patients who developed stercoral ulceration.18 Why a stercoral ulcer develops is unclear, although implicated factors predisposing the left colon to ulceration include dehydrated and hard feces, a narrow-diameter colon with high pressure, and relatively poor blood supply.
Patients with perforated ulcers usually present with peritonitis and findings of an acute abdomen.18 The scybalum sometimes is palpable as an abdominal mass. Plain films of the abdomen might demonstrate pneumoperitoneum, fecal loading, or calcified scybala. Nonperforating ulcers can manifest with lower gastrointestinal bleeding. Caution must be used in performing disimpaction in patients with hard fecal masses in the rectum, because removing the mass can result in severe hemorrhage if the underlying blood vessel in the ulcer base is torn during removal.
The antimesenteric border of the colon is most commonly involved, usually in the sigmoid or proximal rectum. Ulcers usually are large, irregular, and sharply demarcated, and they may be single or multiple. Ulcers conform to the contour of the impacted scybala and result from ischemic pressure necrosis. A rounded or ovoid perforation may be seen in the center of the ulcer. Necrotic colonic mucosa with acute and chronic inflammation is noted on histology.17 Differential diagnosis includes spontaneous colonic perforation, malignancy, ischemia, and infection.
Perforated stercoral ulcers require emergency laparotomy with resection of the affected colonic segment. A Hartmann’s operation is the preferred procedure, and along with extensive peritoneal lavage, it is associated with a lower mortality than other surgical procedures.16,18 Nonperforating stercoral ulcers might respond to antibiotics and aggressive treatment of constipation, although surgical resection remains the only definitive treatment.
ULCERATION INDUCED BY NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
NSAIDs are among the most frequently administered drugs in the world, and their adverse side effects involve not only the stomach and duodenum but also the distal portions of the small intestine and colon. Gastroduodenal risks of NSAIDs are well known, but lower gastrointestinal tract risks of NSAIDs occur with similar frequency.19,20 The gastroduodenal effects of NSAIDs are discussed in Chapter 51.
Clinical Features
NSAIDs cause small intestinal and colonic inflammation and ulceration and have a wide spectrum of manifestations from clinically silent subtle mucosal changes to significant ulceration and overt bleeding or intestinal obstruction.20,21 Inflammation secondary to NSAIDs can cause subtle changes known as NSAID enteropathy, in which there is increased intestinal permeability, inflammation, and subtle bleeding. The clinical picture often is silent and undiagnosed unless it progresses to manifest with anemia and hypoalbuminemia. More-overt presentations include weight loss, anemia, diarrhea, overt bleeding, and perforation.20,22 Symptoms of partial small bowel obstruction such as vomiting and colicky abdominal pain may be seen secondary to the development of diaphragm-like strictures. Laboratory evaluation often is notable for hypoalbuminemia and iron deficiency anemia.
Pathology
Autopsy findings of NSAID users established that small intestinal ulcerations distal to the duodenum were prevalent.22 Of 713 patients studied, NSAIDs had been prescribed to 249 in the six months before death; 8.4% of the NSAID users had ulcerations of the small intestine compared with only 0.6% of the NSAID nonusers.22 Although no information is available regarding morbidity caused by NSAIDs during life, three of the NSAID users died of small intestinal perforation.22
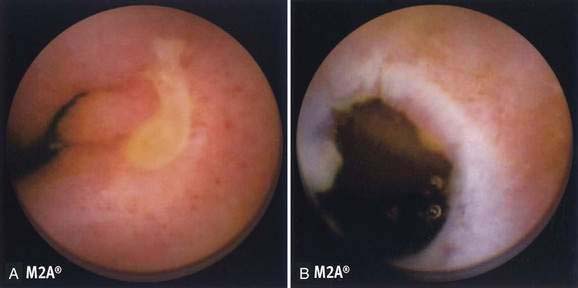
The pathologic appearance of NSAID-induced ulceration is nonspecific: Ulcerations can be single or multiple and range from tiny punched-out ulcers to confluent areas of deep ulcer with stricture formation. Capsule endoscopy has shown abnormalities ranging from reddening of the mucosa to erosions, ulcers, and active bleeding (Fig. 115-3A).23 The intervening mucosa is normal. NSAID ulcers cannot be distinguished from nonspecific or idiopathic intestinal ulcerations on the basis of their gross or microscopic pathologic appearance. Ulcerations rarely are associated with diaphragm-like strictures in patients with long-standing NSAID use, an association referred to as diaphragm disease; diaphragms are thin, 2- to 4-mm thick septae, concentric strictures that comprise mucosa and submucosa with or without submucosal fibrosis (Fig. 115-3B).
Pathogenesis
The mechanisms of NSAID-induced injury to the small intestine are not completely understood, but may involve both systemic and local events (Fig. 115-4). NSAIDs appear to cause a disturbance in the microcirculation of the villi, leading to the loss of epithelial cells.24,25 Initially, endothelial projections develop at the tip of the villus vascular arcade. Blood flow slows and stasis develops in the capillaries at the tip of the villus, occluding vascular lumens. Endothelial cells become vacuolated, with further stasis of blood flow and sloughing of the overlying epithelial cells.24 It is not clear if this is mediated through cyclooxygenase (COX)-1 or COX-2 pathways. The topical effects of NSAIDs appear to require prolonged exposure of the drug to the mucosa, and this may be effected by the enterohepatic circulation of NSAIDs. The initial injury progresses to mucosal barrier dysfunction. Indeed, Bjarnason demonstrated that NSAIDs cause increased intestinal permeability in humans by showing loss of chromium-51–labeled proteins into the intestine.26 In another study, 33 of 49 (67%) patients taking oral NSAIDs were found to have intestinal inflammation, measured by scintigraphic assessment of accumulation of indium-111–labeled white blood cells in the small intestine and by fecal excretion of indium-111.27 Nineteen of the 32 patients who also underwent simultaneous scanning with 99mTc-labeled red blood cells showed blood loss at sites identical to where intestinal inflammation was demonstrated. Loss of mucosal integrity allows luminal contents, including bile acids, pancreatic secretions, bacteria, and food antigens, to enter the mucosa. This process results in neutrophil chemotaxis with nonspecific inflammation and ulceration as a systemic response to the initial injury.
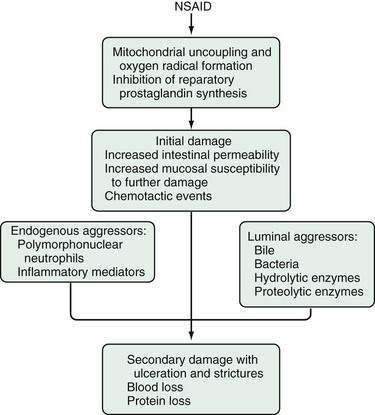
Figure 115-4. Hypothetical sequence of events involved in the pathogenesis of nonsteroidal anti-inflammatory drug (NSAID) enteropathy.
(From Aabakken L. Small-bowel side-effects of non-steroidal anti-inflammatory drugs. Eur J Gastroenterol Hepatol 1999; 11(4):383-388.
Although the role of COX-1 and COX2 has not been clearly defined in NSAID-induced injury to the small intestine, it is clear that COX-2 inhibitors cause less deleterious clinical effects on the small intestine than do COX-1 inhibitors. Using capsule endoscopy, Goldstein and colleagues found significantly less small intestinal injury in healthy subjects taking a selective COX-2 inhibitor (celecoxib) than in those who were taking ibuprofen plus omeprazole.28
Diagnosis
Despite the use of esophagogastroduodenoscopy (EGD), colonoscopy, and barium contrast studies, no source of blood loss is found in one half of patients with iron deficiency anemia who are taking NSAIDs. Capsule and double balloon enteroscopy, however, have significantly changed the diagnosis and management of small intestine ulceration and diaphragm disease (see Fig. 115-3).23,29 Tibble and associates demonstrated that fecal excretion of calprotectin, a nondegraded neutrophil cytosolic protein, can be used to assess intestinal inflammation, and therefore that this test might be practical to diagnose NSAID enteropathy. Fecal calprotectin levels correlate with fecal excretion of indium-111.30 Maiden showed that after two weeks of diclofenac, 75% of volunteers had increased fecal calprotectin levels and 68% showed injury on capsule enteroscopy.23 Thus, NSAIDs can cause injury and ulceration throughout the gastrointestinal tract. This observation has led to the recommendation that NSAIDs be discontinued before extensive evaluation of patients with obscure gastrointestinal bleeding because of the likelihood that these agents are responsible for the blood loss.
Treatment
Although avoidance of NSAIDs is the most effective therapy for NSAID enteropathy, experimental studies show that metronidazole reduces inflammation and occult blood loss without changing intestinal permeability in patients with NSAID enteropathy.31 Sulfasalazine also has been shown to reduce intestinal inflammation, as measured by fecal indium-labeled neutrophil excretion, suggesting that active mediators of inflammation as well as anaerobic organisms normally found in the small intestine might play a role in the pathogenesis of NSAID enteropathy.32 Recommendations on the use of such antibiotics to treat patients with NSAID-induced enteropathy have not been formalized.
DIFFUSE ULCERATIONS
DEFINITIONS
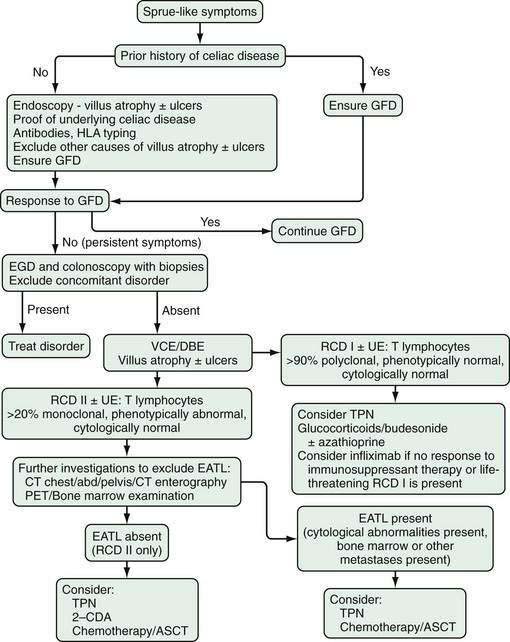
Refractory celiac disease itself encompasses a heterogeneous group of patients and is discussed more fully in Chapter 104. For the purposes of this discussion, refractory celiac disease refers to patients with small intestinal histology or antibodies consistent with celiac disease; severe, persistent malabsorption, often with diffuse small intestinal ulcerations (ulcerative enteritis) despite strict adherence to a gluten-free diet for longer than one year, or when severe symptoms require intervention independent of the duration of a gluten-free diet; and no overt lymphoma. Other causes of villus atrophy, malabsorption, and diarrhea must be excluded.33,34 Two types of refractory celiac disease (RCD) are recognized: RCD I, where there is a normal expression of T-cell lymphocyte surface markers, and RCD II, where the T cells are aberrant. The T cells in RCD II are immunophenotypically abnormal but cytologically normal and do not form tumor masses.35
Continued clonal expansion of the abnormal T-cell population in RCD II eventually leads to enteropathy-associated T-cell lymphoma (EATL). EATL can complicate established celiac disease or can manifest de novo with multiple intestinal ulcerations and malabsorption in patients without previously known underlying celiac disease but with small intestinal biopsies that demonstrate villus atrophy. EATL is recognized by the World Health Organization International Classification Project as a specific disorder and is discussed in greater depth in Chapter 29.
BACKGROUND
The association of malabsorption and lymphoma was reported first in 1937. In the 1980s, evidence in favor of underlying celiac disease as the predisposing factor for ulcerative enteritis, refractory celiac disease, and EATL in many patients emerged from registries, genotyping studies, and literature reviews. As technologic advances occurred, molecular biology studies, polymerase chain reaction (PCR) amplification, immunophenotypical analysis, and other methods were used to link celiac disease, refractory celiac disease, ulcerative enteritis, and EATL via intraepithelial T-cell lymphocyte abnormalities.36,37
The repertoire of intraepithelial and lamina propria lymphocytes is polyclonal before and shortly after birth. Because of interactions with the intestinal microenvironment, including food antigens and microbes, the repertoire becomes oligoclonal by the time adulthood is reached.38 In RCD I, the intraepithelial T-cell phenotype is similar to that of uncomplicated celiac disease: polyclonal with normal expression of cell surface markers. Less than 10% of the T cells are aberrant in intestinal biopsy specimens of RCD I, and when more than 20% of the T cells become immunophenotypically abnormal, RCD I is said to have progressed to RCD II.39
The expansion and hyperplasia of T cells in active celiac disease and RCD is at least partially regulated by interleukin-15 (IL-15). In active celiac disease, IL-15 synthesis is up-regulated in the intestinal mucosa. Similarly in RCD, IL-15 is markedly overexpressed in intestinal epithelial cells and in the lamina propria, inducing clonal expansion of intraepithelial lymphocytes and initiating T-cell cytotoxicity against intestinal epithelial cells.40 IL-15 has been shown to deliver signals that impair SMAD3–dependent transforming growth factor-β (TGF-β) signaling in T lymphocytes, which leads to the promotion and continuation of intestinal inflammation.41
Cytogenetic studies have demonstrated that the intraepithelial T lymphocytes in RCD II possess a partial trisomy of 1q22q44.42 This gain on chromosome 1q, also shown to be present in 16% of cases of EATL, may be an early event on the way to malignant transformation of T cells in RCD. EATL tumor cells have chromosomal imbalances in 87% of cases, 58% showing gains on chromosome 9q and 16% on chromosome 1q; recurrent genetic losses occur on chromosomes 8p, 13q, and 9p. Chromosomes 9q and 8p are of particular importance because 9q has a gene implicated in lymphomagenesis, and 8p has a gene implicated in apoptosis. With gains and losses of important genetic material, increased production and decreased apoptosis of intraepithelial T cells might contribute to the accumulation in RCD and subsequent development of EATL.43
RCD II is said to have developed once more than 20% of the T cells are aberrant. The intraepithelial lymphocytes in RCD II are phenotypically abnormal but not yet cytologically abnormal. Intestinal ulcers often are present, especially in the jejunum, defining the clinical syndrome of ulcerative enteritis, but no tumor masses are seen. RCD II with or without ulcerative enteritis is considered a cryptic lymphoma and represents a continuum with EATL. Monoclonal expansion continues, and once tumor masses are present or there is radiologic or bone marrow evidence of cytologically abnormal T cells, EATL is said to have developed.33
Celiac disease is associated with the HLA-DQ2 and HLA-DQ8 alleles, and both RCD II and EATL have been associated with HLA-DQ2 homogyzosity.44 Genetic variants in the MYO9B gene on chromosome 19 also have been linked to celiac disease, and genetic studies have reported a single nucleotide polymorphism, rs7259292 T allele in the MYO9B gene, to be present more often in patients with RCD II and EATL.45
CLINICAL FEATURES
Physical examination reveals profound weight loss, cachexia and signs of severe malabsorption, steatorrhea, and protein-losing enteropathy. Abdominal tenderness may be mild, diffuse, or severe. Hepatomegaly and splenic atrophy may be present. Peripheral lymphadenopathy is unusual, but patients who have developed lymphoma can have an abdominal mass. Dermatitis herpetiformis, a condition usually associated with celiac disease, rarely may be observed with RCD. Signs and symptoms of anemia can occur as a result of chronic nutritional deficiency or acute gastrointestinal hemorrhage. Intestinal perforation typically leads to signs of peritonitis. Patients with intestinal obstruction might have acute vomiting and abdominal distention.35,37,44,46
DIAGNOSIS
Laboratory
Laboratory abnormalities reflect the diarrhea, severe malabsorption, and complications of the disease. Findings include iron deficiency or macrocytic anemia, prolongation of the prothrombin time, electrolyte abnormalities consistent with the degree of dehydration, hypoalbuminemia, hypocalcemia, hypomagnesemia, hypocholesterolemia, and low serum carotene levels. Stool abnormalities include increased volume, mild to severe steatorrhea, increased fecal α1-antitrypsin excretion, and a positive fecal occult blood test. The d-xylose test usually is abnormal.33,34,37
Except for de novo EATL, where serology typically is negative at the time of diagnosis, serum antibodies for antigliadin and tissue transglutaminase (tTG) usually are present in ulcerative enteritis, RCD, or EATL. A positive serologic assay for tTG antibodies reverts to negative on a gluten-free diet, similar to the typical serologic pattern of uncomplicated celiac disease.33,35
Although not necessary for diagnosis, HLA-DQ2 or DQ8 genes are found in most patients with one of these clinical syndromes. HLA-DQ2 homozygosity has been linked to an increased risk of developing RCD II and EATL.44,45
Radiology
Radiologic abnormalities are common in these clinical entities. Computed tomography (CT) scanning can demonstrate characteristic findings of celiac disease, such as decrease or loss of jejunal folds, thickening and separation of small bowel loops, increased ileal folds or reversal of the jejunoileal fold pattern, small bowel dilatation and stricturing, intussusception, increased number of small mesenteric vessels, enlarged or cavitated mesenteric lymph nodes, hyposplenism, or metastatic disease. Splenomegaly tends to occur in celiac disease and RCD I.47 CT enterography additionally may show jejunal or ileal wall thickening with mucosal hyperenhancement. The tracer fluorine-18 fluorodeoxyglucose (18F-FDG), a glucose analog taken up by rapidly growing tumor cells and used during positron emission tomography (18F-FDG PET), was shown to be more sensitive and specific than CT in detecting EATL in patients with RCD.48
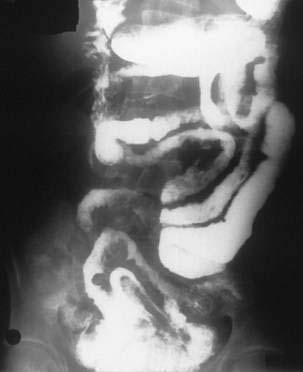
Barium contrast study of the small bowel or enteroclysis can show jejunization of the ileum, separation and thickening of small intestinal loops, intussusceptions, ulcerations, or a mass (Fig. 115-5). Strictures may be single or multiple and appear as areas of luminal narrowing alternating with more dilated portions of small bowel. Abnormalities tend to be more noticeable in the proximal small intestine.37
Endoscopy
Because the ulcers usually are located distal to the duodenum, direct visualization of the jejunum and ileum is necessary and can best be accomplished via videocapsule endoscopy or double-balloon enteroscopy. The major advantages of videocapsule endoscopy are ease of use and visualization of the entire small intestine in 80% to 90% of patients during the eight-hour acquisition time. High-quality videocapsule endoscopy images can demonstrate villus atrophy, loss of folds, scalloping, whitish villi, and small intestinal erosions, ulcerations, nodularity, masses, or strictures (Fig. 115-6). The capsule needs to be used cautiously in patients with known or suspected strictures.49
The major advantage of double-balloon enteroscopy is examination of the entire small intestine with the ability to take biopsies. Double-balloon enteroscopy can detect or exclude RCD I and RCD II, with or without ulcers (ulcerative enteritis), and EATL. Findings are similar to those seen during videocapsule endoscopy.50 During enteroscopy, biopsies should be taken of abnormal as well as endoscopically normal-appearing intervening mucosa.
Occasional patients with ulcerative enteritis, RCD, or EATL also have gastric or colonic ulcers that can be identified on routine endoscopy. Biopsies of normal-appearing gastric and colonic mucosa should be obtained, because abnormal lymphocytes have been demonstrated in these disorders throughout the gastrointestinal tract.51
Pathology
Ulcerative enteritis is more common in RCD II than in RCD I. The ulcers in ulcerative enteritis are diffuse, more commonly located in the jejunum and ileum than in the duodenum, range in size from 1 mm to 3.5 cm, are rarely solitary, and are well circumscribed. Some of the ulcers are superficial, extending only to the muscularis mucosa, but usually they extend to the muscularis propria and occasionally through it, thus causing perforation.35,36 Histologically, they are benign.
Once more than 20% of the intraepithelial T lymphocytes in biopsy specimens are immunophenotypically abnormal, as determined by flow cytometry quantification, RCD II is said to have developed (median 52%, range 27% to 94%).39 In contrast to RCD I, the aberrant T cells in the majority of RCD II patients express intracytoplasmic CD3 and surface CD103, but they lack cell surface expression of the TCR complex, CD3, CD4, and CD8; rarely, the abnormal T cells are surface CD3− and CD8+.39 PCR analysis of the T-cell receptor γ-chain gene further demonstrates monoclonality of the T lymphocytes consistent with RCD II being a cryptic T-cell lymphoma. The T cells in RCD II appear cytologically normal.33,51,52 Once the abnormal intraepithelial clone has been identified, the risk of progression to EATL is very high and should warrant a thorough investigation for EATL, including endoscopic and radiologic studies52 (Fig. 115-7).
When cytologic abnormalities appear, RCD II has progressed onto EATL. Overt T-cell lymphoma can manifest as multiple ulcers, as an ulcerated mass, or as extraintestinal disseminated disease. Tumor cells can be demonstrated in both ulcerated and nonulcerated areas of the intestine. In EATL, the majority of abnormal T lymphocytes are large-to-medium-sized T cells; less commonly they are small and monomorphic. Cell markers are similar to those seen in RCD II.33,43,53
Normal-appearing intestinal mucosa adjacent to or distant from either a benign- or malignant-appearing ulcer or mass can show normal villus architecture or partial or total villus atrophy with crypt hyperplasia and an increased number of intraepithelial lymphocytes with immunophenotypic characteristics as described earlier (Fig. 115-8). Other causes of villus atrophy such as tropical sprue, giardiasis, common variable immunodeficiency syndrome, or autoimmune enteropathy must be excluded.33 Features of inflammatory bowel disease are absent, and there is no evidence of an infectious process.
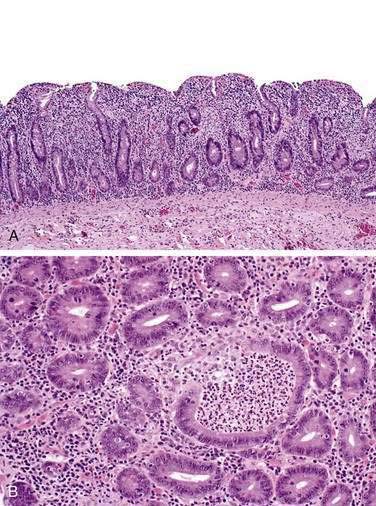
Histology of endoscopically normal gastric and colonic mucosa in patients with RCD II demonstrates widespread distribution of abnormal lymphocytes throughout the gastrointestinal tract; associated lymphocytic gastritis and lymphocytic and collagenous colitis have been reported. In patients with RCD II and concurrent lymphocytic gastritis or lymphocytic colitis, gastric and colonic T cells are monoclonal, immunophenotypically abnormal, and identical to T cells in the duodenum and jejunum. T cells in gastric and colonic biopsies in patients with RCD II without lymphocytic gastritis or colitis tend to have a polyclonal gene rearrangement.51
TREATMENT AND PROGNOSIS
With the recognition that RCD I and II with or without ulcerative enteritis are a continuum on the spectrum of developing aberrancy in T cells that eventually leads to EATL if unchecked, recent therapy has targeted the abnormal T-cell population at an earlier stage with better outcomes over the past five years.35
After gluten withdrawal failure in patients with RCD I with or without ulcerative enteritis that is not life threatening, glucocorticoids achieve a response in 50% to 75% of patients.54 In patients who do not initially respond to glucocorticoids or who become dependent on them, the addition of azathioprine usually is effective therapy to induce or maintain a response35,55 (see Fig. 115-7).
Case reports using infliximab in patients with RCD I and life-threatening symptoms have shown improvement in symptoms, with duodenal histology reverting to near normal.56,57
Once aberrant monoclonal T cells are present, RCD II is considered to have developed. Therapy should be more aggressive and directed against the abnormal T-cell population. Cladribine, 2-chlorodeoxyadenosine (2-CDA), a cytotoxic chemotherapeutic agent with activity against lymphocytes, was used in an open-label, prospective trial in 17 patients with RCD II. Six patients had a clinical improvement, 10 patients (five with ulcerative enteritis) had histologic improvement, and six patients had a significant decrease in aberrant T cells. Seven patients developed EATL and subsequently died. This medication showed promise in stabilizing patients but did not prevent the development of EATL. There also is the additional concern of accelerating the development of EATL or inducing a secondary malignancy.35,58
After conditioning with the chemotherapeutic agents fludarabine and melphalan, seven patients with RCD II underwent autologous stem cell transplant (ASCT) in an open-label, prospective study. After a mean follow-up of 15.5 months, six of the seven patients showed significant improvement in their symptoms along with a reduction in the aberrant T cells in duodenal biopsies. This pilot study suggests that treating RCD II patients with ASCT might result in long-term improvement.46
Once EATL has developed, prognosis is poor. Options are limited to chemotherapy with or without bone marrow transplantation. Chemotherapy is often poorly tolerated secondary to poor nutritional status and complications during treatment. Aggressive chemotherapy followed by ASCT in patients with early-stage disease resulted in better survival59,60 than did treatment of patients with later stage disease.61
Due to widespread T-cell involvement of the gastrointestinal tract in RCD II and EATL, surgical options are limited. Exploratory laparotomy may be necessary if a complication, such as perforation, obstruction, or hemorrhage occurs. Elective surgical resection usually is neither indicated nor possible, especially in a severely malnourished patient. In selected EATL patients with localized involvement, however, surgical resection followed by chemotherapy or ASCT might offer a reasonable chance for survival.35,61
In the future, novel therapeutic options likely will include allogeneic stem cell transplantation, better preconditioning regimens, and biologic agents directed against proinflammatory mediators, such as interleukin-1540 or T-cell markers such as CD52.62
Abdallah H, Leffler D, Dennis M, Kelly CP. Refractory celiac disease. Curr Gastroenterol Rep. 2007;9:401-5. (Ref 34.)
Al-Toma A, Verbeek WH, Hadithi M, et al. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: Retrospective evaluation of single-centre experience. Gut. 2007;56:1373-8. (Ref 35.)
Al-toma A, Visser OJ, van Roessel HM, et al. Autologous hematopoietic stem cell transplantation in refractory celiac disease with aberrant T cells. Blood. 2007;109:2243-9. (Ref 46.)
Biagi F, Lorenzini P, Corazza GR. Literature review on the clinical relationship between ulcerative jejunoileitis, coeliac disease, and enteropathy-associated T-cell. Scand J Gastroenterol. 2000;35:785-90. (Ref 36.)
Bishton MJ, Haynes AP. Combination chemotherapy followed by autologous stem cell transplant for enteropathy-associated T cell lymphoma. Br J Haematol. 2007;136:111-13. (Ref 59.)
Boydstun JSJr, Gaffey TA, Bartholomew LG. Clinicopathologic study of nonspecific ulcers of the small intestine. Dig Dis Sci. 1981;26:911-16. (Ref 1.)
Daum S, Cellier C, Mulder CJ. Refractory coeliac disease. Best Pract Res Clin Gastroenterol. 2005;19:413-24. (Ref 33.)
Daum S, Wahnschaffe U, Glasenapp R, et al. Capsule endoscopy in refractory celiac disease. Endoscopy. 2007;39:455-8. (Ref 49.)
Fortun PJ, Hawkey CJ. Nonsteroidal antiinflammatory drugs and the small intestine. Curr Opin Gastroenterol. 2007;23:134-41. (Ref 21.)
Gekas P, Schuster MM. Stercoral perforation of the colon: Case report and review of the literature. Gastroenterology. 1981;80:1054-8. (Ref 17.)
Hadithi M, Al-toma A, Oudejans J, et al. The value of double-balloon enteroscopy in patients with refractory celiac disease. Am J Gastroenterol. 2007;102:987-96. (Ref 50.)
Laine L, Connors LG, Reicin A, et al. Serious lower gastrointestinal clinical events with nonselective NSAID or coxib use. Gastroenterology. 2003;124:288-92. (Ref 20.)
Maiden L, Thjodleifsson B, Theodors A, et al. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology. 2005;128:1172-8. (Ref 23.)
Sharara AI, Azar C, Amr SS, et al. Solitary rectal ulcer syndrome: Endoscopic spectrum and review of the literature. Gastrointest Endosc. 2005;62:755-62. (Ref 11.)
Tjandra JJ, Fazio VW, Church JM, et al. Clinical conundrum of solitary rectal ulcer. Dis Colon Rectum. 1992;35:227-34. (Ref 2.)
Whittle BJ. Mechanisms underlying intestinal injury induced by anti-inflammatory COX inhibitors. Eur J Pharmacol. 2004;500:427-39. (Ref 24.)
1. Boydstun JSJr, Gaffey TA, Bartholomew LG. Clinicopathologic study of nonspecific ulcers of the small intestine. Dig Dis Sci. 1981;26:911-16.
2. Tjandra JJ, Fazio VW, Church JM, et al. Clinical conundrum of solitary rectal ulcer. Dis Colon Rectum. 1992;35:227-34.
3. Halligan S, Nicholls RJ, Bartram CI. Proctographic changes after rectopexy for solitary rectal ulcer syndrome and preoperative predictive factors for a successful outcome. Br J Surg. 1995;82:314-7.
4. Womack NR, Williams NS, Holmfield JH, Morrison JF. Pressure and prolapse—the cause of solitary rectal ulceration. Gut. 1987;28:1228-33.
5. Eckardt VF, Kanzler G, Remmele W. Anorectal ergotism: another cause of solitary rectal ulcers. Gastroenterology. 1986;91:1123-7.
6. Crowe J, Stellato TA. Radiation-induced solitary rectal ulcer. Dis Colon Rectum. 1985;28:610-12.
7. Jarrett ME, Emmanuel AV, Vaizey CJ, Kamm MA. Behavioural therapy (biofeedback) for solitary rectal ulcer syndrome improves symptoms and mucosal blood flow. Gut. 2004;53:368-70.
8. du Boulay CE, Fairbrother J, Isaacson PG. Mucosal prolapse syndrome—a unifying concept for solitary ulcer syndrome and related disorders. J Clin Pathol. 1983;36:1264-8.
9. Madigan MR, Morson BC. Solitary ulcer of the rectum. Gut. 1969;10:871-81.
10. Torres C, Khaikin M, Bracho J, et al. Solitary rectal ulcer syndrome: clinical findings, surgical treatment, and outcomes. Int J Colorectal Dis. 2007;22:1389-93.
11. Sharara AI, Azar C, Amr SS, et al. Solitary rectal ulcer syndrome: endoscopic spectrum and review of the literature. Gastrointest Endosc. 2005;62:755-62.
12. Martin CJ, Parks TG, Biggart JD. Solitary rectal ulcer syndrome in Northern Ireland. 1971-1980. Br J Surg. 1981;68:744-7.
13. Zargar SA, Khuroo MS, Mahajan R. Sucralfate retention enemas in solitary rectal ulcer. Dis Colon Rectum. 1991;34:455-7.
14. Stoppino V, Cuomo R, Tonti P, et al. Argon plasma coagulation of hemorrhagic solitary rectal ulcer syndrome. J Clin Gastroenterol. 2003;37:392-4.
15. Ortega AE, Klipfel N, Kelso R, et al. Changing concepts in the pathogenesis, evaluation, and management of solitary rectal ulcer syndrome. Am Surg. 2008;74:967-72.
16. Maurer CA, Renzulli P, Mazzucchelli L, et al. Use of accurate diagnostic criteria may increase incidence of stercoral perforation of the colon. Dis Colon Rectum. 2000;43:991-8.
17. Gekas P, Schuster MM. Stercoral perforation of the colon: case report and review of the literature. Gastroenterology. 1981;80:1054-8.
18. Serpell JW, Nicholls RJ. Stercoral perforation of the colon. Br J Surg. 1990;77:1325-9.
19. Wilcox CM, Clark WS. Association of nonsteroidal antiinflammatory drugs with outcome in upper and lower gastrointestinal bleeding. Dig Dis Sci. 1997;42:985-9.
20. Laine L, Connors LG, Reicin A, et al. Serious lower gastrointestinal clinical events with nonselective NSAID or coxib use. Gastroenterology. 2003;124:288-92.
21. Fortun PJ, Hawkey CJ. Nonsteroidal antiinflammatory drugs and the small intestine. Curr Opin Gastroenterol. 2007;23:134-41.
22. Allison MC, Howatson AG, Torrance CJ, et al. Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N Engl J Med. 1992;327:749-54.
23. Maiden L, Thjodleifsson B, Theodors A, et al. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology. 2005;128:1172-8.
24. Whittle BJ. Mechanisms underlying intestinal injury induced by anti-inflammatory COX inhibitors. Eur J Pharmacol. 2004;500:427-39.
25. Adebayo D, Bjarnason I. Is non-steroidal anti-inflammatory drug (NSAID) enteropathy clinically more important than NSAID gastropathy? Postgrad Med J. 2006;82:186-91.
26. Bjarnason I, Williams P, So A, et al. Intestinal permeability and inflammation in rheumatoid arthritis: effects of non-steroidal anti-inflammatory drugs. Lancet. 1984;2:1171-4.
27. Bjarnason I, Zanelli G, Prouse P, et al. Blood and protein loss via small-intestinal inflammation induced by non-steroidal anti-inflammatory drugs. Lancet. 1987;2:711-4.
28. Goldstein JL, Eisen GM, Lewis B, et al. Small bowel mucosal injury is reduced in healthy subjects treated with celecoxib compared with ibuprofen plus omeprazole, as assessed by video capsule endoscopy. Aliment Pharmacol Ther. 2007;25:1211-22.
29. Gross SA, Stark ME. Initial experience with double-balloon enteroscopy at a U.S. center. Gastrointest Endosc. 2008;67:890-7.
30. Tibble JA, Sigthorsson G, Foster R, et al. High prevalence of NSAID enteropathy as shown by a simple faecal test. Gut. 1999;45:362-6.
31. Bjarnason I, Hayllar J, Smethurst P, et al. Metronidazole reduces intestinal inflammation and blood loss in non-steroidal anti-inflammatory drug induced enteropathy. Gut. 1992;33:1204-8.
32. Bjarnason I, Hopkinson N, Zanelli G, et al. Treatment of non-steroidal anti-inflammatory drug induced enteropathy. Gut. 1990;31:777-80.
33. Daum S, Cellier C, Mulder CJ. Refractory coeliac disease. Best Pract Res Clin Gastroenterol. 2005;19:413-24.
34. Abdallah H, Leffler D, Dennis M, Kelly CP. Refractory celiac disease. Curr Gastroenterol Rep. 2007;9:401-5.
35. Al-Toma A, Verbeek WH, Hadithi M, et al. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: retrospective evaluation of single-centre experience. Gut. 2007;56:1373-8.
36. Biagi F, Lorenzini P, Corazza GR. Literature review on the clinical relationship between ulcerative jejunoileitis, coeliac disease, and enteropathy-associated T-cell. Scand J Gastroenterol. 2000;35:785-90.
37. Cellier C, Delabesse E, Helmer C, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet. 2000;356:203-8.
38. Probert CS, Saubermann LJ, Balk S, Blumberg RS. Repertoire of the alpha beta T-cell receptor in the intestine. Immunol Rev. 2007;215:215-25.
39. Verbeek WH, Goerres MS, von Blomberg BM, et al. Flow cytometric determination of aberrant intra-epithelial lymphocytes predicts T-cell lymphoma development more accurately than T-cell clonality analysis in refractory celiac disease. Clin Immunol. 2008;126:48-56.
40. Mention JJ, Ben Ahmed M, Begue B, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology. 2003;125:730-45.
41. Benahmed M, Meresse B, Arnulf B, et al. Inhibition of TGF-β signaling by IL-15: a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology. 2007;132:994-1008.
42. Verkarre V, Romana SP, Cellier C, et al. Recurrent partial trisomy 1q22-q44 in clonal intraepithelial lymphocytes in refractory celiac sprue. Gastroenterology. 2003;125:40-6.
43. Zettl A, Ott G, Makulik A, et al. Chromosomal gains at 9q characterize enteropathy-type T-cell lymphoma. Am J Pathol. 2002;161:1635-45.
44. Al-Toma A, Goerres MS, Meijer JW, et al. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol. 2006;4:315-19.
45. Wolters VM, Verbeek WH, Zhernakova A, et al. The MYO9B gene is a strong risk factor for developing refractory celiac disease. Clin Gastroenterol Hepatol. 2007;5:1399-405. 405 e1-2
46. Al-toma A, Visser OJ, van Roessel HM, et al. Autologous hematopoietic stem cell transplantation in refractory celiac disease with aberrant T cells. Blood. 2007;109:2243-9.
47. Mallant M, Hadithi M, Al-Toma AB, et al. Abdominal computed tomography in refractory coeliac disease and enteropathy associated T-cell lymphoma. World J Gastroenterol. 2007;13:1696-700.
48. Hadithi M, Mallant M, Oudejans J, et al. 18F-FDG PET versus CT for the detection of enteropathy-associated T-cell lymphoma in refractory celiac disease. J Nucl Med. 2006;47:1622-7.
49. Daum S, Wahnschaffe U, Glasenapp R, et al. Capsule endoscopy in refractory celiac disease. Endoscopy. 2007;39:455-8.
50. Hadithi M, Al-toma A, Oudejans J, et al. The value of double-balloon enteroscopy in patients with refractory celiac disease. Am J Gastroenterol. 2007;102:987-96.
51. Verkarre V, Asnafi V, Lecomte T, et al. Refractory coeliac sprue is a diffuse gastrointestinal disease. Gut. 2003;52:205-11.
52. Cellier C, Cerf-Bensussan N. Treatment of clonal refractory celiac disease or cryptic intraepithelial lymphoma: a long road from bench to bedside. Clin Gastroenterol Hepatol. 2006;4:1320-1.
53. Farstad IN, Johansen FE, Vlatkovic L, et al. Heterogeneity of intraepithelial lymphocytes in refractory sprue: potential implications of CD30 expression. Gut. 2002;51:372-8.
54. Brar P, Lee S, Lewis S, et al. Budesonide in the treatment of refractory celiac disease. Am J Gastroenterol. 2007;102:2265-9.
55. Goerres MS, Meijer JW, Wahab PJ, et al. Azathioprine and prednisone combination therapy in refractory coeliac disease. Aliment Pharmacol Ther. 2003;18:487-94.
56. Costantino G, della Torre A, Lo Presti MA, et al. Treatment of life-threatening type I refractory coeliac disease with long-term infliximab. Dig Liver Dis. 2008;40:74-7.
57. Gillett HR, Arnott ID, McIntyre M, et al. Successful infliximab treatment for steroid-refractory celiac disease: a case report. Gastroenterology. 2002;122:800-5.
58. Al-Toma A, Goerres MS, Meijer JW, et al. Cladribine therapy in refractory celiac disease with aberrant T cells. Clin Gastroenterol Hepatol. 2006;4:1322-7.
59. Bishton MJ, Haynes AP. Combination chemotherapy followed by autologous stem cell transplant for enteropathy-associated T cell lymphoma. Br J Haematol. 2007;136:111-13.
60. Rongey C, Micallef I, Smyrk T, Murray J. Successful treatment of enteropathy-associated T cell lymphoma with autologous stem cell transplant. Dig Dis Sci. 2006;51:1082-6.
61. Al-Toma A, Verbeek WH, Visser OJ, et al. Disappointing outcome of autologous stem cell transplantation for enteropathy-associated T-cell lymphoma. Dig Liver Dis. 2007;39:634-41.
62. Vivas S, Ruiz de Morales JM, Ramos F, Suarez-Vilela D. Alemtuzumab for refractory celiac disease in a patient at risk for enteropathy-associated T-cell lymphoma. N Engl J Med. 2006;354:2514-15.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 115 Ulcers of the Small and Large Intestine
ISOLATED ULCERS
NONSPECIFIC OR IDIOPATHIC SMALL INTESTINAL ULCERATION
Solitary ulcers of the small intestine result from a wide variety of causes (Table 115-1). Radiation injury is a known cause of small intestinal ulceration and is discussed in Chapter 39. Solitary ulcers beyond the duodenum that cannot be explained on the basis of any known etiology are referred to as nonspecific or idiopathic intestinal ulcers. Such solitary nonspecific ulcers are rare, with an incidence of 4 per 100,000.1
Table 115-1 Causes of Small Intestinal Ulceration
Clinical Features
In a review of the Mayo Clinic’s experience with 59 cases of small intestinal ulcers over a 22-year period ending in 1979, Boydstun and associates1 showed that 53 (89.8%) patients had no identifiable cause of ulceration. Patients ranged in age from 17 to 77 years, with most presenting in the fifth and sixth decades of life; no gender predominance was found. The most common presenting symptom was intermittent small bowel obstruction (63%). Physical findings ranged from nonspecific abdominal tenderness and distention to an acute abdomen resulting from intestinal perforation. Laboratory evaluation was notable only for anemia in one half of the patients. Radiologic studies localized the ulcer in a minority of patients.
Pathology
In the Mayo Clinic series,1 the ileum was the most common location of nonspecific ulceration (78%), and perforation (13 cases, 22%) occurred most commonly in the jejunum (78%). At surgery, 41 patients were found to have solitary ulcers, five patients had two ulcers, and six patients had more than three ulcers. Ulcer size varied between 0.3 and 5 cm. On pathologic examination, the ulcers were predominantly on the antimesenteric border of the small intestine and, in some cases, were associated with a fibrous scar that narrowed the lumen. Microscopy revealed nonspecific chronic inflammation that ended abruptly at the ulcer edge. The intervening bowel and vasculature were normal.1
SOLITARY RECTAL ULCER SYNDROME
Solitary rectal ulcer syndrome (SRUS) is an uncommon or under-reported disorder of defecation that affects patients of all ages. The term is a misnomer: Patients can present with hyperemic mucosa only, a solitary ulcer, multiple ulcers, or even a polypoid lesion resembling carcinoma.2 Regardless, the histology of SRUS is typical, showing fibromuscular obliteration of the lamina propria and smooth muscle fibers extending from a hypertrophied muscularis mucosa to the lumen.2 The diagnosis of SRUS often is delayed because of its varied endoscopic appearance and a lack of awareness of the disorder.
Pathogenesis
SRUS is a disorder of defecation, but its pathogenesis is uncertain, and it has a spectrum of disease presentations. A large subgroup of patients with SRUS strain excessively during defecation, and some have a behavioral disorder. Occult or overt rectal prolapse with paradoxical contraction of the pelvic floor during defecation appears to be involved in most patients3; evidence of inappropriate pelvic floor contraction has been shown in electromyographic and video-proctographic studies.3 It has been suggested that the rectal mucosa can be traumatized from the pressure of being prolapsed against a closed anal canal4 and that straining during defecation results in prolapse and high fecal voiding pressures that reduce local blood flow, causing ischemia and ulceration.4 The mucosa of the anterior rectal wall, 7 to 10 cm above the anal verge, is the most common area of such prolapse into the anal canal, and this corresponds to the usual location of ulceration in SRUS.
SRUS also has been associated with the use of ergotamine suppositories and is well known after radiotherapy, further supporting a pathogenic role for ischemia.5,6 Successful treatment of SRUS using biofeedback has been associated with an increase in local blood flow, additionally suggesting that SRUS may be associated with reduced rectal blood flow from impaired extrinsic autonomic cholinergic nerve activity.7
The association of SRUS and rectal prolapse, however, is neither pathogenically clear nor universal; the incidence of associated rectal prolapse varies from 13% to 94%.2 It is assumed that the ulcer develops as a result of local trauma to the apex of the prolapse, either because of manual attempts to reduce the prolapse digitally or because of contractions of the external anal sphincter when the mucosa prolapses through the anal canal.4
Du Boulay and colleagues have shown that the histology of the rectal mucosa in patients with SRUS is similar to that seen at other sites of mucosal prolapse, suggesting that prolapse of the mucosa alone rather than the entire rectal wall is important in SRUS pathogenesis.8 Ischemia results in fibromuscular obliteration of the lamina propria and the formation of an ulcer. Once the ulcer is formed, it can further intensify the urge to defecate; this urge combined with straining and changes in local blood flow causes persistent symptoms and chronic ulceration.
Clinical Features
Patients with SRUS present with varied symptoms, but most patients typically complain of passage of mucus and blood per rectum on defecation.9,10 Some patients also complain of tenesmus, straining, altered bowel habits, and the sensation of incomplete evacuation. Men and women are affected equally, and they usually present in the third and fourth decades, respectively.11 The mean duration of symptoms is long, ranging from 3.5 to 5 years, possibly reflecting a delay in diagnosis.
Diagnosis and Pathology
Diagnosis of SRUS is based on clinical symptoms, physical examination, endoscopic findings, and typical histology. Physical examination can demonstrate tenderness in the left lower quadrant. On digital rectal examination, there may be reduced anal sphincter tone and an indurated area, or thickened folds may be palpated.12 Overt rectal prolapse may be demonstrated by having the patient assume a squatting position and straining as if to have a bowel movement. Sigmoidoscopy may demonstrate single or multiple ulcers or a patch of erythematous mucosa on the anterior rectal wall within 10 cm of the anal verge. The lesion has a polypoid appearance (Fig. 115-1) in 25% to 44% of patients.2,12 Differential diagnosis includes inflammatory bowel disease, malignancy, ischemic colitis, stercoral ulcer, medication-induced ulceration, trauma, and infections, including amebiasis and secondary syphilis.11
Biopsy specimens always should be taken from the ulcer margin and from any abnormal-appearing mucosa. In 1969, Madigan and Morson9 first described the histologic features of SRUS. There is fibromuscular obliteration of the lamina propria by collagen from fibroblasts and smooth muscle fibers derived from the muscularis mucosae. The muscularis mucosae is often hypertrophied, and its fibers are in continuity with those in the lamina propria. There is no significant increase in the number of inflammatory cells. The polypoid variant is similar to the ulcerative variant except for regenerative hyperplastic changes, such as cystic dilatation and mucus cell production.11 Epithelial elements and lamina propria can be displaced into the submucosa (Fig. 115-2). This displaced tissue can then undergo cystic dilatation because of mucus retention. The misplaced and dysplastic-appearing glands may be misdiagnosed as adenocarcinoma, especially when the histologic and macroscopic features of SRUS are not recognized; at times SRUS is present in association with an carcinoma, further confusing the issue.
Histology typically provides a definitive diagnosis; however, defecography may be useful to shed light on the pathophysiology of SRUS, especially if surgery is being considered. Defecography may be used to demonstrate mucosal prolapse, intussusception, rectal prolapse, a nonrelaxing puborectalis muscle, and incomplete or delayed evacuation.11 Endorectal ultrasound can demonstrate the presence and components of rectal wall thickening, particularly the muscularis propria, and may be useful to distinguish SRUS from other conditions such as invasive cancer.11
Treatment
Asymptomatic patients might not require any treatment, and in some patients, SRUS resolves spontaneously. Treatment includes improving bowel habits; consuming a high-fiber diet; using bulk laxatives, local agents, and biofeedback; and undergoing surgery. The addition of fiber as a bulking agent along with bowel habit training to reduce straining can improve symptoms in patients with mild disease. Local agents such as topical glucocorticoids and aminosalicylates are not effective. Sucralfate enemas and human fibrin sealant have been effective in small studies.13 Argon plasma coagulation (APC) has been used to treat hemorrhage from SRUS; continued treatment with APC has been associated with symptomatic and endoscopic improvement.14
Behavioral therapy or biofeedback is the first line of therapy for those with more-severe disease, and it improves symptoms in more than 50% of patients; ulcer healing, however, is seen in a minority of patients. Behavioral therapy aims at bowel habit training with normalization of pelvic floor coordination. Jarrett and associates demonstrated that biofeedback resulted in improved rectal blood flow, which was associated with a successful clinical outcome.7
Surgery is indicated in symptomatic patients who have severe disease and who do not respond to medical or biofeedback therapy, but the best surgical option depends upon the underlying anatomic pathology, and every patient must be assessed individually.15 Surgical procedures include excision of the ulcer, low anterior resection, colostomy, or anterior resection with rectopexy.10 It is difficult to compare surgical treatments for SRUS because of the small number of patients in surgical series and the variety of anatomic pathology that underlies SRUS.10
STERCORAL ULCERS OF THE COLON
Stercoral ulcers result from pressure necrosis of the mucosa caused by the direct effect of a hard fecal mass (scybalum). Over time, the pressure of the scybalum results in local ischemic necrosis and ulceration and can eventuate in perforation. These ulcers are rare and usually asymptomatic until they manifest with lower gastrointestinal bleeding or colonic perforation. Fecal disimpaction occasionally precipitates rectal hemorrhage when the scybalum is removed, along with an adherent blood vessel in the subjacent ulcer crater. Maurer and coworkers observed that 3.2% of colonic perforations in their series were caused by stercoral ulcers.16
Chronic constipation is the major risk factor for stercoral ulceration and, although described in patients of all ages, it is more common among elderly patients with clinical features that may be associated with constipation.17 Although constipation and fecal impaction are observed commonly, complications of stercoral ulceration are relatively uncommon. Serpell and Nicholls reviewed 64 cases of stercoral perforation of the colon. The median age of these patients was 60 years, and 23% of them were nursing home residents. Factors that increase constipation and formation of a scybalum, such as antacids containing aluminum hydroxide, use of narcotic analgesics, constipating sedatives and psychiatric medications, and chronic renal failure, were observed in patients who developed stercoral ulceration.18 Why a stercoral ulcer develops is unclear, although implicated factors predisposing the left colon to ulceration include dehydrated and hard feces, a narrow-diameter colon with high pressure, and relatively poor blood supply.
Patients with perforated ulcers usually present with peritonitis and findings of an acute abdomen.18 The scybalum sometimes is palpable as an abdominal mass. Plain films of the abdomen might demonstrate pneumoperitoneum, fecal loading, or calcified scybala. Nonperforating ulcers can manifest with lower gastrointestinal bleeding. Caution must be used in performing disimpaction in patients with hard fecal masses in the rectum, because removing the mass can result in severe hemorrhage if the underlying blood vessel in the ulcer base is torn during removal.
The antimesenteric border of the colon is most commonly involved, usually in the sigmoid or proximal rectum. Ulcers usually are large, irregular, and sharply demarcated, and they may be single or multiple. Ulcers conform to the contour of the impacted scybala and result from ischemic pressure necrosis. A rounded or ovoid perforation may be seen in the center of the ulcer. Necrotic colonic mucosa with acute and chronic inflammation is noted on histology.17 Differential diagnosis includes spontaneous colonic perforation, malignancy, ischemia, and infection.
Perforated stercoral ulcers require emergency laparotomy with resection of the affected colonic segment. A Hartmann’s operation is the preferred procedure, and along with extensive peritoneal lavage, it is associated with a lower mortality than other surgical procedures.16,18 Nonperforating stercoral ulcers might respond to antibiotics and aggressive treatment of constipation, although surgical resection remains the only definitive treatment.
ULCERATION INDUCED BY NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
NSAIDs are among the most frequently administered drugs in the world, and their adverse side effects involve not only the stomach and duodenum but also the distal portions of the small intestine and colon. Gastroduodenal risks of NSAIDs are well known, but lower gastrointestinal tract risks of NSAIDs occur with similar frequency.19,
[/not-level-membership-for-gastroenterology-and-hepatology-category]
