CHAPTER 112 Ulcerative Colitis
Ulcerative colitis (UC) is a chronic idiopathic inflammatory disease of the gastrointestinal tract that affects the large bowel and is a major disorder under the broad group of conditions termed inflammatory bowel diseases (IBDs). Dr. Samuel Wilks is credited with being the first to describe UC in 1859 when he wrote on “idiopathic colitis” and recognized it as distinct from the then more common bacillary dysentery.1 He also reported the pathologic finding of dilated and thinned colon with severe pancolonic inflammation in a patient with this condition.2 In 1909, Hawkins described the chronic and relapsing nature of the disease course and the “stealthy hemorrhage” onset of distal disease, in which bleeding often occurred in the presence of constipation.3 In that same year, Sir Arthur Hurst gave a more complete description of UC, including its sigmoidoscopic appearances and differentiation from bacillary dysentery.4
EPIDEMIOLOGY
In general, there has been a distinct north-south gradient in risk. The areas with the highest rates of reported incidence and prevalence of UC include North America, England, northern Europe, and Australia (Tables 112-1 and 112-2). In North America, incidence rates range from 6.0 to 15.6 cases per 100,000 person-years, and the prevalence ranges from 38 to 246 cases per 100,000 persons.5 In Europe, incidence rates range from 1.5 to 20.3 cases per 100,000 person-years, with prevalence of 21 to 243 cases per 100,000 persons.5 The disease initially had been considered more common in northern Europe, although studies now suggest that the incidence of UC in southern Europe is comparable to that in northern Europe.6 In contrast, studies have reported significantly lower incidence rates of 0.6 to 6 per 100,000 person-years in other parts of the world, including Asia, Africa, and Latin America.5
Table 112-1 Incidence of Ulcerative Colitis in Various Geographic Regions
| REGION | PERIOD OF STUDY | INCIDENCE (PER 100,000 PERSON-YEARS) |
|---|---|---|
| North America | ||
| Alberta | 1981 | 6.0 |
| Manitoba | 1987-1996 | 14.3-15.6 |
| California | 1980-1981 | 10.9 |
| Minnesota | 1984-1993 | 8.3 |
| Europe | ||
| Scandinavia | 1980-1999 | 9.2-20.3 |
| Great Britain | 1985-1994 | 13.9 |
| Northern Europe | 1988-1994 | 3.2-11.8 |
| Southern Europe | 1980-1994 | 1.5-9.6 |
| Asia | ||
| India | 1999-2000 | 6.0 |
| Japan | 1991 | 1.9 |
| Korea | 1992-1994 | 1.2 |
| Africa | ||
| South Africa | 1980-1984 | 0.6-5.0 |
| Central and South America | ||
| All countries | 1987-1993 | 1.2-2.2 |
Adapted from Loftus EV. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004; 126:1504.
Table 112-2 Prevalence of Ulcerative Colitis in Various Geographic Regions
| REGION | PERIOD OF STUDY | PREVALENCE (PER 100,000 PERSONS) |
|---|---|---|
| North America | ||
| Alberta | 1981 | 37.5 |
| Manitoba | 1994 | 169.5 |
| Minnesota | 2001 | 246 |
| Europe | ||
| Scandinavia | 1987 | 161.2 |
| Great Britain | 1995-1996 | 122-243 |
| Northern Europe | 1984 | 24.8 |
| Southern Europe | 1988-1992 | 21.4-121 |
| Asia | ||
| Israel | 1980-1985 | 55.2-70.6 |
| India | 1999 | 44.3 |
| Japan | 1991 | 18.1 |
| Korea | 1997 | 7.6 |
| Singapore | 1985-1996 | 6.0 |
Adapted from Loftus EV. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004; 126:1504.
The overall incidence of UC has remained relatively stable since the 1980s; however, there appears to be geographic variation in this time frame. Although the incidence rates in North America, England, and Sweden have remained unchanged or have declined, those in southern Europe have increased.7 These time trends contrast with those of Crohn’s disease, which has shown an increase in incidence across geographic regions.
There appears to be a marked ethnic variation in the incidence of UC. One ethnic group with high incidence of this disease is the Jewish population. Incidence rates of UC among Jews have been shown to be several-fold higher than in the non-Jewish population across various geographic regions. In the United States, for example, the annual incidence of UC among Jews is 13 cases per 100,000 person-years compared with 3.8 per 100,000 among non-Jewish whites.7 Within Israel, Ashkenazi Jews have a higher incidence than do Sephardic Jews, but they have a lower incidence than in the United States and northern Europe, suggesting that environmental factors also play an etiologic role.
UC traditionally has been considered extremely uncommon in minority populations, but recent studies have challenged this notion. Early studies reported that UC was rare in blacks. Most of these studies, however, were conducted in regions with limited black populations, and more-recent studies suggest an increasing incidence of UC among African Americans. By the late 1970s, incidence rates were comparable between whites and nonwhites in the United States.8,9 An increase in incidence of UC also has been observed among blacks in South Africa, although the incidence rate still remains lower than that for South African whites.10
In Asia, UC is generally is less common than in the Western countries. The prevalence and annual incidence rates for UC in Japan have remained relatively stable at about 5.5 cases per 100,000 persons and 0.36 to 0.5 cases per 100,000 persons, respectively11; this stability is in contrast with the rising incidence of Crohn’s disease in the Japanese population. Limited data also suggest similar findings in the Chinese and Korean populations.12,13 The prevalence of UC in India has been reported to be substantially lower than that among Europeans,14 although an accurate assessment of its epidemiology is hampered by several of the previously mentioned issues. In contrast to the limited data on indigenous South Asians, several studies have demonstrated that South Asian immigrants in England are more likely to have UC than are European natives.14–16 This changing epidemiology with a population emigrating from a low-risk to a high-risk geographic region supports the concept of environmental influences on development of disease.
ETIOLOGY AND PATHOGENESIS
GENETICS
Family History
Genetic factors have been linked to the development of UC, supported largely by the observation that family history is one of the most important risk factors for developing the disease. A familial incidence of UC has been recognized for many years, and although figures vary widely in different studies, about 10% to 20% of patients have at least one other affected family member.17 Familial associations generally occur in first-degree relatives. The relative risk of the same disease in a sibling of a person with UC has been estimated to be between 7% and 17% based on North American and European studies. Parents, offspring, and second-degree relatives appear to be at a lower risk for developing UC than are first-degree relatives. Data from the United States suggest a preponderance of parent-sibling combinations, but in the United Kingdom, the disease is shared more commonly by siblings. Indeed, the strongest evidence of a genetic influence for UC is derived from twin studies. In three large European twin pair studies, approximately 6% to 16% of monozygotic twin pairs had concordant UC compared with 0% to 5% of dizygotic twin pairs.18–20 These concordance rates are substantially lower than those for Crohn’s disease, suggesting that genetic determinants, although important, play a less-significant role for UC than for Crohn’s disease. No twin pair demonstrated both UC and Crohn’s disease, further supporting the genetic basis of these disorders.
Familial association is greater in persons of Jewish descent, a heritage known to have a higher incidence of IBD. The lifetime risk of developing disease is three-fold higher among first-degree relatives of Jewish patients compared with relatives of non-Jewish patients.21 A similar increase in risk also has been observed in relatives of patients who had early onset of disease. This familial association contrasts with the low incidence of UC among spouses of patients with IBD in most series. Although reports of IBD in both spouses are rare, a study of 30 conjugal instances of IBD (17 of which were concordant for Crohn’s disease, 3 for UC, and 10 mixed) found that the majority of these couples developed IBD after cohabitation, thus suggesting a shared environmental exposure.22
For all affected first-degree relatives within a family, there is a high concordance for type of disease (UC vs. Crohn’s disease) and occurrence of extraintestinal manifestations.23 In fact, epidemiologic studies in families with multiple affected members have demonstrated disease-type concordance rates of 75% to 80%, the other 20% to 25% of families being mixed (i.e., having members with both UC and Crohn’s disease).24 The overlap in inheritance patterns among the mixed families suggests that a subset of genes associated with IBD confers susceptibility to both UC and Crohn’s disease, whereas others are specific to one disease or the other.
Numerous studies have examined the effect of family history on disease location. In UC, no consistent correlation has been observed in the studies that examined disease extent namely, left-sided colitis versus extensive colitis and family history of UC.25
Genetic Mutations
The inheritance of UC cannot be described by a simple mendelian genetics model. It is likely that multiple genes are involved and that different genes confer susceptibility, disease specificity, and phenotype. Linkage studies have suggested that there are susceptibility genes for UC on chromosomes 1, 2, 3, 5, 6, 7, 10, 12, and 17.26–29 The IBD2 locus on chromosome 12 appears to have strong linkage demonstrated in studies involving large numbers of families with UC.28 The NOD2/CARD15 gene mutations located on chromosome 16 associated with Crohn’s disease have not been associated with UC, although UC patients from families with a history of Crohn’s disease and UC might possess NOD2 variants.30 In contrast, the C3435T polymorphism for the human multidrug resistance 1 (MDR1) gene is linked to susceptibility for UC but not Crohn’s disease.31 The MDR1 gene product, P-glycoprotein, is highly expressed in intestinal epithelial cells and serves an important barrier function against xenobiotics. In contrast to NOD2/CARD15, the frequency of this polymorphism in patients with Crohn’s disease is similar to that in control subjects.
Genome-wide association studies have looked for overlap of Crohn’s disease genes in patients with UC by using a nonsynonymous single nucleotide polymorphism (SNP) scan.32,33 The Crohn’s disease autophagy genes ATG16L1 on chromosome 2q37 (which encodes autophagy-related 16-like protein) and IRGM on chromosome 5q33 (which encodes immunity-related GTPase [guanosine triphosphatase] family, M), both of which are involved in bacterial processing and the protection of cells from various bacterial pathogens and their toxins (i.e., autophagy), are not seen in patients with UC.32 A number of Crohn’s disease loci or genes (or both) have been identified in UC, however, and include IL-23R on chromosome 1p31, which encodes the interleukin[IL]-23 receptor; chromosome 3p21, which encodes MST1 and other potential genes of interest; IL-12β on chromosome 5q33, which encodes the IL-12 receptor β1 subunit (also known as p40) that constitutes part of both the IL-23 and IL-12 receptors; NKX2-3 on chromosome 10q24, which encodes NK2 transcription factor related, locus 3; and chromosome 17q21, which encodes STAT3 and other potential genes of interest.
Another genome-wide association study has observed a strong association between the gene encoding IL-23R and both Crohn’s disease and UC.33 Several polymorphisms of this gene have been identified, most notably the Arg381Gln polymorphism. Heterozygous carriage of the glutamine allele is associated with a three-fold decreased risk of Crohn’s disease and a more-modest reduction in the risk of UC in non-Jewish populations; this reduction is not seen in UC within the Jewish population. IL-23R is important because it plays a key role in the differentiation of a relatively newly discovered subset of T cells called Th17 cells (see later).
ENVIRONMENTAL FACTORS
Numerous clinical and experimental observations have suggested involvement of intestinal bacterial microbiota in the pathogenesis of IBD. The most obvious observation perhaps is that Crohn’s disease and UC preferentially occur in regions of the bowel that contain the highest concentration of bacteria, namely, the terminal ileum and the colon, where bacterial concentrations approach 1012 organisms per gram of luminal contents. Interestingly, diverting the fecal stream in patients with Crohn’s disease can treat and even prevent disease, whereas reinfusion of ileostomy contents leads to new inflammatory changes within only one week.34 Other human data have shown that antibiotics are useful in the treatment or postoperative prevention of Crohn’s disease and pouchitis. Finally, probiotics (discussed later in this chapter) have been shown to have efficacy in the primary and secondary prevention of pouchitis. The most glaring evidence of the necessary role of bacteria in the pathogenesis of IBD from rodent data is that genetically susceptible mice or rats in a gnotobiotic (germ-free) environment do not have intestinal inflammation; however, these same rodents rapidly develop intestinal inflammation after bacterial colonization.35–38 Just as in humans, rodent gut inflammation can be treated and prevented with antibiotics and probiotics.39,40
With respect to the human gastrointestinal microbiome, much has been learned in the last few years. Complex mathematical models have estimated that the human gastrointestinal microbiome contains at least 1800 genera and between 15,000 and 36,000 species of bacteria41–43; three studies have identified more than 45,000 bacterial small-subunit (SSU) rRNA genes, a technique that captures at most only 50% of the predicted species-level biodiversity.41–43 Of the bacterial genes identified to date, almost all (more than 98%) can be grouped into four phyla: Firmicutes accounts for 64% of the total and includes the family Lachnospiraceae (e.g., Clostridium groups XIVa and IV) and the subgroup Bacillus (e.g., Streptococcaceae and Lactobacillales). Bacteroidetes account for 23% of the total, Proteobacteria account for 8% of the total and include the family Enterobacteriaceae (e.g., Escherichia coli), and Actinobacteria account for 3%.43
Four general mechanisms have been postulated to explain how components of the normal intestinal microbiome might initiate or contribute to the development of the chronic inflammatory state.44 First, microbes can induce intestinal inflammation, either by adhering to or invading intestinal epithelial cells, thereby causing downstream proinflammatory cytokine production or by producing enterotoxins.
Second, a breakdown in the balance between protective and harmful intestinal bacteria, termed dysbiosis, can lead to disease. Most studies comparing the intestinal microbiome in IBD with that in healthy controls show biodiversity in the IBD populations is decreased by 30% to 50%. One study found that this reduction in biodiversity was due to decreased concentrations of Firmicutes (specifically Lachnospiraceae) by 300-fold and Bacteroides by 50-fold.43 The loss of these organisms is important because they are known to produce short-chain fatty acids, such as butyrate, which nourish colonocytes. As a result of their decrease, the relative concentrations of Proteobacteria and Actinobacteria increase in IBD patients relative to controls, although quantitative PCR analysis showed that the absolute numbers of Enterobacteriaceae were not higher in IBD patients than in controls. Loss of protective bacteria, however, could set the stage for overgrowth of pathogenic bacteria. Another interesting finding in this study is that the microbiota of patients with Crohn’s disease and UC were similar to each other. In addition, the study did not identify any individual species that was particularly prevalent in grossly abnormal diseased tissue, and thus no active bacterial etiologic agent was suspected of causing or propagating disease.
In addition to infectious agents, several other environmental factors have been proposed as contributing etiologic factors of UC. The best characterized environmental factor associated with UC is cigarette smoking. Numerous studies have consistently shown that UC is more common among nonsmokers than among current smokers, with the relative risk of UC in nonsmokers ranging from two to six45; this association is independent of genetic background and gender. Furthermore, there may be a dose-response relationship, with the disease more common in current light smokers than in heavy smokers. This relationship is consistent with observations that clinical improvement with nicotine therapy in patients with UC appears to be limited to those treated with high doses of nicotine and not those receiving lower doses. This risk of developing UC with smoking is particularly high for former smokers, especially within the first two years of smoking cessation. The rebound effect also is higher for former heavy smokers than for former light smokers.46 Smokers also appear to have reduced rates of hospitalization for UC and reduced rates of pouchitis following colectomy.47 Studies on the role of passive smoking in UC have yielded conflicting results. A recent meta-analysis of 10 studies examining this issue found no association between passive smoking and future development of UC.48
Several mechanisms have been postulated to account for the apparent protective effect of active smoking on UC. These include modulation of cellular and humoral immunity, changes in cytokine levels, increased generation of free oxygen radicals, and modification of eicosanoid-mediated inflammation. Smoking also might have an effect on mucus production by the colonic mucosa, and might alter colonic mucosal blood flow and intestinal motility. No single mechanism, however, can explain the clinical observation of the beneficial influence of smoking on UC and its adverse effect on Crohn’s disease (see Chapter 111).
Other environmental risk factors that have been suggested to influence the development of UC include diet (wheat, maize, cow’s milk, refined sugar, fruits and vegetables, alcohol), oral contraceptives, food additives (silicon dioxide), toothpaste, and breast-feeding7; none, however, has been shown conclusively to be associated with UC. Studies have suggested an inverse relationship between appendectomy and the subsequent development of UC49,50; the mechanisms for this protective effect of appendectomy on UC are unknown. It is possible that removal of appendiceal-associated lymphoid tissues abrogates certain pathologic alterations in mucosal immune responses and therefore prevents the onset of UC.
IMMUNE FACTORS
Humoral Immunity
Histologic examination of the inflamed colon indicates a marked increase in the number of plasma cells. This increase is not uniform among cells producing different classes of immunoglobulins. The largest proportional increase occurs in immunoglobulin (Ig)G synthesis, which has the highest pathogenic potential among antibody classes. The increase in IgG synthesis in UC is most pronounced in the IgG1 and IgG3 subclasses, in contrast to Crohn’s disease, in which the increase in IgG2 synthesis is more prominent.51,52 This disparity in the local IgG subclass response likely reflects differences in antigenic stimuli or host immunoregulatory responses between the two groups of IBD patients. The increased IgG synthesis in IBD may represent polyclonal stimulation; patients with UC often have circulating antibodies to dietary, bacterial, and self antigens that are mostly of the IgG isotype, usually the IgG1 subclass. Many of these antibodies are thought to be epiphenomena because the serum antibody titers do not correlate with clinical features. Nevertheless, the known cross-reaction between enterobacterial antigens and colonic epithelial epitopes may be an important triggering event, even though, later in the course of the disease, the serum antibody titer to either the bacterial or the colonic antigen may be unimportant.
The concept that UC is an autoimmune disease is supported by its increased association with other autoimmune disorders, including thyroid disease, diabetes mellitus, and pernicious anemia.53 Patients with UC have varying levels of autoantibodies to lymphocytes, ribonucleic acid, smooth muscle, gastric parietal cell, and thyroid; these are specific for neither tissue nor disease. Antibodies to epithelial cell-associated components, which specifically recognize intestinal antigen, also have been described.53 The best-characterized intestinal autoantigen is a 40-kDa epithelial antigen found in normal colonic epithelium.54 This autoantigen is recognized by IgG eluted from the inflamed colonic mucosa of patients with UC and is a component of the tropomyosin family of cytoskeletal proteins.55 The antibody response to this 40-kDa protein appears to be unique to UC and is not found in Crohn’s disease nor in other inflammatory conditions. This autoantigen shares an epitope with antigens found in the skin, bile duct, eyes, and joints, sites often involved in the extraintestinal manifestations of UC. The precise pathogenic significance of this autoantibody in UC, however, remains unclear at present.
An autoantibody that has received significant attention in UC patients is pANCA.56 This autoantibody is present in 60% to 85% of patients with UC.57,58 It is synthesized within the lamina propria and is of the IgG1 subclass. The antigen to which the pANCA is directed has not yet been determined with certainty, and a variety of putative antigens have been proposed, including nuclear histone and nonhistone proteins. The most recent evidence suggests that the antigen is a 50-kd nuclear envelope protein that is specific to myeloid cells.59
Just as with other autoantibodies found in patients with UC, the pathogenic relevance of pANCA in this disorder is unknown. In fact, the prevailing thought is that pANCA has no pathogenic role in UC but that it might serve as a potential marker of susceptibility and genetically distinct subsets of UC. The level of pANCA titer does not correlate with disease activity, but it might decline in patients with long-standing remission or in patients who have had colectomy at least 10 years previously. Studies have suggested that pANCA may be associated with a more-aggressive disease course60 and with the development of pouchitis after ileal pouch-anal anastomosis (IPAA) in patients with UC.61,62 Also of interest is that Crohn’s disease patients with colonic disease have higher rates of pANCA seroreactivity than those without colonic disease, suggesting a possible UC-like Crohn’s disease phenotype.63
Another type of autoantibody commonly seen in IBD, especially in Crohn’s disease, is that against bacterial antigens. Antibodies to bacterial antigens seen in UC include anti-CBir1 and anti-OmpC. Anti-Cbir1 is an antibody to flagellin from Clostridium species; it is found in about 6% of UC patients64 and also appears to be associated with the development of pouchitis.65 Anti-CBir1 also is found in 50% of patients with Crohn’s disease, in which it is associated with more complicated disease. 64 Anti-OmpC (outer membrane porin C of E. coli) is seen more often in UC patients who have a mixed family history of Crohn’s disease and UC rather than those with a family history of only UC.66
Cellular Immunity
Activation of the TLRs and NLRs results in downstream activation of nuclear factor-κB (NF-κB), which then stimulates the transcription of genes coding for various proinflammatory cytokines (including TNF, IL-1, IL-6, and IL-8), chemokines, adhesion molecules, and costimulatory molecules. In addition, activation of NF-κB stimulates the maturation of dendritic cells, which are involved in antigen presentation. Defects in any of the PRR pathways can lead to abnormal bacterial processing and possibly IBD.67
The adaptive immune system, which is governed by T cells and B cells, is specific. Mucosal lymphocytes often are divided into two groups based on location: lamina propria lymphocytes and intraepithelial lymphocytes (IELs). Lamina propria lymphocytes express surface adhesion molecules, α4β7, that provide a homing signal for peripheral immune cells to the mucosal sites.68 Most investigators have found a similar distribution of T-cell subsets (CD4+, CD8+) within the lamina propria in patients with UC compared with that in controls.69 Also, the absolute number of IELs is normal or reduced in UC, most of which are CD8+ cell. The function of IELs has not been well characterized, but it has been suggested that they are cytotoxic and perhaps active in suppressing local immune response. Regardless of their functional status, mucosal T cells within the lamina propria and epithelium, as well as peripheral blood T cells, display a variety of activation markers, suggesting an activated memory phenotype.70 Studies have suggested that the T-cell receptor repertoire is altered in active IBD.71
Although T-cell–mediated immunity has attracted the most attention in the pathogenesis of UC, nonspecific cellular immunity also is altered. In patients with active disease, there is an overproduction of circulating monocytes as well as mucosal macrophages.72 The inflamed mucosa of patients with UC also exhibits infiltration of substantial numbers of granulocytes.
Epithelial Cells
Intestinal epithelial cells serve barrier functions and play a role in enteric immunity. Colonocytes express class II major histocompatibility complex (MHC) antigens and can function as antigen-presenting cells.73 In addition, they also express cytokine receptors, secrete various cytokines and chemokines, and express leukocyte adhesion molecules.74–77 Thus, abnormalities in colonic epithelial cells can contribute to the development of UC.
Patients with UC have an increased turnover rate of colonic epithelium78 and other abnormalities of epithelial cells including reduced metabolism of short-chain fatty acids, especially butyrate, abnormal membrane permeability,79 and altered composition of glycoprotein mucus produced by the colonic epithelium.80 Specifically, the mucus layer in UC appears to be thinner than normal.81 These and other abnormalities can lead to the finding of increased numbers of adherent bacteria, in both the mucus layer and even at the epithelial surface, in patients with UC.82–84 The role of epithelial cells in the pathogenesis of IBD is supported further by animal models of colitis produced by disruption of colonic epithelium.85
Consequences of Immune Activation
The Th1 response is characterized by cell-mediated immunity and is associated with the production of interleukin (IL)-2 and interferon (IFN)-γ. The differentiation of T cells along a Th1 pathway is stimulated by IL-12 generated in response to exposure to infectious agents. The Th2 response is characterized by the production of cytokines IL-4, IL-5, IL-10, and IL-13, which amplify the humoral immune response. Th1 and Th2 subsets reciprocally down-regulate each other through cytokine production.86 Th1 and Th2 pathways can be regulated by unique regulatory T cell (Treg) subsets that produce IL-10 and transforming growth factor (TGF)-β and down-regulate inflammation.87
Historically, the oversimplified view of adaptive immunity in IBD is that Crohn’s disease is mediated by Th1 cells whereas UC is mediated by Th2 cells; there is considerable evidence that the story is much more complex. Macrophages in the inflamed colon of patients with active UC synthesize IL-1β, TNF, and IL-6, whereas lamina propria T cells probably produce IL-2 and IFN-γ. This immune response can be up-regulated further by presentation of antigen to CD4+ lymphocytes by colonic epithelial cells that express HLA class II antigens.73 Studies have implicated a specialized type of T cell, the natural killer (NK) T cell, which seems to mediate the Th2 response in UC.88,89 These NK T cells, which are not classical NK T cells in that they do not express the typical NK T-cell receptors seen with classical NK T cells, secrete large amounts of IL-5 and IL-13 and are actually cytotoxic for intestinal epithelial cells.
A novel T-cell–mediated inflammatory pathway, which appears to be involved in the pathogenesis of both Crohn’s disease and UC, has been discovered and centers on the Th17 cell lineage. Th17 cells have been shown to produce a variety of cytokines, most notably IL-6 and IL-17. IL-17 is a potent proinflammatory cytokine that not only facilitates T-cell activation but also stimulates a variety of cells, including fibroblasts, macrophages, epithelial cells, and endothelial cells, to produce an array of proinflammatory cytokines, including IL-1, IL-6, TNF-α, and chemokines.90 Th17 cell development is inhibited by Th1 and Th2 cells but is promoted by IL-6, TGF-β, IL-21, and IL-23R.91 IL-23R, which is highly expressed by activated Th17 cells, also is expressed by NK cells, NK T cells, other CD4+ T cells, and CD8+ T cells.29 The interaction of IL-23 with its receptor has been shown to have a central role in the development of inflammation in various mouse models of colitis.92,93 Antibodies to IL-23 thus represent another potential therapeutic strategy for the treatment of IBD.
Release of these various cytokines from the T-cell inflammatory pathways also might lead to other abnormalities seen in UC, such as increased epithelial cell permeability and collagen synthesis. Alteration of endothelium by a variety of cytokines can result in local ischemia. Increased expression of endothelial adhesion molecules in response to inflammatory mediators recruits circulating granulocytes and monocytes to the inflamed tissues, thus further perpetuating the inflammatory response. Elevated cytokine levels within the mucosa also stimulate the release of metalloproteinase from fibroblasts with subsequent matrix degradation. Mucosal concentrations of many mediators have been shown to be elevated in patients with active UC, including leukotrienes, thromboxane, platelet-activating factor, nitric oxide, and reactive oxygen metabolites.94 These mediators, which are mostly released from active macrophages and neutrophils, contribute to inflammation and mucosal injury and alter epithelial cell permeability, thereby further contributing to diarrhea. Diarrhea in UC also is caused by complement activation and the release of kinins and other inflammatory mediators from mast cells and eosinophils (see Chapter 2).
PSYCHOGENIC FACTORS
Psychosomatic factors first were implicated in the pathogenesis of UC in the 1930s,95 but there is no good direct evidence to support this concept. Since the introduction of glucocorticoids for the treatment of patients with UC and the focus on immunologic aspects of the pathogenesis of the disease in the 1950s, this previously widely held notion has diminished in popularity.
Experimental studies have helped identify mechanisms of the proinflammatory potential of stress in animal models of colitis.95 When rats are exposed to stress before proinflammatory stimuli are introduced, the severity of colonic inflammation is increased. This particular response has been shown not to be mediated by either vasopressin or corticotropin-releasing factor. In addition, stress has been shown to directly increase intestinal permeability in rats, an action mediated by cholinergic nerves, and to potentiate intestinal inflammation in this particular situation. There are indeed studies reporting that psychosocial stress increases the risk of relapse in patients with quiescent UC.96,97 Conversely, many of the psychological features observed in patients with UC are likely secondary to this chronic disease process, a phenomenon physicians must be aware of when managing these patients.
PATHOLOGY
At the time of initial presentation, approximately 45% of patients with UC have disease limited to the rectosigmoid, 35% have disease extending beyond the sigmoid but not involving the entire colon, and 20% of patients have pancolitis.98 The disease typically is most severe distally and progressively less severe more proximally. In contrast to Crohn’s disease, continuous and symmetrical involvement is the hallmark of UC (Fig. 112-1), with a sharp transition between diseased and uninvolved segments of the colon.
There are a few exceptions to this general rule. First, medical therapy can result in areas of sparing. For example, topical enema therapy can lead to near-complete mucosal healing in the rectum and distal sigmoid colon. Second, up to 75% of patients with left-sided UC have periappendiceal inflammation in the colon and patchy inflammation in the cecum,99 resembling the skip pattern characteristic of Crohn’s disease. These patterns of rectal sparing and skip lesions can lead to a misdiagnosis of Crohn’s disease.
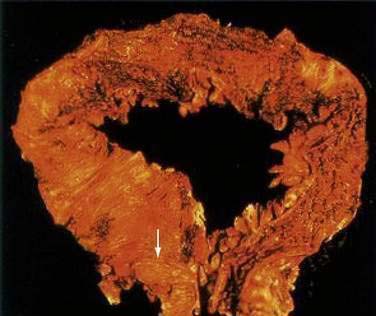
Macroscopically, the mucosa in UC appears hyperemic, edematous, and granular in mild disease. As disease progresses, the mucosa becomes hemorrhagic, with visible punctate ulcers. These ulcers can enlarge and extend into the lamina propria. They often are irregular in shape with overhanging edges or may be linear along the line of the teniae coli. Epithelial regeneration with recurrent attacks results in the formation of pseudopolyps, which is typical of long-standing UC but which also may be seen in acute disease (Fig. 112-2). Another characteristic appearance of long-standing disease is atrophic and featureless colonic mucosa, associated with shortening and narrowing of the colon. Patients with severe disease can develop acute dilatation of the colon, also characterized by thin bowel wall and grossly ulcerated mucosa with only small fragments or islands of mucosa remaining. With perforation of the colon, a fibrinopurulent exudate may be seen on the serosal surface of the bowel.

During the healing phase of UC, the inflammatory infiltrate subsides and epithelial regeneration takes place. Epithelial cells undergoing regenerative changes become cuboidal with eccentric, large nuclei, and prominent nucleoli. These features may be confused with dysplasia. Thus, a diagnosis of dysplasia in UC should be made with caution in the presence of acute inflammation. Accordingly, surveillance colonoscopy (see “Dysplasia and Colorectal Cancer”) should be performed during a period of remission.
A classic histologic feature of chronic quiescent UC is crypt architectural distortion or actual dropout of glands (Fig. 112-3). Architectural changes include branching or bifid glands, wide separation among glands, and shortened glands that do not extend down to the muscularis mucosa. Architectural alteration is a prominent feature of chronic quiescent UC, but the histologic abnormalities can revert to normal after mild flares early in the course of disease. Another characteristic feature of chronic quiescent UC is Paneth cell metaplasia, with Paneth cells located distal to the hepatic flexure, where they normally are absent. Other nonspecific chronic changes seen in UC include neuronal hypertrophy and fibromuscular hyperplasia of the muscularis mucosa. Varying degrees of acute or chronic inflammation of the lamina propria may be present in chronic quiescent disease. A thin band of predominantly lymphocytic inflammation occasionally may be seen deep to the muscularis mucosa, presenting diagnostic challenges.
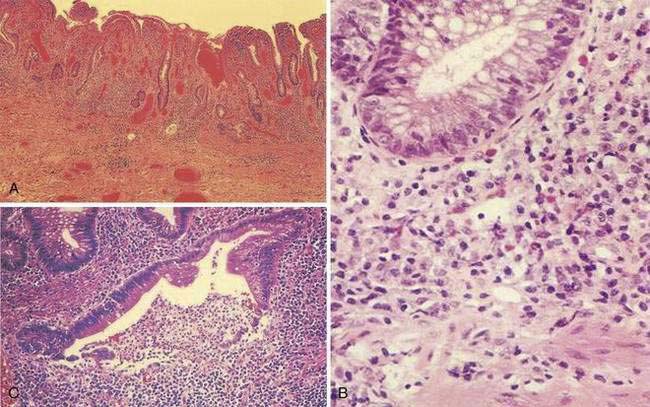
Most of these pathologic findings are not specific for UC. Features that reflect chronicity and thus argue against a diagnosis of infectious or acute self-limited colitis include distorted crypt architecture, crypt atrophy, increased intercrypt spacing to fewer than six crypts per millimeter, an irregular mucosal surface, basal lymphoid aggregates, and a chronic inflammatory infiltrate.100,101 The histologic severity of inflammation does not necessarily correlate with clinical disease activity in patients with UC, because patients may be relatively symptom free although histology reveals significant inflammation.
CLINICAL FEATURES
Patients with UC can present with a variety of symptoms. Common symptoms include diarrhea, rectal bleeding, passage of mucus, tenesmus, urgency, and abdominal pain. In more-severe cases, fever and weight loss may be prominent. The symptom complex tends to differ according to the extent of disease.102 Patients with proctitis often have local symptoms of tenesmus, urgency, mucus, and bleeding, whereas patients with extensive colitis usually have more diarrhea, weight loss, fever, clinically significant blood loss, and abdominal pain. In general, the severity of the symptoms correlates with the severity of the disease; however, active disease may be found at colonoscopy in patients who are otherwise asymptomatic. Additionally, patients with known UC can have severe symptoms that are not necessarily due to UC, such as those caused by bacterial (e.g., Clostridium difficile) or viral (e.g., cytomegalovirus) infections or a host of other similar disorders.
The onset of UC typically is slow and insidious. Symptoms usually have been present for weeks or months by the time the typical patient seeks medical attention. The median interval between the onset of symptoms and diagnosis of UC is approximately nine months.103 Some patients with UC present much more acutely, with symptoms mimicking acute infectious colitis. Indeed, it is not uncommon to find a patient whose UC began after a documented gastrointestinal infection, such as Salmonella or C. difficile. This observation raises the question whether the infection revealed preexisting but silent disease or whether it was actually the initiating factor.
SYMPTOMS
Rectal Bleeding
Rectal bleeding is common in UC, its characteristics determined by the distribution of disease. Patients with proctitis usually complain of passing fresh blood, either separately from the stool or streaked on the surface of a normal or hard stool.104 This symptom often is mistaken for bleeding from hemorrhoids. In contrast to hemorrhoidal bleeding, however, patients with ulcerative proctitis often pass a mixture of blood and mucus and might even be incontinent. Patients with proctitis also often complain of the frequent and urgent need to defecate, only to pass small quantities of blood and mucus without fecal matter. When the disease extends proximal to the rectum, blood usually is mixed with stool or there may be grossly bloody diarrhea. When disease activity is severe, patients typically pass liquid stool containing blood, pus, and fecal matter. This stool often is likened to anchovy sauce, and some patients with this symptom do not actually recognize that they are passing blood. Unless the patient has severe disease, passage of blood clots is unusual and suggests other diagnoses such as a tumor. Active UC that is sufficient to cause diarrhea almost always is associated with macroscopically evident blood. The diagnosis needs to be questioned if visible blood is absent.
Diarrhea
Diarrhea is common but not always present in patients with UC. Up to 30% of patients with proctitis or proctosigmoiditis complain of constipation and hard stools.104 Most patients with active disease complain of frequent passage of loose or liquid stools and may have nocturnal diarrhea. Fecal urgency, a sensation of incomplete fecal evacuation, and fecal incontinence also are common, especially when the rectum is severely inflamed. Diarrhea in this setting often is accompanied by passage of large quantities of mucus, blood, and pus.
The pathophysiology of diarrhea in UC involves several mechanisms, but failure to absorb salt and water is the predominant factor105 and results from reduced Na+,K+-ATPase (adenosine triphosphatase) pump activity, increased mucosal permeability, and altered membrane phospholipids. High mucosal concentrations of lipid inflammatory mediators, which are detected in UC, have been shown to stimulate chloride secretion in normal colon, and it is possible that these mediators also contribute to diarrhea by increasing mucosal permeability. Urgency and tenesmus, which are common symptoms when the rectum is inflamed, are caused by decreased rectal compliance and loss of the reservoir capacity of the inflamed rectum.106 With severe inflammation, the urgency can be sufficiently acute to cause incontinence.
Colonic motility is altered by inflammation, and there is rapid transit through the inflamed colon. With left-sided disease, distal colonic transit is rapid, but there is actual slowing of proximal transit,107 which might help explain the constipation that is commonly seen in patients with distal colitis. Prolonged transit in the small intestine also occurs in the presence of active colonic inflammation.107
LABORATORY FINDINGS
Mild or moderate attacks rarely are associated with any biochemical disturbance. Hypokalemia, metabolic alkalosis, and elevated serum levels of blood urea nitrogen and creatinine may be present in severe flares of UC, reflecting volume depletion. Hypoalbuminemia may be seen with acute and chronic disease. Minor elevations in serum levels of aspartate aminotransferase or alkaline phosphatase also are commonly associated with severe disease, but these changes are transient and return to normal when the disease enters remission; these abnormalities probably reflect a combination of fatty liver, sepsis, and poor nutrition. Persistently elevated liver biochemical tests, especially serum alkaline phosphatase, are seen in about 3% of patients with UC and should lead to further investigation, particularly to exclude primary sclerosing cholangitis (PSC) (see Chapter 68).
NATURAL HISTORY AND PROGNOSIS
Most (80%) patients with UC have a disease course characterized by intermittent flares interposed between variable periods of remission. The duration of relapse-free periods varies greatly from patient to patient. More than 50% of patients present with mild disease at their first attack, and 6% to 19% of patients have severe disease at presentation.108,109 Following the initial flare, 40% to 65% of patients have an intermittent course, and 5% to 10% of patients have a chronic continuous course.110,111 Up to 10% of patients have a severe first attack ultimately requiring colectomy.107 In population-based studies, the proportion of patients with active disease remains relatively constant over years, with approximately 50% of all patients being in remission at any time point during follow-up (Fig. 112-4).98,110 Twenty-five years after the diagnosis of UC, 90% of patients still have a relapsing course (Fig. 112-5)97; however, disease activity in the preceding years predicts the subsequent chance of disease activity. The probability of remaining in remission for one year after a relapse has been estimated at 30%. After being in remission for one year, the risk of relapse decreases to 20% for the following year. Few patients (1%) with UC have only one attack followed by a relapse-free course,110 and they likely have misdiagnosed infectious colitis.
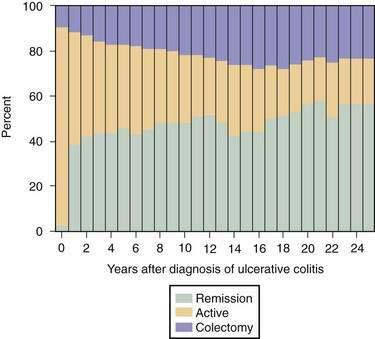
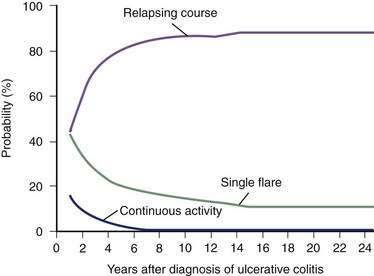
Factors influencing disease relapse and remission include bacterial and viral infections, the use of nonsteroidal anti-inflammatory drugs (NSAIDs) and antibiotics, smoking, seasonality, and psychosocial stress. Both the severity and extent of disease are important prognostic factors for the first attack of UC. In general, patients with disease that is limited to the distal colon do better than those with extensive colitis. UC diagnosed in the elderly generally has been thought to manifest with more-severe initial attacks, but this pattern has not been observed consistently. No one factor has been consistently identified as predicting future disease activity. In patients initially presenting with proctitis or proctosigmoiditis, disease extension occurs in approximately 10% to 30% of patients at 10 years after diagnosis.108,112,113 Less commonly, extensive colitis regresses over time with treatment.
Colectomy rates vary in different studies, in part related to the different proportions of patients with extensive versus limited disease, but rates seem to be highest in the first year of diagnosis. In one Scandinavian study, the colectomy rate was 10% within the first year of diagnosis, 3% in the second year, and approximately 1% per year thereafter.110 In general, the overall colectomy rate is 24% at 10 years and 30% at 25 years (see Fig. 112-4).98 The probability of colectomy was higher in patients with extensive colitis. A more-recent European study reported a lower colectomy rate of 9% at 10 years, with a four-fold higher risk of colectomy in patients with extensive colitis.114
Despite the burden of a chronic illness, more than 90% of patients with UC are able to maintain capacity for work after 10 years of disease, and data suggest that the overall quality of life is not impaired significantly, including marital issues, physical activities, and social function.98,109,115 The disease can affect the quality of life to some degree during acute flares, however, and even during periods of remission, patients might remain anxious for fear of relapse and alter their lifestyle accordingly.
Despite the morbidity of UC, mortality associated with the disease has dropped dramatically since the late 1950s and 1960s. The mortality rate for a severe attack of UC was approximately 35% before the introduction of glucocorticoid therapy and now is less than 2%. Long-term survival does not differ significantly from that expected for age-matched controls, even with the risk of colorectal cancer that attends long-standing colitis. It is now generally believed that patients with UC have life expectancies comparable to those of the general population, although studies have reached conflicting conclusions.108,111,116–119 Mortality risk is greatest in the elderly and in those with extensive colitis, mostly related to postoperative complications within the first two years of disease and to comorbidities.
DIAGNOSIS
Currently, there is no single test that allows the diagnosis of UC with acceptable sensitivity and specificity. Thus, diagnosis relies on a combination of compatible clinical features, endoscopic appearances, and histologic findings. Stool cultures should be obtained to exclude infection with routine bacterial pathogenic organisms; assay for toxins A and B of C. difficile, and examinations for ova and parasites also should be performed. Infection with E. coli O157:H7 should be considered and requires special stool cultures (or molecular probes). Similarly, special cultures for gonococcus or Chlamydia may be necessary in selected cases. In immunosuppressed patients, the possibility of opportunistic infection of the colon must be excluded (see “Differential Diagnosis”). The diagnosis of UC should be questioned if there is only a single episode of acute illness or if the histopathology findings are nonspecific and lack signs of chronicity.
ENDOSCOPY
Multiple biopsy specimens should be taken from throughout the colon to map the histologic extent of disease and to confirm the diagnosis if there is concern about Crohn’s disease.120 In addition, intubation and biopsy of the terminal ileum should be attempted to exclude the presence of Crohn’s disease or other disease states that can mimic IBD.
The hallmark of UC is symmetrical and continuous inflammation that begins in the rectum and extends proximally without interruption for the entire extent of disease. The earliest endoscopic sign of UC is a decrease or loss of the normal vascular pattern, with mucosal erythema and edema (Fig. 112-6); distortion or loss of vascular markings may be the only endoscopic evidence of UC in patients with quiescent disease. As disease progresses, the mucosa becomes granular and friable. With more-severe inflammation, the mucosa may be covered by yellow-brown mucopurulent exudates associated with mucosal ulcerations. In UC, mucosal ulcerations occur in areas of inflammation, vary in size from a few millimeters to several centimeters, and may be punctate, annular, linear, or serpiginous. Finally, severe UC is associated with mucosa that bleeds spontaneously, and, with diffuse colitis, there may be extensive areas of denuded mucosa from severe mucosal ulcerations (see Fig. 112-6). Marked edema can at times lead to narrowing of the lumen.
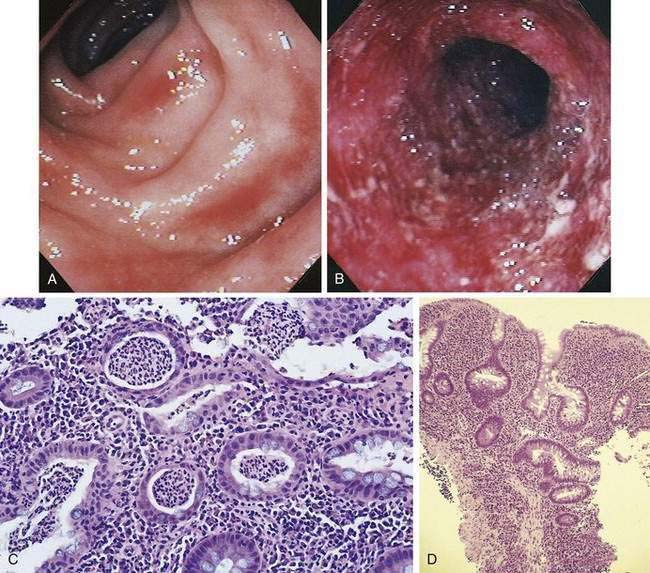
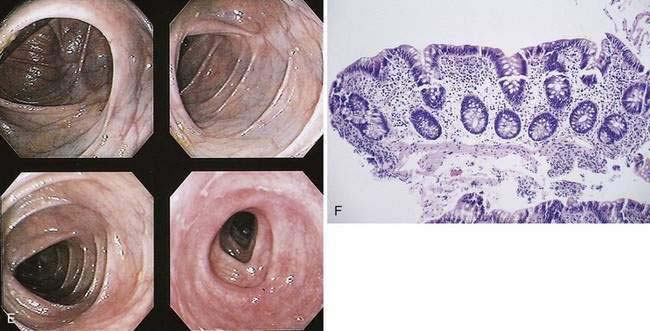
RADIOLOGY
Plain Films
Patients with a severe attack of UC should have a supine plain film of the abdomen. The presence of intraperitoneal air may be missed on plain abdominal films, however, and CT has demonstrated a better diagnostic yield than plain abdominal radiography for detecting disease complications and extent. In the presence of severe disease, the luminal margin of the colon—the interface between the colonic mucosa and the luminal gas—becomes edematous and irregular. Thickening of the colonic wall often is apparent on a plain film, and prognostic signs such as islands of residual mucosa surrounded by extensive deep ulcerations, distention of the small bowel, and dilatation of the colon can be detected (Fig. 112-7).
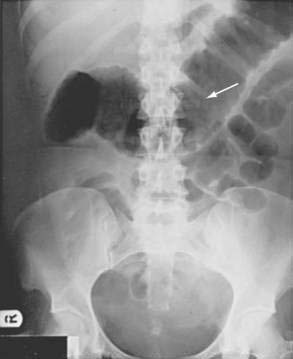
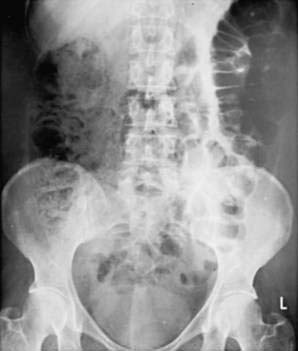
Plain films also are useful for detecting the presence of fecal material. Inflamed colons seldom contains feces, and no fecal material is present when the whole colon is involved. It is common, however, for a patient with left-sided disease to have proximal constipation (Fig. 112-8). Thus, a plain film can give considerable information with respect to the extent of disease. The presence of marked colonic dilatation suggests fulminant colitis or toxic megacolon. A plain abdominal film also can detect unsuspected free air and is especially useful in following the daily progress of a patient on high-dose glucocorticoid therapy in whom such a complication may be masked.
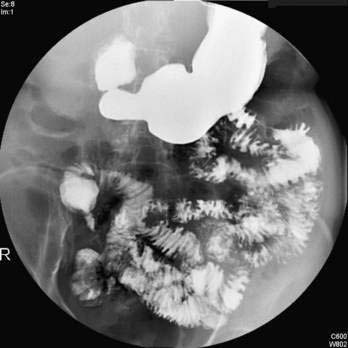
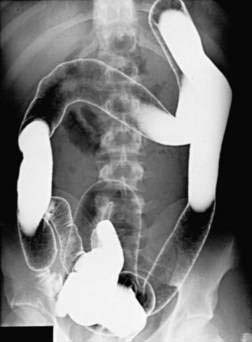
Barium Enema
The earliest radiologic change of UC seen on barium studies is fine mucosal granularity (Fig. 112-9). The mucosal line becomes irregular and is not as sharp as that of a normal colon. With increasing severity, the mucosal line becomes thickened and irregular, and superficial ulcers are well shown en face. Deep ulceration can appear as collar-stud or collar-button ulcers in tangent, which indicates that the ulceration has extended through the mucosa to the muscularis propria (Fig. 112-10).
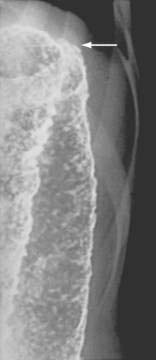
Haustral folds may be normal in mild disease but become edematous and thickened as disease progresses. Loss of haustrations also can occur, especially in patients with long-standing disease (Fig. 112-11). Because the left colon may normally lack haustration, this sign is relevant for only the ascending and transverse colon. With long-standing disease, loss of haustration can lead to a featureless and tubular appearance of the colon.
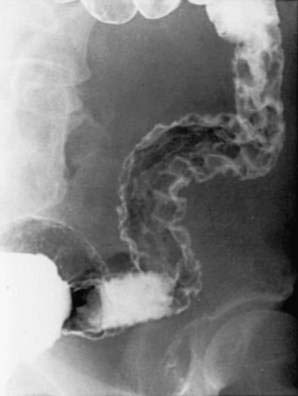
Other chronic changes are shortening of the colon and widening of the presacral (retrorectal) space as seen on a lateral film of the rectum. Pseudopolyps may be present and often are filiform. In the presence of active changes, these pseudopolypoid changes can resemble a cobblestone pattern (see Fig. 112-11).
DIFFERENTIAL DIAGNOSIS
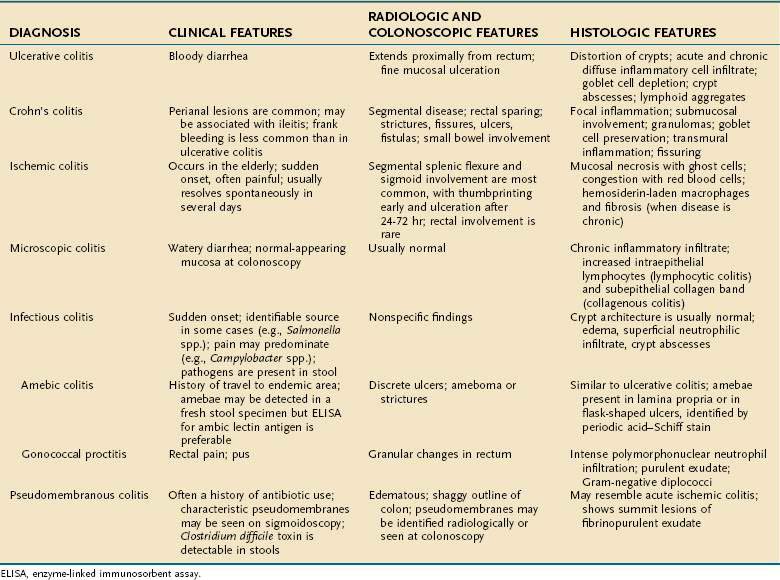
A variety of inflammatory and noninflammatory diseases of the colon can mimic UC and need to be considered in establishing the correct diagnosis. This differential diagnosis can be grouped broadly into three categories: Crohn’s disease, infections, and noninfectious causes (Table 112-3).
| Infectious Causes |
EHEC, enterohemorrhagic E. coli.
CROHN’S DISEASE
Crohn’s disease should be excluded in all patients given a diagnosis of UC, and colonoscopy with multiple biopsies is important in this regard. The presence of skip lesions or granulomas supports the diagnosis of Crohn’s disease. It is important to recognize that muciphage granulomas may be present in patients who do not have Crohn’s disease. Other endoscopic features distinguishing UC from Crohn’s disease are listed in Table 112-4.
Table 112-4 Endoscopic Differentiation of Ulcerative Colitis and Crohn’s Disease
| FEATURE | ULCERATIVE COLITIS | CROHN’S DISEASE |
|---|---|---|
| Distribution | Diffuse inflammation that extends proximally from the anorectal junction | Rectal sparing, frequent skip lesions |
| Inflammation | Diffuse erythema, early loss of vascular markings with mucosal granularity or friability | Focal and asymmetrical, cobblestoning; granularity and friability less commonly |
| Ulceration | Small ulcers in a diffusely inflamed mucosa; deep, ragged ulcers in severe disease | Aphthoid ulcers, linear or serpiginous ulceration; intervening mucosa is often normal |
| Colonic lumen | Often narrowed in long-standing chronic disease; tubular colon; strictures are rare | Strictures are common |
“Indeterminate” colitis is diagnosed in approximately 10% of patients when the distinction between Crohn’s disease and UC cannot be made. These patients generally are initially managed as if they had UC, until more characteristic features of Crohn’s disease appear. The key points in the differential diagnosis between UC and Crohn’s disease are summarized in Tables 112-4 and 112-5.
INFECTION
Another major category of differential diagnosis for UC is infection (see Chapter 107). As mentioned previously, newly diagnosed UC can manifest as part of a well-documented episode of infectious colitis. It is unknown if the infection prompts the UC or simply unmasks underlying UC that previously had subclinical activity. Patients with documented UC in clinical remission also can develop acute infectious colitis and present with symptoms of a flare of UC. Thus, infections need to be excluded with each episode of disease exacerbation.
A history of antibiotic use suggests pseudomembranous colitis associated with C. difficile (Chapter 108). This infectious colitis, which is among the most common infections in UC patients, manifests with diarrhea and may be superimposed on or even lead to a relapse of UC. In a national survey of hospitalized patients in the United States, the prevalence of C. difficile among UC patients (3.7%) was eight times higher than that of either non-IBD gastroenterology patients or all hospital-discharge patients.121 This study also showed that prevalence of C. difficile infection in UC patients has doubled over the last seven years. C. difficile infection can occur in the absence of antibiotics, especially in the elderly and in patients who are taking proton pump inhibitors or who have had placement of a postpyloric tube. Thus, it is important to exclude C. difficile infection in hospitalized patients whose colitis does not respond to medical therapy as expected or who suddenly lose their initial response to treatment.
Patients with C. difficile infections often present with watery diarrhea (usually without rectal bleeding) and have characteristic pseudomembranes at colonoscopy. This infection can cause severe colitis that progresses to toxic megacolon and bowel perforation. Thus, appropriate stool studies for toxin analysis are necessary to exclude superimposed C. difficile infection, even in patients with established UC who present with an exacerbation.122
In patients from endemic areas, certain protozoan and parasitic infections need to be considered (see Chapters 109 and 110). Amebic colitis tends to have a more prolonged course than do most bacterial colitides, but amebiasis is not a cause of chronic colitis. Schistosomal colitis may be chronic and diffuse, exhibit pseudopolyps, and involve the rectum. The presence of characteristic ova in a biopsy specimen confirms the diagnosis.
Other infectious causes of a bloody diarrhea include opportunistic infections of the colon in immunosuppressed patients (see Chapters 33 and 34).
Cytomegalovirus (CMV) infection has been reported in patients with UC, typically those with long-standing disease who are being treated with glucocorticoids or immunosuppressants; the diagnosis of CMV infection should be considered whenever patients who have UC and are taking glucocorticoids either fail to respond as expected or lose their response to treatment. CMV colitis in steroid-naive patients also has been described.123 Patients with CMV colitis often present with abdominal pain and bloody diarrhea and have discrete deep ulcers on colonoscopy; however, CMV colitis can manifest with diffuse inflammation and resemble UC. Because the clinical presentation of CMV colitis may be indistinguishable from a flare of UC, a high index of suspicion is needed to make the diagnosis. Endoscopic biopsies should be obtained from both the ulcer bed and adjacent mucosa; careful histologic examination for giant cells with intranuclear inclusion bodies is important to confirm the diagnosis.
Mycobacterium avium complex usually causes patchy rather than diffuse inflammation.
OTHER CAUSES
Acute diverticulitis most commonly occurs in the sigmoid colon and does not involve the rectum (see Chapter 117). When the inflammation does extend to the rectum, it tends to be patchy and involves only the upper rectum. This appearance is more likely to be confused with Crohn’s disease than UC.
Ischemic colitis usually occurs in the elderly (see Chapter 114). The classic distribution is segmental involvement in the watershed areas around the splenic flexure or sigmoid colon; ischemic proctitis also has been described.
Radiation colitis usually occurs in patients who have been given radiation therapy for uterine, cervical, or prostate cancer. The location of disease depends on the sites irradiated but typically involves the rectosigmoid. The onset of symptoms often temporally corresponds to the radiation therapy but can develop years afterward (see Chapter 39).
Microscopic colitis, including lymphocytic and collagenous colitis, manifests with diarrhea and should be distinguished readily from UC by lack of rectal bleeding, the normal endoscopic appearance, and characteristic histopathology (see Chapter 124).
Patients with UC can present with symptoms similar to those of IBS (Chapter 118), specifically diarrhea, abdominal pain, fatigue, and poor general well-being. Lack of rectal bleeding and laboratory markers of inflammation, as well as a normal endoscopic and histologic appearance, helps to distinguish IBS from active UC. Patients with quiescent UC can have concomitant symptomatic IBS in the absence of active inflammation. The physician must be able to recognize these subtleties in order to provide appropriate management for the patient. Patients with UC also can present with symptoms mimicking colonic neoplasm (Chapter 123), solitary rectal ulcer syndrome (Chapter 124), diverticular disease (Chapter 117), and factitious diarrhea (Chapter 15). These diagnoses also do not give rise to diffuse inflammation in the colon and, therefore, should be distinguished easily from UC on colonoscopy.
ASSESSMENT OF DISEASE ACTIVITY
Although none of these indices is accepted universally as standard, one of the most commonly used is that of Truelove and Witts.124 This purely clinical classification categorizes disease as mild, moderate, or severe based on a combination of clinical findings and laboratory parameters, including frequency of bowel movements, rectal bleeding, fever, tachycardia, anemia, and elevated ESR (Table 112-6). The Truelove and Witts classification is reliable and simple to use in clinical practice, although it is most applicable for patients with extensive colitis and might not adequately reflect disease severity in patients with limited colitis.
Table 112-6 Truelove and Witts Classification of the Severity of Ulcerative Colitis
| Mild |
ESR, erythrocyte sedimentation rate.
Adapted from Truelove SC, Witts LJ. Cortisone in ulcerative colitis: Final report on a therapeutic trial. Br Med J 1955; 2:1041.
A numerical disease activity instrument that is more useful for patients with limited disease and for conducting clinical trials is the Ulcerative Colitis Disease Activity Index (UCDAI, also known as the Sutherland Index).125 This index, which combines clinical and endoscopic assessments, is the sum of scores from four components: stool frequency, rectal bleeding, sigmoidoscopic findings, and physician’s global assessment (Table 112-7). This disease activity index ranges from 0 to 12, with the higher total scores representing more-severe disease. In general, a patient is considered to be in remission if the UCDAI score is 2 or less and to have severe disease if the score is greater than 10. Clinical response generally is accepted to be reflected by a decrease of three points from the patient’s initial baseline score. An index very similar to the UCDAI that has been used extensively in recent randomized, controlled trials (RCTs) is the Mayo score, which incorporates the same four components as the UCDAI.126
Table 112-7 Ulcerative Colitis Disease Activity Index*
| SCORE | CRITERIA |
|---|---|
| Stool Frequency | |
| 0 | Normal |
| 1 | 1-2 stools/day > normal |
| 2 | 3-4 stools/day > normal |
| 3 | >4 stools/day > normal |
| Rectal Bleeding | |
| 0 | None |
| 1 | Streaks of blood |
| 2 | Obvious blood |
| 3 | Mostly blood |
| Mucosal Appearance | |
| 0 | Normal |
| 1 | Mild friability |
| 2 | Moderate friability |
| 3 | Exudation, spontaneous bleeding |
| Physician Global Assessment | |
| 0 | Normal |
| 1 | Mild |
| 2 | Moderate |
| 3 | Severe |
* Sutherland index: Range, 0-12.
(From Sutherland LR, Martin F, Greer S, et al. 5-Aminosalicylic acid enema in the treatment of distal ulcerative colitis, proctosigmoiditis, and proctitis. Gastroenterology 1987; 92:1894.)
Other scales also have been developed, many of which are modifications of the Truelove and Witts classification and the UCDAI. None of these disease activity instruments has ever been formally validated. There also exist many endoscopic and histologic scales for grading the severity of colitis (Table 112-8).127,128 Endoscopic findings do not always correlate with clinical symptoms, and such correlations generally are more consistent within individuals. Thus, although therapeutic decisions are based primarily on clinical status, it may be useful to follow the sigmoidoscopic mucosal appearance over time in an individual patient, if the clinical response to treatment is uncertain.
Table 112-8 Endoscopic and Histologic Assessment of Disease Activity in Ulcerative Colitis
| SCORE | CRITERIA |
|---|---|
| Endoscopic Assessment | |
| 0 | Normal mucosa |
| 1 | Loss of vascular pattern |
| 2 | Granular, nonfriable mucosa |
| 3 | Friability on rubbing |
| 4 | Spontaneous bleeding, ulceration |
| Histologic Assessment | |
| 0 | Normal |
| 1 | No significant inflammation: Possibly architectural changes of chronic disease and small foci of lymphocytes but no acute inflammation, crypt abscesses, or epithelial destruction |
| 2 | Mild to moderate inflammation: Edema, vascularity, increased acute and chronic inflammatory cells but intact epithelium |
| 3 | Severe inflammation: Heavy infiltrate of acute and chronic inflammatory cells, crypt abscesses, ulceration of surface epithelium, purulent exudate |
TREATMENT
MEDICAL
Several factors should be considered in determining optimal therapy for patients with UC (Fig. 112-12). Current therapeutic strategies can be classified broadly, based on disease activity, into those that treat active disease (induction therapy) (Table 112-9) and those that prevent recurrence of disease once remission is achieved (maintenance therapy) (Table 112-10). This concept of induction and maintenance of remission forms the basis of our evaluation of the efficacy of a specific therapy. The extent of disease is an important consideration that helps determine the route of administration of medication. Thus, for example, proctitis may be treated with suppositories or foam preparations as well as with oral therapy, and enema preparations may be used alone or in combination with systemic therapy for patients with left-sided disease. Other important factors to consider are a patient’s prior response to or side effects from a specific medication and compliance with medication. These factors might favor or preclude the use of a specific agent. Given the chronic nature of UC, medications need to be efficacious and well accepted by patients from the standpoints of safety and ease of administration. The mainstay of medical therapy focuses on regimens that alter host response to decrease mucosal inflammation. Therapies that target other aspects of the systemic inflammatory process or manipulate the enteric flora also have been developed to treat UC.
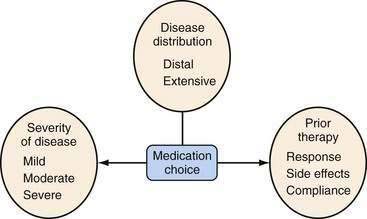
Figure 112-12. Factors that should be considered in the choice of medical therapies for ulcerative colitis.
Table 112-9 Induction Therapy for Ulcerative Colitis Based on Disease Severity
| Mild Disease |
IV, intravenous.
Table 112-10 Maintenance Therapy for Ulcerative Colitis
One important consideration when evaluating the efficacy of a particular medication (e.g., in a RCT that compares a novel therapy to placebo) is the placebo response rate. Even though placebos often are thought of as inert agents, they have been noted to lead to improvement in a variety of both subjective and objective outcome measures in a number of different medical conditions, such as anxiety, depression, insomnia, pain, asthma, obesity, hypertension, and even myocardial infarction.129 In some of these disorders, placebo response rates of up to 40% have been reported. With respect to placebo response rates in UC, a meta-analysis of 40 randomized, placebo-controlled trials, in which the most commonly used activity indices were the Mayo Score or the UCDAI, reported a pooled placebo response rate of 28% and remission rate of 13%.130 Studies using less-stringent outcome definitions were noted to have higher placebo response and remission rates. Univariate analysis suggested that longer follow-up duration, higher number of follow-up visits, longer disease duration, and lower disease severity at study entry were positively associated with the placebo remission rate.
Aminosalicylates
Oral
Sulfasalazine consists of an antibacterial component, sulfapyridine, bonded by an azo bond to a salicylate, 5-aminosalicylic acid (5-ASA, mesalamine) (Fig. 112-13).131 The drug was synthesized by Nana Svartz in 1938-1939 and its benefit for the treatment of IBD was discovered serendipitously in 1941-1942 by her when patients with UC receiving this medication for a presumed diagnosis of rheumatoid arthritis noted improvement in colitis symptoms132; in retrospect, these patients had peripheral arthropathy associated with their IBD. Research subsequently established that 5-ASA is the principal therapeutic moiety of sulfasalazine in IBD and that the sulfapyridine component of the parent drug serves as an inactive carrier, largely preventing absorption of 5-ASA in the small intestine and allowing it to be released in the colon.133,134 Approximately 90% of sulfasalazine reaches the colon, and only a small amount is absorbed in the small intestine. On reaching the colon, the enzyme azoreductase, which is elaborated by colonic bacteria, cleaves the azo bond to release the active constituent moiety, 5-ASA. After 5-ASA is absorbed from the colon, 20% of the compound undergoes hepatic acetylation, forming N-acetyl 5-ASA, and is excreted in the urine. Sulfasalazine is one of several agents in the class of 5-ASA compounds that is considered to be the first line of therapy for inducing remission in patients with mild to moderate UC.131,135 Mesalamine derivatives have not been evaluated in a randomized, controlled fashion in patients with severely active disease. At a dose of 3 to 6 g/day, sulfasalazine induces remission in 39% to 62% of patients with mild to moderate UC, about twice the remission rate of placebo-treated patients.136,137
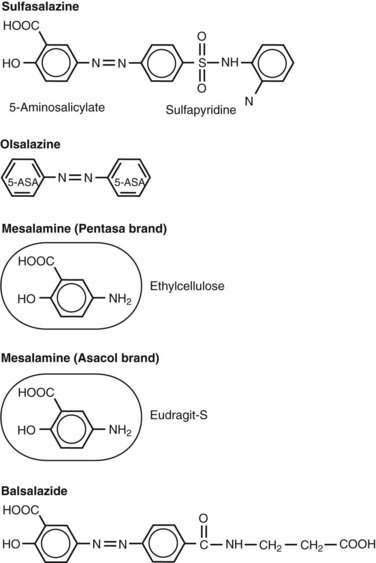
Various formulations and controlled-release systems (Table 112-11; see Fig. 112-13) have been developed to deliver 5-ASA to specific sites of the gastrointestinal tract without the sulfapyridine moiety, which is thought to be responsible for most of the side effects. Olsalazine (Dipentum) is a 5-ASA dimer linked by an azo bond and is formulated in gelatin capsules. Balsalazide (Colazal) consists of a 5-ASA monomer linked to a biologically inactive carrier molecule, 4-aminobenzoyl-β-alanine. Similar to sulfasalazine, 5-ASA is released from olsalazine and balsalazide in the colon upon cleavage of the azo bond via the bacterial enzyme azoreductase. Approximately 99% of the drug is delivered intact to the colon, and its metabolites are cleared rapidly in the urine.
Table 112-11 Oral 5-Aminosalicylic Acid Preparations and Sites of Delivery in the Gastrointestinal Tract
| DRUG | FORMULATION | SITE OF DELIVERY |
|---|---|---|
| Prodrugs | ||
| Sulfasalazine | Sulfapyridine + 5-ASA | Colon |
| Olsalazine | 5-ASA dimer | Colon |
| Balsalazide | 4-aminobenzoyl β-alanine + 5-ASA | Colon |
| Mesalamine Preparations | ||
| Asacol, Claversal, Salofalk | pH sensitive, resin-coated; delayed release | Distal ileum, colon |
| Rowasa | Enema | Distal colon |
| Canasa | Suppository | Rectum |
| Pentasa | Ethylcellulose-coated microgranules; controlled release | Duodenum to colon |
| Lialda | pH sensitive, multi-matrix and polymethacrylate coated; delayed and slow release | Distal ileum, colon |
5-ASA, 5-aminosalicylate.
These oral 5-ASA derivatives (mesalamines) have been shown to be superior to placebo for mildly to moderately active UC.137–142 Meta-analyses have demonstrated that the mesalamines are as efficacious as sulfasalazine, and the various mesalamine preparations appear to be comparable in efficacy.137,140 Balsalazide has been shown to have superior efficacy and a more rapid response compared with traditional mesalamine agents.143,144 In a RCT, balsalazide 6.75 g/day, a dose equivalent to mesalamine 2.4 g daily, achieved higher rates of remission and had better tolerance compared with pH-dependent mesalamine 2.4 g/day.143 It has been suggested that the greatest benefit of balsalazide is in patients with newly diagnosed left-sided UC.144
More important than the specific 5-ASA preparation is the dose-dependent response when 5-ASA is used as an induction therapy for active UC.137,140 For this indication, mesalamine is not effective at doses lower than 2 g daily, and there is an increased response at doses of 4 to 4.8 g daily. The ASCEND I and II trials showed that mesalamine at doses of 2.4 and 4.8 g/day have similar efficacy for patients with mildly active disease, but the higher dose (4.8 g/day) was more efficacious in patients with moderately active disease.145,146 This dose of mesalamine is comparable to 12 g/day of sulfasalazine, which is impractical in clinical practice because of the high probability of intolerance. No RCT has evaluated the use of aminosalicylates for severely active UC, but these agents are generally thought not to be effective in severely active disease.
Once remission is achieved, sulfasalazine and other 5-aminosalicylates are effective in maintaining it.147–150 This benefit appears to be dose dependent for sulfasalazine, with a dose of 2 g/day often used to balance efficacy and adverse side effects.147 Such a dose-dependent response, however, has not been found with the other 5-ASA preparations, and at doses of 1.5 to 4.8 g/day, remission can be maintained in more than 50% of patients.151 One meta-analysis has suggested that sulfasalazine might have a slight but statistically significant therapeutic superiority relative to the newer 5-ASAs in maintaining remission when considering trials of six months’ duration; however, when these trials were combined with those of 12 months’ duration, this statistically significant benefit was lost.151 A double-blind RCT comparing two doses of balsalazide (1.5 g twice daily and 3 g twice daily) with mesalamine 0.5 g three times daily for six months reported a remission rate of 77.5% with the higher dose of balsalazide compared with remission rates of 56.8% and 43.8% with mesalamine and the lower dose of balsalazide, respectively.152 In general, the same dose of 5-ASA derivative that induces remission is recommended for maintenance therapy, although this recommendation has not been formally tested in a randomized, placebo-controlled fashion.
Common side effects of sulfasalazine include fever, rash, nausea, vomiting, and headache (Table 112-12). Other, less-common but important side effects of sulfasalazine include hypersensitivity reactions, reversible sperm abnormalities, and impairment of folate absorption. Approximately 15% of patients taking sulfasalazine develop significant side effects that require discontinuing the medication. Up to 90% of patients who are intolerant to sulfasalazine, however, can tolerate mesalamine.153 In clinical trials, the newer 5-ASA preparations and balsalazide have been shown to be better tolerated than sulfasalazine,140,154,155 although the adverse event profiles during maintenance therapy appear to be similar for 5-ASA preparations and sulfasalazine.151 Sulfasalazine can impair folate absorption (by competitively inhibiting the jejunal enzyme, folate conjugase) thereby contributing to anemia, and folate supplementation should be prescribed to patients receiving sulfasalazine. Olsalazine is associated with drug-induced diarrhea in up to 10% of patients, which often limits its use. It has been noted that if olsalazine is ingested with meals and is continued despite the diarrhea, the incidence of this side effect can be lessened substantially to 3%. A systematic review of oral 5-ASA for maintenance of remission in UC found olsalazine to be significantly inferior to sulfasalazine, and this reduced efficacy was related mostly to a significantly higher rate of withdrawals because of adverse events.151 Oral mesalamine preparations do not appear to have significant dose-dependent toxicity.
Table 112-12 Side Effects of Sulfasalazine and 5-Aminosalicylates
| Dose-Related |
Topical
Topical mesalamine derivatives may be used as an alternative monotherapy or as an adjunctive therapy to oral agents in patients with left-sided colitis or pancolitis. They are effective for inducing remission in patients with mildly to moderately active distal UC, without a clear dose-response effect156,157 in nonrefractory patients. The standard dosing regimens used to induce remission are 1 to 4 g of 5-ASA in the form of an enema nightly, or mesalamine suppositories 1 to 1.5 g either nightly or in divided doses throughout the day. Mesalamine enemas have been shown to be comparable to oral sulfasalazine in the treatment of active distal UC, with fewer side effects.155 Similar efficacies have been demonstrated for mesalamine enemas regardless of whether the 1-, 2-, or 4-g formulation is used for inducing remission in patients with mild to moderate left-sided UC not requiring concurrent glucocorticoids or immunomodulators. In fact, mesalamine enemas are perceived to be even more effective than topical glucocorticoid enemas in this setting.157,159 A combination of topical and oral mesalamine also may be more effective than either agent alone in patients with left-sided colitis or pancolitis, suggesting a dose-response effect.160,161 In patients with proctitis, mesalamine suppositories, 500 mg administered twice daily, have been shown to be beneficial for treating active disease.162 Mesalamine foam has a more uniform distribution and longer persistence in the distal colon compared with mesalamine enemas. The foam preparation has been shown to have better patient acceptance than the enema preparation,163 but mesalamine foams currently are not available in the United States.
Topical mesalamine preparations also are effective for maintaining remission in left-sided UC or proctitis.156,157 The effective maintenance dosing interval ranges from nightly to every three days. Topical mesalamine is as effective as oral mesalamine,164 and the combination of topical and oral mesalamine may be more effective than oral mesalamine alone as a maintenance regimen.165
Glucocorticoids
Systemic
At doses equivalent to 40 to 60 mg/day of oral prednisone, glucocorticoids are effective first-line therapy for moderate or severe flares of UC.124,166–170 The use of doses higher than 60 mg/day is associated with increased side effects without appreciable clinical benefit and thus should be avoided. The addition of sulfasalazine to corticosteroids in moderately to severely active UC does not offer any incremental benefit. Although no study has directly compared the efficacy of oral and parenteral glucocorticoids, the latter commonly are used in severe disease. No adequately designed controlled study has been performed to confirm the clinical impression that continuous infusion of parenteral glucocorticoids is superior to pulse therapy.
The use of adrenocorticotropin (ACTH) has been suggested as an alternative to conventional glucocorticoid therapy of active UC in small studies.171 One double-blind RCT suggested that intravenous ACTH was more effective than intravenous hydrocortisone for the treatment of severely active UC only in steroid-naïve patients172; this observation has not been confirmed. Because most patients with severely active flares have been treated previously with glucocorticoids, ACTH rarely is used in clinical practice. A noteworthy complication of ACTH therapy is bilateral adrenal hemorrhage.
Glucocorticoids have no maintenance benefits in patients with UC. Steroid-dependent patients, or patients who are unable to taper off glucocorticoids without experiencing disease exacerbation, benefit from the addition of steroid-sparing agents. There has been no trial to date assessing mesalamine therapy and its efficacy in maintaining remission induced with glucocorticoids. The long-term remission rate in patients who require parenteral glucocorticoids for severe UC is approximately 50%.173 Immunomodulatory agents, as discussed, should be considered in patients who are dependent on steroids, who require two courses of glucocorticoids for induction of clinical response or remission within one year, or who require parenteral glucocorticoids to induce remission. In addition to the use of immunomodulatory agents, one should consider using infliximab for steroid-dependent patients.
Glucocorticoids are associated with many mild and serious side effects in patients with IBD (Table 112-13). These side effects occur commonly and involve nearly every organ system. Every effort should be made to minimize glucocorticoid use and exposure.
| Cutaneous |
Budesonide is a glucocorticoid preparation that is structurally different from prednisone. The presence of 16α,17α-acetyl side chains allows enhanced topical anti-inflammatory activity and affinity for glucocorticoid receptors compared with prednisone.174 In addition, budesonide has an approximately 90% first-pass metabolism in the liver and erythrocytes and is converted to metabolites that have little or no biological activity. The resultant low systemic bioavailability translates to significantly less toxicity compared with traditional glucocorticoids. Entocort is a controlled-ileal-release oral budesonide preparation consisting of Eudragit-L-100–coated microgranules with an internal ethyl cellulose component; it releases budesonide at pH greater than 5.5, and about 50% to 80% of budesonide is absorbed in the ileocecal region. There currently is no oral formulation of budesonide that provides optimal release characteristics for the entire length of the colon. A small uncontrolled study has suggested that Budenofalk, which is not available in the United States, may be effective for prednisone-dependent UC.175 Controlled studies have not shown the benefit of oral budesonide for the treatment of active UC.176
Topical
Topical glucocorticoids in liquid and foam formulations are effective short-term therapy for active UC distal to the splenic flexure.177,178 Foam preparations often are tolerated better by patients and may be easier to retain than liquid preparations. Topical glucocorticoids have been found to be less effective than topical mesalamine for inducing remission of distal UC159; however, the combination of topical corticosteroids and topical mesalamine has been more efficacious than either alone in the short-term treatment of distal UC.179
Whereas systemic absorption of glucocorticoids with topical therapy is significantly less than that with oral administration, prolonged treatment with topical glucocorticoids still may be associated with steroid-related side effects and should be avoided. As mentioned previously, budesonide is a potent corticosteroid with a rapid first-pass metabolism. Budesonide enemas, which currently are neither available nor approved in the United States, have been shown to be effective for the treatment of active distal UC in several controlled trials. In a double-blind RCT of patients with active distal UC, budesonide, 2 mg/100 mL for six weeks, resulted in a remission rate of 19% compared with 4% in patients receiving placebo therapy (P < 0.05).180 Subsequent trials have shown budesonide enema to be as efficacious as or even superior to prednisolone enema without resultant depression of endogenous cortisol levels.181–183
Budesonide enema perhaps is inferior in efficacy to mesalamine enema,184 but it clearly presents an alternative topical glucocorticoid for treatment of distal UC. The optimal dose for budesonide enema consistently has been shown to be 2 mg/100 mL once daily.180,181,185 Budesonide in foam preparation also has been shown to have comparable efficacy with traditional hydrocortisone foam for the treatment of active proctosigmoiditis.186 Additional studies are needed to determine the effect of longer-term topical budesonide use. As with other glucocorticoid preparations, budesonide enema is not effective for maintaining remission in UC.185
Immunomodulators
Azathioprine and 6-Mercaptopurine
Of the various immunomodulatory agents, the most widely used are azathioprine and 6-MP. These two agents are purine analogs that interfere with nucleic acid metabolism and cell growth and exert cytotoxic effects on lymphoid cells. They are inactive prodrugs with subtle structural differences. Azathioprine is nonenzymatically converted to 6-MP, which is then metabolized through a series of enzymatic pathways to active and inactive metabolites (see Fig. 111-8). The two primary metabolites of 6-MP are 6-thioguanine nucleotides (6-TGNs) and 6-methylmercaptopurine (6-MMP). The 6-TGN metabolites are thought be responsible for the immunomodulatory action of azathioprine and 6-MP and their bone marrow suppression property, whereas hepatotoxicity is thought to be related to 6-MMP. One key enzyme involved in the biotransformation of 6-MP is thiopurine methyltransferase (TPMT), which converts 6-MP to its inactive metabolites, 6-MMP and 6-methylmercaptopurine ribonucleotides.
There is a population polymorphism in the TPMT gene: 89% of the population have homozygous wild-type TPMT, and 11% and 0.3% of the population have heterozygous and homozygous mutations, respectively.187 Persons with heterozygous and homozygous TPMT mutations have decreased to absent enzyme activity. The clinical significance of this genetic polymorphism is that inherited differences in TPMT may be responsible for most of the variability in drug response observed among individual patients.
The efficacy of azathioprine in the treatment of UC is a matter of debate. Four RCTs have evaluated azathioprine for inducting remission in active UC (Table 112-14).188–191 These four studies were small, heterogeneous in design, used different outcome definitions for response, and reached different conclusions. Two of the studies involved steroid-dependent patients,190,191 one other study used steroids for induction,188 and two studies used 5-ASAs as a comparator group rather than placebo.189,191 Only one study showed a significant benefit with azathioprine compared with 5-ASA for induction therapy in steroid-dependent disease.191
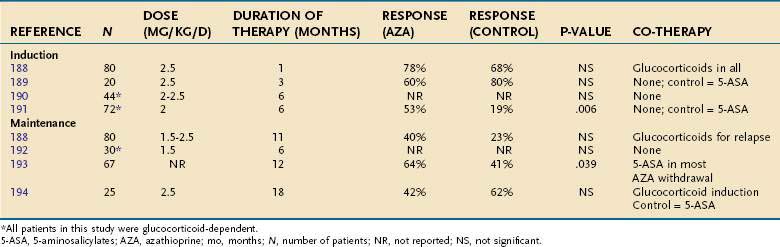
With respect to the use of azathioprine for maintenance of remission in UC, four RCTs have been performed (see Table 112-14).188,192–194 Just as with studies of induction therapy, these four studies also had small sample sizes, used heterogeneous designs with different outcome definitions of response, allowed for various cotherapies, and again reached different conclusions. One of the studies was in steroid-dependent disease,192 another allowed the use of steroids for relapse,188 one study used 5-ASA as a comparator group rather than placebo,194 and another included patients who were mostly taking 5-ASAs and was actually a study of azathioprine withdrawal.193 Only this withdrawal study showed a benefit with continued azathioprine.
The optimal dose of azathioprine or 6-MP for treating UC is unclear, and no formal dose-ranging study has been reported in the literature. The effective doses for 6-MP and azathioprine generally are 1 to 1.5 mg/kg/day and 2 to 3 mg/kg/day, respectively.195 At these doses, however, there still may be nonresponders and, for them, higher doses may be necessary. Induction of leukopenia had been advocated for dose optimization,196 but this practice was not supported by subsequent studies.197–199 Monitoring metabolite levels may be beneficial in determining the optimal dose of azathioprine or 6-MP.
To date, at least 13 studies examining response in IBD with respect to 6-TGN level have been published. A meta-analysis of the first 12 of these studies found that the studies were similar in that they were retrospective and the majority of patients were adults with Crohn’s disease, but they were heterogeneous with respect to sample size, the proportion of patients in remission, and the activity indices used to assess response.200 Of the seven studies that reported data on 6-TGN threshold levels, a pooled analysis of the first six studies showed a three-fold significantly higher rate of remission among patients with a 6-TGN level of greater than 230 to 260 pmol/8 × 108 red blood cells. Incorporation of 6-TGN metabolite measurement into the management regimen of patients receiving azathioprine or 6-MP therapy for IBD is not mandatory and it is a subject of continuing controversy.
Currently, it is recommended in the package insert and by the U.S. Food and Drug Administration (FDA) to determine TPMT genotype or phenotype before initiating therapy. The active metabolites, 6-TGNs, also are responsible for myelosuppression with therapy, and patients with TPMT mutation or decreased TPMT enzyme activity are more likely to experience this toxicity because of preferential shunting of 6-MP metabolism toward the excessive production of 6-TGN.201 Thus, identifying TPMT polymorphism before initiating azathioprine or 6-MP therapy can decrease the risk of myelotoxicity. Patients with homozygous wild-type TPMT or normal (to high) TPMT enzyme activity level may receive these agents starting at the weight-based optimal dose of 2.5 mg/kg/day for azathioprine or 1.5 mg/kg/day for 6-MP. It has been suggested by some investigators that in patients with heterozygous TPMT mutation or intermediate enzyme activity level, 6-MP or azathioprine should be started at 50% of the weight-based optimal dose. Alternative therapy should be considered in patients with homozygous mutations for TPMT. Regardless of whether a patient’s TMPT genotype or phenotype is known, continued frequent monitoring of complete blood counts remains necessary, because only 27% of all patients with leukopenia have TPMT mutations.202 In addition, two studies have reported that TPMT testing may be cost effective.203,204
Azathioprine and 6-MP therapy have a delayed onset of action. The mean time to clinical response with azathioprine or 6-MP therapy in patients with UC has been reported to be three to four months in uncontrolled studies,205,206 a figure that is similar to the 17 weeks’ response time to clinical benefit in placebo-controlled trials of azathioprine or 6-MP therapy for active Crohn’s disease.207 Intravenous loading of azathioprine at 40 mg/kg for 36 hours does not shorten the time required for a therapeutic response in patients with Crohn’s disease.202 Such practice presumably would have the same results if attempted in patients with UC.
Because azathioprine or 6-MP therapy is associated with a number of potentially significant toxicities, its duration of therapy should be determined by weighing clinical benefit against these potential toxicities. The optimal duration of maintenance therapy with azathioprine or 6-MP currently is unknown in patients with UC. In patients with Crohn’s disease, the maintenance benefit of azathioprine or 6-MP can be observed for at least five years.208,209 Based on these data in Crohn’s disease and the paucity of alternative maintenance therapies, in patients with UC in whom remission is maintained with azathioprine or 6-MP, treatment generally is continued indefinitely as long as there is no significant adverse side effect.
Common side effects of azathioprine and 6-MP therapy include nausea, vomiting, bone marrow suppression, pancreatitis, allergic reactions, and infections (Table 112-15).210,211 Bone marrow suppression occurs in 2% to 5% of patients.210,212 It is dose dependent and manifests primarily as leukopenia, although all three cell lines may be affected. This hematologic toxicity can increase with concurrent use of sulfasalazine or mesalamine compounds.198,213–215 It is known that mesalamine can interact with the enzyme TPMT, leading to increased levels of 6-TGN, and that this interaction has been associated with leukopenia. Bone marrow suppression is managed by reducing the dosage of immunomodulator or withdrawing the medication. Routine monitoring of complete blood count with differentials is necessary for patients receiving azathioprine or 6-MP and should be continued for the entire duration of therapy. Allergic reactions to azathioprine or 6-MP usually manifest as fever, rash, and arthralgia and resolve following discontinuation of these medications.212,216 Recurrence of similar reactions occurs with medication challenge, although patients who develop allergic reactions to one agent may be able to tolerate subsequent challenge with the other.217 Pancreatitis also is idiosyncratic and independent of dosage.212,218,219 It usually occurs during the first month of therapy and is reversible upon withdrawal of the drug.
Table 112-15 Side Effects of Azathioprine and 6-Mercaptopurine
Patients using azathioprine or 6-MP therapy can have abnormal liver biochemical tests, but these usually resolve following drug withdrawal.220 Because liver biopsy is not performed routinely in these patients, their pattern of hepatic injury, if any, is unknown. Cholestasis with inflammation, nodular regenerative hyperplasia, and peliosis hepatis have been reported with azathioprine and 6-MP therapy.212,220 As is the case for complete blood counts, routine monitoring of liver biochemical tests is recommended. An increased risk of malignancy, primarily lymphoma, has been reported, but not consistently. A meta-analysis of six studies examining this risk reported a four-fold elevated risk of lymphoma with 6-MP/azathioprine.221 The lymphoma that develops in patients who have IBD and receive these immunomodulatory agents appears to be associated with Epstein-Barr virus.222
Cyclosporine
There has only been one randomized, placebo-controlled trial evaluating the efficacy of intravenous cyclosporine in severe UC. In this study of 20 patients who did not respond to at least seven days of intravenous hydrocortisone, nine (82%) of the 11 patients receiving continuous intravenous infusion of cyclosporine at 4 mg/kg/day responded, compared with none of the nine patients receiving placebo therapy.223 The time to clinical response was rapid, at a mean of seven days. After the intravenous route of therapy was converted to oral cyclosporine, 44% of those patients who responded initially required colectomy during the six-month follow-up period.
Intravenous cyclosporine monotherapy may be as effective as intravenous glucocorticoids in patients with severely active UC; its use thus potentially minimizes the toxicities of combination therapy.224 The addition of azathioprine or 6-MP in patients who have responded to intravenous cyclosporine has been shown in other studies to reduce the rate of relapse or colectomy.225,226 Thus, cyclosporine can be considered a bridge therapy to control active disease in patients with steroid-refractory UC while waiting for elective surgery or the onset of action of azathioprine or 6-MP.
With the addition of azathioprine, long-term remission at one year may be more likely in patients who initially respond to intravenous cyclosporine monotherapy than in those who respond to intravenous glucocorticoids. A European retrospective cohort study of 142 patients who were treated with cyclosporine, 118 of whom responded initially, reported the probability of avoiding colectomy to be 63% at one year, 41% at four years, and 12% at seven years; overall, 54% of patients required colectomy at some point.227 Patients who were already taking 6-MP or azathioprine at the time cyclosporine was initiated continued taking their current dose, and those who were naïve to 6-MP or azathioprine were started at target doses at the time of response to cyclosporine during their hospitalization. The authors found that 59% of patients previously taking 6-MP or azathioprine required eventual colectomy, compared with 31% for patients naïve to these drugs (P < 0.05).
Because most of the serious adverse effects associated with the use of cyclosporine are dose-dependent, intravenous doses lower than 4 mg/kg that still can achieve efficacy are desirable. One RCT has shown that a dose of 2 mg/kg is as effective as 4 mg/kg given intravenously in patients with severely active UC, judged by clinical response rates, time to response, and short-term colectomy rates.228 The mean plasma cyclosporine levels were 237 ng/mL in patients receiving the 2 mg/kg dose and 332 ng/mL in patients receiving the 4 mg/kg dose. Thus, initiating therapy at 2 mg/kg may be reasonable, but regardless of the dose used, careful monitoring of plasma cyclosporine trough levels is necessary.
Cyclosporine has been associated with many adverse effects, including paresthesias, tremors, headache, hypertrichosis, and gingival hyperplasia (Table 112-16). Other potentially serious toxicities include hypertension, seizures, electrolyte and liver biochemistry abnormalities, nephrotoxicity, anaphylaxis, and opportunistic infections. These complications are mostly dose-dependent. Severe complications have been reported with cyclosporine in up to 12% of patients with UC,229 and two large series have reported death rates of 1.8% to 2.8% with cyclosporine, more than half of which were due to infections acquired while taking the drug.227,229
Table 112-16 Side Effects of Cyclosporine
Methotrexate
Methotrexate is a folic acid antagonist and has antimetabolite and anti-inflammatory properties. Although early reports suggested potential benefit of methotrexate administered intramuscularly or orally in UC, the only randomized, placebo-controlled trial failed to demonstrate its efficacy for the treatment of active UC.230 In this study of 67 patients with chronic active UC, oral methotrexate at 12.5 mg/wk for nine months was comparable to placebo therapy in the rate of achieving first remission, time to first remission, relapse following remission, and the mean glucocorticoid dose. It is unknown if methotrexate at higher doses administered intramuscularly or subcutaneously may be beneficial in inducing or maintaining remission in UC. Given the absence of data supporting its efficacy, methotrexate cannot at this time be considered a standard therapy for UC.
Other Immunomodulators
Alternative immunomodulators have been explored for patients who do not tolerate or have not responded to the previously mentioned immunosuppressants. Mycophenolate mofetil has pharmacodynamic properties similar to those of azathioprine and 6-MP but a more rapid onset of action. A pilot study of patients with chronic active UC receiving concomitant prednisolone found azathioprine to be superior to mycophenolate mofetil throughout the one-year study period, with remission rates at one year of 100% and 88%, respectively.231 Uncontrolled studies reported less than 50% remission rates with mycophenolate mofetil therapy in patients with steroid-dependent UC232,233 and the intolerance rate was high.232 A substantial number of patients developed adverse effects necessitating drug withdrawal, including recurrent upper respiratory tract infection, bacterial meningitis, depression, and migraine headache.231,233
Tacrolimus is another immunosuppressant with actions similar to those of cyclosporine. In contrast to cyclosporine, it has a 100-fold greater potency and a more-rapid onset of action. A number of small uncontrolled studies have suggested benefit of oral or intravenous tacrolimus for the treatment of patients with refractory UC. The only randomized, placebo-controlled trial of tacrolimus in UC involved 63 Japanese patients with either steroid-dependent or steroid-refractory disease who were randomized to receive either initial oral tacrolimus at 0.05 mg/kg or placebo twice daily.234 Patients in the high-trough concentration (10 to 15 ng/mL) tacrolimus group had a significantly higher rate of response and nonsignificantly higher rate of remission than those in the placebo group at week two, and a number of patients demonstrated response or remission (or both) after an additional 10 weeks of open-label therapy. As with cyclosporine, tacrolimus can result in a number of toxicities including nephrotoxicity, electrolyte abnormalities, nausea, diarrhea, headache, tremors, paresthesias, insomnia, alopecia, hirsutism, and gingival hyperplasia.235,236 Thus, given the limited data and potential for harmful adverse events, the use of these alternative immunomodulators currently is not incorporated into standard practice.
Antibiotics
Antibiotics have a limited role in the management of UC, and most controlled studies have not demonstrated their benefit either in active disease or maintenance of remission.237–241 The most commonly used antibiotics in this setting are metronidazole and ciprofloxacin. One RCT found oral tobramycin to be superior to placebo as a short-term adjunctive therapy to glucocorticoids for active UC.242 Another RCT reported a modest benefit for the addition of ciprofloxacin for six months in patients with UC refractory to mesalamine and corticosteroids.243 At present, the data showing efficacy of antibiotics for treatment of patients with UC are not as convincing as are the data for antibiotic treatment of Crohn’s disease. Thus, at present the primary role of antibiotics in the treatment of UC is in the management of its suppurative complications.
Probiotics, Prebiotics, and Synbiotics
Probiotics are living organisms in foods and dietary supplements that might beneficially affect the host in a number of ways, including improving its intestinal microbial balance, blocking adhesion sites on colonocytes (which might improve mucosal barrier function), and enhancing local immune response.39,244 A probiotic can be a specific nonpathogenic strain of a bacterial species or a mixture of multiple species and strains, most commonly including Lactobacillus or Bifidobacterium species; sometimes they contain fungal antigens as well. An example of a common probiotic is VSL#3, which contains four strains of Lactobacillus (Lactobacillus acidophilus, Lactobacillus delbrueckii subspecies bulgaricus, Lactobacillus plantarium, and Lactobacillus casei), three strains of Bifidobacterium (Bifidobacterium infantis, Bifidobacterium longum, Bifidobacterium breve), and one strain of Streptococcus (Streptococcus salivarius subspecies thermophilus).
Prebiotics are nondigestible food ingredients that selectively stimulate the growth or activity of one or more organisms of the intestinal microbiota, such as Lactobacillus or Bifidobacterium species, thereby potentially conferring beneficial effects to the host.245,246 The majority of prebiotics are nondigestible oligosaccharides, with galacto-oligosaccharide, fructo-oligosaccharide, lactulose, and inulin being the most commonly used agents.
With respect to the use of these agents for inducing remission in mildly to moderately active UC, four RCTs have been performed using different agents.247–250 Two of three studies that measured rates of remission found no benefit of probiotics (VSL#3 in one study, fermented milk in the other) added to 5-aminosalicylates247,248; the third study found that E. coli Nissle 1917 combined with glucocorticoids had efficacy similar to that of mesalazine combined with glucocorticoids.249 The fourth study, which used a synbiotic, reported a nonsignificant improvement in disease activity when the synbiotic was combined with standard therapy.250 With respect to the use of these agents for the maintenance of remission in mildly to moderately active UC, six RCTs have been published.249,251–255 Two of these studies reported significantly lower rates of relapse for patients receiving a probiotic (Bifidobacterium in one study, fermented milk in the other) after medically induced remission compared with those receiving placebo,251,252 and the other four studies (using E. coli Nissle in three studies and Lactobacillus rhamnosus strain GG in the fourth) found no difference in rates of relapse.250,253–255
Nontraditional probiotic therapies that also have been evaluated include Saccharomyces boulardii and Trichuris suis.256,257 A small, uncontrolled study of 24 patients with mild to moderate active UC suggested a potential benefit of Saccharomyces boulardii when used in addition to mesalamine.256 The use of helminths in active UC was investigated by Weinstock and colleagues, who randomized 54 patients with active disease to receive 2500 T. suis ova or placebo orally every 2 weeks for 12 weeks and reported that rates of improvement were significantly higher in the active treatment group at week 12 (43% vs. 17%, P = 0.04); significant improvement was seen as early as week six.257
Nutritional Therapy
Short-chain fatty acids, especially butyrate, have been shown to be the main energy substrate for colonocytes. Butyrate metabolism accounts for approximately 70% of colonocyte oxygen use. The suggestion that there is an impairment of colonocyte oxidation of short-chain fatty acids in UC led to therapeutic investigations of this form of nutritional therapy. Indeed, placebo-controlled studies have found butyrate enemas to be beneficial in treating mildly active, left-sided colitis.258–260
Fish oil containing eicosapentaenoic acid has been found to attenuate colitis in animal models of colitis, probably by protecting the integrity of colonic mucosa, suppressing the inflammatory response, or both.261–263 In a small, placebo-controlled, crossover study of patients with mild to moderate UC, treatment with fish oil resulted in a 56% reduction in disease activity compared with a 4% reduction in controls (P < 0.05).264 This benefit has not been confirmed in other studies, and a benefit in maintaining remission has not been observed.265–267 Furthermore, compliance is limited because of side effects and the odor of the fish oil preparation.
In contrast to Crohn’s disease, where bowel rest and total parenteral nutrition can improve disease, multiple studies have not found total parenteral nutrition with or without bowel rest to have any therapeutic advantage in patients with UC.268,269 Parenteral nutrition, however, can offer nutritional benefit in these patients. In general it is important to provide adequate nutrition to patients with UC who are about to undergo surgery. Nutrition is no more effective than placebo, however, for use as primary therapy of active UC.
Nicotine
Based on the observation that smoking is associated with a decreased risk of developing UC and that a former smoker with active colitis may gain clinical benefit on resuming smoking, nicotine has been used to treat patients with this disease. RCTs have shown some benefit of transdermal nicotine in the treatment of active UC.270–273 When administered at the highest tolerated dosage of 22 mg/day or less for four weeks in patients with mildly to moderately active UC, transdermal nicotine resulted in clinical improvement in 39% of patients compared with 9% of patients who received placebo therapy (P = 0.007).270 As a single therapy, however, transdermal nicotine was not as effective as low-dose prednisolone.274 Common side effects included nausea, lightheadedness, itching, and tremor. Topical nicotine therapy has fewer side effects and may be an alternative. Pilot studies have shown topical nicotine to be beneficial in patients with distal UC, but no large RCT has been performed,275,276 and transdermal nicotine has not been found to be effective as a maintenance therapy.277 Thus, based on available data on clinical efficacy and the overall poor patient tolerability, nicotine cannot be considered part of the standard treatment for patients with UC.
Heparin
Because of their negative charge, the glycosaminoglycans that constitute heparin have varied biological effects, including significant anti-inflammatory actions and augmentation of the peptide growth factors involved in intestinal mucosal repair and regeneration. Based on reports of fortuitous improvement in patients with UC receiving heparin for treatment of deep venous thromboses, pilot studies have suggested that unfractionated heparin may be effective for inducing remission in patients with severe, refractory UC.278,279 Compared with glucocorticoids as a first-line therapy, however, small RCTs have reported conflicting results.280,281
Intravenous heparin therapy has been associated with substantial bleeding complications. Low-molecular-weight heparin (LMWH) offers advantages over unfractionated heparin in its subcutaneous route of administration, and preliminary studies suggested a benefit of LMWH in the treatment of active UC.282,283 Unfortunately, this finding was not confirmed in a large, placebo-controlled trial of patients with mildly to moderately active UC receiving LMWH for six weeks.284 At this time, the use of either unfractionated heparin or LMWH cannot be advocated as primary therapy for patients with active UC.
Biological Therapy
Anti-Tumor Necrosis Factor Antibodies
TNF is a key proinflammatory cytokine that has been demonstrated to play a role in several disease states, including IBD. Elevated TNF concentrations have been found in inflamed intestine in patients with Crohn’s disease and UC, and stool and mucosal concentrations of TNF in patients with IBD have been shown to correlate with clinical disease activity. Infliximab (Remicade) is a chimeric monoclonal antibody of IgG1 subclass directed against human TNF-α. It consists of 75% human and 25% murine components (Fig. 112-14). The efficacy of infliximab in Crohn’s disease is well established, and it is approved by the FDA to treat Crohn’s disease and UC. Infliximab is thought to operate in Crohn’s disease via a multitude of mechanisms, including antagonizing the activity of TNF-α,285,286 initiating cytotoxicity on immune cells,287 and inducing T-cell apoptosis.288,289
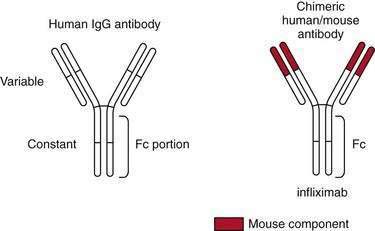
Results from two large, multicenter, randomized, double-blind, placebo-controlled trials (ACT 1 and 2) showed efficacy of infliximab therapy in UC.290 In these two similarly designed trials, 728 patients with moderately to severely active UC who failed conventional therapy with glucocorticoids alone or in combination with thiopurines (ACT 1) or glucocorticoids alone or in combination with thiopurines and 5-aminosalicylates (ACT 2) were randomized to placebo, infliximab 5 mg/kg, or infliximab 10 mg/kg at weeks 0 and 2 and then every eight weeks through week 46 (ACT 1) or week 22 (ACT 2). With respect to clinical response at week 8, in ACT 1 69% and 61% of patients receiving infliximab at 5 and 10 mg/kg, respectively, had a clinical response, compared with 37% of patients receiving placebo (P < 0.001 for both comparisons). In ACT 2 at week 8, 64% and 69% of patients receiving infliximab at 5 mg/kg and 10 mg/kg, respectively, had a clinical response, compared with 29% of patients receiving placebo (P < 0.001 for both comparisons). With respect to clinical remission at week 8 in ACT 1, 39% and 32% of patients receiving infliximab at 5 mg/kg and 10 mg/kg, respectively, attained remission, compared with 15% of patients receiving placebo (P < 0.003 for both comparisons). In ACT 2 at week 8, 34% and 28% of patients receiving infliximab at 5 mg/kg and 10 mg/kg, respectively, attained remission, compared with 6% of patients receiving placebo (P < 0.001 for both comparisons). The results for clinical remission at week 30 (ACT 1 and 2) and week 54 (ACT 1) were very similar for all groups, with highly significant greater than two-fold higher remission rates for the infliximab-treated patients. The proportions of patients with a sustained clinical response or remission also were significantly higher in the infliximab groups. Treatment with infliximab also was shown to have steroid-sparing and mucosal healing properties.
Anti-Adhesion Molecules
Several agents directed at blocking small adhesion molecules have been evaluated for the treatment of UC. These molecules are glycoproteins expressed on the surfaces of endothelial cells and lymphocytes. Adhesion molecules are important in cellular trafficking in IBD and other diseases, in which immune and inflammatory cells from the periphery are recruited into sites of inflammation. Among these, natalizumab is a humanized IgG4 monoclonal antibody against lymphocyte adhesion molecules, α4 integrins. A pilot study of 10 patients with active UC suggested clinical benefit with a single infusion of 3 mg/kg of natalizumab.291 Natalizumab currently is approved for treating patients who have Crohn’s disease and in whom anti-TNF therapy has failed; its use in UC is now undergoing evaluation.
Another anti-adhesion molecule agent is MLN-02 (formerly called LDP-02), a humanized IgG1 monoclonal antibody to α4β7 integrin. In a phase 2 study, two infusions of 0.5 mg/kg of MNL-02 administered 29 days apart were found to be effective in achieving clinical remission and response at six weeks after the initial infusion in patients with moderately active UC.292
Others
Although historically considered more important in the inflammation of Crohn’s disease, as it is produced by Th1 cells, IL-2 also has been implicated in UC inflammation (see earlier). Two agents designed to block the binding of IL-2 to its receptor have been examined for potential efficacy in UC. Daclizumab, a humanized monoclonal antibody against the IL-2 receptor (CD25), was suggested to be beneficial in patients with refractory UC in a small open-label pilot study.293 A potential clinical benefit also has been reported with basiliximab, a chimeric monoclonal antibody to the IL-2 receptor, in a small, uncontrolled study of patients with steroid-refractory UC.294 Along with the emphasis on T-cell–mediated immune response in the pathogenesis of UC, a humanized monoclonal antibody to CD3, visilizumab, has shown promise in an open-label phase 1 study of 32 hospitalized patients with UC whose disease failed to respond to intravenous glucocorticoids.295
Other biological therapies include agents targeted at tissue repair and restitution following mucosal injury. In this regard, epidermal growth factor (EGF) is a potent mitogenic peptide that stimulates cell proliferation in the gastrointestinal tract. A preliminary study showed that EGF enemas at a dose of 5 µg daily for two weeks was effective in treating mild to moderate left-sided UC when administered along with oral mesalamine.296 In contrast, another potent stimulant of intestinal epithelial cells, repifermin (keratinocyte growth factor 2) was not found to be more effective than placebo when administered intravenously in patients with active UC in a phase 2 dose-ranging study.297 Further studies clearly are necessary to confirm some of these early promising findings.
Cytapheresis
Active UC is characterized by activation and infiltration of leukocytes in the colonic mucosa. Because leukocyte-derived inflammatory cytokines play an important role in initiating and perpetuating the inflammatory process, reduction of peripheral blood levels of leukocytes has been proposed as a therapeutic option for treating UC. Several methods of depleting peripheral blood leukocytes have been developed and have been shown to hold promise in the treatment of severely active UC in small controlled and uncontrolled studies.298–305
Granulocyte/monocyte apheresis using the Adacolumn Apheresis System (JIMRO, Ltd., Takasaki, Japan) in patients with moderate to severe active UC was formally evaluated in a large multicenter randomized, double-blind, sham-controlled North American pivotal trial of 168 patients and a smaller identically designed companion trial of 47 patients in Europe and Japan.306 In this study, patients were required to have active disease despite concomitant therapy with 5-aminosalicylates, glucocorticoids, or 6-MP and azathioprine. Neither the pivotal nor the companion study showed a difference in clinical remission or response rates (as defined by the Mayo score) at week 12 between the Adacolumn and sham treatment groups after a total of 10 apheresis sessions over a nine-week period. Thus, at this time, Adacolumn apheresis in similar patients cannot be recommended.
Peroxisome Proliferator Receptor Agonists
Peroxisome proliferator-activated receptor (PPAR)-γ is a nuclear hormone receptor best known for its roles in regulating metabolism and adipocyte differentiation. It also has been shown to have immunomodulatory and anti-inflammatory properties in multiple sites, including the colon.307–310 A randomized double-blind, placebo-controlled trial assessed the efficacy of a 12-week treatment with the PPAR-γ ligand rosiglitazone in 105 patients with mildly to moderately active UC.311 The authors reported a significant benefit of rosiglitazone over placebo with respect to clinical response (44% vs. 23%, P = 0.04) and clinical remission (17% vs. 2%, P = 0.01) at 12 weeks based on the Mayo score. However, given the reports of a potential for increased risk of myocardial infarction and long-bone fractures with rosiglitazone (based on studies in diabetic patients, in whom this medication is mostly used), use of rosiglitazone should be restricted to patients who have failed or cannot tolerate standard medical therapy.
ALGORITHMS FOR THE TREATMENT OF ULCERATIVE COLITIS
An algorithm for treating patients with active UC of mild to moderate severity is outlined in Figure 112-15. The treatment of severely active UC is shown in Figure 112-16.
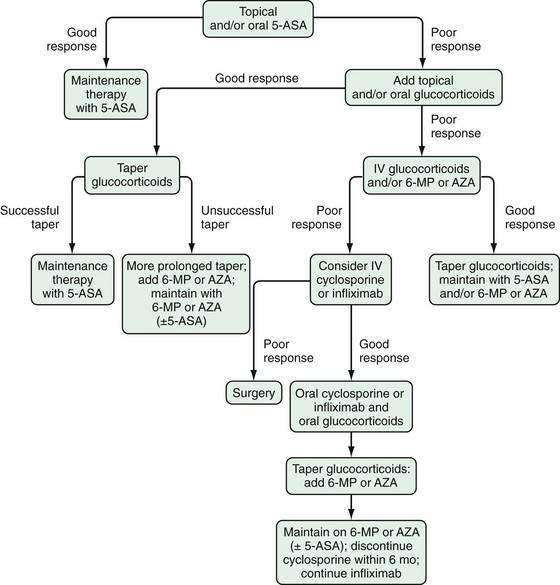
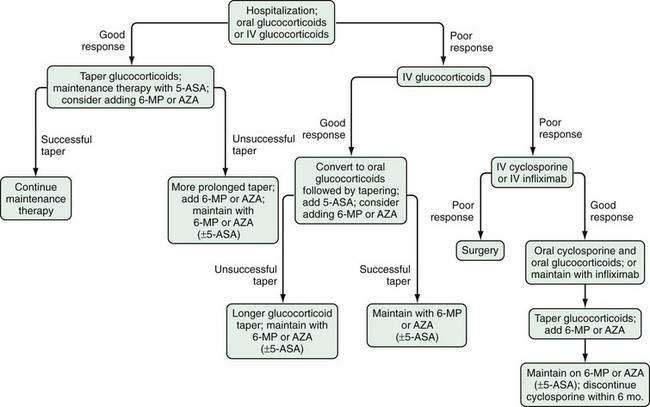
SURGICAL
Removal of the colon and rectum cures UC. Common indications for surgical therapy of UC are medically refractory disease, intractable disease with impaired quality of life, and unacceptable side effects from medical therapy (Table 112-17). Other indications include uncontrolled bleeding, toxic megacolon, perforation, dysplasia or carcinoma, systemic complications, and growth retardation. The goals of surgical treatment are to remove the entire diseased colon while preserving continence and sexual function. Elimination of the risk of colorectal cancer is also important. The role of prophylactic proctocolectomy in patients with long-standing extensive UC is controversial. Whereas most clinicians do not routinely recommend proctocolectomy solely for the purpose of prophylaxis against colorectal cancer, patients should be informed of the limitations of our current colonoscopic surveillance program (see Chapters 122 and 123).
Table 112-17 Indications for Surgery in Patients with Ulcerative Colitis
There are multiple surgical options for UC (see Chapter 113), including subtotal colectomy with ileostomy, colectomy with ileorectal anastomosis, proctocolectomy with Brooke ileostomy, proctocolectomy with continent ileostomy, restorative proctocolectomy with ileal pouch-anal anastomosis (IPAA), and proctocolectomy with ileal pouch-anal transition zone anastomosis (Fig. 112-17). The choice of operation is based on several factors, including the indication for and urgency of surgery, the age and general health of the patient, the status of the patient’s anal function, and the patient’s preference of functional outcome and lifestyle.
Figure 112-17. Schematic diagrams of various surgical options for the management of ulcerative colitis. A, Conventional (Brooke) ileostomy with a subtotal colectomy and a Hartman pouch. B, Subtotal colectomy with ileorectal anastomosis. C, Conventional (Brooke) ileostomy with a total proctocolectomy. D, Continent ileostomy (Koch pouch) with total proctocolectomy. E, Restorative proctocolectomy with ileal pouch anal anastomosis (see Chapter 113).
(A-E, Adapted from Blumberg D, Beck DE. Surgery for ulcerative colitis. Gastroenterol Clin North Am 2002; 31:219.)
Colectomy
Total proctocolectomy with a permanent Brooke end-ileostomy was one of the earliest operations performed for UC. Removal of the entire colon and rectum eliminates any future disease and risk of colorectal cancer. The primary disadvantage of this operation is the presence of the permanent ileostomy, which might not be acceptable from the standpoint of quality of life for some patients. This is the operation of choice for elderly patients, those with anal dysfunction, and those who do not wish to have a restorative proctocolectomy. Proctocolectomy with continent ileostomy (Koch pouch) was developed as an alternative to the conventional end-ileostomy.312 In this operation, loops of small bowel are used to create an intra-abdominal pouch with an intussuscepted (nipple) valve. This pouch allows storage of stool contents and is attached to the abdominal wall with a flush ostomy opening. The stool contents in the pouch are emptied by inserting a suction-catheter through the stoma. Because of technical challenges associated with this operation (e.g., slippage of the nipple valve) and the development of restorative procedures, proctocolectomy with continent ileostomy rarely is performed.
Proctocolectomy with Ileal Pouch-Anal Anastomosis
Restorative proctocolectomy with IPAA currently is the operation of choice for most patients with UC who require elective colectomy. In this procedure, the entire colon and rectum are removed, the anal sphincters are preserved, and a pouch is constructed from approximately 20 cm of the distal ileum (see Fig. 112-16). Bowel continuity is established by anastomosing this pouch with the anal canal. An IPAA usually is performed as a two-stage operation, during the first stage of which a temporary diverting ileostomy is created to allow the ileal pouch to heal. This operation can be performed as a single-stage operation; however, there have been reports suggesting a higher rate of bowel obstruction and sepsis when this is done.313 The ileostomy is reversed after approximately two to four months.
Proctocolectomy with IPAA presents technical challenges and might not be suitable or technically feasible for all patients. Most reports suggest satisfactory quality of life following IPAA surgery.314 Mean stool frequency ranges from four to nine bowel movements per day, including one or two nocturnal stools. Nocturnal seepage occurs in approximately 20% of patients in the early postoperative period but is infrequent after the first year.
Rates of complications following IPAA surgery vary widely. In a series from the Cleveland Clinic of more than 1000 patients undergoing restorative proctocolectomy and IPAA, most of whom had UC, the overall morbidity rate was 63% (early complications 28%, late complications 51%).314 A larger case series from the Mayo Clinic included 1885 patients who had proctocolectomy with IPAA for UC and who were followed for up to 20 years (mean follow-up 11 years).315 Pouch success rates were reported of 96.3%, 93.3%, 92.4%, and 92.1% by 5, 10, 15, and 20 years, respectively, however, complication rates also were high and included pouchitis (48% by 10 years, 70% by 20 years), small bowel obstruction (42% by 20 years), anastomotic stricture (39% by 20 years), abscess (16% by 20 years), and fistula (14% by 20 years), as well as pelvic sepsis. Other complications of proctocolectomy with IPAA are covered in Chapter 113 and include fecal incontinence and sexual and urinary dysfunction.
Laparoscopic approaches to the proctocolectomy with IPAA have become popular among both physicians and patients. Because this modality is relatively new, comparison data versus open proctocolectomy with IPAA are limited. A case-matched series from the Mayo Clinic, however, which included 100 consecutive laparoscopic IPAA case patients matched to 200 open IPAA control patients, reported that although the median operative time was significantly longer for the laparoscopic group by 103 minutes, laparoscopic patients had significantly improved recovery, as witnessed by lower median time to regular diet (three vs. five days), time to ileostomy output (two vs. three days), length of stay (four vs. seven days), and intravenous narcotic use (all P < 0.05).316 In addition, postoperative morbidity and hospital readmission rates were similar between the two groups and no mortalities were observed. Fewer patients required reoperation within three months with the laparoscopic approach (3% vs. 6.5%, P < 0.2). A follow-up survey in this same cohort of patients one year after operation showed that patients reported high cosmetic, body image, quality of life, and sexual function scores irrespective of the type of operation.317
A smaller randomized trial from Amsterdam of 60 patients who underwent laparoscopic or open IPAA found significantly higher operative time for the laparoscopic approach (by 77 minutes) but similar morbidity (20% vs. 17%), length of stay (10 vs. 11 days), time to regular diet, narcotic requirement, and quality of life at three months after surgery in both groups.318 Larger RCTs will need to be conducted before any definitive conclusions can be made regarding the superiority of one approach over another. The experience of the particular surgeon and surgical center is likely to be an important determinant of success rates irrespective of procedure.