CHAPTER 99 TUMORS OF THE SPINAL CORD
Tumors of the spinal cord are a rare but distinct group of central nervous system neoplasms that affect all ages. Often slow growing, they should be considered in any patient who presents with neck or back pain, radiculopathy, or myelopathy. Classification is based on location and cell of origin: Intramedullary tumors most often arise from glial cells; intradural extramedullary tumors may arise from Schwann cells of the nerve roots or meningeal cells covering the spinal cord; and extradural tumors commonly arise from the bony elements of the vertebral column. Magnetic resonance imaging (MRI) plays a central role in anatomical localization and diagnosis. Since the first resection of a spinal cord tumor by Victor Horsley in 1887, advances in microsurgical techniques have allowed physicians to more accurately diagnose and treat spinal cord tumors. However, much remains to be learned about optimal management for this rare but challenging group of neoplasms.
EPIDEMIOLOGY
Spinal cord tumors are rare, accounting for only 4% to 10% of all primary central nervous system tumors. According to several population-based studies, their annual incidence is estimated to be 0.5 to 1.4 per 100,000.1,2 Spinal cord tumors are classified into three groups according to anatomical location: extradural, intradural extramedullary, and intramedullary (Fig. 99-1).
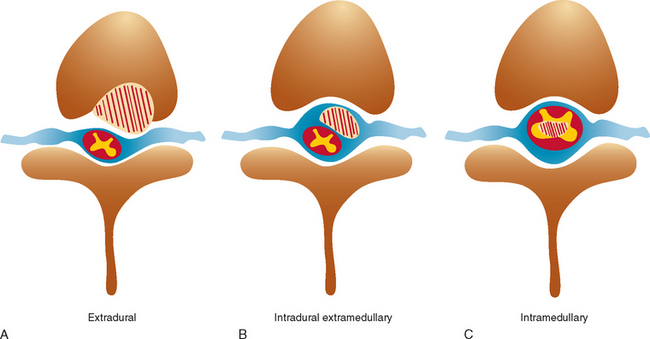
Figure 99-1 Anatomical location of spinal cord tumors. A, Extradural. B, Intradural extramedullary. C, Intramedullary.
Intradural extramedullary tumors represent more than half of the nonextradural spinal cord tumors in adults. Approximately 80% of these tumors are either nerve sheath tumors (neurofibromas and schwannomas) or meningiomas; epidermoids, dermoids, teratomas, and metastases make up much of the remaining fraction (Table 99-1). Nerve sheath tumors most commonly manifest in the fourth decade and are evenly distributed among men and women. Multiple intraspinal neurofibromas are often found in patients with neurofibromatosis type 1. Malignant degeneration of a neurofibroma into a malignant peripheral nerve sheath tumor is a rare but highly lethal complication in patients with neurofibromatosis type 1. Spinal meningiomas most often manifest in the fifth to seventh decades and exhibit a striking female predilection (4 : 1). In women, 80% occur in the thoracic spine. This regional predominance does not hold true in men, in whom meningiomas are found throughout the cervical, thoracic, and, less commonly, lumbosacral cord. Ionizing radiation and trauma are known risk factors for the development of spinal meningiomas.3
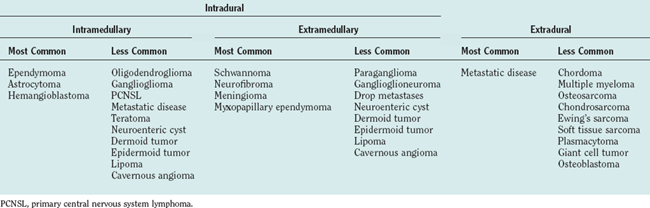
Intramedullary tumors account for less than one third of all spinal tumors in adults, whereas the majority of spinal tumors in children are intramedullary.4 Glial tumors (ependymomas and astrocytomas) predominate in all age groups. Ependymomas are increasingly more common with age, reaching a peak incidence in the fourth decade. They occur more frequently in men than in women. The remainder of the category is composed of rare tumors such as hemangioblastoma, neurocytoma, ganglioglioma, lipoma, primitive neuroectodermal tumor, lymphoma, and metastatic disease. Hemangioblastomas also exhibit a male preponderance and may occur sporadically or in association with von Hippel–Lindau disease. Gangliogliomas, although rare in adults, have been recognized as among the most common intramedullary tumors in children.5
CLINICAL MANIFESTATIONS
Pain from spinal cord tumors manifests in several different forms. Most commonly, patients experience midline or axial spinal pain. It is often worse at night and increased with recumbency, coughing, sneezing, straining, or other maneuvers that increase intrathoracic or intra-abdominal pressure. Paraspinal pain and tenderness may also be present as a result of local muscle spasm. Pain and dysesthesias that follow a radicular distribution are most common with extramedullary lesions that affect the dorsal roots or plexus; painful dysesthesias affecting an entire limb have also been described. A sensation of lancinating pain or electricity down the length of the spine, Lhermitte’s sign, can be caused by compressive cervical cord lesions. Thoracic tumors may cause a bandlike pain around the chest or abdomen, resembling the pain of herpetic neuralgia. Tumors in this location may also cause progressive kyphoscoliosis, particularly in children.6
Weakness, reflex change, sensory loss, and bowel or bladder dysfunction may be present early or late in the course of disease. Intramedullary lesions tend to produce dysfunction earlier than do extramedullary lesions, which often cause deficits by external compression rather than direct invasion of tissue. In general, rapid progression or short duration of symptoms portends a higher grade tumor and a poorer prognosis.7,8 Weakness from spinal cord tumors may be secondary to (1) direct involvement of the anterior horn cells or descending motor tracts within the cord itself, as with intramedullary tumors; (2) compression or infiltration of the ventral roots as they exit the spinal cord, as with nerve sheath tumors; or (3) external compression of the corticospinal tracts, resulting in myelopathy and upper motor neuron weakness, as with many extramedullary tumors, particularly thoracic meningiomas or epidural metastases.9 Sensory loss is often patchy, involving one or more particular modalities more than others. For example, central cord lesions, such as ependymomas, can cause pain and loss of temperature sensation with preserved position and vibratory sensation by affecting the central crossing fibers in the cord. Because of lamellation of the spinothalamic tracts, there may be sacral sparing of pain and temperature sensation from central lesions as well (Fig. 99-2). Compressive lesions often manifest with pain and an ascending sensory level; accompanying signs may include spasticity, upper motor neuron weakness, and bladder dysfunction. In general, sensory loss from intramedullary cord lesions tends to be symmetrical, whereas sensory loss from extramedullary lesions is often asymmetrical as a result of involvement of nerve roots.
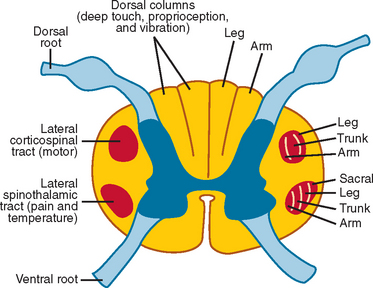
Several well-recognized syndromes may be encountered from spinal cord tumors at various levels. Tumors in the lower lumbar region may produce a conus medullaris syndrome, characterized by early bowel, bladder, and sexual dysfunction; by a mixture of upper and lower motor neuron weakness; and by patchy numbness. Tumors in this region can also produce a cauda equina syndrome, characterized by urinary retention, saddle anesthesia, lower motor neuron weakness, and reflex loss. Brown-Séquard syndrome—with ipsilateral weakness; contralateral pain; and loss of proprioception, vibration sensation, and temperature sensation—can result from eccentric intramedullary or extramedullary lesions. Tumors at the foramen magnum can cause suboccipital pain, lower cranial nerve palsies, bilateral hand atrophy, and loss of position and vibration sensation in the upper extremities more than in the lower extremities. Finally, hydrocephalus can be present in association with spinal cord tumors, particularly those in the high cervical region, perhaps as a result of high cerebrospinal fluid (CSF) protein level, spinal block, or outflow obstruction of the fourth ventricle caused by leptomeningeal thickening.10
The differential diagnosis of spinal cord tumors includes (1) nonneoplastic or dysembryonic lesions, such as lipoma or syringomyelia; (2) vascular malformations, including arterial venous malformation and dural arteriovenous fistula; (3) vascular disease, such as spinal cord infarction or amyloid angiopathy11; (4) demyelinating disease; (5) inflammatory myelitis, as in transverse myelitis or sarcoidosis; (6) infectious disease, including syphilis, human T cell lymphotropic virus type 1, and schistosomiasis; (7) neurodegenerative disease, such as amyotrophic lateral sclerosis; and (8) extrinsic compression from degenerative joint disease, as in spondylosis or hypertrophic arthritis, epidural metastatic disease, extramedullary hematopoiesis, and epidural lipomatosis (Table 99-2).
| Dysembryonic lesions | Lipoma |
| Neuroenteric cyst | |
| Dermoid tumor | |
| Epidermoid tumor | |
| Vascular malformations | AVM |
| Dural AVF | |
| Vascular disease | Spinal cord stroke |
| Amyloid angiopathy | |
| Demyelinating disease | Multiple sclerosis |
| Inflammatory disease | Transverse myelitis |
| Sarcoidosis | |
| Systemic lupus erythematosus | |
| Infectious disease | Syphilis |
| Tuberculosis | |
| HTLV-1 | |
| Schistosomiasis | |
| Degenerative joint disease | Cervical spondylosis |
| Hypertrophic arthritis | |
| Herniated disc | |
| Neurodegenerative disease | ALS |
| Miscellaneous | Post-irradiation myelopathy |
| Syringomyelia | |
| Epidural lipomatosis | |
| Extramedullary hematopoiesis |
ALS, amyotrophic lateral sclerosis; AVF, arteriovenous fistula; AVM, arteriovenous malformation; HTLV-1, human T cell lymphotrophic virus type 1.
DIAGNOSTIC TESTS
Computed Tomography and Computed Tomographic Myelography
Like the plain radiograph, computed tomography (CT) has largely been supplanted by MRI except in patients who cannot tolerate or who have a contraindication to MRI. CT does remain the study of choice for visualization of osseous structures and can demonstrate bony changes surrounding an extramedullary mass in the region of the neural foramen. CT is also useful during preoperative planning to evaluate spinal stability. Intravenous contrast material enhances the sensitivity of CT for detecting soft tissue pathology if MRI cannot be performed. For tumors in the cervical region, computed tomographic angiography may be employed to define the relationship of the tumor with the vertebral arteries. Computed tomographic myelography is superior to standard myelography for demonstrating the rounded filling defects of intradural extramedullary lesions or widening of the spinal cord caused by intramedullary lesions.
Magnetic Resonance Imaging
MRI plays a central role in the imaging of spinal cord tumors and affords superior anatomical localization of soft tissue masses. The majority of tumors can easily be classified as extradural, intradural extramedullary, or intramedullary by MRI alone; this thereby greatly aids in characterization of the tumor. Both sagittal and axial T1- and T2-weighted images should be obtained, in addition to sagittal and axial T1-weighted images after the administration of gadolinium. Diffusion-weighted imaging is occasionally useful for detecting cytotoxic edema and for differentiating tumors from abscesses.12
Astrocytomas usually manifest as focal enlargements of the spinal cord (Fig. 99-3). They are typically hypointense or isointense on T1-weighted images and hyperintense on T2-weighted images. Unlike their intracranial counterparts, nearly all spinal astrocytomas are enhanced after administration of contrast material, regardless of World Health Organization grade. Contrast material helps delineate tumor margins from surrounding edema, cyst, or syrinx. Although both cysts and syrines are hyperintense on T2-weighted images, cysts usually show enhancement along their borders, whereas syrinxes do not.
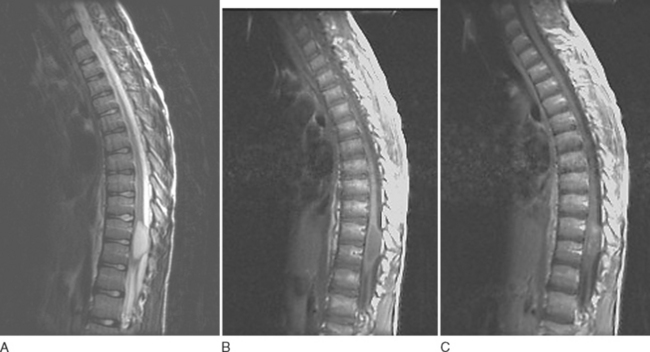
Ependymomas share many imaging characteristics with astrocytomas, but there are several features that may help distinguish the two. Ependymomas typically are enhanced more intensely than are astrocytomas. They are more likely to have heterogeneous signals because of hemorrhagic or cystic components. On T2-weighted images, dark caps, representing hemosiderin deposition, may be seen. Finally, ependymomas tend to be more central on axial cuts and have a greater predilection for the lower spinal cord. Unlike cellular ependymomas, myxopapillary ependymomas are often hyperintense on T1-weighted images, which probably reflects their mucinous nature (Fig. 99-4).
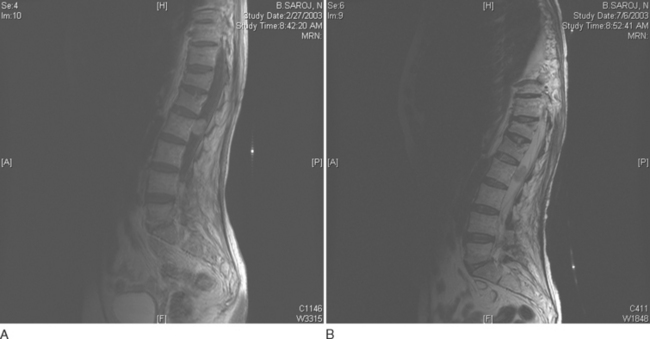
Hemangioblastomas tend to have slightly different MRI characteristics according to size. Smaller lesions (10 mm or less) are isointense on T1-weighted images, hyperintense on T2-weighted images, and uniformly enhanced with gadolinium (Fig. 99-5). Larger lesions are often hypointense or isointense on T1-weighted images, heterogeneous on T2-weighted images, and heterogeneously enhanced after contrast material administration.13 Many hemangioblastomas have associated cysts, which can be quite large and may be hyperintense on T1-weighted images as a result of high protein content. Most lesions have surrounding areas of hypointensity on T1-weighted images and hyperintensity on T2-weighted images, which represent peritumoral edema.14
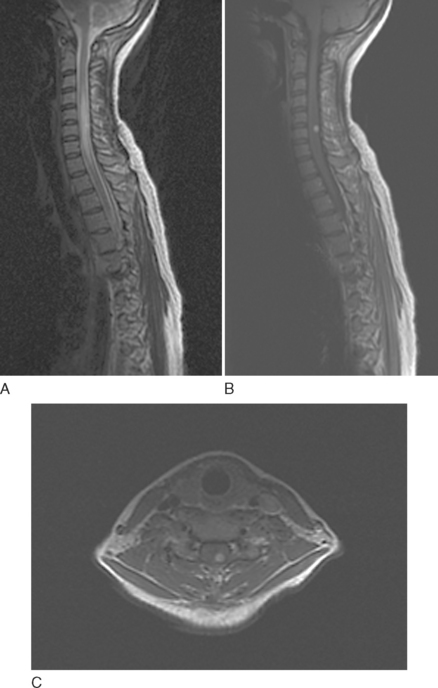
Intramedullary metastases are characterized by hyperintensity on T2-weighted images and homogenous enhancement after gadolinium administration (Fig. 99-6). Additional leptomeningeal deposits are often seen on postcontrast T1-weighted images.
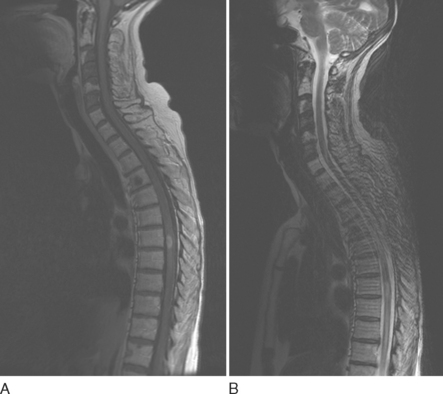
Nerve sheath tumors are characterized by isointense signal on T1-weighted images and markedly strong signal on T2-weighted images with occasional weak signal centrally (the so-called target appearance). Enhancement is variable: Some tumors show uniform enhancement, whereas others may be only peripherally enhanced or nonenhanced (Fig. 99-7). When multiple tumors are present, they are more likely to represent neurofibromas than schwannomas. Malignant degeneration of a nerve sheath tumor can be difficult to recognize radiographically, although malignant lesions tend to be larger and have more irregular, infiltrating borders.15
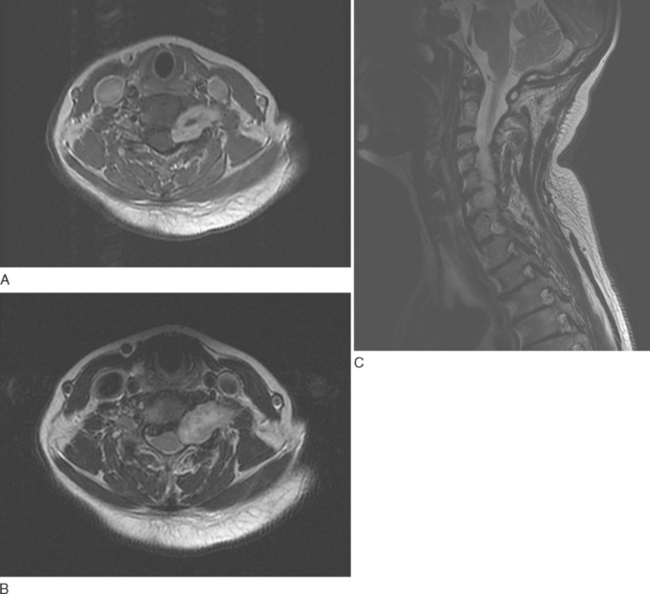
Spinal meningiomas have an isointense or hypointense appearance on T1-weighted images and a slightly hyperintense appearance on T2-weighted images (Fig. 99-8). They are intensely and homogeneously enhanced after contrast material administration, except within areas of calcification. These areas are dark on all sequences and have little associated enhancement.
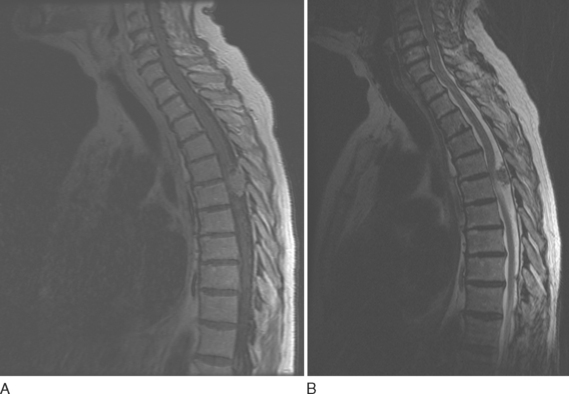
Epidural metastatic lesions are most often isointense on T1-weighted images, hyperintense on T2-weighted images, and enhanced after contrast material administration. In cases of suspected spinal cord compression, the entire spine must be imaged, because 30% of patients have multiple deposits at the time of diagnosis (Fig. 99-9).
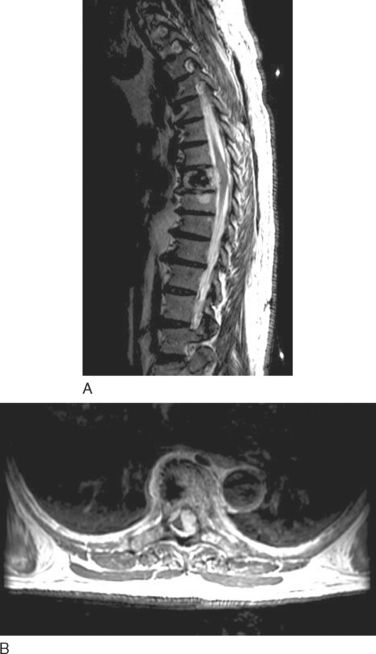
MANAGEMENT
Surgery
Surgery is the mainstay of treatment for most spinal cord tumors, particularly since the advent of the operating microscope, intraoperative ultrasonography, evoked potential monitoring, surgical lasers, and the ultrasonic aspirator. The objectives of surgery range from gross total resection to limited biopsy for pathological diagnosis. Unlike excision of intracranial neoplasms, surgery for spinal tumors must take into account spinal stability, particularly in pediatric patients, and implantation of hardware may be necessary to avoid postoperative deformity or instability. Most tumors are resected with a dorsal approach through a posterior midline incision, laminectomy, and durotomy. Intramedullary lesions, with the exception of pia-based hemangioblastomas, are often completely surrounded by normal tissue. They cannot be visualized from the surface, and a midline myelotomy is required for exposure. In such cases, intraoperative ultrasonography may be used to help delineate the location, length, and depth of the myelotomy.16 Intraoperative motor and/or somatosensory evoked potentials are routinely employed to assess the integrity of the spinal motor and/or sensory pathways during tumor resection and to prevent postoperative functional deterioration.17 The extent of resection is guided by lesion anatomy, surgical experience, results of intraoperative monitoring, and preliminary histological diagnosis obtained from frozen sections at the time of surgery.18 Patients with preoperative functional independence and few medical comorbid conditions and in whom frozen section exhibits low-grade histopathology are generally the best candidates for aggressive surgical resection.
Radiation therapy was traditionally considered the initial treatment of choice for most patients with extradural spinal cord compression. The use of decompressive laminectomy in combination with radiation therapy did not improve outcome.19 Surgery was confined to patients presenting with extradural cord compression without a histological diagnosis, radioresistant tumors, compression of the spinal cord by bone, and recurrent cord compression after radiation therapy.
In a prospective, randomized, controlled trial, Patchell and colleagues compared anterior decompression followed by radiation therapy (30 Gy) with radiation therapy alone.20 The study was stopped after interim analysis at midpoint because of a significant benefit in the patients who received both surgery and radiation therapy. A total of 101 patients were enrolled. Patients treated with surgery retained the ability to walk significantly longer than did those treated with radiotherapy alone (median, 126 days versus 35 days; P = .006). Surgically treated patients also maintained continence and functional Frankel and American Spinal Injury Association scores significantly longer than did patients receiving radiation. The length of survival was not significantly different between the two groups, although there was a trend toward longer survival time in patients who underwent surgery (median, 129 days versus 100 days; P = .08). Of importance was that the median length of hospitalization during which the cord compression was diagnosed and treated was 10 days for both treatment groups (P = .86). This study suggests that patients with extradural cord compression treated with radical direct decompressive surgery plus postoperative radiotherapy regained the ability to walk more often and maintained it longer than did patients treated with radiation alone. Surgery enabled most patients to remain ambulatory and continent for the remainder of their lives, whereas patients treated with radiation alone spent approximately two thirds of their remaining lives unable to walk and incontinent. Whether the findings of this study can be extrapolated to other institutions without the surgical resources to perform emergency anterior decompression in patients with epidural cord compression routinely, and whether the benefit of surgery applies to radiosensitive tumors such as breast cancer, remains to be seen. Nonetheless, this is an important study that could change the management of patients with extradural cord compression.
Radiation
Because there have been no randomized prospective studies examining the efficacy of radiation for primary spinal cord tumors, significant controversy regarding its use remains. Many retrospective series exist, but their usefulness is limited by small sample size, limited follow-up, and lack of matched controls. In general, most practitioners agree that there is no role for radiation in patients with completely resected low-grade gliomas who are monitored with serial surveillance scans. Complete resection is achieved more often with ependymomas than with astrocytomas, but the dominant pattern of failure is local for both. In most cases in which gross total resection has not been achieved, postoperative local radiation therapy is administered up to a dosage of 4000 to 5400 cGy. Dosages higher than 5500 cGy are associated with increased risk of radiationinduced myelopathy. Children’s tolerance may be lower.21,22 Image-guided frameless stereotactic radiosurgery has emerged as a treatment option for primary and metastatic spinal cord tumors.23,24 In this system, high-dose radiation can be delivered to a tumor volume with minimal damage to adjacent normal structures.
For low-grade astrocytomas, surgery and postoperative radiation produce survival rates of 50% to 91% at 5 years and 40% to 91% at 10 years.25 For ependymomas, survival is slightly better: 60% to 100% at both 5 and 10 years. High-grade astrocytomas appear to respond to radiation in the short term, but overall survival rates are dismal. Craniospinal radiation may be employed for cases of ependymoma with central nervous system dissemination. Postoperative radiation therapy is rarely employed for hemangioblastomas, nerve sheath tumors, and spinal meningiomas, except in the case of malignant peripheral nerve sheath tumor15 or malignant meningioma.
Radiation is the mainstay of therapy for patients with epidural spinal cord compression resulting from metastatic disease. Focal radiation therapy is usually administered at a dose of 3000 cGy (30 Gy) administered over 2 weeks.26
OUTCOMES
Postoperatively, patients frequently experience transient worsening of neurological function. The most important predictors of postoperative neurological deterioration are preoperative disability and malignant histology.27 Significant postoperative motor morbidity resulting in functional dependence is lowest in patients with normal preoperative examination findings. This fact argues for early surgery (before neurological deterioration) in patients with intramedullary tumors. Patients with severe or long-standing deficits rarely show improvement after surgery. Manipulation of the posterior columns during a standard dorsal myelotomy often results in transient or permanent proprioceptive impairment, even in patients with normal preoperative examination findings. Other surgical complications include CSF leak, meningitis, transient or permanent bowel or bladder dysfunction, and spinal instability that leads to kyphoscoliosis.
In patients with ependymomas, gross total resection provides the best chance at long-term disease-free survival. Two groups have reported disease-free survival rates of 100% after radical resection without postoperative radiotherapy; seven of these patients were monitored for more than 10 years.28,29 Overall 10-year survival rates are better for spinal ependymomas than for astrocytomas, ranging from 50% to 95%.30,31 The most important prognostic factor is the ability to attain gross total resection; histological grade and preoperative neurological status are also important.32–34 For spinal cord astrocytomas, 5- and 10-year survival rates are 50% to 73% and 23% to 54%, respectively.30 As with ependymomas, important prognostic factors include histological grade (median length of survival, 98 months for World Health Organization grade I disease versus 15 months for grade IV disease); symptom duration before diagnosis (longer duration correlates with increased length of survival); preoperative performance status (5-year survival rate of 75% for Karnofsky Performance Status scores of 80 to 100 versus 51% for scores of <60); and postoperative performance status.7 Factors not shown to be relevant prognostically include sex, age, site and extension of tumor, extent of surgical resection, and dose of postoperative radiotherapy.7,30,35 Hemangioblastomas have the best long-term clinical outcome of the three most common intramedullary tumors. More than 90% of patients remain clinically stable or improve, and complete resection virtually eliminates the risk of recurrence for sporadic tumors.36,37 Poor prognostic factors include significant preoperative deficits, large tumor size, and anterior tumor location.36 For patients with von Hippel–Lindau disease, the risk of developing new hemangioblastomas or continued growth of existing ones warrants routine long-term follow-up with gadolinium-enhanced MRI.
Results of surgery for extramedullary tumors tend to be very good; most patients exhibit significant improvement in neurological function postoperatively. Gross total resection of nerve sheath tumors is curative in most cases. For meningiomas, the recurrence risk is also quite low, except for en plaque lesions, lesions with significant extradural spread, or tumors with malignant histological processes. Patients with neurofibromatosis type 1 and spinal neurofibromas are at increased risk for developing recurrent tumors and therefore require close follow-up. In addition, they have a significantly higher long-term mortality rate than do patients without neurofibromatosis type 1 (10-year survival rates of 60% and ≥90%, respectively).38
For epidural spinal cord compression from metastatic disease, the prognosis depends primarily on pretreatment neurological function. In addition, radiosensitivity of the underlying tumor plays a role. Lymphoma, myeloma, and prostate cancer are the most radiosensitive, whereas renal cell and non–small cell lung cancer lesions tend to be relatively radioresistant. Of the patients who are ambulatory at diagnosis, almost all remain ambulatory after radiation therapy. In contrast, only 2% to 6% of patients who are paraplegic at the onset of radiotherapy regain the ability to walk.26
CONCLUSION
Balmaceda C. Chemotherapy for intramedullary spinal cord tumors. J Neurooncol. 2000;47:293-307.
Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15:679-684.
Isaacson SR. Radiation therapy and the management of intramedullary spinal cord tumors. J Neurooncol. 2000;47:231-238.
1 Sasanelli F, Beghi E, Kurland LT. Primary intraspinal neoplasms in Rochester, Minnesota, 1935–1981. Neuroepidemiology. 1983;2:156-163.
2 Helseth A, Mork SJ. Primary intraspinal neoplasms in Norway, 1955 to 1986. A population-based survey of 467 patients. J Neurosurg. 1989;71:842-845.
3 Harrison MJ, Wolfe DE, Lau TS, et al. Radiationinduced meningiomas: experience at the Mount Sinai hospital and review of the literature. J Neurosurg. 1991;75:564-574.
4 Sloof JL, Minehan KJ, McCarty CS. Primary Intramedullary Tumors of the Spinal Cord and Filum Terminale. Philadelphia: WB Saunders, 1964.
5 Miller DC. Surgical pathology of intramedullary spinal cord neoplasms. J Neurooncol. 2000;47:189-194.
6 Rossitch EJr, Zeidman SM, Burger PC, et al. Clinical and pathological analysis of spinal cord astrocytomas in children. Neurosurgery. 1990;27:193-196.
7 Innocenzi G, Salvati M, Cervoni L, et al. Prognostic factors in intramedullary astrocytomas. Clin Neurol Neurosurg. 1997;99:1-5.
8 Rodrigues GB, Waldron JN, Wong CS, et al. A retrospective analysis of 52 cases of spinal cord glioma managed with radiation therapy. Int J Radiat Oncol Biol Phys. 2000;48:837-842.
9 Solero CL, Fornari M, Giombini S, et al. Spinal meningiomas: review of 174 operated cases. Neurosurgery. 1989;25:153-160.
10 Rifkinson-Mann S, Wisoff JH, Epstein F. The association of hydrocephalus with intramedullary spinal cord tumors: a series of 25 patients.Neurosurgery. 1990;27:749-754. discussion, Neurosurgery. 1990;27:754.
11 Lee M, Epstein FJ, Rezai AR, et al. Nonneoplastic intramedullary spinal cord lesions mimicking tumors.Neurosurgery. 1998;43:788-794. discussion, Neurosurgery. 1998;43:794-795.
12 Van Goethem JW, van den Hauwe L, Ozsarlak O, et al. Spinal tumors. Eur J Radiol. 2004;50:159-176.
13 Chu BC, Terae S, Hida K, et al. MR findings in spinal hemangioblastoma: correlation with symptoms and with angiographic and surgical findings. AJNR Am J Neuroradiol. 2001;22:206-217.
14 Baker KB, Moran CJ, Wippold FJ2nd, et al. MR imaging of spinal hemangioblastoma. AJR Am J Roentgenol. 2000;174:377-382.
15 Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573-1577.
16 Platt JF, Rubin JM, Chandler WF, et al. Intraoperative spinal sonography in the evaluation of intramedullary tumors. J Ultrasound Med. 1988;7:317-325.
17 Morota N, Deletis V, Constantini S, et al. The role of motor evoked potentials during surgery for intramedullary spinal cord tumors. Neurosurgery. 1997;41:1327-1336.
18 Parsa AT, Lee J, Parney IF, et al. Spinal cord and intraduralextraparenchymal spinal tumors: current best care practices and strategies. J Neurooncol. 2004;69:291-318.
19 Young RF, Post EM, King GA. Treatment of spinal epidural metastases: randomized prospective comparison of laminectomy and radiotherapy. J Neurosurg. 1980;53:741-748.
20 Patchell R, Tibbs PA, Regine O, et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomized trial. Lancet. 2005;366:643-648.
21 Marcus RBJr, Million RR. The incidence of myelitis after irradiation of the cervical spinal cord. Int J Radiat Oncol Biol Phys. 1990;19:3-8.
22 Sundaresan N, Gutierrez FA, Larsen MB. Radiation myelopathy in children. Ann Neurol. 1978;4:47-50.
23 Ryu SI, Chang SD, Kim DH, et al. Image-guided hypo-fractionated stereotactic radiosurgery to spinal lesions. Neurosurgery. 2001;49:838-846.
24 Gerszten PC, Ozhasoglu C, Burton SA, et al. CyberKnife frameless stereotactic radiosurgery for spinal lesions: clinical experience in 125 cases.Neurosurgery. 2004;55:89-98. discussion, Neurosurgery. 2004;55:98-99.
25 Isaacson SR. Radiation therapy and the management of intramedullary spinal cord tumors. J Neurooncol. 2000;47:231-238.
26 Prasad D, Schiff D. Malignant spinal-cord compression. Lancet Oncol. 2005;6:15-24.
27 Constantini S, Allen JC, Epstein F. Pediatric and adult primary spinal cord tumors. In: Black PM, Loeffler JS, editors. Cancer of the Nervous System. 1st ed. Cambridge, MA: Blackwell Scientific; 1997:637-649.
28 Epstein FJ, Farmer JP, Freed D. Adult intramedullary spinal cord ependymomas: the result of surgery in 38 patients. J Neurosurg. 1993;79:204-209.
29 McCormick PC, Torres R, Post KD, et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523-532.
30 Abdel-Wahab M, Corn B, Wolfson A, et al. Prognostic factors and survival in patients with spinal cord gliomas after radiation therapy. Am J Clin Oncol. 1999;22:344-351.
31 Waldron JN, Laperriere NJ, Jaakkimainen L, et al. Spinal cord ependymomas: a retrospective analysis of 59 cases. Int J Radiat Oncol Biol Phys. 1993;27:223-229.
32 Hoshimaru M, Koyama T, Hashimoto N, et al. Results of microsurgical treatment for intramedullary spinal cord ependymomas: analysis of 36 cases. Neurosurgery. 1999;44:264-269.
33 Whitaker SJ, Bessell EM, Ashley SE, et al. Postoperative radiotherapy in the management of spinal cord ependymoma. J Neurosurg. 1991;74:720-728.
34 Cooper PR. Outcome after operative treatment of intramedullary spinal cord tumors in adults: intermediate and long-term results in 51 patients. Neurosurgery. 1989;25:855-859.
35 Jyothirmayi R, Madhavan J, Nair MK, et al. Conservative surgery and radiotherapy in the treatment of spinal cord astrocytoma. J Neurooncol. 1997;33:205-211.
36 Lonser RR, Weil RJ, Wanebo JE, et al. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:106-116.
37 Roonprapunt C, Silvera VM, Setton A, et al. Surgical management of isolated hemangioblastomas of the spinal cord.Neurosurgery. 2001;49:321-327. discussion, Neurosurgery. 2001;49:327-328.
38 Seppala MT, Haltia MJ, Sankila RJ, et al. Long-term outcome after removal of spinal neurofibroma. J Neurosurg. 1995;82:572-577.