Chapter 78 Tumors of the liver
Pathologic aspects
Overview
According to their histogenesis, primary intrahepatic tumors are classified into three main categories—hepatocellular, biliary, and mesenchymal tumors—although additional rare entities exist. Liver tumor classification has recently been reviewed, and this chapter will present main pathologic aspects of intrahepatic liver tumors according to the 2010 World Health Organization (WHO) classification (Bosman et al, 2010).
Hepatocellular Tumors
Hepatocellular Carcinoma
Clinical and Epidemiologic Background (See Chapter 80)
Hepatocellular carcinoma (HCC) is the most common primary malignant tumor in the adult liver. It ranks among the most frequent neoplasms—approximately 6% of all new cancer diagnosed worldwide with an estimated incidence of about 500,000 to a million new cases per year. It is a deadly malignancy and the third most frequent cause of cancer death among men, causing 600,000 deaths worldwide per year (McGlynn & London, 2005; Sherman, 2005; Parkin et al, 2005; Bruix et al, 2004).
One of the striking characteristics of HCC is the marked geographic variation in its frequency, which is mainly related to geographic distribution of chronic liver disease risk factors. East Asia and sub-Saharan Africa have a very high incidence, whereas Italy, Spain, and Latin America are at intermediate risk. A relatively low but increasing incidence is found in Western Europe, the United States, Canada, and Scandinavia. In these countries, the incidence rate is 50 times lower than in Southeast Asia (Bosch et al, 2004; Kahn et al, 2002; Seeff & Hoofnagle, 2006).
The association between cirrhosis and HCC has been well established. Indeed, 60% to 90% of HCC arises in cirrhotic livers. Conversely, approximately 1% to 3% of patients with cirrhosis will develop HCC annually (Colombo et al, 1991; Johnson & Williams, 1987; Zaman et al, 1985). Most of the epidemiologic variation in HCC prevalence is thought to reflect differences in etiologic risk factors for liver cirrhosis that encompasses a large group of environmental factors (El-Serag et al, 2008). The major known risk factors for HCC are hepatitis viruses (chronic hepatitis B and C; see Chapter 64), toxins (alcohol, aflatoxins), metabolic diseases (metabolic syndrome, α1 antitrypsin deficiency, Wilson disease), hereditary hemochromatosis, and immune-related diseases (primary biliary cirrhosis, autoimmune hepatitis; see Chapter 70A). Given that the burden of chronic liver diseases is expected to rise along with increasing rates of alcoholism, hepatitis B and C infection, and fatty liver disease, it is expected that the incidence of HCC will also increase in the near future (Nordenstedt et al, 2010; El-Serag, 2008).
HCC is primarily a disease of older men, and its incidence generally increases with age. In Western Europe and the United States, most patients with HCC are between 50 and 75 years of age, although there are geographic differences. It occurs more frequently in men than women, with a male/female ratio ranging from 2 : 1 to 9 : 1, although the reason for this is not clear (El-Serag et al, 2008). Routine liver function tests are variably abnormal, often reflecting the underlying cirrhosis without a consistent pattern of alterations. Serum α-fetoprotein (AFP) is elevated in most symptomatic tumors, but small cancers are associated with lower or even normal levels. Thus serum AFP is not a reliable diagnostic test for HCC screening in patients with cirrhosis (Bruix & Sherman, 2005).
Ultrasound (US) is a sensitive technique for identifying nodules larger than 1.5 cm and is thus often used for detection, whereas magnetic resonance imaging (MRI) is a more accurate procedure for liver mass characterization (Bolondi et al, 2005; see Chapters 13 and 17). Reliable and highly specific imaging criteria now allow confident diagnosis of HCC in cirrhotic patients so that liver biopsy can be avoided when hyperarterialization associated with early washout is present in dynamic imaging (Bruix & Sherman, 2005; Forner et al, 2008). Screening programs have thus been proposed to follow cirrhotic patients by repeat US. These programs have been successful in identifying small and asymptomatic HCCs in high-incidence areas such as Japan, China, and Alaska but have been less effective in regions where the tumor incidence is low (Sangiovanni et al, 2004).
HCC is a peculiar tumor for several reasons. Indeed, its morphologic patterns are various, beyond the usual classification based on growth pattern and tumor differentiation. Furthermore, molecular pathogenesis of HCC is complex, involving different molecular pathways that may reflect both etiologic factors and underlying liver disease (Thorgeirsson & Grisham, 2002; Villanueva et al, 2007).
Gross Pathology
HCC can adopt a wide range of gross configurations, and several macroscopic classifications of varying complexity have been proposed, but their clinical relevance has not yet been proven. Tumor size ranges from less than 1 cm to more than 30 cm in diameter. At the time of diagnosis, mean size of HCC arising in a cirrhotic liver is usually less than those occurring in nonfibrotic liver. HCC tumors less than 2 cm are recognized as small or early HCC (International Working Party [IWP], 1995). Recently, small HCC have been subdivided into vaguely nodular and well-circumscribed HCC, two patterns with differences in prognosis; the vaguely nodular form has a better prognosis (International Consensus Group [ICG], 2009; Hytiroglou et al, 2007). Although the diagnosis of large HCC relies mainly on imaging modalities, biopsy is often requested for the diagnosis of these small nodular lesions. Upon gross examination, HCC may display a nodular, infiltrative, or diffuse macroscopic pattern (Okuda et al, 1984).
Nodular (Expanding) Hepatocellular Carcinoma
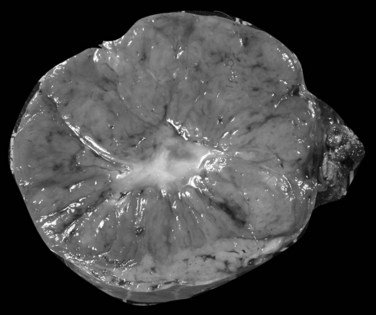
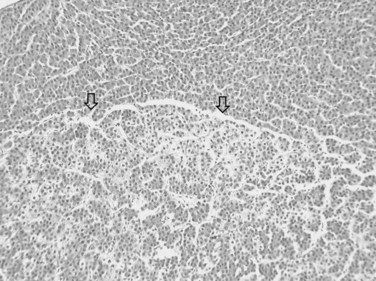
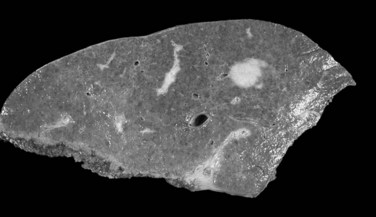
The nodular (expanding) pattern is the most common type of HCC, and it is typically seen in association with cirrhosis. This group of tumors is characterized by a sharp demarcation between the tumor mass and the compressed and partly atrophic surrounding parenchyma (Fig. 78.1). The presence of distorted hepatic vessels, including arteries, that form curved structures on the surface of the tumor mass or are visible on the cut surface support the concept of an expanding growth pattern. The nodule may be solitary or multiple across the liver, when these develop as a complication of cirrhosis. When nodules are multiple, small (<1 cm in diameter), and adjacent to the main tumor nodule, they are known as satellite nodules (Fig. 78.2).
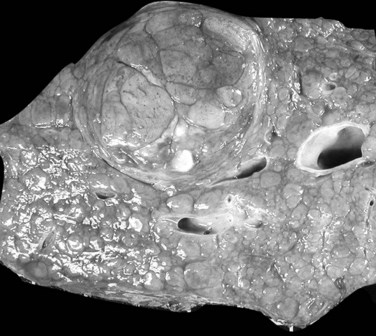
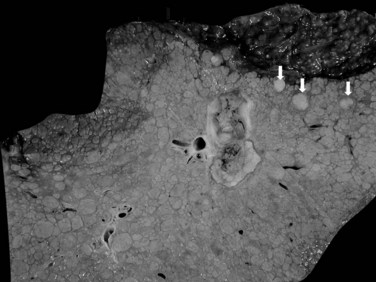
In cases of multiple HCCs, nodules may represent either multifocal independent tumors or intrahepatic metastasis. Such a distinction is quite impossible based on pathologic study alone but could be addressed with surrogate molecular analysis (Paradis et al, 1998; Sakamoto et al, 1989). On cut section, nodular HCC is well delineated from nontumoral liver and circumscribed totally or partially by a fibrous capsule (Okuda et al, 1977). Capsule formation is considered to begin at a tumor diameter of at least 10 mm or greater, because no distinct capsule has been noted in lesions less than 10 mm (Nakanuma et al, 1986). The prognostic significance of capsule formation has not yet been definitively settled. HCC typically forms soft masses that vary in color from gray, light brown, or yellow-green, often punctuated by foci of hemorrhage or necrosis when the tumor reaches a significant volume (Fig. 78.3).
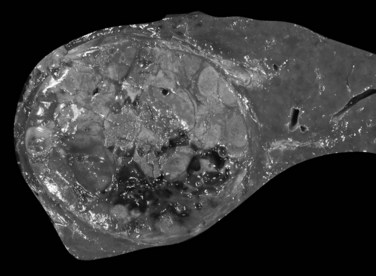
Infiltrative (Massive) Hepatocellular Carcinoma
The infiltrative (massive) pattern is usually characterized by a large, solitary mass that occupies a substantial portion of the liver. The lesion is poorly circumscribed with ill-defined, invasive borders (Fig. 78.4). This pattern has been associated more commonly with noncirrhotic livers (Smalley et al, 1988; Grando-Lemaire et al, 1999). On cut section, the tumor extends into and distorts the adjacent nontumor tissue, interdigitating with surrounding parenchyma. Vessels are incorporated into, not displaced by, the tumor mass so that tortuous vessels around the tumor are not so obvious (Okuda et al, 1982).

Diffuse Hepatocellular Carcinoma
The diffuse pattern is the least common type of HCC and represents a widespread infiltration by numerous small nodules that virtually replace the entire liver. In this pattern, tumor consists of several unconnected small tumors of roughly similar size. Multicentric origin or intrahepatic spread after portal vein invasion have been discussed as pathogenic mechanisms (Fig. 78.5; Nakashima & Kojiro, 1986).
Pedunculated Hepatocellular Carcinoma
Pedunculation is noted in rare instances, presumably reflecting an origin from an accessory hepatic lobe (Horie et al, 1983). The identification of the pedunculated form of HCC is significant, because even in the case of large tumors, limited resection may give excellent results.
Limits of Gross Classification
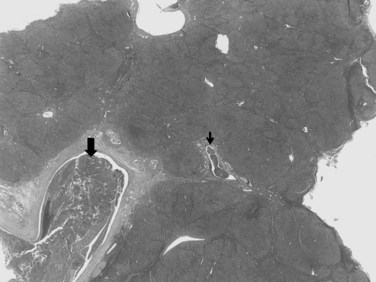
In nodular, advanced, and infiltrative HCC, invasion of large veins is common even at gross examination. The portal vein is more often involved than hepatic veins, inferior vena cava, or right atrium (Fig. 78.6). Portal vein invasion may be associated with thrombosis, and vascular invasion should be determined with care at the initial gross examination (macrovascular invasion). In some instances, intravascular tumor plugs in the close periphery of a large tumor may be difficult to distinguish from satellite tumor nodules. Invasion of large bile ducts may produce biliary obstruction, and hemobilia might be occasionally found.
Histopathology
Histopathologic evaluation is no more systematic, because dynamic imaging has high diagnostic accuracy for tumors larger than 3 cm. When imaging is not diagnostic, US-guided biopsy must be performed. This is often the case for HCC smaller than 2 cm, when biomarkers have low predictive values, and hyperarterialization may be incomplete or absent. For nodules smaller than 1 cm, biopsy is generally not recommended because of its limited performance. These diagnostic criteria have been endorsed by most international liver disease associations (Bruix & Sherman, 2005; Bruix et al, 2001). Nevertheless, despite the major advances in radiologic procedures, the definitive diagnosis continues to be based primarily on accurate examination and interpretation of histologic material for any small or atypical nodule.
Histopathologically, the diagnosis of HCC is based on the distinction between tumor cells and normal hepatocytes (Anthony, 1973). Therefore, the microscopic evaluation entails assessment of cytologic characteristics of tumor cells and evaluation of their architectural pattern.
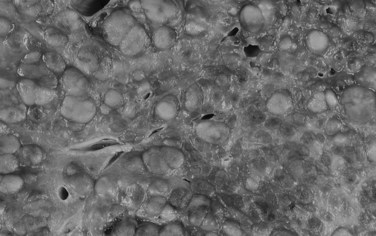
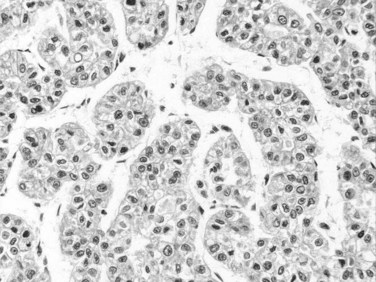
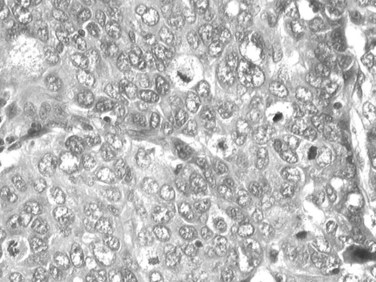
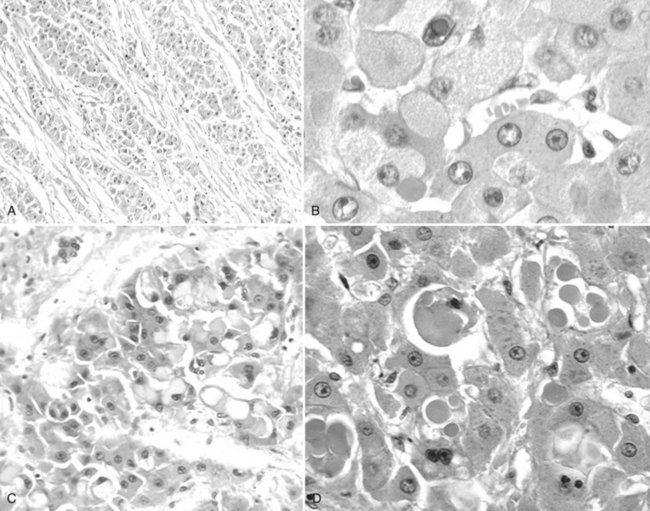
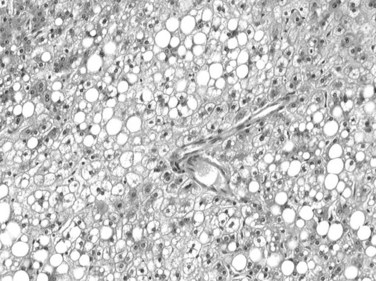
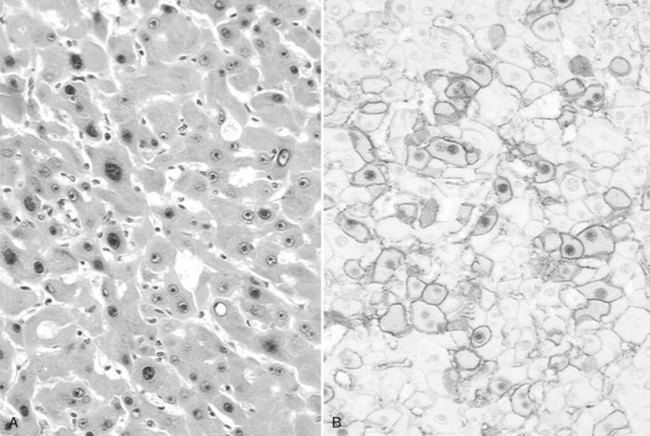
Tumor cells may be seen in varying degrees of hepatocellular differentiation within a single tumor. Nuclei are usually basophilic and often irregular, with prominent nucleoli and a high nuclear/cytoplasmic ratio. In well-differentiated HCC, the tumor cells may closely resemble normal hepatocytes, with a polygonal shape, distinct cell membranes, and eosinophilic granular cytoplasm (Fig. 78.7). Bile canaliculi can often be seen by light microscopy or can be demonstrated by immunostaining. When dilated, they might contain bile pigment, a characteristic feature of hepatocellular differentiation. Accumulation of glycogen or fat in tumor cells may produce a clear-cell appearance (Fig. 78.8). Cytoplasmic fat droplets might be more common in small HCCs. Mallory-Denk bodies, hyaline globules, and eosinophilic or so-called ground-glass cytoplasmic inclusions may also be observed.
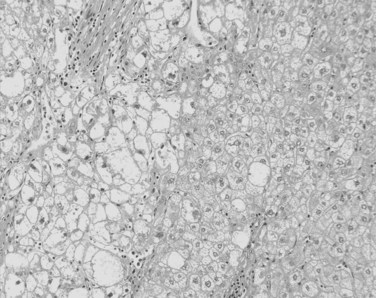
As the tumor evolves to a poorly differentiated phenotype with cell-to-cell heterogeneity, bizarre nuclei or giant tumoral cells may appear. Mitosis and apoptotic bodies can be observed (Fig. 78.9). Different degrees of cellular differentiation are usually present within a single large tumor, although small HCC tend to be more homogeneous.
Growth Patterns
The arrangement of the cells contributes to the variability in the microscopic appearance. On this basis, several types of tumor have been categorized, but it has not yet been fully established whether these variations reflect differences in behavior and influence prognosis (Qin & Tang, 2002). The three main architectural patterns of growth of HCC are trabecular; acinar, or pseudoglandular; and scirrhous.
Trabecular Growth
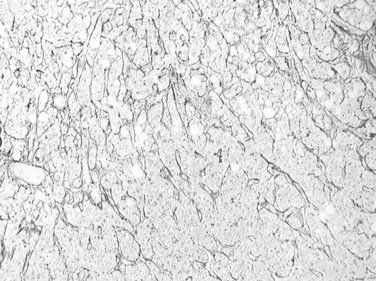
Trabecular growth occurs when tumoral hepatocytes are arranged in plates in varying thicknesses from 2 to more than 20 cells (see Fig. 78.7). This feature resumes the normal trabecular arrangement of liver plates. Neoplastic cells are organized along simplified sinusoids lined by flat endothelial cells with few or no Kupffer cells. Compared with normal liver plates, the reticulin framework is commonly sparse or absent (Fig. 78.10). A compact or solid pattern occurs when the trabeculae are closely aligned, and the sinusoids become compressed and unapparent.
Acinar (Pseudoglandular) Growth
The acinar, or pseudoglandular, pattern results either from glandlike dilation of the canaliculi between tumor cells, in which the lumens can contain bile, or central degeneration of trabeculae, in which the lumens contain mainly degenerative products with fibrin (Fig. 78.11). As with the trabecular pattern of growth, stroma is typically sparse. The lack of a desmoplastic stroma reaction is a helpful diagnostic clue, when other glandular malignant epithelial neoplasms, especially cholangiocarcinomas, are discussed. On occasion, large vascular lakes resembling peliosis can develop within the pseudoglandular formations.
Histologic Variants
Fibrolamellar Hepatocellular Carcinoma
This subset, which differs from other types of HCC in clinical features and prognosis, is the only variant with clinical significance. It was first delineated as a distinct entity in 1980 (Craig et al, 1980). In a population-based study, fibrolamellar HCC constituted 0.85% of all cases of primary liver cancer and 13.4% of all cases in patients younger than age 40 (El-Serag & Davila, 2004). The clinical presentation is similar to other HCCs, except that it occurs in young people with equal gender ratio and has no association with chronic liver disease, cirrhosis, or any other known predisposing risk factors.
On gross examination, fibrolamellar HCC is a firm, mostly well-defined but unencapsulated single nodule that can range from 5 cm to more than 20 cm (Saab & Yao, 1996). On cut section, the tumor is gray to brown with scalloped borders and a solid consistency (Fig. 78.12). Prominent fibrous septa subdivide the mass and may connect with a central zone of scarring. Such features may be confused with focal nodular hyperplasia. In addition, several reports in the literature describe fibrolamellar HCC spatially associated with focal nodular hyperplasia, although filiation between these two lesions has never been convincingly demonstrated (Saul et al, 1987). Calcifications may also be observed.
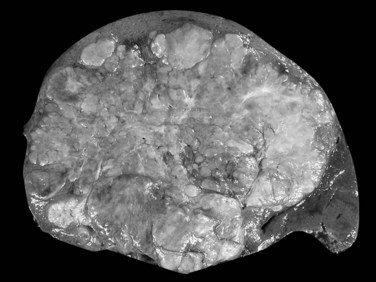
FIGURE 78.12 Fibrolamellar hepatocellular carcinoma. Large lobulated tumor is well delineated from normal surrounding liver.
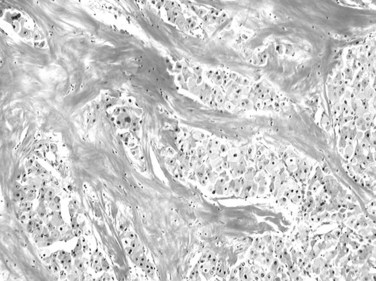
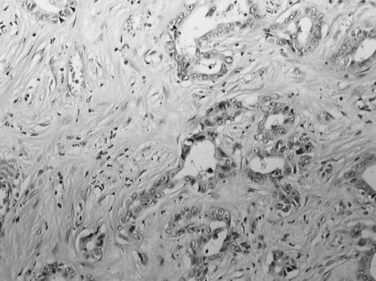
The distinctive histologic features are fibrous stroma and large, eosinophilic tumor cells (Fig. 78.13; Berman & McNeill, 1988). The stroma comprises dense, fibrous bands of varying thickness organized around nests, cords, and sheets of neoplastic cells (Nerlich et al, 1992). The tumor cells are usually larger than normal hepatocytes and display abundant, granular, and deeply eosinophilic cytoplasm with prominent nucleoli. Bile pigment is common, and fat or glycogen accumulation is sometimes seen. Most fibrolamellar carcinomas are histologically low grade, mitoses are usually sparse, and nuclear pleomorphism and multinucleation are infrequent. Cytoplasmic inclusions of various types are common, including ground-glass pale bodies (Fig. 78.14), eosinophilic cytoplasmic globules of variable periodic acid-Schiff positivity, and rarely Mallory-Denk bodies. In contrast to most HCC, fibrolamellar HCC seems to abundantly express type 7 cytokeratin (CK) and, in some cells, biliary-type CK19 (Van Eyken et al, 1990).
Clear-Cell Carcinoma
This predominant tumoral cell type is rich in glycogen or lipid and accordingly has a clear cytoplasm (Buchanan & Huvos, 1974). The clear-cell variant has been associated with a better prognosis, but the survival advantage, if present, is minor and has not been confirmed (Kishi et al, 1983). Clear-cell HCC may be associated with hypoglycemia and hypercholesterolemia, and sudden death from severe hypoglycemia has been reported (Sasaki et al, 1981; Ross & Kurian, 1985).
Sarcomatoid Hepatocellular Carcinoma
This variant is characterized by a sarcomatous-appearing component of spindle-shaped or giant tumor cells (Kojiro et al, 1989). The elongated spindle cells are arranged in bundles, occasionally with interlacing or storiform patterns. The giant cells are multinucleated, markedly pleomorphic, and cytologically anaplastic, and osteoclast-like giant cells are described in some instances (Kuwano et al, 1984). These tumors have often been referred to as carcinosarcomas. The sarcomatoid component varies in its extent, and histologic transitions with the carcinomatous elements are often noted. The spindle-shaped cells are typically immunoreactive for keratin and α-fetoprotein (Kakizoe et al, 1987).
Sclerosing Hepatocellular Carcinoma
Sclerosing HCC is a rare but distinctive variant characterized by abundant, diffusely distributed fibrous stroma and compressed, sometimes elongated malignant hepatocytes. The tumor tends to occur in an older age group, affects men and women equally, and might be associated with hypercalcemia (Omata et al, 1981). A large, firm, gray-white mass is found on gross examination. The hepatocellular nature of the lesion is best appreciated on a cytologic, rather than architectural, basis. Individual tumor cells, especially at the periphery of the mass, correspond to those of the usual HCC. Although generally smaller in size, they exhibit granular eosinophilic cytoplasm, vesicular nuclei, and conspicuous nucleoli. Bile pigment can sometimes be discerned.
Other Morphologic Patterns of Hepatocellular Carcinoma
It is well known that HCCs that arise in hemochromatosis fail to show accumulation of stainable iron, and iron-free foci in siderotic macroregenerative nodules in human cirrhotic livers have been suggested to be a marker for incipient neoplastic lesions (Hirota et al, 1982; Deugnier et al, 1993). Some HCCs accumulate copper, which can be detected using histochemical methods. Accumulation of large amounts of copper and copper-binding protein (orcein-positive) has been found chiefly in the fibrolamellar variant of HCC (Lefkowitch et al, 1983).
There are some unusual instances in which HCC produces substances that occur in some rare metabolic disorders (e.g., black HCC in a patient without Dubin-Johnson syndrome; Roth et al, 1982). A granulomatous sarcoid-like reaction may be encountered within HCC, the granulomas being characterized by epithelioid cells, Langerhans-type giant cells, and varying numbers of lymphocytes (Nakashima & Kojiro, 1986). Massive lymphoid stroma may also circumscribe otherwise typical HCC.
Hepatocellular Carcinoma in Noncirrhotic Liver
HCC in noncirrhotic liver encompasses several entities. It may develop during the evolution of a chronic fibrosing liver disease at the stage of incomplete cirrhosis (septal fibrosis). This is especially common in the context of chronic hepatitis B (Lam et al, 2004). Because of the possible reversibility of cirrhosis, it is not known whether HCC in incomplete cirrhosis develops during an ongoing fibrogenesis or along with the reversion of cirrhosis. In this context, HCC does not show any specific morphologic criteria. HCC may also develop in strictly normal liver or in liver with mild changes, such as steatosis. This group includes the fibrolamellar variant of HCC, that arising from malignant transformation of liver cell adenoma (see below), and a group that develops in the context of other chronic liver disease, including nonalcoholic fatty liver disease (Paradis et al, 2009; Marrero et al, 2002).
Grading and Other Pathologic Prognostic Factors
Grading of HCC relies on the Edmondson and Steiner system, which subdivides HCC into four grades, from I to IV, on the basis of histologic and cytologic resemblance to normal liver (Edmonson & Steiner, 1954). This grading has been shown to correlate with the DNA content and cellular proliferation indices of the tumor (Grigioni et al, 1989). Grade I is the well-differentiated type, in which hepatocyte-like cells are arranged in thin trabeculae. Small HCC tends to be grade I, although these are often not uniform in their differentiation. Grade II HCC is composed of larger tumor cells with abnormal nuclei; glandular structures may also be present. In grade IV, neoplastic cells are much less differentiated, with hyperchromatic nuclei and loss of trabecular pattern. In fact, most HCC seen initially is grade II or III (Kenmochi et al, 1987). Therefore, and as for other carcinomas, the general tendency is to summarize the grading using a three-tiered system comprising well-, moderately, and poorly differentiated HCC.
Nonetheless, tumor grade is a weak independent predictor of the clinical course and conveys little prognostic information (Lai et al, 1979; Chuong et al, 1982). Furthermore, grading of HCC is often heterogeneous alongside the tumor so that liver biopsy might have limited performance for grading HCC. Consequently, the histologic grading of HCC is useful, but rather as a descriptive tool; its practical value is limited (Pawlik et al, 2007).
Besides grading, other histologic features of HCC, such as architectural pattern, have little or no independent prognostic value. However, several staging systems have been proposed for HCC (Llovet et al, 1999; Marrero et al, 2005). The main prognostic factors are related to general health status; tumor stage, specifically the number and size of nodules, presence of vascular invasion, and extrahepatic spread; liver function, defined by the Child-Turcotte-Pugh classification system and by serum bilirubin and serum albumin levels; and portal hypertension. Etiology has not been identified as an independent prognostic factor.
Size is a major prognostic factor: small or minute carcinomas have a better prognosis, although with larger cancers, the tumor size does not directly correlate with outcome. Presence of satellite nodules around the main tumor has been also recognized as a prognostic factor in several studies. Improved survival has been associated with tumors that are encapsulated or fail to invade surrounding hepatic parenchyma (Ohnishi et al, 1987; Arii et al, 2000; Sutton et al, 1988).
Macroscopic and microscopic vascular invasion are important histoprognostic criteria that deserve special mention. Vascular invasion is a known predictor of recurrence and survival, directly associated with histologic differentiation, degree, and size of the main nodule (Nathan et al, 2009; Pawlik et al, 2005). Characteristically, the prevalence of microscopic vascular invasion increases with tumor size, with up to 60% to 90% in nodules larger than 5 cm. Other histologic findings are not consistently correlated with outcome, although occasional series have reported a better prognosis with clear-cell HCC and tumors of low histologic grade.
Using microarray technology, recent studies have shown that a subset of adult HCC displays phenotypic traits of hepatoblasts. These tumors retent stem cells markers and express CK7 and CK19. Interestingly, worse survival was demonstrated for this subgroup (Lee et al, 2004). Although the significance of CK7 and CK19 expression in HCC is still unclear, it might represent a relevant prognostic factor, although a larger study with multidimensional analysis should be performed.
Molecular Genetics
From experimental hepatic carcinogenesis and epidemiologic studies, it appears that liver carcinogenesis follows a multistep process (see Chapter 8C). Although a few cases of HCC arise in normal liver, the vast majority of them develop from the stepwise pathway: normal liver progresses to fibrosis then to cirrhosis and finally to HCC. Therefore, cirrhosis is recognized as a precancerous lesion (IWP, 1995).
As a result of careful follow-up of cirrhotic patients and detailed pathologic analysis of cirrhotic explanted livers, relevant insights have been gained into the early pathogenesis of HCC. Data are less clear when associated molecular events are concerned; however, a consensus supports the idea that HCC results from cumulative genetic and epigenetic events (Thorgeirsson & Grisham, 2002). Although recurrent gene abnormalities have been reported in fully developed HCC, the early molecular events are far less unknown.
Several studies using different approaches have looked for early molecular abnormalities in regular cirrhosis. Several molecular markers have been evaluated in macronodules, and none of the oncogenes or tumor suppressor genes involved in advanced HCC have been repeatedly found altered in the precancerous lesions. By contrast, proliferation markers, neoangiogenesis, telomerase expression, allelic losses, and clonality have been studied with more consistent results. Interestingly, these studies convincingly demonstrate, using clonal analysis, that among cirrhotic micronodules that looked similar on light microscope, some are already monoclonal (neoplastic), and others are polyclonal (regenerative; Paradis et al, 1998, 2000). Telomerase, an enzyme that allows unrestricted cell proliferation and that is specifically expressed in cancer, can be detected in some but not all of these clonal micronodules without any remarkable histopathologic features (Oh et al, 2003). In the same orientation, and using cell-cycle markers, studies have shown that cirrhosis associated with HCC, or that will give rise to HCC, already expresses increased cell-cycle markers. Therefore even in regular cirrhosis with a bland histopathologic appearance, foci of neoplastic clonal proliferation of liver cells with a limitless capacity of proliferation clearly are already present.
Most genomic studies address advanced HCC, and several efforts have been made to provide molecular classification of HCC based on gene expression (Villanueva et al, 2007; Chuma et al, 2003). Global gene-expression profiling may be the most appropriate technology to unravel the pathogenesis of HCC and explore its heterogeneous origin (Lee et al, 2004; Wurmbach et al, 2007; Llovet et al, 2006; Hoshida et al, 2008). In fact, application of gene-expression profiling of HCC has identified subgroups of patients according to etiologic factors, different stages of the disease, recurrence, and survival (Villanueva et al, 2008; Ladeiro et al, 2008; Boyault et al, 2007). The diversity of these tumors is also found at the molecular level, and a large number of genetic and epigenetic alterations have been described, some of them specific of risk factors. Recently, several comprehensive studies have shown that a molecular classification of HCC can be drawn, linking the transcriptomic pattern of gene expression (Boyault et al, 2007), micro-RNA profiling (Ladeiro et al, 2008), promoter methylations, chromosome gains, and deletions (Xue et al, 2008) to mutation of oncogenes and tumor suppressor genes (de la Coste et al, 1998). Major classes of tumors emerging from these comprehensive analyses are also related to important carcinogenesis pathways, such as activation of ß-catenin (de la Coste et al, 1998), AKT/mTOR (Villanueva et al, 2008), or inactivation of TP53 and RB1. Whether this molecular subclassification may provide a clue to personalized, targeted treatment or to prognosis is under active investigation (Villanueva et al, 2008). Interestingly, using genome-wide expression profiling of formalin-fixed, paraffin-embedded tissues, a recent study demonstrated that a reproducible gene-expression signature correlating with survival was only described in liver tissue adjacent to the tumor in patients with HCC (Hoshida et al, 2008).
Premalignant Hepatocellular Lesions and Small Hepatocellular Carcinomas
It is commonly accepted that in the context of cirrhosis, there is a stepwise progression from cirrhotic nodule to HCC. Several terms have been used in the past to define the intermediate lesions, such as adenomatous hyperplasia and atypical adenomatous hyperplasia; but in 1995, the IWP proposed a unified nomenclature that has gained wide acceptance and is still currently used. This classification was recently reviewed and completed by an international group that added definitions of the early and small HCC (ICG, 2009).
Macroregenerative Nodules
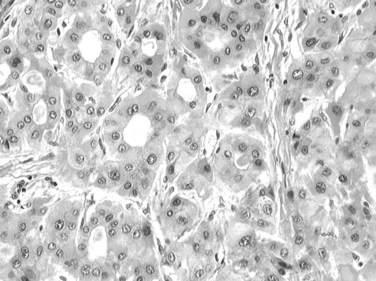
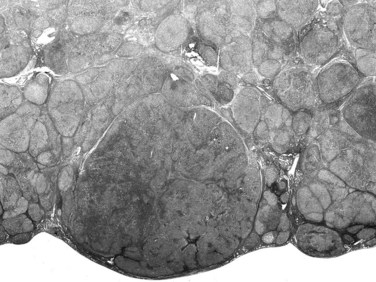
A macroregenerative nodule (MRN) is a tumor-like hepatocellular mass that can arise in the setting of cirrhosis. These lesions have increasingly been detected, the result of both improved radiographic imaging techniques and more widespread screening of cirrhotic patients. Most MRN are large, discrete nodules that range from 1 to 3 cm in diameter. There is no minimum size threshold for definition; MRNs have to be appreciated according to the size of background cirrhotic nodules, which may be twofold to threefold larger than cirrhotic nodules (Theise et al, 1992; Terada et al, 1993). Macronodules are often well demarcated and surrounded by condensed connective tissue. MRNs are common, and they have been found after careful inspection in about 10% of cirrhotic livers at autopsy or at transplantation (Theise et al, 1992; Furuya et al, 1988; Mion et al, 1996). Histologically, most MRNs are indistinguishable from the usual parenchymal nodules seen in cirrhosis: normal-appearing hepatocytes are arrayed in plates one or two cells thick, limited by a regular sinusoid lining and bounded by typical fibrous septa containing blood vessels, bile ductules, and varying degrees of inflammatory infiltration (Fig. 78.15).
Dysplastic Nodules
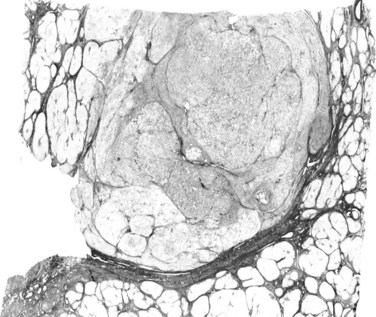
Dysplastic nodules (DNs) are sizable lesions arising in cirrhosis, and they differ from the surrounding liver parenchyma with regard to size, color, texture, and degree of bulging of the cut surface. Based on microscopic features, DNs are further subdivided into low grade (LGDN) and high grade (HGDN), the latter being closer to HCC in the spectrum of hepatocarcinogenesis (IWP, 1995; ICG, 2009). Briefly, LGDNs display features suggestive of a clonal cell population but lack architectural atypia, whereas HGDNs show cytologic and architectural atypia but insufficient for a diagnosis of malignancy.
The premalignant nature of DNs is supported by different clues, including the common association with HCC in resected and explanted end-stage cirrhotic livers (Theise et al, 1992; Furuya et al, 1988; Libbrecht et al, 2005; Sakamoto et al, 1991); the presence of hepatocellular cyto-architectural abnormalities, featuring a lesion on the way to HCC (Ferrell et al, 1992; Ganne-Carrie et al, 1996; Takayama et al, 1990); the morphologic evidence of neoangiogenesis under the form of unpaired arteries supporting the abnormally ongoing vascular supply (Roncalli et al, 1999; Park et al, 2000; El-Assal et al, 1998); the detection of both genetic and epigenetic changes, greater than those in the surroundings but less frequent and consistent than in HCC; and a natural history showing an increased risk for malignant transformation compared with control cirrhotic nodules (Maggioni et al, 2000; Kobayashi et al, 2006; Terasaki et al, 1998; Seki et al, 2000).
Among the various cytologic alterations that characterize DNs are enlarged, crowded, or irregular nuclei with patent nucleoli and an increased nuclear/cytoplasmic ratio. Atypical architectural findings involve expansile proliferative zones, sometimes located within an MRN (nodule-in-nodule formations); focal loss of associated reticulin framework; and foci of abnormal structural patterns that includes irregular thickening of hepatic plates (IWP, 1995; Fig. 78.16). According to Eastern pathologists, stromal invasion, defined as the presence of tumor cells invading into the portal tracts or fibrous septa, is the most relevant feature distinguishing HGDN from early HCC (International Consensus Group for Hepatocellular Neoplasia, 2009). The degree and extent of these features vary greatly among patients, thus forming a histologic continuum that stretches between ordinary MRNs and obvious hepatocellular carcinoma.
Dysplastic nodules must be differentiated from dysplastic foci, microscopic changes incidentally recognized in cirrhotic tissue. According to histopathologic criteria, dysplastic foci are split into two groups: those with large-cell changes or those with small-cell changes. Although large liver cell change, previously called large liver cell dysplasia (Fig. 78.17) consists of abnormal but nonneoplastic hepatocytes that are predictors of HCC development; small liver cell dysplasia is composed of neoplastic cells that are direct precursors of HCC (Lee et al, 1997; Borzio et al, 1995).
Small Hepatocellular Carcinoma
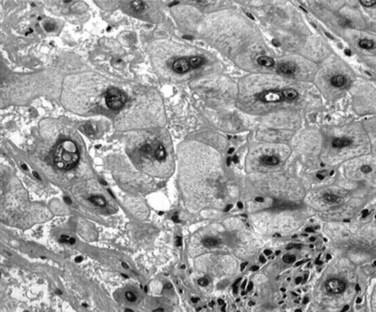
A “small” liver cancer is currently defined as HCC with a maximum diameter of less than 2 cm (Nakashima & Kojiro, 1986). However, the lack of a widely accepted definition has led to confusion, and HCCs smaller than 3 to 5 cm have also been included in this group by others. Small HCCs are usually clinically silent and are often discovered incidentally on explanted liver or at autopsy (Mion et al, 1996). Small HCCs are most often a well-differentiated lesion, sometimes still containing portal triads, growing without substantial destruction of the preexisting hepatic framework and seldom showing angioinvasion (Sakamoto et al, 1991).
Small HCCs have been subdivided into two different entities: those with indistinct margins, called vaguely nodular HCC, and those with distinct margins, called distinctively nodular HCC (ICG, 2009). Whereas vaguely nodular HCC is the earliest, most indolent, and least angioinvasive type of HCC, distinctively nodular HCC is thought to have already acquired an ability to invade the vessels and to metastasize (Hytiroglou, 2007; Kojiro & Roskams, 2005; Theise et al, 2002). Therefore small HCC with distinct margins seems to be a progressed cancer in spite of its small size. On the liver biopsy, the difference between these two entities cannot be appreciated, because it requires the notion of gross features, which are not recognizable in small fragments. Therefore the term well-differentiated HCC may be better used in the biopsy report.
Natural History of Premalignant Lesions and Diagnostic Challenges
Few prospective studies have been conducted in large series of histologically proven nonneoplastic nodules detected by US during surveillance programs in cirrhosis. Taken together, these studies have shown that only a minority of macroregenerative or dysplastic nodules became malignant, those transforming are mostly in the group of HGDN. Furthermore, 40% to 60% stabilized, and a few definitely disappeared during follow-up (Borzio et al, 2003; Kondo et al, 1990). Most of these nodules are 1 to 2 cm, so they seldom display a diagnostic pattern at imaging. Therefore histologic assessment is needed at baseline, and it is worth repeating when sampling is not adequate.
Whether HCC regularly develops from DNs in a low grade–high grade–small and early–small HCC sequence in individual patients is still unclear. This hypothesis is supported by the feature of so-called nodules in nodules, which have been mainly reported in the Eastern literature (Kojiro, 2004; Roncalli et al, 2004, 2007). However, HCC has also been found to originate from outside dysplastic nodules during the surveillance of cirrhotic patients, supporting the notion that HCC can also develop “de novo,” by skipping the gradual transition of the sequence (Borzio et al, 2003).
From a clinical point of view, correct classification of hepatocellular nodules, as well as the exact number of truly malignant nodules, is crucial in order to plan the most appropriate therapy. However, the diagnostic approach to these small nodular lesions is a challenge. The prevalence of HCC is closely dependent on the size of the lesion. Indeed, almost half of those lesions smaller than 1 cm are nonmalignant, whereas the large majority of lesions exceeding 2 cm are HCC, so that in the group of lesions greater than 2 cm, a diagnosis of nonmalignancy should suggest the suspicion of a diagnostic error. It has been well established that, in cirrhosis, the progression from nonmalignant nodules to dysplastic to early and advanced HCC is characterized by the progressive loss of the portal blood with a “de novo” arterial vascularization (Hayashi et al, 2002). However, in the group of lesions sized 1 to 2 cm, about 20% of small HCC is hypovascular, so that histology is required in most of these cases, either at baseline or during follow-up, to confirm the nature of the lesions (Forner et al, 2008).
In clinical practice, liver biopsy is today mostly performed to classify small hepatocellular nodules (Bruix & Sherman, 2005). Diagnosis has to be made on tiny and often fragmented material, and clinicians need a conclusive report as to whether lesions are benign or malignant. Nevertheless, the diagnosis is not always straightforward, and other techniques may be required in addition to standard staining. Fortunately, immunocytochemical tools useful to distinguish malignant from nonmalignant nodules within the group of well-differentiated hepatocellular lesions have been developed recently, such as glypican-3 antibody (Di Tommaso et al, 2007).
Hepatoblastoma
Hepatoblastoma is the most common primary liver tumor of childhood, accounting for 45% of the malignant hepatocellular neoplasms of infants and young children (Stocker, 2001; see Chapter 82). Almost all cases of hepatoblastoma occur during the first 3 years of life, although rare cases have been described in older children (Lack et al, 1982). Hepatoblastoma has been associated with congenital or genetic disorders, including Beckwith-Wiedeman syndrome, Wilms tumor, familial adenomatous polyposis, glycogen storage disease, and various congenital anomalies (Ishak & Glunz, 1967).
Hepatoblastomas are typically solitary, well-circumscribed, and occasionally encapsulated masses mostly located in the right lobe; size ranges from 5 to 20 cm in diameter (Stocker, 2001). The cut surface of hepatoblastoma has a bulging, lobulated contour and a variable tan to light brown to gray-white appearance with occasional foci of hemorrhage, cystic degeneration, necrosis, or calcification (Fig. 78.18; Stocker, 2001; Lack et al, 1982).
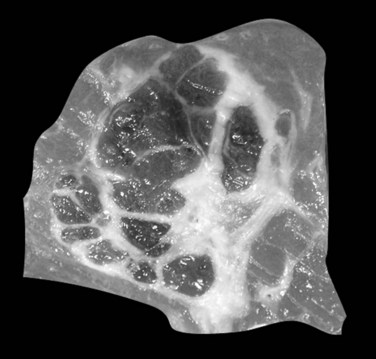
FIGURE 78.18 Hepatoblastoma. Well-delineated tumor with lobulation and hemorraghic foci and areas of cystic degeneration.
(Image courtesy Dr A. Fabre.)
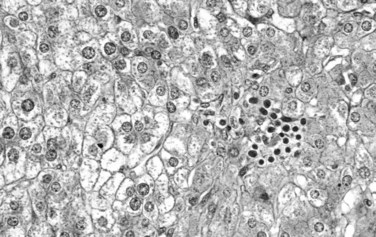
Fetal-type cells are smaller than normal hepatocytes with a uniform monotonous appearance, an abundant polygonal cytoplasm, and round, regular nuclei with inconspicuous nucleoli (Fig. 78.19). The cells are organized in irregular plates; nuclear pleomorphism is minimal, and mitoses are few, but foci of extramedullary hematopoiesis are a common finding. The embryonal-type cell is composed of less mature tumoral cells—dark, angular cells with compact cytoplasm and poorly defined outlines (Fig. 78.20). Scattered mitoses are regularly noted, and foci of necrosis are occasionally seen.
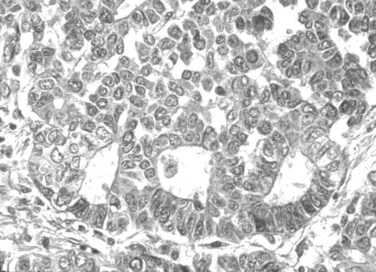
Fetal-type and embryonal-type cells often coexist, and transitions between the two are frequently present. In addition to the two major patterns, other types are described. The anaplastic type consists of small uniform cells similar to those of neuroblastoma with scanty cytoplasm, hyperchromatic nuclei, and frequent mitoses (Haas et al, 2001).
Hepatoblastoma is an aggressive neoplasm that invades locally and spreads to regional lymph nodes, lungs, adrenal glands, brain, and bone marrow. The most important predictor of outcome is the tumor stage at presentation. Once stage is taken into account, the various histologic subtypes become less important prognostic indicators (Conran et al, 1992). Nonetheless, tumors of pure fetal-type histology tend to have the best prognosis (Haas et al, 1989).
Benign Liver Tumors (See Chapter 79A, Chapter 79B )
Benign hepatocellular lesions encompass two main entities that strongly differ in terms of pathogenesis, clinical presentation, and behavior: focal nodular hyperplasia (FNH) and hepatocellular adenoma (HCA). Both groups are mainly observed in young women, usually in the context of oral contraception, and lesions develop in an otherwise normal liver. Whereas HCA is a neoplastic, clonal, and proliferative lesion, there is now consensus on the polyclonal, nonneoplastic, and reactive origin of FNH (Paradis et al, 1997; Bioulac-Sage et al, 2007a).
Focal Nodular Hyperplasia
FNH is ten times more common than HCA and is the second most common benign liver process after hemangioma. It is predominantly diagnosed in women 30 to 50 years of age. Most FNH is found incidentally, but some is revealed by clinical symptoms or biologic alterations such as pain, liver mass, or increased γ-glutamyltransferase (GGT) levels. However, complications such as rupture or bleeding are exceptional, and no evidence of malignant transformation has been reported. In contrast to HCA, the diagnosis of FNH is strongly suggested by imaging techniques, so that histopathologic examination is required for diagnosis in a minority of cases. When performed, fine needle biopsy of FNH can be difficult, because fibrous septa and thick abnormal arteries, the hallmarks of FNH, are usually missing (Fabre et al, 2002; Makhlouf et al, 2005).
On gross examination, FNH appears usually as a solitary, discrete, rounded mass, pale to beige, and well-delineated from background normal liver but without a defined fibrous capsule (Fig. 78.21). On cut surface, FNH displays a variegated and partly nodular organization, and frequently, but not always, a central stellate scar with radiating fibrous cords are also visualized at imaging (Wanless et al, 1985; Nguyen et al, 1999).
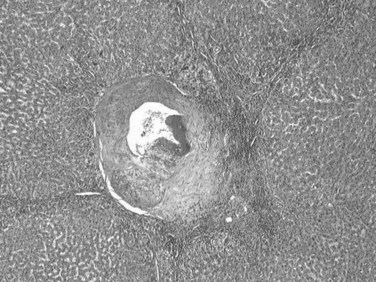
The typical histopathologic features of classical FNH include fibrous septa, which contain large dystrophic arteries with a peripheral ductular reaction, and an absence of interlobular bile ducts (Fig. 78.22). Hepatocytes are usually unremarkable, arranged in normal or mildly thickened plates without cytologic atypia, lined by a well-preserved sinusoidal framework. Steatosis or cholestatic degeneration with Mallory-Denk bodies may be seen.
FNH has been divided into classic and nonclassic forms (Nguyen et al, 1999). In forms of FNH other than the classic one, the diagnosis may prove difficult, because some of the distinguishing pathologic features may be unapparent. Such forms consist of FNH without a central scar, a mixed hyperplastic and adenomatous form, FNH with massive steatosis, and a form with atypia of the large-cell type (Nguyen et al, 1999). Strong evidence has been found, based on clonal features and the angiopoietin gene-expression profile, that the previously so-called telangiectatic variant of FNH may, in fact, belong to the HCA group (see below; Paradis et al, 2004).
Immunohistochemistry may provide interesting information. Immunostaining with glutamine synthetase that shows focal positive hepatocellular areas, usually centered by hepatic veins and described as a maplike pattern, is highly consistent with the diagnosis of FNH and suggests a focal maintenance of lobular organization (Bioulac-Sage et al, 2009a). In addition, the perisinusoidal spaces in FNH exhibit an aberrant extracellular matrix associated with abnormal fibrillin-1 expression (Lepreux et al, 2004). Further study showed that the phenotype of endothelial cells lining the vascular channels in FNH differs from those in the remaining liver, associated with a downregulation of angiotensin I–converting enzyme/CD143 (Grantzdorffer et al, 2004).
FNH and FNH-like nodules are now well known to develop in the context of hepatic venous outflow obstruction, including Budd-Chiari syndrome (Maetani et al, 2002; Rangheard et al, 2002), and in other circulatory disorders of the liver such as portal vein thrombosis, portal vein agenesis, hereditary telangiectasia, and even in cirrhosis; this supports the concept of a vascular trigger in the pathogenesis of FNH (Kondo, 2001; Kondo et al, 2004; Bureau et al, 2004). Evidence supports that FNH is a hyperplastic reaction resulting from an arterial malformation related to increased arterial blood flow (Wanless et al, 1985).
Similar to HCA, FNH has been observed in association with the use of oral contraceptives, although the relationship remains controversial (Scott et al, 1984). FNH may occur together with HCA (Grange et al, 1987), and it has been found to be associated with a variety of nonhepatic tumors and tumorlike conditions, such as HCA, hemangioma of the liver, and several other tumors (e.g., in siblings with glioblastoma; Handra-Luca et al, 2006). The significance of these associations is unclear, but it suggests that the pathogenesis of FNH is probably heterogeneous.
Hepatocellular Adenomas
HCA is a rare, benign liver cell neoplasm strongly associated with oral contraceptive (OC) use and androgen steroid therapy (Coombes et al, 1978; Nime et al, 1979). Its estimated incidence is 0.1 per year per 100,000 in non-OC users and reaches 3 to 4 per 100,000 in long-tem OC users. HCA can also occur spontaneously or in association with underlying metabolic diseases, including type 1 glycogen storage disease, iron overload related to β-thalassemia, and diabetes mellitus. Therefore, HCA represents a heterogeneous group of tumors in which histopathologic features may vary according to the etiologic background (Bioulac-Sage et al, 2007b).
HCA is usually solitary, sometimes pedunculated, with size ranging from a few millimeters to 30 cm. Large subcapsular vessels are commonly observed with large superficial tumors (Fig. 78.23). On analysis of cut sections, the tumor is soft, white to brown, and well delineated with little or no fibrous capsule (Fig. 78.24). Heterogeneous areas of necrosis or hemorrhage may be observed, especially in larger tumors. Histologically, HCA consists of a proliferation of benign hepatocytes of normal size or slightly enlarged with a normal nuclear/cytoplasmic ratio. Hepatocytes are arranged in a trabecular pattern without any residual portal tracts; a pseudoglandular growth pattern is possible but rare. Small, thin, and unpaired vessels without other portal tract elements—including connective tissue, bile ducts, or ductular reaction—are usually found throughout the tumor (Fig. 78.25). The cytoplasm of hepatocytes may be normal, glycogen-rich, or fatty, and cellular atypia can be impressive, especially in patients who have taken steroids for many years. In that context, differential diagnosis with HCC may be difficult. Vascular changes are frequent and include sinusoidal dilation, peliosis, infarcts, and hemorrhage. These changes may leave edematous or fibrotic regions, often with hemosiderin-laden macrophages.
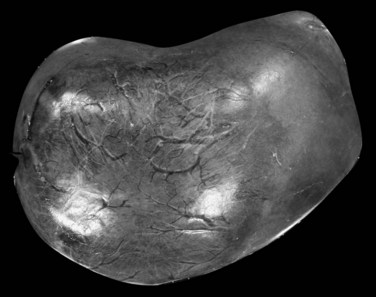
FIGURE 78.23 Hepatocellular adenoma. External view of a superficial tumor with large subcapsular and dilated vessels.
Compared with patients with FNH, those with HCA are more likely to come to medical attention with symptoms such as spontaneous bleeding and hemorrhage, with an increased risk according to size for tumors larger than 5 cm in diameter (Dokmak et al, 2009). The risk for malignant transformation of HCA ranges between 4% to 10%, with a higher rate in males and in large HCA (Bioulac-Sage et al, 2007; Dokmak et al, 2009). In addition, recent evidence suggests that metabolic syndrome may favor development of HCC in a preexisting HCA (Paradis et al, 2009). Increased incidence of metabolic syndrome may partly explain the rising incidence of malignant transformation of HCA, especially in the male population (Farges et al, 2011).
Multiple HCA and so-called adenomatosis are not so rare (Fig. 78.26). Patients with multiple HCAs are predominantly female, but the use of OC appears to be less prevalent (Fléjou et al, 1985). Patients with glycogen storage disease type I are also at risk of developing multiple HCA (Labrune et al, 1997). Nevertheless, these tumors share the same clinical and imaging characteristics independent of their number (Dokmak et al, 2009; Lewin et al, 2006). In addition, a recent study supports that the risk of complications, including bleeding and malignant transformation, is similar to that in patients with solitary HCA, and it is not influenced by the number of tumors (Dokmak et al, 2009; Bioulac-Sage et al, 2009b). The three main morphologic patterns of liver adenomatosis have been described: 1) steatotic, 2) peliotic/telangiectatic, and 3) mixed (Lewin et al, 2006). To note, the proportion of steatotic HCA is higher, and presence of microadenomatous foci in the “nontumoral liver” is more often observed in patients with liver adenomatosis (Dokmak et al, 2009).
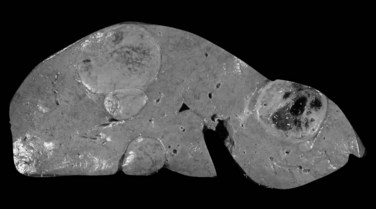
FIGURE 78.26 Adenomatosis. Multiple hepatocellular adenomas of various sizes are dispersed throughout the liver.
Molecular Classification of Hepatocellular Adenomas
Molecular comprehensive studies have recently garnered further insights into the structure of HCA, demonstrating some degree of molecular and histologic heterogeneity among the group of HCAs. Today, HCAs are subdivided into three main subtypes according to phenotypic and molecular features: 1) hepatocyte nuclear factor (HNF) 1α–mutated steatotic, 2) telangiectatic/inflammatory, and 3) β-catenin–mutated (Paradis et al, 2004; Rebouissou et al, 2008; Zucman-Rossi et al, 2006). In addition, a small group of HCA that does not display any specific morphologic or genotypic features remains unclassified.
The first group of HCA displays biallelic mutations of the TCF1 gene, inactivating the HNF-1α transcription factor. This homogeneous group is phenotypically characterized by marked steatosis and absence of cytologic abnormalities or inflammatory infiltrates (Fig. 78.27). Whereas both HNF-1α mutations are somatic in most cases, patients with inherited mutation in one allele of HNF-1α and an additional somatic mutation may develop HCA in the context of maturity-onset diabetes of the young type 3 (MODY3) and are predisposed to familial liver adenomatosis (Bluteau et al, 2002; Bacq et al, 2003).
The second group of HCA displays β-catenin–activating mutations and is characterized by increased risk for malignant transformation into HCC. These HCAs are mostly encountered in male patients and frequently show significant cell atypias and pseudoglandular formations (Fig. 78.28; Zucman-Rossi et al, 2006).
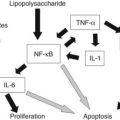
The third group of HCA is the inflammatory/telangiectatic adenoma group. These tumors, previously called telangiectatic variant of FNH, are well-delineated, unencapsulated tumors with areas of vascular changes without any fibrous scar (Fig. 78.29; Paradis et al, 2004; Wanless et al, 1989; Bioulac-Sage et al, 2005). Histologically, the hepatocellular proliferation contains small clusters of arteries embedded in collagen, often associated with an inflammatory infiltrate (lymphocytes and macrophages) and occasionally with ductular proliferation. In addition, foci of sinusoidal dilation and peliotic changes are usually present, and mild or significant steatosis may also be observed within tumoral cells. Although commonly observed in women using OC, telangiectatic/inflammatory HCA is often reported in patients with increased body mass index (BMI) associated with inflammatory syndrome (increased C-reactive protein [CRP] or fibrinogen serum levels; Paradis et al, 2004). Interestingly, in approximately 60% of these lesions, the interleukin (IL)-6 signaling pathway was found to be activated in relation to mutations in the IL6ST gene that encodes the signaling coreceptor gp130. As a matter of fact, the gain-of-function somatic mutations in gp130 may result in the inflammatory phenotype of HCA, which would explain activation of the acute inflammatory phase observed in tumoral hepatocytes (Rebouissou et al, 2009).
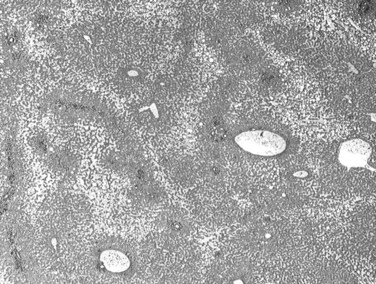
FIGURE 78.29 Telangiectatic/inflammatory adenoma. Extended foci of sinusoidal dilation are visible at low magnification.
Surrogate immunophenotypic markers related to the genetic abnormalities may be used for the classification of the main subtypes of HCA (Bioulac-Sage et al, 2007b). Indeed, expression of liver fatty acid binding protein (LFABP), a protein positively regulated by HNF-1α, is absent in steatotic HNF-1α–mutated HCA, whereas it is expressed in nontumoral liver. Similarly, telangiectatic/inflammatory HCA displays positive immunostaining with acute-phase inflammatory proteins, such as serum amyloid A (SAA) and CRP. Most β-catenin–mutated HCA presents abnormal and cytoplasmic and/or nuclear staining of β-catenin in tumoral hepatocytes, usually with a focal positivity restricted to a few isolated tumoral hepatocytes. Immunostaining with glutamine synthetase, a β-catenin–targeted gene, showing a strong and diffuse cytoplasmic staining increases the sensibility for diagnosis of β-catenin–mutated HCA.
Biliary Cell Tumors
Cholangiocarcinoma
Epidemiology and Clinical Background (See Chapters 50A and 50B)
Cholangiocarcinoma (CC) is a malignancy with biliary differentiation that develops along the biliary tree. It is the second most frequent primary malignant tumor of the liver (5% to 15% of liver primary malignancies; Malhi & Gores, 2006; Welzel et al, 2006). CC is mainly observed in adults with a peak incidence between 60 and 70 years and a male/female ratio of 1.5 : 1 (Parkin et al, 2005). CC is classified according to the anatomic location, either intrahepatic (IH-CC) or extrahepatic (EH-CC; Welzel et al, 2006). CC within the intrahepatic bile ducts is referred to as peripheral cholangiocarcinoma, arising in the bifurcation of the common bile duct as hilar CC, or Klatskin tumor; those in the extrahepatic bile ducts are extrahepatic CC (Klatskin, 1965). The updated WHO classification divides CC into IH-CC, hilar CC, and EH-CC (Bosman et al, 2010).
Hilar tumors are the most common, accounting for 60% to 70% of CC; peripheral lesions and tumors located in the extrahepatic bile duct represent 5% to 10% and 20% to 30% of CC respectively (Malhi & Gores, 2006; Patel, 2006). Although hilar CCs are usually considered an IH-CC, their characteristics in terms of clinical presentation, morphology, and phenotype are closer to EH-CC.
Prevalence of CC shows a wide geographic distribution largely as a result of variations in regional environmental risk factors, with the highest incidence observed in Asian countries where parasitic infections—namely, Opisthorchis viverrini and Clonorchis sinensis—are endemic (Kim et al, 1989; Lim et al, 2006; Watanapa & Watanapa, 2002; see Chapter 45). Importantly, incidence of CC is rising in most countries, even in nonendemic areas. Such an increase is specifically observed for IH-CC, including hilar tumors, whereas incidence of EH-CC is stable or even decreased (Khan et al, 2002; Patel, 2001, 2006; Shaib et al, 2004).
Although several risk factors for CC are well established, most occur in patients without evident predisposing factors (Chapman, 1999). Primary sclerosing cholangitis (PSC) associated with ulcerative colitis is one of the most recognized risk factors (see Chapter 41). Indeed, patients with PSC display a 1.5% cumulative annual risk of developing CC, and 10% to 20% will eventually develop CC (Boberg et al, 2002). Fibropolycystic liver diseases that include choledocal cyst, Caroli syndrome, and congenital hepatic fibrosis are also common risk factors (Yamato et al, 1998; see Chapter 46); infections with parasitic liver flukes, such as Opisthorchis viverrini and Clonorchis sinensis, contribute to the high incidence of CC in Asia (Curado & Shin, 2007; Lim et al, 2006; see Chapter 45). Hepatholithiasis (see Chapters 39 and 44), cholelithiasis, cholangitis, chronic pancreatitis, and Thorotrast exposure are also risk factors (Chapman, 1999; Lipshutz et al, 2002). More recently, associations of CC with obesity, diabetes, human immunodeficiency virus (HIV) and hepatitis C virus (HCV) infection, and alcoholic liver disease have been reported in large population databases (Shaib et al, 2005; Welzel et al, 2007).
Clinical symptoms at diagnosis are dependent on the anatomic location, the growth types of the CC, and the tumor-node-metastasis (TNM) stage (Malhi & Gores, 2006). Although IH-CC is usually discovered at a late stage, hilar CC and EH-CC usually are found earlier with cholestasis (Ebata et al, 2009). The prognosis of CC is dismal, partly related to its propensity to invade directly adjacent liver parenchyma and to spread along the portal pedicles. Presence of perineural and vascular invasion is frequent in the hilar tumors. In the intraductal growth type, tumor often spreads intraluminally along the ducts, and intrahepatic metastases develop in nearly all advanced cases. The incidence of metastases to regional lymph nodes is higher than in HCC. Blood-borne spread occurs later, particularly to the lungs (Jiang et al, 2009; Patel, 2001; Suzuki et al, 2002).
Pathology (See Chapter 47)
Most CC develops in noncirrhotic liver. The Liver Cancer Study Group proposed a classification of CC based on growth patterns, and three main types emerged: 1) mass forming, 2) periductal infiltrating, and 3) intraductal growing ( Yamasaki et al, 2003). These patterns are associated with different clinical evolution, the intraductal growing and the periductal infiltrating showing the best and worse prognoses, respectively (65% survival at 5 years vs. <5%; Jiang et al, 2009; Suzuki et al, 2002).
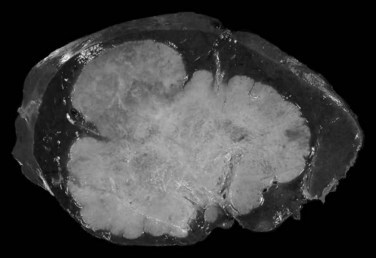
The mass-forming variation is the main type of IH-CC, accounting for approximately two thirds of cases. It is a solitary, nodular lesion developed in the liver parenchyma. The tumor is usually well delineated, not encapsulated, and gray to white with a firm and solid consistency (Fig. 78.30). In cases with large tumors, adjacent satellite tumoral nodules are commonly observed. The periductal infiltrating type spreads along the portal tracts with stricture of the involved ducts, potentially leading to obstructive dilation and cholangitis of the peripheral bile ducts (Fig. 78.31). The intraductal growing type is a polypoid or papillary tumor developed within dilatated bile duct lumen (Fig. 78.32). This type represents malignant progression of biliary papillomatosis or intraductal papillary neoplasm (IPN) of the bile duct (see below). These three patterns may overlap in the same tumor. Although most CC occurs in a background normal liver, recent reports suggest that independently to its origin, cirrhosis increases the risk 10-fold for patients to develop CC (Fig. 78.33; Shaib et al, 2005; Kobayashi et al, 2000).
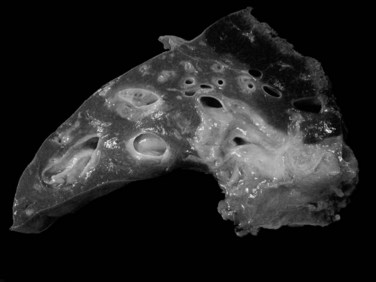
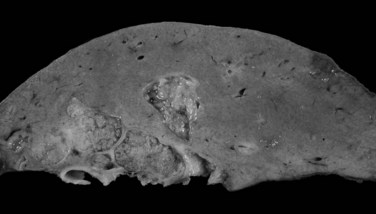
The next most common form is mixed mass forming and periductal infiltrating CC, observed in 25% of cases. The pure peri-infiltrating type and the intraductal growth pattern are seldom observed (Guglielmi et al, 2009). CC arising in the intrahepatic small bile ducts or ductules is usually the mass-forming type, whereas CC arising in the intrahepatic large bile ducts (hilar CC) may be any of the three types (Nakanuma et al, 2008).
Most CCs are adenocarcinomas with a prominent, fibrous stroma. CC can be graded as well-, moderately, or poorly differentiated adenocarcinoma according to morphology. Most CCs are well-differentiated tumors with a glandular or tubular pattern of growth, although micropapillary, acinar, or cordlike patterns also occur. Similar to their normal counterparts, tumor cells are small or medium sized, cuboidal or columnar, with small nuclei and nucleoli (Fig. 78.34). Cytoplasm is usually pale, slightly eosinophilic, and sometimes more abundant and clear. Mucus secretion may be highlighted by Alcian blue staining, though the amount is usually small. Less differentiated tumors may show cribriform formations and/or a cordlike pattern, and poorly differentiated cancers are characterized by marked cellular pleomorphism. Hilar and EH-CC are more often well differentiated compared with peripheral CC (Guedj et al, 2009). Presence of dense, hyaline, fibrous stroma is a key feature of CC. Usually, the center of the tumor is densely sclerotic and hypocellular with focal calcifications, whereas the periphery of the tumors is more cellular. CC frequently infiltrates into portal tracts, invading portal vessels, and perineural invasion is a frequent finding in hilar CC (Fig. 78.35; Guedj et al, 2009).
Because CC originates from biliary epithelial cells, tumor cells display immunostaining with anti–CK7 and anti–CK19, carcinoembryonic antigen (CEA), and epithelial membrane antigen (EMA). The keratin profile is particularly useful for the differentiation of IH-CC from metastatic carcinoma derived from colorectal origin: CK7 is constantly expressed in CC, and CK20 is constantly expressed in metastatic colon carcinoma (Rullier et al, 2000). The diagnosis of IH-CC may be a challenging main diagnostic issue concerning the differential diagnosis between CC and metastatic carcinoma. In that setting, fine needle aspiration, and especially cytologic smears, have been shown as performant as core biopsy with a specificity of 100%, a sensitivity of 84%, and no false-positive cases of CC (Pupulim et al, 2008). In contrast, performance of biopsy is much more limited in cases of extrahepatic strictures. Additional approaches, including evaluation of ploidy and fluorescence in situ hybridization (FISH), have been shown to increase sensitivity up to 34% and specificity up to 98% (Kipp et al, 2004; Levy et al, 2008).
Biliary Papillomatosis and Biliary Intraepithelial Neoplasia
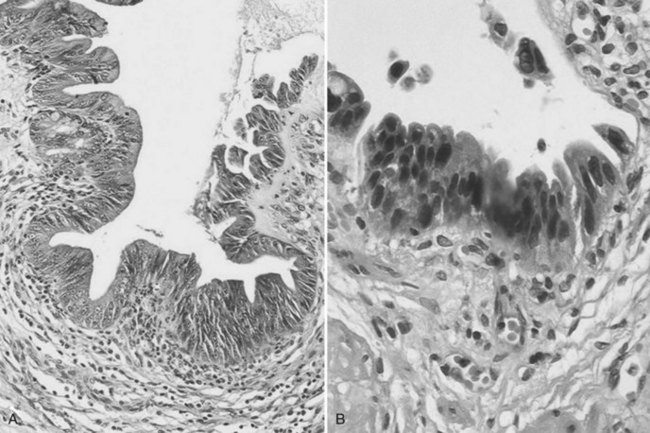
Biliary papillomatosis is closely associated with the intraductal growing pattern of CC. It is a rare condition characterized by the multicentric proliferation of columnar epithelium within the large bile ducts, although it can diffuse anywhere in the large intrahepatic or extrahepatic bile ducts (Padfield et al, 1988; Mercadier et al, 1984; Gouma et al, 1984). Biliary papillomatosis is a disease of middle-aged or older adults, and men are affected about twice as often as women. Grossly, the neoplasm invades the bile duct lumen, which is filled with soft pink to tan papillary excrescences, although skipped areas may be present (Zen et al, 2006). The surface epithelial cells covering these papillae are pancreatobiliary-, gastric-, or intestinal-type cells (Fig. 78.36). According to the WHO classification, and in similarity with pancreatic lesions, biliary papillomatosis has been renamed biliary intraepithelial papillary neoplasm (IPN). Although often cytologically benign, the cells can display greater degrees of nuclear atypia; in situ or invasive carcinoma is occasionally noted (Gouma et al, 1984; Neumann et al, 1976). Specific differentiation of the epithelial lining may be observed, including that of the pancreatobiliary type, intestinal type, oncocytic type, and gastric type. This invasive tumor may be a conventional, tubular-type adenocarcinoma, or in 10% to 15% of cases, it may be a mucinous (colloid) adenocarcinoma (Zen et al, 2006; Lee et al, 2004). Oncocytic variants of biliary papillomatosis have also been reported that share similar clinical features with their nononcocytic counterparts. The invasive tumors associated with these oncocytic papillary lesions are also oncocytic, as a result of the presence of abundant cytoplasmic mitochondria (Rouzbahman et al, 2007).
A precancerous lesion may also develop on flat bile duct epithelium. This lesion is now termed biliary intraepithelial neoplasia (BilIN). BilINs are characterized by abnormal epithelial cells with multilayering of nuclei and micropapillary projections into the duct lumen (Zen et al, 2007). The abnormal cells have an increased nuclear/cytoplasmic ratio, partial loss of nuclear polarity, and nuclear hyperchromasia. They are divided according to degrees of atypia into low-grade (BilIN-1), moderate (BilIN-2), and high-grade dysplastic lesions (BilIN-3) (Fig. 78.37; Zen et al, 2005).
Hepatocholangiocarcinoma
Hepatocholangiocarcinoma—also known as combined hepatocellular cholangiocarcinoma, or mixed HCC-CC—is a rare but increasingly recognized neoplasm of the liver, accounting for approximately 5% of primary liver malignancies (Allen & Lisa, 1949; Aoki et al, 1993; Maeda et al, 1995). It shares unequivocal features of both HCC and CC as defined by the WHO classification. Depending on various investigations, patients with combined HCC-CC share similar clinical and pathologic features with patients with HCC (Maeda et al, 1995; Ng et al, 1998) or CC (Tickoo et al, 2002; Jarnagin et al, 2002), or their tumors are clinicopathologically different from those of CC or HCC. Caution must be exercised when interpreting the results of these studies, regarding how combined HCC-CCs were defined in the studies, and there may have been population and etiology differences as well. Taken together, it appears that combined HCC-CCs lack a specific clinical parameter, and this may be explained by various studies across different geographic regions, etiologies, and populations.
Although a unified histopathologic criteria for combined HCC-CC is still not available, it is generally accepted that a firm diagnosis of combined HCC-CC requires evidence of HCC differentiation—a trabecular growth pattern, bile production, or bile canaliculi—and clear evidence of CC, such as true glandular structures formed by biliary-type epithelium, mucin production, or prominent desmoplastic stroma (Fig. 78.38; Maeda et al, 1995; Yano et al, 2003; Goodman et al, 1985). Separate HCC and CC coincidentally found in the same liver is generally considered a collision tumor and is excluded by the WHO classification of combined HCC-CC. This has been further supported by the genetic findings that two independent neoplastic clones develop at close proximity, hence no histologic transitions exist (Fujii et al, 2000).
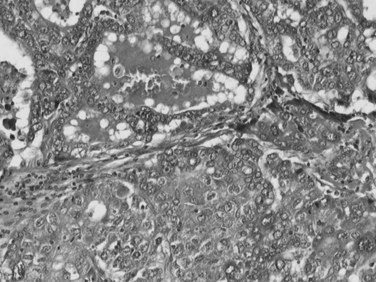
The cell of origin of combined HCC-CC has been a matter of debate. The fact that the HCC and CC elements intermingle with each other in a transitional area strongly supports that both components of the cancer derive from hepatic progenitor cells. Recent molecular investigations support this hypothesis, because single clonal tumors with a homogeneous genetic background in both HCC and CC components of combined HCC-CC have been demonstrated, suggesting that histologic diversity of HCC-CC is a phenotypic expression of the divergent differentiation potential of a single clone (Fujii et al, 2000; Theise, 2002; Cazals-Hatem et al, 2004).
Cholangiolocellular Carcinoma
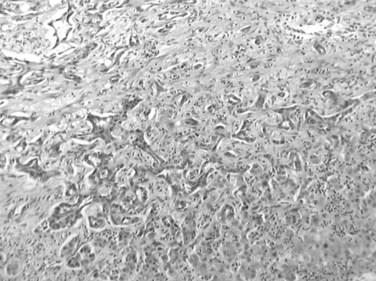
Cholangiolocellular carcinoma is a very rare neoplasm, accounting for less than 1% of primary liver cancers (Komuta et al, 2008; Steiner & Higginson, 1959). In this tumor, more than 90% of the neoplasm is composed of small, monotonous glands that represent residual ductular proliferation with an antler-like anastomosing pattern (Fig. 78.39). In addition, HCC and CC components may also be present, altogether comprising less than 10% of the neoplasm, although these three histologic components showed transitions among each other (Komuta et al, 2008). The relationship between cholangiocellular carcinoma and combined HCC-CC remains to be clarified, however, they may overlap to some degree and belong to a spectrum of the primary liver neoplasms that arise from the hepatic progenitor cells.
Biliary Cystadenoma and Cystadenocarcinoma (See Chapter 79B)
Biliary Cystadenoma
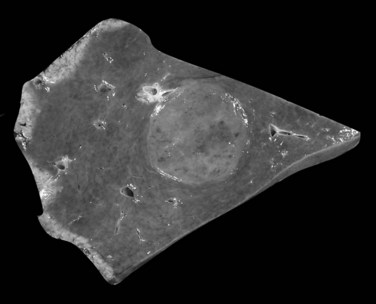
Biliary cystadenoma is an uncommon cystic neoplasm that accounts for less than 5% of all intrahepatic biliary cysts (Ishak et al, 1977; Van Roekel et al, 1982). The histogenesis of biliary cystadenoma remains uncertain, although an origin from embryonic foregut rests has been advanced (Akwari et al, 1990). Almost all of these tumors occur in middle-aged women, with a peak incidence in the fifth decade (Devaney et al, 1994).
Biliary cystadenomas are characteristically large (2.5 to 28 cm), multiloculated cysts without communication with the intrahepatic biliary system (Fig. 78.40). The tumor is solitary and spherical and contains white to yellow to brown mucinous or gelatinous material. Individual locules vary in size, and the internal surface is typically smooth with occasional trabeculations or papillations (Ishak et al, 1977). If solid areas are present, concern should be raised for an invasive component (Devaney et al, 1994; Buetow et al, 1995).
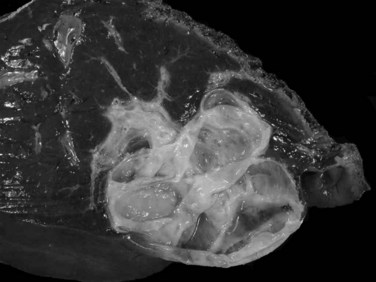
FIGURE 78.40 Cystadenoma. The tumor is cystic, solitary, and spherical with a smooth, white internal surface.
Underneath is an ovarian type stroma, which is absent in cases arising in men (Devaney et al, 1994). Typically, this stroma is densely cellular and composed of closely packed spindle cells reminiscent of ovarian stroma. This stroma stains with antibodies to estrogen and progesterone receptor, and growth can occur during hormone replacement therapy and pregnancy (Daniels et al, 2006). As dysplasia may be patchy, and invasive tumors may arise in as many as 25% of cystadenomas (Ishak et al, 1977), the gross specimens of cystic tumors need to be carefully examined for suspicious areas, and extensive sampling of the cyst should be performed.
Biliary Cystadenocarcinoma
This rare malignancy generally develops as a complication of a biliary cystadenoma, which may or may not demonstrate the distinctive mesenchymal stroma (Ishak et al, 1977; Wheeler & Edmondson, 1985b; Woods, 1981). Most patients are between 45 and 70 years of age, and men and women are equally affected. Cysts are generally multilocular and range in size from 5 cm in diameter to more than 20 cm. Although the gross appearance can be difficult to distinguish from a biliary cystadenoma, the suspicion of malignancy should be raised if areas of large, solid, thickening papillary masses are present (Ishak et al, 1977).
Histologically, cystadenocarcinomas are usually well-differentiated adenocarcinomas, often with an intracystic papillary component, and they are composed of malignant epithelial cells with varying degrees of nuclear stratification, pleomorphism, and hyperchromasia; often, the benign epithelium of the preexisting cystadenoma can be identified. Transitions can sometimes be discerned with varying degrees of epithelial dysplasia (Woods, 1981). The tumor infiltrates the underlying cyst wall, and vascular invasion and extension into adjoining hepatic parenchyma or adjacent organs are characteristic of malignancy. These tumors tend to grow slowly, but they eventually invade adjacent structures and metastasize to distant sites. In rare cases the carcinoma demonstrates adenosquamous, oncocytic, or spindle-cell (pseudosarcomatous) differentiation (Moore et al, 1984; Unger et al, 1987; Wolf et al, 1992).
Other Benign Cystic and Bile Duct Lesions: Ciliated Hepatic Foregut Cyst, Biliary Adenoma, and Biliary Hamartoma
Ciliated Hepatic Foregut Cysts
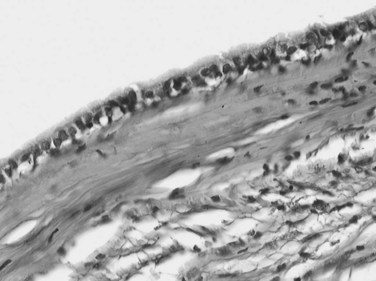
A ciliated hepatic foregut cyst is a rare lesion that is generally solitary and unilocular (Terada et al, 1990). This lesion is smaller than 3 cm and lined by a characteristic ciliated pseudostratified columnar epithelium with mucous cells and an underlying fibrous wall that contains abundant smooth muscle (Fig. 78.41; Vick et al, 1999a). Occasional cases of squamous carcinoma arising in a ciliated hepatic foregut cyst have been reported (Vick et al, 1999b). Because these features are similar to those seen in bronchial and esophageal cysts, an analogous origin from the embryonic foregut is suggested (Wheeler & Edmondson, 1984).
Bile Duct Adenoma
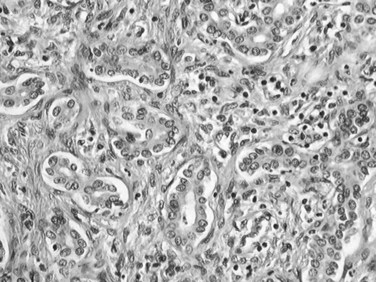
Bile duct adenoma is an incidental finding, often discovered at surgery, because most develop superficially under the Glisson capsule (Gold et al, 1978; Allaire et al, 1988). It is an inconsequential lesion that may be mistaken for bile duct hamartoma or metastatic carcinoma (Govindarajan & Peters, 1984). Almost all bile duct adenomas are less than 1 cm in diameter, although cases up to 4 cm have been recorded (Govindarajan & Peters, 1984). The lesion typically appears as a discrete white nodule, usually solitary and subcapsular in location, with a firm consistency and unencapsulated margin. Histologically, bile duct adenoma is made of a compact proliferation of small ductules with small or inapparent lumens lined by cuboidal epithelium with slightly basophilic cytoplasm and bland, regular nuclei (Fig. 78.42). Mitoses, cytologic atypia, and nuclear pleomorphism are absent. Ductular structures are surrounded by a fibrotic, hyalinized stroma that can distort ductal structures, portal tracts are often entrapped within the tumor, and scattered inflammatory cells are sometimes seen. Malignant transformation has not been clearly documented.
Biliary Microhamartoma
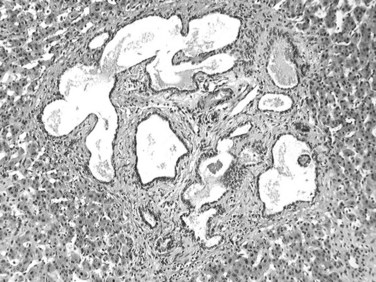
Biliary microhamartoma, also known as a von Meyenburg complex, is a developmental malformation resulting from the aberrant remodeling of the ductal plate. The lesion is relatively common; it occurs in about 3% of all autopsied patients and is generally found incidentally. Microhamartomas are often multiple, but when numerous, this raises the possibility of early autosomal dominant polycystic disease or congenital hepatic fibrosis (Desmet, 1992). The lesions appear as small, subcapsular white or green nodules that seldom exceed 0.5 cm in diameter, often located within or close to portal tracts. Microscopically, biliary microhamartomas are composed of numerous bile duct–like structures with an angulated or branching configuration with varying degrees of dilation. These structures are lined by a single layer of low columnar or elongated epithelium, and their lumens contain bile plugs or eosinophilic proteinaceous debris; ducts are embedded within a dense, fibrous stroma (Fig. 78.43). In rare cases, microhamartomas have been complicated by the development of cholangiocarcinoma (Burns et al, 1990; Honda et al, 1986).
Mesenchymal Tumors
Benign Mesenchymal Tumors
Hemangioma
Hemangioma is the most common primary tumor of the liver, with a prevalence in autopsy surveys that ranges from 0.4% up to 20% (Karhunen, 1986). This lesion is discovered in all ages and both sexes, although hemangiomas are more frequent in older age groups, and in most series they favor women (Bioulac-Sage et al, 2008).
Most hemangiomas are clinically silent and are discovered incidentally during radiologic examination, surgery, or autopsy. However, larger tumors can occasionally become clinically evident with abdominal discomfort, hepatomegaly, or a palpable abdominal mass (Schnelldorfer et al, 2010). Rare reports describe spontaneous rupture with hemoperitoneum or a bleeding diathesis with hypofibrinogenemia or platelet sequestration and consequent thrombocytopenia (Kasabach-Merritt syndrome; Gandolfi et al, 1991). Hemangiomas can be correctly identified in most instances by radiographic imaging (see Chapter 17). Needle biopsy is not necessary and is, in fact, contraindicated because of the risk of severe hemorrhage (Trastek et al, 1983).
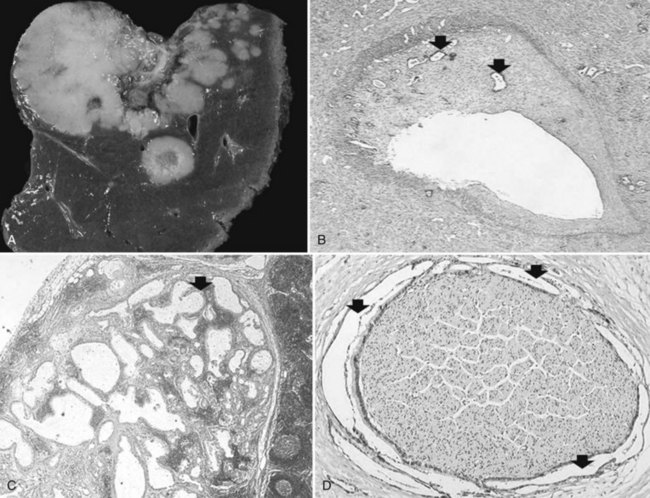
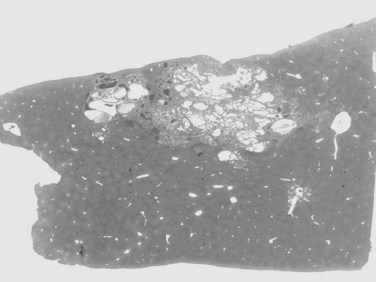
Most hemangiomas are solitary lesions less than 4 cm in diameter (Fig. 78.44). Multiple tumors are seen in up to 25% of cases, and hemangiomas up to 30 cm have been recorded. The tumors can occur anywhere in the liver but are frequently located immediately beneath the hepatic capsule. The cut surface demonstrates a soft, dark red, spongy mass with blood-filled cavities and occasional foci of thrombosis, scarring, or calcification. Most are well demarcated from the surrounding liver. Histologically, hemangiomas are composed of dilated vascular spaces lined by flattened endothelial cells and supported by connective tissue septa. The septa consist of poorly cellular fibrous bands with varying degrees of myxoid change and scarring. Thick-walled blood vessels and scattered bile ducts are sometimes found in larger septa, and the vascular spaces are frequently the sites of thrombi in various stages of organization. Hemangiomas may also demonstrate involutional changes with extensive hyalinization, obliteration of the vascular channels, and sometimes calcification.
Infantile Hemangioendothelioma
Infantile hemangioendothelioma is a benign pediatric vascular proliferation of the liver (see Chapter 82). Although uncommon, it is nonetheless the predominant hepatic neoplasm of the first year of life, and only a few cases have been reported in adults. Most cases manifest clinically with hepatomegaly, diffuse abdominal enlargement, or a palpable abdominal mass. The prognosis is generally good, because the tumors usually undergo spontaneous involution and can completely regress (Dachman et al, 1983); however, a potentially life-threatening feature is not uncommon because of arteriovenous shunting through the tumor that may result in cardiac failure, with a reported mortality rate of about 25% (Vorse et al, 1983).
Some hemangioendotheliomas may demonstrate atypical histologic features with atypical crowded and multilayered endothelial cells that show hyperchromatic nuclei and occasional mitoses. This atypical pattern has been referred to as type 2, in contrast with the more common type 1 pattern (Dehner, 1978). Most of the type 2 tumors pursue a benign course, but some exhibit more aggressive behavior with extensive local growth and, in some instances, metastases. Consequently such tumors have been considered by some investigators as a low-grade childhood form of angiosarcoma (Dehner & Ishak, 1971).
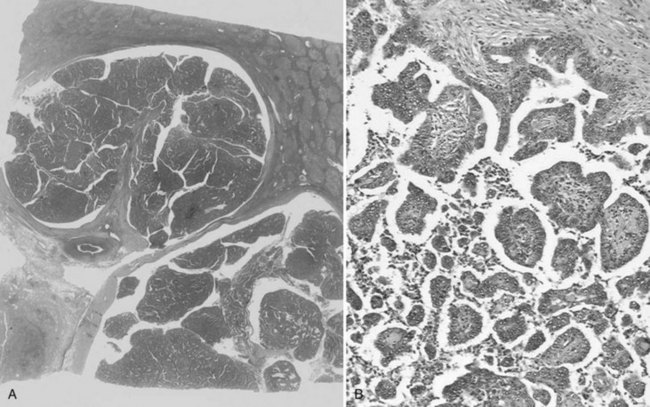
Mesenchymal Hamartoma
Mesenchymal hamartoma is a tumor-like lesion arising in young children, usually those younger than 2 years, although sporadic examples are described in adolescents and adults (see Chapter 82; Dehner, 1978; Gramlich et al, 1988). It is thought to represent a developmental anomaly, probably arising from the aberrant formation of primitive portal mesenchyme together with secondary degenerative changes (Siddiqui & McKenna, 2006). Although some cases are discovered incidentally, most are clinically evident because of their large size, causing progressive abdominal distension or a palpable abdominal mass (Srouji et al, 1978).
Mesenchymal hamartoma appears grossly as a solitary mass that ranges from 3 to 30 cm in diameter. Typically, the tumor is well demarcated from the adjacent liver, although an infiltrative border and satellite lesions are not so rare (Stocker & Ishak, 1983). On cut surface, the lesion is composed of multiple small cysts filled with clear or mucoid fluid, lined by a smooth surface, and surrounded by a myxoid tissue with fibrous bands (Dehner et al, 1975).
Histologically, mesenchymal hamartoma is characterized by the association of immature myxoid mesenchymal tissue, bile ducts, blood vessels, and mature hepatocytes. The mesenchymal component consists of elongated bland cells within an edematous myxoid stroma that contains varying amounts of collagen. Focally, this stroma undergoes cystic degeneration, yielding a lymphangiomatous appearance and giving rise to the grossly visible cysts. In addition, numerous thick-walled veins, foci of extramedullary hematopoiesis, and scattered bile ducts can be observed. The ducts often exhibit branching or cystically dilated lumina. Bile duct cells can be atrophic or hyperplastic, and small clusters of unremarkable hepatocytes that lack a lobular arrangement are interspersed within the tumor (Stocker & Ishak, 1983).
Angiomyolipoma
Angiomyolipoma (AML) is a rare tumor that occurs at a median age of about 45 years; it is predominant in women by a ratio of 5 : 1 (Goodman & Ishak, 1984; see Chapter 79A), and 10% of cases occur in the setting of tuberous sclerosis, usually associated with coexisting renal AML (Petrolla & Xin, 2008). Size ranges widely, from 1 cm in diameter to more than 20 cm. Tumors are usually single, but multiple tumors in the liver and multiorgan involvement have been described (Nonomura et al 1994, 1995). Only one malignant degeneration in a liver AML has been reported in the literature (Dalle et al, 2000).
Most AMLs are well circumscribed but not encapsulated (Fig. 78.45). AMLs have three key components: 1) tortuous vessels, 2) fat, and 3) myoid cells. Liver AMLs demonstrate solid growth with a variable amount of fat and myoid elements. Hematopoietic cells may also be seen in some cases. Myoid cells may be epithelioid or spindled. The fat component is typically mature, but lipoblast-like cells may be found. A hallmark of this lesion is the existence of tortuous, thick-walled vessels that are usually lacking in elastic lamina and are often rimmed by epithelioid myoid cells (Fig. 78.46; Tsui et al, 1999). The diagnosis of AML may be confirmed by immunohistochemistry, because myoid cells are reactive to HMB-45 (Sturtz & Dabbs, 1994).
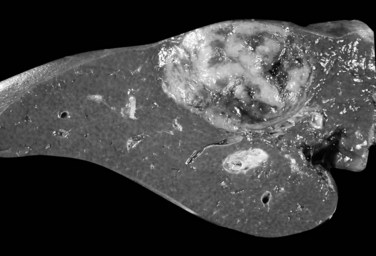
FIGURE 78.45 Angiomyolipoma. A well-circumscribed, soft, white nodular lesion with foci of hemorrhage.
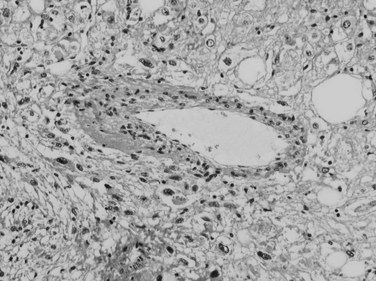
FIGURE 78.46 Angiomyolipoma, light microscopy. The lesion is composed of myoid cells with fat droplets and thick-walled vessels.
The histogenesis of AML is unclear, but recent studies suggest AML is a clonal proliferation that favors a neoplastic process (Flemming et al, 2000). Strong arguments suggest that the three cell types are derived from a single precursor cell in the perivascular space, the perivascular epithelioid cell (PEC; Tsui et al, 1999; Hornick & Fletcher, 2006). AMLs composed of a large number of epithelioid cells have been termed PEComas (perivascular epithelioid cell tumors; Bonetti et al, 1997).
Lipoma
Lipomatous tumors are rarely found in the liver. Most occur in adults as asymptomatic lesions, incidentally detected by radiographic imaging (Roberts et al, 1986). Typically, the tumors are solitary masses ranging from 1 to 20 cm. Microscopically, they are composed of mature adipose tissue, either pure or accompanied by a vascular, hematopoietic, or myeloid component. Depending on the constituents, the tumor is designated a pure lipoma, angiolipoma, or myelolipoma (Nishizaki et al, 1989; Peters et al 1983).
Coelomic fat ectopia consists of a rounded nodule of fat attached to the hepatic capsule, usually on the anterior diaphragmatic aspect of the right lobe (Komori et al, 2008). The histologic features accordingly entail mature adipose tissue with various degenerative changes, including calcification (Wheeler & Edmonson, 1985b).
Fibrous Tumors
Localized fibrous tumors of the liver have been reported under a variety of names, including fibroma, solitary fibrous mesothelioma, and benign mesothelioma. Similar to their pleural counterparts, these tumors arise from submesothelial connective tissue and form large masses, from 5 cm in diameter to more than 25 cm, that protrude from the hepatic surface (Komori et al, 2008). The histologic features entail interlacing bundles of bland spindle cells with variable cellularity and intermixed collagen fibers (Kim & Damjanov, 1983; Kottke-Marchant et al, 1989).
Malignant Mesenchymal Tumors
Epithelioid Hemangioendothelioma
Epithelioid hemangioendothelioma is a low-grade malignant tumor of endothelial origin that can occur in several sites, including the liver. At diagnosis, most patients are in their fourth or fifth decade of life, and about 60% are women (Ishak et al, 1984). Unlike hepatic angiosarcoma, no strong etiologic associations or predisposing factors have been identified, although a relationship with long-term OC use has been suggested (Dean et al, 1985).
Epithelioid hemangioendothelioma tends to be an indolent and slowly growing tumor, but the natural history can vary considerably ( Woodal et al, 2008). Most patients have a prolonged survival, whereas others suffer rapid deterioration with early death from hepatic failure or extensive tumor spread (Dean et al, 1985; Dietze et al, 1989). Metastases commonly involve the lung, spleen, abdominal lymph nodes, omentum, and peritoneum. Even with metastatic disease, some patients can survive for many years, although the distinction between metastatic spread and multicentric disease is sometimes difficult to make (Mehrabi et al, 2006).
In about 80% of cases, the tumor is multifocal with involvement of both lobes by tumor nodules of various sizes. Nodules are white to tan and have a firm consistency (Fig. 78.47), and the background liver is generally normal (Ishak et al, 1984).
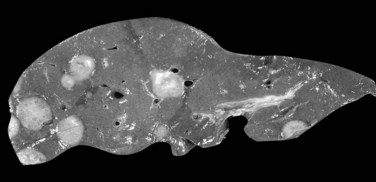
Histologically the tumor comprises small groups of tumor cells surrounded by a distinctive and often abundant stroma of varying myxoid or sclerotic character. The neoplastic cells display abundant eosinophilic cytoplasm and large vesicular and hyperchromatic nuclei with variable nucleoli. These cells reveal their endothelial origin by displaying one or more intracytoplasmic vacuoles that represent intracellular vascular lumina. In addition, they can form small capillary-like lumina, which sometimes contain red blood cells (Makhlouf et al, 1999).
At the actively proliferating margin of the tumor, the neoplastic cells infiltrate the sinusoids, resulting in ductular transformation, atrophy, and eventual obliteration of the adjacent hepatic plates. The underlying lobular architecture is frequently preserved, and remnants of portal tracts can be identified. The tumor also invades the central veins, portal vein branches, and larger blood vessels. This intravascular growth appears as intraluminal papillary clusters or tuftlike projections (Bioulac-Sage et al, 2008). Infiltrating tumor cells are accompanied by a prominent stromal matrix that is initially fibrillar or hyaline, and an attendant mixed inflammatory infiltrate is often also present. Toward its center, the tumor ultimately becomes densely sclerotic, often with focal calcification, and the tumor cells can be difficult to discern (Weiss et al, 1986).
Angiosarcoma
Hepatic angiosarcoma is the most common primary sarcoma of the liver (Brady et al, 1977). Association with exposure to Thorotrast, vinyl chloride monomer, or inorganic arsenic compounds have been identified in 25% to 40% of angiosarcomas (Brady et al, 1977; Falk et al, 1981a; Kojiro et al, 1985; Sherman, 2009). Although these agents either have been discontinued or are under stricter controls, the prolonged latency period suggests that associated angiosarcomas will continue to be encountered for many years (Falk et al, 1981a). The peak incidence is in the sixth and seventh decades of life, and approximately 75% of the patients are men. Rare cases have been reported in children, most in association with infantile hemangioendothelioma (Strate et al, 1984).
The prognosis of patients with hepatic angiosarcoma is very poor. Most patients deteriorate rapidly and die within 6 months of diagnosis, usually from hepatic failure, extensive tumor burden, or tumor rupture. Distant metastases are noted in more than half the patients, most commonly to the lungs, regional lymph nodes, spleen, or bone marrow (Locker et al, 1979).
On gross examination, angiosarcomas typically appear as multiple gray or hemorrhagic masses scattered throughout both liver lobes (Fig. 78.48). The size varies greatly, between 0.1 and 5 cm, and tumors have a spongy consistency with irregular, ill-defined borders. Less often, the tumor is represented by a large solitary mass, typically more than 15 cm in diameter.
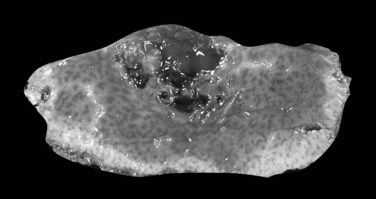
The histologic appearance is characterized by a proliferation of malignant endothelial cells with pale, ill-defined cytoplasm and enlarged pleomorphic and hyperchromatic nuclei (Fig. 78.49). The cells usually assume an elongated orientation, but they can also appear rounded or polygonal with conspicuous cytoplasm and prominent nucleoli. Mitoses, although in variable number, are often numerous and bizarre: in some cases the cells show evidence of phagocytic behavior (Bioulac-Sage et al, 2008; Ludwig & Hoffman, 1975).
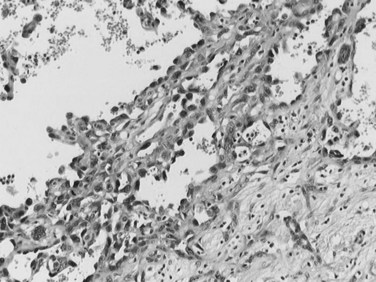
The tumor infiltrates preexisting vascular structures and eventually destroys the hepatic parenchyma. With early involvement, the malignant cells line the sinusoids and separate the hepatic plates. With progression, the hepatocytes atrophy and are ultimately replaced by fibrosis, and the sinusoids dilate, producing irregular pelioid-like vascular spaces of varying size. These spaces are lined by multilayered tumor cells, sometimes in a papillary arrangement, and contain blood and necrotic debris. Extramedullary hematopoiesis is often present (Kojiro et al, 1985). The tumor also invades central veins or portal vein branches, which can lead to luminal occlusion with subsequent hemorrhage, necrosis, and infarction.
In angiosarcomas associated with Thorotrast, vinyl chloride, and arsenic exposure, an initial precursor stage that precedes the development of overt tumor has been recognized, with hypertrophy and hyperplasia of endothelial cells together with reactive hepatocyte alterations (Tamburro et al 1984).
Undifferentiated (Embryonal) Sarcoma
Embryonal sarcoma of the liver is a highly malignant childhood neoplasm (Stocker & Ishak, 1978; see Chapter 82). It is the third most common hepatic malignancy in the pediatric population after hepatoblastoma and hepatocellular carcinoma. The peak incidence is between 6 and 10 years with an age range that extends from newborn infants to almost 30 years of age (Stocker & Ishak, 1983, 1978; Pachera et al, 2008). The prognosis of undifferentiated sarcoma is very poor. Death often occurs because of massive local tumor growth with direct extension into contiguous structures and even through the diaphragm.
On gross examination, the tumors are large, well-demarcated masses, usually 10 to 30 cm in diameter with a soft gelatinous consistency. Most of the tumors occupy the right lobe of the liver. The cut surface is variegated with areas of solid gray-white tumor interrupted by foci of cystic degeneration, hemorrhage, and necrosis (Stocker & Ishak, 1978).
Histologically, the tumors are composed of malignant spindle-shaped cells separated by myxoid stroma. The neoplastic cells are grouped into closely packed sheets or fascicles or scattered loosely in the stroma. They vary greatly in size and have pleomorphic, hyperchromatic nuclei; inconspicuous nucleoli; and poorly defined borders. Mitoses are often numerous and bizarre, and multinucleated tumor giant cells can be seen. A characteristic finding is the presence of periodic acid-Schiff–positive, diastase-resistant eosinophilic globules within tumor cells and in the extracellular stroma (Ma et al, 2008). The myxoid stroma dominates the less cellular portions of the tumor and harbors numerous thin-walled veins and often contains foci of extramedullary hematopoiesis. Areas of hemorrhage and necrosis are abundant, and bands of dense, hyalinized collagen are occasionally noted. The periphery of the tumor typically entraps bile ducts that are cystically dilated and lined by a cuboidal epithelium that often displays hyperplastic or atypical features. The tumor lacks histologic evidence of cellular differentiation, and its histogenesis remains uncertain (Lack et al, 1991). Embryonal sarcoma represents a primitive mesenchymal neoplasm capable of divergent differentiation (Choi et al, 2010).
Other Sarcomas
Most primary hepatic sarcomas occur in adults between 40 and 60 years of age who are seen with signs and symptoms indicative of a hepatic mass. These primary hepatic sarcomas should be distinguished from metastatic involvement by sarcomas of other organs and from sarcomatoid hepatocellular carcinoma (Kakizoe et al, 1987). The overall prognosis is poor, and most patients die within a year of diagnosis.
Hepatic sarcomas histologically resemble their soft-tissue counterparts. Among the types reported are fibrosarcoma, follicular dendritic cell sarcoma, desmoplastic nested single-cell tumor, malignant fibrous histiocytoma, hemangiopericytoma, osteosarcoma, malignant schwannoma, and dedifferentiated liposarcoma (Choi et al, 2010; Hill et al, 2005; Li et al, 2008; Kim et al, 1987; Lederman et al, 1987). Leiomyosarcomas are described arising within the hepatic parenchyma, from the ligamentum teres, and from the hepatic vein, where they have been associated with Budd-Chiari syndrome (Shamseddine et al, 2010; Kuma et al, 2008; Fong & Ruebner, 1974; Hilliard et al, 2005).
Kaposi sarcoma is seen primarily in the setting of the acquired immunodeficiency syndrome (AIDS). Histologically, the tumor is characterized by multifocal nodules composed of proliferated spindle cells with slit-like spaces that contain extravasated erythrocytes. These lesions are predominantly centered on portal tracts, but they also spread into and destroy the parenchyma (Bioulac-Sage et al, 2008; Friedman, 1988).
Hepatobiliary rhabdomyosarcoma is a rare neoplasm that usually arises from the common bile duct (Ali et al, 2009). Most patients are young children, with exceptional cases reported in adults (Stocker, 2001; Lack et al, 1981). Histologically the tumor resembles botryoid-type embryonal rhabdomyosarcomas that occur elsewhere in the body. Soft polypoid masses protrude into the ductal lumen and are covered by intact, ulcerated, or inflamed biliary epithelium. The tumor is composed of small hyperchromatic cells with variable eosinophilic cytoplasm and rare cross-striations. Beneath the biliary epithelium, the cells are densely packed and form a so-called cambium layer, whereas deeper in the tumor, they are separated by a loose myxoid stroma. The diagnosis is secured by immunohistochemical demonstration of desmin, muscle-specific actin, or myoglobin expression or with the ultrastructural finding of the characteristic myofilaments.
Miscellaneous Tumors
Inflammatory pseudotumors of the liver are nonneoplastic masses of unknown etiology (see Chapter 48). They occur at any age and clinically manifest with abdominal pain together with various systemic manifestations that include low-grade fever, weight loss, leukocytosis, anemia, and inflammatory syndrome (Tang et al, 2010). Although the cause is unknown, an as yet unidentified infectious or immunoglobulin G4 autoimmune-related process have been suggested (Horiuchi et al, 1990; Yamamoto et al, 2009).
Neuroendocrine Tumor
Neuroendocrine tumors in the liver are almost always metastatic, and a primary lesion in the gastrointestinal tract or pancreas is usually identified (see Chapter 81B). In rare cases, however, primary hepatic endocrine tumors have been described in which no extrahepatic site of involvement can be identified (Sioutos et al, 1991; Miura & Shirasawa, 1988). Because of the normal presence of endocrine cells in biliary epithelium, an origin from intrahepatic bile ducts has often been suggested. Histologically, the tumor is composed of small, uniform tumor cells with regular round nuclei and moderate granular cytoplasm arranged in nests, cords, and pseudoglandular patterns. Dense core neurosecretory granules are found on immunostaining.
Other Tumors
Several examples of malignant rhabdoid tumors of the liver have been recognized. These tumors are highly aggressive neoplasms with a distinctive histologic appearance (Hunt & Anderson, 1990). Rare cases of primary hepatic germ-cell tumors of several types—including teratomas, yolk sac tumors, and choriocarcinoma—have been described. The combination of yolk sac tumor and hepatoblastoma has also been described (Cross & Variend, 1992); these tumors exhibit malignant behavior, although occasional long-term survival has been obtained with cancer chemotherapy.
Squamous carcinoma of the liver is uncommon, with most cases developing within simple hepatic cysts, in various forms of fibropolycystic liver disease, or in teratomas (Gresham & Rue, 1985). Adrenal rest tumor and pancreatic heterotopia have also been described and might be misleading (Wolf et al, 1990; Contreras et al, 1985).
Hematopoietic Neoplasms
Primary Hepatic Lymphoma
The liver may represent the sole site of involvement by malignant lymphoma, but these primary hepatic lymphomas are uncommon, although hepatitis C infection may be a risk factor (Santos et al, 2003).
On gross examination, most cases are characterized by a single large mass or, less often, by multiple discrete nodules (Anthony et al, 1990). Rarely, the liver is diffusely infiltrated without a well-defined mass lesion. The tumors are predominantly classified as non-Hodgkin lymphomas of diffuse large cell type, with occasional examples of small cleaved or uncleaved varieties such as mucosa-associated lymphoid tissue (MALT) tumors (Deshald et al, 2010; Doi et al, 2008). Most of the tumors are phenotypically of B-cell lineage, although occasional T-cell lymphomas have also been described (Noronha et al, 2005; Andreola et al, 1988). The neoplastic lymphocytes expand the portal tracts and invade into adjacent parenchyma from large infiltrative nodules. The lobules are otherwise little affected, although occasional cases, particularly with T-cell lymphomas, demonstrate sinusoidal infiltration (Farcet et al, 1990).
Secondary Involvement
All varieties of lymphomas, leukemias, and hematopoietic proliferations can secondarily affect the liver, usually as a manifestation of disseminated disease and often in conjunction with splenic involvement. As a general rule, the low-grade non-Hodgkin lymphomas tend to produce multiple portal-based infiltrates, whereas the intermediate- and high-grade types give rise to large, irregular tumor masses. Sinusoidal involvement is uncommon except with peripheral T-cell lymphomas (Farcet et al, 1990; Gaulard et al, 1986).
Hepatic involvement in Hodgkin disease can manifest as large, irregular masses or as small, diffusely distributed nodules. The characteristic infiltrate, which predominantly affects portal tracts, consists of a polymorphous collection of Reed-Sternberg cells or variants together with small lymphocytes, large mononuclear cells, plasma cells, eosinophils, and neutrophils in varying numbers (Dich et al, 1989).
Leukemias of both acute and chronic types tend to permeate the sinusoids with malignant cells, with involvement of the portal tracts additionally noted with the lymphoid varieties. Hepatic involvement is especially common and conspicuous in hairy cell leukemia (Zafrani et al, 1987).
Langerhans cell histiocytosis involves the liver in about 40% of cases (Iwai et al, 1988). The most important clinical manifestations include jaundice and portal hypertension, resulting in part from infiltration of large bile ducts to produce a sclerosing cholangitis-like picture.
Metastasis
Metastatic cancers are by far the most common form of hepatic malignancy, accounting for more than 95% of the total (see Chapter 81A, Chapter 81B, Chapter 81C ). In autopsy series, liver metastases are noted in about 40% of patients with malignant tumors, and the sites of origin include virtually every organ in the body (Schulz & Hort, 1981). The more frequent primary sites include carcinomas of the colon, pancreas, lung, and breast in adults and neuroblastoma, Wilms tumor, and rhabdomyosarcoma in pediatric patients (Strohmeyer & Schultz, 1986). Although the origin of the tumor is often known when metastases are biopsied, carcinomas of the pancreas, stomach, and lung are particularly notorious for being occult primary neoplasms.
Grossly, metastatic tumors in the liver are almost always multiple, but they may appear as solitary nodules, confluent masses, or as small, diffusely infiltrative lesions that can mimic cirrhosis (Fig. 78.50; Borja et al, 1975). The size range varies considerably. Typically these tumors are gray to yellow nodules, sometimes complicated by central necrosis, umbilication, or cystic change, and they can occur anywhere within the parenchymal mass (Strohmeyer & Schultz, 1986). Some metastases may display prominent fibrous changes.

Akwari OE, et al. Hepatobiliary cystadenoma with mesenchymal stroma. Ann Surg. 1990;211:18-27.
Ali S, et al. Biliary rhabdomyoscarcoma mimicking choledochal cyst. J Gastrointestin Liver Dis. 2009;18:95-97.
Allaire GS, et al. Bile duct adenoma: a study of 152 cases. Am J Surg Pathol. 1988;12:708-715.
Allen RA, Lisa JR. Combined liver cell and bile duct carcinoma. Am J Pathol. 1949;25:647-655.
Andreola S, et al. Primary lymphoma of the liver showing immunohistochemical evidence of T-cell origin: successful management by right trisegmentectomy. Dig Dis Sci. 1988;33:1632-1636.
Anthony PP. Primary carcinoma of the liver: a study of 282 cases in Ugandan Africans. J Pathol. 1973;110:37-48.
Anthony PP, et al. Primary lymphoma of the liver: clinical and pathological features of 10 patients. J Clin Pathol. 1990;43:1007-1013.
Aoki K, et al. Combined hepatocellular carcinoma and cholangiocarcinoma: clinical features and computed tomographic findings. Hepatology. 1993;18:1090-1095.
Arii S, et al. Results of surgical and nonsurgical treatment for small-sized hepatocellular carcinomas: a retrospective and nationwide survey in Japan. The Liver Cancer Study Group of Japan. Hepatology. 2000;32:1224-1229.
Bacq Y, et al. Familial liver adenomatosis associated with hepatocyte nuclear factor 1 alpha inactivation. Gastroenterology. 2003;125:1470-1475.
Berman JJ, McNeill RE. Cirrhosis with atypia: a potential pitfall in the interpretation of liver aspirates. Acta Cytol. 1988;32:11-14.
Bioulac-Sage P, et al. Clinical, morphologic, and molecular features defining so-called telangiectatic focal nodular hyperplasias of the liver. Gastroenterology. 2005;128:1211-1218.
Bioulac-Sage P, et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology. 2007;46:740-748.
Bioulac-Sage P, et al. Pathological diagnosis of liver cell adenoma and focal nodular hyperplasia: Bordeaux update. J Hepatol. 2007;46:521-527.
Bioulac-Sage P, et al. Benign and malignant vascular tumors of the liver in adults. Semin Liver Dis. 2008;28:302-314.
Bioulac-Sage P, et al. Over-expression of glutamine synthetase in focal nodular hyperplasia: a novel easy diagnostic tool in surgical pathology. Liver Int. 2009;29:459-465.
Bioulac-Sage P, et al. Hepatocellular adenoma management and phenotypic classification: the Bordeaux experience. Hepatology. 2009;50:481-489.
Bluteau O, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002;32:312-315.
Boberg KM, et al. Cholangiocarcinoma in primary sclerosing cholangitis: risk factors and clinical presentation. Scand J Gastroenterol. 2002;37:1205-1211.
Bolondi L, et al. Characterization of small nodules in cirrhosis by assessment of vascularity: the problem of hypovascular hepatocellular carcinoma. Hepatology. 2005;42:27-34.
Bonetti F, et al. The perivascular epithelioid cell and related lesions. Adv Anat Pathol. 1997;4:343-358.
Borja ER, et al. Metastatic carcinomatosis of the liver mimicking cirrhosis: case report and review of the literature. Cancer. 1975;35:445-449.
Borzio M, et al. Liver cell dysplasia is a major risk factor for hepatocellular carcinoma in cirrhosis: a prospective study. Gastroenterology. 1995;108:812-817.
Borzio M, et al. Impact of large regenerative, low-grade and high-grade dysplastic nodules in hepatocellular carcinoma development. J Hepatol. 2003;39:208-214.
Bosch FX, et al. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5-S16.
Bosman FT, et al. WHO Classification of Tumours of the Digestive System, 4th ed. 2010.
Boyault S, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42-52.
Brady J, et al. Angiosarcoma of the liver: an epidemiologic survey. J Natl Cancer Inst. 1977;59:1383-1385.
Buchanan TF, Huvos AG. Clear-cell carcinoma of the liver: a clinicopathologic study of 13 patients. Am J Clin Pathol. 1974;61:529-539.
Buetow PC, et al. Biliary cystadenoma and cystadenocarcinoma: clinical imaging pathologic correlations with emphasis on the importance of ovarian stroma. Radiology. 1995;196:805-810.
Bureau C, et al. Liver nodules ressembling focal nodular hyperplasia after portal vein thrombosis. J Hepatol. 2004;41:499-500.
Burns CD, et al. Cholangiocarcinoma in association with multiple biliary microhamartomas. Arch Pathol Lab Med. 1990;114:1287-1289.
Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208-1236.
Bruix J, et al. Clinical management of hepatocellular carcinoma: conclusions of the Barcelona-2000 EASL Conference. European Association for the Study of the Liver. J Hepatol. 2001;35:421-430.
Bruix J, et al. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215-219.
Cazals-Hatem D, et al. Clinical and molecular analysis of combined hepatocellular-cholangiocarcinomas. J Hepatol. 2004;41:292-298.
Chapman RW. Risk factors for biliary tract carcinogenesis. Ann Oncol. 1999;10(Suppl 4):308-311.
Choi BS, et al. Follicular dendritic cell sarcoma: a case report and review of the literature. Cancer Res Treat. 2010;42:121-124.
Chuma M, et al. Expression profiling in multistage hepatocarcinogenesis: identification of HSP70 as a molecular marker of early hepatocellular carcinoma. Hepatology. 2003;37:198-207.
Chuong JJ, et al. The histopathologic and clinical indicators of prognosis in hepatoma. J Clin Gastroenterol. 1982;4:547-552.
Colombo M, et al. Hepatocellular carcinoma in Italian patients with cirrhosis. N Engl J Med. 1991;325:675-680.
Conran RM, et al. Hepatoblastoma: the prognostic significance of histologic type. Pediatr Pathol. 1992;12:167-183.
Contreras P, et al. Adrenal rest tumor of the liver causing Cushing’s syndrome: treatment with ketoconazole preceding an apparent surgical cure. J Clin Endocrinol Metab. 1985;60:21-28.
Coombes GB, et al. An androgen-associated hepatic adenoma in a trans-sexual. Br J Surg. 1978;65:869-870.
Craig JR, et al. Fibrolamellar carcinoma of the liver: a tumor of adolescents and young adults with distinctive clinico-pathologic features. Cancer. 1980;46:372-379.
Cross SS, Variend S. Combined hepatoblastoma and yolk sac tumor of the liver. Cancer. 1992;69:1323-1326.
Curado MPE, Shin A. Cancer Incidence in Five Continents, vol 9. Lyon: IARC Scientific. 2007.
Dachman AH, et al. Infantile hemangioendothelioma of the liver: a radiologic-pathologic-clinical correlation. AJR Am J Roentgenol. 1983;140:1091-1096.
Dalle I, et al. Malignant angiomyolipoma of the liver: a hitherto unreported variant. Histopathology. 2000;36:443-450.
Daniels JA, et al. Biliary cystadenomas: hormone receptor expression and clinical management. Dig Dis Sci. 2006;51:623-628.
Dean PJ, et al. Malignant epithelioid hemangioendothelioma of the liver in young women: relationship to oral contraceptive use. Am J Surg Pathol. 1985;9:695-704.
Dehner LP. Hepatic tumors in the pediatric age group: a distinctive clinicopathologic spectrum. Perspect Pediatr Pathol. 1978;4:217-268.
Dehner LP, Ishak KG. Vascular tumors of the liver in infants and children: a study of 30 cases and review of the literature. Arch Pathol. 1971;92:101-111.
Dehner LP, et al. Infantile mesenchymal hamartoma of the liver: histologic and ultrastructural observations. Arch Pathol. 1975;99:379-382.
de La Coste A, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847-8851.
Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation.”. Hepatology. 1992;16:1069-1083.
Deugnier YM, et al. Preneoplastic significance of hepatic iron-free foci in genetic hemochromatosis: a study of 185 patients. Hepatology. 1993;18:1363-1369.
Devaney K, et al. Hepatobiliary cystadenoma and cystadenocarcinoma: a light microscopic and immunohistochemical study of 70 patients. Am J Surg Pathol. 1994;18:1078-1091.
Dich NH, et al. Hepatic involvement in Hodgkin’s disease: clues to histologic diagnosis. Cancer. 1989;64:2121-2126.
Dietze O, et al. Malignant epithelioid haemangioendothelioma of the liver: a clinicopathological and histochemical study of 12 cases. Histopathology. 1989;15:225-237.
Di Tommaso L, et al. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology. 2007;45:725-734.
Doi H, et al. Primary hepatic marginal zone B cell lymphoma of mucosa-associated lymphoid tissue type: case report and review of the literature. Int J Hematol. 2008;88:418-423.
Dokmak S, et al. A single-center surgical experience of 122 patients with single and multiple hepatocellular adenomas. Gastroenterology. 2009;137:1698-1705.
Ebata T, et al. The concept of perihilar cholangiocarcinoma is valid. Br J Surg. 2009;96:926-934.
Edmondson HA, Steiner PE. Primary carcinoma of the liver: a study of 100 cases among 48,900 necropsies. Cancer. 1954;7:462-503.
El-Assal ON, et al. Clinical significance of microvessel density and vascular endothelial growth factor expression in hepatocellular carcinoma and surrounding liver: possible involvement of vascular endothelial growth factor in the angiogenesis of cirrhotic liver. Hepatology. 1998;27:1554-1562.
El-Serag HB, Davila JA. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatology. 2004;39:798-803.
El-Serag HB, et al. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752-1763.
Fabre A, et al. Histologic scoring of liver biopsy in focal nodular hyperplasia with atypical presentation. Hepatology. 2002;35:414-420.
Falk H, et al. Arsenic-related hepatic angiosarcoma. Am J Ind Med. 1981;2:43-50.
Falk H, et al. Epidemiology of hepatic angiosarcoma in the United States: 1964-1974. Environ Health Perspect. 1981;41:107-113.
Farcet JP, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75:2213-2219.
Farges O, et al. Changing trends in malignant transformation of hepatocellular adenoma. Gut. 2011;60:85-89.
Ferrell L, et al. Incidence and diagnostic features of macroregenerative nodules vs. small hepatocellular carcinoma in cirrhotic livers. Hepatology. 1992;16:1372-1381.
Fléjou JF, et al. Liver adenomatosis: an entity distinct from liver adenoma? Gastroenterology. 1985;89:1132-1138.
Flemming P, et al. Common and epithelioid variants of hepatic angiomyolipoma exhibit clonal growth and share a distinctive immunophenotype. Hepatology. 2000;32:213-217.
Fong JA, Ruebner BH. Primary leiomyosarcoma of the liver. Hum Pathol. 1974;5:115-119.
Forner A, et al. Diagnosis of hepatic nodules 20 mm or smaller in cirrhosis: prospective validation of the noninvasive diagnostic criteria for hepatocellular carcinoma. Hepatology. 2008;47:97-9104.
Fried RH, et al. Benign cartilaginous tumor (chondroma) of the liver. Gastroenterology. 1992;103:678-680.
Friedman SL. Gastrointestinal and hepatobiliary neoplasms in AIDS. Gastroenterol Clin North Am. 1988;17:465-486.
Fujii H, et al. Genetic classification of combined hepatocellular cholangiocarcinoma. Hum Pathol. 2000;31:1011-1017.
Furuya K, et al. Macroregenerative nodule of the liver: a clinicopathologic study of 345 autopsy cases of chronic liver disease. Cancer. 1988;61:99-105.
Gandolfi L, et al. Natural history of hepatic haemangiomas: clinical and ultrasound study. Gut. 1991;32:677-680.
Ganne-Carrie N, et al. Predictive score for the development of hepatocellular carcinoma and additional value of liver large cell dysplasia in Western patients with cirrhosis. Hepatology. 1996;23:1112-1118.
Gaulard P, et al. Peripheral T-cell lymphoma presenting as predominant liver disease: a report of three cases. Hepatology. 1986;6:864-868.
Gold JH, et al. Benign tumors of the liver: pathologic examination of 45 cases. Am J Clin Pathol. 1978;70:6-17.
Goodman ZD, Ishak KG. Angiomyolipomas of the liver. Am J Surg Pathol. 1984;8:745-750.
Goodman ZD, et al. Combined hepatocellular-cholangiocarcinoma: a histologic and immunohistochemical study. Cancer. 1985;55:124-135.
Gouma DJ, et al. Intrahepatic biliary papillomatosis. Br J Surg. 1984;71:72-74.
Govindarajan S, Peters RL. The bile duct adenoma: a lesion distinct from Meyenburg complex. Arch Pathol Lab Med. 1984;108:922-924.
Gramlich TL, et al. Mesenchymal hamartoma of the liver: report of a case in a 28-year-old. Hum Pathol. 1988;19:991-992.
Grando-Lemaire V, et al. Hepatocellular carcinoma without cirrhosis in the West: epidemiological factors and histopathology of the non-tumorous liver. Groupe d’Etude et de Traitement du Carcinome Hepatocellulaire. J Hepatol. 1999;31:508-513.
Grange JD, et al. Liver adenoma and focal nodular hyperplasia in a man with high endogenous sex steroids. Gastroenterology. 1987;93:1409-1413.
Grantzdorffer I, et al. Angiotensin I–converting enzyme (CD143) is down-regulated in focal nodular hyperplasia of the liver. Am J Surg Pathol. 2004;28:84-88.
Gresham GA, Rue LW3rd. Squamous cell carcinoma of the liver. Hum Pathol. 1985;16:413-416.
Grigioni WF, et al. Primary liver neoplasms: evaluation of proliferative index using MoAb Ki67. J Pathol. 1989;158:23-29.
Guedj N, et al. Comparative protein expression profiles of hilar and peripheral hepatic cholangiocarcinomas. J Hepatol. 2009;51:93-101.
Guglielmi A, et al. Intrahepatic cholangiocarcinoma: prognostic factors after surgical resection. World J Surg. 2009;33:1247-1254.
Haas JE, et al. Histopathology and prognosis in childhood hepatoblastoma and hepatocarcinoma. Cancer. 1989;64:1082-1095.
Haas JE, et al. Small cell undifferentiated histology in hepatoblastoma may be unfavorable. Cancer. 2001;92:3130-3134.
Handra-Luca A, et al. Multiple mixed adenoma–focal nodular hyperplasia of the liver associated with spontaneous intrahepatic porto-systemic shunt: a new type of vascular malformation associated with the multiple focal nodular hyperplasia syndrome? Histopathology. 2006;48:309-311.
Hayashi M, et al. Progression to hypervascular hepatocellular carcinoma: correlation with intranodular blood supply evaluated with CT during intraarterial injection of contrast material. Radiology. 2002;225:143-149.
Hill DA, et al. Desmoplastic nested spindle cell tumor of liver: report of four cases of a proposed new entity. Am J Surg Pathol. 2005;29:1-9.
Hilliard NJ, et al. Leiomyosarcoma of the inferior vena cava: three case reports and review of the literature. Ann Diagn Pathol. 2005;9:259-266.
Hirota N, et al. Resistance to iron accumulation and presence of hepatitis B surface antigen in preneoplastic and neoplastic lesions in human hemochromatotic livers. Hepatogastroenterology. 1982;29:49-51.
Honda N, et al. Bile duct carcinoma associated with multiple von Meyenburg complexes in the liver. Hum Pathol. 1986;17:1287-1290.
Horie Y, et al. Pedunculated hepatocellular carcinoma: report of three cases and review of literature. Cancer. 1983;51:746-751.
Horiuchi R, et al. Inflammatory pseudotumor of the liver: clinicopathologic study and review of the literature. Cancer. 1990;65:1583-1590.
Hornick JL, Fletcher CD. PEComa: what do we know so far? Histopathology. 2006;48:75-82.
Hoshida Y, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995-2004.
Hunt SJ, Anderson WD. Malignant rhabdoid tumor of the liver: a distinct clinicopathologic entity. Am J Clin Pathol. 1990;94:645-648.
Hytiroglou P, et al. Hepatic precancerous lesions and small hepatocellular carcinoma. Gastroenterol Clin North Am. 2007;36:867-887.
International Consensus Group for Hepatocellular Neoplasia. Pathologic diagnosis of early hepatocellular carcinoma: a report of the International Consensus Group for Hepatocellular Neoplasia. Hepatology. 2009;49:658-664.
International Working Party. Terminology of nodular hepatocellular lesions. Hepatology. 1995;22:983-993.
Ishak KG, Glunz PR. Hepatoblastoma and hepatocarcinoma in infancy and childhood: report of 47 cases. Cancer. 1967;20:396-422.
Ishak KG, et al. Biliary cystadenoma and cystadenocarcinoma: report of 14 cases and review of the literature. Cancer. 1977;39:322-338.
Ishak KG, et al. Epithelioid hemangioendothelioma of the liver: a clinicopathologic and follow-up study of 32 cases. Hum Pathol. 1984;15:839-852.
Iwai M, et al. Cholestatic liver disease in a 20-yr-old woman with histiocytosis X. Am J Gastroenterol. 1988;83:164-168.
Jarnagin WR, et al. Combined hepatocellular and cholangiocarcinoma: demographic, clinical, and prognostic factors. Cancer. 2002;94:2040-2046.
Jiang BG, et al. Retrospective analysis of histopathologic prognostic factors after hepatectomy for intrahepatic cholangiocarcinoma. Cancer. 2009;15:257-261.
Johnson PJ, Williams R. Cirrhosis and the aetiology of hepatocellular carcinoma. J Hepatol. 1987;4:140-147.
Kakizoe S, et al. Hepatocellular carcinoma with sarcomatous change: clinicopathologic and immunohistochemical studies of 14 autopsy cases. Cancer. 1987;59:310-316.
Karhunen PJ. Benign hepatic tumours and tumour-like conditions in men. J Clin Pathol. 1986;39:183-188.
Kenmochi K, et al. Relationship of histologic grade of hepatocellular carcinoma (HCC) to tumor size, and demonstration of tumor cells of multiple different grades in single small HCC. Liver. 1987;7:18-26.
Khan SA, et al. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J Hepatol. 2002;37:806-813.
Kim H, Damjanov I. Localized fibrous mesothelioma of the liver: report of a giant tumor studied by light and electron microscopy. Cancer. 1983;52:1662-1665.
Kim YI, et al. Dedifferentiated liposarcoma of the liver. Cancer. 1987;60:2785-2790.
Kim YI, et al. Intraductal variant of peripheral cholangiocarcinoma of the liver with Clonorchis sinensis infection. Cancer. 1989;63:1562-1566.
Kipp BR, et al. A comparison of routine cytology and fluorescence in situ hybridization for the detection of malignant bile duct strictures. Am J Gastroenterol. 2004;99:1675-1681.
Kishi K, et al. Hepatocellular carcinoma: a clinical and pathologic analysis of 57 hepatectomy cases. Cancer. 1983;51:542-548.
Klatskin G. Adenocarcinomas of the hepatic duct at its bifurcation within the porta hepatis: an unusual tumor with distinctive clinical and pathological features. Am J Med. 1965;38:241-256.
Kobayashi M, et al. Incidence of primary cholangiocellular carcinoma of the liver in Japanese patients with hepatitis C virus–related cirrhosis. Cancer. 2000;88:2471-2477.
Kobayashi M, et al. Dysplastic nodules frequently develop into hepatocellular carcinoma in patients with chronic viral hepatitis and cirrhosis. Cancer. 2006;106:636-647.
Kojiro M. Focus on dysplastic nodules and early hepatocellular carcinoma: an Eastern point of view. Liver Transpl. 2004;10:3-8.
Kojiro M, Roskams T. Early hepatocellular carcinoma and dysplastic nodules. Semin Liver Dis. 2005;25:133-142.
Kojiro M, et al. Thorium dioxide–related angiosarcoma of the liver: pathomorphologic study of 29 autopsy cases. Arch Pathol Lab Med. 1985;109:853-857.
Kojiro M, et al. Hepatocellular carcinoma with sarcomatous change: a special reference to the relationship with anticancer therapy. Cancer Chemother Pharmacol. 1989;23:4-8.
Komori K, et al. Mesothelial cyst of the liver in a neonate. Pediatr Surg Int. 2008;24:463-465.
Komuta M, et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology. 2008;47:1544-1556.
Kondo F. Benign nodular hepatocellular lesions caused by abnormal hepatic circulation: etiological analysis and introduction of a new concept. J Gastroenterol Hepatol. 2001;16:1319-1328.
Kondo F, et al. Histological features and clinical course of large regenerative nodules: evaluation of their precancerous potentiality. Hepatology. 1990;12:592-598.
Kondo F, et al. Nodular lesions associated with abnormal liver circulation. Intervirology. 2004;47:277-287.
Kottke-Marchant K, et al. Localized fibrous tumor (localized fibrous mesothelioma) of the liver. Cancer. 1989;64:1096-1102.
Kuma S, et al. Leiomyosarcoma originating from the superior mesenteric vein: a case report and review of the literature. Ann Vasc Surg. 2008;22:453-455.
Kuwano H, et al. Hepatocellular carcinoma with osteoclast-like giant cells. Cancer. 1984;54:837-842.
Labrune P, et al. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr. 1997;24:276-279.
Lack EE, et al. Botryoid rhabdomyosarcoma of the biliary tract. Am J Surg Pathol. 1981;5:643-652.
Lack EE, et al. Hepatoblastoma: a clinical and pathologic study of 54 cases. Am J Surg Pathol. 1982;6:693-705.
Lack EE, et al. Undifferentiated (embryonal) sarcoma of the liver: clinical and pathologic study of 16 cases with emphasis on immunohistochemical features. Am J Surg Pathol. 1991;15:1-16.
Ladeiro Y, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955-1963.
Lai CL, et al. Histologic prognostic indicators in hepatocellular carcinoma. Cancer. 1979;44:1677-1683.
Lam CM, et al. Different presentation of hepatitis B–related hepatocellular carcinoma in a cohort of 1863 young and old patients: implications for screening. Aliment Pharmacol Ther. 2004;19:771-777.
Lederman SM, et al. Hepatic neurofibromatosis, malignant schwannoma, and angiosarcoma in von Recklinghausen’s disease. Gastroenterology. 1987;92:234-239.
Lee JS, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667-676.
Lee RG, et al. Large cell change (liver cell dysplasia) and hepatocellular carcinoma in cirrhosis: matched case-control study, pathological analysis, and pathogenetic hypothesis. Hepatology. 1997;26:1415-1422.
Lee SS, et al. Clinicopathologic review of 58 patients with biliary papillomatosis. Cancer. 2004;100:783-793.
Lefkowitch JH, et al. Copper and copper-binding protein in fibrolamellar liver cell carcinoma. Cancer. 1983;51:97-9100.
Lepreux S, et al. Expression of fibrillin-1 in focal nodular hyperplasia of the liver: a role in microcirculation adaptability. Comp Hepatol. 2004;3(Suppl 1):S57.
Levy MJ, et al. Prospective evaluation of advanced molecular markers and imaging techniques in patients with indeterminate bile duct strictures. Am J Gastroenterol. 2008;103:1263-1273.
Lewin M, et al. Liver adenomatosis: classification of MR imaging features and comparison with pathologic findings. Radiology. 2006;241:433-440.
Li YR, et al. Primary hepatic malignant fibrous histiocytoma: clinicopathologic characteristics and prognostic value of ezrin expression. Am J Surg Pathol. 2008;32:1144-1158.
Libbrecht L, et al. Preneoplastic lesions in human hepatocarcinogenesis. Liver Int. 2005;25:16-27.
Lim MK, et al. Clonorchis sinensis infection and increasing risk of cholangiocarcinoma in the Republic of Korea. Am J Trop Med Hyg. 2006;75:93-96.
Lipshutz GS, et al. Thorotrast-induced liver neoplasia: a collective review. J Am Coll Surg. 2002;195:713-718.
Llovet JM, et al. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis. 1999;19:329-338.
Llovet JM, et al. A molecular signature to discriminate dysplastic nodules from early hepatocellular carcinoma in HCV cirrhosis. Gastroenterology. 2006;131:1758-1767.
Locker GY, et al. The clinical features of hepatic angiosarcoma: a report of four cases and a review of the English literature. Medicine (Baltimore). 1979;58:48-64.
Ludwig J, Hoffman HN. Hemangiosarcoma of the liver: spectrum of morphologic changes and clinical findings. Mayo Clin Proc. 1975;50:255-263.
Ma L, et al. Undifferentiated embryonal sarcoma of liver in an old female: case report and review of the literature. World J Gastroenterol. 2008;14:7267-7270.
Maeda T, et al. Combined hepatocellular and cholangiocarcinoma: proposed criteria according to cytokeratin expression and analysis of clinicopathologic features. Hum Pathol. 1995;26:956-964.
Maetani Y, et al. Benign hepatic nodules in Budd-Chiari syndrome: radiologic-pathologic correlation with emphasis on the central scar. AJR Am J Roentgenol. 2002;178:869-875.
Maggioni M, et al. Molecular changes in hepatocellular dysplastic nodules on microdissected liver biopsies. Hepatology. 2000;32:942-946.
Makhlouf HR, et al. Epithelioid hemangioendothelioma of the liver: a clinicopathologic study of 137 cases. Cancer. 1999;85:562-582.
Makhlouf HR, et al. Diagnosis of focal nodular hyperplasia of the liver by needle biopsy. Hum Pathol. 2005;36:1210-1216.
Malhi H, Gores GJ. Cholangiocarcinoma: modern advances in understanding a deadly old disease. J Hepatol. 2006;45:856-867.
Marrero JA, et al. NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology. 2002;36:1349-1354.
Marrero JA, et al. Prognosis of hepatocellular carcinoma: comparison of 7 staging systems in an American cohort. Hepatology. 2005;41:707-716.
McGlynn KA, London WT. Epidemiology and natural history of hepatocellular carcinoma. Best Pract Res Clin Gastroenterol. 2005;19:3-23.
Mehrabi A, et al. Primary malignant hepatic epithelioid hemangioendothelioma: a comprehensive review of the literature with emphasis on the surgical therapy. Cancer. 2006;107:2108-2121.
Mercadier M, et al. Papillomatosis of the intrahepatic bile ducts. World J Surg. 1984;8:30-35.
Moore S, et al. Adenosquamous carcinoma of the liver arising in biliary cystadenocarcinoma: clinical, radiologic, and pathologic features with review of the literature. J Clin Gastroenterol. 1984;6:267-275.
Mion F, et al. Adult cirrhotic liver explants: precancerous lesions and undetected small hepatocellular carcinomas. Gastroenterology. 1996;111:1587-1592.
Miura K, Shirasawa H. Primary carcinoid tumor of the liver. Am J Clin Pathol. 1988;89:561-564.
Nakanuma Y, et al. Incidental solitary hepatocellular carcinomas smaller than 1 cm in size found at autopsy: a morphologic study. Hepatology. 1986;6:631-635.
Nakanuma Y, et al. Pathology of peripheral intrahepatic cholangiocarcinoma with reference to tumorigenesis. Hepatol Res. 2008;38:325-334.
Nakashima T, Kojiro M. Pathologic characteristics of hepatocellular carcinoma. Semin Liver Dis. 1986;6:259-266.
Nathan H, et al. Predictors of survival after resection of early hepatocellular carcinoma. Ann Surg. 2009;249:799-805.
Nerlich AG, et al. Excessive collagen formation in fibrolamellar carcinoma of the liver: a morphological and biochemical study. Mod Pathol. 1992;5:580-585.
Neumann RD, et al. Adenocarcinoma in biliary papillomatosis. Gastroenterology. 1976;70:779-782.
Ng IO, et al. Combined hepatocellular cholangiocarcinoma: a clinicopathological study. J Gastroenterol Hepatol. 1998;13:34-40.
Nguyen BN, et al. Focal nodular hyperplasia of the liver: a comprehensive pathologic study of 305 lesions and recognition of new histologic forms. Am J Surg Pathol. 1999;23:1441-1454.
Nime F, et al. The histology of liver tumors in oral contraceptive users observed during a national survey by the American College of Surgeons Commission on Cancer. Cancer. 1979;44:1481-1489.
Nishizaki T, et al. Myelolipoma of the liver: a case report. Cancer. 1989;63:930-934.
Nonomura A, et al. Angiomyolipoma of the liver: a collective review. J Gastroenterol. 1994;29:95-105.
Nonomura A, et al. Multiple angiomyolipoma of the liver. J Clin Gastroenterol. 1995;20:248-251.
Nordenstedt H, et al. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl 3):S206-S214.
Noronha V, et al. Primary non-Hodgkin’s lymphoma of the liver. Crit Rev Oncol Hematol. 2005;53:199-207.
Oh BK, et al. Telomere shortening and telomerase reactivation in dysplastic nodules of human hepatocarcinogenesis. J Hepatol. 2003;39:786-792.
Ohnishi K, et al. Prognosis of hepatocellular carcinoma smaller than 5 cm in relation to treatment: study of 100 patients. Hepatology. 1987;7:1285-1290.
Okuda K, et al. Clinicopathologic features of encapsulated hepatocellular carcinoma: a study of 26 cases. Cancer. 1977;40:1240-1245.
Okuda K, et al. Hepatocellular carcinoma arising in noncirrhotic and highly cirrhotic livers: a comparative study of histopathology and frequency of hepatitis B markers. Cancer. 1982;49:450-455.
Okuda K, et al. Gross anatomic features of hepatocellular carcinoma from three disparate geographic areas: proposal of new classification. Cancer. 1984;54:2165-2173.
Omata M, et al. Sclerosing hepatic carcinoma: relationship to hypercalcemia. Liver. 1981;1:33-49.
Pachera S, et al. Undifferentiated embryonal sarcoma of the liver: case report and literature survey. J Hepatobiliary Pancreat Surg. 2008;15:536-544.
Padfield CJ, et al. Mucinous biliary papillomatosis: a tumour in need of wider recognition. Histopathology. 1988;13:687-694.
Paradis V, et al. Evidence for the polyclonal nature of focal nodular hyperplasia of the liver by the study of X-chromosome inactivation. Hepatology. 1997;26:891-895.
Paradis V, et al. Clonal analysis of macronodules in cirrhosis. Hepatology. 1998;28:953-958.
Paradis V, et al. Clonal analysis of micronodules in virus C–induced liver cirrhosis using laser capture microdissection (LCM) and HUMARA assay. Lab Invest. 2000;80:1553-1559.
Paradis V, et al. Telangiectatic focal nodular hyperplasia: a variant of hepatocellular adenoma. Gastroenterology. 2004;126:1323-1329.
Paradis V, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology. 2009;49:851-859.
Park YN, et al. Increased expression of vascular endothelial growth factor and angiogenesis in the early stage of multistep hepatocarcinogenesis. Arch Pathol Lab Med. 2000;124:1061-1065.
Parkin DM, et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74-108.
Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001;33:1353-1357.
Patel T. Cholangiocarcinoma. Nat Clin Pract Gastroenterol Hepatol. 2006;3:33-42.
Pawlik TM, et al. Tumor size predicts vascular invasion and histologic grade: implications for selection of surgical treatment for hepatocellular carcinoma. Liver Transpl. 2005;11:1086-1092.
Pawlik TM, et al. Preoperative assessment of hepatocellular carcinoma tumor grade using needle biopsy: implications for transplant eligibility. Ann Surg. 2007;245:435-442.
Peters WM, et al. Angiomyelolipoma of the liver. Histopathology. 1983;7:99-106.
Petrolla AA, Xin W. Hepatic angiomyolipoma. Arch Pathol Lab Med. 2008;132:1679-1682.
Pupulim LF, et al. Algorithm for immediate cytologic diagnosis of hepatic tumors. AJR Am J Roentgenol. 2008;190:W208-W212.
Qin LX, Tang ZY. The prognostic significance of clinical and pathological features in hepatocellular carcinoma. World J Gastroenterol. 2002;8:193-199.
Rangheard AS, et al. Focal nodular hyperplasia inducing hepatic vein obstruction. AJR Am J Roentgenol. 2002;179:759-762.
Rebouissou S, et al. Molecular pathogenesis of focal nodular hyperplasia and hepatocellular adenoma. J Hepatol. 2008;48:163-170.
Rebouissou S, et al. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature. 2009;457:200-204.
Roberts JL, et al. Lipomatous tumors of the liver: evaluation with CT and US. Radiology. 1986;158:613-617.
Rojiani AM, et al. Hepatic hemangioblastoma: an unusual presentation in a patient with von Hippel-Lindau disease. Am J Surg Pathol. 1991;15:81-86.
Roncalli M. Hepatocellular nodules in cirrhosis: focus on diagnostic criteria on liver biopsy: a Western experience. Liver Transpl. 2004;10:9-15.
Roncalli M, et al. The vascular profile of regenerative and dysplastic nodules of the cirrhotic liver: implications for diagnosis and classification. Hepatology. 1999;30:1174-1178.
Roncalli M, et al. Hepatocellular dysplastic nodules. Hepatol Res. 2007;37(Suppl 2):125-134.
Ross JS, Kurian S. Clear cell hepatocellular carcinoma: sudden death from severe hypoglycemia. Am J Gastroenterol. 1985;80:188-194.
Roth JA, et al. A black hepatocellular carcinoma with Dubin–Johnson-like pigment and Mallory bodies: a histochemical and ultrastructural study. Am J Surg Pathol. 1982;6:375-382.
Rouzbahman M, et al. Oncocytic papillary neoplasms of the biliary tract: a clinicopathological, mucin core and Wnt pathway protein analysis of four cases. Pathology. 2007;39:413-418.
Rullier A, et al. Cytokeratin 7 and 20 expression in cholangiocarcinomas varies along the biliary tract but still differs from that in colorectal carcinoma metastasis. Am J Surg Pathol. 2000;24:870-876.
Saab S, Yao F. Fibrolamellar hepatocellular carcinoma: case reports and a review of the literature. Dig Dis Sci. 1996;41:1981-1985.
Sakamoto M, et al. Multicentric independent development of hepatocellular carcinoma revealed by analysis of hepatitis B virus integration pattern. Am J Surg Pathol. 1989;13:1064-1067.
Sakamoto M, et al. Early stages of multistep hepatocarcinogenesis: adenomatous hyperplasia and early hepatocellular carcinoma. Hum Pathol. 1991;22:172-178.
Sangiovanni A, et al. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterology. 2004;126:1005-1014.
Santos ES, et al. Primary hepatic non-Hodgkin’s lymphomas: case report and review of the literature. Am J Gastroenterol. 2003;98:2789-2793.
Santos I, et al. Primary hepatic leiomyoma: case report. Abdom Imaging. 2010;36:315-317.
Sasaki K, et al. Hepatic clear cell carcinoma associated with hypoglycemia and hypercholesterolemia. Cancer. 1981;47:820-822.
Saul SH, et al. The fibrolamellar variant of hepatocellular carcinoma: its association with focal nodular hyperplasia. Cancer. 1987;60:3049-3055.
Schnelldorfer T, et al. Management of giant hemangioma of the liver: resection versus observation. J Am Coll Surg. 2010;211:724-730.
Schulz W, Hort W. The distribution of metastases in the liver: a quantitative postmortem study. Virchows Arch A Pathol Anat Histol. 1981;394:89-96.
Scott LD, et al. Oral contraceptives, pregnancy, and focal nodular hyperplasia of the liver. JAMA. 1984;251:1461-1463.
Seeff LB, Hoofnagle JH. Epidemiology of hepatocellular carcinoma in areas of low hepatitis B and hepatitis C endemicity. Oncogene. 2006;25:3771-3777.
Seki S, et al. Outcomes of dysplastic nodules in human cirrhotic liver: a clinicopathological study. Clin Cancer Res. 2000;6:3469-3473.
Shaib YH, et al. Rising incidence of intrahepatic cholangiocarcinoma in the United States: a true increase? J Hepatol. 2004;40:472-477.
Shaib YH, et al. Risk factors of intrahepatic cholangiocarcinoma in the United States: a case-control study. Gastroenterology. 2005;128:620-626.
Shamseddine A, et al. Unusually young age distribution of primary hepatic leiomyosarcoma: case series and review of the adult literature. World J Surg Oncol. 2010;8:56.
Sherman M. Hepatocellular carcinoma: epidemiology, risk factors, and screening. Semin Liver Dis. 2005;25:143-154.
Sherman M. Vinyl chloride and the liver. J Hepatol. 2009;51:1074-1081.
Siddiqui MA, McKenna BJ. Hepatic mesenchymal hamartoma: a short review. Arch Pathol Lab Med. 2006;130:1567-1569.
Sioutos N, et al. Primary hepatic carcinoid tumor: an electron microscopic and immunohistochemical study. Am J Clin Pathol. 1991;95:172-175.
Smalley SR, et al. Hepatoma in the noncirrhotic liver. Cancer.. 1988;62:1414-1424.
Srouji MN, et al. Mesenchymal hamartoma of the liver in infants. Cancer. 1978;42:2483-2489.
Steiner PE, Higginson J. Cholangiolocellular carcinoma of the liver. Cancer. 1959;12:753-759.
Stocker JT. Hepatic tumors in children. Clin Liver Dis. 2001;5:259-281.
Stocker JT, Ishak KG. Undifferentiated (embryonal) sarcoma of the liver: report of 31 cases. Cancer. 1978;42:336-348.
Stocker JT, Ishak KG. Mesenchymal hamartoma of the liver: report of 30 cases and review of the literature. Pediatr Pathol. 1983;1:245-267.
Strate SM, et al. Delayed development of angiosarcoma in multinodular infantile hepatic hemangioendothelioma. Arch Pathol Lab Med. 1984;108:943-944.
Strohmeyer T, Schultz W. The distribution of metastases of different primary tumors in the liver. Liver. 1986;6:184-187.
Sturtz CL, Dabbs DJ. Angiomyolipomas: the nature and expression of the HMB45 antigen. Mod Pathol. 1994;7:842-845.
Sutton FM, et al. Factors affecting the prognosis of primary liver carcinoma. J Clin Oncol. 1988;6:321-328.
Suzuki S, et al. Clinicopathological prognostic factors and impact of surgical treatment of mass-forming intrahepatic cholangiocarcinoma. World J Surg. 2002;26:687-693.
Takayama T, et al. Malignant transformation of adenomatous hyperplasia to hepatocellular carcinoma. Lancet. 1990;336:1150-1153.
Tamburro CH, et al. Early hepatic histologic alterations among chemical (vinyl monomer) workers. Hepatology. 1984;4:413-418.
Tang L, et al. Inflammatory myofibroblastic tumor of the liver: a cohort study. World J Surg. 2010;34:309-313.
Terada T, et al. Ciliated hepatic foregut cyst: a mucus histochemical, immunohistochemical, and ultrastructural study in three cases in comparison with normal bronchi and intrahepatic bile ducts. Am J Surg Pathol. 1990;14:356-363.
Terada T, et al. A clinicopathologic study of adenomatous hyperplasia of the liver in 209 consecutive cirrhotic livers examined by autopsy. Cancer. 1993;72:1551-1556.
Terasaki S, et al. Histological features predicting malignant transformation of nonmalignant hepatocellular nodules: a prospective study. Gastroenterology. 1998;115:1216-1222.
Theise ND. New principles of cell plasticity. C R Biol. 2002;325:1039-1043.
Theise ND, et al. Macroregenerative nodules and hepatocellular carcinoma in forty-four sequential adult liver explants with cirrhosis. Hepatology. 1992;16:949-955.
Theise ND, et al. Dysplastic nodules and hepatocarcinogenesis. Clin Liver Dis. 2002;6:497-512.
Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339-346.
Tickoo SK, et al. Combined hepatocellular cholangiocarcinoma: a histopathologic, immunohistochemical, and in situ hybridization study. Am J Surg Pathol. 2002;26:989-997.
Trastek VF, et al. Cavernous hemangiomas of the liver: resect or observe? Am J Surg. 1983;145:49-53.
Tsui WM, et al. Hepatic angiomyolipoma: a clinicopathologic study of 30 cases and delineation of unusual morphologic variants. Am J Surg Pathol. 1999;23:34-48.
Unger PD, et al. Pseudosarcomatous cystadenocarcinoma of the liver. Hum Pathol. 1987;18:521-523.
Van Eyken P, et al. Abundant expression of cytokeratin 7 in fibrolamellar carcinoma of the liver. Histopathology. 1990;17:101-107.
Van Roekel V, et al. Cystadenoma of the liver. J Clin Gastroenterol. 1982;4:167-172.
Van Steenbergen W, et al. Hepatic lymphangiomatosis: report of a case and review of the literature. Gastroenterology. 1985;88:1968-1972.
Vick DJ, et al. Ciliated hepatic foregut cyst: a study of six cases and review of the literature. Am J Surg Pathol. 1999;23:671-677.
Vick DJ, et al. Squamous cell carcinoma arising in a ciliated hepatic foregut cyst. Arch Pathol Lab Med. 1999;123:1115-1117.
Villanueva A, et al. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55-76.
Villanueva A, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972-1983.
Vorse HB, et al. Hepatic hemangiomatosis of infancy. Am J Dis Child. 1983;137:672-673.
Wanless IR, et al. On the pathogenesis of focal nodular hyperplasia of the liver. Hepatology. 1985;5:1194-1200.
Wanless IR, et al. Multiple focal nodular hyperplasia of the liver associated with vascular malformations of various organs and neoplasia of the brain: a new syndrome. Mod Pathol. 1989;2:456-462.
Watanapa P, Watanapa WB. Liver fluke–associated cholangiocarcinoma. Br J Surg. 2002;89:962-970.
Weiss SW, et al. Epithelioid hemangioendothelioma and related lesions. Semin Diagn Pathol. 1986;3:259-287.
Welzel TM, et al. Impact of classification of hilar cholangiocarcinomas (Klatskin tumors) on the incidence of intra- and extrahepatic cholangiocarcinoma in the United States. J Natl Cancer Inst. 2006;98:873-875.
Welzel TM, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based case-control study. Clin Gastroenterol Hepatol. 2007;5:1221-1228.
Wheeler DA, Edmondson HA. Ciliated hepatic foregut cyst. Am J Surg Pathol. 1984;8:467-470.
Wheeler DA, Edmondson HA. Coelomic fat ectopia in the liver. Arch Pathol Lab Med. 1985;109:783-785.
Wheeler DA, Edmondson HA. Cystadenoma with mesenchymal stroma (CMS) in the liver and bile ducts: a clinicopathologic study of 17 cases, 4 with malignant change. Cancer. 1985;56:1434-1445.
Wolf HK, et al. Exocrine pancreatic tissue in human liver: a metaplastic process? Am J Surg Pathol. 1990;14:590-595.
Wolf HK, et al. Oncocytic differentiation in intrahepatic biliary cystadenocarcinoma. Mod Pathol. 1992;5:665-668.
Woodall CE, et al. Hepatic malignant epithelioid hemangioendothelioma: a case report and review of the literature. Am Surg. 2008;74:64-68.
Woods GL. Biliary cystadenocarcinoma: case report of hepatic malignancy originating in benign cystadenoma. Cancer. 1981;47:2936-2940.
Wurmbach E, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45:938-947.
Xue W, et al. DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 2008;22:1439-1444.
Yamato T, et al. Intrahepatic cholangiocarcinoma arising in congenital hepatic fibrosis: report of an autopsy case. J Hepatol. 1998;28:717-722.
Yamamoto H, et al. Inflammatory myofibroblastic tumor versus IgG4-related sclerosing disease and inflammatory pseudotumor: a comparative clinicopathologic study. Am J Surg Pathol. 2009;33:1330-1340.
Yano Y, et al. Combined hepatocellular and cholangiocarcinoma: a clinicopathologic study of 26 resected cases. Jpn J Clin Oncol. 2003;33:283-287.
Zafrani ES, et al. The hepatic sinusoid in hairy cell leukemia: an ultrastructural study of 12 cases. Hum Pathol. 1987;18:801-807.
Zaman SN, et al. Risk factors in development of hepatocellular carcinoma in cirrhosis: prospective study of 613 patients. Lancet. 1985;1:1357-1360.
Zen Y, et al. Proposal of histological criteria for intraepithelial atypical/proliferative biliary epithelial lesions of the bile duct in hepatolithiasis with respect to cholangiocarcinoma: preliminary report based on interobserver agreement. Pathol Int. 2005;55:180-188.
Zen Y, et al. Biliary papillary tumors share pathological features with intraductal papillary mucinous neoplasm of the pancreas. Hepatology. 2006;44:1333-1343.
Zen Y, et al. Biliary intraepithelial neoplasia: an international interobserver agreement study and proposal for diagnostic criteria. Mod Pathol. 2007;20:701-709.
Zucman-Rossi J, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43:515-524.