[level-membership-for-neurosurgery-category]
CHAPTER 308 Tumors of the Craniovertebral Junction
The craniovertebral junction is a biomechanical and anatomic unit that comprises the clivus, foramen magnum, and upper two cervical vertebrae. The neoplasms that arise within the structures are osseous in nature or extensions from the soft tissue that surround the craniovertebral junction, or they are neoplasms that arise from the neural structures contained within the bony anatomy.1–4 The diagnosis of such lesions has been greatly facilitated by modern neurodiagnostic imaging.
There is no single symptom or neurological finding that is pathognomonic for a lesion in this location.5,6 Because of the generous size of the subarachnoid spaces at the cervicomedullary junction, symptoms arise only after a lesion has achieved large proportions. These patients have a fluctuating neurological course, and an erroneous diagnosis is common owing to the anatomic complexities of the decussation of the sensory and motor tracts.1,6
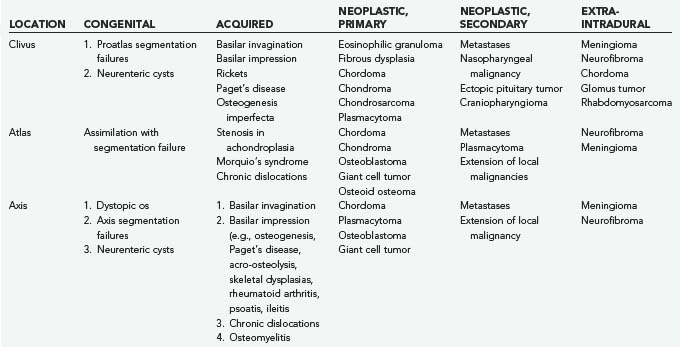
The first systematic evaluation of foramen magnum tumors was performed by Elsberg and Strauss.7 Several authors have since reported extra-axial lesions affecting the region, such as meningiomas and neurinomas.8–12 Osseous neoplastic involvement of the craniovertebral junction may be due to chordoma, chondrosarcoma, plasmacytoma, osteoblastoma, fibrous dysplasia, metastatic tumor, and giant cell tumor. Table 308-1 summarizes my experience.
Common Clinical Manifestations of Craniovertebral Junction Tumors
Tumors of the craniovertebral junction, whether extracranial with secondary involvement of the intracranial and intraspinal structures or primarily intracranial with secondary extension into the spinal canal, have characteristics that reflect compression of neighboring structures or traction. They also may have distal effects such as hydrocephalus, syringohydromyelia, and vascular compromise.9,13,14 Chordoma often involves the cranial base and upper cervical spine extensively and may be associated with only minimal complaints of headache and neck pain for several years. Unfortunately, this hiatus is followed by a rapid progression of brainstem and cervical spinal cord dysfunction that brings the lesion to light. In the report of Meyer and coworkers, the time from the onset of symptoms to the diagnosis of extramedullary tumor at the foramen magnum was 2.5 years.2
The clinical presentation of craniovertebral junction tumors can be divided into intracranial lesions, “straddle lesions,” and those affecting the high cervical spinal cord.7 The effects of vascular compromise and alterations of cerebrospinal fluid (CSF) circulation add to the constellation of symptoms and signs.
Patients with intracranial lesions present with involvement of the lower cranial nerves, brainstem dysfunction, and occasionally cerebellar symptoms. Patients with straddle lesions have a paucity of cranial nerve dysfunction and a predominance of high cervical myelopathy. High cervical lesions do not produce cranial nerve and cerebellar signs, except for involvement of the spinal accessory nerve and sometimes the descending tracts of the trigeminal nerve and the lower decussations of the motor and sensory tracks.15
The most common presentation is pain referred to the second cervical dermatome.1 The head is held flexed, and the condition may resemble torticollis. The pain is described as an aching sensation that is aggravated by neck and head motion and referred to the suboccipital region. Unfortunately, the symptom of pain alone may predate other clinical findings for many years. Paresthesias or dysesthesias of the face, hands, and limbs are frequently reported. An abnormal cold sensation of the lower extremities was described by Elsberg and Strauss7 and by Beatty13 as being pathognomonic of lesions of the high cervical cord. Most frequently, pain and temperature sensation is affected, followed by loss of joint sensation. This finding is seen in the upper extremities and may then proceed clockwise around the limbs. A suspended sensory loss with patches of preservation of sensation in the upper extremities may confuse the presentation. “Dissociated” sensory loss has been described in about one fourth of extracranial lesions of the cervicomedullary junction, even though this finding has been considered to reflect an intramedullary process.2
Spastic weakness of the extremities is a prominent feature in patients with tumors in this region. The weakness may begin in the ipsilateral limb and progress to the lower limb of the same side, followed by weakness of the contralateral lower limb; finally, weakness becomes apparent in the contralateral upper limb. This distinct progression of motor symptoms is an important characteristic of lesions of the cervicomedullary junction.15 Localized wasting of the intrinsic muscles of the hand may develop ipsilateral to the lesion. Taylor and Byrnes postulate venous stagnation of the anterior horn cells and the lower cervical cord as a result of decreased venous drainage, which typically occurs rostral to the lower portion of the cervical spinal cord.16 Other proposed mechanisms include anterior spinal artery compression, hydromyelia secondary to CSF obstruction, venous obstruction with spinal cord edema, and spinal cord rotation with contralateral traction.
A tumor at the foramen magnum may produce a mixture of upper motor neuron findings in the upper and lower extremities. This pattern reflects the pyramidal decussation that begins just below the obex and ends near the uppermost cervical spinal cord. The more medial fibers of the pyramidal tract carry impulses to the upper extremities and cross superior to the lateral fibers that serve the lower extremities. Similarly, the sensory decussation of the medial lemniscus may produce a varied pattern of sensory abnormalities.17 Thus, a tumor situated at the ventral aspect of the cervicomedullary junction can cause sensory aberrations in the lower extremities first. The syndrome of cruciate paralysis has been associated with trauma as well as tumors with basilar invagination.18
Transient symptoms may be due to both vascular changes and instability at the craniovertebral junction.1,19 Such symptoms can manifest as paralysis, paresthesias, drop attacks, and vertebrobasilar syndromes such as migraine and visual loss in the homonymous visual fields. Cranial nerve palsies may be the result of nuclear involvement in the brainstem, traction, compression of the subarachnoid segments, or interosseous disease.1,15 The most common cranial nerves affected are the vagus, glossopharyngeal, and hypoglossal. Their involvement leads to dysphagia, slurred speech, and repeated episodes of aspiration into the tracheobronchial tree, resulting in pneumonia and weight loss. Tumors of the upper cervical canal can present with involvement of the spinal root of the accessory nerve, manifesting as torticollis and weakness of the trapezius and sternocleidomastoid muscles. About 15% to 20% of patients develop tinnitus, vertigo, and hearing loss related to involvement of the vestibulocochlear nerve.15
The differential diagnoses considered most often by Meyer and coworkers in their initial evaluation of 102 documented cases of benign extramedullary tumors of the foramen magnum included cervical spondylosis (25%), multiple sclerosis (18%), syringomyelia (17%), intramedullary tumor (15%), Chiari’s malformation (5.5%), and carpal tunnel syndrome (5.5%).2 Other erroneous diagnoses in patients with lesions at the craniovertebral junction are intramedullary tumors of the brainstem and upper cervical cord, amyotrophic lateral sclerosis, and subacute combined degeneration. Cervical spondylosis may be difficult to differentiate when it coexists. The presentation of restless legs syndrome in patients with craniovertebral compressive pathology has been well documented by Glasauer and Egnatchick.20
Neurodiagnostic Imaging
The complex anatomy and pathology of tumors in this region demand precise definition and depiction of the tumor’s extent and its relationship to the vital structures of the brainstem and spinal cord, lower cranial nerves, and vascular structures. Thus, complementary multimodality imaging includes plain radiography, magnetic resonance imaging (MRI), magnetic resonance angiography, computed tomography (CT), and three-dimensional CT angiography. The sensitivity of MRI is greatly enhanced by the addition of intravenous gadolinium and by performing separate magnetic resonance venography and angiography.5
Cerebral angiography is useful in understanding the dynamics of collateral circulation and tumor vascularity. Temporary balloon occlusion is a means of assessing a patient’s tolerance of vascular occlusion of the carotid or vertebral circulation before surgery.15 Such information is especially useful when lesions are encased by tumor. These two tests provide information about the resectability of difficult lesions.
Electrophysiologic Tests
Intraoperatively, we monitor somatosensory brainstem evoked potentials and perform facial electromyography when lesions extend to the cerebellopontine angle and when using the preauricular infratemporal fossa approach.15,21,22 The intraoperative evaluation of cranial nerve IX and cranial nerve X function is accomplished by placing electrodes in the soft palate and against the true vocal cords, incorporating these latter electrodes in the endotracheal tube for the anesthetic. Hypoglossal nerve electromyography supplements the evaluation with an electrode placed directly into the tongue. Brainstem monitoring still yields a significant number of false-negative and false-positive results, but improved techniques make these adjuncts useful in the intraoperative assessment of the function of the cervicomedullary junction.
Surgical Approaches and Decision Making
The goal of treating benign osseous pathology differs from that of treating malignant disease, in which complete excision is the objective. Benign lesions create a space among the neurovascular structures, thereby allowing surgical debulking and resection “from within.” Malignant disease, however, requires a much more radical resection with clean margins. In most instances, benign lesions such as chordomas are radioresistant; hence, gross total resection should be the aim. Craniovertebral stability, both before and after operative intervention, must be considered in the development of approaches.14,19,23 Thus, the factors that influence the specific treatment of tumors arising in this region are (1) mechanism of compression and direction of encroachment, (2) whether the lesion is benign or malignant, (3) whether the lesion has an associated vascular or intramedullary component (e.g., syringomyelia), (4) craniospinal stability, and (5) patient age. Lesions of the craniovertebral junction do affect the pediatric population, although to a lesser extent than they affect adults.3,5,24 In children, the potential for growth, concerns about stability, and the patient’s size are critical. From this perspective, midface growth centers are the nasal septum and the pterygoid plates. Hence, in a child, a transpalatal approach to the clivus and the sella would be considered before a sublabial transsphenoidal or a maxillary drop-down procedure, with the goal of avoiding damage to the growth centers.25
Advances in neurodiagnostic imaging and microsurgical instrumentation have allowed the development of extensive surgical approaches based on an understanding of the complex anatomy, the craniovertebral dynamics, and the site of encroachment. Consequently, the entire circumference of the foramen magnum is within the neurosurgeon’s reach (Fig. 308-1). However, several surgical limitations and considerations must be appreciated when designing an approach to the craniovertebral junction (Table 308-2). The most frequently used anterior route is the transoral-transpalatopharyngeal approach.1,25–27 Laterally, this approach is limited by the pterygoid plates, the hypoglossal canal, the eustachian tubes, and the width between the vertebral arteries. The limitation imposed by the pterygoid plates can be overcome by using a transmaxillary route with down-fracture of the maxilla and an extended maxillotomy if needed.1,28 A combined exposure with mandibulotomy and midline glossotomy lowers the midline exposure to C5.5,29,30 The disadvantages of the ventral approaches are overshadowed by their easy access, ability to cross the midline, and vertical extensions.31,32 The ventral and dorsal midline approaches permit the anterior and posterior 90 degrees of the craniovertebral junction to be scanned, respectively.
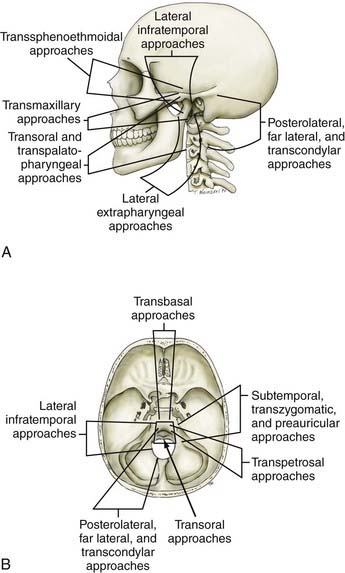
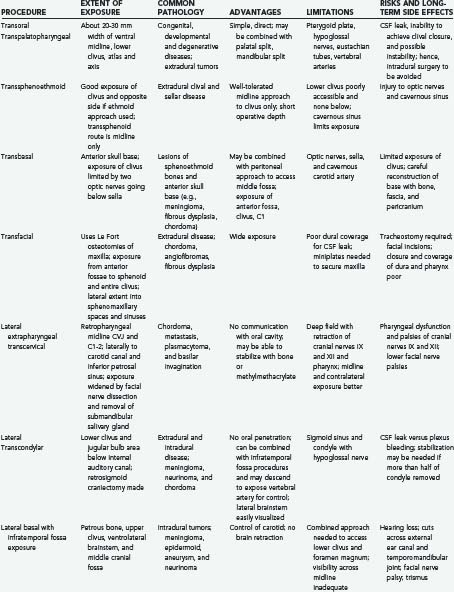
As a general rule, midline extra-axial tumors are best approached in an extra-axial fashion without retracting the brain or violating the dura. A lateral avenue should be used for tumors that are situated laterally.33–35 The transsphenoidal-transsphenoethmoidal approach provides a shallow operative depth and is well tolerated as a route to the upper two thirds of the clivus.36–38 This exposure provides better access to the contralateral side; hence, at times, a bilateral approach may be necessary. The transfacial routes provide wide access to the anterior and midline skull base and include the ventral aspect of the clivus.39,40 This approach is ideal for treating extradural lesions such as chordomas, angiofibromas, and fibrous dysplasias. Dural coverage must be provided by vascularized muscle flaps obtained from the temporalis muscle.
The transbasal approach provides access to the anterior skull base. Unless combined with removal of the supraorbital bar, this approach is limited by the distance between the two optic nerves and the need to work beneath the sella turcica.23 Nonetheless, we have used this approach in several patients and have achieved good reconstruction of the floor.
A lateral extrapharyngeal route is effective and safe for reaching the upper cervical spine.41 At the clivus, however, access becomes difficult because of the pyramidal narrowing of the exposure at the depths of the wound. In an effort to correct this problem, we reroute the facial nerve with upward displacement of the angle of the mandible,25 thereby exposing the lower clivus. We believe that this approach should be limited to metastases, chordomas, and plasmacytomas that affect the axis and atlas vertebrae. Its use for treatment of basilar invagination and intradural pathology is limited.
The true lateral-transcondylar approach to the ventral aspect of the lower brainstem and clivus, as well as the upper cervical cord, demands resection of a portion of the lateral atlantal mass and the occipital condyle. Exposure of the lower clivus and the ventral brainstem is enhanced when combined with a retrosigmoid craniectomy.34 It is useful in the treatment of both extradural and intradural lesions such as chordomas, meningiomas, and neurofibromas.4,14,33 When combined with infratemporal procedures, it allows both anterior and posterior extensions that overcome the limitations of the sigmoid sinus and the hypoglossal nerve.1,34,42 The risk for CSF leak and the possibility of destabilization are high. Troublesome bleeding is routinely encountered from the paravertebral venous plexuses.
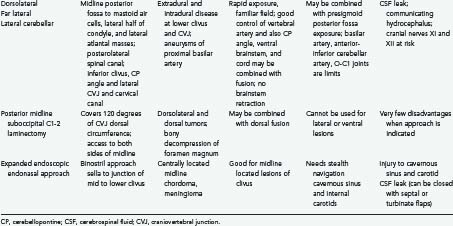
The posterolateral route has been recognized for many years. It enables scanning of the foramen magnum to the 90 degrees available with the posterior midline approach as well as the 90 degrees available with the lateral transcondylar approach.1,43 The latter uses a standard midline posterior exposure with a lateral cerebellar approach and includes partial resection of the mastoid process and the posterior third to half of the occipital condyle to provide exposure of the jugular bulb and the medial aspect of the lateral atlantal mass. This provides access to the vertebral artery, which can be rerouted and displaced from the foramen transversarium, thus allowing for control from the C2 level upward. The ability to create a fusion makes this an ideal approach for posterior, lateral, and ventrolateral lesions. Thus, the entire circumference of the foramen magnum is accessible.
The expanded endonasal endoscopic approach is a valid, minimally invasive alternative for treatment of centrally located clivus chordomas or any such lesions or could be an adjunct for the central part of a lesion with lateral extension.36,44–46 A standard nasal access is used with the binostril approach, with the endoscope typically positioned in the right superior nasal cavity by one surgeon who is responsible for maintaining the visual field. This arrangement allows a second surgeon to use both hands and an instrument in each nostril for the resection of the tumor. Dissection of both middle turbinates can expose a large working field, although this is not essential. The mucoperiosteal flaps from the middle turbinate can be used to close off a CSF leak.38 The medial wall of the clival carotids represents the lateral limits of the clival exposure. The floor of the sphenoid is further drilled to get access to the mid and lower clivus. Resection of medial pterygoid process can be performed for exposure of lateral extension of the lower part of the tumor. A total ethmoidectomy can augment exposure into the upper clivus. Closure is performed with multilayered reconstruction technique. In the series by Dehdashti and associates,46 12 patients underwent such operation over a 3-year span. There were four CSF leaks. One patient developed hydrocephalus and one new deficit with clot and one pneumocephalus. Cranial deficits improved over time. A gross resection was accomplished in 7 of the 12 patients. In the series by Frank and associates,45 nine chordomas were approached in this fashion, with a total removal in three. An internal carotid injury occurred in one patient, with two persistent CSF leaks.
Extradural Tumors of the Craniovertebral Junction
Clival and Craniovertebral Junction Chordomas
Chordomas are rare, aggressive, locally destructive tumors of presumed notochordal origin that arise along the vertebral axis and show a proclivity for the spheno-occipital and sacral regions.1,9,37,47 Chordomas of the clivus and craniovertebral junction are the most common extradural neoplasms in this region. The overall incidence of chordomas is 0.2 to 0.5 per 100,000 persons per year, and they account for about 0.15% of all intracranial tumors. About 25% of chordomas occur at the base of the skull, arising from the clivus. Although usually midline, the notochord may have distal projections that extend to the clinoid processes of the petrous bones. Most patients experience symptoms referable to the tumor for more than a year before diagnosis.
Pathology
Chordomas have been divided into classic chordomas, chondroid chordomas, and atypical chordomas.48 Classic chordomas are lobulated, pinkish gray, gelatinous tumors that infiltrate bone but may appear grossly as somewhat demarcated; they account for 80% to 85% of all chordomas. Histologically, they exhibit a variable mix of sheets and cords or clusters of small polygonal cells with eosinophilic cytoplasm and hyperchromatic nuclei. A myxoid matrix is present. Cytologic atypia is absent or minimal.
A subpopulation, the chondroid chordoma, arises in the spheno-occiput and exhibits cartilaginous differentiation. Some authors dispute the existence of chondroid chordomas, preferring to regard these cartilage-containing neoplasms as chondrosarcomas. Chondroid chordomas have a more indolent clinical course; the survival rate is 15.8 years, compared with 4.1 years for typical classic chordomas.47,48 Chondroid chordomas account for 5% to 15% of all chordomas.
Atypical chordomas have a sarcomatoid appearance, with round cells and epithelial or spindle cells present with large areas of necrosis. These solid tumors are aggressive and account for 1.3% to 8% of all chordomas. In the series by Heffelfinger and coworkers, only one patient with an atypical chordoma survived more than 10 years, whereas almost 50% of those with chondroid chordomas survived more than 10 years.48 The frequency of mitotic figures, nuclear pleomorphology, and hyperchromatism does not appear to affect the ultimate outcome.49,50 In rare circumstances, chordomas may dedifferentiate into malignant chondrosarcomas, fibrosarcomas, and even osteosarcomas.51
The immunohistochemical profile of reactivity with antibodies to vimentin cytokeratin, epithelium membrane antigen, and S-100 protein tend to distinguish chordomas from other sarcomatoid round cell or myxoid neoplasms.49,50 Chondrosarcomas are negative for cytokeratin, epithelium membrane antigen, and carcinoembryonic antigen. Vimentin and S-100 protein are present in both chondrosarcomas and chordomas. Immunostaining from keratin has no prognostic value regarding the aggressiveness of the tumor. Chondrosarcomas have been lumped with chordomas because of supposed parallel lines of occurrence, location, and aggressive behavior.
Imaging Characteristics
Cranial-based chordomas are clearly defined by the use of high-resolution CT, on which they appear as solitary or multiple areas of decreased attenuation within the clivus. T2-weighed MRI reveals a bright signal from the marrow in the skull base, signifying replacement of bone by tumor.9,25 At times, calcification is noted in the abnormal areas in the retropharyngeal space, representing sequestered bone fragments in tumor. Chordomas enhance with intravenous contrast and are well visualized by MRI. Chordomas are isointense on T1-weighted MRI (Fig. 308-2).
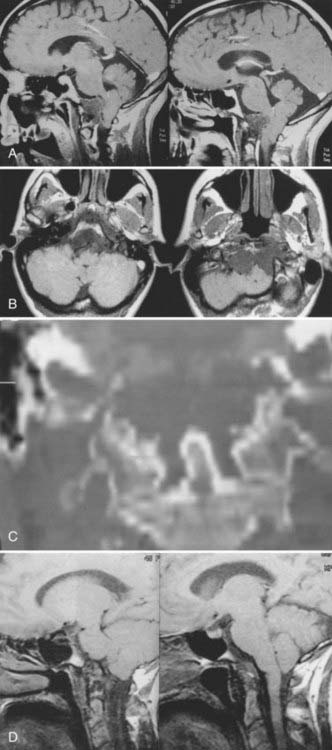
Presentation
Chordomas usually occur in adults, with a peak incidence occurring in the fourth decade of life.9,47,52 Less than 5% of these tumors arise in children, and they have a predilection for the spheno-occipital region.5 Headaches are often occipitocervical in location and aggravated by changes in craniovertebral positioning. In my series, 55% of patients benefited from craniovertebral stabilization in addition to tumor resection, owing to involvement of the occipital condyles.15,25 Lateral extension of these tumors can give rise to unilateral symptoms, such as hypoglossal nerve palsy. Larger tumors have the potential to cause both upper and lower cranial nerve palsies and a variety of problems related to brainstem compression. Chordomas often cause symptoms from local growth into the nasal cavity, pharynx, and paranasal sinuses.
Surgical Series
Forsyth and colleagues reviewed 51 intracranial chordomas treated surgically between 1960 and 1984 at the Mayo Clinic.52 The median age at presentation was 46 years, and 19 tumors were classified as chondroid chordomas. Eleven patients (22%) underwent biopsy, and 40 patients (78%) had subtotal resection. The survival rates for patients who underwent biopsy were 36% and 0% at 5 and 10 years, respectively, whereas survival rates for those with subtotal resections were 55% and 45% at 5 and 10 years, respectively. Patients who underwent postoperative radiation therapy tended to have longer disease-free survival times. Disease-free survival was the same for patients with chondroid chordomas as for those with typical chordomas.
Watkins and associates described 38 patients treated at the National Hospital of Neurology and Neurosurgery in London between 1958 and 1988.53 Craniotomies were used in 28 patients, and transoral or transmaxillary routes were used in 10 others. All patients underwent postoperative external-beam radiotherapy of 50 to 60 Gy. Recurrence developed in 23 patients, and 13 died within 5 years. Twelve patients were lost to follow-up. The authors concluded that two groups existed: one with indolent disease and another with aggressive growth and poor outcome.
In a more recent publication in 2001, Crockard and associates described a multidisciplinary approach to skull base chordomas.54 A primary first time surgery was done in 24 and redo surgery (initial surgery elsewhere) in 18. A total removal was made in 2, radical 30 and subtotal or partial in the remainder. However, morbidity was significant, with CSF leak in 15, meningitis in 3, new cranial nerve abnormalities in 6 and persistent dysphonia in 4 patients. Three patients died.
Gay and colleagues reviewed the management of 46 chordomas and 14 chondrosarcomas involving the cranial base between 1984 and 1993 at the University of Pittsburgh.47 They recommended an aggressive approach to achieve long-term recurrence-free survival. Fifty percent of patients had undergone previous surgery before referral, and 22% had undergone previous external-beam radiation therapy. The surgical approach was a subtemporal-infratemporal fossa approach, sometimes combined with a transpetrous approach. In other instances, an extended subfrontal approach was used, and in a few cases, the lateral transcondylar approach was used. There was a high tendency to stay between the subtemporal-infratemporal fossa approach and the extended subfrontal approach. Using this technique, the rate of total or near-total resection was 67%. Eighteen patients had died by the 5-year follow-up. Postoperatively, 20% of patients underwent external-beam, proton-beam, or gamma radiation therapy. In patients who had total resection, the overall 5-year recurrence-free survival rate was 84%, compared with 64% in those with partial resection. However, the rate of morbidity was high. Thirty percent developed CSF leaks, 10% experienced meningitis, and 80% had an immediate new cranial nerve deficit. Using the Karnofsky performance score, 40% of patients had permanent functional deterioration. Based on this experience, the authors advocated aggressive initial surgical resection, with the sparing application of radiation therapy.
In 1997, Al-Mefty and Borba reported their results with an aggressive surgical approach combined with postoperative proton-beam therapy in 25 patients treated between 1990 and 1996.9 Radical or subtotal (>90%) removal was achieved in 84% of patients undergoing multiple procedures, when necessary, and extensive drilling of bone beyond the limits of tumor involvement. Postoperatively, 68% of the patients received a mean of 68-cGy–equivalent proton-beam radiotherapy. The postoperative mortality rate was 4%. The postoperative rate of morbidity, however, was 48%, although only 8% suffered permanent neurological deficits. Eighteen percent of the patients treated with proton-beam therapy developed radiation necrosis. The mean follow-up was only 25 months, however, making conclusions about outcome and survival difficult.
In a subsequent follow-up, Colli reviewed the series of Al-Mefty published in 2001.51 There were 55 chordomas and 10 chondrosarcomas. Follow-up of 1 to 150 months was reported, with a median of 38 months. A radical or subtotal removal was performed in 77.8% of patients. At 5-year follow-up, the mortality rate was 14.3%. The recurrence-free survival rates were 100% for chondrosarcomas and 50.7% for chordomas. Of those who received proton-beam therapy, 90.9% survived, as opposed to only 19.4% who received conventional postoperative radiation therapy. However, the complication rate was 60%, and 28.6% had a permanent worsening in neurological deficit. A transient deficit was present in 22.8%, which included new cranial nerve palsy, CSF leak, and hydrocephalus. The authors concluded that the survival was best with radical resection followed by proton-beam therapy.
Maira and coworkers37 achieved total tumor removal in 7 of 10 patients undergoing repeated transsphenoidal procedures for chordoma. They reported no evidence of disease in these patients at a mean of 38 months after surgery and encountered a cranial nerve complication in only 1 patient.
Couldwell and colleagues reported on a variation of the standard transsphenoidal approach to the sella with emphasis on extended approaches and parasellar approaches.36 This series of 105 patients reported in 2004 included 18 clivus chordomas. Total removal was accomplished in 66%. A hemiparesis was seen in 1 patient and new cranial nerve deficits in 2 patients, and an internal carotid occlusion was also found. Three patients had internal carotid artery hemorrhage, and 1 patient had a CSF leak, which was persistent. No deaths were reported.
During the past 15 years, cranial-based exposures of the clivus and upper cervical-craniovertebral chordomas have become accepted. Thus, it is important to compare recent series with at least a 5- to 10-year follow-up. Since 1985, 25 males and 18 females with cranial-base chordomas have been treated at the University of Iowa Hospitals and Clinics.15,25 Fourteen (34%) had undergone previous surgery; 11 (25%) had undergone previous radiation therapy, 5 of whom had proton-beam therapy. The 43 patients underwent 49 skull base procedures, and 9 required stabilization. Fifteen patients underwent a transoral-transpalatopharyngeal approach, 8 underwent a transmaxillary approach, 6 underwent a transsphenoethmoidal approach, 4 underwent an infratemporal fossa primary approach, 4 underwent a lateral extrapharyngeal approach, 6 underwent a transcondylar approach, 5 underwent a transfacial approach, and 1 underwent a transbasal approach. Gross total resection was possible in 12 of the 43 individuals (Fig. 308-3). Subtotal resection (>90%), which was documented on postoperative MRI, was achieved in 14 individuals. Seven patients died during the 15-year follow-up period. All patients had typical chordomas and underwent detailed histologic and immunohistochemical analysis of the tumor. Of the 15 individuals who underwent a transpalatopharyngeal approach, 5 died, 3 patients within 2 years of the transoral procedure at my institution. Each of these 3 patients had previously undergone more than four operative approaches to the tumor and had also undergone proton-beam therapy. The time from proton-beam radiation to the recurrence presenting at my institution was less than 3 years. There were no cases of postoperative CSF leakage, meningitis, or new cranial nerve deficits.
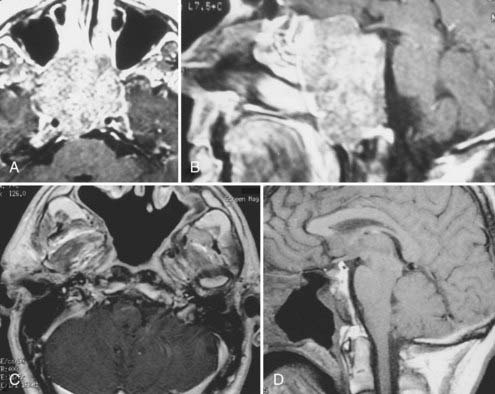
Samii and associates,55 in a recent review of 49 patients who had undergone resective surgeries at a single institution, came to the conclusion that complete tumor removal was accomplished in 49% and subtotally in 51%. The 5- and 10-year survival rates were 65% and 39%, respectively.
Chordomas in children are rare.3,5,56–59 Classic chordoma is seen in 28% and atypical chordoma in 72%. Thus, the prognosis reflects atypical lesions. All children with atypical lesions died within a mean of 6 months. Patients with classic chordomas treated within the past 10 years have survived a mean of 17 months. Metastasis occurs in about 60% of children younger than 5 years.
In a clinical pathologic study of 73 cases of children and adolescents referred to the Massachusetts General Hospital for proton-beam therapy for skull base chordoma between 1981 and 2003, good control with proton-beam therapy was achieved in children.56 Metastasis occurred in 58% of children younger than 5 years, whereas only 8.5% of those older than 5 years had subsequent metastasis. In an earlier combined study from Loma Linda, California, chondrosarcomas fared better over the long term than chordomas, and tumor recurrences were thought to be higher in patients who had tumor volume of more than 25 mL.60
Radiation Therapy
The role of radiation therapy in the management of skull base chordomas is now fairly well established. Amendola and colleagues concluded that conventional radiation therapy provided better local control when administered postoperatively than when delivered for a recurrence after surgical resection.61 Patients receiving more than 80 Gy had an 80% local control rate, whereas those receiving 40 to 60 Gy had a 20% success rate. There has been a consistent movement toward achieving a higher dosage with conventional external-beam radiotherapy. Radiation therapy itself has severe risks, including tumor recurrence, brain radiation necrosis, and radiation vasculitis.
In 1993, Forsyth and coworkers reported the results of 39 patients treated with surgery and conventional radiation therapy.52 The overall survival rates were 51% at 5 years and 35% at 10 years. Fractionated proton-beam therapy exploits favorable dose-localization characteristics and can deliver between 70 and 80 Gy to the tumor, whereas nearby nervous structures receive much lower doses. Researchers at both Harvard and Berkley Laboratory have published results with this modality.62–64 Their patients had an impressive actuarial control rate of 82% at 5 years and 58% at 10 years. Of the relapses, 95% were local recurrences, and 20% had distant metastasis. A poor prognosis was associated with tumors with a volume greater than 75 mL, more than 10% tumor necrosis, and involvement of the cervical spine. There was no histologic correlation with poor prognosis. Under this same protocol, 18 children, ranging in age from 4 to 18 years, were treated with proton-beam therapy. The median tumor dose was 69 Gy, with a 72-month median follow-up. Over a 5-year period, the actuarial survival was 68%, and the 5-year disease-free survival rate was 63%.
In summary, the data strongly support the view that radiation therapy, particularly proton-beam therapy combined with surgery, represents a substantial improvement over the natural history of chordoma (see Fig. 308-3C and D). Gamma-radiation therapy may deliver the benefits of proton therapy with fewer complications, but this remains to be demonstrated.65
Surgical Intervention
It is difficult to cure a patient with chordoma by surgical resection alone. No single operative approach can be used for all craniocervical chordomas.15,47,51,54,55 I believe that most cases are best approached with the initial intent to resect rather than just biopsy the tumor. Location of the tumor is the single most important factor in determining the approach. In some cases, several different approaches are necessary for adequate resection (Fig. 308-4). The consensus is that gross surgical resection or near-total resection should be the goal. The overall rate of morbidity associated with management must be weighed against the rate of recurrence and the extent of tumor resection. A close follow-up with postoperative imaging is essential and may be needed every 3 months for the first year, especially for aggressive lesions. I reoperate on patients with gross tumor recurrence before subjecting them to radiation therapy. In subtotal resections, atypical lesions, and children, proton-beam therapy has been offered after immediate reduction of the tumor’s volume.
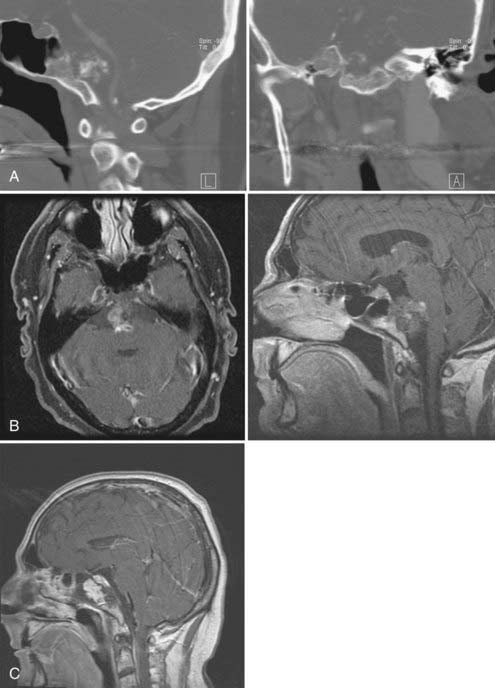
Plasmacytoma
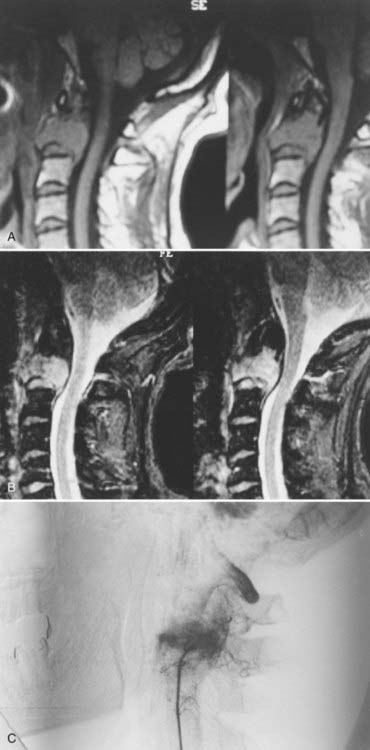
Solitary plasmacytomas are clinical entities distinct from multiple myeloma, even though both are manifestations of a continuum of B-cell lymphoproliferative diseases.5,41,66–68 The clinical distinction between the two is in the long-term prognosis. In a review of 84 patients with solitary plasmacytomas of the spine, McLain and Weinstein found that 44% of patients had developed disseminated disease at the end of 5 years, and the 5-year survival rate was 60%.69 In contrast, the 5-year mortality rate of disseminated myeloma (multiple myeloma) of the spinal column is 82%. Despite this improved outlook for solitary plasmacytomas, once a solitary plasmacytoma disseminates, the disease behaves much like multiple myeloma.67 The primary abnormality in multiple myeloma is slow, uncontrolled proliferation of immature and mature plasma cells in the bone marrow.66,70 This monoclonal population of cells produces a homogeneous immunoglobulin composed of a single class of heavy chain and one type of light chain referred to as the M protein. Multiple myeloma is associated with plasmacytosis of the bone marrow and the presence of immunoglobulin A-X monoclonal immunoglobulin in the cytoplasm of the tumor cell as well as in the serum. These serum and bone marrow findings are diagnostic. In contrast, plasmacytoma does not appear in the bone marrow and is localized to the tissue involved. I have seen plasmacytomas in the craniovertebral junction involving the occipital bone, the axis, and the atlas vertebrae.1,41,71
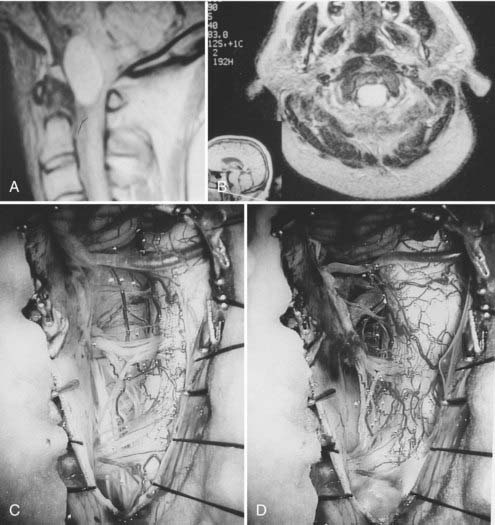
On gross examination, plasmacytomas are soft, grayish, moderately vascular lesions. They often reside within the diploic bone and only initially respect the cortices. When they expand, the cortex is destroyed. The tumor invades the vertebral bodies of the skull base and engulfs the vertebral vessels. When such tumors are present in the spinal canal, 20% involve the pedicles. The treatment of plasmacytoma involves diagnostic biopsy and stabilization. Because surgical attempts at resection can be fairly bloody, I advocate preoperative tumor embolization41 (Fig. 308-5). In my experience, surgery requires internal and external fixation with instrumentation, followed by radiation therapy. Occasionally, decompression of the cervicomedullary junction is essential and may be performed from a transoral-transpalatopharyngeal route or from a lateral extrapharyngeal-transcervical approach. Careful follow-up, including evaluation of serum, urine, and bone marrow, is essential.
Eosinophilic Granulomas
Eosinophilic granuloma affecting the spine is a childhood disease. The age of symptomatic involvement is 11 to 12 years. The most common area of occurrence is the thoracic and lumbar spine, although the upper cervical spine and clivus can be involved. The incidence of solitary eosinophilic granuloma of the spine is 8%. In children, multiple lesions occur in 38% of cases.72–74
Treatment of eosinophilic granuloma with low-dose radiation has been advocated and reported to be effective.15,74,75 Simple biopsy and immobilization usually lead to reconstitution of the vertebral height24 because eosinophilic granulomas spare the enchondral ossifications within the vertebral bodies. Multiple lesions with systemic conditions such as Letterer-Siwe disease and Hand-Schüller-Christian disease are the progressive form of this entity and require chemotherapy. Chemotherapy is not indicated for solid vertebral body lesions.
Calcium Pyrophosphate Masses or Pseudogout
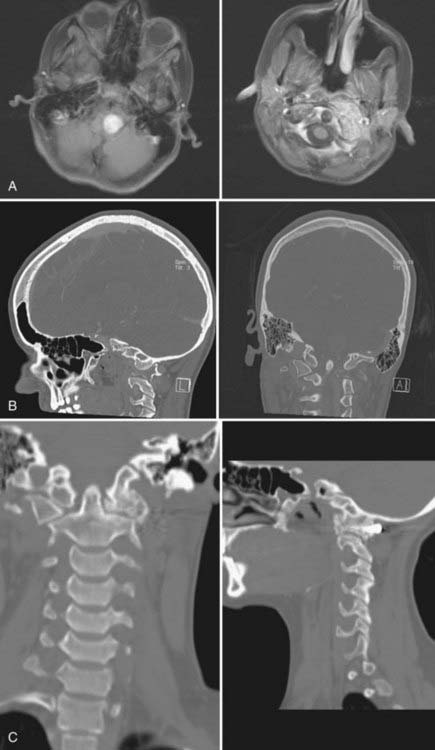
Calcium pyrophosphate dihydrate deposition (CPPD) is a rare cause of retro-odontoid mass lesions in elderly individuals.15 This condition is severely underdiagnosed. There is an increasing geriatric population that presents with neurological deficits secondary to a large mass at the ventral foramen magnum and C1. In our series, 48 patients presented with an average age of 75 years.76 The male-to-female ratio is equal. The duration of symptoms was 21 months, and pain was a major symptom in 90%. Paresthesias occurred in 44%, and cranial nerve deficits were seen in 44%. Neck pain occurred in 85% of individuals and paresthesias in 61%. Calcification of the mass or the transverse cruciate ligament was seen on CT in all patients. The mass may extend laterally into the joints between the occipital condyle and the lateral masses of C1 and C2.
A gross total resection was accomplished in every patient using the transoral-transpharyngeal route (Fig. 308-6). There were no recurrences, and improvement or resolution of preoperative neurological deficits occurred in 78% of individuals. A dorsal occipitoatlantoaxial fusion was necessary in 82%. We felt that fusion was not mandatory because concomitant ligamentous calcification was present and atlantoaxial joint ankylosis was seen in 18% of patients.77
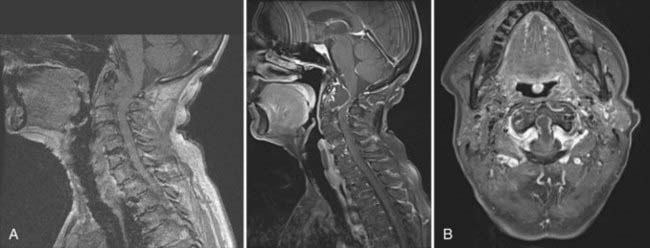
The diagnosis of pseudogout should be suspected before surgery and confirmed at frozen section. The recognition of abnormal crystals on birefringence is well documented.78 Unfortunately, these are older individuals who have significant comorbidities that must be taken into consideration before planning operative intervention.
Osteoid Osteomas and Osteoblastomas
These lesions are osteoblastic, with a propensity to involve the posterior elements of the cervical spine.41 Osteoblastomas and osteoid osteomas are differentiated from each other primarily on the basis of their size.1 Osteoid osteomas are more common in males and are usually smaller than 1 to 1.5 cm. They are limited to the cortical framework and have no paraspinal extension. Patients with osteoid osteomas usually present in the second or third decade of life.68,79,80 They commonly complain of neck pain that worsens at night and is relieved by salicylates.76 Radiographically, these lesions have a central circular nidus that appears within a radiolucent area surrounded by a sclerotic border.68 The lesion is well outlined by CT, and the bone scan is invariably positive.79,80
In contrast, osteoblastomas have been called giant osteoids or osteofibromas. These highly vascular lesions are predominantly lytic; they expand into the spinal canal and creep into the paravertebral space. Osteoblastomas tend to involve the pedicles and laminae as well as the spinous processes.73 The neoplasms expand the medullary bone and appear on radiographs as lytic lesions with expansion of the cortical margin. Less peripheral sclerosis is seen than with osteoid osteomas. Like osteoid osteomas, osteoblastomas are well localized on isotope bone scan.
The treatment of both osteoblastoma and osteoid osteoma is resection. Curettage and bone grafting of vertebral osteoblastomas have provided good long-term results.76 Complete excision is desirable but may not be possible at the craniovertebral junction (Fig. 308-7). George and coworkers, in their manuscript reporting on bone tumors of the craniovertebral junction, had encountered eight osteoid osteomas and three osteoblastomas.68 These had been located at the C1-2 level. The preoperative symptoms were pain and neck stiffness, and they improved or disappeared after surgical resection. They believed that CT-guided biopsies proved to be poor because an erroneous diagnosis was made on such biopsy, whereas the pathology at surgery changed to solitary plasmacytoma or fibromyosarcoma. In several other individuals being treated for abnormalities of the bone at the atlantoaxial level, CT-guided biopsies proved to be similarly erroneous until the final pathology was made at total excision.68 Radiation therapy may be considered in individuals who have undergone subtotal resection and bone grafting. Radiation alone may provide reconstitution of bone and preserve spinal stability if immobilization is adequate. A very small number of such lesions may undergo malignant transformation. Long-term follow-up of treated patients is mandatory.
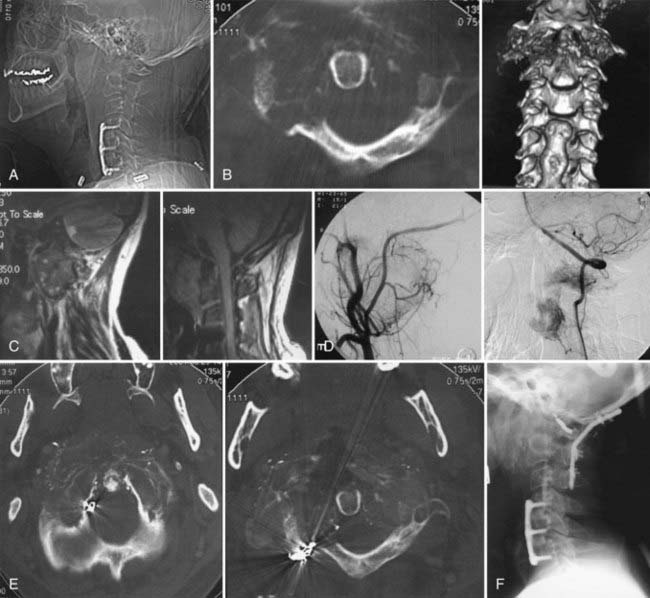
Aneurysmal Bone Cyst at the Craniovertebral Junction
These bone cysts are benign, highly vascular lesions that are expansile and most often occur in long bones, flat bones, and less often the spinal column. They can present as primary bone lesions in association with other bony lesions such as giant cell tumor, osteoblastoma, chondroid myxofibroma, fibrous dysplasia, and osteosarcoma.5 Lesions that involve the atlas vertebra make up about 1% of all spinal aneurysmal bone cysts. The treatment of choice for accessible lesions is en bloc excision or curettage.81,82 On CT, an expansile cystic mass with a thin rim of calcification is usually seen. Bony trabeculae within the cyst may be present. Unfortunately, this appearance can occur with osteoblastoma as well as giant cell tumor and bone. Fluid-filled levels identified on MRI, however, are characteristic of aneurysmal bone cyst. Although benign, aneurysmal bone cysts can grow rapidly and cause destruction of bone.
Histologically, aneurysmal bone cysts are composed of cavernous blood-filled spaces separated by bony trabeculae or osteoid tissue. The expansile nature of the lesion causes swelling, bone destruction, pain, and fractures.83–85
Superselective embolization of arterial feeders has been used as a primary modality of treatment in long bone and thoracic spine areas.86 The disadvantage of this is that treatment of aneurysmal bone cysts of the atlas includes the risk for embolizing the spinal cord and embolic complications. In addition, multiple embolization procedures are necessary to achieve clinical and radiographic healing of the cyst. Percutaneous intralesional injection of calcitonin and methylprednisolone as sclerosing agents into the aneurysmal bone cyst at the atlas has been reported.53
Aneurysmal bone cysts have been recorded as occurring after radiation therapy such as for pontine and brainstem gliomas. In my series, a 15-year-old girl was seen 18 months after radiation for a primary brainstem glioma.5 The brainstem tumor responded to the radiation as well as chemotherapy. However, significant neck pain with moderate degree of rotary luxation of C1 on C2 and secondary odontoid invagination prompted further evaluation and care (Fig. 308-8). She was treated with superselective embolization made in three sittings. She was followed with brace immobilization, and at 5 months, the CT scan showed reconstitution of the entire lateral mass of C1 as well as calcification of the soft tissues that had come out of the bony shell.
Intradural Tumors
Foramen Magnum Meningiomas
Meningiomas are common at the foramen magnum and account for 2% to 3% of all meningiomas.2,5,9,35,43 Females represent between 66% and 73% of cases. Typically, patients become symptomatic between 35 and 60 years old, although meningiomas occasionally occur at the foramen magnum in children. Foramen magnum meningiomas may occur in the setting of neurofibromatosis.6,87,88
Typically, the lesions are attached to the anterior rim of the foramen magnum and frequently invade the region of the entrance around the vertebral artery and the exit of the cervical nerve roots.5,51,89 These globoid and often fibrous lesions usually extend above and below the foramen magnum equally (Fig. 308-9). Occasionally, they show a marked predilection for either intracranial or intraspinal development. Many subtypes of meningioma have been identified at the foramen magnum, including meningothelial, transitional, fibrous, xanthomatous, clear cell, and lymphoplasmacytic-rich meningiomas.90,91
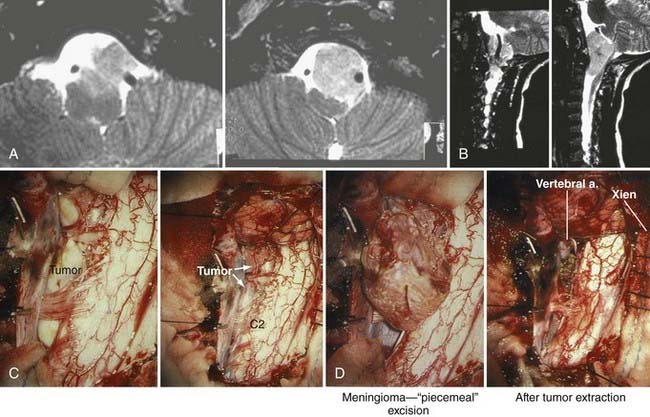
MRI provides excellent high-resolution information about the tumor, vascular encasement, its relationship to the brainstem, and CSF spaces. Typically, meningiomas are isointense on T1-weighted images and isointense to hypointense on T2-weighted images and enhance intensely after gadolinium administration.76 Magnetic resonance angiography provides critical arterial detail, including vertebral artery patency. It is used routinely as an alternative to conventional angiography.51 Magnetic resonance venography can be used to evaluate the vein of Labbé, sigmoid sinus, and dominance of the transverse sinus; thus, it can help plan the surgical approach in some patients.
Treatment
The mainstay of treatment for foramen magnum meningioma is surgical resection. Samii and colleagues89 reported complete removal in 63% of cases and subtotal removal in 30%, and their patients’ mean Karnofsky performance scores increased from 63 to 73. George and associates4 reported complete removal in 86% and subtotal removal in 11% of their patients. Clinical grade improved in 90% of patients, stabilized in 2.5%, and worsened in 7.5%—the latter corresponding to the three deaths in their series.4
In a recent review of the manuscript of Bassiouni and associates,43 George reports on his experience of more than 90 foramen magnum meningiomas operated on up to 2006. He believed that a retrocondylar route was sufficient in 95% of cases but did feel that vertebral artery mobilization was necessary. Bassiouni and associates, in 2006, reported on a series of 25 foramen magnum meningiomas approached through a posterolateral suboccipital retrocondylar route.43 The dural attachment of the meningioma was anterior in 36% and anterolateral in 64% of cases. The occipital condyle was not resected in these individuals. Tumors were said to be completely removed in 23 of 25, with about 40% incidence of postoperative morbidity. CSF leak occurred in 5 of 25 patients. Simpson grade 2 resection was achieved in 89.3%. The transient complication rate was 40%, with a permanent rate of 7.1%.
In 2005, we reported on our series of 41 patients with foramen magnum meningiomas, four cases of which occurred in the pediatric population.15 The average age at diagnosis was 50.1 years, and 78% were in females. The symptom duration was an average of 12.4 months. Pain was the major presenter in 68%, with motor deficit seen in 26%. Gross involvement of the vertebral artery occurred in 32% of patients. The surgical treatment consisted of posterolateral approach to foramen magnum and the clivus. In five individuals, a combined presigmoid and retrosigmoid craniotomy allowed for gross total removal. Gross total resection was accomplished in 87% of foramen magnum meningiomas. Partial resection was made in 10% of patients, and a repeat surgical procedure was required in 3% of patients. Surgery was combined with radiation therapy in 13% of individuals with a diagnosis of clear cell or angioblastic meningioma.5 However, surgical complications with a new deficit were seen in 32% of individuals, and a resolution of symptoms occurred in half of these individuals. A fusion procedure was required in two individuals after a posterolateral transcondylar approach. Tracheostomy was required in two individuals with new hypoglossal or vagal nerve dysfunction that was not present preoperatively.
Despite these difficulties, Arnautovic and colleagues92 reported 67% gross total resections, 11% near-total resections, and 22% subtotal resections in patients harboring ventral foramen magnum meningiomas. These results are similar to the surgical results for all foramen magnum meningiomas. Further, Karnofsky performance scores increased postoperatively in 15 patients. Statistical analysis demonstrated that gross total or near-total resection and a higher preoperative Karnofsky score indicated a significantly improved outcome. These series demonstrate that complete resection of foramen meningiomas can be obtained in most cases, particularly during the first resection, and that resection can improve patients’ postoperative performance scores.
Postoperative lower cranial palsies are the most frequent complication. These palsies are significant because they are associated with longer periods of hospitalization.89,92 Lower cranial nerve palsies resulting in aspiration pneumonia and death accounted for the mortality rates in several series. Multiple regression analysis indicated that recurrent tumor, arachnoid scarring, craniovertebral meningioma, and no preoperative cranial nerve deficits were predictors of postoperative aspiration. Aggressive, comprehensive treatment is recommended after these deficits are discovered, including speech therapy, possible vocal cord medialization or injection, tracheostomy, or gastric tube placement.
Severe preoperative deficits and poor Karnofsky performance scores are also predictors of postoperative morbidity and mortality. Weakness and poor mobility predispose patients to pneumonia, deep venous thrombosis, and pulmonary embolism, which may account for up to 50% of postoperative mortality.4,89,92
Other postoperative complications include CSF leak, meningitis, and hydrocephalus that may require insertion of a ventriculoperitoneal shunt. Other cranial nerve palsies can occur, but most resolve. Occipitocervical instability can occur, but this complication can be eliminated with judicious resection of no more than one third to one half of the occipital condyle. In recent series, the overall morbidity rate has been about 30%, and perioperative mortality rates have ranged from 0% to 7.5%. Incomplete tumor removal and aggressive recurrence of invasive meningioma have responded to conventional radiation.93–95
Muthukumar and colleagues reported their experience treating five foramen magnum meningiomas using stereotactic radiosurgery.96 Patients with tumors less than 35 mm in diameter and with a reasonable performance status were selected for treatment. At a median follow-up of 36 months, no tumor growth had occurred, and cranial nerve deficits had not worsened, but the series was small, and follow-up data were limited. Furthermore, their population represented ideal surgical candidates, whose results were likely to be more favorable than those of nonideal patients for whom nonsurgical treatment would be preferable. Much more experience in patient selection is required before stereotactic radiosurgery can be considered a primary treatment for foramen magnum meningiomas. Others have performed surgical resection with low thresholds for leaving adherent tumor and used postoperative Gamma Knife radiosurgery to treat the residual tumor.97 Therefore, stereotactic radiosurgery may represent a valuable adjunctive therapy.
Intradural Extramedullary Tumors at C1 and C2
The most common extramedullary intradural spinal tumors are meningiomas, neurofibromas, and schwannomas.98,99 Together, these tumors represent about 55% of spinal tumors.2,4,77,88,92 Other intradural tumors, such as hemangioblastoma, dermoids, epidermoids, neurenteric cysts, and mixed tumors, are uncommon.1,11,100–102 Many of these lesions are benign and resectable, and the outlook after surgical therapy is excellent.
Neurofibromas produce symptoms in patients between 20 and 60 years old.1,2,11 Patients with neurofibromatosis type 1 tend to develop symptoms at a younger age5,10 and have multiple or bilateral tumors. Neurinomas occur more frequently at C2 than at C1. The median time from onset of symptoms to diagnosis is 24 months. Although neurinomas are associated with nerve roots, symptoms tend to manifest distally with weakness and poor coordination or, less commonly, in a radicular pattern.98,103,104 Clinical signs also mirror those of foramen magnum meningiomas; however, hypesthesia in the second cervical division is an important localizing sign.
Although findings on plain radiography may be normal or demonstrate cervical spondylosis, the presence of an enlarged intervertebral foramen is highly suggestive of a nerve root tumor. Neurinomas are usually isointense to hypointense on T1-weighted MRI and hyperintense on T2-weighted MRI (Fig. 308-10).99 Intratumoral hemorrhage and cyst formation can affect the postcontrast appearance of neurinomas (homogeneous, heterogeneous, and even ring enhancement). If necessary, magnetic resonance angiography can evaluate the vertebrobasilar arterial tree. CT provides detailed bony definition. If MRI is contraindicated, CT myelography may improve intradural visualization of the tumor.
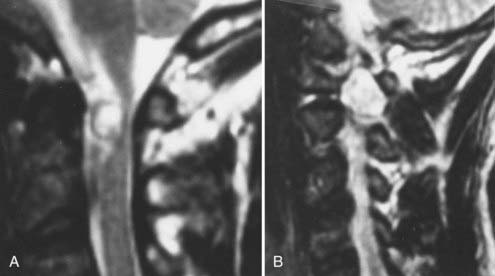
Neurenteric cysts are a developmental abnormality resulting in a cystic endodermal structure that can impinge on the spinal cord. Although most common in the cervical and thoracic regions, neurenteric cysts rarely appear in the region of the foramen magnum, typically in an anterior location. Their clinical presentation is similar to that of other foramen magnum lesions, with the exception that the cysts can lead to repeated episodes of meningitis.102 Plain radiographs can demonstrate vertebral segmentation failure. CT is useful for fully delineating bony abnormalities. MRI demonstrates a cystic structure with T1- and T2-weighted imaging characteristics similar to those of proteinaceous CSF (Fig. 308-11).101
Treatment
Preoperative cardiopulmonary assessment is essential, as is discussion with the patient and family about the possibility of prolonged postoperative tracheal intubation or tracheostomy. Patients with large lesions within the spinal canal require preoperative evaluation of cervical motion to assess stability and determine whether neurological deficits develop during flexion due to compression of the brainstem by the tumor.1,105 This evaluation allows positioning under general anesthesia.
Although posterior approaches are most commonly used to resect neurinomas, use of the anterolateral4 and transoral exposures has been reported.26 In rare instances, schwannomas of the craniovertebral junction may be ventral to the dentate ligament; in such cases, section of the ligament or even of the spinal accessory nerve may be required. Dumbbell neurofibromas can cause craniovertebral instability, and this possibility must be kept in mind at the time of reconstruction. The surgical outcome of neurinoma resection is excellent. Patients with myelopathy and quadriparesis typically improve significantly, or their symptoms resolve completely. Resection rarely causes new radicular symptoms and usually improves existing radicular symptoms.
Al-Mefty O, Borba LAB. Skull base chordomas: a management challenge. J Neurosurg. 1997;86:182-189.
Arnautovic KI, Al-Mefty O, Husain M. Ventral foramen magnum meningiomas. J Neurosurg (Spine 1). 2000;92:71-80.
Cocke EWJr, Robertson JH, Robertson JT, et al. The extended maxillotomy and subtotal maxillectomy for excision of skull base tumors. Arch Otolaryngol Head Neck Surg. 1990;116:92-104.
Coffin CM, Swanson PE, Wick MR, Dehner LP. Chordoma in childhood and adolescence. A clinicopathologic analysis of 12 cases. Arch Pathol Lab Med. 1993;117:927-933.
Colli BO, Al-Mefty O. Chordomas of the craniocervical junction: follow-up review and prognostic factors. J Neurosurg. 2001;95:933-943.
Forsyth PA, Cascino TL, Shaw EG, et al. Intracranial chordomas: a clinicopathological and prognostic study of 51 cases. J Neurosurg. 1993;78:741-747.
Frank G, Sciarretta V, Calbucci F, et al. The endoscopic transnasal transsphenoidal approach for the treatment of cranial base chordomas and chondrosarcomas. Neurosurgery. 2006;59:ONS50-ONS57.
George B, Lot G, Boissonnet H. Meningioma of the foramen magnum: a series of 40 cases. Surg Neurol. 1997;47:364-370.
Hug EB, Loredo LN, Slater JD, et al. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91:432-439.
Lot G, George B. Cervical neuromas with extradural components: surgical management in a series of 57 patients. Neurosurgery. 1997;41:813-822.
Menezes AH. Craniovertebral junction neoplasms in the pediatric population. Childs Nerv Syst. 2008;24:1173-1186.
Menezes AH, Traynelis VC, Fenoy AJ, et al. Honored Guest Presentation. Surgery at the Crossroads: craniocervical neoplasms. Clin Neurosurg. 2005;52:218-228.
Mohit AA, Eskridge J, Ellenbogen R, Shaffrey C. Aneurysmal bone cyst of the atlas: successful treatment through selective arterial embolization: Case report. Neurosurgery. 2004;55:E1001-E1005.
Passacantilli E, Santoro A, Pichierri A, et al. Anterolateral approach to the craniocervical junction. J Neurosurg Spine. 2005;3:123-128.
Sen CN, Sekhar LN. An extreme lateral approach to intradural lesions of the cervical spine and foramen magnum. Neurosurgery. 1990;27:197-206.
Taylor AR, Byrnes DP. Foramen magnum and high cervical cord compression. Brain. 1974;97:473-480.
Uttley D, Moore A, Archer DJ. Surgical management of midline skull base tumors: a new approach. J Neurosurg. 1989;71:705-710.
Zunkeler B, Schelper R, Menezes AH. Periodontoid calcium pyrophosphate dihydrate deposition disease. “Pseudogout” mass lesions of the craniocervical junction. J Neurosurg. 1996;85:803-809.
1 Menezes AH, Traynelis VC. Tumors of the craniovertebral junction. In: Youmans J, editor. Neurological Surgery. Philadelphia: Saunders; 1995:3041-3072.
2 Meyer FB, Ebersold MJ, Reese DF. Benign tumors of the foramen magnum. J Neurosurg. 1984;61:136-142.
3 Tsai EC, Santoreneos S, Rutka JT. Tumors of the skull base in children: review of tumor types and management strategies. Neurosurg Focus. 2002;12:1-13.
4 George B, Lot G, Boissonnet H. Meningioma of the foramen magnum: A series of 40 cases. Surg Neurol. 1997;47:364-370.
5 Menezes AH. Craniovertebral junction neoplasms in the pediatric population. Childs Nerv Syst. 2008;24:1173-1186.
6 Howe JR, Taren JA. Foramen magnum tumors. Pitfalls in diagnosis. JAMA. 1973;225:1061-1066.
7 Elsberg CA, Strauss I. Tumors of the spinal cord which project into the posterior cranial fossa. Arch Neurol Psychiatry. 1929;21:261-273.
8 Bassiouni H, Ntoukas V, Asgari S, et al. Foramen magnum meningiomas: clinical outcome after microsurgical resection via a posterolateral suboccipital retrocondylar approach. Neurosurgery. 2006;59:1177-1185.
9 Al-Mefty O, Borba LAB. Skull base chordomas: a management challenge. J Neurosurg. 1997;86:182-189.
10 Yasuoka S, Okazaki H, Daube JR, et al. Foramen magnum tumors. Analysis of 57 cases of benign extramedullary tumors. J Neurosurg. 1978;49:828-838.
11 Leal Filho MB, Borges G, Ferreira A, et al. Schwannoma of the craniocervical junction: surgical approach of two cases. Arq Neuropsiquiatr. 2003;61:639-641.
12 Rousselin B, Helenon O, Zingraff J, et al. Pseudotumor of the craniocervical junction during long-term hemodialysis. Arthritis Rheum. 1990;33:1567-1573.
13 Beatty RA. Cold dysesthesia: a symptom of extramedullary tumors of the spinal cord. J Neurosurg. 1970;33:75-78.
14 Menezes AH. Tumors of the craniocervical junction. In: Menezes AH, Sonntag VKH, editors. Principles of Spinal Surgery. New York: McGraw-Hill; 1996:1335-1353.
15 Menezes AH, Traynelis VC, Fenoy AJ, et al. Honored Guest Presentation. Surgery at the crossroads: craniocervical Neoplasms. Clin Neurosurg. 2005;52:218-228.
16 Taylor AR, Byrnes DP. Foramen magnum and high cervical cord compression. Brain. 1974;97:473-480.
17 Cohen L, McCrae D. Tumors in the region of the foramen magnum. J Neurosurg. 1962;19:462-469.
18 Bell HS. Paralysis of both arms from injury of the upper portion of the pyramidal decussation: “cruciate paralysis.”. J Neurosurg. 1970;33:376-380.
19 Shin H, Barrenechea IJ, Lesser J, et al. Occipitocervical fusion after resection of craniovertebral junction tumors. J Neurosurg Spine. 2006;4:137-144.
20 Glasauer FE, Egnatchick JE. Restless legs syndrome: an unusual cause for a perplexing syndrome. Spinal Cord. 1999;37:862-865.
21 Forbes HJ, Allen PW, Waller CS, et al. Spinal cord monitoring in scoliosis surgery. Experience with 1168 cases. J Bone Joint Surg Br. 1991;73:487-491.
22 May DM, Jones SJ, Crockard HA. Somatosensory evoked potential monitoring in cervical surgery: identification of pre- and intraoperative risk factors associated with neurological deterioration. J Neurosurg. 1996;85:566-573.
23 Derome PJ. Surgical management of tumours invading the skull base. Can J Neurol Sci. 1985;12:345-347.
24 Sherk HH, Nicholson JT, Nixon JE. Vertebra plana and eosinophilic granuloma of the cervical spine in children. Spine. 1978;3:116-121.
25 Menezes AH, Gantz BJ, Traynelis VC, et al. Cranial base chordomas. Clin Neurosurg. 1998;44:491-509.
26 Crockard HA, Bradford R. Transoral transclival removal of a schwannoma anterior to the craniocervical junction. Case report. J Neurosurg. 1985;62:293-295.
27 Seifert V, Laszig R. Transoral transpalatal removal of a giant premesencephalic clivus chordoma. Acta Neurochir (Wien). 1991;112:141-146.
28 Cocke EWJr, Robertson JH, Robertson JT, et al. The extended maxillotomy and subtotal maxillectomy for excision of skull base tumors. Arch Otolaryngol Head Neck Surg. 1990;116:92-104.
29 Hall JE, Denis F, Murray J. Exposure of the upper cervical spine for spinal decompression by a mandible and tongue splitting approach. J Bone Joint Surg Am. 1977;59:121-123.
30 Moore LJ, Schwartz HC. Median labiomandibular glossotomy for access to the cervical spine. J Oral Maxillofac Surg. 1985;43:909-912.
31 DeMonte F, Diaz EJr, Callender D, Suk I. Transmandibular, circumglossal, retropharyngeal approach for chordomas of the clivus and upper cervical spine. Neurosurg Focus. 2001;10:1-5.
32 Kaibara T, Hurlbert RJ, Sutherland GR. Transoral resection of axial lesions augmented by intraoperative magnetic resonance imaging. J Neurosurg (Spine). 2001;95:239-242.
33 Sen CN, Sekhar LN. An extreme lateral approach to intradural lesions of the cervical spine and foramen magnum. Neurosurgery. 1990;27:197-206.
34 Siwanuwatn R, Deshmukh P, Figueiredo EG, et al. Quantitative analysis of the working area and angle of attack for the retrosigmoid, combined petrosal, and transcochlear approaches to the petroclival region. J Neurosurg. 2006;104:137-142.
35 Passacantilli E, Santoro A, Pichierri A, et al. Anterolateral approach to the craniocervical junction. J Neurosurg Spine. 2005;3:123-128.
36 Couldwell WT, Weiss MH, Rabb C, et al. Variations on the standard transsphenoidal approach to the sellar region, with emphasis on the extended approaches and parasellar approaches: surgical experience in 105 cases. Neurosurgery. 2004;55:539-547.
37 Maira G, Pallini R, Anile C, et al. Surgical treatment of clival chordomas: the transsphenoidal approach revisited. J Neurosurg. 1996;85:784-792.
38 Fortes FSG, Carrau RL, Snyderman CH, et al. The posterior pedicle inferior turbinate flap: a new vascularized flap for skull base reconstruction. Laryngoscope. 2007;117:1329-1332.
39 Janecka IP, Sen CN, Sekhar LN, et al. Facial translocation: a new approach to the cranial base. Otolaryngol Head Neck Surg. 1990;103:413-419.
40 Uttley D, Moore A, Archer DJ. Surgical management of midline skull base tumors: a new approach. J Neurosurg. 1989;71:705-710.
41 Piper JG, Menezes AH. Management of strategies for tumors of the axis vertebra. J Neurosurg. 1996;84:543-551.
42 Hakuba A, Nishimura S, Jang BJ. A combined retroauricular and preauricular transpetrosal-transtentorial approach to clivus meningiomas. Surg Neurol. 1988;30:108-116.
43 Bassiouni H, Ntoukas V, Asgari S, et al. Foramen magnum meningiomas: clinical outcome after microsurgical resection via a posterolateral suboccipital retrocondylar approach. Neurosurgery. 2006;59:1177-1187.
44 Kassam AB, Gardner P, Snyderman C, et al. Expanded endonasal approach. Fully endoscopic, completely transnasal approach to the middle third of the clivus, petrous bone, middle cranial fossa, and infratemporal fossa. Neurosurg Focus. 2005;19:E6.
45 Frank G, Sciarretta V, Calbucci F, et al. The endoscopic transnasal transsphenoidal approach for the treatment of cranial base chordomas and chondrosarcomas. Neurosurgery. 2006;59:ONS50-ONS57.
46 Dehdashti AR, Karabatsou K, Ganna A, et al. Expanded endoscopic endonasal approach for treatment of clival chordomas: early results in 12 patients. Neurosurgery. 2008;63:299-309.
47 Gay E, Sekhar LN, Rubinstein E, et al. Chordomas and chondrosarcomas of the cranial base: results and follow-up of 60 patients. Neurosurgery. 1995;36:887-897.
48 Heffelfinger MJ, Dahlin DC, MacCarty CS, et al. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973;32:410-420.
49 Bottles K, Beckstead JH. Enzyme histochemical characterization of chordomas. Am J Surg Pathol. 1984;8:443-447.
50 Rich TA, Schiller A, Suit HD, et al. Clinical and pathologic review of 48 cases of chordoma. Cancer. 1985;56:182-187.
51 Colli BO, Al-Mefty O. Chordomas of the craniocervical junction: follow-up review and prognostic factors. J Neurosurg. 2001;95:933-943.
52 Forsyth PA, Cascino TL, Shaw EG, et al. Intracranial chordomas: a clinicopathological and prognostic study of 51 cases. J Neurosurg. 1993;78:741-747.
53 Watkins L, Khudodas ES, Kaleoglu M, et al. Skull base chordomas: a review of 38 patients, 1958-1988. Br J Neurosurg. 1993;7:241-248.
54 Crockard HA, Steel T, Plowman N, et al. A multidisciplinary team approach to skull base chordomas. J Neurosurg. 2001;95:175-183.
55 Samii A, Gerganov VM, Herold C, et al. Chordomas of the skull base: surgical management and outcome. J Neurosurg. 2007;107:319-324.
56 Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE. Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol. 2006;30:811-818.
57 Sibley RK, Day DL, Dehner LP, Trueworthy RC. Metastasizing chordoma in early childhood: a pathological and immunohistochemical study with review of the literature. Pediatr Pathol. 1987;7:287-301.
58 Coffin CM, Swanson PE, Wick MR, Dehner LP. Chordoma in childhood and adolescence. A clinicopathologic analysis of 12 cases. Arch Pathol Lab Med. 1993;117:927-933.
59 Borba LA, Al-Mefty O, Mrak RE, Suen J. Cranial chordomas in children and adolescents. J Neurosurg. 1996;84:584-591.
60 Hug EB, Loredo LN, Slater JD, et al. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91:432-439.
61 Amendola BE, Amendola MA, Oliver E, et al. Chordoma: role of radiation therapy. Radiology. 1986;158:839-843.
62 Austin JP, Urie MM, Cardenosa G, et al. Probable causes of recurrence in patients with chordoma and chondrosarcoma of the base of skull and cervical spine. Int J Radiat Oncol Biol Phys. 1993;25:439-444.
63 Berson AM, Castro JR, Petti P, et al. Charged particle irradiation of chordoma and chondrosarcoma of the base of the skull and cervical spine. The Lawrence Berkeley Laboratory experience. Int J Radiat Oncol Biol Phys. 1988;15:559-565.
64 Castro JR, Lindstadt DE, Bahary JP, et al. Experience in charged particle irradiation of tumors of the skull base: 1977-92. Int J Radiat Oncol Biol Phys. 1994;29:647-655.
65 Kondziolka D, Lunsford LD, Flickinger JC. The role of radiosurgery in the management of chordoma and chondrosarcoma of the cranial base. Neurosurgery. 1991;29:38-46.
66 Corwin J, Lindberg RD. Solitary plasmacytoma of bone versus extramedullary plasmacytoma and their relationship to multiple myeloma. Cancer. 1979;43:1007-1013.
67 Dimopoulos MA, Goldstein J, Fuller L, et al. Curability of solitary bone plasmacytoma. J Clin Oncol. 1992;10:587-590.
68 George B, Archilli M, Cornelius JF. Bone tumors at the cranio-cervical junction. Surgical management and results from a series of 41 cases. Acta Neurochir (Wein). 2006;148:741-749.
69 McLain RF, Weinstein JN. Solitary plasmacytomas of the spine: a review of 84 cases. J Spinal Disord. 1989;2:69-74.
70 Alexanian R, Dimopoulos M. The treatment of multiple myeloma. N Engl J Med. 1994;17:448-489.
71 Miyachi S, Negoro M, Sato K, et al. Myeloma manifesting as a large jugular tumor. Case report. Neurosurgery. 1990;27:971-977.
72 Osenbach RK, Youngblood LA, Menezes AH. Atlantoaxial instability secondary to solitary eosinophilic granuloma of C2 in a 12-year-old girl. J Spinal Disord. 1990;3:408-412.
73 Bohlman HH, Sachs BL, Carter JR, et al. Primary neoplasms of the cervical spine. J Bone Joint Surg Am. 1986;68:483-494.
74 Green NE, Robertson WWJr, Kilroy AW. Eosinophilic granuloma of the spine with associated neural deficit. Report of three cases. J Bone Joint Surg Am. 1980;62:1198-1202.
75 Sanchez RL, Llovet J, Morena A. Symptomatic eosinophilic granuloma of the spine. Report of two cases and review of the literature. Orthopaedics. 1984;7:1721-1726.
76 Menezes AH, Traynelis VC. Tumors of the craniovertebral junction. In: Youmans J, editor. Neurological Surgery. 4th ed. Philadelphia: Saunders; 1995:3041-3072.
77 Fenoy AJ, Menezes AH, Donovan KA, Kralik SF. Calcium pyrophosphate dihydrate crystal deposition in the craniovertebral junction. J Neurosurg Spine. 2008;8:22-29.
78 Zunkeler B, Schelper R, Menezes AH. Periodontoid calcium pyrophosphate dihydrate deposition disease. “Pseudogout” mass lesions of the craniocervical junction. J Neurosurg. 1996;85:803-809.
79 DiLorenzo N, Palatinsky E, Bardella L, et al. Benign osteoblastoma of the clivus removed by a transoral approach: case report. Neurosurgery. 1987;20:52-55.
80 Marsh BW, Bonfiglio M, Brady LP, et al. Benign osteoblastoma. Range of manifestations. J Bone Joint Surg Am. 1975;57:1-9.
81 Anderson BJ, Goldhagen P, Cahill DW. Aneurysmal bone cyst of the odontoid process: case report. Neurosurgery. 1991;28:592-594.
82 Bongioanni F, Assadurian E, Polivka M, George B. Aneurysmal bone cyst of the atlas: operative removal through an anterolateral approach. A case report. J Bone Joint Surg Am. 1996;78:1574-1577.
83 Leithner A, Windhager R, Lang S, et al. Aneurysmal bone cyst: a population-based epidemiologic study and literature review. Clin Orthop Relat Res. 1999;363:176-179.
84 Szendroi M, Cser I, Konya A, Renyi-Vamos A. Aneurysmal bone cyst: a review of 52 primary and 16 secondary cases. Arch Orthop Trauma Surg. 1992;111:318-322.
85 Vergel De Dios AM, Bond JR, Shives TC, et al. Aneurysmal bone cyst: a clinicopathologic study of 238 cases. Cancer. 1992;69:2921-2931.
86 Mohit AA, Eskridge J, Ellenbogen R, Shaffrey C. Aneurysmal bone cyst of the atlas: Successful treatment through selective arterial embolization: case report. Neurosurgery. 2004;55:E1001-E1005.
87 Harada H, Kumon Y, Hatta N, et al. Neurofibromatosis type 2 with multiple primary brain tumors in monozygotic twins. Surg Neurol. 1999;51:528-535.
88 Barber DB, Quattrone BE, Lomba ME, Able AC. Neurofibromatosis: an unusual cause of cervical myopathy. J Spinal Cord Med. 1998;21:148-150.
89 Samii M, Klekamp J, Carvalho G. Surgical results for meningiomas of the craniocervical junction. Neurosurgery. 1996;39:1086-1095.
90 Castellano F, Ruggiero G. Meningiomas of the posterior fossa. Acta Radiol Suppl (Stockh). 1953;104:1-177.
91 Yamaki T, Ikeda T, Sakamoto Y, et al. Lymphoplasmacyte-rich meningioma with clinical resemblance to inflammatory pseudotumor. J Neurosurg. 1997;86:898-904.
92 Arnautovic KI, Al-Mefty O, Husain M. Ventral foramen magnum meningiomas. J Neurosurg. 2000;92(Spine 1):71-80.
93 Forbes AR, Goldberg ID. Radiation therapy in the treatment of meningioma: The Joint Center of Radiation Therapy experience 1970 to 1982. J Clin Oncol. 1984;2:1139-1143.
94 Mirimanoff RO, Dosoretz DE, Linggood RM, et al. Meningioma: analysis of recurrence and progression following neurosurgical resection. J Neurosurg. 1985;62:18-24.
95 Petty AM, Kun LE, Meyer GA. Radiation therapy for incompletely resected meningiomas. J Neurosurg. 1985;62:502-507.
96 Muthukumar N, Kondziolka D, Lunsford LD, Flickinger JC. Stereotactic radiosurgery for anterior foramen magnum meningiomas. Surg Neurol. 1999;51:268-273.
97 Liu C, Node Y, Teramoto A. Treatment of posterior skull base tumors. J Nippon Med Sch. 1998;65:316-319.
98 Bucci MN, McGillicuddy JE, Taren JA, Hoff JT. Management of anteriorly located C1-C2 neurofibromata. Surg Neurol. 1990;33:15-18.
99 Hu HP, Huang QL. Signal intensity correlation of MRI with pathological findings in spinal neurinomas. Neuroradiology. 1992;34:98-102.
100 Breeze RE, Nichols P, Segal JT. Intradural epithelial cyst at the craniovertebral junction. J Neurosurg. 1990;73:788-791.
101 Koksel T, Revesz T, Crockard HA. Craniospinal neurenteric cyst. Br J Neurosurg. 1990;4:425-428.
102 Menezes AH, Ryken TC. Craniocervical intradural neurenteric cysts. Pediatr Neurosurg. 1995;22:88-95.
103 Ohba S, Akiyama T, Kanai R, et al. Endodermal cyst of the cranio-cervical junction. Acta Neurochir (Wein). 2008;150:257-263.
104 Lot G, George B. Cervical neuromas with extradural components: surgical management in a series of 57 patients. Neurosurgery. 1997;41:813-822.
105 Stein BM. Spinal intradural tumors. In: Wilkins RE, Rengachary S, editors. Neurosurgery. New York: McGraw-Hill; 1985:1048-1061.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 308 Tumors of the Craniovertebral Junction
The craniovertebral junction is a biomechanical and anatomic unit that comprises the clivus, foramen magnum, and upper two cervical vertebrae. The neoplasms that arise within the structures are osseous in nature or extensions from the soft tissue that surround the craniovertebral junction, or they are neoplasms that arise from the neural structures contained within the bony anatomy.1–4 The diagnosis of such lesions has been greatly facilitated by modern neurodiagnostic imaging.
There is no single symptom or neurological finding that is pathognomonic for a lesion in this location.5,6 Because of the generous size of the subarachnoid spaces at the cervicomedullary junction, symptoms arise only after a lesion has achieved large proportions. These patients have a fluctuating neurological course, and an erroneous diagnosis is common owing to the anatomic complexities of the decussation of the sensory and motor tracts.1,6
The first systematic evaluation of foramen magnum tumors was performed by Elsberg and Strauss.7 Several authors have since reported extra-axial lesions affecting the region, such as meningiomas and neurinomas.8–12 Osseous neoplastic involvement of the craniovertebral junction may be due to chordoma, chondrosarcoma, plasmacytoma, osteoblastoma, fibrous dysplasia, metastatic tumor, and giant cell tumor. Table 308-1 summarizes my experience.
Common Clinical Manifestations of Craniovertebral Junction Tumors
Tumors of the craniovertebral junction, whether extracranial with secondary involvement of the intracranial and intraspinal structures or primarily intracranial with secondary extension into the spinal canal, have characteristics that reflect compression of neighboring structures or traction. They also may have distal effects such as hydrocephalus, syringohydromyelia, and vascular compromise.9,13,14 Chordoma often involves the cranial base and upper cervical spine extensively and may be associated with only minimal complaints of headache and neck pain for several years. Unfortunately, this hiatus is followed by a rapid progression of brainstem and cervical spinal cord dysfunction that brings the lesion to light. In the report of Meyer and coworkers, the time from the onset of symptoms to the diagnosis of extramedullary tumor at the foramen magnum was 2.5 years.2
The clinical presentation of craniovertebral junction tumors can be divided into intracranial lesions, “straddle lesions,” and those affecting the high cervical spinal cord.7 The effects of vascular compromise and alterations of cerebrospinal fluid (CSF) circulation add to the constellation of symptoms and signs.
Patients with intracranial lesions present with involvement of the lower cranial nerves, brainstem dysfunction, and occasionally cerebellar symptoms. Patients with straddle lesions have a paucity of cranial nerve dysfunction and a predominance of high cervical myelopathy. High cervical lesions do not produce cranial nerve and cerebellar signs, except for involvement of the spinal accessory nerve and sometimes the descending tracts of the trigeminal nerve and the lower decussations of the motor and sensory tracks.15
The most common presentation is pain referred to the second cervical dermatome.1 The head is held flexed, and the condition may resemble torticollis. The pain is described as an aching sensation that is aggravated by neck and head motion and referred to the suboccipital region. Unfortunately, the symptom of pain alone may predate other clinical findings for many years. Paresthesias or dysesthesias of the face, hands, and limbs are frequently reported. An abnormal cold sensation of the lower extremities was described by Elsberg and Strauss7 and by Beatty13 as being pathognomonic of lesions of the high cervical cord. Most frequently, pain and temperature sensation is affected, followed by loss of joint sensation. This finding is seen in the upper extremities and may then proceed clockwise around the limbs. A suspended sensory loss with patches of preservation of sensation in the upper extremities may confuse the presentation. “Dissociated” sensory loss has been described in about one fourth of extracranial lesions of the cervicomedullary junction, even though this finding has been considered to reflect an intramedullary process.2
Spastic weakness of the extremities is a prominent feature in patients with tumors in this region. The weakness may begin in the ipsilateral limb and progress to the lower limb of the same side, followed by weakness of the contralateral lower limb; finally, weakness becomes apparent in the contralateral upper limb. This distinct progression of motor symptoms is an important characteristic of lesions of the cervicomedullary junction.15 Localized wasting of the intrinsic muscles of the hand may develop ipsilateral to the lesion. Taylor and Byrnes postulate venous stagnation of the anterior horn cells and the lower cervical cord as a result of decreased venous drainage, which typically occurs rostral to the lower portion of the cervical spinal cord.16 Other proposed mechanisms include anterior spinal artery compression, hydromyelia secondary to CSF obstruction, venous obstruction with spinal cord edema, and spinal cord rotation with contralateral traction.
A tumor at the foramen magnum may produce a mixture of upper motor neuron findings in the upper and lower extremities. This pattern reflects the pyramidal decussation that begins just below the obex and ends near the uppermost cervical spinal cord. The more medial fibers of the pyramidal tract carry impulses to the upper extremities and cross superior to the lateral fibers that serve the lower extremities. Similarly, the sensory decussation of the medial lemniscus may produce a varied pattern of sensory abnormalities.17 Thus, a tumor situated at the ventral aspect of the cervicomedullary junction can cause sensory aberrations in the lower extremities first. The syndrome of cruciate paralysis has been associated with trauma as well as tumors with basilar invagination.18
Transient symptoms may be due to both vascular changes and instability at the craniovertebral junction.1,19 Such symptoms can manifest as paralysis, paresthesias, drop attacks, and vertebrobasilar syndromes such as migraine and visual loss in the homonymous visual fields. Cranial nerve palsies may be the result of nuclear involvement in the brainstem, traction, compression of the subarachnoid segments, or interosseous disease.1,15 The most common cranial nerves affected are the vagus, glossopharyngeal, and hypoglossal. Their involvement leads to dysphagia, slurred speech, and repeated episodes of aspiration into the tracheobronchial tree, resulting in pneumonia and weight loss. Tumors of the upper cervical canal can present with involvement of the spinal root of the accessory nerve, manifesting as torticollis and weakness of the trapezius and sternocleidomastoid muscles. About 15% to 20% of patients develop tinnitus, vertigo, and hearing loss related to involvement of the vestibulocochlear nerve.15
The differential diagnoses considered most often by Meyer and coworkers in their initial evaluation of 102 documented cases of benign extramedullary tumors of the foramen magnum included cervical spondylosis (25%), multiple sclerosis (18%), syringomyelia (17%), intramedullary tumor (15%), Chiari’s malformation (5.5%), and carpal tunnel syndrome (5.5%).2 Other erroneous diagnoses in patients with lesions at the craniovertebral junction are intramedullary tumors of the brainstem and upper cervical cord, amyotrophic lateral sclerosis, and subacute combined degeneration. Cervical spondylosis may be difficult to differentiate when it coexists. The presentation of restless legs syndrome in patients with craniovertebral compressive pathology has been well documented by Glasauer and Egnatchick.20
Neurodiagnostic Imaging
The complex anatomy and pathology of tumors in this region demand precise definition and depiction of the tumor’s extent and its relationship to the vital structures of the brainstem and spinal cord, lower cranial nerves, and vascular structures. Thus, complementary multimodality imaging includes plain radiography, magnetic resonance imaging (MRI), magnetic resonance angiography, computed tomography (CT), and three-dimensional CT angiography. The sensitivity of MRI is greatly enhanced by the addition of intravenous gadolinium and by performing separate magnetic resonance venography and angiography.5
Cerebral angiography is useful in understanding the dynamics of collateral circulation and tumor vascularity. Temporary balloon occlusion is a means of assessing a patient’s tolerance of vascular occlusion of the carotid or vertebral circulation before surgery.15 Such information is especially useful when lesions are encased by tumor. These two tests provide information about the resectability of difficult lesions.
Electrophysiologic Tests
Intraoperatively, we monitor somatosensory brainstem evoked potentials and perform facial electromyography when lesions extend to the cerebellopontine angle and when using the preauricular infratemporal fossa approach.15,21,22 The intraoperative evaluation of cranial nerve IX and cranial nerve X function is accomplished by placing electrodes in the soft palate and against the true vocal cords, incorporating these latter electrodes in the endotracheal tube for the anesthetic. Hypoglossal nerve electromyography supplements the evaluation with an electrode placed directly into the tongue. Brainstem monitoring still yields a significant number of false-negative and false-positive results, but improved techniques make these adjuncts useful in the intraoperative assessment of the function of the cervicomedullary junction.
Surgical Approaches and Decision Making
The goal of treating benign osseous pathology differs from that of treating malignant disease, in which complete excision is the objective. Benign lesions create a space among the neurovascular structures, thereby allowing surgical debulking and resection “from within.” Malignant disease, however, requires a much more radical resection with clean margins. In most instances, benign lesions such as chordomas are radioresistant; hence, gross total resection should be the aim. Craniovertebral stability, both before and after operative intervention, must be considered in the development of approaches.14,19,23 Thus, the factors that influence the specific treatment of tumors arising in this region are (1) mechanism of compression and direction of encroachment, (2) whether the lesion is benign or malignant, (3) whether the lesion has an associated vascular or intramedullary component (e.g., syringomyelia), (4) craniospinal stability, and (5) patient age. Lesions of the craniovertebral junction do affect the pediatric population, although to a lesser extent than they affect adults.3,5,24 In children, the potential for growth, concerns about stability, and the patient’s size are critical. From this perspective, midface growth centers are the nasal septum and the pterygoid plates. Hence, in a child, a transpalatal approach to the clivus and the sella would be considered before a sublabial transsphenoidal or a maxillary drop-down procedure, with the goal of avoiding damage to the growth centers.25
Advances in neurodiagnostic imaging and microsurgical instrumentation have allowed the development of extensive surgical approaches based on an understanding of the complex anatomy, the craniovertebral dynamics, and the site of encroachment. Consequently, the entire circumference of the foramen magnum is within the neurosurgeon’s reach (Fig. 308-1). However, several surgical limitations and considerations must be appreciated when designing an approach to the craniovertebral junction (Table 308-2). The most frequently used anterior route is the transoral-transpalatopharyngeal approach.1,25–27 Laterally, this approach is limited by the pterygoid plates, the hypoglossal canal, the eustachian tubes, and the width between the vertebral arteries. The limitation imposed by the pterygoid plates can be overcome by using a transmaxillary route with down-fracture of the maxilla and an extended maxillotomy if needed.1,28 A combined exposure with mandibulotomy and midline glossotomy lowers the midline exposure to C5.5,29,30 The disadvantages of the ventral approaches are overshadowed by their easy access, ability to cross the midline, and vertical extensions.31,32 The ventral and dorsal midline approaches permit the anterior and posterior 90 degrees of the craniovertebral junction to be scanned, respectively.
As a general rule, midline extra-axial tumors are best approached in an extra-axial fashion without retracting the brain or violating the dura. A lateral avenue should be used for tumors that are situated laterally.33–35 The transsphenoidal-transsphenoethmoidal approach provides a shallow operative depth and is well tolerated as a route to the upper two thirds of the clivus.36–38 This exposure provides better access to the contralateral side; hence, at times, a bilateral approach may be necessary. The transfacial routes provide wide access to the anterior and midline skull base and include the ventral aspect of the clivus.39,40 This approach is ideal for treating extradural lesions such as chordomas, angiofibromas, and fibrous dysplasias. Dural coverage must be provided by vascularized muscle flaps obtained from the temporalis muscle.
The transbasal approach provides access to the anterior skull base. Unless combined with removal of the supraorbital bar, this approach is limited by the distance between the two optic nerves and the need to work beneath the sella turcica.23 Nonetheless, we have used this approach in several patients and have achieved good reconstruction of the floor.
A lateral extrapharyngeal route is effective and safe for reaching the upper cervical spine.41 At the clivus, however, access becomes difficult because of the pyramidal narrowing of the exposure at the depths of the wound. In an effort to correct this problem, we reroute the facial nerve with upward displacement of the angle of the mandible,25 thereby exposing the lower clivus. We believe that this approach should be limited to metastases, chordomas, and plasmacytomas that affect the axis and atlas vertebrae. Its use for treatment of basilar invagination and intradural pathology is limited.
The true lateral-transcondylar approach to the ventral aspect of the lower brainstem and clivus, as well as the upper cervical cord, demands resection of a portion of the lateral atlantal mass and the occipital condyle. Exposure of the lower clivus and the ventral brainstem is enhanced when combined with a retrosigmoid craniectomy.34 It is useful in the treatment of both extradural and intradural lesions such as chordomas, meningiomas, and neurofibromas.4,14,33 When combined with infratemporal procedures, it allows both anterior and posterior extensions that overcome the limitations of the sigmoid sinus and the hypoglossal nerve.1,34,42 The risk for CSF leak and the possibility of destabilization are high. Troublesome bleeding is routinely encountered from the paravertebral venous plexuses.
The posterolateral route has been recognized for many years. It enables scanning of the foramen magnum to the 90 degrees available with the posterior midline approach as well as the 90 degrees available with the lateral transcondylar approach.1,43 The latter uses a standard midline posterior exposure with a lateral cerebellar approach and includes partial resection of the mastoid process and the posterior third to half of the occipital condyle to provide exposure of the jugular bulb and the medial aspect of the lateral atlantal mass. This provides access to the vertebral artery, which can be rerouted and displaced from the foramen transversarium, thus allowing for control from the C2 level upward. The ability to create a fusion makes this an ideal approach for posterior, lateral, and ventrolateral lesions. Thus, the entire circumference of the foramen magnum is accessible.
The expanded endonasal endoscopic approach is a valid, minimally invasive alternative for treatment of centrally located clivus chordomas or any such lesions or could be an adjunct for the central part of a lesion with lateral extension.36,44–46 A standard nasal access is used with the binostril approach, with the endoscope typically positioned in the right superior nasal cavity by one surgeon who is responsible for maintaining the visual field. This arrangement allows a second surgeon to use both hands and an instrument in each nostril for the resection of the tumor. Dissection of both middle turbinates can expose a large working field, although this is not essential. The mucoperiosteal flaps from the middle turbinate can be used to close off a CSF leak.38 The medial wall of the clival carotids represents the lateral limits of the clival exposure. The floor of the sphenoid is further drilled to get access to the mid and lower clivus. Resection of medial pterygoid process can be performed for exposure of lateral extension of the lower part of the tumor. A total ethmoidectomy can augment exposure into the upper clivus. Closure is performed with multilayered reconstruction technique. In the series by Dehdashti and associates,46 12 patients underwent such operation over a 3-year span. There were four CSF leaks. One patient developed hydrocephalus and one new deficit with clot and one pneumocephalus. Cranial deficits improved over time. A gross resection was accomplished in 7 of the 12 patients. In the series by Frank and associates,45 nine chordomas were approached in this fashion, with a total removal in three. An internal carotid injury occurred in one patient, with two persistent CSF leaks.
Extradural Tumors of the Craniovertebral Junction
Clival and Craniovertebral Junction Chordomas
Chordomas are rare, aggressive, locally destructive tumors of presumed notochordal origin that arise along the vertebral axis and show a proclivity for the spheno-occipital and sacral regions.1,9,37,47 Chordomas of the clivus and craniovertebral junction are the most common extradural neoplasms in this region. The overall incidence of chordomas is 0.2 to 0.5 per 100,000 persons per year, and they account for about 0.15% of all intracranial tumors. About 25% of chordomas occur at the base of the skull, arising from the clivus. Although usually midline, the notochord may have distal projections that extend to the clinoid processes of the petrous bones. Most patients experience symptoms referable to the tumor for more than a year before diagnosis.
Pathology
Chordomas have been divided into classic chordomas, chondroid chordomas, and atypical chordomas.48 Classic chordomas are lobulated, pinkish gray, gelatinous tumors that infiltrate bone but may appear grossly as somewhat demarcated; they account for 80% to 85% of all chordomas. Histologically, they exhibit a variable mix of sheets and cords or clusters of small polygonal cells with eosinophilic cytoplasm and hyperchromatic nuclei. A myxoid matrix is present. Cytologic atypia is absent or minimal.
A subpopulation, the chondroid chordoma, arises in the spheno-occiput and exhibits cartilaginous differentiation. Some authors dispute the existence of chondroid chordomas, preferring to regard these cartilage-containing neoplasms as chondrosarcomas. Chondroid chordomas have a more indolent clinical course; the survival rate is 15.8 years, compared with 4.1 years for typical classic chordomas.47,48 Chondroid chordomas account for 5% to 15% of all chordomas.
Atypical chordomas have a sarcomatoid appearance, with round cells and epithelial or spindle cells present with large areas of necrosis. These solid tumors are aggressive and account for 1.3% to 8% of all chordomas. In the series by Heffelfinger and coworkers, only one patient with an atypical chordoma survived more than 10 years, whereas almost 50% of those with chondroid chordomas survived more than 10 years.48 The frequency of mitotic figures, nuclear pleomorphology, and hyperchromatism does not appear to affect the ultimate outcome.49,50 In rare circumstances, chordomas may dedifferentiate into malignant chondrosarcomas, fibrosarcomas, and even osteosarcomas.51
The immunohistochemical profile of reactivity with antibodies to vimentin cytokeratin, epithelium membrane antigen, and S-100 protein tend to distinguish chordomas from other sarcomatoid round cell or myxoid neoplasms.49,50 Chondrosarcomas are negative for cytokeratin, epithelium membrane antigen, and carcinoembryonic antigen. Vimentin and S-100 protein are present in both chondrosarcomas and chordomas. Immunostaining from keratin has no prognostic value regarding the aggressiveness of the tumor. Chondrosarcomas have been lumped with chordomas because of supposed parallel lines of occurrence, location, and aggressive behavior.
Imaging Characteristics
Cranial-based chordomas are clearly defined by the use of high-resolution CT, on which they appear as solitary or multiple areas of decreased attenuation within the clivus. T2-weighed MRI reveals a bright signal from the marrow in the skull base, signifying replacement of bone by tumor.9,25 At times, calcification is noted in the abnormal areas in the retropharyngeal space, representing sequestered bone fragments in tumor. Chordomas enhance with intravenous contrast and are well visualized by MRI. Chordomas are isointense on T1-weighted MRI (Fig. 308-2).
Presentation
Chordomas usually occur in adults, with a peak incidence occurring in the fourth decade of life.9,47,52 Less than 5% of these tumors arise in children, and they have a predilection for the spheno-occipital region.5 Headaches are often occipitocervical in location and aggravated by changes in craniovertebral positioning. In my series, 55% of patients benefited from craniovertebral stabilization in addition to tumor resection, owing to involvement of the occipital condyles.15,25 Lateral extension of these tumors can give rise to unilateral symptoms, such as hypoglossal nerve palsy. Larger tumors have the potential to cause both upper and lower cranial nerve palsies and a variety of problems related to brainstem compression. Chordomas often cause symptoms from local growth into the nasal cavity, pharynx, and paranasal sinuses.
Surgical Series
Forsyth and colleagues reviewed 51 intracranial chordomas treated surgically between 1960 and 1984 at the Mayo Clinic.52 The median age at presentation was 46 years, and 19 tumors were classified as chondroid chordomas. Eleven patients (22%) underwent biopsy, and 40 patients (78%) had subtotal resection. The survival rates for patients who underwent biopsy were 36% and 0% at 5 and 10 years, respectively, whereas survival rates for those with subtotal resections were 55% and 45% at 5 and 10 years, respectively. Patients who underwent postoperative radiation therapy tended to have longer disease-free survival times. Disease-free survival was the same for patients with chondroid chordomas as for those with typical chordomas.
Watkins and associates described 38 patients treated at the National Hospital of Neurology and Neurosurgery in London between 1958 and 1988.53 Craniotomies were used in 28 patients, and transoral or transmaxillary routes were used in 10 others. All patients underwent postoperative external-beam radiotherapy of 50 to 60 Gy. Recurrence developed in 23 patients, and 13 died within 5 years. Twelve patients were lost to follow-up. The authors concluded that two groups existed: one with indolent disease and another with aggressive growth and poor outcome.
In a more recent publication in 2001, Crockard and associates described a multidisciplinary approach to skull base chordomas.54 A primary first time surgery was done in 24 and redo surgery (initial surgery elsewhere) in 18. A total removal was made in 2, radical 30 and subtotal or partial in the remainder. However, morbidity was significant, with CSF leak in 15, meningitis in 3, new cranial nerve abnormalities in 6 and persistent dysphonia in 4 patients. Three patients died.
Gay and colleagues reviewed the management of 46 chordomas and 14 chondrosarcomas involving the cranial base between 1984 and 1993 at the University of Pittsburgh.47 They recommended an aggressive approach to achieve long-term recurrence-free survival. Fifty percent of patients had undergone previous surgery before referral, and 22% had undergone previous external-beam radiation therapy. The surgical approach was a subtemporal-infratemporal fossa approach, sometimes combined with a transpetrous approach. In other instances, an extended subfrontal approach was used, and in a few cases, the lateral transcondylar approach was used. There was a high tendency to stay between the subtemporal-infratemporal fossa approach and the extended subfrontal approach. Using this technique, the rate of total or near-total resection was 67%. Eighteen patients had died by the 5-year follow-up. Postoperatively, 20% of patients underwent external-beam, proton-beam, or gamma radiation therapy. In patients who had total resection, the overall 5-year recurrence-free survival rate was 84%, compared with 64% in those with partial resection. However, the rate of morbidity was high. Thirty percent developed CSF leaks, 10% experienced meningitis, and 80% had an immediate new cranial nerve deficit. Using the Karnofsky performance score, 40% of patients had permanent functional deterioration. Based on this experience, the authors advocated aggressive initial surgical resection, with the sparing application of radiation therapy.
In 1997, Al-Mefty and Borba reported their results with an aggressive surgical approach combined with postoperative proton-beam therapy in 25 patients treated between 1990 and 1996.9 Radical or subtotal (>90%) removal was achieved in 84% of patients undergoing multiple procedures, when necessary, and extensive drilling of bone beyond the limits of tumor involvement. Postoperatively, 68% of the patients received a mean of 68-cGy–equivalent proton-beam radiotherapy. The postoperative mortality rate was 4%. The postoperative rate of morbidity, however, was 48%, although only 8% suffered permanent neurological deficits. Eighteen percent of the patients treated with proton-beam therapy developed radiation necrosis. The mean follow-up was only 25 months, however, making conclusions about outcome and survival difficult.
In a subsequent follow-up, Colli reviewed the series of Al-Mefty published in 2001.51 There were 55 chordomas and 10 chondrosarcomas. Follow-up of 1 to 150 months was reported, with a median of 38 months. A radical or subtotal removal was performed in 77.8% of patients. At 5-year follow-up, the mortality rate was 14.3%. The recurrence-free survival rates were 100% for chondrosarcomas and 50.7% for chordomas. Of those who received proton-beam therapy, 90.9% survived, as opposed to only 19.4% who received conventional postoperative radiation therapy. However, the complication rate was 60%, and 28.6% had a permanent worsening in neurological deficit. A transient deficit was present in 22.8%, which included new cranial nerve palsy, CSF leak, and hydrocephalus. The authors concluded that the survival was best with radical resection followed by proton-beam therapy.
Maira and coworkers37 achieved total tumor removal in 7 of 10 patients undergoing repeated transsphenoidal procedures for chordoma. They reported no evidence of disease in these patients at a mean of 38 months after surgery and encountered a cranial nerve complication in only 1 patient.
Couldwell and colleagues reported on a variation of the standard transsphenoidal approach to the sella with emphasis on extended approaches and parasellar approaches.36
[/not-level-membership-for-neurosurgery-category]
