CHAPTER 98 TUMORS OF THE BRAIN
Tumors of the brain are regarded as one of the most devastating group of neurological diseases—they are associated with significant neurological morbidity, they lead to progressive physical, cognitive and emotional dysfunction and are frequently fatal. The term brain tumor is used to describe both primary tumors that originate from the brain, cranial nerves, pituitary gland. or meninges and secondary tumors (metastases) that arise from organs outside the nervous system. These tumors present in many different ways dependent on their location, their rate of growth, and their effect on healthy neural tissue. Diagnosis requires careful history and examination, imaging, and histological examination, and management is best determined in a multidisciplinary team environment comprising neurologists, neurosurgeons, oncologists, neuropathologists, neuroradiologists, and clinical nurse specialists.
EPIDEMIOLOGY
The incidence of primary brain tumors is considerably higher than tumor registry figures suggest. Based on a study from the southwest of England ascertaining data mainly from radiology records, the crude annual incidence for primary tumors was found to be 21 in 100,000.1 The annual incidence in the United States as ascertained from the Central Brain Tumor Registry is lower at 6.7 in 100,000 persons.2 There is increasing evidence that the incidence of gliomas and lymphomas is increasing, particularly in elderly patients, although this is more likely to be due to increased case ascertainment, with the increasing availability of modern imaging techniques.3
ETIOLOGY
Numerous epidemiological studies have been carried out to investigate etiological factors, but no clear risk factors have emerged apart from therapeutic ionizing irradiation. Cranial radiotherapy, even at low doses, has been shown to increase the relative risk of meningiomas by a factor of 10 and gliomas by a factor of 3.4 Other radiotherapy-induced tumors include cranial osteosarcomas, soft tissue sarcomas, schwannomas, and peripheral nerve sheath tumors. They have been described following radiotherapy for tinea capitis, craniopharyngioma, and pituitary adenomas and prophylactic cranial irradiation for acute lymphoblastic leukemia. Second tumors tend to lie within the radiation field, usually in lower dose regions, and develop from a few years to many decades after irradiation. The reported median time to the development of gliomas is 7 years. Sarcomas develop with a longer lag time and meningiomas may be seen 30 or 40 years later. The histology is identical to spontaneous tumors, although meningiomas are more likely to contain atypical features and have a worse prognosis.
No other environmental exposure has been clearly identified as a risk factor. There is widespread concern about the possible risks of cellular telephones, but case-control studies have not shown any increased risk in respect of any subtype of brain tumor using measures of the type of telephone, duration and frequency of use, and cumulative hours of use.5 So far, the consensus of opinion based on four studies is that mobile telephone use does not increase the risk of developing a brain tumor. However, with the exponential increase in the ownership and duration of use of these hand-held devices, it is important to continue surveillance of brain tumor trends in order to detect a latent period of several decades for the development of a tumor.
CLINICAL FEATURES
Raised Intracranial Pressure
As a brain tumor grows, there is displacement of cerebrospinal fluid into the spinal compartment and a reduction of blood volume. Eventually, the intracranial pressure rises because the skull behaves as a rigid box. Headache is the most common symptom of brain tumors, occurring in 23% of patients at initial presentation and 46% by the time of hospital admission. Headache alone, however, is an extremely rare presenting symptom, occurring in only 1.9% of patients.6 Because headache is such a common symptom in the population as a whole, it accounts for a disproportionate number of referrals of patients to neurology clinics concerned about the possibility of a brain tumor. Most brain tumor headaches are intermittent and nonspecific and may be indistinguishable from tension headaches.7 They may occasionally indicate the side of the tumor. Certain features of a headache are suggestive but not pathognomic of raised intracranial pressure. These include headaches that wake the patient at night or are worse on waking and improve shortly after rising, as well as headache associated with visual obscurations (transient fogging associated with changes in posture). Supratentorial tumors typically produce frontal headaches, whereas posterior fossa tumors usually result in occipital headache or neck pain. Nausea and vomiting may be a feature of raised pressure but may also occur as an early symptom of fourth ventricular tumors.
Brain tumors cause increased intracranial pressure by a variety of different mechanisms. They may have grown so large in a relatively short space of time that they cause stretching of pain-sensitive intracranial structures by a direct mass effect or by an effect on the microvasculature leading to cerebral edema. Smaller tumors, particularly those located in the posterior fossa, may cause headaches by obstructing cerebrospinal fluid circulation and producing obstructive hydrocephalus. Tumors may also cause raised intracranial pressure by producing large cysts. Occasionally, meningeal-based tumors cause localized headache through stretching of overlying dura. As a general rule, headaches with migrainous features are rarely due to an underlying tumor, although occasionally occipital tumors produce occipital seizures that are similar in many respects to migraine.
Seizure Disorder
Temporal and frontal tumors are more likely to cause seizures than are occipital or parietal tumors, particularly when cortically based. The characteristics of the seizure depend on the location of the tumor. Frontal lobe tumors cause typically brief, frequent, and nocturnal seizures, which tend to spread rapidly and may become generalized. Common manifestations of a frontal lobe seizure include bicycling movements of the legs at night, turning of the head and eyes to the side away from the tumor (frontal adversive seizure), speech arrest, and hemiclonic spasms with a jacksonian march (posterior frontal tumors) in clear consciousness. In contrast, mesial temporal tumors can begin with olfactory or gustatory hallucinations, an epigastric rising sensation, or psychic experiences such as déjà vu or depersonalization. Once the seizures progress to a loss of awareness, the patients may stare blankly, speak unintelligibly, or exhibit lip smacking, picking at clothing, or other automatisms. Secondary generalized tonic-clonic seizures may follow on from partial seizures, more frequently in untreated patients. The presence of seizures is a favorable prognostic factor for survival, possibly due to lead-time bias in diagnosis and possibly due to the slow growth of epileptogenic tumors compared with more high-grade destructive tumors. In a study of patients with low-grade astrocytomas, the 5-year survival for patients with epilepsy as the only sign of tumor was 63% compared with 27% among the whole group.8
DIAGNOSIS
The diagnosis of a brain tumor is made by a combination of contrast-enhanced computed tomography scanning/magnetic resonance imaging and pathological classification of either a biopsy or resection specimen. Over the past decade or so, there have been a number of newer techniques introduced to complement conventional structural imaging, including proton magnetic resonance spectroscopy, functional metabolic imaging (single photon and positron emission tomography), and advanced magnetic resonance techniques, such as perfusion imaging (measuring blood flow and blood volume), diffusion weighted imaging (measuring cellularity), and diffusion tensor imaging (assessing integrity of white matter pathways). These are being gradually integrated into the routine preoperative evaluation of a brain tumor but add little to the conventional sequences in terms of refining diagnostic certainty.9
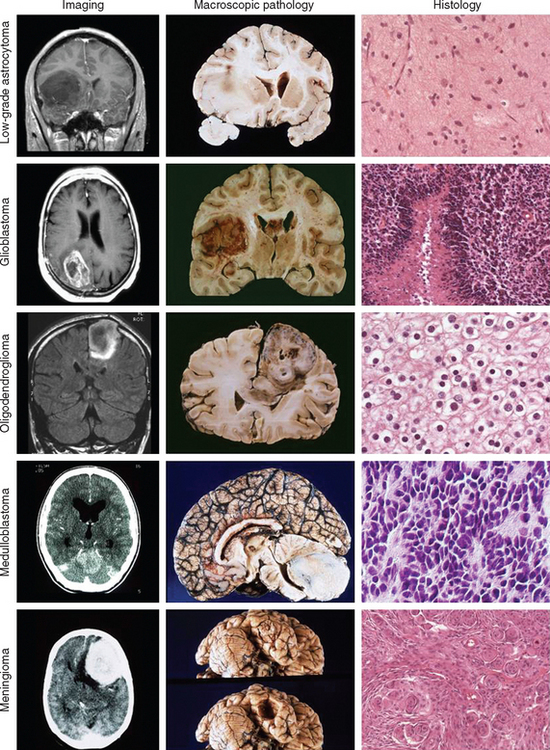
Just as there is clinicopathological correlation between World Health Organization (WHO) grade and prognosis, there is also a radiological/pathological correlation, specifically with respect to the degree of contrast enhancement seen within the tumor. Most grade II gliomas do not enhance, unlike grades III and IV, where there is usually irregular ring enhancement and, in the case of grade IV tumors, central necrosis. However, as mentioned, certain grade I gliomas, particularly juvenile pilocytic astrocytomas, also enhance, and this can occasionally give rise to diagnostic confusion, particularly in adults, in whom juvenile pilocytic astrocytomas are much less common than malignant gliomas. The key imaging characteristics of common types of tumors are summarized in Table 98-1; see also Figure 98-3 for radiological/pathological correlations.
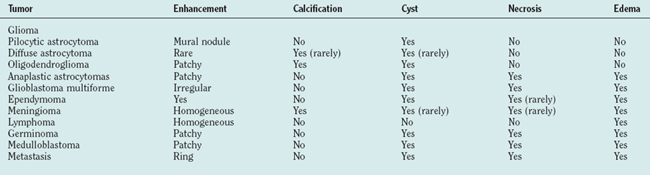
PATHOLOGY
The WHO published a landmark classification in 1993 and, in 2000, further refined their classification system (Table 98-2).10 The key to the WHO classification is the stratification of tumors according to their biological activity so that the lower the WHO grade, the better the overall prognosis. As a general rule, the category of grade I tumors is reserved for neoplasms that have a stable histology and that are potentially curable by surgical removal alone. In contrast, tumors that appear histologically “benign,” yet are known to progressively transform over time into higher grade lesions, are categorized as grade II neoplasms. Those tumors with anaplastic histology are regarded as grade III high-grade neoplasms, and the most malignant phenotype is classified as grade IV. The peak of age of incidence is proportional to the most common histological grade; that is, grade I tumors usually present in childhood, grade II in young adulthood, grade III in middle age, and grade IV in older age. The exception to this rule is the grade IV primitive neuroectodermal tumors, which occur most frequently in childhood.
TABLE 98-2 World Health Organization Classification and Grading of Tumors of the Nervous System
| Tumors of Neuroepithelial Tissue | WHO Grade |
|---|---|
| Astrocytic tumors | |
| Pilocytic astrocytoma | I |
| Pleomorphic xanthoastrocytoma | I, II |
| Subependymal giant cell astrocytoma | I |
| Desmoplastic infantile astrocytoma | I |
| Diffuse astrocytoma | II |
| Anaplastic astrocytoma | III |
| Glioblastoma multiforme | IV |
| Gliosarcoma | IV |
| Oligodendroglial tumors | |
| Oligodendroglioma | II |
| Anaplastic oligodendroglioma | III |
| Mixed astrocytic and oligodendroglial tumors | |
| Oligoastrocytoma | II |
| Anaplastic oligoastrocytomas | III |
| Ependymal tumors | |
| Subependymoma | I |
| Myxopapillary ependymoma | I |
| Ependymoma | II |
| Anaplastic ependymoma | III |
| Choroid plexus tumors | |
| Choroid plexus papilloma | I |
| Choroid plexus carcinoma | IV |
| Neuronal and mixed neuronal-glial tumors | |
| Gangliocytoma | I |
| Ganglioglioma | I, II |
| Desmoplastic infantile ganglioglioma | I |
| Dysembryoplastic neuroepithelial tumor | I |
| Central neurocytoma | I |
| Pineal parenchymal tumors | |
| Pineocytoma | I |
| Pineoblastoma | IV |
| Embryonal tumors | |
| Primitive neuroectodermal tumors | IV |
| Medulloblastoma | IV |
| Meningeal tumors | |
| Meningioma | I |
| Atypical meningioma | II |
| Anaplastic meningioma | III |
| Hemangiopericytoma | |
| Melanocytic tumor of the meninges | |
| Tumors of vascular origin | |
| Cavernous angioma | |
| Hemangioblastoma | |
| Germ cell tumors | |
| Germinoma | |
| Embryonal carcinoma | |
| Yolk sac tumor (endodermal sinus tumor) | |
| Choriocarcinoma | |
| Teratoma | |
| Tumors of the sellar region | |
| Pituitary adenoma | |
| Pituitary carcinoma | |
| Craniopharyngioma | |
| Primary central nervous system lymphomas | |
| Peripheral nerve sheath tumors | |
| Vestibular schwannoma | |
| Trigeminal schwannoma | |
| Facial nerve schwannoma | |
| Malignant peripheral nerve sheath tumor | |
| Metastatic tumors | |
Gliomas
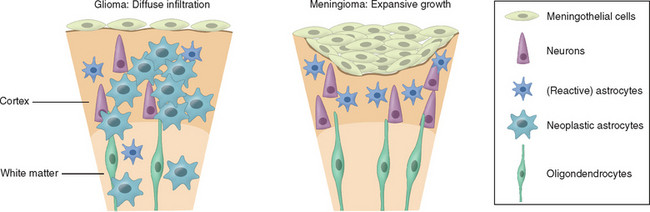
Gliomas are the most common type of primary brain tumor and are so called because they share morphological and immunohistochemical features with astrocytes and oligodendroglial and ependymal cells. Astrocytic neoplasms are the most common of the three and include tumors of all WHO grades. Low-grade gliomas can be subdivided into WHO grade I tumors (e.g., pilocytic astrocytomas, pleomorphic xanthoastrocytomas, and subependymal giant cell astrocytomas [usually but not invariably associated with tuberose sclerosis]) and WHO grade II tumors (e.g., diffuse and gemistocytic astrocytomas, oligodendrogliomas, and oligoastrocytomas [which contain elements of both astrocytic and oligodendroglial lineage, otherwise known as mixed gliomas]). These two grades should be regarded as distinctive groups in that grade I tumors never progress into grade II tumors, unlike grade II tumors, which frequently transform into grade III and grade IV tumors (as discussed later). Therefore, grade II tumors should be thought of as part of a biological continuum that extends through to grade IV tumors.
Grade I gliomas are usually well circumscribed and potentially curable by surgical resection alone. The most common type is the juvenile pilocytic astrocytoma, so called because it presents in childhood and is characterized by the presence of astrocytes with hairlike (pilocytic) processes. The tumor has a narrow zone of microscopic infiltration and appears radiologically as an enhancing mural nodule, which is supplied by capillaries that lack a complete blood-brain barrier surrounded by a cyst. This leads to the contrast enhancement seen on computed tomography scans or magnetic resonance images (Fig. 98-1).
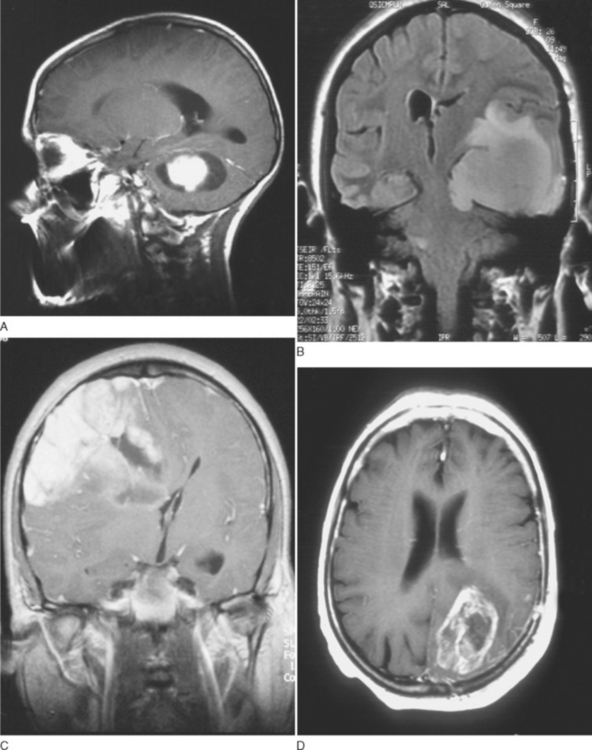
GENETICS
The molecular classification of gliomas has become increasingly important in therapeutic decision making, particularly the distinction between tumors of astrocytic lineage and oligodendroglial lineage, with the latter being characterized by losses of part of chromosomes 1 and 19.
Astrocytomas can be distinguished from oligodendrogliomas on the basis of histological features, as described earlier. During the past decade, the specific association between tumors of oligodendroglial lineage, particularly tumors with classic histology, and loss of the short arm of chromosome 1 (1p) and the long arm of chromosome 19 (19q) has been recognized. Loss of 1p/19q is found in 40% to 90% of oligodendrogliomas compared with less than 1% of astrocytomas. Nearly all tumors with 1p loss also have 19q loss, suggesting that inactivation of one or more genes on each of these arms is a fundamental event in oligodendroglioma oncogenesis. This chromosomal fingerprint is tightly associated with chemosensitivity and prolonged survival and has taken molecular classification out of the basic science laboratory and into the clinical arena.11 Candidate tumor suppressor genes have still not been identified. Other genetic alterations less frequently encountered in oligodendroglioma include losses of chromosomes 4, 6, 14, 11p, and 22q. Neuropathology laboratories are increasingly offering genotyping for 1p and 19q; the results will influence the prognosis, as 70% to 80% of long-term survivors have 1p/19q loss.
The correlation with chemosensitivity is particularly high in patients, with 75% of patients with anaplastic oligodendrogliomas responding to treatment with procarbazine, CCNU, and vincristine and 50% achieving a prolonged and durable survival.11 Similar but less dramatic responses have also been observed in patients with low-grade oligodendrogliomas.
TREATMENT
The use of each therapeutic modality therefore should be dictated by the location of the tumor within the brain and within the skull, specifically, whether it is intra-axial (e.g., glioma) or extra-axial (e.g., meningioma), the likely histology, and the patient’s age and general condition. The distinction between intra- and extra-axial tumors is important, as the effect on surrounding brain tissue and the likely plane of dissection are critical to the success of radical surgery (Fig. 98-2). When considering surgery for intrinsic tumors, patient factors, particularly age and performance status, are far more important in terms of prognosis than any specific treatments used, and this needs to be borne in mind when evaluating the efficacy of any treatment and claims that survival is significantly improved.
Surgery
There have been a number of recent advances in tumor neurosugery including computerized neuronavigation techniques, improved preoperative mapping of eloquent brain areas using functional magnetic resonance imaging, and assessment of white matter pathways by diffusion tractography. Frameless stereotaxy now enables the surgeon to delineate the tumor boundaries seen on preoperative magnetic resonance imaging with the surface markings of the brain in three dimensions. However, this may be affected by brain shift as the cranial cavity is opened and therefore becomes less accurate at deeper levels of the brain. In this respect, intraoperative magnetic resonance imaging now allows the surgeon to view the results of his or her resection while the patient is still on the table and remove further tissue as necessary according to the scan appearances of the tumor. These have all contributed to improving the morbidity and mortality of neurosurgery, but an effect on overall survival has yet to be demonstrated. Nevertheless, the ability to remove more and more abnormal tissue with less and less risk to the patient and eventually to resect almost an entire intrinsic tumor remains the Holy Grail of oncological neurosurgery.
Chemotherapy
Chemotherapy may be delivered before surgery or radiotherapy, known as neoadjuvant treatment, simultaneously with radiotherapy, known as concomitant treatment, or after surgery or radiotherapy, known as adjuvant treatment. It may given as part of the primary treatment or at progression, particularly for malignant gliomas.
Temozolomide is increasingly being used for brain tumor treatment. It is an imidazotetrazine derivative of dacarbazine with good oral bioavailablity and is rapidly metabolized to an active derivative. It works by methylating the O6 position on guanidine and depletes the drug resistance enzyme methylyguanine methyltransferase (MGMT) enzyme. It is relatively well tolerated and so has become the chemotherapeutic agent of choice for many patients with gliomas, although it has yet to be compared head-to-head with the conventional combination regimen of procarbazine, CCNU, and vincristine (PCV).
SUPPORTIVE TREATMENT
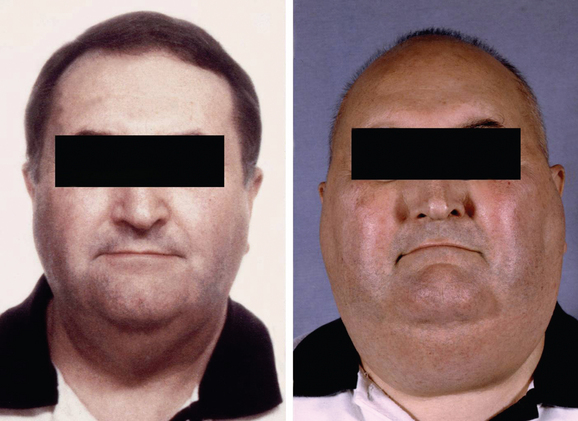
Steroids are used extensively in neuro-oncology to reduce peritumoral edema, to improve symptoms and quality of life, and to reduce the morbidity of brain tumor surgery. Dexamethasone is the most frequently used steroid and is 10 times more potent than prednisolone. The main side effects include weight gain, fluid retention, impaired glucose tolerance, skin changes, proximal myopathy, and increased susceptibility to infections. Not uncommonly, the patient becomes cushingoid leading to dramatic changes in body habitus and facial appearance (Fig. 98-4). As with all steroids, the aim is to use the lowest dose possible that controls symptoms. Steroids have a lympholytic effect and are therefore cytotoxic to primary central nervous system lymphomas. If there is a clinical suspicion of lymphoma based on the results of a scan, then steroids should be avoided prior to biopsy as they may cause temporary disappearance of the tumor.
INTRA-AXIAL TUMORS
Low-Grade Gliomas
The management of low-grade gliomas is one of the most controversial issues in neuro-oncology. These are typically diffusely infiltrating tumors, often invading but not destroying eloquent regions of the brain, and cannot be removed completely in the majority of cases by surgical resection alone. Although some patients may survive for decades, the majority of these tumors eventually become high-grade gliomas and these are usually fatal. There are a large number of surgical series advocating the benefits of complete resection over partial resection but these are all retrospective and include patients with a variety of tumor histologies, specifically pilocytic and non-pilocytic astrocytomas. They are by definition subject to selection bias, and therefore reports of better outcomes for patients with larger resections need to be interpreted in light of the known prognostic factors which include tumor size.12 Despite these reservations, surgery is the only certain way of removing a large volume of tumor tissue and is widely used in young, fit patients with tumors in noneloquent regions (e.g., nondominant frontal or temporal lobes). Some surgeons will resect tumors in dominant frontal temporal lobes, using the techniques of awake craniotomy to monitor language perioperatively, but there are no comparative data published comparing this approach with a more conservative policy. This lack of clear benefit is supported in part by a population-based study of nearly 1000 patients with low-grade glioma that showed a relatively small and nonsignificant difference in overall survival between patients who had had a biopsy only (6.4 years), a subtotal tumor resection (6.8 years), or a gross-total tumor resection (7.6 years).13
The benefit of early radiation in low-grade gliomas is similarly unclear. The European Organisation for Research and Treatment of Cancer (EORTC) reported interim results from a trial comparing early radiotherapy (at diagnosis) with radiotherapy at progression. This was a prospective trial and followed patients for a median of 5 years. Patients who had early radiotherapy showed a significant improvement in time to progression compared with patients irradiated at tumor progression but there was no difference in overall survival. The 5-year estimate was 63% versus 66% (overall survival) and 44% versus 37% (time to progression) for the treated and control arms respectively.14 On the basis of these data, the majority of oncologists irradiate patients with low-grade glioma only at progression or rarely for control of intractable seizures. The standard dosage schedule is 54 Gy in 33 fractions over 6.5 weeks.
Chemotherapy is being increasingly used for low-grade gliomas, particularly those with oligodendroglial elements. There are strong indications that a significant percentage of low-grade oligodendrogliomas respond favorably to PCV chemotherapy with improvements in seizure control and cognitive function more readily apparent than shrinkage of the tumors in magnetic resonance images.15 Temozolomide has also been found to be useful in oligodendrogliomas both as primary treatment and in recurrent disease after radiotherapy, particularly in those tumors with loss of chromosome 1p and 19q.
Malignant Gliomas
Relative contraindications to surgery include poor performance status, significant medical comorbidity, and tumors in eloquent or inaccessible locations. For these patients, the risks of surgery may be outweighed by the potential benefits. The overall morbidity rate for untreated malignant gliomas is 24% with a mortality rate of 1.5%. The chances of neurological improvement with surgery are just over 20% with less than 10% of patients deteriorating.16
In contrast, there is unanimity of opinion about the benefits of radiotherapy for malignant gliomas as it is the only treatment that has been proved to extend survival in this patient group. Radiotherapy is indicated for the treatment of grades III and IV astrocytomas and oligodendrogliomas, and treatment with radical intent is given to patients under the age of 70 years who have a good performance status, that is, a Karnofsky performance scale of at least 70, implying the ability to self-care. The radiation field encompasses the contrast-enhanced T1-weighted target with a margin of between 2 and 3 cm to sterilize “satellite” tumor cells. The total dose is usually 60 Gy, delivered over 6 weeks in 30 fractions. In a landmark study, the median survival of patients with malignant gliomas increased from 14 weeks with supportive treatment alone to 36 weeks with radical radiotherapy.17 Lower-dose schedules, such as 30 Gy in 10 fractions, are used mainly in the palliative setting, particularly for patients over the age of 70 in poor general condition.
Chemotherapy
Chemotherapy for malignant gliomas has traditionally been used as adjuvant treatment following radiotherapy (mainly in the United States) or at recurrence. The common drugs used are nitrosoureas such as BCNU (as adjuvant treatment) and PCV or temozolomide at recurrence. There is no clear benefit for the use of adjuvant chemotherapy over radiotherapy alone, although a meta-analysis based on 12 randomized trials suggested a small benefit of chemotherapy compared with radiotherapy alone (a 5% increase in 2-year survival).18 A trial is under way comparing PCV with temozolomide in recurrent disease, and the results will not be available until 2008.
A trial has shown that the use of concomitant temozolomide with radical radiotherapy followed by six cycles of adjuvant temozolomide in patients with glioblastoma multiforme offered a significant survival advantage over radiotherapy alone with minimal additional toxicity. Although the increase in median survival from 12.1 months with radiotherapy alone to 14.6 months in the concomitant temozolomide group was relatively modest, the 2-year survival rate increased from 10.4% to 26.5%.19 These results represent a significant improvement in the outlook of patients with glioblastoma multiforme, although it remains to be seen whether these data can be extrapolated to patients with anaplastic astrocytomas. Further trials to confirm these findings are unlikely to occur given that the sample size was large (573 patients from 85 centers), that prognostic factors were well matched between the two groups, and that 85% of patients in the concomitant arm completed both radiotherapy and temozolomide as planned. Furthermore, an exploratory subgroup analysis defined according to known prognostic factors demonstrated a survival benefit in nearly all subgroups. However, in a parallel study on the same patient group investigating the role of genetic silencing of the MGMT (O6-methylguanine-DNA methyltransferase) DNA-repair gene by promoter methylation, there was a striking survival benefit in those patients who received temozolomide and whose tumors contained a methylated MGMT promoter compared with those who did not have a methylated MGMT promoter.20
Chemotherapy is being increasingly used for patients with anaplastic oligodendrogliomas following a landmark study from the National Cancer Institute of Canada study reporting a 75% response rate in patients with anaplastic oligodendrogliomas treated with PCV.11 Subsequently, temozolomide (TMZ) has also been found to have activity with high response rates and durable responses. Because of the increasing interest in chemotherapy for anaplastic oligodendrogliomas, trials are being carried out investigating the role of neoadjuvant and adjuvant chemotherapy. Rather surprisingly, a randomized controlled clinical trial of neoadjuvant intensive PCV chemotherapy followed by radiotherapy versus radiotherapy alone in patients with pure and mixed anaplastic oligodendrogliomas showed no benefit in terms of overall survival between the two groups. Although there was a slight prolongation of progression-free survival in the combined treatment group, this was at the expense of considerable acute toxicity in the PCV group. As predicted, patients with 1p and 19q loss lived longer than other patients irrespective of treatment.21 An abstract, just presented at the time of writing, has also failed to demonstrate a survival benefit for PCV therapy when used as adjuvant treatment to radiotherapy in patients with anaplastic oligodendrogliomas.
PROGNOSIS
Extra-axial Tumors
Meningiomas
Surgery is the only curative treatment, but for tumors around the base of the skull and foramen magnum, a policy of watch and wait or stereotactic radiotherapy may be more appropriate. The recurrence rate depends on the extent of tumor resection and the histological grade. In order to achieve complete excision, the whole tumor and associated dura must be exposed and removed or coagulated. Even for seemingly completely excised tumors, the recurrence rate is 3% at 5 years and 10% at 10 years. Tumors with atypical or malignant histology are associated with 5-year recurrence rates of 40% and 80%, respectively. For this reason, all malignant meningiomas should have adjuvant radiotherapy. For atypical meningiomas, the decision to irradiate postoperatively will depend on the completeness of resection, the accessibility of the tumor, and the age of the patient. Fractionated radiotherapy at doses of around 54 Gy offers control rates between 80% and 95%. Stereotactic radiotherapy can be given to small tumors where the meningioma is in an inaccessible location, such as the skull base, and close to vital structures, such as the optic nerve. Radiosurgery with doses of approximately 15 Gy is probably equally effective for tumors less than 3 cm in diameter. Stereotactic radiotherapy is now the treatment of choice for optic nerve sheath meningiomas.
ACKNOWLEDGMENTS
The author would like to acknowledge the contribution of Professor Sebastian Brandner for Figures 98-2 and 98-3 and for the constructive suggestions about the pathology and genetics of brain tumors.
Grant R. Overview: brain tumor diagnosis and management/Royal College of Physicians guidelines. J Neurol Neurosurg Psychiatry. 2004;75(Suppl II):ii37-ii42.
Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003.
Ries LAG, Eisner MP, Kosary CL, et al, editors. SEER cancer statistics review 1975–2001. Bethesda: National Cancer Institute, 2004.
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996.
1 Pobereskin LH, Chadduck JB. Incidence of brain tumors in two English counties: a population based study. J Neurol Neurosurg Psychiatry. 2000;69:464-471.
2 Ries LAG, Eisner MP, Kosary CL, et al, editors. SEER cancer statistics review 1975–2001. Bethesda: National Cancer Institute, 2004.
3 Werner MH, Phuphanich S, Lyman GH. The increasing incidence of malignant gliomas and primary central nervous system lymphomas in the elderly. Cancer. 1995;76:1634-1642.
4 Ron E, Modan B, Boice J, et al. Tumors of the brain and nervous system following radiotherapy in childhood. N Engl J Med. 1988;319:1033-1039.
5 Inskip PD, Tarone RE, Hatch EE, et al. Cellular-telephone use and brain tumors. N Engl J Med. 2001;344:79-86.
6 Grant R. Overview: brain tumor diagnosis and management/Royal College of Physicians guidelines. J Neurol Neurosurg Psychiatry. 2004;75(Suppl II):ii37-ii42.
7 Forsyth PA, Posner JB. Headaches in patients with brain tumors: a study of 111 patients. Neurology. 1993;43:1678-1683.
8 van Veelen MLC, Avezaat CJJ, Kros JM, et al. Supratentorial low grade astrocytoma: prognostic factors, dedifferentiation, and the issue of early versus late surgery. J Neurol Neurosurg Psychiatry. 1998;64:581-587.
9 Rees JH. Advances in MR imaging of brain tumors. Curr Opinion Neurol. 2003;16:643-650.
10 Kleihues P, Cavenee WK, editors. Pathology and genetics of tumors of the nervous system. In World Health Organisation Classification of Tumors. Lyon, France: IARC Press, 2000.
11 Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473-1479.
12 Keles GE, Lamborn KR, Berger MS. Low-grade hemispheric gliomas in adults: a critical review of extent of resection as a factor influencing outcome. J Neurosurg. 2001;95:735-745.
13 Johannesen TB, Langmark F, Lote K. Progress in long-term survival in adult patients with supratentorial low-grade gliomas: a population-based study of 993 patients in whom tumors were diagnosed between 1970 and 1993. J Neurosurg. 2003;99:854-862.
14 Karim ABMF, Afra D, Cornu P, et al. Randomized trial on the efficacy of radiotherapy for cerebral low-grade glioma in the adult: European Organization for Research and Treatment of Cancer Study 22845 with the Medial Research Council BR04: an interim analysis. Int J Radiat Oncol Biol Phys. 2002;52:316-324.
15 Streffer J, Schabet M, Bamberg M, et al. A role for preirradiation PCV chemotherapy for oligodendroglial brain tumors. J Neurol. 2000;247:297-302.
16 Chang SM, Parney IF, McDermott M, et al. Perioperative complications and neurological outcomes of first and second craniotomies among patients enrolled in the Glioma Outcome Project. J Neurosurg. 2003;98:1175-1181.
17 Walker MD, Alexander EJr, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas: a co-operative clinical trial. J Neurosurg. 1978;49:333-343.
18 Stewart LA. Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet. 2002;359:1011-1018.
19 Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996.
20 Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003.
21 Cairncross G, Seiferheld W, Shaw E, et al: An Intergroup randomized controlled clinical trial of chemotherapy plus radiation (RT) versus RT alone for pure and mixed anaplastic oligodendrogliomas: Initial report of RTOG 94–02 [abstract]. Presented at the 2004 ASCO annual meeting, New Orleans, La.






