CHAPTER 69 Tumors of the Bile Ducts, Gallbladder, and Ampulla
Biliary malignancies comprise the vast majority of biliary neoplasms and are divided into the following three categories: (1) carcinomas of the intra- and extrahepatic bile ducts; (2) carcinoma of the gallbladder; and (3) carcinoma of the ampulla of Vater.1 In the United States and other Western nations, biliary malignancies are rare. In certain parts of the world, however, their prevalence rates are high, making them leading causes of cancer death. Biliary cancers are highly aggressive with dismal prognoses. Often these cancers are diagnosed at an advanced stage. In general, they are resistant to chemotherapy. Advances in the understanding of the molecular pathogenesis of these tumors has allowed the development of new experimental models and targeted therapies, and advances in diagnosis and surgical treatment have resulted in improved outcomes for subsets of patients.
CHOLANGIOCARCINOMA
Cholangiocarcinoma is an epithelial carcinoma with differentiated features of biliary epithelium that arises from the intra- and extrahepatic biliary tree.2 It is the most common bile duct tumor and second most common primary hepatic malignancy (after hepatocellular carcinoma, see Chapter 94). Since the 1970s, the incidence has increased substantially in Western societies.3
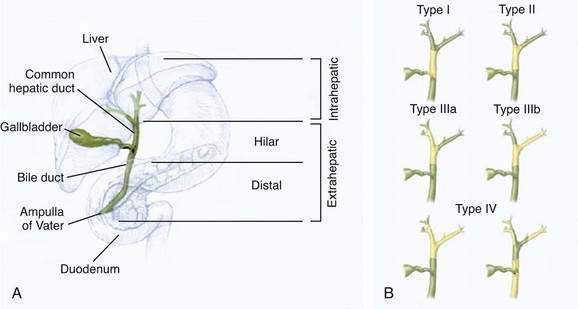
The classification of cholangiocarcinomas into intra- and extrahepatic cancers is based on differences in anatomy, etiology, pathogenesis, molecular signature, and treatment. The second-order bile ducts are the anatomic margin for the distinction between these two subsets (see Chapter 62). Extrahepatic cholangiocarcinomas account for 80% to 90% of these cancers and can be further subclassified into hilar or distal bile duct cancers. Hilar cholangiocarcinoma, also referred to as Klatskin tumors, are described clinically according to the Bismuth-Corlette classification as types I to IV (Fig. 69-1). Type I cholangiocarcinomas involve the common hepatic duct distal to the union of the right and left hepatic ducts; type II tumors involve the union of the right and left hepatic ducts; type IIIa tumors involve the union of the right and left hepatic ducts and extend up the right hepatic duct; type IIIb tumors involve the union of the right and left hepatic ducts and extend up the left hepatic duct; and type IV tumors are multifocal or involve the biliary confluence and extend up the right and left hepatic ducts. The natural course of cholangiocarcinoma is aggressive, with a median survival of less than 24 months following diagnosis.4 The only potentially curative treatment is surgical. Unfortunately, the majority of patients are diagnosed at an advanced stage that precludes curative surgery.
EPIDEMIOLOGY
Global incidence rates for cholangiocarcinoma are heterogeneous. The highest incidence is observed in Southeast Asia, with rates up to 96 per 100,000 population, and the lowest incidence is observed in Australia, with rates as low as 0.1 per 100,000 population.5 In the United States, incidence rates are 0.95 per 100,000 population for intrahepatic cholangiocarcinoma and 0.82 per 100,000 for extrahepatic cholangiocarcinoma. Ethnic differences in the incidence of cholangiocarcinoma have been observed in the United States, with the highest rates observed in patients of Hispanic descent and the lowest in African Americans. Since the 1980s, age-adjusted incidence rates for intrahepatic cholangiocarcinoma have increased, whereas those for extrahepatic cholangiocarcinoma have remained stable; the cause of the overall increase in incidence is unknown.3 Fifty-two percent to 54% of patients with cholangiocarcinoma are male. Globally, the average age at diagnosis is older than 50 years. In Western industrialized nations, most cases are diagnosed around age 65, and cholangiocarcinoma is uncommon before age 40 except in patients with primary sclerosing cholangitis (see Chapter 68).
ETIOLOGY
In the majority of cases, the etiology of cholangiocarcinoma is unknown. Several risk factors have been identified (Table 69-1). These risk factors are characterized by their association with inflammation and cholestasis. Primary sclerosing cholangitis (PSC) is one of the most common risk factors (see Chapter 68). In patients with PSC, the annual frequency rate of cholangiocarcinoma is 0.6% to 1.5%, and the overall prevalence rate of cholangiocarcinoma is 5% to 15%. In the majority of patients with PSC in whom cholangiocarcinoma develops, the diagnosis of cholangiocarcinoma is made within two-and-a-half years of the diagnosis of PSC.6 Other risk factors for cholangiocarcinoma include biliary infections with Opisthorchis viverrini and Clonorchis sinensis, which are endemic in East Asia (see Chapter 82).7 Biliary malformations such as Caroli’s disease and choledochal cysts are associated with a 10% to 15% risk for the development of cholangiocarcinoma (see Chapter 62),8 whereas hepatolithiasis (“recurrent pyogenic cholangitis”) carries a 10% risk for the development of cholangiocarcinoma (see Chapter 68).9 Recurrent bacterial cholangitis in the setting of biliary-enteric drainage procedures has also been associated with the development of cholangiocarcinoma.10 Carcinogens such as thorotrast (used as a radiologic contrast agent in the past) and dioxins have been associated with an increased risk of cholangiocarcinoma.11 Hepatitis C and cirrhosis are considered possible risk factors for cholangiocarcinoma (see Chapter 79).5
Table 69-1 Risk Factors for Cholangiocarcinoma
PATHOLOGY
Cholangiocarcinoma is a paucicellular, highly desmoplastic tumor. Macroscopically, it can be described according to its growth characteristics as mass forming, periductal-infiltrating, or intraductal-papillary. Intrahepatic cholangiocarcinomas are typically mass forming, whereas ductal carcinomas can present in any of the three growth forms. Histologically, 90% of cholangiocarcinomas are adenocarcinomas. Other histologic types include intestinal-type adenocarcinoma, clear cell adenocarcinoma, signet-ring cell carcinoma, adenosquamous carcinoma, squamous cell carcinoma, and small cell carcinoma.12 Intraductal-papillary adenocarcinomas spread superficially along the biliary mucosa without deep invasion of the fibromuscular wall layers and have a better prognosis than nonpapillary cancers.13 Metastases to regional and peripancreatic lymph nodes are frequently observed with this growth type.
PATHOGENESIS
As noted earlier, etiologic associations as well as experimental data provide evidence for inflammation and bile acids as key factors in the molecular pathogenesis of cholangiocarcinoma. Inflammation provides a microenvironment that promotes malignant transformation of bile duct–associated cells such as cholangiocytes, biliary stem cells, or epithelial cells within peribiliary glands (see Chapter 62). In subsets of cholangiocarcinoma and mixed hepatocellular-cholangiocarcinoma, hepatic stem/progenitor cells have been suggested to be the cells of origin.14 On a molecular level, increased tissue concentrations of cytokines, bile acids, and growth factors can lead to functional inactivation of mismatch DNA repair genes and tumor suppressor genes and promote the expression of proto-oncogenes. Cytokines stimulate expression of inducible nitric oxide synthase (iNOS) in epithelial cells, thereby resulting in increased intracellular levels of nitric oxide (NO) and reactive nitrogen oxide species (RNOS). NO and RNOS interact with cellular DNA and proteins and thus result in DNA mutations and strand breaks, with inactivation of DNA repair proteins.15 A variety of different oncogenic mutations have been described in cholangiocarcinoma (Table 69-2). Their frequencies differ depending on the anatomic location, stage, type, and etiology of the tumor and the ethnicity of the patient.
| MALIGNANT PHENOTYPE | DYSREGULATED GENE PRODUCTS AND PATHWAYS |
|---|---|
| Proliferation |
Growth factors, dysregulated signaling pathways, and tyrosine kinases promote further tumor cell proliferation, as well as inhibition of senescence and apoptosis. Key signaling pathways in cholangiocarcinoma involve interleukin (IL)-6, cyclooxygenase-2 (COX-2), epidermal growth factor receptor (EGFR), ErbB2 (also known as human epidermal growth factor receptor 2 [HER 2]), hepatocyte growth factor (HGF), and vascular endothelial cell growth factor receptor (VEGFR). The IL-6 signaling axis in particular has been shown to be critical to the pathogenesis of cholangiocarcinoma.16,17 Increased IL-6 signaling is sustained in cholangiocarcinoma cells by the following mechanisms: (1) autocrine secretion of high levels of IL-6; (2) COX-2-mediated up-regulation of the IL-6 receptor subunit gp130; and (3) inactivation of the negative feedback loop through epigenetic silencing of suppressor of cytokine signaling 3 (SOCS3). Increased IL-6 signaling constitutively activates the PI3K pathway, Janus Kinase (JAK) signal transducers and activators of transcription (STAT) pathway, and p38- and p42/44-mitogen-activated protein kinase (MAPK) pathways, which in turn mediate cell survival, cell proliferation, and evasion of cell senescence. The EGFR pathway is also constitutively activated, resulting in further activation of p38- and p42/44-MAPK.18 The EGFR homolog ErbB2 is overexpressed in many cholangiocarcinomas, resulting in downstream induction of Raf/MAPK pathways. In addition to its oncogenic effects, MAPK induces overexpression of COX-2, which is further stimulated by increased concentrations of bile acids, oxysterols, and iNOS.19,20 COX-2 also mediates cell survival and proliferation. Sustained cell proliferation is further mediated by increased HGF secretion and overexpression of its receptor, c-Met.21 The multiple pathways form a complex interacting network.
CLINICAL FEATURES AND DIAGNOSIS
The diagnosis of cholangiocarcinoma is challenging because of the paucicellular character of the malignancy and requires a multidisciplinary approach that includes clinical evaluation and laboratory, endoscopic, and radiologic studies (Fig. 69-2 and Table 69-3). Intrahepatic cholangiocarcinoma manifests predominantly with abdominal pain and systemic symptoms such as cachexia, malaise, and fatigue. Extrahepatic cholangiocarcinoma manifests in most cases with painless jaundice secondary to malignant biliary obstruction. In 10% of patients, bacterial cholangitis is the initial presenting symptom. An “atrophy-hypertrophy” complex can be documented by physical examination as palpable hypertrophy of the contralateral, unaffected lobe of the liver, with atrophy of the affected lobe as a result of vascular encasement and bile ductal obstruction.
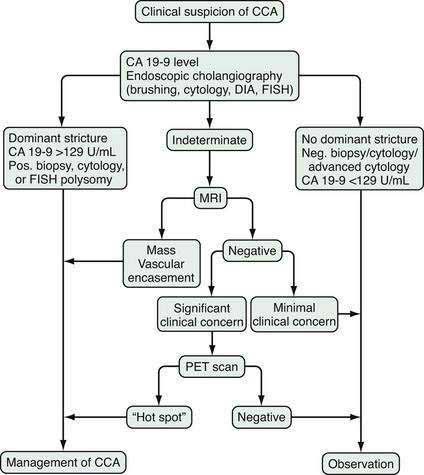
Table 69-4 Sloan-Kettering Cancer Staging System for Cholangiocarcinoma
| STAGE | CRITERIA |
|---|---|
| T1 |
Laboratory analysis may reveal evidence of obstructive cholestasis (e.g., elevation of serum alkaline phosphatase and bilirubin levels). Serum levels of several serum tumor markers (CA 19-9, carcinoembryonic antigen [CEA], and Ca-125) may be elevated in patients with cholangiocarcinoma; however, none of these serum markers is specific, and they each can be elevated in other gastroenterologic or gynecologic malignancies and in the setting of biliary inflammation or infection.22 The most commonly used marker is CA 19-9. In patients with PSC, the sensitivity and specificity of CA 19-9 for the diagnosis of cholangiocarcinoma is 79% and 98%, respectively, when the cutoff value is 129 U/mL. In patients without PSC, the sensitivity is 53% with a cutoff value of 100 U/mL.23,24 Importantly, patients with a negative Lewis antigen status do not express CA 19-9, and significant CA 19-9 elevations can occasionally be observed in patients with bacterial cholangitis and choledocholithiasis.
An essential diagnostic tool for cholangiocarcinoma is cholangiography, which provides both anatomic information and material for a tissue diagnosis. Several approaches can be used, including endoscopic retrograde cholangiopancreatography (ERCP), percutaneous transhepatic cholangiography (THC), and magnetic resonance cholangiography (MRCP) (Figs. 69-3 and 69-4). The choice of ERCP versus percutaneous THC depends on the location of a suspicious biliary stricture, local expertise, and accessibility of the stricture to the technique. Either technique provides information on intrabiliary tumor extension and allows cytologic sampling and therapeutic intervention for malignant biliary obstruction. Although such interventions are not possible with MRCP, this technique is noninvasive and provides additional information on the extent of extrabiliary tumor, vascular encasement, the relation of the primary tumor to surrounding structures, and intra- as well as extrahepatic metastases.2
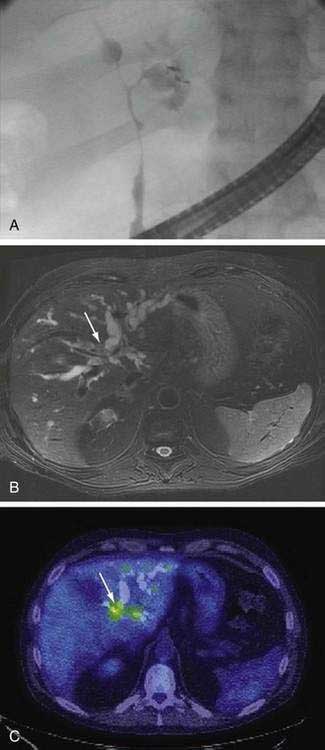
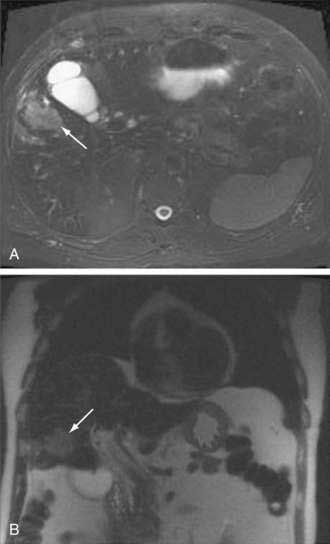
Computed tomography (CT) is used primarily for preoperative planning; it provides information on vascular and other anatomic structures that affect surgical decision making. The sensitivity of CT for detection of nodal N2 metastases (see later), however, is only 50%, and its accuracy in the evaluation of resectability is only 60% to 75%. Magnetic resonance imaging (MRI) is currently the best imaging technique for cholangiocarcinoma; its sensitivity and tumor tissue depiction are improved by the use of ferumoxide as a contrast agent.25 Cross-sectional imaging should be performed prior to cholangiography to help guide decisions regarding biliary drainage (see later). The use of positron emission tomography (PET)/CT should be limited to cases that are indeterminate with regard to extent and other features (including malignancy) after the other studies are performed. Its sensitivity for regional lymph node metastases is only 12%. Also, the sensitivity of CT and PET for the detection of cholangiocarcinoma do not differ. PET/CT achieves sensitivity and specificity rates of 93% and 80% for the detection of intrahepatic cholangiocarcinoma but only 55% and 33% for the detection of extrahepatic cholangiocarcinoma. False-positive PET results can be observed in the setting of inflammation.26
Endoscopic ultrasound (EUS) can contribute additional information about tumor dimension and anatomic location in relation to surrounding structures. In addition, it provides information about regional lymphadenopathy and allows sampling of lymph nodes by fine-needle aspiration. Biopsy of the primary lesion via EUS is highly discouraged, however, because of the potential for tumor cell spread.2
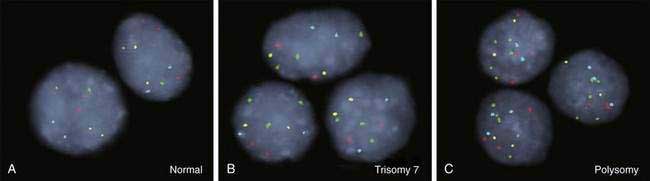
As for most malignancies, a tissue diagnosis of cholangiocarcinoma is desirable; however, a pathologic diagnosis is challenging because of the tumor’s paucicellular character. Tissue is difficult to obtain, and cellular reactive changes resulting from inflammation often complicate the diagnosis. The most common technique for collecting tumor tissue is brush cytology during ERCP, but conventional cytology is limited because of low specificity. Fluorescence in situ hybridization (FISH) has been demonstrated to increase the sensitivity and specificity of cytology for diagnosing cholangiocarcinoma in patients with and without PSC.27 Typical findings include (1) trisomy 7; (2) tetrasomy or duplication of all labeled chromosomes; or (3) polysomy or amplification of at least three chromosomes (Fig. 69-5). Polysomy is diagnostic of cholangiocarcinoma in the appropriate clinical setting, whereas trisomy 7 is not diagnostic but increases a patient’s risk for the development of cholangiocarcinoma. Tetrasomy has to be interpreted with caution because it can be observed during the M phase of the cell cycle during mitosis (see Chapter 3).2
STAGING
Cholangiocarcinoma has been classified according to the American Joint Committee on Cancer (AJCC)/ International Union Against Cancer (UICC) TNM (tumor node metastasis) system.28 This system is based on hepatocellular carcinoma and is therefore only of limited use in cholangiocarcinoma. It is especially suboptimal for hilar cholangiocarcinoma. An improved, preoperative staging system for extrahepatic cholangiocarcinoma is needed. In oncologic surgery, negative surgical margins (R0) correlate with lack of postoperative recurrence and with prognosis. A staging system for hilar cholangiocarcinoma that incorporates resectability requires that the extent of biliary disease, vascular encasement, and hepatic lobar atrophy be assessed, in addition to the information provided by a clinical TNM system. Unilobar atrophy is clinically important, because it indicates unilateral biliary obstruction with ipsilateral vascular encasement (atrophy-hypertrophy complex). This finding does not indicate unresectability unless it occurs in combination with contralateral vascular or biliary tumor involvement.29 Such a staging system has been proposed at Memorial Sloan-Kettering Cancer Center (MSKCC) (Table 69-4). Resectability, likelihood of curative (R0) resection, metastatic spread to N2-level lymph nodes (where N1 signifies metastasis to lymph nodes within the hepatoduodenal ligament and N2 signifies metastasis to peripancreatic, periduodenal, celiac, superior mesenteric, or posterior pancreaticoduodenal lymph nodes), and survival correlate with the tumor stage in this classification system.29
Table 69-5 Exclusion Criteria for Resection of Hilar Cholangiocarcinoma
TREATMENT
The only curative treatment option for cholangiocarcinoma is surgical extirpation. Surgical outcomes have improved substantially in the 2000s because of careful patient selection with lower surgical mortality rates and higher rates of R0 (negative surgical margin) resection.30 Surgical treatment options include resection and liver transplantation (see Chapter 95).
Solitary intrahepatic cholangiocarcinoma can be resected by hepatic segmentectomy or lobectomy. Five-year survival rates after resection of intrahepatic cholangiocarcinoma range from 22% to 42%. Survival is positively correlated with early tumor stage (localized tumor), younger age, and better performance status.30 Liver transplantation is not an option for intrahepatic cholangiocarcinoma because of disappointing five-year survival rates of 0% to 18%.31
Resection is also the treatment of choice for extrahepatic cholangiocarcinoma in the absence of PSC (Table 69-5). Five-year survival rates for extrahepatic CCA after R0 resection are 11% to 41% for hilar and 27% to 37% for distal cholangiocarcinoma. Unfortunately, R0 resectability rates are less than 50%.32 Occasionally, increased resectability can be achieved by preoperative portal vein embolization, resulting in compensatory hyperplasia of the contralateral hepatic lobe. This technique allows extended partial hepatectomy because of the increased volume of the remnant liver.33 Neither adjuvant nor neoadjuvant chemotherapy or radiation therapy has been shown to be effective and is therefore not recommended in patients with cholangiocarcinoma who undergo resection.
Table 69-3 Diagnostic Criteria for Cholangiocarcinoma
* Serum CA 19-9 >100 U/mL in patients with primary sclerosing cholangitis.
In the past, liver transplantation was not an option for patients with extrahepatic cholangiocarcinoma because five-year survival rates were only 23% to 26%.34 Subsequently, new liver transplantation protocols were developed and led to 5-year survival rates of 76% in carefully selected patients with hilar cholangiocarcinoma.35 The selection criteria are strict (see Table 69-3 and Chapter 95). The treatment protocol consists of neoadjuvant external beam radiation therapy with concurrent systemic 5-fluorouracil chemotherapy, followed by brachytherapy and oral maintenance chemotherapy with capecitabine until transplantation.
At the current time, no curative medical therapies for cholangiocarcinoma are available. A variety of chemotherapeutic agents such as gemcitabine, other antimetabolites, taxanes, platinum analogs, anthracyclines, and mitomycin have been evaluated as single or combination therapies. The only chemotherapeutic drug approved by the U.S. Food and Drug Administration (FDA) for cholangiocarcinoma is gemcitabine; however, no randomized, controlled phase III trials have shown a significant survival benefit with chemotherapy in patients with cholangiocarcinoma. Available studies have been statistically flawed or underpowered or have shown poor response rates. Therefore, chemotherapy is not a treatment option for these malignancies unless used in the palliative (see below) or neoadjuvant setting. Similarly, the use of radiation therapy remains controversial.2
Palliative Treatments
Cholangiocarcinoma is associated with substantial morbidity. Commonly, patients experience cholestasis, abdominal pain, and cachexia, which limit the quality of life. Therefore, palliative treatments are essential in the management of patients with cholangiocarcinoma. Options for restoration of biliary drainage include endoscopic, percutaneous, and surgical techniques. Endoscopic and percutaneous methods are based on placement of biliary stents (see Chapter 70), whereas surgical approaches create a bypass via a choledocho- or hepaticojejunostomy. The efficacies of similar endoscopic and surgical approaches are similar, but the mortality rate, frequency of procedure-related complications, and duration of hospital stay are higher for surgical palliation.36 The decision to pursue endoscopic or percutaneous biliary stent deployment is based on the anatomic location of the malignant stricture (see Chapter 70). In general, unilateral restoration of bile flow is sufficient. Cross-sectional imaging is critical before a stent is placed to avoid attempts at endoscopic drainage of an atrophic lobe or a lobe in which adequate drainage is not feasible. Retrograde injection of dye without drainage carries a high risk of iatrogenic bacterial cholangitis, which can be severe. Early intervention in a patient with malignant biliary obstruction is recommended because the time to normalization of the serum bilirubin level doubles from three to six weeks when the total bilirubin serum level is greater than 10 mg/dL.37
Most recently, photodynamic therapy (PDT) has been shown to be a feasible palliative treatment option that reduces cholestasis, improves quality of life, and possibly provides a survival benefit.38,39 PDT involves the systemic application of a photosensitizing agent (e.g., hematoporphyrin), followed by localized illumination of the tumor at a specific wavelength. Tumor cell cytotoxicity is achieved through reactive oxygen species-mediated cell death, tumor-vessel thrombosis, and tumor-specific immune reactions. The procedure is well tolerated and characterized by a low complication rate.
GALLBLADDER CARCINOMA
Gallbladder carcinoma is the second most common primary biliary malignancy and the fifth most common malignancy of the gastrointestinal tract. Like other biliary malignancies, gallbladder carcinoma is diagnosed at an advanced stage in the majority of cases. In only one third of the cases is the specific diagnosis of gallbladder carcinoma made prior to surgical exploration.40 The growth kinetics of gallbladder carcinoma are faster than those for cholangiocarcinoma, and, in general, gallbladder carcinoma is diagnosed at a later stage than ampullary carcinoma (see later). Gallbladder carcinoma is not amenable to medical or radiation therapy, and surgical resection is the only potentially curative treatment. Unfortunately, only a minority of patients are surgical candidates at the time of diagnosis. The prognosis of gallbladder carcinoma is dismal, with five-year survival rates of 0% to 10% and median survival of less than six months. In the 2000s, more aggressive surgical approaches have been advocated to improve outcomes.
EPIDEMIOLOGY
The distribution of gallbladder carcinoma is geographically heterogeneous, with the highest incidence rates (up to 21.5 per 100,000 population) observed in India. Incidence rates are also high in South America, Asia, and certain Eastern European countries such as Poland.41 Gallbladder carcinoma is rare in Western European countries and the United States, where the National Cancer Institute reported an age-adjusted incidence rate of 1.2 per 100,000 in 2007. Global incidence rates of gallbladder carcinoma parallel the incidence rates of cholelithiasis. With the exception of Japan, where the incidence has increased, age-adjusted incidence rates of gallbladder carcinoma have remained relatively stable in most countries since the 1970s.41 Ethnic differences in the incidence of gallbladder carcinoma have been described within the United States, with higher rates in whites than in African Americans. Mortality rates also vary globally. The age-adjusted mortality rate in the United States between 2000 and 2005 was 0.7 per 100,000, with an overall decrease since 1990. The highest mortality rate (35 per 100,000) was reported in southern Chile.42 The average age at diagnosis is 65 years, and the peak incidence is observed in the seventh and eighth decades of life. Globally, there is a female predisposition to gallbladder carcinoma.41
ETIOLOGY
The cause of gallbladder carcinoma is not well understood but is thought to be multifactorial. Several risk factors for gallbladder carcinoma have been described (Table 69-6). The primary risk factor for gallbladder carcinoma is cholelithiasis (see Chapter 65). Gallstones are found in 65% to 90% of patients with gallbladder carcinoma. Populations with high rates of cholelithiasis also have high rates of gallbladder carcinoma. Autopsy-based studies from Chile have suggested a seven-fold increased risk of gallbladder carcinoma in patients with cholelithiasis, whereas epidemiologic studies in the United States have observed only a marginally significant three-fold increased risk of gallbladder carcinoma in men with cholelithiasis.43,44 Gallbladder carcinoma actually develops in only 1% to 3% of patients with cholelithiasis, and 20% of patients with gallbladder carcinoma do not have evidence of cholelithiasis. Therefore, a prophylactic cholecystectomy in an asymptomatic patient with gallstones to prevent gallbladder carcinoma cannot be recommended. A positive correlation between the risk of gallbladder carcinoma and the size and number of gallstones has been reported but reflects the duration of cholelithiasis.45 No differences in the risk of gallbladder carcinoma have been observed with different types of gallstones. Porcelain gallbladder (extensive calcification of the gallbladder wall) is a classic, although controversial, risk factor for gallbladder carcinoma.46 Although an increased risk of gallbladder carcinoma has been reported in patients with a porcelain gallbladder, the risk may be limited to patients with selective mucosal calcification (types II and III porcelain gallbladder) rather than those with diffuse mucosal calcification (type I).47
Table 69-6 Risk Factors for Gallbladder Carcinoma
* Methylcholanthrene, O-aminoazotoluene, nitrosamines, possibly others.
Adenomatous polyps of the gallbladder constitute another risk factor for gallbladder carcinoma (see Chapter 67). The risk correlates positively with the size, type, and growth rate of the polyps. Patients with polyps that are greater than 1 cm in size, sessile, and associated with gallstones, exhibit a rapid increase in size, demonstrate arterial flow on Doppler ultrasonography, or are symptomatic are at increased risk of malignant transformation, and patients with such polyps warrant prophylactic cholecystectomy.46,48
Anomalous union of the pancreaticobiliary ductal system (AUPBD) has been associated with the development of gallbladder carcinoma. In this congenital defect, the pancreatic and bile ducts unite outside the duodenal wall in a long common channel. The anomaly is found incidentally in 1.5% to 2% of patients who undergo ERCP and leads to cholestasis and reflux of pancreatic secretions into the gallbladder with resulting chronic inflammation of the mucosa. Approximately 10% of patients with gallbladder carcinoma have coexisting AUPBD, and gallbladder carcinoma develops in 15% to 40% of those with AUPBD. Patients with an associated choledochal cyst have a lower frequency of gallbladder carcinoma than those without a choledochal cyst.49 Patients with AUPBD are usually 10 years younger at the time of diagnosis of gallbladder carcinoma and have a lower frequency of cholelithiasis than those without AUPBD. On the basis of a significantly increased risk of gallbladder carcinoma, several Japanese hepatobiliary oncology associations have recommended an aggressive, prophylactic surgical approach to patients with AUPBD.46
PSC has been associated with gallbladder carcinoma, and studies have reported that adenocarcinoma of the gallbladder develops in up to 20% of patients with PSC and that 40% to 60% of gallbladder masses in patients with PSC are malignant.50,51 Therefore, patients with PSC and a gallbladder mass of any size should undergo prophylactic cholecystectomy or be monitored closely for gallbladder carcinoma.
Adenomyomatosis of the gallbladder, which is characterized by microscopic invaginations (Rokitansky-Aschoff sinuses) of the mucosa with cyst formation in the muscularis propria, has been linked to gallbladder carcinoma (see Chapter 67). The magnitude of the malignant potential of adenomyomatosis has not been clearly defined and appears to depend on morphologic features and the patient’s age.52 Adenomyomatosis generally is viewed as a benign condition.
Other conditions associated with gallbladder carcinoma include inflammatory bowel disease, intrahepatic biliary dysplasia, and cholangiocarcinoma.51 Chronic carriers of Salmonella typhi or paratyphi have been shown to be at increased risk for the development of gallbladder carcinoma.53 Other bacteria such as Escherichia coli and Helicobacter pylori also have been associated with gallbladder carcinoma, but the data are not conclusive. First-degree relatives of patients with gallbladder carcinoma have a relative risk of 13.9 for developing this malignancy.54 Carcinogens, including methylcholanthrene, O-aminoazotoluene, and nitrosamines, have been identified in animal models of gallbladder carcinoma. Other potential carcinogens include mustard oil, products of free radical oxidation, and secondary bile acids.55
PATHOLOGY
From 80% to 95% of gallbladder carcinomas are adenocarcinomas; the majority of these are moderately-to-well differentiated.42 Less common types, in order of frequency, include undifferentiated or anaplastic carcinoma, squamous cell carcinoma, and adenosquamous carcinoma. Rare types include carcinoids, small cell carcinomas, malignant melanomas, lymphomas, and sarcomas.55 Sixty percent of gallbladder carcinomas are located in the gallbladder fundus, 30% in the body, and 10% in the gallbladder neck.56 Analogous to cholangiocarcinoma, the papillary form of gallbladder carcinoma has a lower potential for invasion and metastatic spread to lymph nodes.57 Gallbladder carcinoma spreads via direct invasion, lymphogenic or hematogenic metastasis, perineural invasion, and intraperitoneal or intraductal invasion. Lymphatic tumor cell spread is determined by the physiologic gallbladder lymphatic plexus, including the first-level lymph nodes along the biliary tract (cystic duct, bile duct, and hepatic duct) followed by pancreaticoduodenal lymph nodes as well as lymph nodes along the common hepatic artery and celiac axis. Lymph node metastases are described in 54% to 64% of patients and correlate with the depth of invasion. Gallbladder carcinoma has a predisposition to involve the liver bed because of venous drainage, predominantly into hepatic segments IVb and V (see Chapter 71), and the anatomic approximation that allows direct hepatic invasion. Perineural spread is observed in 24% and intraductal spread in 19% of cases.
PATHOGENESIS
Gallbladder carcinoma can develop from foci of mucosal dysplasia or carcinoma in situ (CIS) that progress to adenocarcinoma or from an adenoma-carcinoma sequence similar to that seen with colon cancer (see Chapter 123).58 Foci of dysplasia and CIS are frequently found adjacent to gallbladder carcinoma in surgically resected gallbladder specimens and are thought to be precursors of invasive adenocarcinoma.42 The time of progression of dysplasia to carcinoma is estimated to be 10 to 15 years.59 Like cholangiocarcinoma, the major pathogenic factor is inflammation. Increased iNOS and COX-2 expression has been demonstrated immunohistochemically in gallbladder carcinoma samples as well as in hyperplastic gallbladder mucosa from patients with AUPBD and also has been associated with TP53 tumor suppressor gene mutations in patients with gallbladder carcinoma. High expression and mutation rates of the TP53 gene have been demonstrated in 35% to 92% of gallbladder carcinomas, 86% of carcinomas in situ, and 28% of dysplastic foci, supporting an early role for TP53 mutation in the dysplasia-carcinoma progression sequence.60,61 In up to 60% of patients with gallbladder carcinoma, mutations of the K-ras oncogene have been detected; the frequency is highest in patients with AUPBD.62 Single studies have reported up-regulation of ErbB2 and the nm23 metastasis suppressor protein.63,64 Mutations and increased expression of the nuclear oncogene that encodes p16INK4 have been shown in several studies.65 Other studies have demonstrated loss of heterozygosity or microsatellite instabilities in chromosomal regions that harbor known or putative tumor suppressor genes.66 Therefore, available data indicate pathogenetic mechanisms similar to those of other biliary malignancies, but the molecular pathogenesis appears to depend on the specific etiology and mode of progression.
CLINICAL FEATURES AND DIAGNOSIS
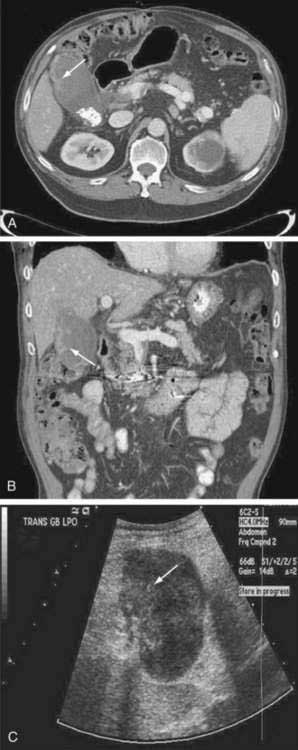
Abdominal ultrasound is often one of the first imaging studies performed in a patient who presents with the aforementioned symptoms. The sensitivity and accuracy of ultrasound for gallbladder carcinoma are 85% and 80%, respectively; early cancers, especially sessile polyps, can be missed. Typical imaging presentations of gallbladder carcinoma include focal or diffuse mural thickening of the gallbladder, an intraluminal mass greater than 2 cm in size that originates in the gallbladder wall, and a subhepatic mass that replaces or obscures the gallbladder and often invades adjacent organs (Fig. 69-6). Findings indicative of the malignant nature of a gallbladder lesion include irregular, asymmetrical mural thickening greater than 1 cm in depth and a nodular or smooth intraluminal mass greater than 1 cm in size, with fixation to the gallbladder wall, that is not displaced by the patient’s movements and has no acoustic shadow. In indeterminate cases, Doppler ultrasound can be attempted to differentiate a malignant from a benign gallbladder lesion on the basis of the pattern of the color signal, blood flow velocity, and resistive index (a measure of resistance to arterial blood flow).67 MRI and CT can be helpful in the diagnosis if the ultrasound findings are indeterminate. The role of PET in gallbladder carcinoma is evolving, and currently PET is not part of the routine evaluation. The sensitivity of PET for detecting gallbladder carcinoma is only 75% to 78%.68,69 Its main impact is in the detection of distant metastases that result in a change in management.26
STAGING
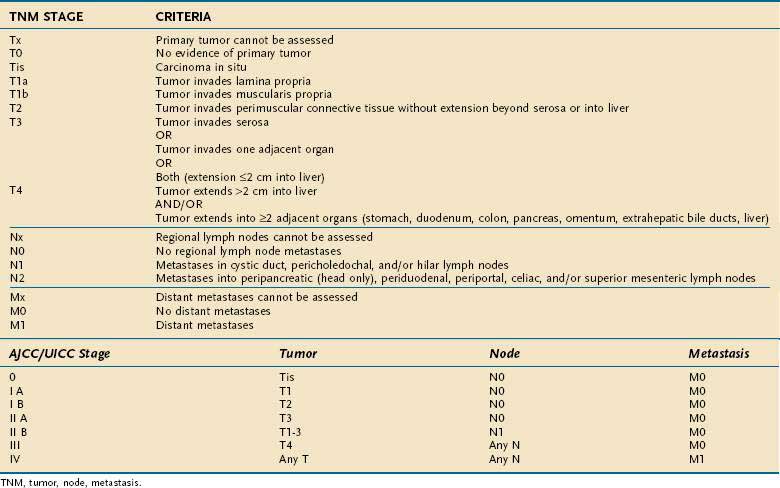
The most commonly used staging system is the TNM system described by the AJCC and UICC. The TNM-based staging system correlates with survival. Reported five-year survival rates for patients with stages 0, I, II, III, and IV gallbladder carcinoma are 60%, 39%, 15%, 5%, and 1%, respectively (Table 69-7).
TREATMENT
Surgery is the only potential curative therapeutic option for gallbladder carcinoma. Only 15% to 47% of patients are candidates for surgery at the time of diagnosis because of the advanced stage of their disease. Contraindications to resection include multiple hepatic or peritoneal metastases, malignant ascites, distant metastases, extensive involvement of the hepatoduodenal ligament, encasement or occlusion of major vessels, and poor performance status. Direct involvement of the colon, duodenum, or liver is not considered an absolute contraindication to surgical resection, however. The goal of surgical treatment is an R0 resection, defined as negative margins and nodal dissection one level past microscopically involved lymph nodes. R0 resection in gallbladder carcinoma has been shown to correlate with survival and with significantly increased five-year survival rates.70
Surgical procedures with curative extent include (1) simple cholecystectomy; (2) extended or radical cholecystectomy with additional resection of greater than 2 cm of the gallbladder bed plus lymphadenectomy of the hepatoduodenal ligament behind the second part of the duodenum, head of the pancreas, and celiac axis; (3) extended cholecystectomy with hepatic, segmental, or lobar resection; (4) extended cholecystectomy with extensive para-aortic lymph node resection; and (5) extended cholecystectomy with bile duct resection or pancreaticoduodenectomy. The surgical approach is dictated by the extent of tumor. Stage Tis and T1a (see Table 69-7) gallbladder cancer can be treated with simple cholecystectomy, with five-year survival rates of 85% to 100%. A few reports favor simple cholecystectomy also for stage 1b gallbladder carcinoma and report similar survival rates after either simple or radical cholecystectomy.71,72 Up to 15% of patients with stage 1b gallbladder carcinoma, however, are positive for lymph node metastases, compared with 2.5% of patients with stage 1a gallbladder carcinoma. Also, higher recurrence rates have been observed after simple (versus radical) cholecystectomy; therefore, radical cholecystectomy is preferred for stage 1b gallbladder carcinoma.73,74 Invasion of the muscularis propria, as in stage T2 or higher tumors, requires radical cholecystectomy. The extent and benefit of partial hepatectomy in stage T2 tumors are controversial, and partial hepatectomy has not been shown to prolong survival. Also, the surgical approach to stage T3 and T4 tumors is controversial. Some studies show no five-year survival benefit after radical cholecystectomy for stage T3 and T4 tumors, but other studies report five-year survival rates of 15% to 63% and 7% to 25%, respectively. Because of the poor prognosis of gallbladder carcinoma and the possibility of a survival benefit, as well as prolongation of survival until recurrence, a radical surgical approach to these advanced-stage gallbladder carcinomas is recommended by many centers.
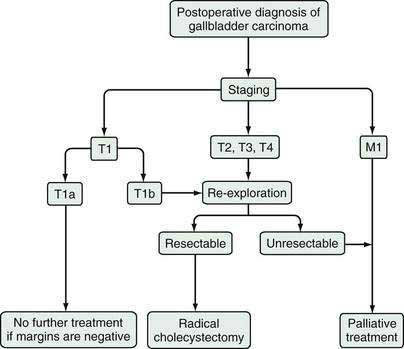
When gallbladder carcinoma is diagnosed during laparoscopy, the procedure should be converted to an open procedure, and the laparoscopic port sites should be resected, because tumor may recur at these sites secondary to iatrogenic dissemination.75 Further surgical management then depends on the tumor stage, as outlined above and in Figure 69-7. When gallbladder carcinoma is diagnosed postoperatively, further management depends on the tumor stage and the presence or absence of tumor at the margins of the surgical specimen.
AMPULLARY CARCINOMA
EPIDEMIOLOGY
Ampullary carcinomas are rare, accounting for fewer than 1% of all gastrointestinal cancers and 4% to 8% of periampullary carcinomas. The annual incidence has been estimated to be 0.6 per 100,000 population.76,77 Peak incidence is in the seventh decade of life. There is a slight male predominance, with a male-to-female ratio of 1.48 : 1.77 Racial heterogeneity has been observed; the vast majority of patients are white, followed by patients of Hispanic and Asian descent. African Americans have the lowest incidence rates in the United States.76
ETIOLOGY
Although the etiology of ampullary carcinomas is unknown in the majority of cases, several conditions have been associated with this malignancy, mostly in case reports or small series. Familial adenomatous polyposis (FAP) is an important risk factor for the development of ampullary carcinomas, with a relative risk of 124 (see Chapter 122).78 Periampullary carcinoma is the second most common cause of death (after colon cancer) in patients with FAP. Usually, periampullary carcinoma arises later than colorectal carcinoma in this patient group but earlier in comparison with sporadic ampullary carcinomas.77 Screening for upper gastrointestinal lesions (polyps or carcinoma) at regular intervals of six months to four years, depending on the degree of duodenal polyposis, is therefore recommended in patients with FAP. Similarly, increased rates of ampullary carcinoma have been described in patients with Gardner’s syndrome, a variant of FAP (see Chapter 122).79 Hereditary nonpolyposis colorectal cancer does not appear to predispose to ampullary carcinoma.80 Other genetic diseases reported to predispose to the development of ampullary carcinoma include neurofibromatosis type I and Muir-Torre syndrome.81,82 As for cholangiocarcinoma, chronic liver fluke infection has been reported to be a risk factor for ampullary carcinoma (see Chapter 82).77
PATHOLOGY
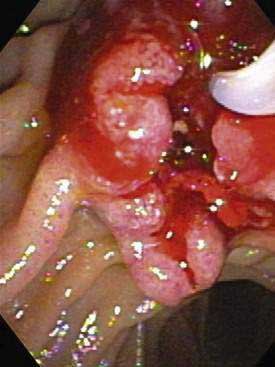
The ampulla of Vater is an anatomically complex area that consists of the papilla, common pancreaticobiliary channel, distal bile duct, and distal main pancreatic duct. Macroscopically, ampullary carcinomas are classified into the following three subtypes: (1) intramural protruding (intra-ampullary), (2) extramural protruding (periampullary), and (3) ulcerating ampullary77 (Fig. 69-8). The ulcerating type is usually diagnosed at an advanced stage and has the highest rates of lymph node metastases. Consistent with its anatomic heterogeneity, the ampulla includes several different histologic cell types, such as epithelia of the common pancreaticobiliary channel, bile duct, pancreatic duct, or duodenal mucosa, Brunner’s glands, and aberrant pancreatic acini in the wall of the bile duct. The most common site of cellular atypia is found in the area of the common pancreaticobiliary channel, followed by the pancreatic duct, duodenal epithelium, and Brunner’s glands.83 Seventy-five percent of ampullary neoplasias are adenocarcinomas, 20% are benign adenomas, and 5% are neuroendocrine tumors.84 Adenocarcinomas account for 90% of ampullary malignancies; the rest include unusual types, such as mucinous, signet-ring cell, and undifferentiated carcinomas.84 Histopathologically, 90% of ampullary adenocarcinomas can be classified into pancreaticobiliary or intestinal types.85,86 Immunohistochemically, the two types can be distinguished by high cytokeratin 7 expression and a lack of intestinal apomucin (MUC2) in the pancreaticobiliary type and cytokeratin 20 as well as MUC2 expression in the intestinal type.87 The frequencies of the two different histologic types, as well as their correlation with invasion, lymph node metastases, and prognosis, differ in various studies, with some studies indicating that the frequency is higher and the prognosis is worse for the pancreaticobiliary type and other studies finding the opposite results.85–87
PATHOGENESIS
The majority of ampullary carcinomas follow an adenoma-carcinoma sequence. In 30% to 91% of ampullary carcinomas, residual adenomatous tissue is found.77 Although precursor lesions can develop from intestinal as well as pancreaticobiliary-type tissue, carcinomas of the intestinal type typically develop from adenomas, whereas pancreaticobiliary and ulcerating carcinomas often lack a precursor lesion.77 On the molecular level, an increased frequency of K-ras mutations has been observed in 24% to 47% of tumors and is more common in the intestinal than pancreaticobiliary type.88 Also, p53 overexpression has been observed in 46% of tumors and is thought to be associated with ulcerating ampullary carcinomas. In an immunohistochemical study, aberrant expression of cell cycle regulators (i.e., p21WAF1/CIP1, p27Kip1, p16INK4, cyclin D1, type 8, and cyclin E, and the retinoblastoma protein [pRb]), was observed.89 These changes are similar to those seen in colorectal and pancreatic carcinomas, yet are distinctive. More research is necessary to understand the development of these tumors on a molecular level.
CLINICAL FEATURES AND DIAGNOSIS
Like the other periampullary and biliary malignancies, ampullary carcinomas present initially with obstructive jaundice in 70% to 82% of cases. Pancreaticobiliary ampullary carcinomas in particular have been reported to present initially with obstructive jaundice.85 Because of their anatomic location, cholestasis develops at an earlier stage than in other periampullary and biliary malignancies, and the resectability rate is therefore higher at the time of diagnosis. Nonicteric patients may present with bacterial cholangitis. Rare patients have “silver stools” as a result of the combination of acholic stools that result from bile duct obstruction and bleeding of the tumor. When obstructive cholangitis is suspected, further diagnostic evaluation is similar to that for other biliary malignancies. Immunohistochemical analysis has shown high expression of CEA and CA 19-9 in the tumor,90 but no studies have evaluated the serum concentrations of these markers in patients with ampullary carcinoma.
Usually, ampullary carcinomas are diagnosed by endoscopy on the basis of their macroscopic appearance and findings on biopsy specimens (see Fig. 69-8). Subsequent diagnostic tests are directed toward an assessment of resectability and detection of metastases. As for other biliary and periampullary carcinomas, radiologic techniques such as CT and MRI are used commonly in this setting. On MRI/MRCP, ampullary carcinoma usually is seen as a discrete, hypodense mass on T2-weighted images. Occasionally, the tumor can present as irregular thickening around the bile duct or bulging into the duodenum. Frequently, dilatation of both the bile and pancreatic ducts (“double-duct sign”) or only the bile duct is seen; dilatation of the pancreatic duct alone is seen rarely.91 Often, EUS is used in the preoperative evaluation. Its accuracy for detecting invasion of adjacent organs is 80% to 90%, and its sensitivity and specificity for detecting vascular invasion are 73% and 90%, respectively.92,93
STAGING
Ampullary carcinomas are classified according to the AJCC/UICC TNM classification.28 The T stage was shown to be predictive of survival in a univariate analysis but not in a multivariate analysis.85,87 Independent predictors of survival by multivariate analysis are lymphovascular invasion, perineural invasion, stage greater than or equal to III, and pancreaticobiliary subtype.85 Several studies have confirmed the significance of nodal involvement in predicting survival.87,94
TREATMENT
As for other biliary malignancies, surgical resection is the only curative treatment for ampullary carcinomas. In contrast to the other biliary malignancies, however, 77% to 88% of ampullary carcinomas are resectable at the time of diagnosis.95 The standard surgical approach is pancreaticoduodenectomy. Outcomes are excellent in the absence of lymph node metastases, with five-year survival rates of 68% to 78%.96,97 In the presence of lymph node–positive disease, the prognosis worsens significantly, with five-year survival rates of 16% to 25%.96,97 Extracapsular lymph node involvement results in further worsening of the prognosis, with a five-year survival rate of only 9%.90 Nodal microinvolvement has been reported to be an adverse prognostic factor, and immunohistochemical analysis of resected nodes has been recommended.98 Limited surgical or endoscopic papillectomy has been reported but is not recommended, because recurrence rates are higher than with pancreaticoduodenectomy.1 Chemotherapy and radiation therapy have not been evaluated in randomized controlled trials. A few studies have used 5-fluorouracil and radiation as adjuvant therapy in patients with node-positive disease and reported improved survival.99,100 In the absence of large, randomized controlled trials, however, such treatment is not standard. Palliative treatment should be directed at alleviating tumor-associated complications with the goal of optimizing the patient’s quality of life. Obstructive cholestasis is a major cause of morbidity and can usually be treated palliatively either by endoscopic or percutaneous placement of a biliary stent or by a surgical bypass similar to that carried out for other biliary or periampullary malignancies.
OTHER TUMORS OF THE BILE DUCTS AND GALLBLADDER
Other neoplastic diseases may involve the biliary tract (Table 69-8). Their inclusion in the differential diagnosis of biliary tumors is essential, because management differs depending on the tumor type. Tumors of neuroectodermal origin such as carcinoids (see Chapter 31) and paragangliomas are rare and typically nonfunctioning.101 They are located most commonly in the ampulla of Vater. Occasionally, carcinoids develop in the extrahepatic biliary tree, predominantly in the bile duct. Patients are usually female and young. Primary carcinoids of the biliary tract constitute less than 1% of all gastrointestinal carcinoids and usually are not associated with the carcinoid syndrome.102,103 Approximately one third of patients have metastases at diagnosis. The treatment of choice is surgical resection, and the prognosis is generally good.104–106 Patients with paragangliomas often present with gastrointestinal bleeding; only 25% present with jaundice. Their malignant potential has been estimated to be 33%, and some investigators recommend pancreaticoduodenectomy as the treatment of choice.107 Granular cell tumors, which are of neuronal derivation, are extremely rare; only a few cases have been described. Usually, they are located in the extrahepatic biliary tree, particularly at the junction of the cystic duct and the bile duct.108 Occasionally, they can cause biliary obstruction, as occurs when they are located in the hepatic hilum.108 Because of their benign character, resection is usually curative.109 Rarely, neuromas of the extrahepatic biliary tree develop after cholecystectomy.110
| GALLBLADDER | |
|---|---|
| Benign | Adenoma |
| Granular cell tumor | |
| Mesenchymal tumor (lipoma, leiomyoma, hemangioma, lymphangioma) | |
| Paraganglioma | |
| Malignant | Adenosquamous carcinoma |
| Carcinoid | |
| Small cell carcinoma | |
| Spindle cell sarcomatoid carcinoma | |
| Others (angiosarcoma, carcinosarcoma, Kaposi’s sarcoma, leiomyosarcoma, malignant fibrous histiocytoma, melanoma, metastatic tumors, non-Hodgkin’s lymphoma, rhabdomyosarcoma) | |
| Tumor-like lesions | Adenomyoma/adenomyomatosis |
| Cholesterol polyp | |
| Heterotopia (gastric, pancreatic, liver, adrenal, thyroid) | |
| Inflammatory polyp |
| BILE DUCTS | |
|---|---|
| Benign | Adenomyoma |
| Bile duct adenoma | |
| Biliary adenofibroma | |
| Biliary cystadenoma and cystadenocarcinoma | |
| Biliary hamartoma | |
| Ciliated hepatic foregut cyst | |
| Granular cell tumor | |
| Neuroma | |
| Serous cystadenoma | |
| Solitary or multiple cysts | |
| Malignant | Carcinoid |
| Embryonal (botryoid) rhabdosarcoma | |
| Leukemia | |
| Lymphoma | |
| Melanoma | |
| Metastatic tumor | |
| Paraganglioma | |
| Precursor lesions | Biliary dysplasia (intraepithelial neoplasia/atypical hyperplasia) |
| Intraductal papillary neoplasia |
Mesenchymal tumors, such as lipomas, leiomyomas, hemangiomas, and lymphangiomas, have been described in the gallbladder. In general, mesenchymal tumors are extremely rare and restricted to case reports. Lymphangiomas are often asymptomatic and only incidentally detected; however, they can increase in size and result in abdominal pain or jaundice. Ultrasound, CT, and MRI/MRCP aid in the preoperative diagnosis. Usually, they manifest as a multilocular, fluid-filled, cystic mass with thin walls and septa and show enhanced signal density with contrast administration.111 Most of the reported cases have been treated successfully with surgical resection, including cholecystectomy if the tumor is located within the gallbladder, or endoscopic resection if the tumor is in the area of the ampulla of Vater.112–115 Hamartomas also have been reported in the area of the ampulla of Vater and have been resected successfully by endoscopy.116
Heterotopia of the gallbladder may be caused by gastric, pancreatic, hepatic, adrenal, or thyroid tissue. Clinical complications such as hemorrhage are extremely rare.117,118 Benign bile duct lesions include adenomas, cystadenomas, adenofibromas, cysts, and granular cell tumors. Adenomyomas are found more commonly in the ampulla of Vater. Cystadenomas are more common in women and manifest primarily with abdominal pain. They are found predominantly in the intrahepatic biliary tree and are characterized on ultrasound by papillary extrusions of the wall and septa. They are considered premalignant because of their potential to transform into cystadenocarcinomas; hence, the treatment of choice is complete resection (see Chapter 94).119–121
Malignant tumors of the biliary tree other than cholangiocarcinoma include cystadenocarcinomas, lymphomas, and malignant melanomas. These malignancies arise primarily in the extrahepatic bile ducts. Cystadenocarcinomas can be distinguished morphologically from cholangiocarcinomas by their cystic character.122 They are rarely located in the gallbladder.123 Symptoms are nonspecific, and CT and MRI can be helpful in making the diagnosis. The treatment of choice is surgical resection.123 Malignant melanoma of the biliary tree is uncommon and should prompt investigation for a cutaneous melanoma, because cases of metastatic spread to the bile ducts have been described.124,125 Lymphomas can occasionally involve the extrahepatic biliary tree and often are mistaken for cholangiocarcinoma.126,127 In general, biliary lymphomas are very rare and account for less than 1% of lymphomas.127,128 Embryonal rhabdomyosarcoma of the biliary tree is extremely rare in adults but is the most common malignant tumor at this anatomic location in children.129 Frequently, it is misdiagnosed preoperatively as a choledochal cyst.130 Complete surgical resection is rarely possible, and a multidisciplinary approach to treatment is recommended. The prognosis of biliary rhabdosarcomas is good, with reported five-year survival rates of up to 78%.131 Few reports exist of follicular lymphomas originating in the extrahepatic biliary tree and gallbladder. Often, these tumors are diagnosed after resection.
Blechacz B, Gores GJ. Cholangiocarcinoma: Advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48:308-21. (Ref 2.)
Buckles DC, Lindor KD, Larusso NF, et al. In primary sclerosing cholangitis, gallbladder polyps are frequently malignant. Am J Gastroenterol. 2002;97:1138-42. (Ref 50.)
Csendes A, Becerra M, Rojas J, Medina E. Number and size of stones in patients with asymptomatic and symptomatic gallstones and gallbladder carcinoma: A prospective study of 592 cases. J Gastrointest Surg. 2000;4:481-5. (Ref 45.)
Gores GJ, Nagorney DM, Rosen CB. Cholangiocarcinoma: Is transplantation an option? For whom? J Hepatol. 2007;47:455-9. (Ref 35.)
Isomoto H, Kobayashi S, Werneburg NW, et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42:1329-38. (Ref 16.)
Ito H, Matros E, Brooks DC, et al. Treatment outcomes associated with surgery for gallbladder cancer: A 20-year experience. J Gastrointest Surg. 2004;8:183-90. (Ref 70.)
Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184-90. (Ref 15.)
Kimura W, Futakawa N, Yamagata S, et al. Different clinicopathologic findings in two histologic types of carcinoma of papilla of Vater. Jpn J Cancer Res. 1994;85:161-6. (Ref 86.)
Levy C, Lymp J, Angulo P, et al. The value of serum CA 19-9 in predicting cholangiocarcinomas in patients with primary sclerosing cholangitis. Dig Dis Sci. 2005;50:1734-40. (Ref 23.)
Moreno Luna LE, Kipp B, Halling KC, et al. Advanced cytologic techniques for the detection of malignant pancreatobiliary strictures. Gastroenterology. 2006;131:1064-72. (Ref 27.)
O’Connell JB, Maggard MA, Manunga J, et al. Survival after resection of ampullary carcinoma: A national population-based study. Ann Surg Oncol. 2008;15:1820-7. (Ref 76.)
Petrowsky H, Wildbrett P, Husarik DB, et al. Impact of integrated positron emission tomography and computed tomography on staging and management of gallbladder cancer and cholangiocarcinoma. J Hepatol. 2006;45:43-50. (Ref 26.)
van der Gaag NA, ten Kate FJ, Lagarde SM, et al. Prognostic significance of extracapsular lymph node involvement in patients with adenocarcinoma of the ampulla of Vater. Br J Surg. 2008;95:735-43. (Ref 94.)
Yoon JH, Canbay AE, Werneburg NW, et al. Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: Implications for biliary tract carcinogenesis. Hepatology. 2004;39:732-8. (Ref 19.)
Yoon JH, Gwak GY, Lee HS, et al. Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J Hepatol. 2004;41:808-14. (Ref 18.)
1. Kondo S, Takada T, Miyazaki M, et al. Guidelines for the management of biliary tract and ampullary carcinomas: Surgical treatment. J Hepatobiliary Pancreat Surg. 2008;15:41-54.
2. Blechacz B, Gores GJ. Cholangiocarcinoma: Advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48:308-21.
3. Welzel TM, McGlynn KA, Hsing AW, et al. Impact of classification of hilar cholangiocarcinomas (Klatskin tumors) on the incidence of intra- and extrahepatic cholangiocarcinoma in the United States. J Natl Cancer Inst. 2006;98:873-5.
4. Farley DR, Weaver AL, Nagorney DM. “Natural history” of unresected cholangiocarcinoma: Patient outcome after noncurative intervention. Mayo Clin Proc. 1995;70:425-9.
5. Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Semin Liver Dis. 2004;24:115-25.
6. Burak K, Angulo P, Pasha TM, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol. 2004;99:523-6.
7. Watanapa P, Watanapa WB. Liver fluke-associated cholangiocarcinoma. Br J Surg. 2002;89:962-70.
8. Chapman RW. Risk factors for biliary tract carcinogenesis. Ann Oncol. 1999;10(Suppl 4):308-11.
9. Lesurtel M, Regimbeau JM, Farges O, et al. Intrahepatic cholangiocarcinoma and hepatolithiasis: An unusual association in Western countries. Eur J Gastroenterol Hepatol. 2002;14:1025-7.
10. Tocchi A, Mazzoni G, Liotta G, et al. Late development of bile duct cancer in patients who had biliary-enteric drainage for benign disease: A follow-up study of more than 1,000 patients. Ann Surg. 2001;234:210-14.
11. Walker NJ, Crockett PW, Nyska A, et al. Dose-additive carcinogenicity of a defined mixture of “dioxin-like compounds.”. Environ Health Perspect. 2005;113:43-8.
12. Blechacz BR, Gores GJ. Cholangiocarcinoma. Clin Liver Dis. 2008;12:131-50.
13. Lim JH, Park CK. Pathology of cholangiocarcinoma. Abdom Imaging. 2004;29:540-7.
14. Nomoto K, Tsuneyama K, Cheng C, et al. Intrahepatic cholangiocarcinoma arising in cirrhotic liver frequently expressed p63-positive basal/stem-cell phenotype. Pathol Res Pract. 2006;202:71-6.
15. Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184-90.
16. Isomoto H, Kobayashi S, Werneburg NW, et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42:1329-38.
17. Kobayashi S, Werneburg NW, Bronk SF, et al. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology. 2005;128:2054-65.
18. Yoon JH, Gwak GY, Lee HS, et al. Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J Hepatol. 2004;41:808-14.
19. Yoon JH, Canbay AE, Werneburg NW, et al. Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: Implications for biliary tract carcinogenesis. Hepatology. 2004;39:732-8.
20. Yoon JH, Higuchi H, Werneburg NW, et al. Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology. 2002;122:985-93.
21. Lai GH, Radaeva S, Nakamura T, Sirica AE. Unique epithelial cell production of hepatocyte growth factor/scatter factor by putative precancerous intestinal metaplasias and associated “intestinal-type” biliary cancer chemically induced in rat liver. Hepatology. 2000;31:1257-65.
22. Chen CY, Shiesh SC, Tsao HC, Lin XZ. The assessment of biliary CA 125, CA 19-9 and CEA in diagnosing cholangiocarcinoma—the influence of sampling time and hepatolithiasis. Hepatogastroenterology. 2002;49:616-20.
23. Levy C, Lymp J, Angulo P, et al. The value of serum CA 19-9 in predicting cholangiocarcinomas in patients with primary sclerosing cholangitis. Dig Dis Sci. 2005;50:1734-40.
24. Patel AH, Harnois DM, Klee GG, et al. The utility of CA 19-9 in the diagnoses of cholangiocarcinoma in patients without primary sclerosing cholangitis. Am J Gastroenterol. 2000;95:204-7.
25. Raman SS, Lu DS, Chen SC, et al. Hepatic MR imaging using ferumoxides: Prospective evaluation with surgical and intraoperative sonographic confirmation in 25 cases. AJR Am J Roentgenol. 2001;177:807-12.
26. Petrowsky H, Wildbrett P, Husarik DB, et al. Impact of integrated positron emission tomography and computed tomography on staging and management of gallbladder cancer and cholangiocarcinoma. J Hepatol. 2006;45:43-50.
27. Moreno Luna LE, Kipp B, Halling KC, et al. Advanced cytologic techniques for the detection of malignant pancreatobiliary strictures. Gastroenterology. 2006;131:1064-72.
28. Greene FL, Page DL, Fleming ID, editors. AJCC Cancer Staging Manual, 6th ed. 2002. Springer, New York: 421
29. Jarnagin WR, Fong Y, DeMatteo RP, et al. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann Surg. 2001;234:507-17.
30. Nathan H, Pawlik TM, Wolfgang CL, et al. Trends in survival after surgery for cholangiocarcinoma: A 30-year population-based SEER database analysis. J Gastrointest Surg. 2007;11:1488-96.
31. Weimann A, Varnholt H, Schlitt HJ, et al. Retrospective analysis of prognostic factors after liver resection and transplantation for cholangiocellular carcinoma. Br J Surg. 2000;87:1182-7.
32. Nagorney DM, Kendrick ML. Hepatic resection in the treatment of hilar cholangiocarcinoma. Adv Surg. 2006;40:159-71.
33. Abdalla EK, Barnett CC, Doherty D, et al. Extended hepatectomy in patients with hepatobiliary malignancies with and without preoperative portal vein embolization. Arch Surg. 2002;137:675-80.
34. Meyer CG, Penn I, James L. Liver transplantation for cholangiocarcinoma: Results in 207 patients. Transplantation. 2000;69:1633-7.
35. Gores GJ, Nagorney DM, Rosen CB. Cholangiocarcinoma: Is transplantation an option? For whom? J Hepatol. 2007;47:455-9.
36. Smith AC, Dowsett JF, Russell RC, et al. Randomised trial of endoscopic stenting versus surgical bypass in malignant low bile duct obstruction. Lancet. 1994;344:1655-60.
37. Weston BR, Ross WA, Wolff RA, et al. Rate of bilirubin regression after stenting in malignant biliary obstruction for the initiation of chemotherapy: How soon should we repeat endoscopic retrograde cholangiopancreatography? Cancer. 2008;112:2417-23.
38. Shim CS, Cheon YK, Cha SW, et al. Prospective study of the effectiveness of percutaneous transhepatic photodynamic therapy for advanced bile duct cancer and the role of intraductal ultrasonography in response assessment. Endoscopy. 2005;37:425-33.
39. Zoepf T, Jakobs R, Arnold JC, et al. Palliation of nonresectable bile duct cancer: Improved survival after photodynamic therapy. Am J Gastroenterol. 2005;100:2426-30.
40. Sheth S, Bedford A, Chopra S. Primary gallbladder cancer: Recognition of risk factors and the role of prophylactic cholecystectomy. Am J Gastroenterol. 2000;95:1402-10.
41. Randi G, Franceschi S, La Vecchia C. Gallbladder cancer worldwide: Geographical distribution and risk factors. Int J Cancer. 2006;118:1591-602.
42. Lazcano-Ponce EC, Miquel JF, Munoz N, et al. Epidemiology and molecular pathology of gallbladder cancer. CA Cancer J Clin. 2001;51:349-64.
43. Maringhini A, Moreau JA, Melton LJ3rd, et al. Gallstones, gallbladder cancer, and other gastrointestinal malignancies. An epidemiologic study in Rochester, Minnesota. Ann Intern Med. 1987;107:30-5.
44. Nervi F, Duarte I, Gomez G, et al. Frequency of gallbladder cancer in Chile, a high-risk area. Int J Cancer. 1988;41:657-60.
45. Csendes A, Becerra M, Rojas J, Medina E. Number and size of stones in patients with asymptomatic and symptomatic gallstones and gallbladder carcinoma: A prospective study of 592 cases. J Gastrointest Surg. 2000;4:481-5.
46. Miyazaki M, Takada T, Miyakawa S, et al. Risk factors for biliary tract and ampullary carcinomas and prophylactic surgery for these factors. J Hepatobiliary Pancreat Surg. 2008;15:15-24.
47. Stephen AE, Berger DL. Carcinoma in the porcelain gallbladder: A relationship revisited. Surgery. 2001;129:699-703.
48. Myers RP, Shaffer EA, Beck PL. Gallbladder polyps: Epidemiology, natural history and management. Can J Gastroenterol. 2002;16:187-94.
49. Funabiki T, Matsubara T, Miyakawa S, Ishihara S. Pancreaticobiliary maljunction and carcinogenesis to biliary and pancreatic malignancy. Langenbecks Arch Surg. 2009;394:159-69.
50. Buckles DC, Lindor KD, Larusso NF, et al. In primary sclerosing cholangitis, gallbladder polyps are frequently malignant. Am J Gastroenterol. 2002;97:1138-42.
51. Lewis JT, Talwalkar JA, Rosen CB, et al. Prevalence and risk factors for gallbladder neoplasia in patients with primary sclerosing cholangitis: Evidence for a metaplasia-dysplasia-carcinoma sequence. Am J Surg Pathol. 2007;31:907-13.
52. Nabatame N, Shirai Y, Nishimura A, et al. High risk of gallbladder carcinoma in elderly patients with segmental adenomyomatosis of the gallbladder. J Exp Clin Cancer Res. 2004;23:593-8.
53. Shukla VK, Singh H, Pandey M, et al. Carcinoma of the gallbladder—is it a sequel of typhoid? Dig Dis Sci. 2000;45:900-3.
54. Fernandez E, La Vecchia C, D’Avanzo B, et al. Family history and the risk of liver, gallbladder, and pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 1994;3:209-12.
55. Misra S, Chaturvedi A, Misra NC, Sharma ID. Carcinoma of the gallbladder. Lancet Oncol. 2003;4:167-76.
56. Reid KM, Ramos-De la Medina A, Donohue JH. Diagnosis and surgical management of gallbladder cancer: A review. J Gastrointest Surg. 2007;11:671-81.
57. Gore RM, Shelhamer RP. Biliary tract neoplasms: Diagnosis and staging. Cancer Imaging. 2007;7(Spec No A):S15-23.
58. Roa I, de Aretxabala X, Araya JC, Roa J. Preneoplastic lesions in gallbladder cancer. J Surg Oncol. 2006;93:615-23.
59. Roa I, Araya JC, Villaseca M, et al. Preneoplastic lesions and gallbladder cancer: An estimate of the period required for progression. Gastroenterology. 1996;111:232-6.
60. Wee A, Teh M, Raju GC. Clinical importance of p53 protein in gall bladder carcinoma and its precursor lesions. J Clin Pathol. 1994;47:453-6.
61. Fujii K, Yokozaki H, Yasui W, et al. High frequency of p53 gene mutation in adenocarcinomas of the gallbladder. Cancer Epidemiol Biomarkers Prev. 1996;5:461-6.
62. Sasatomi E, Tokunaga O, Miyazaki K. Precancerous conditions of gallbladder carcinoma: Overview of histopathologic characteristics and molecular genetic findings. J Hepatobiliary Pancreat Surg. 2000;7:556-67.
63. Fujii K, Yasui W, Shimamoto F, et al. Immunohistochemical analysis of nm23 gene product in human gallbladder carcinomas. Virchows Arch. 1995;426:355-9.
64. Kim YW, Huh SH, Park YK, et al. Expression of the c-erb-B2 and p53 protein in gallbladder carcinomas. Oncol Rep. 2001;8:1127-32.
65. Lynch BC, Lathrop SL, Ye D, et al. Expression of the p16(INK4a) gene product in premalignant and malignant epithelial lesions of the gallbladder. Ann Diagn Pathol. 2008;12:161-4.
66. Yoshida T, Sugai T, Habano W, et al. Microsatellite instability in gallbladder carcinoma: Two independent genetic pathways of gallbladder carcinogenesis. J Gastroenterol. 2000;35:768-74.
67. Hirooka Y, Naitoh Y, Goto H, et al. Differential diagnosis of gall-bladder masses using colour Doppler ultrasonography. J Gastroenterol Hepatol. 1996;11:840-6.
68. Anderson CD, Rice MH, Pinson CW, et al. Fluorodeoxyglucose PET imaging in the evaluation of gallbladder carcinoma and cholangiocarcinoma. J Gastrointest Surg. 2004;8:90-7.
69. Koh T, Taniguchi H, Yamaguchi A, et al. Differential diagnosis of gallbladder cancer using positron emission tomography with fluorine-18-labeled fluoro-deoxyglucose (FDG-PET). J Surg Oncol. 2003;84:74-81.
70. Ito H, Matros E, Brooks DC, et al. Treatment outcomes associated with surgery for gallbladder cancer: A 20-year experience. J Gastrointest Surg. 2004;8:183-90.
71. Wakai T, Shirai Y, Yokoyama N, et al. Early gallbladder carcinoma does not warrant radical resection. Br J Surg. 2001;88:675-8.
72. You DD, Lee HG, Paik KY, et al. What is an adequate extent of resection for T1 gallbladder cancers? Ann Surg. 2008;247:835-8.
73. Mekeel KL, Hemming AW. Surgical management of gallbladder carcinoma: A review. J Gastrointest Surg. 2007;11:1188-93.
74. Miller G, Jarnagin WR. Gallbladder carcinoma. Eur J Surg Oncol. 2008;34:306-12.
75. Paolucci V, Schaeff B, Schneider M, Gutt C. Tumor seeding following laparoscopy: International survey. World J Surg. 1999;23:989-95.
76. O’Connell JB, Maggard MA, Manunga J, et al. Survival after resection of ampullary carcinoma: A national population-based study. Ann Surg Oncol. 2008;15:1820-7.
77. Fischer HP, Zhou H. Pathogenesis of carcinoma of the papilla of Vater. J Hepatobiliary Pancreat Surg. 2004;11:301-9.
78. Offerhaus GJ, Giardiello FM, Krush AJ, et al. The risk of upper gastrointestinal cancer in familial adenomatous polyposis. Gastroenterology. 1992;102:1980-2.
79. Mao C, Huang Y, Howard JM. Carcinoma of the ampulla of Vater and mesenteric fibromatosis (desmoid tumor) associated with Gardner’s syndrome: Problems in management. Pancreas. 1995;10:239-45.
80. Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829-34.
81. Costi R, Caruana P, Sarli L, et al. Ampullary adenocarcinoma in neurofibromatosis type 1. Case report and literature review. Mod Pathol. 2001;14:1169-74.
82. Matthews JJ, Roberts R, O’Reilly DA, et al. Muir-Torre syndrome: A case for surveillance of the ampulla of Vater. Dig Surg. 2002;19:65-6.
83. Kimura W, Ohtsubo K. Incidence, sites of origin, and immunohistochemical and histochemical characteristics of atypical epithelium and minute carcinoma of the papilla of Vater. Cancer. 1988;61:1394-1402.
84. Schirmacher P, Buchler MW. Ampullary adenocarcinoma—differentiation matters. BMC Cancer. 2008;8:251.
85. Carter JT, Grenert JP, Rubenstein L, et al. Tumors of the ampulla of Vater: Histopathologic classification and predictors of survival. J Am Coll Surg. 2008;207:210-18.
86. Kimura W, Futakawa N, Yamagata S, et al. Different clinicopathologic findings in two histologic types of carcinoma of papilla of Vater. Jpn J Cancer Res. 1994;85:161-6.
87. Zhou H, Schaefer N, Wolff M, Fischer HP. Carcinoma of the ampulla of Vater: Comparative histologic/immunohistochemical classification and follow-up. Am J Surg Pathol. 2004;28:875-82.
88. Zhao B, Kimura W, Futakawa N, et al. p53 and p21/Waf1 protein expression and K-ras codon 12 mutation in carcinoma of the papilla of Vater. Am J Gastroenterol. 1999;94:2128-34.
89. Li X, Hui AM, Shi YZ, et al. Deregulation of G1/S transition is a common event in carcinoma of the ampulla of Vater. Hepatogastroenterology. 2002;49:1239-44.
90. Kamisawa T, Fukayama M, Koike M, et al. Carcinoma of the ampulla of Vater: Expression of cancer-associated antigens inversely correlated with prognosis. Am J Gastroenterol. 1988;83:1118-23.
91. Kim JH, Kim MJ, Chung JJ, et al. Differential diagnosis of periampullary carcinomas at MR imaging. Radiographics. 2002;22:1335-52.
92. Puli SR, Singh S, Hagedorn CH, et al. Diagnostic accuracy of EUS for vascular invasion in pancreatic and periampullary cancers: A meta-analysis and systematic review. Gastrointest Endosc. 2007;65:788-97.
93. Tio TL, Sie LH, Kallimanis G, et al. Staging of ampullary and pancreatic carcinoma: Comparison between endosonography and surgery. Gastrointest Endosc. 1996;44:706-13.
94. van der Gaag NA, ten Kate FJ, Lagarde SM, et al. Prognostic significance of extracapsular lymph node involvement in patients with adenocarcinoma of the ampulla of Vater. Br J Surg. 2008;95:735-43.
95. Talamini MA, Moesinger RC, Pitt HA, et al. Adenocarcinoma of the ampulla of Vater. A 28-year experience. Ann Surg. 1997;225:590-9.
96. Bucher P, Chassot G, Durmishi Y, et al. Long-term results of surgical treatment of Vater’s ampulla neoplasms. Hepatogastroenterology. 2007;54:1239-42.
97. Brown KM, Tompkins AJ, Yong S, et al. Pancreaticoduodenectomy is curative in the majority of patients with node-negative ampullary cancer. Arch Surg. 2005;140:529-32.
98. Bogoevski D, Chayeb H, Cataldegirmen G, et al. Nodal microinvolvement in patients with carcinoma of the papilla of Vater receiving no adjuvant chemotherapy. J Gastrointest Surg. 2008.
99. Krishnan S, Rana V, Evans DB, et al. Role of adjuvant chemoradiation therapy in adenocarcinomas of the ampulla of Vater. Int J Radiat Oncol Biol Phys. 2008;70:735-43.
100. Mehta VK, Fisher GA, Ford JM, et al. Adjuvant chemoradiotherapy for “unfavorable” carcinoma of the ampulla of Vater: Preliminary report. Arch Surg. 2001;136:65-9.
101. Caceres M, Mosquera LF, Shih JA, O’Leary JP. Paraganglioma of the bile duct. South Med J. 2001;94:515-18.
102. Kumar S, Agarwal S, Bhargava SK, Minocha VR. Malignant carcinoid tumor of the gallbladder: A case report and review of literature. Trop Gastroenterol. 1992;13:78-84.
103. Yokoyama Y, Fujioka S, Kato K, et al. Primary carcinoid tumor of the gallbladder: Resection of a case metastasizing to the liver and analysis of outcomes. Hepatogastroenterology. 2000;47:135-9.
104. Ferrone CR, Tang LH, D’Angelica M, et al. Extrahepatic bile duct carcinoid tumors: Malignant biliary obstruction with a good prognosis. J Am Coll Surg. 2007;205:357-61.
105. Gusani NJ, Marsh JW, Nalesnik MA, et al. Carcinoid of the extra-hepatic bile duct: A case report with long-term follow-up and review of literature. Am Surg. 2008;74:87-90.
106. Jimenez R, Beguiristain A, Ruiz-Montesinos I, et al. Intrahepatic biliary carcinoid. Am J Clin Oncol. 2008;31:521-2.
107. Bucher P, Mathe Z, Buhler L, et al. Paraganglioma of the ampulla of Vater: A potentially malignant neoplasm. Scand J Gastroenterol. 2004;39:291-5.
108. Lochan R, Balupuri S, Bennett MK, Manas DM. Granular cell tumor as an unusual cause of obstruction at the hepatic hilum: Report of a case. Surg Today. 2006;36:934-6.
109. Hoda RS, Minamiguchi S, Lewin DN, et al. Granular cell tumor of the biliary system: A report of 2 cases with cytologic diagnosis on endoscopic brushing. Acta Cytol. 2005;49:199-203.
110. Wysocki A, Papla B, Budzynski P. Neuromas of the extrahepatic bile ducts as a cause of obstructive jaundice. Eur J Gastroenterol Hepatol. 2002;14:573-6.
111. Choi JY, Kim MJ, Chung JJ, et al. Gallbladder lymphangioma: MR findings. Abdom Imaging. 2002;27:54-7.
112. Kim JK, Yoo KS, Moon JH, et al. Gallbladder lymphangioma: A case report and review of the literature. World J Gastroenterol. 2007;13:320-3.
113. Yang HR, Jan YY, Huang SF, et al. Laparoscopic cholecystectomy for gallbladder lymphangiomas. Surg Endosc. 2003;17:1676.
114. Artaza T, Potenciano JM, Legaz M, et al. Lymphangioma of Vater’s ampulla: A rare cause of obstructive jaundice. Endoscopic therapy. Scand J Gastroenterol. 1995;30:804-6.
115. Sriram PV, Weise C, Seitz U, et al. Lymphangioma of the major duodenal papilla presenting as acute pancreatitis: Treatment by endoscopic snare papillectomy. Gastrointest Endosc. 2000;51:733-6.
116. Chahal P, Prasad GA, Sanderson SO, et al. Endoscopic resection of nonadenomatous ampullary neoplasms. J Clin Gastroenterol. 2007;41:661-6.
117. Boyle L, Gallivan MV, Chun B, Lack EE. Heterotopia of gastric mucosa and liver involving the gallbladder. Report of two cases with literature review. Arch Pathol Lab Med. 1992;116:138-42.
118. Hamazaki K, Fujiwara T. Heterotopic gastric mucosa in the gallbladder. J Gastroenterol. 2000;35:376-81.
119. Delis S, Triantopoulou C, Kyzas PA, Dervenis C. Multiple primary liver lipomas in a patient with chronic hepatitis B: A case report. Eur J Gastroenterol Hepatol. 2007;19:807-9.
120. Yu FC, Chen JH, Yang KC, Wu CC, Chou YY. Hepatobiliary cystadenoma: A report of two cases. J Gastrointestin Liver Dis. 2008;17:203-6.
121. Vogt DP, Henderson JM, Chmielewski E. Cystadenoma and cystadenocarcinoma of the liver: A single center experience. J Am Coll Surg. 2005;200:727-33.
122. Devaney K, Goodman ZD, Ishak KG. Hepatobiliary cystadenoma and cystadenocarcinoma. A light microscopic and immunohistochemical study of 70 patients. Am J Surg Pathol. 1994;18:1078-91.
123. Waldmann J, Zielke A, Moll R, et al. Cystadenocarcinoma of the gallbladder. J Hepatobiliary Pancreat Surg. 2006;13:594-9.
124. Bejarano Gonzalez N, Garcia Moforte N, Darnell Martin A, et al. Primary malignant melanoma of the common bile duct: A case report and literature review. Gastroenterol Hepatol. 2005;28:382-4.
125. Garas G, Bramston B, Edmunds SE. Malignant melanoma metastatic to the common bile duct. J Gastroenterol Hepatol. 2000;15:1348-51.
126. Eliason SC, Grosso LE. Primary biliary malignant lymphoma clinically mimicking cholangiocarcinoma: A case report and review of the literature. Ann Diagn Pathol. 2001;5:25-33.
127. Sugawara G, Nagino M, Oda K, et al. Follicular lymphoma of the extrahepatic bile duct mimicking cholangiocarcinoma. J Hepatobiliary Pancreat Surg. 2008;15:196-9.
128. Zampieri N, Camoglio F, Corroppolo M, et al. Botryoid rhabdomyosarcoma of the biliary tract in children: A unique case report. Eur J Cancer Care (Engl). 2006;15:463-6.
129. Sanz N, de Mingo L, Florez F, Rollan VV. Rhabdomyosarcoma of the biliary tree. Pediatr Surg Int. 1997;12:200-1.
130. Spunt SL, Lobe TE, Pappo AS, et al. Aggressive surgery is unwarranted for biliary tract rhabdomyosarcoma. J Pediatr Surg. 2000;35:309-16.
131. Ferluga D, Luzar B, Gadzijev EM. Follicular lymphoma of the gallbladder and extrahepatic bile ducts. Virchows Arch. 2003;442:136-40.