CHAPTER 94 Tumors and Cysts of the Liver
PRIMARY MALIGNANT TUMORS
Among primary malignant tumors of the liver, hepatocellular carcinoma is by far the most common.
HEPATOCELLULAR CARCINOMA
Epidemiology
Hepatocellular carcinoma is the commonest primary malignant tumor of the liver. It is the fifth most common cancer in men and the eighth most common in women, and it ranks fourth in annual cancer mortality rates.1,2 Information on incidence is derived from an increasing but still limited number of cancer registries, and it is possible to classify countries into broad risk categories only. Moreover, in low-income (developing) countries, especially in sub-Saharan Africa, hepatocellular carcinoma is underdiagnosed and underreported, in some cases by as much as 50%. Despite these sources of inaccuracy, hepatocellular carcinoma clearly has an unusual geographic distribution (Fig. 94-1). Moreover, the tumor is not necessarily uniformly common throughout countries with a high incidence, such as China3 and Mozambique.4 The incidence of hepatocellular carcinoma has increased considerably in Japan since the 1980s, and lesser increases have been recorded in developed Western countries, including North America and Western Europe.5 Interestingly, a study from Japan has shown that the rate of hepatocellular carcinoma began to decline in 2000, presumably because of the aging of the cohort of persons infected with hepatitis C virus (HCV).6 A similar downward trend has been noted in some European countries, including France and Italy.7 By contrast, in the United States, hepatocellular carcinoma is the cancer that has been increasing in incidence most rapidly since 2000, at a time when other major cancers such as cancers of the lung, breast, prostate, and colon are decreasing.8 Considerable racial and ethnic variation exits in the incidence of hepatocellular carcinoma in the United States. The incidence among Asians is highest, almost double that of white Hispanics and more than four times higher than that of whites.9
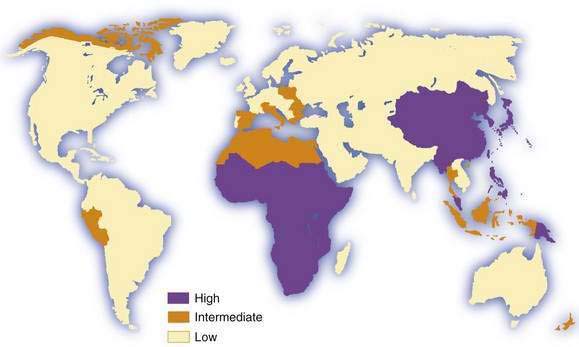
Migrants from countries with a low incidence to areas with a high incidence of hepatocellular carcinoma usually retain the low risk of their country of origin, even after several generations in the new environment. The consequences for migrants from countries with a high incidence to those with a low incidence differ, depending on the major risk factors for the tumor in their country of origin and whether chronic hepatitis B virus (HBV) infection, if this is the major risk factor, is acquired predominantly by the perinatal or horizontal route (see later and Chapter 78).2,10,11
Men are generally more susceptible than women to hepatocellular carcinoma. Male predominance is, however, more obvious in populations at high risk for the tumor (mean male-to-female ratio, 3.7 : 1.0) than in those at low or intermediate risk (2.4 : 1.0).1,2 In industrialized countries, the number of men and number of women with hepatocellular carcinoma in the absence of cirrhosis are almost equal.
The incidence of hepatocellular carcinoma increases progressively with advancing age in all populations, although it tends to level off in the oldest age groups.1,2 In Chinese and particularly in black African populations, however, the mean age of patients with the tumor is appreciably younger than in other populations. This finding is in sharp contrast to the age distribution in Japan, where the incidence of hepatocellular carcinoma is highest in the cohort of men ages 70 to 79 years.6 Hepatocellular carcinoma is rare in children.12,13
Clinical Features
Although the typical clinical features of hepatocellular carcinoma are well recognized (including abdominal pain and weight loss in patients with cirrhosis), more patients are now being diagnosed at an early stage, when they have no specific symptoms or signs. This trend toward earlier diagnosis is probably the result of surveillance programs in patients with chronic liver disease (see later). In far-advanced disease, patients with hepatocellular carcinoma usually present with typical symptoms and signs, and diagnosis is easy. In addition, hepatocellular carcinoma often coexists with cirrhosis,14 and the onset of hepatocellular carcinoma is marked by a sudden unexplained change in the patient’s condition.
Patients with hepatocellular carcinoma often are unaware of its presence until the tumor has reached an advanced stage. The most common, and frequently first, symptom is right hypochondrial or epigastric pain. Other symptoms are listed in Table 94-1.
| SYMPTOM | FREQUENCY (%) |
|---|---|
| Abdominal pain | 59-95 |
| Weight loss | 34-71 |
| Weakness | 22-53 |
| Abdominal swelling | 28-43 |
| Nonspecific gastrointestinal symptoms | 25-28 |
| Jaundice | 5-26 |
| SIGN | |
| Hepatomegaly | 54-98 |
| Ascites | 35-61 |
| Fever | 11-54 |
| Splenomegaly | 27-42 |
| Wasting | 25-41 |
| Jaundice | 4-35 |
| Hepatic bruit | 6-25 |
Physical findings vary with the stage of disease. Early in the course, evidence of cirrhosis alone may be present, or abnormal findings may be absent (see Table 94-1). When the tumor is advanced at the time of the patient’s first medical visit, the liver is almost always enlarged, sometimes massively. Hepatic tenderness is common and may be profound, especially in the later stages. The surface of the enlarged liver is smooth, irregular, or frankly nodular. An arterial bruit may be heard over the tumor15; the bruit is heard in systole, rough in character, and not affected by changing the position of the patient. Although not pathognomonic, a bruit is a useful clue to the diagnosis of hepatocellular carcinoma. Less often, a friction rub may be heard over the tumor, but this sign is more characteristic of hepatic metastases or abscesses.
Ascites may be present when the patient is first seen or may appear with progression of the tumor. In most patients, ascites is the result of long-standing cirrhosis and portal hypertension (see Chapter 91), but in some cases it is caused by invasion of the peritoneum by the primary tumor or metastases. The ascitic fluid may be blood-stained. In a small proportion of patients, hepatocellular carcinoma invades the hepatic veins, thereby causing Budd-Chiari syndrome, and tense ascites results (see Chapter 83).16 Splenomegaly, if present, reflects coexisting cirrhosis and portal hypertension.
Physical evidence of cirrhosis may also be noted. Severe pitting edema of the lower extremities extending up to the groins occurs when hepatocellular carcinoma has invaded the hepatic veins and propagates into and obstructs the inferior vena cava.16 A Virchow-Trosier (supraclavicular) node, Sister Mary Joseph’s (periumbilical) nodule, or enlarged axillary lymph node is rarely present.
Paraneoplastic Manifestations
Some of the deleterious effects of hepatocellular carcinoma are not caused by local effects of the tumor or metastases (Table 94-2). Each of the paraneoplastic syndromes in hepatocellular carcinoma is rare or uncommon. One of the more important is type B hypoglycemia, which occurs in less than 5% of patients, manifests as severe hypoglycemia early in the course of the disease,16 and is believed to result from the defective processing by malignant hepatocytes of the precursor to insulin-like growth factor II (pre-IGF II).17 By contrast, type A hypoglycemia is a milder form of glycopenia that occurs in the terminal stages of hepatocellular carcinoma (and other malignant tumors of the liver). It results from the inability of a liver extensively infiltrated by tumor, and often cirrhotic, to satisfy the demands for glucose by a large, often rapidly growing tumor and by the other tissues of the body.
Table 94-2 Paraneoplastic Syndromes Associated with Hepatocellular Carcinoma
Another important paraneoplastic syndrome is polycythemia (erythrocytosis), which occurs in less than 10% of patients with hepatocellular carcinoma.18 This syndrome appears to be caused by the synthesis of erythropoietin or an erythropoietin-like substance by malignant hepatocytes.
Patients with hepatocellular carcinoma, especially the sclerosing variety, may present with hypercalcemia in the absence of osteolytic metastases. When hypercalcemia is severe, it may result in the typical complications of hypercalcemia, including drowsiness and lethargy. The probable cause is secretion of parathyroid hormone–related protein (PTHrP) by the tumor.19
Cutaneous paraneoplastic manifestations of hepatocellular carcinoma are rare except for pityriasis rotunda (circumscripta), which may be a useful marker of the tumor in black Africans. The rash consists of single or multiple, round or oval, hyperpigmented, scaly lesions on the trunk and thighs that range in diameter from 0.5 to 25 cm.20
Diagnosis
The gold standard for the diagnosis of hepatocellular carcinoma is pathology. For practical purposes (i.e., to apply treatment), hepatocellular carcinoma can only be diagnosed in the presence of an abnormality on imaging of the liver. The development of hepatocellular carcinoma is thought to occur as a result of a multistep sequential process from a dysplastic focus of hepatocytes to a low-grade dysplastic nodule to a high-grade dysplastic nodule to early well-differentiated hepatocellular carcinoma, and then to less differentiated states.21,22 In early hepatocellular carcinoma, particularly when a needle biopsy specimen is examined, controversy may exist among pathologists as to whether a particular specimen is consistent with dysplasia or carcinoma. Dysplastic nodules and even regenerative cirrhotic nodules can be seen on imaging studies and are potentially confused with hepatocellular carcinoma. Although there are specific imaging features based on the enhancement patterns with dynamic imaging of dysplastic nodules and hepatocellular carcinoma (see later), some overlap occurs.23,24 Nevertheless, there is a growing consensus, based on guidelines from the major European and American liver societies and now backed up by published experience, that the diagnosis of hepatocellular carcinoma can be made in the appropriate clinical setting based on specific imaging characteristics, with or without an elevated serum alpha fetoprotein (AFP) level.24–27
Serum Tumor Markers
Many of the substances synthesized and secreted by hepatocellular carcinoma are not biologically active. Nevertheless, a few are produced by a sufficiently large proportion of tumors to warrant their use as serum markers for hepatocellular carcinoma. The most helpful of these markers is AFP (Table 94-3).
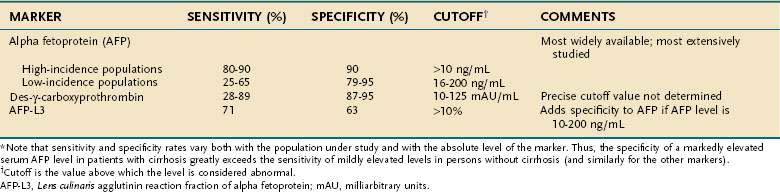
Alpha Fetoprotein
AFP is an α1-globulin normally present in high concentrations in fetal serum but in only minute amounts thereafter. Reappearance of high serum levels of AFP strongly suggests the presence of hepatocellular carcinoma (or hepatoblastoma [see later]),28 especially in populations in which hepatocellular carcinoma is most prevalent: The great majority of Chinese and black African patients have a raised serum concentration of AFP (>10 ng/mL), and approximately 75% have a diagnostic level (>500 ng/mL). These percentages are lower in populations at low or intermediate risk for the tumor, in which the sensitivity ranges from 25% to 65%, with a specificity of 79% to 95% and cutoff values for an elevated and diagnostic level of 16 and 200 ng/mL, respectively.29–35 With higher levels of AFP, the confidence in the diagnosis of hepatocellular carcinoma is greater. Although levels higher than 500 ng/mL usually indicate hepatocellular carcinoma, they sometimes can be seen in patients with active viral hepatitis. In the setting of a cirrhotic patient with a hepatic mass lesion larger than 2 cm in diameter and suggestive features of hepatocellular carcinoma, an AFP level higher than 200 ng/mL is considered diagnostic for hepatocellular carcinoma.25,26,33,36,37 The mean serum value of AFP in affected patients in regions with a high incidence of hepatocellular carcinoma is 60,000 to 80,000 ng/mL, compared with approximately 8,000 ng/mL in regions with a low or intermediate incidence of the tumor. Raised serum values range over six orders of magnitude, although an AFP concentration higher than 1 million ng/mL is rare. False-positive results also may occur in patients with tumors of endodermal origin, nonseminomatous germ cell tumors, and pregnancy. A progressively rising serum AFP concentration is highly suggestive of hepatocellular carcinoma.
AFP is not essential to hepatocarcinogenesis, and thus not all hepatocellular carcinomas produce AFP. The levels of AFP appear to be affected by ethnicity, underlying cause of liver disease, and tumor stage.30,33 Synthesis of AFP by a tumor is permanent and age-related; the younger the patient, the more likely the serum value is to be raised and the higher the level attained. According to the American Association for the Study of Liver Diseases (AASLD) guidelines, hepatocellular carcinoma can be diagnosed with confidence in patients with a serum AFP level higher than 200 ng/mL and a mass in the liver.25 An AFP level higher than about 500 ng/mL predicts worse outcomes with liver transplantation compared with lower levels.38 Attempts to correlate the degree of differentiation of hepatocellular carcinoma with production of AFP have produced conflicting results.
Fucosylated Alpha Fetoprotein
AFP is heterogeneous in structure. Its microheterogeneity results from differences in the oligosaccharide side chain and accounts for the differential affinity of the glycoprotein for lectins. AFP secreted by malignant hepatocytes contains unusual and complex sugar chains that are not found in AFP produced by nontransformed hepatocytes. One variant, Lens culinaris agglutinin reactive fraction (AFP-L3), appears to improve the specificity of AFP, particularly AFP serum levels from 10 to 200 ng/mL.39,40 The recommended cutoff value for AFP-L3 to diagnose hepatocellular carcinoma is higher than 10%, although the specificity varies depending on the absolute level of AFP. A Western series has suggested that a cutoff value of 35% is necessary to achieve 100% specificity.40 Therefore, AFP-L3 is not sufficiently validated to confirm the diagnosis of hepatocellular carcinoma without other supporting findings, such as suggestive imaging.
Des-γ-Carboxy Prothrombin
Serum concentrations of des-γ-carboxy prothrombin (DCP) (also known as prothrombin produced by vitamin K absence or antagonist II [PIVKA II]) are raised in most patients with hepatocellular carcinoma.41 DCP is an abnormal prothrombin that is thought to result from a defect in the post-translational carboxylation of the prothrombin precursor in malignant cells.42 In Western populations, DCP may be a better marker than, or at least a complementary marker to, AFP.43–45 In black Africans, however, DCP is less sensitive and less specific than AFP.46 The appropriate cutoffs are not well established, and thus the precise role of DCP in the diagnosis of hepatocellular carcinoma requires validation.
Other Markers
Multiple other potential serum markers for hepatocellular carcinoma are in the exploratory phase of evaluation, including glypican 3, Golgi protein 73, hepatocyte growth factor, insulin growth factor 1, transforming growth factor-β1, and proteomic profiling using surface-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry.47–51 All these novel markers have been shown to be elevated in patients with hepatocellular carcinoma compared with those with only chronic liver disease, but clear cutoff values and comparisons with other markers have not been established. Some of these markers may be complementary to established markers, although none of them has an established high throughput method of measurement, as required for a clinical test. The roles of these markers in the diagnosis of hepatocellular carcinoma await further study.
Imaging
The diagnosis of hepatocellular carcinoma generally requires imaging evidence of a focal lesion in the liver, although large infiltrating lesions can also be diagnostic. Arterial hyperenhancement, particularly seen on dynamic contrast imaging of the liver, is observed because the blood supply of hepatocellular carcinoma comes from newly formed abnormal arteries (neoangiogenesis).23,52,53 As a nodule transforms from low- to high-grade dysplasia and then to hepatocellular carcinoma, the primary blood supply shifts from portal to arterial—especially new abnormal arterial branches that produce characteristic findings on dynamic contrast imaging of the liver.27
Ultrasonography
Ultrasonography detects most hepatocellular carcinomas but may not distinguish this tumor from other solid lesions in the liver. As with all imaging methods, the sensitivity increases with increasing size of the lesion. A systematic review of eight studies using histologic reviews of liver explants has shown that ultrasound has fair sensitivity (pooled estimate, 48%; 95% confidence interval [CI], 34% to 62%) with good specificity, estimated at 97% (95% CI, 95% to 98%).24 Advantages of ultrasonography include safety, availability, and cost-effectiveness, although it has the drawbacks of being nonstandardized and examiner-dependent. Body habitus, particularly obesity, may limit the sensitivity of this test. Approximately two thirds of symptomatic hepatocellular carcinomas are uniformly hyperechoic, whereas the remainder are partly hyperechoic and partly hypoechoic.54 Small tumors are uniformly hypoechoic. The ultrasonographic appearance is influenced by the presence of fat, calcium, and necrosis. Tumors located immediately under the right hemidiaphragm may be difficult to detect. In Japanese patients in particular, hepatocellular carcinoma may have a well-defined, even thick capsule, which can be seen on ultrasonography. Ultrasonography with Doppler technology is useful for assessing the patency of the inferior vena cava, portal vein and its larger branches, hepatic veins, and biliary tree.
Dynamic contrast-enhanced Doppler ultrasonography with intra-arterial infusion of CO2 microbubbles and intravenous enhanced color Doppler ultrasonography are refinements that, by characterizing hepatic arterial and portal venous flow in tumorous nodules, facilitate the diagnosis of malignant and benign hepatic nodules.55 These techniques are not often performed in the United States.
Computed Tomography
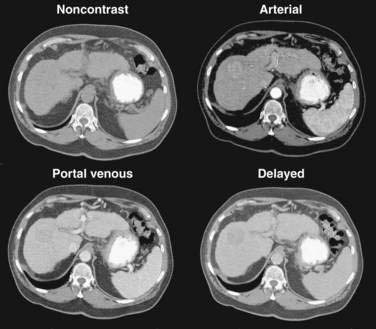
Multiphase, also called dynamic, helical computed tomography (CT) is the imaging technique of choice for the diagnosis of hepatocellular carcinoma.24,54,55 CT during arterial portography is also helpful but rarely done because it is invasive. Phases in dynamic contrast-enhanced CT can include noncontrast, arterial, portal venous, and delayed phases. The classic and most diagnostic pattern for hepatocellular carcinoma is a combination of enhancement in the arterial phase (with the uninvolved liver lacking enhancement), loss of central nodule enhancement compared with the uninvolved liver (washout), and capsular enhancement in the portal-venous and delayed phases (Fig. 94-2).25,56 When the lesion is larger than 2 cm in diameter, this pattern has almost 100% specificity for hepatocellular carcinoma.36,37,56 When the nodule is 1 to 2 cm, guidelines recommend a second type of dynamic imaging (magnetic resonance imaging [MRI] or contrast ultrasonography) to confirm the diagnosis of hepatocellular carcinoma, although the specificity of one dynamic study is higher than 90%.57 CT often finds so-called hypervascular-only lesions, which enhance in the arterial phase and become isodense to the surrounding liver in the portal-venous and delayed phases. These lesions may be dysplastic nodules, arterial portal shunts, atypical hemangiomas, hepatocellular carcinoma, confluent fibrosis, or aberrant venous drainage. When less than 2 cm in diameter, only about 30% are hepatocellular carcinomas, which grow over time. Other causes disappear or remain stable on follow-up studies. Current guidelines recommend biopsy of lesions larger than 1 cm if the serum AFP level is less than 200 ng/mL and serial imaging for lesions smaller than 1 cm.58 Hepatocellular carcinoma may also have other patterns on CT, such as washout only on delayed imaging, a hypovascular nodule, or a fat-containing nodule.27,58 Overall, the pooled estimate of sensitivity and specificity for detecting hepatocellular carcinoma by CT is 67.5% (95% CI, 55% to 80%) and 92.5% (95% CI, 89% to 96%), respectively. Dynamic CT is also useful for detecting invasion into the portal or hepatic veins and identifying the location and number of tumors; these findings are critical for planning treatment.
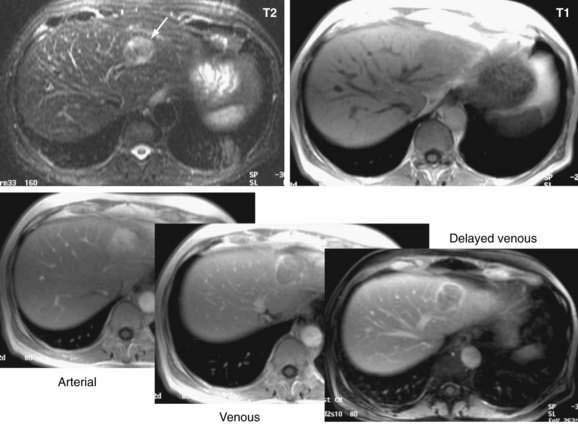
Magnetic Resonance Imaging
Dynamic MRI using gadolinium contrast agents provides another way of distinguishing hepatocellular carcinoma from normal liver tissue. The performance of MRI and the findings on multiphase contrast enhancement are similar to those described for CT (Fig. 94-3). Typically, the signal intensity on T1-weighted images is low.27,54 The pooled estimate of sensitivity and specificity for detecting hepatocellular carcinoma by MRI is 80.6% (95% CI, 70% to 91%) and 84.8% (95% CI, 77% to 93%), respectively.24 MRI may be slightly superior overall to CT, although local expertise should dictate the choice of imaging technique.
Hepatic Angiography
Since the advent of CT and MRI, the diagnostic role of hepatic angiography has decreased. Digital subtraction angiography is helpful for recognizing small hypervascular hepatocellular carcinomas but may miss early, well-differentiated hypovascular tumors. Hepatocellular carcinomas often are densely vascular, although multinodular tumors may be relatively avascular.59 The arteries in the tumor are irregular in caliber and do not taper in the usual way, and the smaller branches may show a bizarre pattern. The hepatic veins fill early, and retrograde filling of the portal veins results from the presence of arteriovenous anastomoses within the tumor. An additional finding is a delay in capillary emptying, which is seen as a blush. The center of some large tumors may be avascular as a result of necrosis or, less often, hemorrhage. Angiography is essential for delineating the hepatic arterial anatomy in planning embolization or chemoembolization of the tumor or infusion of cytotoxic drugs directly into the hepatic artery or its branches (see later).
Pathology
Microscopic Appearance
Hepatocellular carcinoma is classified histologically into well-differentiated, moderately differentiated, and undifferentiated (pleomorphic) forms.60
Progenitor Cell Hepatocellular Carcinoma
A class of primary liver cancer appears to have its origins in progenitor cells, the stem cells of the liver, located in association with the canals of Hering. Progenitor cell activation is seen in association with chronic viral hepatitis and cirrhosis, presumably relegated to senescence of hepatocytes. These tumors may appear morphologically like typical hepatocellular carcinoma or mixed cholangiohepatocellular carcinoma. Tumor cells stain positively for cytokeratin 19, and the tumor appears to have a more aggressive course than typical hepatocellular carcinoma.61
Metastases
Extrahepatic metastases are present at autopsy in 40% to 57% of patients with hepatocellular carcinomas.62 The most common sites are the lungs (up to 50% in some reports) and regional lymph nodes (approximately 20%). The adrenal glands are frequently involved.
Fibrolamellar Hepatocellular Carcinoma
The fibrolamellar variant of hepatocellular carcinoma typically occurs in young patients, has an approximately equal gender distribution, does not secrete AFP, is not caused by chronic hepatitis B or C, and almost always arises in a noncirrhotic liver.63–65 Fibrolamellar hepatocellular carcinoma is more often amenable to surgical treatment and therefore generally carries a better prognosis than that for conventional hepatocellular carcinoma. It does not, however, respond to chemotherapy any better than other forms of hepatocellular carcinoma. The hepatocytes are characteristically plump, deeply eosinophilic, and encompassed by abundant fibrous stroma composed of thin, parallel fibrous bands that separate the cells into trabeculae or nodules. The cytoplasm is packed with swollen mitochondria and, in approximately half of the tumors, contains pale or hyaline bodies. Nuclei are prominent, and mitoses are rare.
Staging
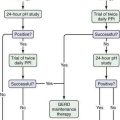
Accurate staging of hepatocellular carcinoma is necessary for prognostication and also to assist with selection of therapy. Determining the optimal staging system for hepatocellular carcinoma has been controversial, in part because it needs to take into account both the severity of the underlying liver disease and the size and degree of spread of the tumor. As with all cancers, the TNM (tumor-node-metastasis) system can be used to stage hepatocellular carcinoma, but this system does not factor in the underlying liver disease. A study66 comparing the usefulness of seven staging systems, including the Okuda, TNM, Cancer of the Liver Italian Program (CLIP), Barcelona Clinic Liver Cancer (BCLC), Chinese University Prognostic Index (CUPI), Japanese Integrated Staging (JIS), and Group d’Etude et Traitement du Carcinome Hépatocellulaire (GETCH) systems in a cohort of patients from the United States, has found the BCLC staging system to have the best independent predictive power for survival. The BCLC system has been adopted by the AASLD for use in its practice guidelines on management of hepatocellular carcinoma.25 This staging classification also includes a treatment schedule based on stage (Fig. 94-4).67
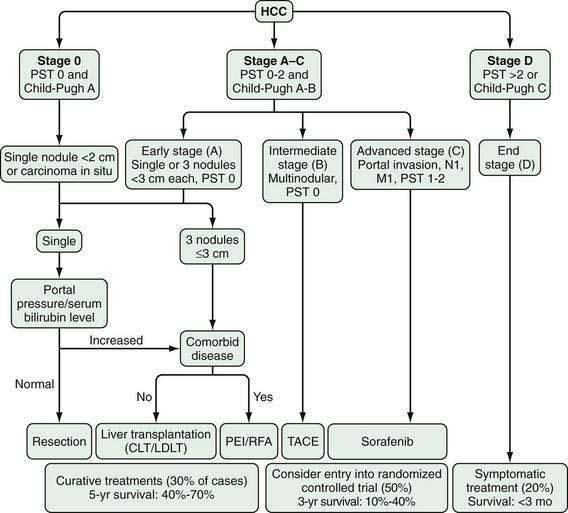
Figure 94-4. Barcelona Clinic Liver Cancer (BCLC) staging classification and treatment schedule. Staging is based on tumor size and spread, the patient’s performance status (PST) on a scale of 0 (good) to >2 (poor), and liver function as assessed by the Child-Pugh class (see Chapter 90). Patients with very early (stage 0) hepatocellular carcinoma (HCC) are optimal candidates for surgical resection. Patients with early (stage A) HCC are candidates for radical therapy (resection, cadaveric liver transplantation [CLT] or live-donor liver transplantation [LDLT], or local ablation via percutaneous ethanol injection [PEI] or radiofrequency ablation [RFA]). Patients with intermediate (stage B) HCC benefit from transarterial chemoembolization (TACE). Patients with advanced HCC, defined as the presence of macroscopic vascular invasion, extrahepatic spread, or cancer-related symptoms (PST 1 or 2) (stage C), benefit from sorafenib. Patients with end-stage disease (stage D) should receive symptomatic treatment. The treatment strategy will transition from one stage to another when treatment fails or is contraindicated. M, metastasis stage; N, nodal stage.
(Adapted from Llovet J, Di Bisceglie A, Bruix J, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst 2008; 100:698-711.)
Causes and Pathogenesis
In contrast to many other malignancies, for which risk factors can only sometimes be identified, the immediate cause of hepatocellular carcinoma can usually be identified and is most commonly chronic viral hepatitis or cirrhosis. Hepatocellular carcinoma is multifactorial in cause and complex in pathogenesis. Four major causative factors have been identified (Table 94-4). The differing blend of risk factors in various parts of the world may explain, in part, the diverse biologic characteristics of hepatocellular carcinoma in various populations.68
| Major Risk Factors |
Hepatitis B Virus
Some 387 million carriers of HBV exist in the world today, and hepatocellular carcinoma will develop in as many as 25% of them (see Chapter 78). HBV accounts for up to 80% of hepatocellular carcinomas, which occur with high frequency in East Asian and African populations.68,69 Persistent HBV infection antedates the development of hepatocellular carcinoma by several to many years, an interval commensurate with a cause and effect relationship between the virus and the tumor. Indeed, in at-risk populations, the HBV carrier state is largely established in early childhood by perinatal or horizontal infection.70,71 Approximately 90% of children infected at this stage of life become chronic carriers of the virus, and these early-onset carriers face a lifetime relative risk for developing hepatocellular carcinoma of more than 100, compared with uninfected controls.72
An effective vaccine against HBV has been available since the early 1980s and, in countries in which this vaccine has been included in the expanded program of immunization for a sufficient length of time, the HBV carrier rate among children has decreased by 10-fold or more. Studies in Taiwan, where universal immunization was started in 1984 and where the rate of HBV carriage among children has decreased by more than 10-fold, have already shown a 70% reduction in the mortality rate from hepatocellular carcinoma in children in the vaccinated age groups.73 This finding gives promise for the ultimate eradication of HBV-induced hepatocellular carcinoma and provides further evidence of the causal role of the virus in the development of this tumor.
HBV DNA is integrated into cellular DNA in approximately 90% of HBV-related hepatocellular carcinomas.74 The sites of chromosomal insertion appear to be random, and whether viral integration is essential for hepatocarcinogenesis is still uncertain. The virus appears to be directly and indirectly carcinogenic.75 Possible direct carcinogenic effects include cis-activation of cellular genes as a result of viral integration, changes in the DNA sequences flanking the integrated viral DNA, transcriptional activation of remote cellular genes by HBV-encoded proteins, particularly the X protein, and effects resulting from viral mutations. The transcriptional activity of the HBV X protein may be mediated by interaction with specific transcription factors, activation of the mitogen-activated protein (MAP) kinase and Janus kinase–signal transducer and activator of transcription (JAK/STAT) pathways, an effect on apoptosis, and modulation of DNA repair. Studies have shown a clear link between the amount of HBV replication (measured as serum level of HBV DNA [viral load]) and subsequent risk of hepatocellular carcinoma. The long-term risk of hepatocellular carcinoma increases markedly in patients with serum HBV DNA levels higher than 104 copies/mL.76 A randomized controlled trial of antiviral therapy has also shown a reduction in the incidence of hepatocellular carcinoma in association with reductions in serum levels of HBV DNA on therapy.77
Indirect carcinogenic effects are the result of the chronic necroinflammatory hepatic disease, in particular cirrhosis, induced by the virus. The increased hepatocyte turnover rate resulting from continuous or recurring cycles of cell necrosis and regeneration acts as a potent tumor promoter. In addition, the distorted architecture characteristic of cirrhosis contributes to the loss of control of hepatocyte growth, and hepatic inflammation generates mutagenic reactive oxygen species. The transgenic mouse model of Chisari and coworkers has provided indirect support for the role of prolonged hepatocyte injury in hepatocarcinogenesis.78
Hepatitis C Virus
Approximately 170 million people in the world today are chronically infected with HCV and are at greatly increased risk for the development of hepatocellular carcinoma. In Japan, Italy, and Spain, HCV is the cause of about 75% of hepatocellular carcinomas, and, in other industrialized countries, HCV infection, often in combination with alcohol abuse, is emerging as a major cause of the tumor.68,79 Patients with HCV-induced hepatocellular carcinoma generally are older than those with HBV-related tumors, and it is likely that the HCV infection is acquired mainly in adult life.
Cirrhosis
In all parts of the world, hepatocellular carcinoma frequently coexists with cirrhosis.80 All causative forms of cirrhosis may be complicated by tumor formation. A long-term follow-up study of 2126 U.S. military veterans with cirrhosis found that hepatocellular carcinoma developed in 100 (4.7%) over an average period of 3.6 years.80 The calculated rate was 1.3/100 patient-years. Risk factors for hepatocellular carcinoma included obesity, low platelet count, and the presence of antibody to hepatitis B core antigen. A similar study from Italy found an incidence of hepatocellular carcinoma of 3.7/100 patient-years among persons with HCV infection and 2.0/100 patient-years among persons with HBV infection. Older age and male gender were confirmed as risk factors among patients with cirrhosis.81
Aflatoxin B1
Dietary exposure to aflatoxin B1, derived from the fungi Aspergillus flavus and Aspergillus parasiticus, is an important risk factor for hepatocellular carcinoma in parts of Africa and Asia. These molds are ubiquitous in nature and contaminate a number of staple foodstuffs in tropical and subtropical regions (see Chapter 87). Epidemiologic studies have shown a strong correlation between the dietary intake of aflatoxin B1 and incidence of hepatocellular carcinoma.82 Moreover, aflatoxin B1 and HBV interact synergistically in the pathogenesis of hepatocellular carcinoma. Heavy dietary exposure to aflatoxin B1 may contribute to hepatocarcinogenesis through an inactivating mutation of the third base of codon 249 of the TP53 tumor suppressor gene.83,84
Other Liver Conditions
Hepatocellular carcinoma develops in as many as 45% of patients with hemochromatosis (see Chapter 74).85 Malignant transformation was thought previously to occur only in the presence of cirrhosis (and is certainly more likely to do so), but this complication also has been reported in patients without cirrhosis.86 Excessive free iron in tissues may be carcinogenic, perhaps by generating mutagenic reactive oxygen species.87 Further support for this theory comes from the observations that black Africans with dietary iron overload are at increased risk of hepatocellular carcinoma88 and that rats fed a diet high in iron develop iron-free dysplastic foci and hepatocellular carcinoma in the absence of cirrhosis.89 Hepatocellular carcinoma develops occasionally in patients with Wilson disease, but only in the presence of cirrhosis (see Chapter 75).90 Malignant transformation has been attributed to the cirrhosis but also may result from oxidant stress secondary to the accumulation of copper in the liver.91 Hepatocellular carcinoma also may develop in patients with other inherited metabolic disorders that are complicated by cirrhosis, such as α1-antitrypsin deficiency and type 1 hereditary tyrosinemia, and in patients with certain inherited diseases in the absence of cirrhosis—for example, type 1 glycogen storage disease (see Chapter 76). Hepatocellular carcinoma develops in approximately 40% of patients with membranous obstruction of the inferior vena cava, a rare congenital or acquired anomaly (see Chapter 83). Continuous cycles of hepatocyte necrosis followed by regeneration resulting from the severe and unremitting hepatic venous congestion render the cells susceptible to environmental mutagens, as well as to spontaneous mutations.92
More recently, the role of obesity, diabetes mellitus, and fatty liver disease have come to be recognized in the causation of hepatocellular carcinoma,93–95 although the mechanisms whereby these overlapping conditions contribute to causing hepatocellular carcinoma is unknown. Certainly, cirrhosis caused by nonalcoholic steatohepatitis may give rise to hepatocellular carcinoma, but it appears that these risk factors may also be additive to chronic viral hepatitis and cirrhosis.
A statistically significant correlation between the use of oral contraceptive steroids and the occurrence of hepatocellular carcinoma has been demonstrated in countries in which the incidence of hepatocellular carcinoma is low and no overriding risk factor for development of the tumor is present.96 Epidemiologic evidence of a link between cigarette smoking and the occurrence of hepatocellular carcinoma is conflicting, although most of the evidence suggests that smoking is a minor risk factor.97 Heavy smokers have an approximately 50% higher risk than nonsmokers. The incidence of hepatocellular carcinoma is increased in patients with human immunodeficiency virus (HIV) infection compared with controls in the general population, presumably because of the increased rate of chronic viral hepatitis in the HIV-positive population.98
Although the aforementioned risk factors have been identified, the precise mechanisms whereby they lead to hepatocellular carcinoma still need to be elucidated. Multiple cellular pathways are involved in causing unconstrained proliferation of hepatocytes and increased angiogenesis against a background of chronic liver disease. These pathways have become the targets for new molecular therapies against hepatocellular carcinoma (Table 94-5).99
Table 94-5 Key Molecular Pathways Involved in Hepatocarcinogenesis
JAK/STAT, janus kinase/signal transducers and activators of transcription; mTOR, mammalian target of rapamycin.
Adapted from Roberts L. Emerging experimental therapies for hepatocellular carcinoma: What if you can’t cure? In: McCullough A, ed. AASLD Postgraduate Course, 2007. Boston: AASLD, 2007; p 185.
Natural History and Prognosis
Symptomatic hepatocellular carcinoma carries a grave prognosis; in fact, the annual incidence and mortality rates for the tumor are almost identical. The main reasons for the poor outcome are the extent of tumor burden when the patient is first seen and the frequent presence of coexisting cirrhosis and hepatic dysfunction. The natural history of hepatocellular carcinoma in its florid form is one of rapid progression, with increasing hepatomegaly, abdominal pain, wasting, and deepening jaundice, and with death ensuing in two to four months. In industrialized countries, however, the tumor appears to run a more indolent course with longer survival times.100 Rare cases of spontaneous tumor regression have been reported.
Treatment
Important advances in the treatment of hepatocellular carcinoma have occurred since the 1980s; these advances include randomized controlled trials that support the benefits of certain treatments such as chemoembolization and the multikinase inhibitor sorafenib. Overwhelming evidence supports the superiority of liver transplantation over other therapies for patients with portal hypertension and cirrhosis. Because hepatocellular carcinoma is usually a combination of two diseases—the underlying liver disease (usually cirrhosis with varying degrees of decompensation) and the cancer itself—both factors must be taken into account when selecting treatment. When presented with a patient with hepatocellular carcinoma, the clinician should decide which is the best initial therapy: surgical resection or liver transplantation, if the patient is a candidate for either; ethanol or radiofrequency ablation, if possible based on the size of the tumor; chemoembolization; and, if the tumor is too advanced, sorafenib or a clinical trial. Table 94-6 describes the treatment options for hepatocellular carcinoma. The BCLC staging classification and treatment schedule can help guide the clinician in choosing the most appropriate treatment (see Fig. 94-4).
| MODALITY | COMMENTS |
|---|---|
| Surgical resection | Curative but limited to noncirrhotic patients and cirrhotic patients without portal hypertension |
| May be technically difficult | |
| High recurrence rate | |
| Liver transplantation | Successful in selected patients (Milan criteria; see text and Chapter 95) |
| Requires lifelong immunosuppression | |
| Expensive and not available worldwide | |
| Alcohol injection and radiofrequency ablation | Potentially curative for small tumors, including multiple tumors |
| High recurrence rate | |
| Chemoembolization | Prolongs survival in unresectable tumors if hepatic function is preserved |
| Chemotherapy | Palliative only; can be used as an adjunct to surgical resection or transplantation |
| Drug toxicity common | |
| Targeted molecular therapies | Sorafenib is the first such agent shown to improve patient survival |
Surgical Resection
Surgical therapy, whether by tumor resection or liver transplantation, offers the best chance of cure for hepatocellular carcinoma. For resection to be considered, the tumor should be confined to one lobe of the liver, favorably located, and, ideally, the nontumorous liver tissue should not be cirrhotic. Expert surgical centers can achieve five- and ten-year survival rates of 40% and 26%, respectively, with a mean tumor diameter of 8.8 cm in noncirrhotic patients.101 Unfortunately, these patients represent less than 5% of Western cases.102,103 Resection is also effective if the tumor is limited to the left lobe or a portion of the right lobe, thereby permitting a segmental resection if the patient has Child (Child-Pugh) class A cirrhosis, the serum bilirubin level is normal, and portal hypertension is not present (based on imaging, a normal platelet count, and lack of varices on endoscopy or on direct measurement of the hepatic venous pressure gradient). Using these criteria, five-year survival rates of 50% can be achieved. In parts of the world where liver transplantation is not available, surgical resection is a viable option, particularly for Child class A patients without portal hypertension and with a Model for End-stage Liver Disease (MELD) score of 9 (see Chapter 90). All the tumor nodules need to be removed with negative margins, and the patient needs to be left with enough functional liver volume (usually defined as ≅40%) to survive the postoperative period.104–106 Overall, resection is feasible in only approximately 15% of patients. Resection performed at expert surgical centers carries an operative mortality rate of less than 5%, but at low volume centers the mortality rate is almost three times greater.107 Unfortunately, recurrence after resection occurs in more than 50% in the long term, and salvage liver transplantation is rarely possible.108
Liver Transplantation
Liver transplantation is performed in patients in whom the tumor is not resectable but is confined to the liver or in whom advanced cirrhosis and poor liver function preclude resection (see Chapter 95).25 Liver transplantation is the ideal therapy for hepatocellular carcinoma because it provides the largest possible resection margin, removes the remaining liver, which is at high risk for de novo tumors, and replaces the dysfunctioning liver. Liver transplantation can fail in patients with extrahepatic tumor, which tends to grow rapidly under the influence of post-transplantation immunosuppression. Because the availability of donor livers is limited, the consensus is that the outcomes of liver transplantation for hepatocellular carcinoma should be similar to those for other indications for liver transplantation and superior to those for other treatments for hepatocellular carcinoma. Several large series have demonstrated that if one selects candidates based on the Milan criteria—a single lesion up to 5 cm in size or two to three lesions, each up to 3 cm, with no large-vessel vascular invasion or metastasis—the five-year survival rate is 70% to 75%, and the tumor recurrence rate is 10% to 15%.102,109–111 These criteria led to the hepatocellular carcinoma MELD exception pathway, which was adopted in the United States in 2002. As a result of the change, the frequency of hepatocellular carcinoma as an indication for liver transplantation rose from 4.6% to 26% of the total adult liver transplant population. Additionally, progression of the tumor beyond the Milan criteria before a patient undergoes transplantation has largely been eliminated.38,112 In other parts of the world, waiting times before transplantation remain critical, and when the waiting time increases to one year, as many as one half of patients will not receive a transplant.102 An analysis of four-year survival rates for all patients transplanted in the United States has confirmed that overall outcomes for those transplanted with hepatocellular carcinoma are only minimally worse than for those transplanted for other indications.38 Certain subgroups of patients do worse, including those with nodules 3 to 5 cm in diameter, a MELD score of 20, and a serum AFP level of ≥455 ng/mL.
Some authorities have advocated a modest expansion of the Milan criteria, with use of the so-called University of California, San Francisco (UCSF) criteria (a single lesion up to 6.5 cm in diameter or two or three lesions up to 4.5 cm each, with a total tumor diameter of 8 cm), based on excellent prospective outcomes from a small, single-center series, but these patients generally need a special exception from the regional review board in the United States.113 Other groups who use similar criteria have shown similarly good results.114 A larger multicenter study is needed before these criteria can be widely adopted.
Local Ablation
Local ablative therapies are potentially curative treatments for patients with small tumors, usually smaller than 3 to 5 cm in diameter, that are not amenable to resection or liver transplantation because of patient preference, the number and location of lesions, or significant hepatic dysfunction (Child class B or C; see Fig. 94-4).25,115 The first of these techniques available was percutaneous ethanol injection (PEI), a relatively effective and safe method that is still used and is most effective for lesions smaller than 2 to 3 cm in diameter.116 PEI requires multiple sessions and, in patients with small tumors and intact hepatic function, can lead to survival rates similar to those for surgical resection, although no randomized studies have been performed to demonstrate equivalent outcomes.117 Complications are rare but include tumor seeding of the needle track. More recently, radiofrequency ablation (RFA) has supplanted PEI, because it is more effective, particularly with larger tumors (up to 3 to 5 cm), requires fewer sessions, and has similar complication rates.118 RFA can be performed percutaneously or by a laparoscopic or open surgical approach. Survival rates are similar to those for surgical resection, although recurrence rates are higher and complications are uncommon.117,119 PEI is generally favored over RFA for lesions adjacent to a major vessel or large bile ducts. A randomized study has suggested that a combination of RFA and chemoembolization for tumors larger than 3 cm in diameter produces a survival benefit when compared with either treatment alone.120 PEI and RFA have been used to stabilize tumor growth in patients awaiting liver transplantation, but their use for this purpose is controversial and probably unnecessary, unless the waiting time for transplantation is more than six months or the tumor burden is near the limits of acceptability for transplantation.102,121–123
Chemoembolization
Transarterial chemoembolization (TACE) is a palliative treatment reserved for patients with relatively intact hepatic function (Child class A or B) and tumors not amenable to local ablative treatments because of size, number, or location (see Fig. 94-4).25,121 Six randomized trials and a meta-analysis have compared embolization or chemoembolization with supportive care and have shown overall improved survival with treatment.124–130 The effectiveness of TACE before liver transplantation has not been fully elucidated, but TACE can be considered if the waiting time for transplantation is more than six months or the tumor size is near the acceptable limit.131,132 Theoretically, TACE can be used to reduce the size of the tumor to make resection or transplantation possible (downstaging) or to allow a more conservative resection, although study results are mixed as to whether this approach is effective.133,134
Chemotherapy
A large number of anticancer drugs, including alkylating agents, antitumor antibiotics, antimetabolites, plant alkaloids, platinum derivatives, procarbazine, estrogen receptor modulators, and somatostatin, have been tried alone and in various combinations and by different routes of administration for the treatment of hepatocellular carcinoma, but response rates have invariably been less than 20%.126,135 Several small-molecule, targeted anticancer agents have been developed and studied for the treatment of hepatocellular carcinoma. Sorafenib, an inhibitor of Raf kinase and the tyrosine kinase activity of vascular endothelial growth factor receptors (VEGFRs) and platelet-derived growth factor receptor (PDGFR), is the first of these new agents to show modest improvement in survival compared with supportive care.136 The drug should be considered for patients with intact hepatic function (Child class A) and portal vein thrombosis, extrahepatic tumor, or failures of other therapies (see Fig. 94-4). Other targeted agents, alone and in combination with each other and with traditional chemotherapy, are being studied. Patients with advanced hepatic dysfunction (Child class C) or advanced tumor symptoms (Eastern Cooperative Oncology Group [ECOG] performance status > 2) have such a poor prognosis that only supportive care should be offered (see Fig. 94-4).25
Screening
Because symptomatic hepatocellular carcinoma seldom is amenable to surgical cure and responds poorly to conservative treatments, a pressing need exists to prevent the tumor or detect it at a presymptomatic stage when surgical intervention is still possible. Programs for detecting subclinical hepatocellular carcinomas are of two types: (1) screening whole populations with a high incidence of the tumor; and (2) case finding and long-term surveillance of persons at high risk for the development of hepatocellular carcinoma. Mass population screening has rarely been attempted, whereas case finding and surveillance of high-risk persons are more feasible25 and have been shown to be cost-effective in countries with a high incidence of the tumor.
An AASLD practice guideline published in 2005 provides recommendations for screening (Table 94-7).25 Briefly, patients at high risk of developing hepatocellular carcinoma should be entered into a surveillance program. Surveillance for hepatocellular carcinoma should be performed using ultrasonography at 6- to 12-month intervals, but AFP alone should not be used for screening unless ultrasound is not available. Although CT and MRI are effective imaging modalities for the diagnosis of hepatocellular carcinoma, they are not recommended for routine use in surveillance but may be considered if adequate ultrasound images cannot be obtained because of body habitus. Growing evidence suggests that surveillance for hepatocellular carcinoma in patients with cirrhosis improves outcome.137
Table 94-7 High-Risk Groups for Whom Surveillance for Hepatocellular Carcinoma Is Recommended
| HIGH-RISK GROUP | FACTORS THAT ADD TO RISK |
|---|---|
| Hepatitis B viral carriers | Africans, age > 20 yr |
| Asian men, age > 40 yr | |
| Asian women, age > 50 yr | |
| Family history of hepatocellular carcinoma | |
| Patients with cirrhosis | |
| Patients with high serum HBV DNA level and ongoing hepatic injury | |
| Patients with cirrhosis | Alcoholic cirrhosis |
| α1-Antitrypsin deficiency* | |
| Autoimmune hepatitis* | |
| Hemochromatosis | |
| Hepatitis B | |
| Hepatitis C | |
| Nonalcoholic steatohepatitis* | |
| Primary biliary cirrhosis |
HBV DNA, hepatitis B viral deoxyribonucleic acid.
* No data on efficacy of surveillance available.
Adapted from Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology 2005; 42:1208-36.
Prevention
Although great progress has been achieved in the primary prevention of HBV-induced hepatocellular carcinoma with universal infant vaccination against HBV in many countries, the full impact of universal HBV vaccination on the occurrence of the tumor will not be realized for many years. A significant reduction has already been noted in childhood hepatocellular carcinoma in Taiwan, where universal infant vaccination was adopted in the mid-1980s.138 In the meantime, the huge numbers of existing HBV carriers worldwide remain at risk for hepatocellular carcinoma, and little progress has been made in preventing malignant transformation in persons with chronic viral hepatitis, nor has much progress has been made on other fronts. A vaccine against HCV will not be available in the near future, and prevention of aflatoxin-induced tumors is far from a becoming a reality, despite ongoing trials of chemopreventive agents.
Considerable interest has been expressed in the impact of antiviral therapy against HBV and HCV in reducing the incidence of hepatocellular carcinoma. One randomized controlled trial of long-term therapy of lamivudine versus placebo in patients with chronic hepatitis B has shown a significant decrease in the frequency of clinical events in the treated group, including a decrease in the frequency of hepatocellular carcinoma.77 Several large retrospective studies have shown a decrease in frequency of hepatocellular carcinoma in patients successfully treated for chronic hepatitis C with interferon-based regimens.139
INTRAHEPATIC CHOLANGIOCARCINOMA
Cholangiocarcinoma is a malignant neoplasm arising from the biliary duct epithelium. It often carries different names based on the particular portion of the biliary tree involved—small intrahepatic bile ducts (peripheral cholangiocarcinoma), hepatic duct bifurcation (perihilar cholangiocarcinoma, or Klatskin tumor), and extrahepatic bile ducts (bile duct carcinoma). The location of the tumor has a major impact on the presenting symptoms and treatment approach. Perihilar cholangiocarcinoma is classified with the intrahepatic group based on International Classification of Diseases, 9th revision (ICD-9) codes even though it is extrahepatic in origin and is the most common form.140,141 This section will be limited to a discussion of intrahepatic cholangiocarcinoma; extrahepatic cholangiocarcinoma is discussed in Chapter 69.
Epidemiology
Intrahepatic cholangiocarcinoma represents approximately 10% to 20% of all primary liver cancers and 20% to 25% of cholangiocarcinomas. The geographic variation in prevalence rates is marked, ranging from 0.2 to 96/100,000 in men and from 0.1 to 38/100,000 in women, because of differences in the frequencies of known risk factors in various populations.142 The highest prevalence rates are found in parts of Asia, most notably certain regions of Thailand, Hong Kong, China, Japan, and Korea. Chronic infestation of the biliary tree with one of the liver flukes is thought to be the cause of these high rates.143 The overall prevalence rate in the United States is 0.85/100,000, with a 1.5-fold higher rate in men than women. The rate in whites is about equal to that in African Americans and about half that in Asians.
Although the underlying predisposing factor for most cases of cholangiocarcinoma is unknown, a number of risk factors have been recognized. The strongest association is with Opisthorchis viverrini, a liver fluke endemic in parts of Southeast Asia and acquired by ingestion of raw or uncooked fish.142,144,145 The association with Clonorchis sinensis, a related liver fluke, is weaker.146 An association with the radiographic contrast agent thorium dioxide (Thorotrast), which was banned in the 1950s, has been well established.147 Primary sclerosing cholangitis is linked to a diagnosis of cholangiocarcinoma at a young age, with a lifetime risk of 8% to 20% (see Chapter 68).148–150 Congenital and acquired abnormalities of the biliary tract that may result in bile stasis, chronic inflammation, and infection, as in biliary atresia,151 von Meyenburg complexes,152 Caroli’s disease,153 choledochal cyst,153 and intrahepatic cholelithiasis, have been associated with the development of cholangiocarcinoma. Cirrhosis, particularly caused by HCV, also has been associated with cholangiocarcinoma.154
Intrahepatic cholangiocarcinoma is rare before the age of 40 years, and historically the worldwide approximate average age at presentation is 50 years. Epidemiologic data indicate that the age at presentation has shifted to more than 65 years. Additionally, the incidence and mortality rates are increasing worldwide.140 Surveillance, Epidemiology and End Results (SEER) registry data from the United States have shown a 165% increase between the late 1970s and the late 1990s.142 This increase may be a result, in part, of the increased prevalence of cirrhosis, particularly HCV-associated cirrhosis.154
Molecular Pathogenesis
Malignant transformation of the bile duct cells generally occurs in an environment of inflammation or cholestasis (or both), usually as a result of one of the known risk factors. The proposal has been made that a combination of these environmental factors and genetic predisposition—for example, defects in oncogenes or bile salt transporters—leads to an accumulation of genetic defects that results in carcinoma.140,155 A polymorphism in the gene for the natural killer cell receptor G2D (NKG2D) has been associated with an increased risk of cholangiocarcinoma in patients with primary sclerosing cholangitis.156 At the molecular level, numerous changes have been described, including mutations of the K-ras gene, the gene for interleukin-6, and allelic loss or mutations of TP53 and p16, as well as many others (see Chapter 69).
Clinical Features
Peripheral cholangiocarcinoma seldom produces symptoms until the tumor is advanced. The clinical features are then similar to those of hepatocellular carcinoma, including malaise, weight loss, abdominal pain, and jaundice, which may be more frequent and prominent than with hepatocellular carcinoma.153,157 The clinical presentation of perihilar and extrahepatic cholangiocarcinoma is with progressive painless jaundice, acholic stools, pruritus with or without weight loss, and, rarely, cholangitis.158 Patients with primary sclerosing cholangitis may present with worsening jaundice caused by a dominant bile duct stricture, weakness, and weight loss.
Diagnosis
In patients with perihilar cholangiocarcinoma and extrahepatic cholangiocarcinoma, obstructive jaundice is evident, with elevated serum levels of bilirubin, alkaline phosphatase, gamma glutamyl transpeptidase (GGTP), and often aminotransferases. In patients with peripheral cholangiocarcinoma, often only the alkaline phosphatase level is elevated. CA 19-9 is the most frequently used serum tumor marker for cholangiocarcinoma but has significant limitations because CA 19-9 levels are also elevated in pancreatic, colorectal, gastric, and gynecologic cancers and in acute bacterial cholangitis.159 CA 19-9 is always undetectable in the 7% of the population that is Lewis blood group–negative. In patients with unexplained biliary obstruction without primary sclerosing cholangitis, the sensitivity of CA 19-9 is 53%, and the negative predictive value is 72% to 92%, for a cutoff value of 100 U/mL. In patients with primary sclerosing cholangitis, the sensitivity ranges from 38% to 89% and specificity from 50% to 98%. The addition of carcinoembryonic antigen (CEA) probably does not improve the performance of CA 19-9 in the setting of primary sclerosing cholangitis.
Initial imaging with ultrasound helps identify biliary obstruction. Dynamic contrast-enhanced CT or MRI further aids in localizing the lesion and determining the possibility of resection.140,160 MRI with magnetic resonance cholangiography (MRCP) is a superior modality because of a higher sensitivity than CT for detecting lesions and localizing biliary obstruction. The tumor is hypodense on T1-weighted images and moderately intense on T2-weighted images. Endoscopic retrograde cholangiopancreatography (ERCP) or transhepatic cholangiography allows for localization of the tumor, sampling of tissue and bile, and relief of biliary obstruction if the tumor is unresectable. A perihilar tumor may demonstrate a classic appearance on ERCP, but for other biliary strictures, particularly in patients with primary sclerosing cholangitis, determining whether cholangiocarcinoma is present may be difficult. Cytology has a 30% sensitivity, which improves to 40% to 70% with the addition of brushings and biopsies. Newer cytologic techniques that assess the cells for aneuploidy and chromosomal aberrations may improve the diagnostic yield but are not widely available (see Chapter 69). Endoscopic ultrasound (EUS) with fine-needle aspiration (FNA) in patients without primary sclerosing cholangitis has the advantage of improving sensitivity and specificity for diagnosis of the primary lesion and nodal metastasis but the disadvantage of causing peritoneal seeding, and thus should be avoided if surgical resection is contemplated. Percutaneous biopsies also carry the risk of peritoneal seeding and are generally avoided if the tumor is potentially resectable.
Pathology
Peripheral cholangiocarcinoma usually is a large and solitary tumor, but it may be multinodular.161 It is grayish-white, firm, and occasionally umbilicated and can produce a focal hepatic mass, a tumor growing along and infiltrating the bile ducts, or an intraductal papillary lesion.160 The tumor is poorly vascularized and rarely bleeds internally or ruptures. Perihilar cholangiocarcinoma may take the form of a firm intramural tumor that encircles the bile duct, a bulky mass centered on the duct or hilar region that radiates into the hepatic tissue, or a spongy friable mass within the lumen of the duct. Metastatic nodules may be distributed irregularly throughout the liver. The bile ducts peripheral to the tumor may be dilated, resulting in some cases in biliary cirrhosis. Metastases in regional lymph nodes occur in about 50% of cases.
Microscopically, cholangiocarcinoma exhibits acinar or tubular structures that resemble those of other adenocarcinomas.161 Most tumors are well differentiated. Secretion of mucus may be demonstrable, but bile production is not seen. The tumor cells provoke a variable desmoplastic reaction, and, in many tumors, the collagenized stroma may be the most prominent feature. Distinguishing the tumor from metastatic adenocarcinoma may be difficult, and some experts have advocated assuming that an adenocarcinoma in the liver is cholangiocarcinoma if no primary tumor can be found elsewhere.162
Treatment and Prognosis
Early diagnosis of intrahepatic cholangiocarcinoma is unusual, and the annual mortality rate is almost identical to the annual incidence of the tumor.153,157 Long-term survival after diagnosis in the United States based on the SEER database is dismal, with a one-year survival rate of 28% and a five-year survival rate less than 5%. The five-year survival rate has not improved since the late 1980s.142
In a person with suspected or proven intrahepatic cholangiocarcinoma, staging is recommended to determine surgical resectability, which is the only opportunity for cure. The staging evaluation usually includes dynamic MRI and MRCP of the abdomen (or dynamic helical CT, if MRI is unavailable) and a chest x-ray or CT.140 Positron emission tomography (PET) has been assessed in small series and does not clearly add to other modalities. EUS with FNA of suspicious lymph nodes may detect otherwise unrecognized metastasis in up to 20% of cases.160 Surgical resectability of intrahepatic cholangiocarcinoma should be determined in conjunction with an experienced hepatobiliary surgeon and requires the ability to achieve clear surgical margins, which usually necessitates a major hepatectomy. Criteria for resection include absence of all the following: evidence of extrahepatic metastasis; main portal vein or hepatic artery invasion or encasement; bilateral segmental bile duct involvement; and contralateral hepatic lobar atrophy. Additionally, the patient must be medically fit to undergo surgery and have sufficient hepatic reserve. Without clear margins of resection, surgery provides benefits similar to those of endoscopic or biliary drainage. Patients well selected for surgical resection achieve a one- to two-year median survival and a 29% to 36% five-year survival rate.
If resection is not possible and major biliary obstruction is present, biliary drainage, either endoscopic or percutaneous, should be performed, because drainage appears to improve symptoms and survival (see Chapter 70).162 Placement of an expandable metal stent is generally preferred to plastic stents if the expected survival of the patient is more that three to six months.140 The rates of response and survival following radiation therapy and chemotherapy are modest, as suggested by predominantly small uncontrolled series, so these modalities generally should be restricted to clinical trials. Photodynamic therapy in addition to biliary stent placement may provide benefit. Liver transplantation alone results in unacceptably high recurrence rates and limited survival.160 In a single center report, aggressive preoperative therapy of unresectable hilar cholangiocarcinoma with external beam radiation, brachytherapy, and chemosensitization, followed by liver transplantation in patients who survived the treatment and had contained disease, produced a five-year survival rate of 82%.163
HEPATOBLASTOMA
Epidemiology
In children, hepatoblastoma is the third most common malignant tumor and the most common malignant hepatic tumor. It occurs almost exclusively in the first three years of life; boys are affected twice as often as girls.164,165
Clinical Features
Most children with hepatoblastoma come to medical attention because of abdominal swelling.166 Other reasons include failure to thrive, weight loss, poor appetite, abdominal pain, irritability, and intermittent vomiting and diarrhea. The tumorous liver almost always is enlarged and firm and may be tender. Its surface is smooth or nodular. Hepatoblastomas rarely rupture. Distant metastases are evident, usually in the lung, in 20% of patients at the initial visit.167 The tumor occasionally causes isosexual precocity in boys as a result of the ectopic production of human chorionic gonadotropin.168
Diagnosis
AFP is present in high concentrations in the serum of 80% to 90% of patients with hepatoblastoma and is a useful clue to diagnosis.169 The few patients with a low serum AFP level appear to have a worse prognosis.170 Anemia is common, as is thrombocytosis, which is attributed to raised serum thrombopoietin levels. Pulmonary metastases and, rarely, mottled calcification in the tumor may be seen on plain radiography. Ultrasonography is the most widely used initial imaging technique, although the findings are not specific. CT and MRI are used to define the extent of the tumor and plan definitive surgery. The tumor is seen as an avascular mass on hepatic arteriography.171
Pathology
Hepatoblastomas are the malignant derivatives of incompletely differentiated hepatocyte precursors. Their constituents are diverse, reflecting both the multipotentiality of their mesodermal origin and the progressive stages of embryonic and fetal development. Hepatoblastomas are classified morphologically into an epithelial type, composed predominantly of epithelial cells of varying maturity, and a mixed epithelial and mesenchymal type, which contains, in addition, tissues of mesenchymal derivation.166,169 The tumors usually are solitary, ranging in diameter from 5 to 25 cm, and always well circumscribed (about half are encapsulated). They vary in color, ranging from tan to grayish-white, and contain foci of hemorrhage, necrosis, and calcification. Vascular channels may be prominent on the capsular surface. Epithelial hepatoblastomas are solid, whereas tumors of the mixed variety often are separated into lobules by white bands of collagen tissue.
Two types of epithelial cells are present in the tumor.172 Cells of the first type resemble fetal hepatocytes and are arranged in irregular plates, usually two cells thick, with bile canaliculi between individual cells and sinusoids between plates. Cells of the second type are embryonal and are less differentiated than the fetal type. Mixed hepatoblastomas contain mesenchymal tissue consisting of areas of a highly cellular primitive type of mesenchyme intimately admixed with epithelial elements. Cartilage and striated muscle may be present. Hepatoblastomas may show foci of squamous cells, with or without keratinization, and foreign body–type giant cells. Vascular invasion may be evident. Metastases most commonly involve lung, abdominal lymph nodes, and brain.
Cause and Pathogenesis
Hepatoblastoma may occur sporadically or in association with hereditary syndromes such as familial adenomatous polyposis (FAP) and Beckwith-Wiedemann syndrome, suggesting a possible role for chromosomes 5 and 11 in the genesis of the tumor. The FAP tumor suppressor gene down-regulates β-catenin. Sporadic hepatoblastoma is not associated with any known environmental risk factor, and its pathogenesis is unclear. Most patients with hepatoblastoma have mutations of the FAP gene, and a similar number have activating mutations of the β-catenin gene, raising the possibility that the wnt signaling pathway plays a role in the development of the tumor.173
Treatment and Prognosis
Hepatoblastomas are rapidly progressive. If the lesion is solitary and sufficiently localized to be resectable, surgery often is curative, with five-year survival rates as high as 75%.166 The current practice is to pretreat the patient with cisplatin and doxorubicin. When the tumor is judged to be inoperable, neoadjuvant chemotherapy may reduce the size of the tumor sufficiently to permit resection. Encouraging results also have been obtained with liver transplantation in patients with bilobar multifocal tumors without extrahepatic extension.174 If surgery is not possible or the tumor recurs after surgery, the prognosis generally is poor.
HEMANGIOSARCOMA
Epidemiology
Although rare, angiosarcoma is the most common malignant mesenchymal tumor of the liver.175,176 It occurs almost exclusively in adults and is most prevalent in the sixth and seventh decades of life.177,178 Men are affected four times as often as women.
Pathogenesis
Despite its rarity, hepatic angiosarcoma is of special interest because specific risk factors have been identified, although no cause is discerned in most cases. In early reports, the tumor became evident approximately 20 years after the patient had been exposed to thorium dioxide (see Chapter 87).179 Angiosarcoma also has occurred in German vintners who used arsenic-containing insecticides and drank wine adulterated with arsenic.180 A few patients with angiosarcoma had taken potassium arsenite (Fowler’s solution) for many years to treat psoriasis.181 Hepatic angiosarcoma in workers exposed to vinyl chloride monomer (VCM) was first reported in 1974.177,182,183 The monomer is converted by enzymes of the endoplasmic reticulum to reactive metabolites that form DNA adducts and guanosine-to-adenine transitions in the K-ras and TP53 genes. Angiosarcomas have occurred after exposures of 11 to 37 years (or after shorter periods with a heavy initial exposure). The mean age of patients at diagnosis is 48 years. In addition to angiosarcoma, persons exposed to VCM may be at increased risk of hepatocellular carcinoma and soft tissue sarcoma.
Clinical Features
The most common presenting symptom is upper abdominal pain. Other frequent complaints are abdominal swelling, rapidly progressing liver failure, malaise, weight loss, poor appetite, and nausea.176,177 Vomiting occurs occasionally. The duration of symptoms generally ranges from one week to six months, but a few patients have had symptoms for as long as two years before seeking medical attention.
Diagnosis
A rising serum bilirubin level and other evidence of progressive hepatic dysfunction may be present, especially in the later stages of the tumor. Plain radiography may show pulmonary metastases, a raised right hemidiaphragm, or, rarely, skeletal metastases. In patients who received thorium dioxide, radiopaque deposits of the material may be evident in the liver and spleen.179 One or more mass lesions may be demonstrated on ultrasonography, CT, or MRI, but diffusely infiltrating tumor may not be visualized. Hepatic arteriography reveals a characteristic appearance.184 The hepatic arteries are displaced by the tumor, which shows a blush and “puddling” during the middle of the arterial phase that persist for many seconds, except in the central area, which may be hypovascular.
Complications and Prognosis
Hepatic angiosarcomas grow rapidly, and the prognosis is poor; death ensues within six months. Patients may have thrombocytopenia resulting from entrapment of platelets within the tumor (Kasabach-Merritt syndrome), disseminated intravascular coagulation with secondary fibrinolysis,185 or microangiopathic hemolytic anemia as a result of fragmentation of erythrocytes within the tumor circulation.186
Pathology
Angiosarcomas usually are multicentric.187 Their hallmark is the presence of blood-filled cysts, although solid growth also is seen. The lesions are fairly well circumscribed but not encapsulated. Larger masses are spongy and bulge beneath Glisson’s capsule.
Treatment
Operative treatment usually is precluded by the advanced stage of the tumor. Even when surgery is undertaken, the patient commonly survives only one to three years, although long-term survival may be achieved in the few patients with a solitary tumor.176 The results of irradiation and chemotherapy are poor.
EPITHELIOID HEMANGIOENDOTHELIOMA
Epidemiology
Epithelioid hemangioendothelioma is a rare tumor whose incidence is not known. A case series of 137 cases has been collected at a specialized referral center.188 Two thirds of patients were female, and the tumor occurred at all ages in adulthood.
Clinical Features
Patients typically present with nonspecific symptoms, such as abdominal pain and weight loss.
OTHER PRIMARY MALIGNANT TUMORS OF THE LIVER
Undifferentiated (embryonal) sarcoma is a rare primary malignancy of the liver that occurs in both children and adults.189,190 The tumor tends to be aggressive, but long-term survival can be achieved with radical surgery and chemotherapy. Other rare sarcomas arising in the liver include liposarcoma,191 lymphoma,176,192 and rhabdomyosarcoma.190
HEPATIC METASTASES
The liver is the most frequent target for metastatic spread of tumors. Hepatic metastases occur in 40% to 50% of adult patients with extrahepatic primary malignancies.193 Foremost among the reasons for the high frequency of hepatic metastases are the double blood supply of the liver and the presence of fenestrations in the sinusoidal endothelium that facilitate penetration of malignant cells into the hepatic parenchyma.194 Hepatic metastases commonly originate from primary sites in the distribution of the portal venous system, including the pancreas, stomach, and colon. Outside this distribution, tumors of the lung and breast are the most common origins of hepatic metastases.
Diagnosis
CT is the most useful imaging technique.195 Multiphase helical CT and CT during arterial portography are more sensitive than conventional CT. Dynamic contrast-enhanced Doppler ultrasonography with intra-arterial infusion of CO2 microbubbles also is useful for the diagnosis of hepatic metastases.54 T1-weighted MRI also may be helpful, and iron oxide–enhanced MRI is even better.
Pathology
Macroscopic Appearance
Hepatic metastases almost always are multiple.193 Their pathologic features vary, depending on the site of origin. Metastases are expansive, when they are discrete, or infiltrative. Individual metastases may reach a large size, and, with multiple metastases, the liver may be greatly enlarged. Metastases commonly are gray-white and may show scattered hemorrhages or central necrosis. Individual metastases may be surrounded by a zone of venous stasis. Subcapsular lesions often are umbilicated. The dictum that cirrhotic livers are less likely than noncirrhotic livers to harbor metastatic deposits remains to be verified.
Microscopic Appearance
The microscopic features, including the degree of stromal growth, of most hepatic metastases duplicate those of the tumor of origin. Metastatic deposits usually are easily delineated from the surrounding liver tissue. Invasion of portal or hepatic veins may be seen, although less often than with hepatocellular carcinoma.193 It may be difficult to distinguish metastatic adenocarcinoma from primary cholangiocarcinoma.162
Treatment and Prognosis
The extent of replacement of liver tissue by metastases generally determines the patient’s prognosis. The greater the tumor burden, the worse the outlook, with only approximately 50% of patients surviving three months after the onset of symptoms and less than 10% surviving more than one year.196 Improved imaging modalities, advances in surgical techniques for resection and transplantation, and new chemotherapeutic agents and regional therapies have made it possible to achieve long-term survival in individual patients. Long-term survival has been accomplished most often by resection of hepatic metastases in patients with colorectal cancer, a substantial number of whom have been cured or have obtained up to 20 years of disease-free survival.196–198 Survival for five years can be achieved in up to 60% who undergo resection of a solitary colon cancer metastasis to the liver.199 If the primary tumor has been removed completely and metastases are confined to the liver, resection of hepatic metastases should be considered. Liver transplantation, with or without chemotherapy, has been undertaken in a few patients but is generally not considered. RFA is a valid therapy for colorectal metastases in patients who are unable to tolerate or refuse surgical resection. Other invasive methods of destroying metastases, such as ethanol injection, freezing with cryoprobes, and laser vaporization, warrant further study. Radiation therapy and intra-arterial infusion of cytotoxic drugs have limited roles.
BENIGN TUMORS
HEPATOCELLULAR ADENOMA
Epidemiology and Pathogenesis
Hepatocellular adenomas (or hepatic adenomas) were extremely rare before the use of oral contraceptive steroids became widespread. They are still rare in men, and the development of this tumor in women who are taking or have taken contraceptive steroids strongly implies a cause and effect relationship.200,201 Nevertheless, in light of the large number of women who use this form of contraception, the risk of hepatocellular adenoma is small, and its occurrence implies some form of genetic predisposition (see later). The association is particularly strong with prolonged use of oral contraceptive steroids; the estimated risk for women who use oral contraceptive agents continuously for five to seven years is five times the normal rate, and this risk increases to 25 times with use for longer than nine years. Using preparations with high hormone potency further increases the risk. Contraceptive pill–associated hepatocellular adenomas are more likely to develop in older than younger women.
Hepatocellular adenomas have been linked to both types of synthetic estrogen and all forms of progestogen contained in oral contraceptive preparations.202 Current evidence favors estrogens as the culprit, although progestogens may contribute through their enzyme-inducing properties. The growth of hepatocellular adenomas appears to be hormone-dependent, as evidenced by an increase in size during pregnancy and occasional cases of regression (and even disappearance) of the tumor after cessation of oral contraceptive use. Although hormonal replacement therapy has not proved to be associated with the development of hepatic adenoma, avoidance of this form of therapy in women with a history of oral contraceptive–related hepatic adenoma is prudent, unless compelling reasons to the contrary exist.
Hepatocellular adenomas and adenomatosis also may occur in those receiving long-term anabolic androgenic steroids63 and in those with certain inherited metabolic disturbances, especially type 1 glycogen storage disease, in which one or more hepatocellular adenomas occur in approximately 60% of patients (see Chapter 76).201
Genetic alterations have been identified in hepatocellular adenomas. Bilallelic mutations of the TCF1 gene that codes for hepatocyte nuclear factor 1α (HNF-1α) have been identified in up to 60% of patients with adenoma.203 A second pathway, the wnt pathway, which also is activated in about 25% of patients with hepatocellular carcinoma, is activated in at least some cases of adenoma.204 β-Catenin activation via this pathway appears to confer a higher risk of malignant transformation.205 The third identified pathway for formation of hepatocellular adenomas includes acute inflammatory responses demonstrable by histologic examination of the tumor205 and associated with obesity and alcohol (Fig. 94-5).
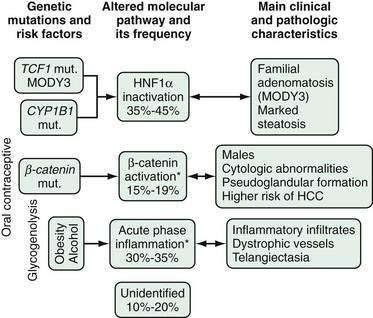
Clinical Features
Hepatocellular adenomas manifest in a number of ways. They may produce no symptoms and be discovered during routine physical examination, if large, or during imaging of the upper abdomen for other reasons, if small. Approximately 25% of patients experience pain in the right hypochondrium or epigastrium. The pain usually is mild and ill-defined but may be severe as a result of bleeding into or infarction of the tumor. If the liver is enlarged, the surface usually is smooth, and the liver may be slightly tender. The most alarming presentation is with an acute hemoperitoneum following rupture of the adenoma. This complication is not uncommon, especially with tumors linked to oral contraceptive use, and carries an appreciable mortality rate.206,207 Tumors that rupture generally are large and solitary, although the most important determinant of rupture is a superficial location. Often, the affected woman is menstruating at the time; rupture also may occur during pregnancy.208
Diagnosis
Serum AFP concentrations are normal. Ultrasonography is used for initial imaging. Multiphase helical CT or MRI may also be used.209,210 The tumor has a clearly defined margin and, often, has almost parallel vessels entering it from the periphery (spoke wheel appearance). Alternatively, the lesion may contain tortuous vessels coursing irregularly through it. The adenoma may have focal avascular areas as a result of hemorrhage or necrosis. Because adenomas mimic normal liver tissue microscopically, needle biopsy and FNA may be of limited diagnostic value because the cytologic appearance of the cells is similar to those of normal hepatocytes, although the tumor does not have normal portal structures.
Pathology
Hepatocellular adenoma generally occurs as a solitary, relatively soft, light brown to yellow tumor. It is sharply circumscribed but does not have a true capsule, although a pseudocapsule is formed by compression of the surrounding liver tissue (Fig. 94-6A).211 Hepatocellular adenomas arise in an otherwise normal liver. Occasionally, two or more tumors are present. Adenomas range in diameter from 1 to 30 cm and are commonly 8 to 15 cm in diameter. They are larger on average in women taking contraceptive steroids than in those not taking contraceptive steroids; they usually occupy a subcapsular position and project slightly from the surface of the liver. A pedunculated variety is seen occasionally. The cut surface of the tumor may show ill-defined lobulation but is never nodular or fibrotic. Foci of hemorrhage or necrosis are frequent, and bile staining may be evident.
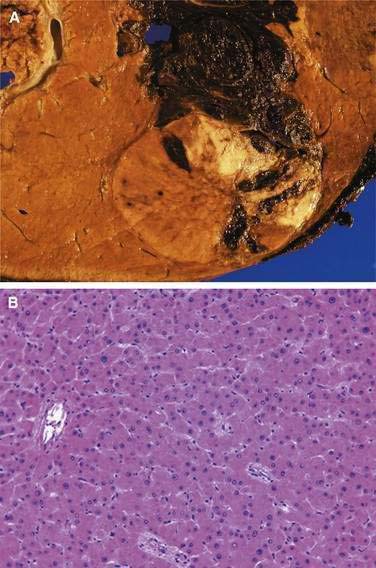
Microscopically, hepatocellular adenoma may mimic normal liver tissue to an astonishing degree (see Fig. 94-6B).211 The tumor is composed of sheets or cords of normal-looking or slightly atypical hepatocytes that show no features of malignancy. Few or no portal tracts or central veins are present, and bile ducts are conspicuously absent. Only an infrequent fibrous or vascular septum traverses the lesion. An essentially normal reticulin pattern is demonstrable throughout the adenoma. The walls of arteries and veins are thickened. Some areas with vascular abnormalities are infarcted, and thrombi may be seen. Peliosis hepatis may be found in relation to the tumor.
Treatment and Prognosis
Because of the danger that a hepatocellular adenoma may rupture, surgical treatment is recommended.211,212 Resection usually is feasible in an uncomplicated case. When rupture has occurred, emergency resection should be performed if possible. If resection cannot be accomplished, the hepatic artery should be ligated. Arterial embolization has also been used successfully to control hemorrhage from a ruptured adenoma, either in preparation for surgery or when surgery is not possible.206 Whether or not the tumor is removed, the patient must refrain from taking oral contraceptive steroids. If the adenoma is not resected, pregnancy should be avoided.
Hepatocellular carcinoma occurs in a small number of women taking oral contraceptive steroids, and speculation has been raised that hepatocellular adenomas might undergo malignant transformation. Indeed, this sequence has been documented in a few instances. Therefore, managing hepatocellular adenomas merely by discontinuing the use of contraceptive steroids carries the risk that malignant transformation may still occur. The management of hepatic adenomatosis is problematic.213 Often, in these cases, the number of tumors is large, and they cannot be resected entirely. The role of liver transplantation for adenomatosis is not clear at present.
CAVERNOUS HEMANGIOMA
Epidemiology
Cavernous hemangioma is the most common benign tumor of the liver and is found in as many as 7% of autopsies.63,211 The lesion is thought to be a congenital malformation or hamartoma that increases in size, initially with growth of the liver and thereafter by ectasia. Cavernous hemangiomas affect persons of all ages, although they manifest most often in the third, fourth, and fifth decades of life. Women are predominantly affected (4 : 1 to 6 : 1) and often present at a younger age and with larger tumors in comparison with men. Cavernous hemangiomas may increase in size with pregnancy or the administration of estrogens and are more common in multiparous than in nulliparous women.
Clinical Features
The great majority of cavernous hemangiomas are small and asymptomatic and are discovered incidentally during imaging of the liver for another reason, at autopsy, or at laparotomy. Larger or multiple lesions produce symptoms.214 Those larger than 4 cm in diameter are called giant cavernous hemangiomas, which may be as large as 27 cm. Upper abdominal pain is the most common complaint and results from partial infarction of the lesion or pressure on adjacent tissues. Early satiety, nausea, and vomiting also may occur. Cavernous hemangiomas occasionally rupture. The only physical finding may be an enlarged liver. Occasionally, an arterial bruit is heard over the tumor. Arteriovenous shunting has been described with cavernous hemangiomas.
Diagnosis
The ultrasonographic appearance is variable and nonspecific, although the lesion usually is echogenic.215,216 Provided that the cavernous hemangioma is larger than 3 cm in diameter, single photon emission computed tomography (SPECT) with colloid 99mTc-labeled red blood cells shows the tumor to be highly vascular and has a sensitivity and accuracy similar to those of MRI.217 Almost all cavernous hemangiomas can be diagnosed by bolus-enhanced CT with sequential scans.218 The center of the lesion remains hypodense, whereas the peripheral zone, which varies in thickness and may have a corrugated inner margin, is enhanced. MRI has a high degree of specificity and a central role in the diagnosis of small hemangiomas (Fig. 94-7).219,220 With small hemangiomas, the contrast material may assume a ring-shaped or C-shaped configuration, with an avascular center resulting from fibrous obliteration; this appearance is pathognomonic.
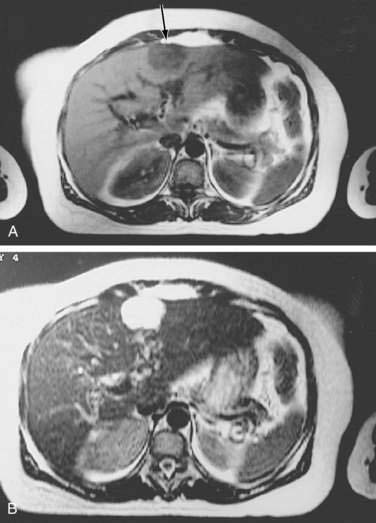
Thrombocytopenia resulting from sequestration and destruction of platelets in large hemangiomas (Kasabach-Merritt syndrome) is seen occasionally in infants but rarely in adults.214,221 Malignant transformation has not been reported.
Pathology
Cavernous hemangiomas usually are solitary lesions, although multiple tumors occur in 10% of patients.63,211 Reddish-purple or bluish masses are seen under Glisson’s capsule or deep in the substance of the liver. The larger lesions may be pedunculated. Cavernous hemangiomas are well circumscribed but seldom encapsulated. They may show central necrosis and, in some cases, the whole tumor is firm in consistency and grayish-white in appearance. Microscopically, hemangiomas are composed of multiple vascular channels of varying sizes lined by a single layer of flat epithelium and supported by fibrous septa. The vascular spaces may contain thrombi. The demonstration of mast cells within hemangiomas suggests that they may have a role in pathogenesis.222 Sclerosing cavernous hemangiomas may sometimes be seen and probably represent natural involution of these lesions.
Occasionally, cavernous hemangiomas are associated with hemangiomas in other organs. They also may coexist with cysts in the liver or pancreas,223 von Meyenburg complexes (see later),224 or focal nodular hyperplasia (see later).225
Treatment
The great majority of cavernous hemangiomas can safely be left untreated. A cavernous hemangioma that is large but localized, and the cause of incapacitating symptoms, should be resected.214 If resection is not feasible, reduction in the size of the tumor with relief of symptoms is rarely achieved with irradiation, arterial ligation, arteriographic embolization, or systemic glucocorticoids.226,227 RFA has been used with some success. If a cavernous hemangioma has ruptured, it may be necessary to embolize or clamp the hepatic artery to stop bleeding before proceeding with resection, although this complication is exceedingly rare.
INFANTILE HEMANGIOENDOTHELIOMA
Epidemiology and Clinical Features
Although rare, infantile hemangioendothelioma is the most common tumor of the liver in infants. Its importance stems from the high incidence of congestive heart failure in infants with this tumor and the resulting high mortality rate. The tumor almost invariably manifests in the first six months of life and is twice as common in girls as in boys.164,228 Hepatic hemangioendothelioma often coexists with hemangiomas in other organs, especially the skin (in approximately 50% of patients).
Small hemangioendotheliomas are usually asymptomatic. The presence of a large lesion is recognized clinically by the diagnostic triad of an enlarged liver, high-output cardiac failure, and multiple cutaneous hemangiomas.201,228 The liver is larger than expected on the basis of the severity of the cardiac failure, and hepatomegaly persists after the heart failure has been treated successfully. When hemangioendotheliomas occur diffusely throughout the liver, as they usually do, their combined effect is to act as a large peripheral arteriovenous shunt. Shunts of this size are responsible for the cardiac failure. Approximately one third of patients have jaundice. Patients may be anemic, partly because of the dilutional effect of the increased circulating plasma volume that develops with large peripheral arteriovenous fistulas. A microangiopathic hemolytic anemia may contribute. In addition, thrombocytopenia may be present (Kasabach-Merritt syndrome). Malignant change is a rare complication.
Diagnosis
Ultrasonography may show one or more echogenic masses in the liver. Hepatic angiography is particularly helpful in diagnosis and shows stretching, but not displacement, of the intrahepatic arteries.229 Abnormal vessels arise from the hepatic arteries and promptly opacify the liver, thereby giving rise to the characteristic blush of an arteriovenous shunt. The circulation time through the liver is short. Focal avascular areas may be evident when hemorrhage into or necrosis of the tumor has occurred. CT with enhancement and MRI are as specific as hepatic arteriography for the diagnosis of hemangioendotheliomas.230 Percutaneous biopsy is contraindicated because of the danger of bleeding.
Pathology
Two types of infantile hemangioendothelioma are recognized. Type I lesions are often calcified and have a fibrous stromal separation (with bile ductules) between channels. Type II lesions have a more malignant and disorganized-appearing endothelial cell lining and no stromal bile ductules.164,231 Infantile hemangioendotheliomas typically are multifocal and produce a nodular deformity of the entire liver. The nodules range in size from a few millimeters to many centimeters and are well demarcated but not encapsulated. At laparotomy, the nodules can be seen to pulsate. They are reddish purple, although large tumors are gray to tan. They may show hemorrhages, fibrosis, or calcification.
Treatment and Prognosis
The course of infantile hemangioendothelioma is characterized by tumor growth during the early months of life, followed by gradual involution.228 If the child survives, the tumor involutes completely. Life-threatening aspects of the disorder are intractable congestive heart failure and, to a lesser extent, consumptive coagulopathy or rupture of the tumor. Cardiac failure should be treated by conventional means initially, but if these measures fail, more aggressive treatment of the tumor, such as embolization, ligation of the hepatic artery, surgical resection, or liver transplantation, should be considered.232,233 Use of glucocorticoids has been successful in many (but not all) patients,234 whereas irradiation has seldom been beneficial. When the tumor is confined to one lobe, surgical resection is curative, even in the presence of cardiac failure.228
TUMOR-LIKE HEPATIC LESIONS
FOCAL NODULAR HYPERPLASIA
Focal nodular hyperplasia is a circumscribed, usually solitary lesion composed of nodules of benign hyperplastic hepatocytes surrounding a central stellate scar.238
Epidemiology and Pathogenesis
Focal nodular hyperplasia is more common than hepatocellular adenoma. The lesion is seen more often in women than in men, although the gender difference is much less striking than that for hepatocellular adenoma. Focal nodular hyperplasia occurs at all ages, but most patients present in the third and fourth decades of life201; the age distribution is similar to that of hepatocellular adenomas and the two lesions may coexist.
The cause of focal nodular hyperplasia is unknown. Abnormalities in arteries in small and medium-sized portal tracts have been described, suggesting a role of vascular malformation in its pathogenesis.239,205 It has been described to occur with other vascular lesions, such as cavernous hemangioma, epithelioid hemangioendothelioma, and hereditary hemorrhagic telangiectasia.240,241 A role for oral contraceptive steroids in the development of the lesion was suggested but has been disputed.201 Nevertheless, some evidence suggests that focal nodular hyperplasia may be hormone-dependent.242,243 Contraceptive steroids may accentuate the vascular abnormalities in focal nodular hyperplasia and cause the lesion to enlarge, become more symptomatic, and, rarely, rupture.
Clinical Features
Most of these lesions do not produce symptoms and are often discovered during upper abdominal imaging for other reasons or because an enlarged liver is felt on routine examination or found during abdominal surgery or at autopsy.201,244,245 Patients may experience mild pain, particularly with bleeding into or necrosis of the lesion.
Diagnosis
Serum AFP levels are normal. The mass lesion seen on ultrasonography and CT is not specific for focal nodular hyperplasia,246,247 unless the central scar and feeding artery are seen (Fig. 94-8). MRI may be useful for the diagnosis of focal nodular hyperplasia.248
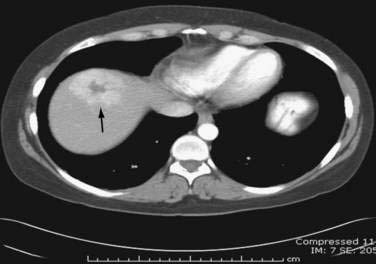
Pathology
Focal nodular hyperplasia manifests as a firm, coarsely nodular, light brown or yellowish-gray mass of variable size with a dense, central stellate scar and radiating fibrous septa that divide the lesion into lobules.249 The nodule may be small, resembling a cirrhotic nodule, or extremely large. The lesion of focal nodular hyperplasia usually occupies a subcapsular position and may be pedunculated. It generally is solitary. Larger lesions may show foci of hemorrhage or necrosis, although these features are seen less frequently than in hepatocellular adenomas. The fibrous septa sometimes are poorly developed, and the central scar may be absent. The lesion is sharply demarcated from the surrounding liver tissue, which is normal, but a true capsule is absent. Focal nodular hyperplasia is associated with hepatic hemangiomas in as many as 20% of cases.
OTHER NODULAR DISORDERS
Nodular regenerative hyperplasia is characterized by nodularity of the liver without fibrosis (see Chapter 35).250 This rare condition may be associated with a number of diseases, such as rheumatoid arthritis and Felty’s syndrome. Although generally diffuse, the nodularity occasionally is focal, in which case the lesion may be mistaken for a tumor. Patients with nodular regenerative hyperplasia typically present clinically with portal hypertension. Partial nodular transformation is characterized by nodules that are limited to the perihilar region of the liver. These patients also present with portal hypertension.
Macroregenerative nodules may occur in advanced cirrhosis or after massive hepatic necrosis. In the presence of cirrhosis, they are believed to be premalignant and may, in addition, be mistaken for hepatic tumors during hepatic imaging.60
Inflammatory pseudotumor is a rare entity, resulting from focal infection, that may be mistaken for a hepatic tumor (see Chapter 82).251 It occurs particularly in young men, who present with intermittent fever, abdominal pain, jaundice, vomiting, and diarrhea. Leukocytosis, an elevated erythrocyte sedimentation rate, and polyclonal hyperglobulinemia are present in approximately 50% of patients. The lesion may be solitary or multiple and shows a mixture of chronic inflammatory cells, with plasma cells predominating. Focal fatty infiltration, or focal fatty sparing in the presence of diffuse fatty infiltration, may also be mistaken for a hepatic tumor (see Chapter 85).252
HEPATIC CYSTS
Hepatic cysts are abnormal fluid-filled spaces in the hepatic parenchyma and biliary tree. They are categorized into three main types—fibrocystic diseases of the liver, cystadenomas and cystadenocarcinomas, and hydatid cysts. Cystadenomas and cystadenocarcinomas are discussed in Chapter 69. Hydatid cysts are discussed in Chapter 82.
FIBROCYSTIC DISEASES OF THE LIVER
Fibrocystic diseases of the liver originate from abnormal persistence or defects in the progressive remodeling of the ductal plate during development, resulting in dilated fluid-filled spaces, including hepatic and choledochal cysts, portal fibrosis, and ductal plate malformations (see Chapter 62).253,254 Fibrocystic disorders of the liver described here include simple hepatic cysts, polycystic liver disease (PCLD), von Meyenburg complexes, and Caroli’s disease (type V choledochal cyst). (The other diseases are congenital hepatic fibrosis and type IV choledochal cysts; see Chapter 62.)
Simple Hepatic Cysts
Simple hepatic cysts are thought to be congenital in origin and have a frequency of about 2.5% of the population.255 They generally are smaller than 5 cm in diameter and can number up to three before being considered part of PCLD.256 The cysts usually are asymptomatic and discovered incidentally during upper abdominal imaging. They occur more often in women than in men, and their prevalence increases with age. When symptomatic, they can produce complications similar to those of PCLD, including intracystic bleeding, infection, rupture, or compression of adjacent organs.
Typically, initial imaging with ultrasound, CT, or MRI provides an accurate diagnosis and distinguishes a simple cyst from a hydatid cyst and cystadenoma. Septations, papillary projects, or calcification should raise the suspicious for an alternative diagnosis.257 Asymptomatic solitary hepatic cysts should be left alone. If intervention is required because of symptoms, percutaneous aspiration and sclerosis with alcohol or doxycycline will almost always ablate the cyst, but recurrence is frequent.256 An alternative approach is laparoscopic (or, rarely, open surgical) fenestration, which is seldom followed by recurrence but has greater morbidity.
Polycystic Liver Disease
PCLD is a rare condition in which multiple cysts form in the hepatic parenchyma; usually it comes to clinical attention in adulthood (Fig. 94-9). PCLD usually presents in association with autosomal dominant polycystic kidney disease (ADPKD)258,259 but can appear as isolated polycystic liver disease.260,261
The cysts range in diameter from a few millimeters to 10 cm or more. They contain clear, colorless, or straw-colored fluid and are lined by a single layer of cuboidal or columnar epithelium, resembling that of bile ducts.258–262 Rarely, the cysts may be lined by squamous epithelium; these cysts may be complicated by the development of squamous cell carcinoma. In addition to the nature of the lining epithelium, evidence for a biliary origin of these cysts is suggested by the composition of the cystic fluid, which has a low glucose content and contains secretory immunoglobulin A and gamma glutamyl transpeptidase. The cysts are thought to arise as a result of ductal plate malformation. This process gives rise to von Meyenburg complexes (see later), which become disconnected from the biliary tree during development and growth and dilate progressively to form cysts.
PCLD is fairly common in patients with ADPKD. It occurs in approximately 24% of patients in the third decade of life to 80% in the sixth decade of life, but the kidney disease usually dominates the clinical course.263 Cysts also may be present in the pancreas, spleen and, less often, other organs. Symptomatic liver disease correlates with advancing age, severity of renal cysts, and renal dysfunction.264 Women tend to have larger and more numerous cysts, and a correlation with the number of pregnancies has been found. The use of exogenous female sex hormones may accelerate the rate of growth and size of the cysts. PCLD may coexist with other fibrocystic liver diseases, such as congenital hepatic fibrosis (in which the patient is likely to present with portal hypertension), Caroli’s disease, or von Meyenburg complexes.258–262 PCLD also is associated with other conditions such as berry aneurysms, mitral valve prolapse, diverticular disease, and inguinal hernias.
Isolated PCLD not associated with ADPKD is rare, representing 7% of all PCLD in autopsy series.265 It usually is asymptomatic.266 Like ADPKD-associated PCLD, isolated PCLD is associated with pregnancy and appears to be more symptomatic in women than men.
ADPKD is a common genetic disease with a frequency of 1:1,000 in whites.267 Two genes are responsible. The gene affected in ADPKD1 is PKD-1, which is located on chromosome 16q13-q23 and expresses a ubiquitous protein, polycystin-1.268,269 The gene responsible for ADPKD2 is PKD-2, which is located on chromosome 4 and expresses polycystin-2. The two polycystins are transmembrane glycoproteins that complex and localize in the primary cilium, a microtubule-based structure found on renal and biliary tubule epithelium and thought to act as a flow sensor and regulator of Ca2+ influx.270 Although the mutation is inherited as an autosomal dominant trait, a second somatic mutation is thought to be necessary to produce the monoclonally derived cysts.271
Isolated PCLD has been shown in North American and Finnish families to be linked to the gene PRKCSH (also known as protein kinase C substrate 80K-H) on chromosome 19 locus p13.2-13.1 and to SEC63 on chromosome 6q21, although other associated genes undoubtedly exist.268,272,273 The gene products hepatocystin and SEC63p are thought to be involved in the folding and quality control of glycoproteins and protein translocation in the endoplasmic reticulum, respectively.274 The genes appear to be autosomal dominant, and a second somatic mutation is thought to be needed to cause disease.
The hepatic cysts in polycystic liver disease, whether or not they occur in association with renal cysts, rarely cause morbidity, and many affected persons are asymptomatic.258–262 The livers of these patients contain only a few cysts or cysts smaller than 2 cm in diameter. Symptoms occur in patients with more numerous and larger cysts (10% to 15% of patients, usually women), generally with markedly enlarged livers. Abdominal discomfort or pain, postprandial fullness, awareness of an upper abdominal mass, a protuberant abdomen, and shortness of breath may be present. Severe pain may be experienced with rupture or infection of a cyst, bleeding into a cyst, or torsion of a pedunculated cyst. Jaundice is evident in approximately 5% of patients and is caused by compression of the major intrahepatic or extrahepatic bile ducts. Ascites, if present, is the result of portal hypertension, which generally is caused by associated congenital hepatic fibrosis but occasionally by compression of the hepatic veins by the cysts. Gastroesophageal variceal bleeding has rarely been reported.275
Liver biochemical test results generally are normal, with intact hepatic function, although serum alkaline phosphatase and GGTP levels may be increased. A raised right hemidiaphragm may be evident on a plain x-ray of the chest in patients with severe PCLD. The diagnosis of PCLD is confirmed by ultrasound, CT, or MRI (see Fig. 94-9).
On the rare occasions when cysts require treatment, fenestration (unroofing) should be performed.258,276 Cyst fenestration originally was done at laparotomy but is now performed laparoscopically. A high recurrence rate is observed for cysts treated in this way. Cysts also have been treated by percutaneous injection of a sclerosing substance such as alcohol or doxycycline, but most patients have too many small cysts to warrant this approach. Patients who fail to respond to cyst fenestration may be considered for partial hepatic resection. Liver transplantation (sometimes combined with renal transplantation) is generally reserved for patients with hepatic failure or severe symptoms that interfere with activities of daily living.277
Fibrocystic Disease Associated with Autosomal Recessive Polycystic Kidney Disease
Fibrocystic liver disease may manifest in childhood as an autosomal recessive disorder that is usually rapidly fatal as a consequence of associated autosomal recessive polycystic kidney disease (ARPKD).258,259 A proportion of patients maintain renal function into adulthood, however, and complications of the associated liver disease then predominate. The liver cysts are microscopic, rather than macroscopic, and present a clinical picture of congenital hepatic fibrosis. Complications of portal hypertension are the usual hepatic manifestations of the disease. The gene responsible for this disease, PKHD1, has been identified at chromosomal locus 6p21-cen, and the ARPKD protein, fibrocystin, is predicted to be an integral receptor-like protein.278 Many different mutations throughout the gene have been identified in patients with ARPKD.
Von Meyenburg Complexes
Von Meyenburg complexes (also known as biliary microhamartomas) are common and do not produce symptoms; they are small and usually multiple. Each complex is composed of cystically dilated intra- and interlobular bile ducts embedded in a fibrous stroma.175,279 The cysts are lined by cuboidal or flat epithelium. They occur in almost all patients with congenital hepatic fibrosis and may coexist with Caroli’s disease or ADPKD. Von Meyenburg complexes are found in or adjacent to portal tracts and are believed to arise as a result of malformation of the ductal plate (see Chapter 62), and they may be complicated by the development of peripheral cholangiocarcinoma.280
Caroli’s Disease
Caroli’s disease is a rare disorder characterized by congenital nonobstructive gross dilatation of the segmental intrahepatic bile ducts.256 The disease has been included in the classification of choledochal cysts (as type V)256,258 and may occur in association with medullary sponge kidney (in 60% to 80% of patients) or congenital hepatic fibrosis (see Chapter 62). Caroli’s disease is believed to be caused by an intrauterine event that arrests ductal plate remodeling at the level of the larger intrahepatic bile ducts.253 The resulting bile duct ectasia may be diffuse or localized. Both autosomal recessive and autosomal dominant modes of inheritance have been proposed. Caroli’s disease affects men and women equally and usually becomes symptomatic in early adulthood; more than 80% of patients present with symptoms before the age of 30 years.
Patients typically present with recurrent episodes of fever and abdominal pain caused by cholangitis. The liver often is enlarged. Ductal ectasia predisposes to bile stagnation, which in turn may lead to cholangitis, abscess formation, and septicemia.281 Gallstones form in the ectatic ducts in one third of patients. The result of these complications may be cholangiocarcinoma, which develops in less than 10% of patients.
Attacks of cholangitis require treatment with antibiotics. Endoscopic retrograde cannulation of the biliary system may be used to facilitate removal of sludge or stones from the accessible part of the biliary system, and the cysts may be drained by an endoscopic or percutaneous route. Liver resection for unilobar Caroli’s disease and liver transplantation for diffuse Caroli’s disease are associated with excellent long-term patient survival and a low rate of complications.282
APPROACH TO THE PATIENT WITH A HEPATIC MASS LESION
The approach to the diagnosis of a mass lesion in the liver will be influenced by the age and gender of the patient and the presence or absence of symptoms (Fig. 94-10). Making a definitive diagnosis of a mass lesion in the liver solely on clinical grounds is seldom possible. Nevertheless, detailed history taking will provide important clues about the probable benign or malignant nature of the lesion.
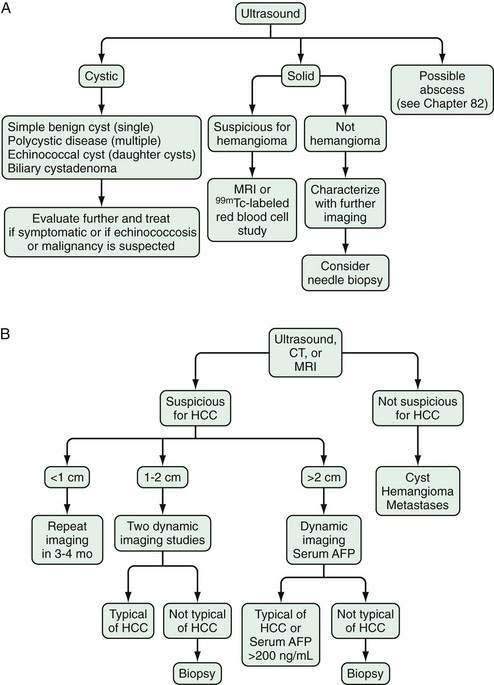
The approach to a mass in the liver differs, depending on whether or not cirrhosis is present. In a noncirrhotic liver, masses or tumors may be found in the liver incidentally or because of symptoms; the main concern is cancer metastatic from elsewhere. Initial imaging studies such as ultrasonography, CT, or MRI will indicate if the lesion is cystic. Cystic lesions should be investigated further and treated only if echinococcal cysts are suspected (see Chapter 82). Possible solid lesions in a noncirrhotic liver include a hemangioma, which can be confirmed by MRI or a 99mTc-labeled red blood cell scan. Alternatively, a biopsy should be considered to exclude malignancy, unless the lesion has the characteristic radiologic appearance of focal nodular hyperplasia. In a patient known to have cirrhosis, the presence of a nodular or mass lesion should be presumed to be hepatocellular carcinoma until proven otherwise. The AASLD practice guidelines provide criteria for the noninvasive diagnosis of hepatocellular carcinoma based on the vascularity of the tumor (see earlier). Contrast enhancement during the arterial phase of a multiphase CT or MRI study with subsequent washout during the venous phase is considered diagnostic of hepatocellular carcinoma if the lesion is larger than 2 cm in diameter. For lesions between 1 and 2 cm in diameter, this characteristic vascularity should be demonstrated on two imaging modalities; biopsy of the lesion may be required. For a lesion smaller than 1 cm, biopsy may be difficult to accomplish technically, and the lesion should be observed; if it becomes larger than 1 cm in diameter, the diagnosis of hepatocellular carcinoma is essentially confirmed. If ascertaining whether a patient has underlying cirrhosis is difficult on clinical and imaging grounds, a biopsy of the nontumorous liver may be done.
Bismuth H, Chiche L, Castaing D. Surgical treatment of hepatocellular carcinomas in noncirrhotic liver: Experience with 68 liver resections. World J Surg. 1995;19:35-41. (Ref 101.)
Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208-36. (Ref 25.)
Colli A, Fraquelli M, Casazza G, et al. Accuracy of ultrasonography, spiral CT, magnetic resonance, and alpha-fetoprotein in diagnosing hepatocellular carcinoma: A systematic review. Am J Gastroenterol. 2006;101:513-23. (Ref 24.)
Desmet VJ. Congenital diseases of intrahepatic bile ducts: Variations on the theme “ductal plate malformation.”. Hepatology. 1992;16:1069-83. (Ref 253.)
El-Serag H, Mason A. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745-50. (Ref 5.)
Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1993;22:520-5. (Ref 262.)
Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD. Cholangiocarcinoma. Lancet. 2005;366:1303-14. (Ref 162.)
Lazaridis KN, Gores GJ. Cholangiocarcinoma. Gastroenterology. 2005;128:1655-67. (Ref 160.)
Lencioni R, Cioni D, Crocetti L, et al. Early-stage hepatocellular carcinoma in patients with cirrhosis: Long-term results of percutaneous image-guided radiofrequency ablation. Radiology. 2005;234:961-7. (Ref 117.)
Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521-31. (Ref 77.)
Llovet J, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology. 2003;37:429-42. (Ref 126.)
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90. (Ref 136.)
Marrero J, Fontana R, Fu S, Conjeevaram H, Su G, Lok A. Alcohol, tobacco and obesity are synergistic risk factors for hepatocellular carcinoma. J Hepatol. 2005;42:218-24. (Ref 93.)
Nguyen MH, Garcia RT, Simpson PW, et al. Racial differences in effectiveness of alpha-fetoprotein for diagnosis of hepatocellular carcinoma in hepatitis C virus cirrhosis. Hepatology. 2002;36:410-17. (Ref 33.)
Ribero D, Curley SA, Imamura H, et al. Selection for resection of hepatocellular carcinoma and surgical strategy: Indications for resection, evaluation of liver function, portal vein embolization, and resection. Ann Surg Oncol. 2008;15:986-92. (Ref 105.)
Zucman-Rossi J. Genetic alterations in hepatocellular adenomas: Recent findings and new challenges. J Hepatol. 2004;40:1036-9. (Ref 204.)
1. Parkin D, Muir C, Whelan S. Cancer incidence in five continents. IARC publication no. 120. Lyon, France: IARC; 1997.
2. Bosch FX, Ribes J, Borras J. Epidemiology of primary liver cancer. Semin Liver Dis. 1999;19:271-85.
3. Terry F. Primary Cancer of the Liver. New York: Alan Liss; 1978.
4. Harington JS, McGlashan ND, Bradshaw E, et al. A spatial and temporal analysis of four cancers in African gold miners from Southern Africa. Br J Cancer. 1976;31:665-78.
5. El-Serag H, Mason A. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745-50.
6. Tanaka H, Imai Y, Hiramatsu N, et al. Declining incidence of hepatocellular carcinoma in Osaka, Japan, from 1990 to 2003. Ann Intern Med. 2008;148:820-6.
7. Bosetti C, Levi F, Boffetta P, et al. Trends in mortality from hepatocellular carcinoma in Europe, 1980-2004. Hepatology. 2008;48:137-45.
8. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71-96.
9. Wong R, Corley D. Racial and ethnic variations in hepatocellular carcinoma incidence in the United States. Am J Med. 2008;121:525-31.
10. Kew MC, Kassianides C, Hodkinson J, et al. Hepatocellular carcinoma in urban born blacks: Frequency and relation to hepatitis B virus infection. Br Med J Clin Res Ed. 1986;293:1339-41.
11. Kew MC, Kassianides C, Berger EL, et al. Prevalence of chronic hepatitis B virus infection in pregnant black women living in Soweto. J Med Virol. 1987;22:263-8.
12. Pham TH, Iqbal CW, Grams JM, et al. Outcomes of primary liver cancer in children: An appraisal of experience. J Pediatr Surg. 2007;42:834-9.
13. Austin MT, Leys CM, Feurer ID, et al. Liver transplantation for childhood hepatic malignancy: A review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg. 2006;41:182-6.
14. Mazzanti R, Gramantieri L, Bolondi L. Hepatocellular carcinoma: Epidemiology and clinical aspects. Mol Aspects Med. 2008;29:130-43.
15. Kew MC, Geddes EW. Hepatocellular carcinoma in rural southern African blacks. Medicine. 1982;61:98-108.
16. Kew MC, Paterson AC. Unusual clinical presentations of hepatocellular carcinoma. Trop Gastroenterol. 1985;6:10-22.
17. Zapf J, Futo E, Peter M, et al. Can “big” insulin-like growth factor II in serum of tumor patients account for the development of extrapancreatic tumor hypoglycemia? J Clin Invest. 1992;90:2574-84.
18. Kew MC, Fisher JW. Serum erythropoietin concentrations in patients with hepatocellular carcinoma. Cancer. 1986;58:2485-8.
19. Yen TC, Hwang SJ, Wang CC, et al. Hypercalcemia and parathyroid hormone–related protein in hepatocellular carcinoma. Liver. 1993;13:311-15.
20. DiBisceglie AM, Hodkinson HJ, Berkowitz I, et al. Pityriasis rotunda. A cutaneous marker of hepatocellular carcinoma in South African blacks. Arch Derm. 1986;122:802-4.
21. Party IW. Terminology of nodular hepatocellular lesions. Hepatology. 1995;22:983-93.
22. Wanless IR. International consensus on histologic diagnosis of early hepatocellular neoplasia. Hepatol Res. 2007;37:S139-41.
23. Efremidis SC, Hytiroglou P. The multistep process of hepatocarcinogenesis in cirrhosis with imaging correlation. Eur Radiol. 2002;12:753-64.
24. Colli A, Fraquelli M, Casazza G, et al. Accuracy of ultrasonography, spiral CT, magnetic resonance, and alpha-fetoprotein in diagnosing hepatocellular carcinoma: A systematic review. Am J Gastroenterol. 2006;101:513-23.
25. Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208-36.
26. Bruix J, Sherman M, Llovet J. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona–2000 EASL conference. J Hepatol. 2001;35:421-30.
27. Willatt JM, Hussain HK, Adusumilli S, Marrero JA. MR Imaging of hepatocellular carcinoma in the cirrhotic liver: Challenges and controversies. Radiology. 2008;247:311-30.
28. Kew MC. Tumour markers of hepatocellular carcinoma. J Gastroenterol Hepatol. 1989;4:373-84.
29. Trevisani F, D’Intino P, Morselli-Labate A, et al. Serum alpha-fetoprotein for diagnosis of hepatocellular carcinoma in patients with chronic liver disease: Influence of HBsAg and anti-HCV status. J Hepatol. 2001;34:570-5.
30. Cedrone A, Covino M, Caturelli E, et al. Utility of alpha-fetoprotein (AFP) in the screening of patients with virus-related chronic liver disease: Does different viral etiology influence AFP levels in hepatocellular carcinoma? A study in 350 Western patients. Hepatogastroenterology. 2000;47:1654-8.
31. Soresi M, Magliarisi C, Campagna P, et al. Usefulness of alpha-fetoprotein in the diagnosis of hepatocellular carcinoma. Anticancer Res. 2003;23:1747-53.
32. Peng YC, Chan CS, Chen GH. The effectiveness of serum alpha-fetoprotein level in anti-HCV–positive patients for screening hepatocellular carcinoma. Hepatogastroenterology. 1999;46:3208-11.
33. Nguyen MH, Garcia RT, Simpson PW, et al. Racial differences in effectiveness of alpha-fetoprotein for diagnosis of hepatocellular carcinoma in hepatitis C virus cirrhosis. Hepatology. 2002;36:410-17.
34. Hyosuk Lee YHC, Chung Yong Kim. Specificities of serum alpha-fetoprotein in HBsAg+ and HBsAg− patients in the diagnosis of hepatocellular carcinoma. Hepatology. 1991;14:68-72.
35. Marrero JA. Screening tests for hepatocellular carcinoma. Clin Liver Dis. 2005;9:235-51.
36. Torzilli G, Minagawa M, Takayama T, et al. Accurate preoperative evaluation of liver mass lesions without fine-needle biopsy. Hepatology. 1999;30:889-93.
37. Levy I, Greig PD, Gallinger S, et al. Resection of hepatocellular carcinoma without preoperative tumor biopsy. Ann Surg. 2001;234:206-9.
38. Ioannou GN, Perkins JD, Carithers RLJr. Liver transplantation for hepatocellular carcinoma: Impact of the MELD allocation system and predictors of survival. Gastroenterology. 2008;134:1342-51.
39. Sterling RK, Jeffers L, Gordon F, et al. Clinical utility of AFP-L3% measurement in North American patients with HCV-related cirrhosis. Am J Gastroenterol. 2007;102:2196-205.
40. Leerapun A, Suravarapu S, Bida J, et al. The utility of lens culinaris agglutinin-reactive α-fetoprotein in the diagnosis of hepatocellular carcinoma: Evaluation in a United States referral population. Clin Gastroenterol Hepatol. 2007;5:394-402.
41. Marrero J, Su G, Wei W, et al. Des-gamma carboxyprothrombin can differentiate hepatocellular carcinoma from nonmalignant chronic liver disease in American patients. Hepatology. 2003;37:1114-21.
42. Ono M, Ohta H, Ohhira M, et al. Measurement of immunoreactive prothrombin precursor and vitamin-K-dependent gamma-carboxylation in human hepatocellular carcinoma tissues: Decreased carboxylation of prothrombin precursor as a cause of des-gamma-carboxyprothrombin synthesis. Tumour Biol. 1990;11:319-26.
43. Tsai SL, Huang GT, Yang PM, et al. Plasma des-gamma-carboxyprothrombin in the early stage of hepatocellular carcinoma. Hepatology. 1990;11:481-7.
44. Grazi GL, Mazziotti A, Legnani C, et al. The role of tumor markers in the diagnosis of hepatocellular carcinoma, with special reference to the des-gamma-carboxy prothrombin. Liver Transplant Surg. 1995;1:249-55.
45. Mita Y, Aoyagi Y, Yanagi M, et al. The usefulness of determining des-gamma-carboxy prothrombin by sensitive enzyme immunoassay in the early diagnosis of patients with hepatocellular carcinoma. Cancer. 1998;82:1643-8.
46. King MA, Kew MC, Kuyl JM, et al. A comparison between des-gamma-carboxy prothrombin and alpha-fetoprotein as markers of hepatocellular carcinoma in southern African blacks. J Gastroenterol Hepatol. 1989;4:17-24.
47. Capurro M, Wanless IR, Sherman M, et al. Glypican-3: A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125:89-97.
48. Marrero JA, Romano PR, Nikolaeva O, et al. GP73, a resident Golgi glycoprotein, is a novel serum marker for hepatocellular carcinoma. J Hepatol. 2005;43:1007-12.
49. Yamagamim H, Moriyama M, Matsumura H, et al. Serum concentrations of human hepatocyte growth factor is a useful indicator for predicting the occurrence of hepatocellular carcinomas in C-viral chronic liver diseases. Cancer. 2002;95:824-34.
50. Mazziotti G, Sorvillo F, Morisco F, et al. Serum insulin-like growth factor I evaluation as a useful tool for predicting the risk of developing hepatocellular carcinoma in patients with hepatitis C virus–related cirrhosis—a prospective study. Cancer. 2002;95:2539-45.
51. Song BC, Chung HY, Kim JA, et al. Transforming growth factor-beta 1 marker of small hepatocellular as a useful serologic carcinoma. Cancer. 2002;94:175-80.
52. Matsuia O. Imaging of multistep human hepatocarcinogenesis by CT during intra-arterial contrast injection. Intervirology. 2004;47:271-6.
53. Park YN, Yang CP, Fernandez GJ, et al. Neoangiogenesis and sinusoidal “capillarization” in dysplastic nodules of the liver. Am J Surgical Pathol. 1998;22:656-62.
54. Yu SC, Yeung DT, So NM. Imaging features of hepatocellular carcinoma. Clin Radiol. 2004;59:145-56.
55. Kudo M. Imaging diagnosis of hepatocellular carcinoma and premalignant/borderline lesions. Semin Liver Dis. 1999;19:297-309.
56. Luca A, Milazzo M, Caruso S, et al. Hepatic nodules detected in cirrhotic patients using high-performance multidetector computed tomography (MDCT) and magnetic resonance imaging (MRI): A radiological-pathological correlation on explanted livers. J Hepatol. 2007;46(Suppl 1):S37.
57. Forner A, Vilana R, Ayuso C, et al. Diagnosis of hepatic nodules 20 mm or smaller in cirrhosis: Prospective validation of the noninvasive diagnostic criteria for hepatocellular carcinoma. Hepatology. 2008;47:97-104.
58. Taouli B, Goh JS, Lu Y, et al. Growth rate of hepatocellular carcinoma: Evaluation with serial computed tomography or magnetic resonance imaging. J Comput Assist Tomogr. 2005;29:425-9.
59. Takayasu K. Hepatic angiography. In: Okuda K, Tabor E, editors. Liver Cancer. New York: Churchill Livingstone; 1997:347-59.
60. Kojiro M. Histopathology of liver cancers. Best Pract Res Clin Gastroenterol. 2005;19:39-62.
61. Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818-22.
62. Yuki K, Hirohashi S, Sakamoto M, et al. Growth and spread of hepatocellular carcinoma. A review of 240 consecutive autopsy cases. Cancer. 1990;66:2174-9.
63. Craig J, Peters R, Edmundson H, Omata M. Fibrolamellar carcinoma of the liver. A tumor of adolescents and young adults with distinctive clinicopathologic features. Cancer. 1980;46:372-9.
64. Torbenson M. Review of the clinicopathologic features of fibrolamellar carcinoma. Adv Anat Pathol. 2007;14:217-23.
65. El-Serag HB, Davila JA. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A U.S. population-based study. Hepatology. 2004;39:798-803.
66. Marrero J, Fontana R, Barrat A, et al. Prognosis of hepatocellular carcinoma: Comparison of 7 staging systems in an American cohort. Hepatology. 2005;41:707-16.
67. Llovet J, Di Bisceglie A, Bruix J, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:698-711.
68. Raza SA, Clifford GM, Franceschi S. Worldwide variation in the relative importance of hepatitis B and hepatitis C viruses in hepatocellular carcinoma: A systematic review. Br J Cancer. 2007;96:1127-34.
69. Di Bisceglie A, Lyra A, Schwartz M, et al. Hepatitis C-related hepatocellular carcinoma in the United States: Influence of ethnic status. Am J Gastroenterol. 2003;98:2060-3.
70. Stevens CE, Szmuness W. Vertical transmission of hepatitis B and neonatal hepatitis B. In: Bianchi L, Gerok W, Sickinger K, editors. Virus and the Liver. Lancaster, England: MTP Press; 1980:285-91.
71. Botha JF, Ritchie MJ, Dusheiko GM, et al. Hepatitis B virus carrier state in black children in Ovamboland: Role of perinatal and horizontal infection. Lancet. 1984;1:1210-12.
72. Beasley RP, Hwang LY. Hepatocellular carcinoma and hepatitis B virus. Semin Liver Dis. 1984;4:113-21.
73. Chang MH, Chang M-H. Decreasing incidence of hepatocellular carcinoma among children following universal hepatitis B immunization. Liver Int. 2003;23:309-14.
74. Kew MC. Clinical, pathologic, and etiologic heterogeneity in hepatocellular carcinoma: Evidence from southern Africa. Hepatology. 1981;1:366-9.
75. Arbuthnot P, Kew M. Hepatitis B virus and hepatocellular carcinoma. Int J Exp Pathol. 2001;82:77-100.
76. Chen C-J, Yang H-I, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295:65-73.
77. Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521-31.
78. Chisari FV, Filippi P, Buras J, et al. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc Natl Acad Sci. 1987;84:6909-13.
79. Blum HE, Moradpour D. Viral pathogenesis of hepatocellular carcinoma. J Gastroenterol Hepatol. 2002;17(Suppl 3):S413-20.
80. Ioannou G, Splan M, Weiss N, et al. Incidence and predictors of hepatocellular carcinoma in patients with cirrhosis. Clin Gastroenterol Hepatol. 2007;5:938-45.
81. Chiaramonte M, Stroffolini T, Vian A, et al. Rate of incidence of hepatocellular carcinoma in patients with compensated viral cirrhosis. Cancer. 1999;85:2132-7.
82. Wogan GN. Aflatoxin exposure as a risk factor in the etiology of hepatocellular carcinoma. In: Okuda K, Tabor E, editors. Liver Cancer. New York: Churchill Livingstone; 1997:51-8.
83. Hsu IC, Metcalf RA, Sun T, et al. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350:427-8.
84. Soini Y, Chiq S, Bennett W, et al. An aflatoxin-associated, mutational hotspot at codon 249 in the p53 tumor suppressor gene occurs in hepatocellular carcinomas from Mexico. Carcinogenesis. 1996;17:1007-12.
85. Deugnier YM, Guyader D, Crantock L, et al. Primary liver cancer in genetic hemochromatosis: A clinical, pathological, and pathogenetic study of 54 cases. Gastroenterology. 1993;104:228-34.
86. Kew M. Pathogenesis of hepatocellular carcinoma in hereditary hemochromatosis: Occurrence in non-cirrhotic patients. Hepatology. 1990;11:1806-7.
87. Loeb LA, James EA, Waltersdorph AM, et al. Mutagenesis by the autoxidation of iron with isolated DNA. Proc Natl Acad Sci U S A. 1988;85:3918-22.
88. Mandishona E, MacPhail AP, Gordeuk VR, et al. Dietary iron overload as a risk factor for hepatocellular carcinoma in black Africans. Hepatology. 1998;27:1563-6.
89. Asare GA, Paterson AC, Kew MC, et al. Iron-free neoplastic nodules and hepatocellular carcinoma without cirrhosis in Wistar rats fed a diet high in iron. J Pathol. 2006;208:82-90.
90. Polio J, Enriquez RE, Chow A, et al. Hepatocellular carcinoma in Wilson’s disease. Case report and review of the literature. J Clin Gastroenterol. 1989;11:220-4.
91. Tokol R, Twedt T, McKim J, et al. Oxidant injury to mitochondria in patients with liver disease and Bedlington terriers with copper toxicosis. Gastroenterology. 1994;107:1788.
92. Kew MC, McKnight A, Hodkinson J, et al. The role of membranous obstruction of the inferior vena cava in the etiology of hepatocellular carcinoma in Southern African blacks. Hepatology. 1989;9:121-5.
93. Marrero J, Fontana R, Fu S, et al. Alcohol, tobacco and obesity are synergistic risk factors for hepatocellular carcinoma. J Hepatol. 2005;42:218-24.
94. Hassan M, Hwang L-Y, Hatte C, et al. Risk factors for hepatocellular carcinoma: Synergism with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206-13.
95. El-Serag HB, Richardson PA, Everhart JE. The role of diabetes in hepatocellular carcinoma: A case-control study among United States Veterans. Am J Gastroenterol. 2001;96:2462-7.
96. Thomas D. Exogenous steroid hormones and hepatocellular carcinoma. In: Tabor E, Di Bisceglie AM, Purcell RH, editors. Etiology, Pathology, and Treatment of Hepatocellular Carcinoma in North America. Houston: Gulf; 1991:77-89.
97. Austin A. The role of tobacco use and alcohol consumption in the etiology of hepatocellular carcinoma. In: Tabor E, Di Bisceglie AM, Purcell RH, editors. Etiology, Pathology, and Treatment of Hepatocellular Carcinoma in North America. Houston: Gulf; 1991:57-75.
98. Patel P, Hanson D, Sullivan P, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992-2003. Ann Intern Med. 2008;148:728-36.
99. Roberts L. Emerging experimental therapies for hepatocellular carcinoma: What if you can’t cure? In: McCullough A, editor. AASLD Postgraduate Course, 2007. Boston: AASLD; 2007:185.
100. Llovet J, Bruix J, Fuster J, et al. Liver transplantation for small hepatocellular carcinoma: The tumor-node-metastasis classification does not have prognostic power. Hepatology. 1998;27:1572-7.
101. Bismuth H, Chiche L, Castaing D. Surgical treatment of hepatocellular carcinomas in noncirrhotic liver: Experience with 68 liver resections. World J Surg. 1995;19:35-41.
102. Llovet J, Fuster J, Bruix J. Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: Resection versus transplantation. Hepatology. 1999;30:1434-40.
103. Bolondi L, Sofia S, Siringo S, et al. Surveillance programme of cirrhotic patients for early diagnosis and treatment of hepatocellular carcinoma: A cost-effectiveness analysis. Gut. 2001;48:251-9.
104. Cucchetti A, Ercolani G, Vivarelli M, et al. Impact of model for end-stage liver disease (MELD) score on prognosis after hepatectomy for hepatocellular carcinoma on cirrhosis. Liver Transpl. 2006;12:966-71.
105. Ribero D, Curley SA, Imamura H, et al. Selection for resection of hepatocellular carcinoma and surgical strategy: Indications for resection, evaluation of liver function, portal vein embolization, and resection. Ann Surg Oncol. 2008;15:986-92.
106. Teh SH, Christein J, Donohue J, et al. Hepatic resection of hepatocellular carcinoma in patients with cirrhosis: Model of end-stage liver disease (MELD) score predicts perioperative mortality. J Gastrointest Surg. 2005;9:1207-15.
107. Dimick JB, Cowan JA, Knol JA, Upchurch GR. Hepatic resection in the United States—indications, outcomes, and hospital procedural volumes from a nationally representative database. Arch Surg. 2003;138:185-91.
108. Ng KK, Vauthey JN, Pawlik TM, et al. Is hepatic resection for large or multinodular hepatocellular carcinoma justified? Results from a multi-institutional database. Ann Surg Oncol. 2005;12:364-73.
109. Bismuth H, Majno PE, Adam R, et al. Liver transplantation for hepatocellular carcinoma. Semin Liver Dis. 1999;19:311-22.
110. Jonas S, Bechstein W, Steinmuller T, et al. Vascular invasion and histopathologic grading determine outcome after liver transplantation for hepatocellular carcinoma in cirrhosis. Hepatology. 2001;33:1080-6.
111. Mazzaferro V, Regalia E, Doci R, et al. Liver transplantation of small hepatocellular carcinoma in patients with cirrhosis. N Engl J Med. 1996;334:693-9.
112. Freeman RB, Edwards EB, Harper AM. Waiting list removal rates among patients with chronic and malignant liver diseases. Am J Transplant. 2006;6:1416-21.
113. Yao FY, Xiao L, Bass NM, et al. Liver transplantation for hepatocellular carcinoma: Validation of the UCSF-Expanded criteria based on preoperative imaging. Am J Transplant. 2007;7:2587-96.
114. Herrero J, Sangro B, Quiroga J, et al. Influence of tumor characteristics on the outcome of liver transplantation among patients with liver cirrhosis and hepatocellular carcinoma. Liver Transpl. 2001;7:631-6.
115. Okada S. Local ablation therapy for hepatocellular carcinoma. Semin Liver Dis. 1999;19:323-8.
116. Livraghi T. Ethanol injection for the treatment of hepatocellular carcinoma. In: Okuda K, Tabor E, editors. Liver Cancer. New York: Churchill Livingstone; 1997:497-510.
117. Lencioni R, Cioni D, Crocetti L, et al. Early-stage hepatocellular carcinoma in patients with cirrhosis: Long-term results of percutaneous image-guided radiofrequency ablation. Radiology. 2005;234:961-7.
118. Shiina S, Teratani T, Obi S, et al. A randomized controlled trial of radiofrequency ablation with ethanol injection for small hepatocellular carcinoma. Gastroenterology. 2005;129:122-30.
119. Curley SA, Marra P, Beaty K, et al. Early and late complications after radiofrequency ablation of malignant liver tumors in 608 patients. Ann Surg. 2004;239:450-8.
120. Cheng BQ, Jia CQ, Liu CT, et al. Chemoembolization combined with radiofrequency ablation for patients with hepatocellular carcinoma larger than 3 cm—a randomized controlled trial. JAMA. 2008;299:1669-77.
121. Befeler A, Hayashi P, Di Bisceglie A. Liver transplantation for hepatocellular carcinoma. Gastroenterology. 2005;128:1752-64.
122. Mazzaferro V, Battiston C, Perrone S, et al. Radiofrequency ablation of small hepatocellular carcinoma in cirrhotic patients awaiting liver transplantation: A prospective study. Ann Surg. 2004;240:900-9.
123. Yao F, Bass N, Nikolai B, et al. A follow-up analysis of the pattern and predictors of dropout from the waiting list for liver transplantation in patients with hepatocellular carcinoma: Implications for the current organ allocation policy. Liver Transpl. 2003;9:684-92.
124. Bruix J, Llovet JM, Castells A, et al. Transarterial embolization versus symptomatic treatment in patients with advanced hepatocellular carcinoma: Results of a randomized, controlled trial in a single institution. Hepatology. 1998;27:1578-83.
125. Lin D-Y, Liaw Y-F, Lee T-Y, Lai C-M. Hepatic arterial embolization in patients with unresectable hepatocellular carcinoma: A randomized controlled trial. Gastroenterology. 1988;94:453-6.
126. Llovet J, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology. 2003;37:429-42.
127. Llovet J, Real M, Montanya X, et al. Arterial embolization, chemoembolization versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: A randomized controlled trial. Lancet. 2002;359:1734-9.
128. Lo CM, Ngan H, Tso WK, et al. Randomized controlled trial of transarterial lipiodol chemoembolization for unresectable hepatocellular carcinoma. Hepatology. 2002;35:1164-71.
129. Oberti F, Ruget O, Cales P, et al. Comparison of lipiodol chemoembolization and conservative treatment for unresectable hepatocellular carcinoma. N Engl J Med. 1995;332:1256-61.
130. Pelletier G, Ducreux M, Gay F, et al. Treatment of unresectable hepatocellular carcinoma with lipiodol chemoembolization: A multicenter randomized trial. J Hepatol. 1998;29:129-34.
131. Graziadei I, Sandmueller K, Waldenberger P, et al. Chemoembolization followed by liver transplantation for hepatocellular carcinoma impedes tumor progression while on the waiting list and leads to excellent outcome. Liver Transpl. 2003;9:557-63.
132. Majno P, Adam R, Bismuth H, et al. Influence of preoperative transarterial lipiodol chemoembolization on resection and transplantation for hepatocellular carcinoma in patients with cirrhosis. Ann Surg. 1997;226:688-701.
133. Wu CC, Ho YZ, Ho WL, et al. Preoperative transcatheter arterial chemoembolization for resectable large hepatocellular carcinoma: A reappraisal. Br J Surg. 1995;82:122-6.
134. Yamashiki N, Tateishi R, Yoshida H, et al. Ablation therapy in containing extension of hepatocellular carcinoma: A simulative analysis of dropout from the waiting list for liver transplantation. Liver Transpl. 2005;11:508-14.
135. Falkson G, Falkson CI. Current approaches in the management of patients with hepatocellular carcinoma. Oncol Res. 1992;4:87-9.
136. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90.
137. Stravitz R, Heuman D, Chand N, et al. Surveillance for hepatocellular carcinoma in patients with cirrhosis improves outcome. Am J Med. 2008;121:119-26.
138. Chang MH, Chen CJ, Lai MS, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336:1855-9.
139. Kasahara A, Hayashi N, Mochizuki K, et al. Risk factors for hepatocellular carcinoma and its incidence after interferon treatment in patients with chronic hepatitis C. Osaka Liver Disease Study Group. Hepatology. 1998;27:1394-402.
140. Khan SA, Davidson BR, Goldin R, et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: Consensus document. Gut. 2002;51(Suppl 6):S1-9.
141. Welzel TM, McGlynn KA, Hsing AW, et al. Impact of classification of hilar cholangiocarcinomas (Klatskin tumors) on the incidence of intra- and extrahepatic cholangiocarcinoma in the United States. J Natl Cancer Inst. 2006;98:873-5.
142. Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Semin Liver Dis. 2004;24:115-25.
143. Srivatanakul P, Parkin DM, Jiang YZ, et al. The role of infection by Opisthorchis viverrini, hepatitis B virus, and aflatoxin exposure in the etiology of liver cancer in Thailand. A correlation study. Cancer. 1991;68:2411-17.
144. Abdel-Rahim AY. Parasitic infections and hepatic neoplasia. Dig Dis. 2001;19:288-91.
145. Srinivasan P, McCall J, Pritchard J, et al. Orthotopic liver transplantation for unresectable hepatoblastoma. Transplantation. 2002;74:652-5.
146. Watanapa P, Watanapa WB. Liver fluke–associated cholangiocarcinoma. Br J Surg. 2002;89:962-70.
147. Wogan GN. The induction of liver cancer by chemicals. In: Cameron HM, Linsell GP, Warwick GP, editors. Liver Cell Cancer. Amsterdam: Elsevier; 1976:121-52.
148. Aadland E, Schrumpf E, Fausa O, et al. Primary sclerosing cholangitis: A long-term follow-up study. Scand J Gastroenterol. 1987;22:655-64.
149. Broome U, Olsson R, Loof L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996;38:610-15.
150. Kornfeld D, Ekbom A, Ihre T. Survival and risk of cholangiocarcinoma in patients with primary sclerosing cholangitis—a population-based study. Scand J Gastroenterol. 1997;32:1042-5.
151. Kulkarni PB, Beatty EJr. Cholangiocarcinoma associated with biliary cirrhosis due to congenital biliary atresia. Am J Dis Child. 1977;131:442-4.
152. Dekker A, Ten Kate FJ, Terpstra OT, et al. Cholangiocarcinoma associated with multiple bile duct hamartomas of the liver. Dig Dis Sci. 1989;34:952-8.
153. Yalcin S. Diagnosis and management of cholangiocarcinomas: A comprehensive review. Hepatogastroenterology. 2004;51:43-50.
154. Shaib YH, El-Serag HB, Davila JA, et al. Risk factors of intrahepatic cholangiocarcinoma in the United States: A case-control study. Gastroenterology. 2005;128:620-6.
155. Berthiaume EMD, Wands JMD. The molecular pathogenesis of cholangiocarcinoma. Semin Liver Dis. 2004:127-37.
156. Melum E, Karlsen TH, Schrumpf E, et al. Cholangiocarcinoma in primary sclerosing cholangitis is associated with NKG2D polymorphisms. Hepatology. 2008;47:90-6.
157. Nakanuma Y, Hoso M, Terada T. Clinical and pathologic features of cholangiocarcinoma. In: Okuda K, Tabor E, editors. Liver Cancer. New York: Churchill Livingstone; 1997:279-90.
158. Klatskin G. Adenomacarcinoma of the hepatic duct at its bifurcation within the porta hepatis: An unusual tumor with distinctive clinical and pathological features. Am J Med. 1965;38:241-56.
159. Nehls O, Gregor M, Klump B. Serum and bile markers for cholangiocarcinoma. Semin Liver Dis. 2004;24:139-54.
160. Lazaridis KN, Gores GJ. Cholangiocarcinoma. Gastroenterology. 2005;128:1655-67.
161. Sugihara S, Kojiro M. Pathology of cholangiocarcinoma. In: Okuda K, Ishak KG, editors. Neoplasms of the Liver. Tokyo: Springer-Verlag; 1987:143-58.
162. Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD. Cholangiocarcinoma. Lancet. 2005;366:1303-14.
163. Rea DJ, Heimbach JK, Rosen CB, et al. Liver transplantation with neoadjuvant chemoradiation is more effective than resection for hilar cholangiocarcinoma. Ann Surg. 2005;242:451-61.
164. Isaacs HJr. Fetal and neonatal hepatic tumors. J Pediatr Surg. 2007;42:1797-803.
165. Di Bisceglie AM. Tumors of the Liver, 4th ed. Philadelphia: Springer; 2006.
166. Herzog CE, Andrassy RJ, Eftekhari F. Childhood cancers: Hepatoblastoma. Oncologist. 2000;5:445-53.
167. Exelby PR, Filler RM, Grosfeld JL. Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular carcinoma: American Academy of Pediatrics Surgical Section Survey—1974. J Pediatr Surg. 1975;10:329-37.
168. McArthur JW, Toll GD, Russfield AB, et al. Sexual precocity attributable to ectopic gonadotropin secretion by hepatoblastoma. Am J Med. 1973;54:390-403.
169. Stocker JT, Ishak KG. Hepatoblastoma. In: Okuda K, Ishak KG, editors. Neoplasms of the Liver. Tokyo: Springer-Verlag; 1987:127-36.
170. De Ioris M, Brugieres L, Zimmermann A, et al. Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: The SIOPEL group experience. Eur J Cancer. 2008;44:545-50.
171. Roebuck DJ, Olsen O, Pariente D. Radiological staging in children with hepatoblastoma. Pediatr Radiol. 2006;36:176-82.
172. Rowland JM, Rowland JM. Hepatoblastoma: Assessment of criteria for histologic classification. Med Pediatr Oncol. 2002;39:478-83.
173. Curia M, Zuckermann M, De Lellis L, et al. Sporadic childhood hepatoblastomas show activation of beta-catenin, mismatch repair defects and p53 mutations. Mod Pathol. 2008;21:7-14.
174. Stringer MD, Stringer MD. The role of liver transplantation in the management of paediatric liver tumours. Ann R Coll Surg Engl. 2007;89:12-21.
175. Anthony PP. Tumors and tumor-like lesions of the liver and biliary tract. In: MacSween RN, Anthony PP, Scheuer PJ, et al, editors. Pathology of the Liver. Edinburgh: Churchill Livingstone; 1994:635-711.
176. Levy AD. Malignant liver tumors. Clin Liver Dis. 2002;6:147-64.
177. Tamburro CH. Relationship of vinyl monomers and liver cancers: Angiosarcoma and hepatocellular carcinoma. Semin Liver Dis. 1984;4:158-69.
178. Locker GY, Doroshow JH, Zwelling LA, et al. The clinical features of hepatic angiosarcoma: A report of four cases and a review of the English literature. Medicine. 1979;58:48-64.
179. Visfeldt J, Poulsen H. On the histopathology of liver and liver tumours in thorium dioxide patients. Acta Pathol Microbiol Scand A. 1972;80:97-108.
180. Roth F. Arsen-leber-tumoren hemangioendotheliom. Arch Pathol. 1955;60:493-501.
181. Regelson W, Kim U, Ospina J, et al. Hemangioendothelial sarcoma of liver from chronic arsenic intoxication by Fowler’s solution. Cancer. 1968;21:514-22.
182. Creech JLJr, Johnson MN. Angiosarcoma of liver in the manufacture of polyvinyl chloride. J Occup Med. 1974;16:150-1.
183. Kielhorn J, Melber C, Wahnschaffe U, et al. Vinyl chloride: Still a cause for concern. Environ Health Perspect. 2000;108:579-88.
184. Whelan JGJr, Creech JL, Tamburro CH. Angiographic and radionuclide characteristics of hepatic angiosarcoma found in vinyl chloride workers. Radiology. 1976;118:549-57.
185. Truell JE, Peck SD, Reiquam CW. Hemangiosarcoma of the liver complicated by disseminated intravascular coagulation. A case report. Gastroenterology. 1973;65:936-42.
186. Alpert LI, Benisch B. Hemangioendothelioma of the liver associated with microangiopathic hemolytic anemia. Report of four cases. Am J Med. 1970;48:624-8.
187. Ishak KG. Malignant mesenchymal tumors of the liver. In: Okuda K, Ishak KG, editors. Neoplasms of the Liver. Tokyo: Springer Verlag; 1987:159-76.
188. Makhlouf HR, Ishak KG, Goodman ZD. Epithelioid hemangioendothelioma of the liver: A clinicopathologic study of 137 cases. Cancer. 1999;85:562-82.
189. Lenze F, Birkfellner T, Lenz P, et al. Undifferentiated embryonal sarcoma of the liver in adults. Cancer. 2008;112:2274-82.
190. Horowitz M, Etcubanas E, Webber B, et al. Hepatic undifferentiated (embryonal) sarcoma and rhabdomyosarcoma in children. Cancer. 1987;59:396-402.
191. Gajda M, Hommann M, Bottcher J, et al. Primary liposarcoma of the liver—a rare mesenchymal tumor. Z Gastroenterol. 2007;45:1241-4.
192. Lisker Melman M, Pittaluga S, Pluda J, et al. Primary hepatic lymphoma of the liver in a patient with acquired immunodeficiency syndrome and chronic hepatitis B. Am J Gastroenterol. 1989;84:1445-8.
193. Pickren JW, Tsukada Y, Lane WW. Liver metastasis: Analysis of autopsy data. In: Weiss L, Gilber HA, editors. Liver Metastases. Boston: CK Hall; 1982:2-18.
194. Dingemans KP, Roos E. Ultrastructural aspects of the invasion of the liver by cancer cells. In: Weiss L, Gilber HA, editors. Liver Metastases. Boston: CK Hall; 1982:51-76.
195. Ward BA, Miller DL, Frank JA, et al. Prospective evaluation of hepatic imaging studies in the detection of colorectal metastases: Correlation with surgical findings. Surgery. 1989;105(Pt 1):180-7.
196. Sheiner PA, Brower ST. Treatment of metastatic cancer to the liver. Semin Liver Dis. 1994;14:169-77.
197. Barr LC, Skene AI, Thomas JM. Metastasectomy. Br J Surg. 1992;79:1268-74.
198. Hughes K, Scheele J, Sugarbaker PH. Surgery for colorectal cancer metastatic to the liver. Optimizing the results of treatment. Surg Clin North Am. 1989;69:339-59.
199. Abdalla EK, Vauthey JN, Ellis LM, et al. Recurrence and outcomes following hepatic resection, radiofrequency ablation, and combined resection/ablation for colorectal liver metastases. Ann Surg. 2004;239:818-25.
200. Herman P, Pugliese V, Machado M, et al. Hepatic adenoma and focal nodular hyperplasia: Differential diagnosis and treatment. World J Surg. 2000;24:372-6.
201. Nagorney DM. Benign hepatic tumors: Focal nodular hyperplasia and hepatocellular adenoma. World J Surg. 1995;19:13-18.
202. Knowles DM2nd, Casarella WJ, Johnson PM, et al. The clinical, radiologic, and pathologic characterization of benign hepatic neoplasms. Alleged association with oral contraceptives. Medicine. 1978;57:223-37.
203. Bluteau O, Jeannot E, Bioulac-Sage P, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002;32:312-15.
204. Zucman-Rossi J. Genetic alterations in hepatocellular adenomas: Recent findings and new challenges. J Hepatol. 2004;40:1036-9.
205. Rebouissou S, Bioulac-Sage P, Zucman-Rossi J. Molecular pathogenesis of focal nodular hyperplasia and hepatocellular adenoma. J Hepatol. 2008;48:163-70.
206. Stoot JH, van der Linden E, Terpstra OT, et al. Life-saving therapy for haemorrhaging liver adenomas using selective arterial embolization. Br J Surg. 2007;94:1249-53.
207. Capussotti L, Ferrero A, Sgotto E, et al. Right hepatectomy with anterior approach for ruptured liver cell adenoma. Hepatogastroenterology. 2007;54:1557-9.
208. Stoot JH, van Roosmalen J, Terpstra OT, et al. Life-threatening hemorrhage from adenomas in the liver during pregnancy. Dig Surg. 2006;23:155.
209. Winterer JT, Kotter E, Ghanem N, et al. Detection and characterization of benign focal liver lesions with multislice CT. Eur Radiol. 2006;16:2427-43.
210. Hussain SM, van den Bos IC, Dwarkasing RS, et al. Hepatocellular adenoma: Findings at state-of-the-art magnetic resonance imaging, ultrasound, computed tomography and pathologic analysis. Eur Radiol. 2006;16:1873-86.
211. Goodman ZD. Benign tumors of the liver. In: Okuda K, Ishak KG, editors. Neoplasms of the Liver. Toyko: Springer Verlag; 1987:105-25.
212. Terkivatan T, de Wilt JH, de Man RA, et al. Treatment of ruptured hepatocellular adenoma. Br J Surg. 2001;88:207-9.
213. Grazioli L, Federle MP, Ichikawa T, et al. Liver adenomatosis: Clinical, histopathologic, and imaging findings in 15 patients. Radiology. 2000;216:395-402.
214. Ibrahim S, Chen CL, Wang SH, et al. Liver resection for benign liver tumors: Indications and outcome. Am J Surg. 2007;193:5-9.
215. Blonski W, Reddy KR, Blonski W, Reddy KR. Evaluation of nonmalignant liver masses. Curr Gastroenterol Rep. 2006;8:38-45.
216. Jelinek J. Imaging of liver hemangiomas. AJR Am J Roentgenol. 1996;166:459.
217. Krause T, Hauenstein K, Studier-Fischer B, et al. Improved evaluation of technetium-99m-red blood cell SPECT in hemangioma of the liver. J Nucl Med. 1993;34:375-80.
218. Freeny PC, Vimont TR, Barnett DC, et al. Cavernous hemangioma of the liver: Ultrasonography, arteriography, and computed tomography. Radiology. 1979;132:143-8.
219. Mastropasqua M, Kanematsu M, Leonardou P, et al. Cavernous hemangiomas in patients with chronic liver disease: MR imaging findings. Magn Reson Imaging. 2004;22:15-18.
220. Yu JS, Kim KW, Park MS, et al. Transient peritumoral enhancement during dynamic MRI of the liver: Cavernous hemangioma versus hepatocellular carcinoma. J Comput Assist Tomogr. 2002;26:411-17.
221. Cooper WH, Martin JF. Hemangioma of the liver with thrombocytopenia. Am J Roentgenol Radium Ther Nucl Med. 1962;88:751-5.
222. Makhlouf HR, Ishak KG, Makhlouf HR, Ishak KG. Sclerosed hemangioma and sclerosing cavernous hemangioma of the liver: A comparative clinicopathologic and immunohistochemical study with emphasis on the role of mast cells in their histogenesis. Liver. 2002;22:70-8.
223. Feldman M. Hemangioma of the liver. Special reference to its association with cysts of the liver and pancreas. Am J Clin Pathol. 1958;29:160.
224. Chung EB, Chung EB. Multiple bile-duct hamartomas. Cancer. 1970;26:287-96.
225. Banz EJ, Baggenstoss AH. Focal cirrhosis of the liver: its relation to the so-called hamartoma (adenoma, benign hepatoma). Cancer. 1953;6:743-55.
226. Park WC, Rhillips R, Park WC, Rhillips R. The role of radiation therapy in the management of hemangiomas of the liver. JAMA. 1970;212:1496-8.
227. Schwartz SI, Husser WC, Schwartz SI, Husser WC. Cavernous hemangioma of the liver. A single institution report of 16 resections. Ann Surg. 1987;205:456-65.
228. Holcomb GW3rd, O’Neill JAJr, Mahboubi S, et al. Experience with hepatic hemangioendothelioma in infancy and childhood. J Pediatr Surg. 1988;23:661-6.
229. Mortensen W, Pettersson H. Infantile hepatic haemangioendothelioma. Angiographic considerations. Acta Radiol. 1979;20:161-9.
230. Halefoglu AM. Magnetic resonance imaging of infantile hemangioendothelioma. Turk J Pediatr. 2007;49:77-81.
231. Zenge JP, Fenton L, Lovell MA, et al. Case report: Infantile hemangioendothelioma. Curr Opin Pediatr. 2002;14:99-102.
232. Rake MO, Liberman MM, Dawson JL, et al. Ligation of the hepatic artery in the treatment of heart failure due to hepati haemangiomatosis. Gut. 1970;11:512-15.
233. Walsh R, Harrington J, Beneck D, et al. Congenital infantile hepatic hemangioendothelioma type II treated with orthotopic liver transplantation. J Pediatr Hematol Oncol. 2004;26:121-3.
234. Frost NC, Esterley NB. Successful treatment of juvenile hemangiomas with prednisone. J Pediatr. 1968;72:351-7.
235. Goodman Z, Ishak KG. Angiomyolipomas of the liver. Am J Surg Pathol. 1984;8:745-50.
236. Brunt E. Benign tumors of the liver. Clin Liver Dis. 2001;5:1-15.
237. Allaire G, Rabin L, Ishak KG, Sesterhenn I. Bile duct adenoma. Am J Surg Pathol. 1988;12:708-15.
238. Kojiro M. Pathology of hepatocellular carcinoma. In: Okuda K, Tabor E, editors. Liver Cancer. New York: Churchill Livingstone; 1997:165-87.
239. Wanless I, Lentz J, Roberts E. Partial nodular transformation of liver in an adult with persistent ductus venosus. Arch Pathol Lab Med. 1985;109:427-32.
240. Wanless IR. Epithelioid hemangioendothelioma, multiple focal nodular hyperplasias, and cavernous hemangiomas of the liver. Arch Pathol Lab Med. 2000;124:1105-7.
241. Buscarini E, Danesino C, Plauchu H, et al. High prevalence of hepatic focal nodular hyperplasia in subjects with hereditary hemorrhagic telangiectasia. Ultrasound Med Biol. 2004;30:1089-97.
242. Ross D, Pina J, Mirza M, et al. Regression of focal nodular hyperplasia after discontinuation of oral contraceptives [letter]. Ann Intern Med. 1976;85:203-4.
243. Mathieu D, Kobeiter H, Maison P, et al. Oral contraceptive use and focal nodular hyperplasia of the liver. Gastroenterology. 2000;118:560-4.
244. Prentice RL, Thomas DB. On the epidemiology of oral contraceptives and disease. Adv Cancer Res. 1987;49:285-401.
245. Vessey MP, Kay CR, Baldwin JA, et al. Oral contraceptives and benign liver tumours. Br Med J. 1977;1:1064-5.
246. Rettenbacher T. Focal liver lesions: Role of contrast-enhanced ultrasound. Eur J Radiol. 2007;64:173-82.
247. Heiken JP. Distinguishing benign from malignant liver tumours. Cancer Imaging. 2007;7(Spec No A):S1-14.
248. Frohlich J. MRI of focal nodular hyperplasia (FNH) with gadobenate dimeglumine (Gd-BOPTA) and SPIO (ferumoxides): An intra-individual comparison. J Magn Reson Imaging. 2004;19:375-6.
249. Bioulac-Sage P, Balabaud C, Bedossa P, et al. Pathological diagnosis of liver cell adenoma and focal nodular hyperplasia: Bordeaux update. J Hepatol. 2007;46:521-7.
250. Reshamwala PA, Kleiner DE, Heller T, et al. Nodular regenerative hyperplasia: not all nodules are created equal. Hepatology. 2006;44:7-14.
251. Tsou YK, Lin CJ, Liu NJ, et al. Inflammatory pseudotumor of the liver: Report of eight cases, including three unusual cases, and a literature review. J Gastroenterol Hepatol. 2007;22:2143-7.
252. Wang SS, Chiang JH, Tsai YT, et al. Focal hepatic fatty infiltration as a cause of pseudotumors: Ultrasonographic patterns and clinical differentiation. J Clin Ultrasound. 1990;18:401-9.
253. Desmet VJ. Congenital diseases of intrahepatic bile ducts: Variations on the theme “ductal plate malformation.”. Hepatology. 1992;16:1069-83.
254. Jorgensen MJ. The ductal plate malformation. Acta Pathol Microbiol Scand Suppl. 1977;257:1-87.
255. Gaines PA, Sampson MA. The prevalence and characterization of simple hepatic cysts by ultrasound examination. Br J Radiol. 1989;62:335-7.
256. Cowles RA, Mulholland MW. Solitary hepatic cysts. J Am Coll Surg. 2001;191:311-21.
257. Mergo P, Ros P. Benign lesions of the liver. Radiol Clin North Am. 1998;36:319-31.
258. D’Agata ID, Jonas MM, Perez-Atayde AR, et al. Combined cystic disease of the liver and kidney. Semin Liver Dis. 1994;14:215-28.
259. Summerfield JA, Nagafuchi Y, Sherlock S, et al. Hepatobiliary fibropolycystic diseases. A clinical and histological review of 51 patients. J Hepatol. 1986;2:141-56.
260. Karhunen PJ, Tenhu M, Karhunen PJ, Tenhu M. Adult polycystic liver and kidney diseases are separate entities. Clin Genet. 1986;30:29-37.
261. Pirson Y, Lannoy N, Peters D, et al. Isolated polycystic liver disease as a distinct genetic disease, unlinked to polycystic kidney disease 1 and polycystic kidney disease 2. Hepatology. 1996;23:249-52.
262. Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1993;22:520-5.
263. Gabow PA, Johnson AM, Kaehny WD, et al. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033-7.
264. Milutinovic J, Fialkow PJ, Rudd TG, et al. Liver cysts in patients with autosomal dominant polycystic kidney disease. Am J Med. 1980;68:741-4.
265. Kwok MK, Lewin KJ. Massive hepatomegaly in adult polycystic liver disease. Am J Surg Pathol. 1988;12:321-4.
266. Qian Q, Li AR, King BF, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164-71.
267. Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332-42.
268. Li A, Davila S, Furu L, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72:691-703.
269. Tahvanainen P, Tahvanainen E, Reijonen H, et al. Polycystic liver disease is genetically heterogeneous: Clinical and linkage studies in eight Finnish families. J Hepatol. 2003;38:39-43.
270. Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129-37.
271. Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40:774-82.
272. Davila S, Furu L, Gharavi AG, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575-7.
273. Waanders E, te Morsche RH, de Man RA, et al. Extensive mutational analysis of PRKCSH and SEC63 broadens the spectrum of polycystic liver disease. Hum Mutat. 2006;27:830.
274. Drenth JPH, Martina JA, van de Kerkhof R, et al. Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol Med. 2005;11:37-42.
275. Srinivasan R. Polycystic liver disease: An unusual cause of bleeding varices. Dig Dis Sci. 1999;44:389-92.
276. Que F, Nagorney DM, Gross JBJr, Torres VE. Liver resection and cyst fenestration in the treatment of severe polycystic liver disease. Gastroenterology. 1995;108:487-94.
277. Washburn WK, Johnson LB, Lewis WD, Jenkins RL. Liver transplantation for adult polycystic liver disease. Liver Transpl Surg. 1996;2:17-22.
278. Harris PC, Rossetti S, Harris PC, Rossetti S. Molecular genetics of autosomal recessive polycystic kidney disease. Mol Genet Metab. 2004;81:75-85.
279. Johnson PJ. Benign and malignant tumors of the liver. In: Bacon BR, DiBisceglie AM, editors. Liver Disease: Diagnosis and Management. New York: Churchill Livingstone; 2000:310-20.
280. Sane S. Primary carcinoma of the liver: Cholangiocarcinoma in hepatolithiasis. Am J Pathol. 1942;18:675.
281. Ulrich F, Steinmuller T, Settmacher U, et al. Therapy of Caroli’s disease by orthotopic liver transplantation. Transplant Proc. 2002;34:2279-80.
282. Ulrich F, Pratschke J, Pascher A, et al. Long-term outcome of liver resection and transplantation for Caroli disease and syndrome. Ann Surg. 2008;247:357-64.