[level-membership-for-surgery-category]27
Treacher Collins Syndrome
Evaluation and Treatment
• Inheritance, Genetic Markers, and Testing
• Considerations during Infancy and into Early Childhood
• Dysmorphology with Treacher Collins Syndrome
• Facial Growth Potential with Treacher Collins Syndrome
• Classification of Temporomandibular Joint and Mandibular Malformation
• Staging of Skeletal Reconstruction: Timing and Techniques
• Indications for First-Stage Mandibular Reconstruction in the Newborn
• Indications for First-Stage Mandibular Reconstruction during the Mixed Dentition
• Facial Soft-Tissue Reconstruction
Treacher Collins syndrome (TCS), which is also known as mandibulofacial dysostosis, is an autosomal dominant condition with variable expressivity.* It is generally characterized by bilaterally symmetric abnormalities of the structures within the first and second branchial arches. Early descriptions are attributed to Berry,10 Treacher Collins,51 and Franceschetti and Klein.41 To date, the physical findings of many hundreds of individuals and families with TCS have been published in the world literature.
The adult patient with fully expressed TCS has a convex horizontally deficient facial profile with a prominent nasal dorsum above a retrusive lower jaw and chin (Fig. 27-1). The eye region is characterized by an antimongoloid slant of the palpebral fissure as a result of colobomata and hypoplasia of the lower lids and the lateral canthi, including the partial absence of the eyelid cilia and inferolateral orbital hypoplasia and dystopia. Tongue-shaped processes of hair frequently extend into the preauricular region. The external ears are absent, malformed, or malposed and hearing is impaired as a result of variable degrees of hypoplasia of the external auditory canals and the ossicles of the middle ears. A characteristic finding is hypoplasia of the malar bones, often with clefting through the arches and limited formation of the residual zygomas, including the glenoid fossa component. The maxilla and mandible bones are also characteristically hypoplastic, with variable effects on the temporomandibular joints (TMJs), the muscles of mastication, and the muscles of facial expression. Interestingly, hypoplasia of the centrally located soft tissues of the face is not seen with TCS. In general, there is an Angle Class II anterior open-bite malocclusion and a steep clockwise-rotated maxillomandibular complex. The posterior facial height is short, and the anterior lower facial height is long. The A-point–to–B-point relationship in profile is consistent with the clockwise rotation of the jaws. The presence of cleft palate (with or without cleft lip and choanal atresia of the nasal cavity) is variable. Dental anomalies are present in 60% of individuals; these include tooth agenesis (33.3%), enamel opacities (20%), and the ectopic eruption of the maxillary first molars (13.3%).
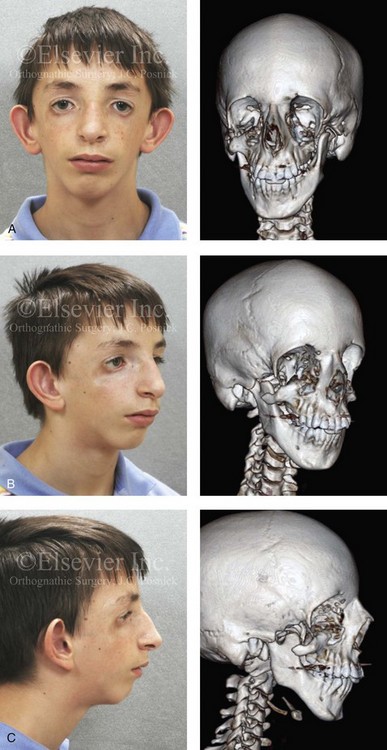
Figure 27-1 An 11-year-old boy with fully expressed Treacher Collins syndrome. His eyes are characterized by an antimongoloid slant of the palpebral fissures as a result of colobomata and hypoplasia of the lower lids and the lateral canthi, including the partial absence of the eyelid cilia and inferolateral orbital dystopia. The appearance of the malpositioned adnexal structures is a reflection of both zygomatic–orbital dystopia and hypoplasia of the soft tissues. The maxilla and the mandible are also deficient and malformed. The patient demonstrates a Type IIA mandibular malformation. The external ears are malformed and lack antihelical folds. A, Frontal facial and three-dimensional computed tomography scan views. B, Oblique facial and three-dimensional computed tomography scan views. C, Profile facial and three-dimensional computed tomography scan views.
Individuals with TCS have a reduced cranial base angle (i.e., basilar kyphosis). Craniosynostosis is not a feature of TCS, but the neurocranium may have an abnormal shape (i.e., decreased anteroposterior length and diminished bitemporal width) that is evident during childhood and remains through adulthood. 110 The degree of craniofacial malformation present at birth is believed to be relatively stable and non-progressive with age (this is discussed in more detail later in this chapter). TCS should not be confused with similar entities such as oculo–auriculo–vertebral dysplasia or Goldenhar syndrome. Distinguishing facial features of patients with TCS often include coloboma of the lower eyelids; the upper eyelids are more likely affected among patients with Goldenhar syndrome.
Inheritance, Genetic Markers, and Testing
The occurrence of TCS is in the range of 1 in 25,000 to 1 in 50,000 live births. Inheritance is autosomal dominant, and male and female individuals are equally affected.51 About 60% of probands with TCS have the disorder as the result of a de novo gene mutation. Each child of an individual with TCS has a 50% chance of inheriting the mutation (Fig. 27-2). Teber and colleagues identified TCOF1 mutations in 28 out of 36 (78%) individuals with a clinically unequivocal diagnosis of TCS.147 The most frequent clinical findings were downward-slanting palpebral fissures, hypoplasia of the zygomatic complex, hypoplasia of the mandible, conductive deafness, any degree of microtia, and atresia of the external ear canal. Although there were interfamilial and intrafamilial variations that ranged from mild to severe, there were no genotype or phenotype correlations. Four clinically unaffected parents were heterozygous for the TCOF1 mutation. The authors concluded that modifying factors are important for phenotypic expression.
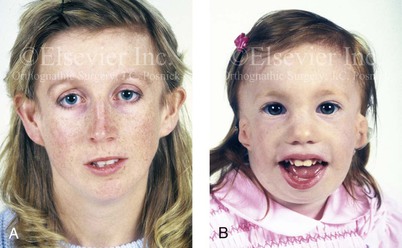
Figure 27-2 This mother and daughter demonstrate the extent of variation in the expression of Treacher Collins syndrome within a family. The mother was not aware that she carried the Treacher Collins gene until after the birth of her daughter. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997.
Genetic counseling for TCS is the process of providing individuals and families with information about the nature, inheritance, and implications of the syndrome to help them make informed medical and personal decisions. Molecular testing should be considered for any individual who presents with at least two major features or three minor features of TCS.30,32,34,62,63,72,85,88,89,154,161,165 Until recently, penetrance in TCS was believed to be complete. Marres and colleagues confirmed the absence of clinical and radiographic findings in an individual with a pathogenic TCOF1 mutation. For this reason, testing is acceptable for individuals with any degree of severity of TCS features to confirm or refute the diagnosis.
Considerations during Infancy and into Early Childhood
At the time of the birth of a child with TCS, concerns will center on the adequacy of the airway, swallowing, feeding, hearing, vision (corneal protection), the presence of cleft palate (with or without cleft lip), any other associated malformations, and the psychosocial well-being of the family.8,17,27,31,77,94,95,101,103,104,120,160 The airway may be compromised as a result of multiple factors. The first is maxillary hypoplasia, which occurs with a degree of choanal stenosis or atresia with the blockage of the nasal airway. Second, mandibular micrognathia with a retropositioned tongue that at least partially obstructs the oropharyngeal and hypopharyngeal spaces will be present. In addition, tracheomalacia, vocal cord anomalies, or neuromotor involvement may also negatively affect the upper airway. Depending on the severity of the anomalies, a spectrum of upper airway compromise may necessitate, at a minimum, special infant positioning, an extended hospital stay, and pulse oximetry monitoring. Surgical considerations for the management of airway compromise in the infant or young adult may include tongue–lip adhesion, the use of a custom-made oral appliance, immediate or delayed tracheostomy, or an urgent mandibular advancement procedure.
When a cleft of the secondary palate is documented, potential additive effects on the airway before and after repair must be considered. When Nager syndrome is present, the soft palate is both clefted and severely hypoplastic to the extent that the achievement of a functioning palate is not feasible.65,96 A primary pharyngeal flap—although helpful for velopharyngeal function—is rarely indicated, because further obstruction of the airway would likely result. Evaluation by a medical geneticist is indicated to clarify the diagnosis and all presenting anomalies, to discuss family planning issues, and to analyze chromosomal studies.
Dysmorphology with Treacher Collins Syndrome
Cranio–Orbito–Zygomatic Region
In earlier published studies, 14 reproducible cranio-orbito-zygomatic measurements taken from each of 26 standard axial CT scans of individuals with symmetric forms of TCS who had not been operated on were compared with the measurements of age-matched controls.110 The interorbital measurements (i.e., medial and lateral orbital wall separations) of the patients with TCS were at the mean when compared with their cohort group (i.e., no measurable orbital hypertelorism or hypotelorism), whereas the zygomatic measurements were significantly less than normal, thereby confirming the extent of malar hypoplasia. The congenitally deficient lateral aspect of the orbits in individuals with TCS was confirmed by the greater than normal values measured for globe protrusion and medial orbital wall protrusion in conjunction with diminished lateral orbital wall lengths. These measurements use the lateral orbital rim as a reference point and confirm the limited depth of the lateral orbits as a result of hypoplasia of the lateral orbital rims. The lateral orbital rims are components of the hypoplastic zygomatic complex. The abnormal shape of the anterior cranial vault in patients with TCS was documented as a diminished intercoronal (bitemporal) distance (width) and decreased cephalic length as compared with values for normal age-matched controls. In general, the CT measurements documented in the described study agree with the clinically observed morphology of TCS. The measurements carried out in the zygomatic structures confirmed the extent of hypoplasia of the zygomas for the group as a whole and in each individual patient; they also documented the extent of globe protrusion and decreased upper face width in patients with TCS. It appears as though the bones immediately adjacent to the involved zygomatic complex also experience a degree of hypoplasia or at least distortion.4,28,39,87,110,113,125,158,159,167,169
Maxillomandibular Region
In 1975, Roberts and colleagues completed a radiographic cephalometric study of TCS. Serial cephalometric radiographs were available for eight patients who presented with the full range of features that are characteristic of this syndrome.124 The expected facial convexity in patients with TCS was substantiated by cephalometric measurements. The extent of convexity was attributed to the severity of mandibular retrognathia (i.e., a decreased sella–nasion–B-point angle). Interestingly, the horizontal projection of the maxilla to the cranial base (i.e., the sella–nasion–A-point angle) remained within normal limits in the patients that were measured. Over time and with growth, the facial convexity remained relatively constant, thus confirming that the facial profile morphology of the infant with TCS was remarkably similar to that of the adult. Anterior facial heights were measured as “upper facial height” (i.e., the nasion to the anterior nasal spine) and “total facial height” (i.e., the nasion to the menton). The upper facial height was found to be relatively normal, whereas the total facial height was often excessive. This is the result of a combination of anterior open-bite malocclusion, mandibular retrognathism, and chin dysplasia characterized by increased vertical length and horizontal retrusion. The angle between the sella–nasion plane and the mandibular plane was obtuse, which confirmed the steepness of the clockwise rotation of the mandibular plane. The angle between the sella–nasion plane and the palatal plane was also obtuse, thus confirming the steepness of the clockwise rotation of the maxillary plane. The posterior facial height was markedly shorter for patients with TCS, with values in five of the eight patients found to be a minimum of four standard deviations less than those of the normal cohort.
The effective horizontal length of the mandible was markedly reduced, with values in most of the patients falling below four standard deviations as compared with those in the normal cohort. The decreased mandible size was evident in both the ramus height and the body length. The gonial angle was confirmed to be obtuse, with values of two standard deviations from the mean as compared with control values. The extent of antigonial notching was also documented in patients with TCS and was found to be similar to that observed among patients with condylar arrest early during their lives (e.g., as a result of juvenile rheumatoid arthritis or TMJ trauma at a young age). The steepness of the clockwise rotation of the maxillary and mandibular planes combined with the anterior and posterior vertical height disproportions and severe horizontal deficiency of the mandible is also indirectly seen in the A-point–to–B-point discrepancy. All of these morphologic findings explain the observed facial dysmorphology.4, 39,49,78,79,87
Facial Soft Tissues
Soft-tissue anomalies in individuals with TCS are manifested primarily in four clinical regions: the external ears; the eyelid–adnexal structures; the preauricular–cheek skin; and the temporal fossa (e.g., temporalis muscle hypoplasia).10,11,22,35,36,64,76,86,132,133,143,148,156 The extent of deficiency within the soft-tissue envelope is variable and, to large extent, reflective of the skeletal involvement. Each patient is unique and must be evaluated individually. The extent of soft-tissue anomalies continues to represent the most difficult obstacle to a favorable reconstruction.
External Auditory Canal, Middle and Inner Ear Structures, and Audiologic Findings
Relatively symmetric (i.e., from side to side) outer and middle ear malformations are characteristic of TCS. Middle-ear abnormalities (e.g., hypoplastic or absent cavities and ossicles) are an almost universal finding.18,55,58,66,118 The majority of patients have a unilateral or bilateral moderate or greater conductive hearing loss, with a flat or reverse sloping configuration on the audiogram. Most of the hearing loss in these patients is attributable to middle-ear malformations. In 1993, Pron and colleagues reported the results of a detailed CT radiologic and audiologic investigation of a large series of pediatric patients with TCS.118 The objectives of the study were 1) to determine the degree and symmetry of the external auditory canal and middle-ear abnormalities and evaluate hearing loss; and 2) to explore the relationship between hearing loss and external auditory canal and middle-ear abnormalities in patients with TCS. Data were available from 29 children who had not yet undergone any ear interventions and who had undergone audiometric testing and focused standardized petrous temporal bone CT scans. A review of the CT scans allowed for the evaluation of the external ear canal and the middle-ear anatomy. The majority of patients (88%) had largely symmetric external auditory canal abnormalities that were either stenotic (31%), atretic (54%), or normal (15%). Middle-ear cavity ossicular deformities were generally symmetric (96%), and cavities were either hypoplastic (85%) or missing (4%). Cavities that were hypoplastic were also deformed and assumed a rectangular rather than an oval shape. Ossicle deformities were generally completely symmetric and consisted largely of hypoplastic (46%) or missing (46%) ossicles. Ossicles (both the malleus and the incus) that were hypoplastic also tended to be ankylosed (82%) to either the lateral or medial wall of the tympanic recess. The structures of the external auditory canal and the middle ear are generally related. Increasing degrees of external ear canal malformations (i.e., normal, stenotic, or atretic) are directly associated with ossicle and cavity malformations. Of the patients with atretic canals, 67% had missing ossicles, and 33% had small or ankylosed ossicles. No abnormalities were noted in the inner-ear structures (i.e., cochlea, vestibule, canals, and internal auditory meatus) in the patients with TCS who were studied. According to the study by Pron and colleagues, hearing loss for the patient with TCS ranges from mild to severe (average, 58 dB of hearing loss).118 The majority of individuals (96%) have a moderate or greater degree of hearing loss.
Relationships exist between the external auditory canal, the middle-ear ossicles, and the degree and configuration of hearing loss.18,55,58,66,118 There is an association between the degree of external ear canal malformation and the degree of hearing loss in patients with TCS. In addition, hypoplastic, ankylosed, and missing ossicles appear to be associated with increasing levels of hearing loss; on average, the hearing loss is 52 dB, 56 dB, and 60 dB of hearing loss, respectively.
Facial Growth Potential with Treacher Collins Syndrome
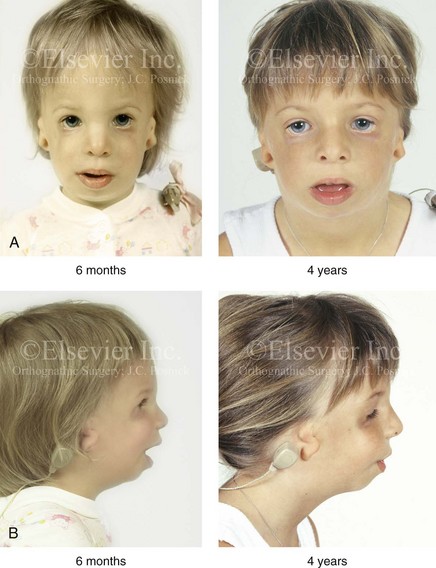
The degree of malformation present at the time of birth in a newborn with TCS is believed to be relatively stable and non-progressive with age (Figs. 27-3 through 27-6).53,102,109,114,115,124 Roberts and associates reviewed serial cephalometric radiographs from individuals who were known to have TCS.124 They documented the stability of the mandible, which continued to grow but not to catch up. Through independent research, Garner reported findings of the cephalometric analysis of three patients of varied ages who were confirmed to have TCS.49 He also documented the relative stability of the anomalies without the progression of deformity at various ages.
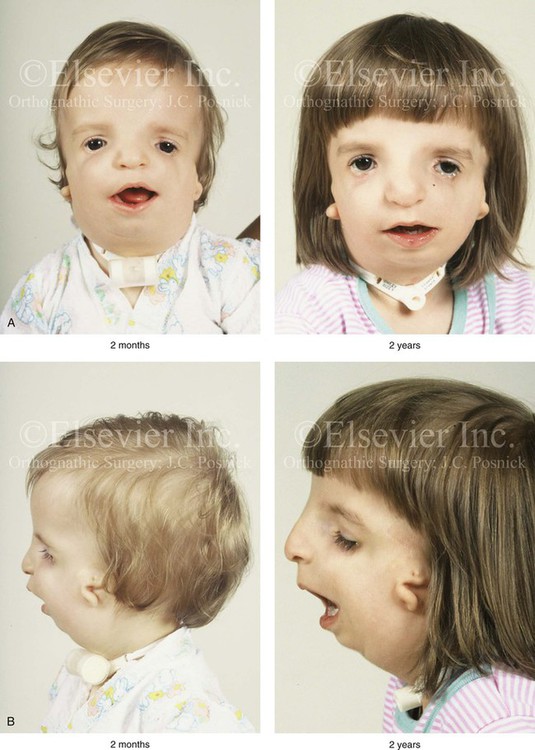
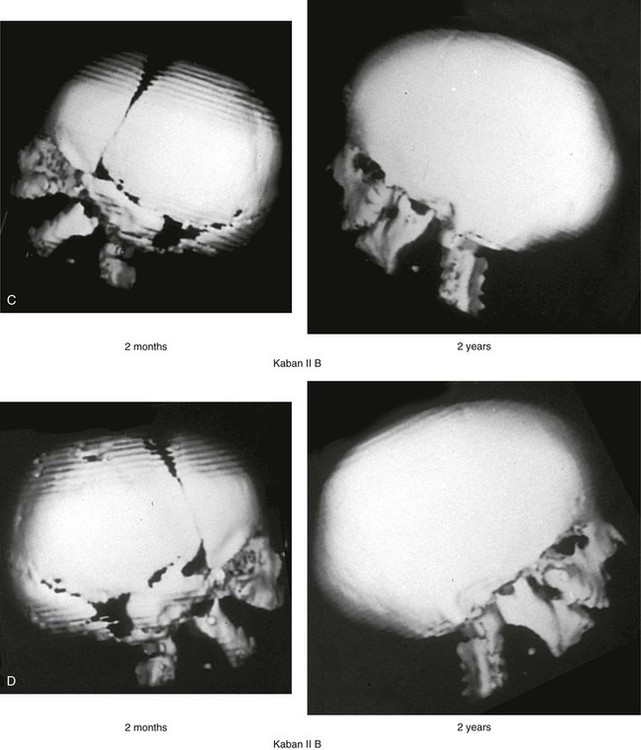
Figure 27-3 A child with a severe form of Treacher Collins syndrome was followed up longitudinally without treatment intervention. No progression of the deformity was seen during clinical or computed tomography scan examination. A, Frontal and B, profile views at 2 months and 2 years of age. C and D, Computed tomography scan views of the craniofacial region at 2 months and 2 years, respectively, demonstrating the consistent growth of the malformed condyles, the coronoid processes, and the ascending rami of the mandible. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997.
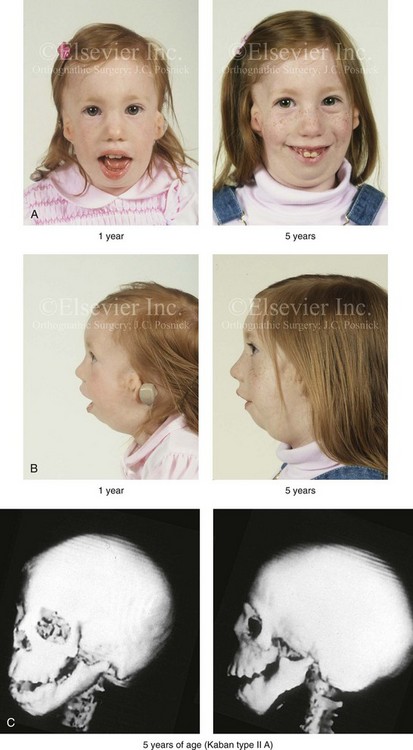
Figure 27-4 A girl who was born with Treacher Collins syndrome was followed up longitudinally at 1 and 5 years of age without treatment intervention. There is consistent growth of the craniofacial region without progression of the deformity. As a result of chronic nasal obstruction and an open-mouth posture, excess vertical facial growth can be seen. A, Frontal views at 1 and 5 years of age. B, Profile views at 1 and 5 years of age. C, Computed tomography scan views at 5 years of age. There is severe hypoplasia of the zygomatic complex with lateroinferior orbital deficiency. The mandible demonstrates a Type IIA malformation.
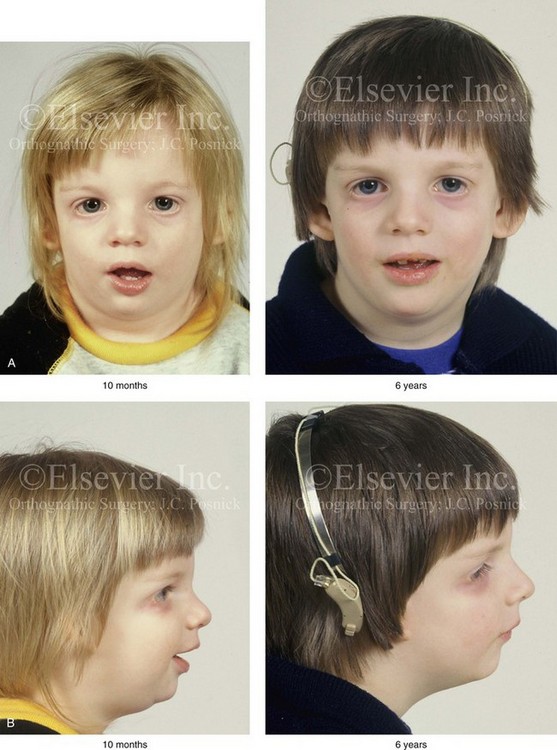
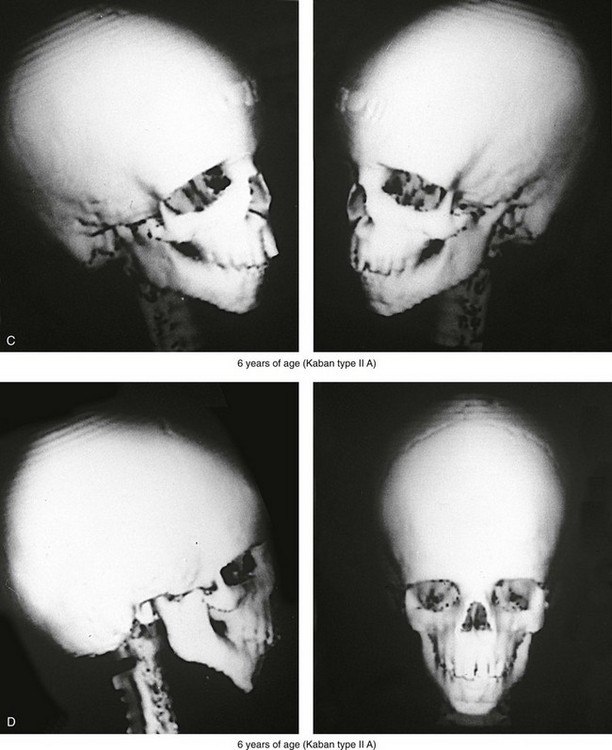
Figure 27-5 A child who was born with Treacher Collins syndrome was followed up longitudinally without treatment intervention. No progression of the facial deformity occurred with growth. A, Frontal views at 10 months and 6 years of age. B, Profile views at 10 months and 6 years of age. C, and D, Computed tomography scans.
Classification of Temporomandibular Joint and Mandibular Malformation
The extent of TMJ–mandibular malformation in patients with TCS will influence the timing and techniques of reconstruction.69,137 A lack of clinically relevant definitions of the presenting glenoid fossa–condyle–ascending ramus malformations has resulted in confusion and miscommunication. Kaban and colleagues revised and clarified the previously described Pruzansky classification system for the degree of TMJ–mandibular malformation observed in patients with hemifacial microsomia.69,119 This classification is also useful to define the presenting anomalies and then to direct reconstruction in the patient with TCS (Fig. 27-7).
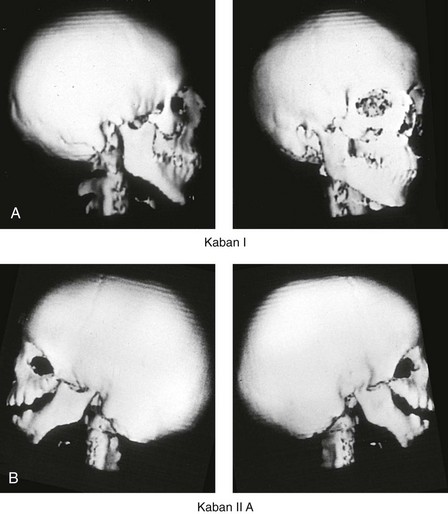
Figure 27-7 Kaban and colleagues described a classification system to define the degree of temporomandibular joint–mandibular malformation observed in patients with hemifacial microsomia. By definition, a Type IIB condylar deficiency will require the construction of a neo-condyle. The Type III malformation will also require neo-glenoid fossa construction. The same classification system is used for Treacher Collins syndrome. Craniofacial computed tomography (CT) scans demonstrate the classification system. A, CT scan views of a Type I mandibular malformation. B, C, and D, CT scan views of a Type IIA mandibular malformation. E and F, CT scan views of a Type III glenoid fossa–mandibular malformation.
A Type I TMJ–mandibular malformation involves minimal hypoplasia of the glenoid fossa and the condyle–ascending ramus on each side. All of the skeletal components are present (see Fig 27-7, A). Mandibular retrognathia and a mild anterior open bite are likely to be present, but TMJ function is essentially normal. The muscles of mastication are intact, and the condyle and the glenoid fossa are also fully intact. There is no need or advantage to constructing a neo-condyle or neo-glenoid fossa.
A Type IIA TMJ–mandible malformation involves a moderate degree of glenoid fossa and condyle–ascending ramus hypoplasia (see Fig. 27-7, B, C, and D). The anatomic location of each joint is anterior and medial, but TMJ function remains satisfactory. One or more of the paired muscles of mastication are likely to be hypoplastic. By definition, in Type IIA malformations, the condyle and the glenoid fossa have function that is at a level deemed better than could reliably be achieved through TMJ replacement. Even in patients with more severe Type IIA malformations, when applying superior and posterior pressure to the chin, the condyles can be seated into a consistent location against the skull base–glenoid fossa. By completing ramus osteotomies, the proximal segments can be positioned into a stable location against the skull base, and the distal mandible can then be reoriented into the preferred location. This will avoid the need to construct neo-condyles (e.g., costochondral grafts). Coronoidectomies may be required to accommodate the short condyles.
The Type III glenoid fossa–mandibular malformation involves deficiency to the extent that no posterior stop of the lower jaw against the cranial base is possible (see Fig. 27-7, E). The condyles and the ascending rami are not present. The disc, the TMJ capsule, and the glenoid fossa on each side are also not developed. Severe mandibular dysmorphology is present, including horizontal retrognathia and decreased posterior ramus height. A tracheostomy is generally required shortly after birth to manage the airway. Eventual construction of both a neo-glenoid fossa and a neo-condyle ascending ramus will be required on each side. Reconstruction with bone grafts (e.g., cranial grafts for the zygomatic complex and either costochondral or a vascularized fibula flap for the mandible) will be required (see discussion to follow).
Staging of Skeletal Reconstruction: Timing and Techniques
The reconstruction and rehabilitation of the Treacher Collins malformation must address unique and specific components of the anomalies, including the following: 1) the zygomatic and orbital region; 2) the maxillomandibular region; 3) the nasal region; 4) the soft-tissue envelope; 5) the external ears; 6) the external auditory canals; and 7) the middle-ear structures (Figs. 27-8 through 27-10).*
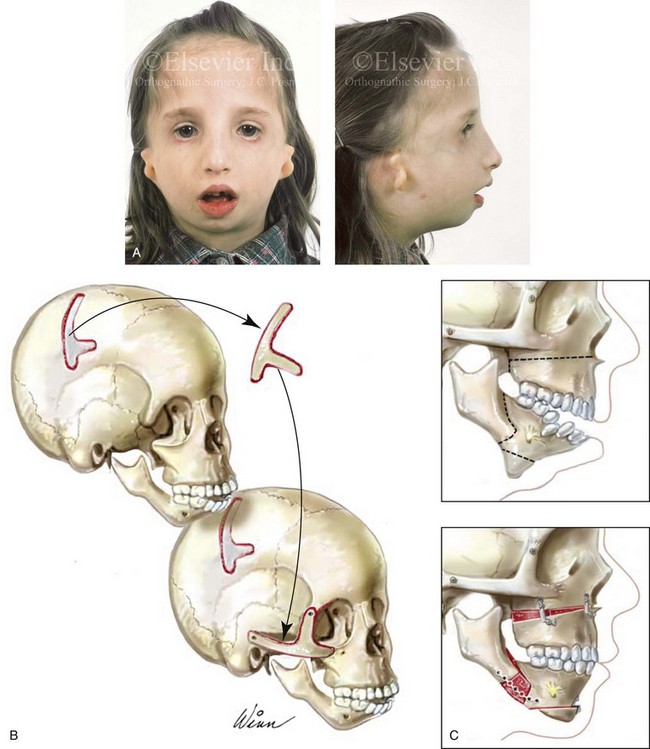
Figure 27-8 A, A child who was born with Treacher Collins syndrome is shown during the mixed dentition. She has severe zygomatic complex and orbital deficiency as well as a Type IIA condylar malformation. B, Illustration of the location of the full-thickness cranial bone graft donor site for zygomatic complex reconstruction. The zygomatic complex graft is also shown inset at the recipient site. C, Illustrations of a maxillofacial complex seen in a teenager with Treacher Collins syndrome (Type IIA mandibular malformation). The cheekbone orbital reconstruction has previously been completed. The proposed maxillary, mandibular, and chin osteotomies are marked out and then completed. Counterclockwise rotation of the maxillomandibular complex is demonstrated as part of the reconstruction.
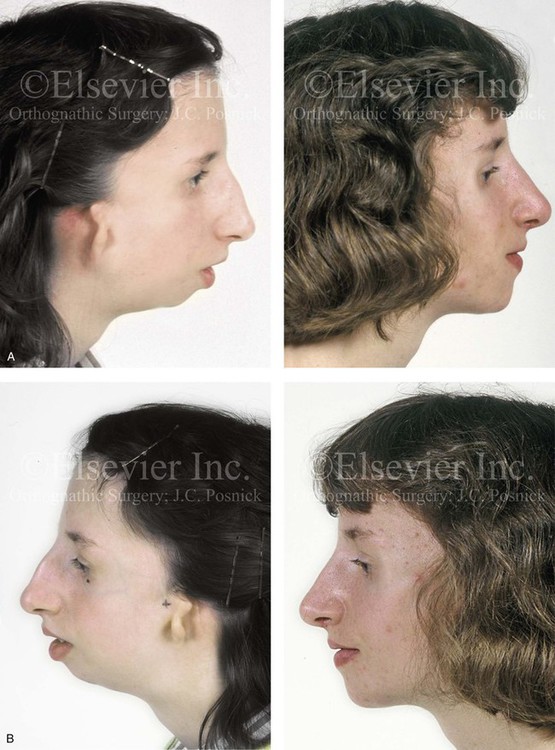
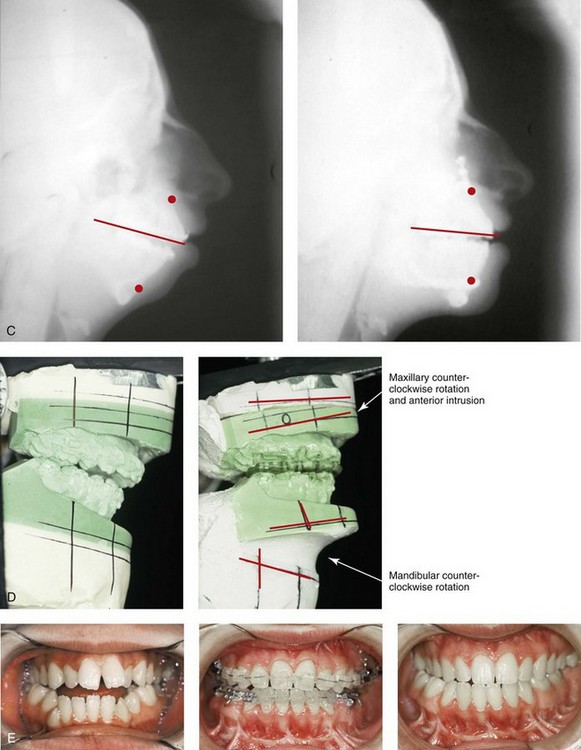
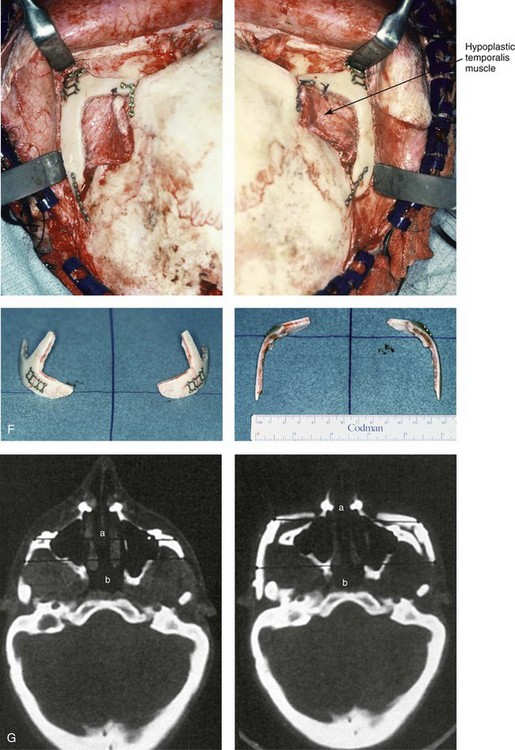
Figure 27-9 Additional images of the patient from Fig. 27-8 showing the stigmata of Treacher Collins syndrome. At the age of 17 years, the patient was referred to this surgeon and agreed to a staged reconstruction that included three operations. The initial preoperative orthodontic treatment included the extraction of mandibular first premolars with the retraction of the anterior dentition to remove compensations. Orthognathic surgery included a Le Fort I osteotomy (counterclockwise rotation and anterior maxillary intrusion); bilateral sagittal split ramus osteotomies (advancement and counterclockwise rotation); and an osseous genioplasty (vertical reduction and advancement). During a second operation, the patient underwent zygomatic complex reconstruction with fixed full-thickness autogenous cranial bone grafts as described in this chapter and as illustrated in Fig. 27-8. This was followed a third operation: septorhinoplasty (open approach) to in-fracture the nasal bones, reduce the dorsal hump (bone and cartilage), and reshape the tip, including septal (caudal strut) grafting (see Chapter 38). A, Right profile views before and after reconstruction. B, Left profile views before and after reconstruction. C, Lateral cephalometric radiographs before and after reconstruction. D, Articulated dental casts that indicate analytic model planning. E, Occlusal views before treatment, early after reconstruction, and 3 years after reconstruction. A degree of maxillary relapse (skeletal versus dental) has occurred but with a retained functional occlusion and favorable facial aesthetics. F, Crafted autogenous cranial grafts shown before and then after inset into the zygomatic defects. The advancement of the hypoplastic temporalis muscles is also demonstrated. The soft-tissue hypoplasia involves multiple layers (i.e., skin, subcutaneous tissue, fascia, and fat tissue) and therefore can not be fully corrected simply by advancing the hypoplastic temporalis muscle. G, Axial computed tomography scan views shown through the zygomas before and after malar reconstruction indicating the improved interzygomatic buttress width, interzygomatic arch width, and zygomatic arch length. A, From Posnick JC, Treacher Collins syndrome: Perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997. F (bottom left, bottom right), G, From Posnick JC, Goldstein JA, Waitzman A: Surgical correction of Treacher Collins malar deficiency: quantitative CT scan analysis of long-term results, Plast Reconstr Surg 92:12, 1993.
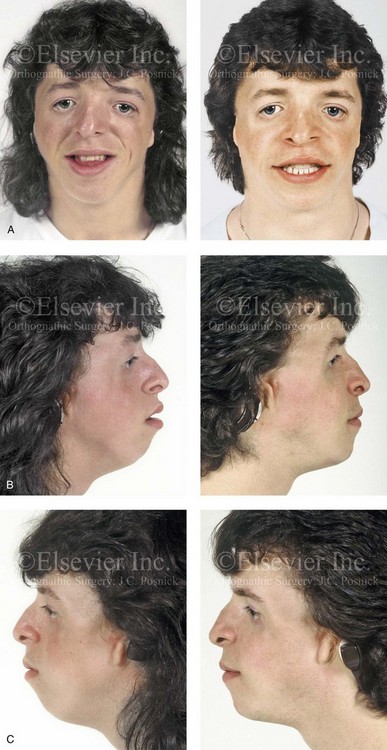
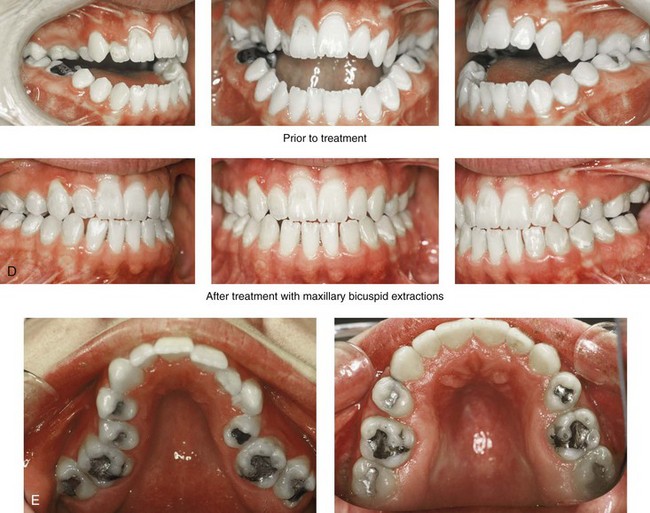
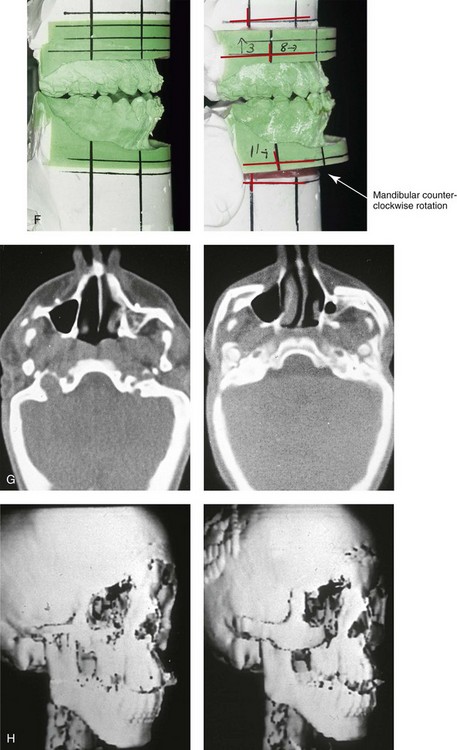
Figure 27-10 A 16-year-old boy born with Treacher Collins syndrome with a Type I mandibular malformation was followed up longitudinally to confirm no progression of the deformity. He then underwent two operations, including orbitozygomatic and orthognathic reconstruction, as described previously. In preparation for orthognathic surgery, orthodontic treatment included maxillary first bicuspid extractions with the retraction of the anterior dentition to correct the arch form. The mandibular second molars are congenitally absent. The patient’s orthognathic surgery included Le Fort I osteotomy (vertical intrusion and horizontal advancement); bilateral sagittal split ramus osteotomies (horizontal advancement); and an osseous genioplasty (horizontal advancement). He is shown before and after orbitozygomatic and orthognathic surgery. A, Frontal views before and after orthognathic surgery. B, Left profile views before and after orthognathic surgery. C, Right profile views before and after reconstruction. D, Occlusal views before and after orthognathic surgery. Note that, with the upper bicuspid extractions and the absent mandibular second molars, the maxillary second molars are no longer in occlusion. E, Palatal views before and after orthognathic surgery. F, Articulated dental casts that indicate analytic model planning. G, Axial computed tomography slices before and after malar reconstruction. H, Three-dimensional computed tomography scans before and after malar reconstruction (i.e., before orthognathic surgery).
Zygomatic and Orbital Reconstruction
In previously published studies, we have documented the efficacy of reconstructing the malar and orbital deficiencies for moderate to severe forms of TCS with the use of full-thickness calvarial bone grafts that have been contoured three-dimensionally to form a new zygomatic complex (see Figs. 27-8 and 27-9 through 27-13).113 The neo-zygoma is then inset and stabilized with microplate and screw fixation. Exposure for the reconstruction is provided by a coronal scalp incision without the need for additional periorbital incisions. Orbital floor defects are reconstructed with fixed split-thickness autogenous cranial bone. Cranial vault donor sites are repaired either with autogenous split cranial grafts or artificial material. For non-autogenous reconstruction of the small skull donor sites, we prefer the use of a titanium mesh base fixed to the adjacent skull. The mesh is then filled in with a bone substitute. Lateral canthopexies are completed through the coronal scalp incision and secured to each new lateral orbital rim. In our study, computed tomography scanning that was performed before surgery and immediately afterward demonstrated significant increases in lateral orbital wall length (depth), lateral orbital (rim) width, interzygomatic arch width, and zygomatic arch length for patients with TCS. The late (i.e., ≥1 year) postoperative scans document that these changes were maintained. No complications occurred during surgery, and repaired skull donor sites healed without clinical defect. Zygomatic and orbital morphologic improvement was achieved in all patients, with follow-up intervals ranging from 24 to 50 months (mean, 35 months) at the close of the study. Unfortunately, over time, I have observed that partial resorption of the zygomatic complex graft occurs in some patients.
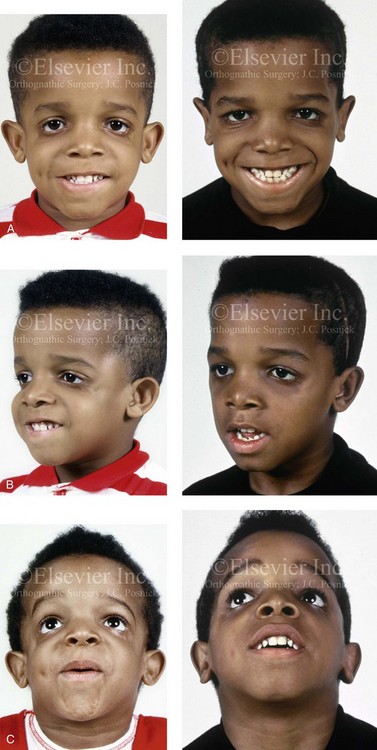
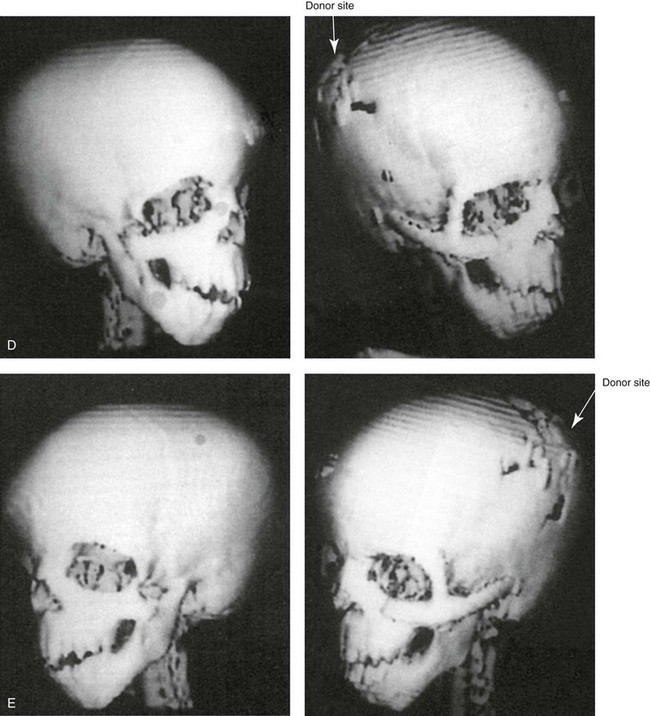
Figure 27-11 A 6-year-old boy who was born with Treacher Collins syndrome and a Type IIA mandibular malformation is shown before and after zygomatico–orbital reconstruction carried out by the technique described. He also underwent bilateral otoplasty to set the ears back (see Chapter 39). A, Frontal views with smile before and after reconstruction. B, Oblique views before and after reconstruction. C, Worm’s-eye views before and after reconstruction. D and E, Computed tomography scan views before and after reconstruction.
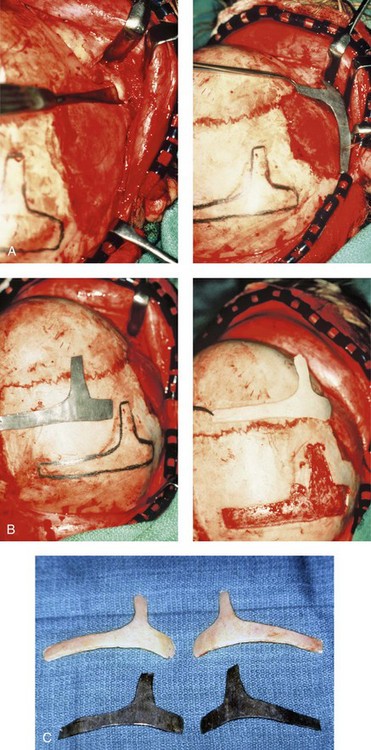
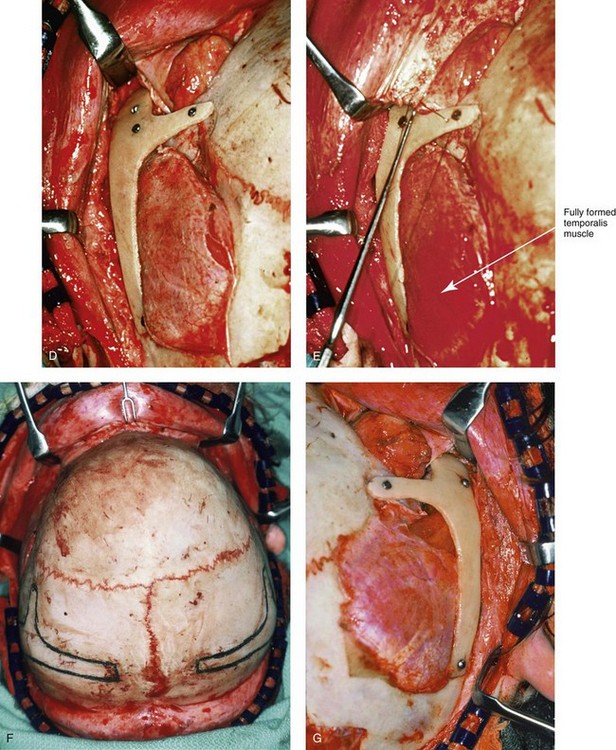
Figure 27-12 Intraoperative views demonstrate the technique for zygomatico–orbital reconstruction in a 5-year-old child as it would be carried out for a patient with Treacher Collins syndrome. Working through a coronal (scalp) incision, the rudimentary zygomatic and orbital structures are exposed. Full-thickness cranial bone grafts are harvested, and each zygoma is crafted from such a graft. Each zygoma graft is inset and stabilized with microplates and screws. The orbital defects are reconstructed with cranial bone graft, and the orbital rims are reshaped with a rotary drill. Lateral canthopexies are completed through the coronal scalp incision and fixed to the new frontozygomatic suture regions. A, A view of the dissected rudimentary zygomatic remnant; the periosteal elevation is deep to the remnant. A template of the proposed zygomatic complex has been made. B, The template is taken to the mid-skull region, and its outline is marked out for craniotomy. The location for graft harvesting is posterior to the coronal suture, lateral to the sagittal suture, and anterior to the lambdoid suture. Through a craniotomy, the full-thickness cranial bone graft is harvested for zygomatic complex reconstruction. The exact same procedure is completed on the contralateral side. C, The two full-thickness cranial bone grafts are then crafted. The two templates and the two full-thickness crafted cranial bone graft–zygomatic complexes are shown. D, The full-thickness cranial bone graft is fixed in place with microscrews. E, The lateral canthus is identified through the coronal incision and a 28-gauge stainless steel wire is passed through. In the patient shown here, the temporalis muscle has minimal hypoplasia. Thus, minimal temporal hallowing is expected after zygomatic reconstruction. The lateral canthus is pexed to the new frontozygomatic region. F, The locations for graft harvesting are outlined. G, The right side cranial graft is shown in place. A,B, From Posnick JC, Goldstein JA, Waitzman A: Surgical correction of Treacher Collins malar deficiency: Quantitative CT scan analysis of long–term results. Plast Reconstr Surg 92:12, 1993.

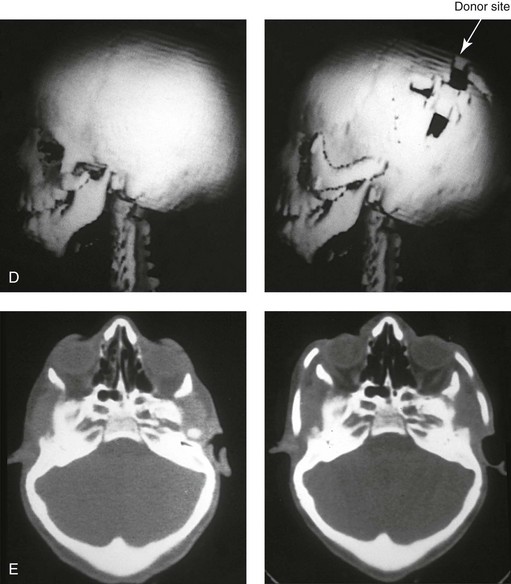
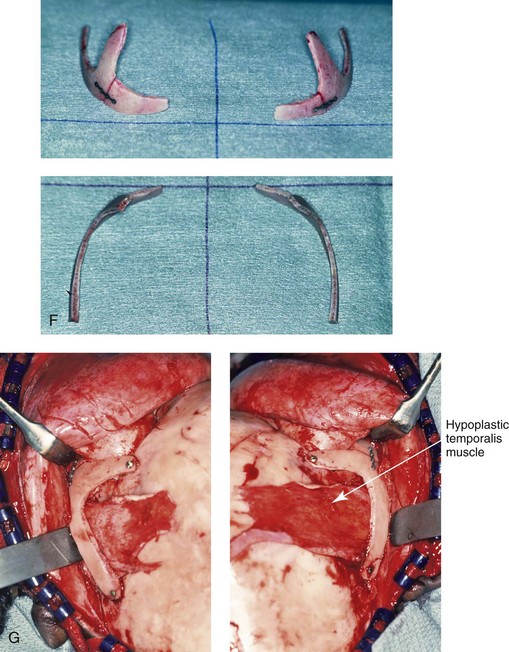
Figure 27-13 A 7-year-old girl who was born with Treacher Collins syndrome is shown before and after zygomatico–orbital reconstruction that was carried out via the technique described in this chapter. A, Frontal views before and after reconstruction. B, Oblique views before and after reconstruction. C, Worm’s-eye views before and after reconstruction. D, Computed tomography scan views before and after zygomatico–orbital reconstruction. E, Axial computed tomography scan views through the mid orbits before and after cranial graft reconstruction. F, Intraoperative views of full-thickness crafted zygomatic complex before inset. G, Left and right intraoperative views of zygomatic complex reconstruction fixed with microplates and screws. The hypoplastic temporalis muscles will be rotated anteriorly as part of the reconstruction. A degree of temporal hallowing is to be expected as a result of the congenital soft-tissue hypoplasia. It is important to not over correct the zygomatic–orbital reconstruction, because this would accentuate the soft-tissue deficiency. A (left), From Posnick JC, Treacher Collins syndrome: Perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997. A (right), From Posnick JC, Goldstein JA, Waitzman A: Surgical correction of Treacher Collins malar deficiency: quantitative CT scan analysis of long-term results, Plast Reconstr Surg 92:12, 1993.
We believe that reconstructing the malar and orbital deficiencies before the patient is 7 years old is not advisable unless severe corneal exposure problems occur and skeletal deficiency is believed to be the cause. By this age, the cranio–orbito–zygomatic bony maturation is nearly complete. Therefore, adult-sized cheekbones may be constructed and placed with limited concern about how further growth will effect the initial result. In addition, skull donor-site reconstruction is simpler than it is at a younger age, because the presence of bicortical cranial bone in those who are more than 7 years old allows for the splitting of adjacent bone to assist with reconstruction (see Figs. 27-11 and 27-12).
Other surgeons have recommended various methods and time frames for the zygomatic and orbital deformities associated with TCS.20,45,60,113,125,149,150,152,167,169 We believe that the construction of the complete zygomatic complex from full-thickness autogenous cranial bone maintains volume and shape better than onlay grafts, which have been universally disappointing. Over time, onlay autogenesis bone grafts that have been placed in the craniofacial region (e.g., supraorbital ridge, zygoma, anterior maxilla, angle of mandible, chin) from all tried donor sites (e.g., skull, hip, rib) have demonstrated significant and unpredictable resorption. Furthermore, no commensurate growth (i.e., volume expansion) of the graft with the underlying bone (e.g., zygoma) should be expected. The belief that “even if the graft partially resorbs, at least something has been gained” should be abandoned; surface irregularities routinely result, which further detract from the baseline malformation.
Maxillomandibular Reconstruction
The basic dysmorphologies of the maxillomandibular complex that require reconstruction in patients with TCS are as follows: 1) altered facial heights (increased anterior lower facial height and decreased posterior facial height); 2) horizontal jaw deficiency (primarily in the mandible but also in the maxilla); and 3) chin dysplasia (increased vertical length and horizontal retrusion).29,43,44,54,61,70,150,152,168 These malformations result in an excessive clockwise rotation of the maxillomandibular complex and an unfavorable A-point–to–B-point relationship when viewed in profile. In general, there is an Angle class II anterior open-bite malocclusion. Interestingly, some individuals will present with either a Class I or Class III relationship. These aspects must be addressed with the incorporation of counterclockwise rotation of the maxillomandibular complex to increase the posterior airway space, to correct the malocclusion, and to restore facial balance.
The most favorable aesthetic results are generally achieved in patients who undergo definitive reconstructive jaw surgery only after early maxillofacial skeletal maturity (i.e., after the age of 13 to 15 years) and when these procedures are carried out in combination with effective definitive orthodontic treatment during the permanent dentition (see Figs. 27-8 through 27-10). Mandibular and possibly maxillary premolar extractions are often required to orthodontically unravel dental root crowding, to normalize the inclination of the anterior teeth, and to fully decompensate the occlusion in preparation for the effective aesthetic and airway repositioning of the jaws (see Fig. 27-7, A, B, C, and D). The surgical technique involves sagittal ramus osteotomies of the mandible in combination with a Le Fort I (maxillary) osteotomy and an oblique osteotomy of the chin. The incorporation of counterclockwise rotation of the maxillomandibular complex is generally required to successfully accomplish certain objectives. Any intranasal airway obstructions are simultaneously managed (e.g., septoplasty, inferior turbinate reduction, recontouring of the nasal floor, widening of the aperture).
The Type III TMJ–mandibular malformation will require surgical construction of the congenitally missing glenoid fossa and condyle on each side (see Fig. 27-7, E). Construction of the neo-glenoid fossa is required and best carried out as part of the zygomatic and orbital reconstruction (see the discussion of zygomatic–orbital reconstruction earlier in this chapter). For the Type III condyle-ascending ramus malformation, construction of a neo-condyle on each side is also required. Mandibular osteotomies alone, with the immediate repositioning or gradual distraction of skeletal segments, will not be adequate. Despite limitations, neo-condylar construction with a costochondral graft remains the preferred alternative when feasible. Occasionally the combined extent of the deficiency of the skeleton and the soft tissues will require the microvascular transfer of a crafted fibula composite flap for each side (i.e., bone and soft tissue). The advantages of a vascularized fibular composite flap are obvious but the donor site morbidity (i.e., need for bilateral lower extremity harvesting) and recipient site requirements (e.g., need for bilateral neck microvascular anastomosis) are significant. In either case, at operation, the distal mandible is repositioned anteriorly to the preferred position of the maxilla. This is assisted by a prefabricated interocclusal acrylic splint, with the distal mandible secured by intermaxillary fixation. The proximal mandible on each side is then constructed with an autogenous costochondral graft or a fibula composite microvascular flap. Contouring of the native mandible with a rotary drill before placing the rib or fibula graft on the lateral side of or adjacent to the edge of the distal mandible is generally required to avoid lateral flaring. Fixation of the graft to the native distal mandible is done with an extended titanium plate from the graft forward along the inferior border of the body of the distal mandible on each side. Graft placement and fixation are generally carried out through an extraoral neck incision. The limited use of intraoral incisions in the ramus regions during this procedure is preferred to minimize the incidence of infection and wound breakdown with subsequent graft loss or TMJ ankylosis.
Indications for First-Stage Mandibular Reconstruction in the Newborn
TCS is a clefting syndrome that involves both sides of the face (i.e., the first and second branchial arches) in which upper airway anomalies occur with both regularity and variation. Hypoplasia of the mandible is one of the constant but variable components of the syndrome.* Individuals with TCS are known to be at risk for sleep apnea, which has been reported to occur in more than 25% of affected individuals. For those with OSA who are tracheostomy dependent, an alternative method of treatment may include the sagittal advancement of the mandible to bring the tongue forward, to create space in the oral cavity, and to open up the retromandibular airway. DO techniques to gradually advance and then hold the mandibular segments until consolidation occurs have been used to improve the airway for almost two decades.* The use of this technique has been reported by several authors as an effective method of decannulating otherwise tracheostomy-dependent individuals with TCS. The implications of this early mandibular advancement approach for long-term airway success and as a definitive jaw reconstruction option for patients with TCS have undergone little critical analysis.37
Anderson and colleagues reported the long-term results of mandibular lengthening with a DO approach that was carried out during the patient’s childhood for the management of airway obstruction in an individual with TCS.2 They documented that the airway improved in a 6-year-old patient with TCS after DO of the mandible to the extent that decannulation and the early resolution of airway obstruction were achieved. They showed that the minimum cross-sectional airway in this patient increased during the period of distraction but did not reach age-adjusted normal values. Subsequent serial scans over 10 progressive years of growth demonstrated that the minimal cross-sectional airway did not increase further. Specifically, measurements of the cross-sectional area that were taken at a fixed point of the upper airway (i.e., the base of the C2 vertebral body) demonstrated no increase in the cross-sectional area during growth. The published longitudinal case study from these authors demonstrates a failure of a sustained satisfactory cross-sectional area of the upper airway with chronologic age. They also used three-dimensional CT scans to document that mandibular growth continued in the same dysmorphic TCS pattern as it had before the distraction intervention. This resulted in an abnormally steep (clockwise rotated) mandibular plane angle. The combination of an increasingly steep mandibular plane angle and the failure of the distraction procedure to produce long-term positive horizontal projection was believed to have created the skeletal basis for ongoing upper airway obstruction. The authors conclude that a mandibular DO procedure carried out in a child with TCS who is experiencing OSA maybe useful for opening the retromandibular airway. However, the initial improvement in the upper airway did not continue during chronologic growth. The clinical consequences were the reemergence of the symptoms of OSA that required further treatment (i.e., continuous positive pressure airway mask at night or tracheostomy). It is also known that a mandibular lengthening procedure carried out during early childhood will have other detrimental effects, including injury to the developing teeth; deformational growth of the mandible; facial soft-tissue scarring; injury to the inferior alveolar nerve; a significant incidence of TMJ ankylosis (both bony and fibrous); perioperative cardiovascular and respiratory complications in some patients; expense to the family and medical health care system; and the need for definitive jaw reconstruction at maturity.7 Despite almost two decades of accumulated worldwide experience, the role of mandibular DO in the treatment protocol for infants and children with TCS remains to be determined through long-term outcome studies. In the absence of sound data, the use of mandibular osteotomies and lengthening procedures—by whatever means—during infancy and childhood should be used cautiously and selectively.
Indications for First-Stage Mandibular Reconstruction during the Mixed Dentition
When a first-stage mandibular reconstruction during the mixed dentition for the Type IIB condylar malformation is felt to be indicated, it is preferably carried out after the child is 7 to 10 years old and only after the eruption of the mandibular permanent first molars.105–110,112,114,115 Unfortunately, a first-stage early mandibular reconstruction will not provide a long-term resolution of the mandibular and other maxillofacial deformities.* Problems with costochondral graft overgrowth, undergrowth, and asymmetrical growth as well as with infection and TMJ ankylosis continue to plague the reconstructive surgeon (see Chapter 26). The final orthognathic correction of the upper jaw, the lower jaw, and the chin region will be required later, after early skeletal maturity (i.e., after 13 to 15 years of age).
The rationale for postponing the correction of the maxillomandibular deformity until the time of early skeletal maturity (i.e., 13 to 15 years of age) is the same as that for patients with standard dentofacial deformities, for those with other associated syndromes, and for those with a cleft lip and palate jaw deformity. The reasons that mandibular surgery during the mixed dentition is generally not preferred include the following: 1) to avoid injury to the developing permanent dentition and the inferior alveolar nerves7,164; 2) to avoid soft-tissue (cutaneous) scarring; 3) to limit perioperative airway and infection complications; 4) to limit the patient’s negative psychosocial memories; and (5) to avoid any iatrogenic deformity of the TMJs and the mandible that may add difficulty to the achievement of a long-term favorable reconstruction.
In individuals with TCS, a Le Fort I osteotomy will be required to correct vertical, horizontal, and transverse maxillary deformities. The maxillomandibular complex invariably benefits from counterclockwise rotation for the correction of pitch orientation. Intrusion of the anterior maxilla is generally needed to establish a more normal upper-lip–to–tooth relationship during smile and in repose. The posterior maxillary height may require lengthening, but more frequently it can remain close to a neutral position. The planned maxillary plane change (i.e., pitch orientation) is carried out to achieve appropriate counterclockwise rotation of the mandible and the chin. The objectives are 1) to normalize the anterior facial height; 2) to open the upper airway; and 3) to establish an aesthetically improved A-point–to–B-point relationship when the patient is viewed in profile. In 1993, Rosen documented the long-term stability of the counterclockwise rotation of the lower jaw when this is required to improve function and enhance the facial aesthetics in patients with mandibular micrognathia. Others have carried out similar clinical studies that have confirmed the long-term skeletal stability of mandibular counterclockwise rotation as part of an orthognathic correction.127,168
For the Type I and IIA mandibular malformations, bilateral intraoral sagittal split osteotomies are generally the preferred ramus procedures, because they allow for effective horizontal advancement, counterclockwise rotation of the mandible (without the need to actually lengthen the posterior facial height); and sufficient bone contact across each osteotomy site (see Fig. 27-8, C). Stabilization at each mandibular osteotomy site is accomplished with titanium plates and screws as needed. Interposed (e.g., autogenous iliac) corticocancellous bone graft is used when required.
For the Type IIB and Type III rib or fibula graft-constructed mandible that was created earlier during the patient’s life, splitting in the ramus region at the time of definitive reconstruction is preferred when feasible. The degree of bone contact across the osteotomy site after the advancement of the distal mandible is generally limited in these circumstances. The need for an interpositioned corticocancellous bone graft (e.g., autogenous iliac) should be anticipated (see Chapter 28).
For most patients with TCS, DO osteogenesis techniques have failed to demonstrate lower morbidity or improved soft-tissue response as compared with standard osteotomies.* Data have indicated significant relapse during the consolidation phase of DO techniques and no ongoing jaw growth when the procedure is carried out during childhood. Kaban and colleagues and others continue to refine the application of curvilinear DO for the correction of the complex mandibular deformities of severe forms of TCS.69 Despite their best efforts when using DO techniques, reliable three-dimensional vector control to achieve preferred proportions and symmetry of the osteotomized skeletal segments remains a work in progress. For these reasons, until DO technology and its clinical applications improve, we prefer the standard orthognathic correction of the maxillomandibular imbalance in patients with TCS when feasible.
Nasal Reconstruction
The bridge of the nose in the adult with TCS often has mild to moderate increased width, and a mid-dorsal hump is always present. The length of the nose is usually normal, but it seems long as a result of the imbalance with the upper and lower skeletal thirds of the face; Also, the tip often droops and lacks the preferred projection. Most patients benefit from a rhinoplasty that includes osteotomies with in-fracture; the conservative reduction of the skeletal and cartilaginous dorsal hump; the removal of the cephalic portions of the lower lateral cartilages; and septal cartilage grafting (i.e., a caudal strut) sutured to the anterior septum to improve tip projection and definition. To achieve the most favorable aesthetic results, it is best to postpone the rhinoplasty until after effective orthognathic procedures have been performed. Exposure is provided through an open (i.e., columella-splitting incision) technique (see Chapter 38 and Fig. 27-9).
Facial Soft-Tissue Reconstruction
Despite several decades of well-intentioned surgical attempts to improve the soft-tissue deficiencies of the eyelid–adnexal regions, few aesthetically pleasing soft-tissue eyelid reconstructive results in patients with TCS have been reported.64,82,83,129,135,143,148,156 The transposition of pedicled upper-eyelid-skin–muscle flaps to deficient regions of the lower eyelid is not technically difficult; unfortunately, the adnexal scarring that inevitably occurs often results in an “operated” look that generally detracts from any positive advantages of the procedure. The placement of full-thickness skin grafts to the lower eyelids is another frequently mentioned option. It predictably leaves a “patchy” look, and it should be reserved only for patients with recalcitrant corneal exposure problems. The lateral canthi are inferiorly displaced as a result of both orbital dystopia and hypoplasia of the lateral canthi and other components of the eyelid structures. Although the correction of the orbital dystopia and direct lateral canthopexies are helpful and should be carried out, they rarely normalize the multilevel soft-tissue hypoplasia of the specialized adnexal structures (i.e., skin, cartilage, eyelashes, ligament, fascia, and tendon). For these reasons, it is best to consult with an experienced oculoplastic surgeon when considering even minor eyelid procedures for patients with TCS.
When the procedure is meticulously designed and expertly executed, the transfer of soft-tissue flaps from other body regions, with microvascular reanastomosis, should be considered for reconstruction of TCS.64,82,83,129,135,143,148,156 When this level of facial soft tissue replacement is required, parascapular free flaps have become the workhorse since they were first described by Dos Santos. Siebert and colleagues found that the parascapular flap allows for the reasonable correction of contour defects in the lateral aspects of the face; this yields improved aesthetic results in selected patients with TCS by providing vascularized soft tissue in the subcutaneous plane while minimizing scarring.135 Fortunately, hypoplasia of the centrally located soft tissues of the face (i.e., forehead, nose, upper and lower lips, chin, and submental region) is not seen in patients with TCS. Dermal fat grafts, autogenous fat injections, and other forms of soft-tissue augmentation have gained popularity. The results of autogenous fat injection are variable and considered technique sensitive; they are generally not harmful, and they are clearly promising. Tanna and colleagues investigated the use of serial autologous fat grafting to restore soft-tissue contour in patients with hemifacial microsomia.144 Patients with moderate to severe hemifacial microsomia were divided into two groups: Group I included those undergoing microvascular free-flap reconstruction (i.e., inframammary extended circumflex scapular flaps; n = 10); Group II included those undergoing multiple staged autogenous fat grafting (n = 21). The two patient groups had similar OMENS scores (2.4 and 2.3, respectively) and similar pre-reconstruction facial symmetry scores (74% and 75%, respectively). At the final evaluation, facial symmetry scores were 121% for the microvascular free-flap group and 99% for the fat-grafting group. Furthermore, no statistically significant difference between the microvascular and fat-graft groups in either patient or physician rating of overall satisfaction was noted. The extrapolation of these study results to the individual with TCS is appropriate.52,73,144
External Ear Reconstruction
The surgical reconstruction of the auricle can be satisfactorily achieved through a staged approach in the hands of a few experts.12–15,19,40,74,75,86,111,130,140,145,146,155,162 The methods described by Brent represent the reference standard for external ear reconstruction.12–15 The successful grafting of a well-sculpted cartilage framework is the foundation for sound auricular repair. According to Brent, most children have adequate rib cartilage for the repair by 6 years of age. At that time, the synchondrotic region of ribs 6 and 7 will provide an ample cartilage block to form the framework of the ear; this is taken from the side contralateral to the ear being constructed. With the use of the preoperatively determined measurements and a prefabricated template, the position of the ear is determined, and a small preauricular incision is made. Unusable vestigial cartilage is excised. A thin-skinned pocket is developed, with the dissection carried beyond the marked auricular outline to recruit sufficient tension-free skin coverage. For patients with bilateral microtia, Brent prefers that the harvesting of cartilage grafts for each ear be separated by several months. Simultaneous bilateral auricular reconstruction would necessitate bilateral chest wounds with severe splinting and the potential for respiratory distress. Other stages of the auricular construction include lobule transposition; the detachment of the auricle with a skin graft; the management of the hairline; and tragus construction. Brent suggests that bilateral microtia repair be completed with five procedures that are carried out at 3-month intervals.
External Auditory Canal and Middle-Ear Reconstruction
Although some of the hearing loss in the patient with TCS is attributable to external auditory canal stenosis or atresia, most of the loss (i.e., an average of 44 dB) is attributable to hypoplastic middle-ear cavities and ankylotic or missing ossicles.18,58,66 Ankylosed or non-functioning ossicles appear to limit hearing conduction to the same extent as if they were absent.55 Attempting reconstruction of the middle ear in patients with TCS can be expected to result in gains in hearing. However, even if the ossicles are mobilized, middle-ear dysmorphology remains; this results in residual hearing loss and leaves the patient dependent on some form of amplification. Jahrsdoerfer reported on a group of patients with TCS who underwent surgery for hearing rehabilitation.66 He confirmed the generally unsuccessful attempts to satisfactorily improve “non-aided” long-term hearing. A form of bone-conduction hearing aid is required for proper amplification in most patients, including those who have undergone reconstruction as well as those who have not. The bone-conduction hearing aid may be a traditional unit, a modified amplification system, or a bone-anchored conduction unit.
According to Brent, for the gains from any middle-ear surgery to outweigh the risks and complications of the procedure, it should be reserved for selected patients with bilateral and unilateral microtia in whom personal motivation is high, in whom the favorable radiologic evidence of middle-ear development is present, and in whom a favorable outcome is expected according to an experienced otologist.12–15 Furthermore, it must be planned through a team approach with an otologist and an auricular/reconstructive surgeon who are competent and experienced with ear-canal atresia, middle-ear malformations, and microtia repair. At the time of middle-ear surgery, the auricular/reconstructive surgeon initiates the procedure by lifting the reconstructed ear from its bed while carefully preserving both the connective tissue on the framework’s undersurface and the overall circulation of the flap. The otologist proceeds by drilling a bony external auditory canal, completing ossiculoplasty, and then repairing the tympanum with a temporal fascia graft. The auricular/reconstructive surgeon then excises the soft tissues to exteriorize the meatus through the chondral region and harvests a skin graft that the otologist uses to line the new external auditory canal.
The Bone-Anchored Hearing Aid Option
Colquitt and colleagues completed a systematic review and an economic evaluation of BAHAs for people who are bilaterally deaf.23 Previous prospective studies of adults or children with bilateral hearing loss were eligible for review. Twelve studies were included; there were seven cohort pre-post studies and five cross-sectional audiologic comparison studies. Comparisons were made between BAHAs and the following: 1) conventional hearing aids, which are also known as air-conduction hearing aids (ACHAs); 2) bone-conduction hearing aids (BCHAs); and 3) unaided hearing and ear surgery. The study also looked at 4) unilateral versus bilateral BAHAs. Outcomes reviewed included hearing measures, validated measures of quality of life, adverse events, and measures of cost-effectiveness.
Janssen and colleagues completed a systematic review of bilateral BAHAs for bilateral permanent conductive hearing loss.68 This was a review of the literature from between 1977 and 2011, and it included studies in which subjects of any age had permanent conductive hearing loss and bilateral implanted BAHAs. Eleven studies met the criteria for data analysis. Bilateral BAHAs were found to provide audiologic benefit as compared with unilateral BAHAs (i.e., improved thresholds for tones, speech in quiet and in noise, improved localization/lateralization, and patient’s perceived subjective benefit). Disadvantages of bilateral BAHAs included 1) listening in noise in some condition; 2) presumed additional cost; and 3) presumed increasing adverse event risk. The authors concluded that bilateral BAHAs provided additional objective and subjective benefit as compared with unilateral BAHAs.
References
1. Acosta, H, Stelnicki, E, Boyd, B, et al. Vertical mesenchymal distraction and bilateral free fibula transfer for severe Treacher Collins syndrome. Plast Reconstr Surg. 2004; 113:1209.
2. Anderson, P, Netherway, D, Abbott, A, et al. Mandibular lengthening by distraction for airway obstruction in Treacher-Collins syndrome: The long-term results. J Craniofac Surg. 2004; 15:47.
3. Argenta, LC, Iacobucci, JJ. Treacher Collins syndrome: Present concepts of the disorder and their surgical correction. World J Surg. 1989; 13:401.
4. Arvystas, M, Shprintzen, RJ. Craniofacial morphology in Treacher Collins syndrome. Cleft Palate J. 1991; 28:226.
5. Arvytas, AH, Netherway, DJ, David, DJ, Brown, T. Application and comparison of techniques for three-dimensional analysis of craniofacial anomalies. J Craniofac Surg. 1990; 1:119–134.
6. Arystas, M, Shprintzen, RJ. Craniofacial morphology in Treacher Collins syndrome. Cleft Palate Craniofac J. 1991; 28:226.
7. Baas, EM, Horsthuis, RBG, Lange, JD. Subjective alveolar nerve function after bilateral sagittal split osteotomy or distraction osteogenesis of mandible. J Oral Maxillofac Surg. 2012; 70:910–918.
8. Beaune, L, Forrest, CR. Adolescents’ perspectives on living and growing up with Treacher Collins syndrome: A qualitative study. Cleft Palate Craniofac J. 2004; 41:343.
9. Behrents, RG, McNamara, JA, Avery, JK. Prenatal mandibulofacial dysostosis (Treacher Collins syndrome). Cleft Palate J. 1977; 14:13.
10. Berry, GA. Note on a congenital defect (coloboma) of the lower lid. R Lond Ophthalmic Hosp Rep. 1889; 12:255.
11. Bregeat, P. Thirty-six years after a mandibulofacial dysostosis operation. Ophthalmic Paediatr Genet. 1985; 6:313.
12. Brent, B. Auricular repair using autogenous rib cartilage grafts: Two decades of experience with 600 cases. Plast Reconstr Surg. 1992; 90:355.
13. Brent, B. Advances in ear reconstruction with autogenous rib cartilage grafts: Personal experience with 1200 cases. Plast Reconstr Surg. 1999; 104:319.
14. Brent, B. The team approach to treating the microtia atresia patient. Otolaryngol Clin North Am. 2000; 33:1353.
15. Brent, B. Microtia repair with rib cartilage grafts: A review of personal experience with 1000 cases. Clin Plast Surg. 2002; 29:257.
16. Brevi, B, Lagana, F, Piazza, F, Sesenna, E. Mandibular distraction osteogenesis with a small semiburied device in neonates: Report of 2 cases. Ear Nose Throat J. 2006; 85:102.
17. Burstein, FD, Cohen, SR, Scott, PH, et al. Surgical therapy for severe refractory sleep apnea in infants and children: Application of the airway zone concept. Plast Reconstr Surg. 1995; 96:34.
18. Caldarelli, DD, Hutchinson, JG, Jr., Pruzansky, S, et al. A comparison of microtia and temporal bone anomalies in hemifacial microsomia and mandibulofacial dysostosis. Cleft Palate J. 1980; 17:103.
19. Cao, Y, Vacanti, JP, Paige, KT, et al. Transplantation of chondrocytes utilizing a polymer-cell construct to produce tissue-engineered cartilage in the shape of a human ear. Plast Reconstr Surg. 1997; 100:297.
20. Clauser, L, Curioni, C, Spanio, S. The use of the temporalis muscle flap in facial and craniofacial reconstructive surgery: A review of 182 cases. J Craniomaxillofac Surg. 1995; 23:203.
21. Codvilla, A. On the means of lengthening, in the lower limbs, the muscles and tissues which are shortened through deformity. Am J Orthop Surg. 1905; 2:353.
22. Collins, ET. Cases with symmetrical congenital notches in the outer part of each lower lid and defective development of malar bones. Trans Ophthalmol Soc UK. 1900; 20:190.
23. Colquitt, JL, Jones, J, Harris, P, et al. Bone anchored hearing aids (BAHAs) for people who are bilaterally deaf: a systematic review and economic evaluation. Health Technol Assess. 2011; 15(26):1–200.
24. Converse, JM, Wood-Smith, D, McCarthy, JG, et al. Bilateral facial microsomia. Plast Reconstr Surg. 1974; 54:413.
25. Cope, JB, Samchukov, ML, Cherkashin, AM. Mandibular distraction osteogenesis: A historic perspective and future directions. Am J Orthod Dentofacial Orthop. 1999; 115:448–460.
26. Da Silva Dalben, G, Costa, B, Gomide, MR. Prevalence of dental anomalies, ectopic eruption and associated oral malformations in subjects with Treacher Collins syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006; 101:588.
27. Da Silva Dalben, G, Teixeira das Neves, L, Ribeiro Gomide, M. Oral health status of children with Treacher Collins syndrome. Spec Care Dentist. 2006; 26:71–75.
28. Dahl, E, Kreiborg, S, Bjork, A. A morphologic description of a dry skull with mandibulofacial dysostosis. Scand J Dent Res. 1975; 83:257.
29. Daniels, S, Ellis, E, III., Carlson, DS. Histologic analysis of costochondral and sternoclavicular grafts in the TMJ of the juvenile monkey. J Oral Maxillofac Surg. 1987; 45:675.
30. Dixon, J, Edwards, SJ, Anderson, I, et al. Identification of the complete coding sequence and genomic organization of the Treacher Collins syndrome gene. Genome Res. 1997; 7:223.
31. Ebata, T, Nishiki, S, Masuda, A, et al. Anaesthesia for Treacher Collins syndrome using a laryngeal mask airway. Can J Anaesth. 1991; 38:1043.
32. Edwards, S, Gladwin, A, Dixon, MJ. The mutational spectrum in Treacher Collins syndrome reveals a predominance of mutations that create a premature-termination codon. Am J Hum Genet. 1997; 60:515.
33. Ellis, E, III., Carlson, DS. Histologic comparison of the costochondral, sternoclavicular, and temporomandibular joints during growth in Macaca mulatta. J Oral Maxillofac Surg. 1986; 44:312.
34. Ellis, PE, Dawson, M, Dixon, MJ. Mutation testing in Treacher Collins syndrome. J Orthod. 2002; 29:293.
35. Farkas, LG, Posnick, JC. Detailed morphology of the nose in patients with Treacher Collins syndrome. Ann Plast Surg. 1989; 22:211.
36. Farkas, LG, Posnick, JC, Winemaker, MJ. Orbital protrusion in Treacher Collins syndrome: a tool for determining the degree of soft tissue damage. Dtsch Z Mund Kiefer Gesichtschir. 1989; 13(6):429–432.
37. Fan, K, Andrews, BT, Liao, E, et al. Protection of the temporomandibular joint during syndromic neonatal mandibular distraction using condylar unloading. Plast Reconstr Surg. 2012; 129:1151–1161.
38. Fernandez, AO, Ronis, ML. The Treacher Collins syndrome patient. Arch Otolaryngol. 1964; 80:505.
39. Figueroa, AA, Peterson-Falzone, SJ, Friede, H, et al. Neurocranial morphology in mandibulofacial dysostosis (Treacher Collins syndrome). Cleft Palate Craniofac J. 1993; 30:369.
40. Firmin, F. Ear reconstruction in cases of typical microtia: Personal experience based on 352 microtic ear corrections. Scand J Plast Reconstr Surg Hand Surg. 1998; 32:35.
41. Franceschetti, A, Klein, D. The mandibulofacial dysostosis: A new hereditary syndrome. Acta Ophthalmol. 1949; 27:143.
42. Francis-West, PH, Robson, L. Craniofacial development: The tissue and molecular interactions that control development of the head. Adv Anat Embryol Cell Biol. 2003; 169:1.
43. Freihofer, HPM. Four-step mandibular lengthening to correct a bird-face deformity. Acta Stomatol Belg. 1990; 87:189.
44. Freihofer, HPM. Variations in the correction of Treacher Collins syndrome. Plast Reconstr Surg. 1997; 99:647.
45. Freihofer, HPM, Borstlap, WA. Reconstruction of the zygomatic area. J Craniomaxillofac Surg. 1989; 17:43.
46. Fryns, JP, Bonhomme, A, Van Den Berge, H. Nager acrofacial dysostosis: An adult male with severe neurologic deficit. Genet Couns. 1996; 7:147.
47. Fuente del Campo, A, Martinez Elizondo, M, Arnaud, E. Treacher Collins syndrome (mandibulofacial dysostosis). Clin Plast Surg. 1994; 21:613.
48. Garcia-Cimbrelo, E, Olsen, B, Ruiz-Yague, M, et al. Ilizarov technique: Results and difficulties. Clin Orthop. 1992; 283:116.
49. Garner, LD. Cephalometric analysis of Berry-Treacher Collins syndrome. Oral Surg. 1967; 23:320.
50. Giugliani, R, Pereira, CH. Nager acrofacial dysostosis with thumb duplication: Report of a case. Clin Genet. 1984; 26:228.
51. Gorlin, RJ, Cohen, MM, Jr., Levin, LS. Syndromes of the head and neck, ed 3. New York: Oxford University Press; 1990.
52. Grahovac, TL, Rubin, JP. Discussion: An analysis of the experiences of 62 patients with moderate complications after full-face fat injection for augmentation. Plast Reconstr Surg. 2012; 129:1369.
53. Heller, JB, Gabbay, JS, Kwan, D, et al. Genioplasty distraction osteogenesis and hyoid advancement for correction of upper airway obstruction in patients with Treacher Collins and Nager syndromes. Plast Reconstr Surg. 2006; 117:2389.
54. Hennig, TB, Ellis, E, III., Carlson, DS. Growth of the mandible following replacement of the mandibular condyle with the sternal end of the clavicle: An experimental investigation in Macaca mulatta. J Oral Maxillofac Surg. 1992; 50:1196.
55. Herberts, G. Otological observations on the “Treacher Collins syndrome. ”. Acta Otolaryngol. 1962; 54:457.
56. Herman, TE, Siegel, MJ. Special imaging case-book: Nager syndrome. J Perinatol. 1998; 18:85.
57. Herring, SW, Rowlett, UF, Pruzansky, S. Anatomical abnormalities in mandibulofacial dysostosis. Am J Med Genet. 1979; 3:225.
58. Holborow, JC, Jr., Caldarelli, DD, Valvassori, GE, et al. The otologic manifestations of mandibulofacial dysostosis. Trans Sect Otolaryngol Am Acad Ophthalmol Otolaryngol. 1977; 84:520.
59. Hopper, RA, Altug, AT, Grayson, BH, et al. Cephalometric analysis of the consolidation phase following bilateral pediatric mandibular distraction. Cleft Palate Craniofac J. 2003; 40:233.
60. Hurwitz, DJ. Long-term results of vascularized cranial bone grafts. J Craniofac Surg. 1994; 5:237.
61. Ilizarov, GA. The principles of the Ilizarov method. Bull Hosp Joint Dis Orthop Inst. 1988; 48:1.
62. Jabs, EW, Li, X, Lovett, M, et al. Genetic and physical mapping of the Treacher Collins syndrome locus with respect to loci in the chromosome 5q3 region. Genomics. 1993; 18:7.
63. Jabs, EW, Xiang, L, Coss, CA, et al. Mapping the Treacher Collins syndrome locus to 5q31. 3–q33. 3. Genomics. 1991; 11:193.
64. Jackson, IT. Reconstruction of the lower eyelid defect in Treacher Collins syndrome. Plast Reconstr Surg. 1981; 67:365.
65. Jackson, IT, Bauer, B, Salech, J, et al. A significant feature of Nager syndrome: Palatal agenesis. Plast Reconstr Surg. 1989; 84:219.
66. Jahrsdoerfer, RA, Aquilar, EA, Yeakley, JW, et al. Treacher Collins syndrome: An otologic challenge. Ann Otol Rhinol Laryngol. 1989; 98:807.
67. James, D, Ma, L. Mandibular reconstruction in children with obstructive sleep apnea due to micrognathia. Plast Reconstr Surg. 1997; 100:1131.
68. Janssen, RM, Hong, P, Chadha, NK. Bilateral bone-anchored hearing aids for bilateral permanent conductive hearing loss: a systematic review. Otolaryngol Head Neck Surg. 2012; 147:412–422.
69. Kaban, LB, Moses, ML, Mulliken, JB. Surgical correction of hemifacial microsomia in the growing child. Plast Reconstr Surg. 1980; 82:9.
70. Kaban, LB, Seldin, EB, Kikinis, R, et al. Clinical application of curvilinear distraction osteogenesis for correction of mandibular deformities. J Oral Maxillofac Surg. 2009; 67:996–1008.
71. Karp, NS, Thorne, CHM, McCarthy, JG, et al. Bone lengthening in the craniofacial skeleton. Ann Plast Surg. 1990; 24:231.
72. Katsanis, S, Jabs, EW. Treacher Collins syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. SourceGeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle, 1993-2004.
73. Kim, SM, Kim, YS, Hong, JW, et al. An analysis of the experiences of 62 patients with moderate complications after full-face fat injection for augmentation. Plast Reconstr Surg. 2012; 129:1359.
74. Kobus, K, Szczyt, M, Latkowski, I, Wojcicki, P. Reconstruction of the auricle. Br J Plast Surg. 2002; 55:645.
75. Kobus, K, Wojcicki, P. Surgical treatment of Treacher Collins syndrome. Ann Plast Surg. 2006; 56:549.
76. Kolar, JC, Farkas, LG, Munro, IR. Surface morphology in Treacher Collins syndrome: An anthropometric study. Cleft Palate J. 1985; 22:266.
77. Kovacs, AL. Use of the Augustine stylet anticipating difficult tracheal intubation in Treacher Collins syndrome. J Clin Anaesth. 1992; 4:409.
78. Kreiborg, S. The skeletal anatomy of mandibulofacial dysostosis (Treacher Collins syndrome) [discussion]. Plast Reconstr Surg. 1986; 78:469.
79. Kreiborg, S, Dahl, E. The cranial base and face in mandibulofacial dysostosis. Am J Med Genet. 1993; 47:753.
80. Gui, L, Zhang, Z, Zang, M, et al. Restoration of facial symmetry in hemifacial microsomia with mandibular outer cortex bone grafting combined with distraction osteogenesis. Plast Reconstr Surg. 2011; 127:1997–2004.
81. Longacre, JJ, deStefano, GA, Holmstrand, KE. The surgical management of the first and second branchial arch syndromes. Plast Reconstr Surg. 1963; 31:507.
82. Longaker, MT, Flynn, A, Siebert, JW. Microsurgical correction of bilateral facial contour deformities. Plast Reconstr Surg. 1996; 98:951.
83. Longaker, MT, Siebert, J. Microsurgical correction of facial contour in congenital craniofacial malformations: The marriage of hard and soft tissue. Plast Reconstr Surg. 1996; 98:942.
84. Maran, AGD. The Treacher Collins syndrome patient. J Laryngol. 1964; 78:135.
85. Marres, HA, Cremers, CW, Dixon, MJ, et al. The Treacher Collins syndrome: A clinical, radiological, and genetic linkage study on two pedigrees. Arch Otolaryngol Head Neck Surg. 1995; 121:509.
86. Marres, HA, Cremers, CW, Marres, EH. Treacher Collins syndrome: Management of major and minor anomalies of the ear. Rev Laryngol Otol Rhinol. 1995; 116:105.
87. Marsh, JL, Celin, SE, Vannier, MW, et al. The skeletal anatomy of mandibulofacial dysostosis (Treacher Collins syndrome). Plast Reconstr Surg. 1986; 78:460.
88. Marsh, K, Dixon, J, Dixon, MJ. Mutations in the Treacher Collins syndrome gene lead to mislocalization of the nucleolar protein treacle. Hum Mol Genet. 1998; 7:1795.
89. Marszalek, B, Wojckicki, P, Kazimierz, K, et al. Clinical features, treatment and genetic background of Treacher Collins syndrome. J Appl Genet. 2002; 43:223.
90. McCarthy, JG, Schreiber, J, Karp, N, et al. Lengthening the human mandible by gradual distraction. Plast Reconstr Surg. 1992; 89:1.
91. Molina, F. A CT scan technique for quantitative volumetric assessment of the mandible after distraction osteogenesis [discussion]. Plast Reconstr Surg. 1997; 99:1248.
92. Molina, F, Ortiz-Monasterio, F. Mandibular elongation and remodeling by distraction: A farewell to major osteotomies. Plast Reconstr Surg. 1995; 96:825.
93. Moore, MH, Guzman-Stein, G, Proudman, TW, et al. Mandibular lengthening by distraction for airway obstruction in Treacher Collins syndrome. J Craniofac Surg. 1994; 5:22.
94. Moos, KF. Mandibular reconstruction in children with obstructive sleep apnea due to micrognathia [discussion]. Plast Reconstr Surg. 1997; 100:1138.
95. Mukhopadhyay, P, Mukherjee, P, Adhikary, M. Problems in the anaesthetic management of Pierre Robin and Treacher Collins syndrome. Indian Pediatr. 1992; 29:1120.
96. Nager, FR, deRaynier, JP. Das gehororgan bei den angeborenen kopf-missbildungen. Pract Otorhinolaryngol. 1948; 10(Suppl 2):1.
97. Obwegeser, HL. Zur korrektur der dysostosis otomandibularis. Schweiz Monatsschr Zahnheilkd. 1970; 80:331.
98. Obwegeser, HL. Correction of skeletal anomalies of oromandibular dysostosis. J Maxillofac Surg. 1974; 2:73.
99. Obwegeser, HL, Hadjiianghelou, O. Two ways to correct bird face deformity. Oral Surg. 1987; 64:507.
100. Opitz, C, Ring, P, Stoll, C. Orthodontic and surgical treatment of patients with congenital unilateral and bilateral mandibulofacial dysostosis. J Orofac Orthop. 2004; 65:150.
101. Perkins, JA, Sie, KCY, Milczuk, H, et al. Airway management in children with craniofacial anomalies. Cleft Palate Craniofac J. 1997; 34:135.
102. Peterson-Falzone, S, Figueroa, AA. Longitudinal changes in cranial base angulation in mandibulofacial dysostosis. Cleft Palate J. 1989; 26:31.
103. Pope, AW, Speltz, ML. Research on psychosocial issues of children with craniofacial anomalies: Progress and challenges. Cleft Palate Craniofac J. 1997; 34:371.
104. Pope, AW, Ward, J. Self-perceived facial appearance and psychosocial adjustment in preadolescents with craniofacial anomalies. Cleft Palate Craniofac J. 1997; 34:396.
105. Posnick, JC. Occlusal plane rotation: aesthetic enhancement in mandibular micrognathia [discussion]. Plast Reconstr Surg. 1993; 91:1241.
106. Posnick, JC. Treacher Collins syndrome. In: Aston SJ, Beasley RW, Thorne CAM, eds. Grabb and Smith plastic surgery, ed 5. Philadelphia: Lippincott-Raven; 1997:313–319.
107. Posnick, JC. Treacher Collins syndrome: Perspectives in evaluation and treatment. J Maxillofac Surg. 1997; 155(10):1120–1133.
108. Posnick, JC. Discussion: Midface growth after costochondral graft reconstruction of the mandibular ramus in hemifacial microsomia. J Oral Maxillofac Surg. 1998; 56:127–128.
109. Posnick, JC, Treacher Collins syndrome: Evaluation and treatment. Craniofacial and maxillofacial surgery in children and young adults. Posnick, JC, eds. Craniofacial and maxillofacial surgery in children and young adults; Vol I. WB Saunders Co, Philadelphia, 2000:391–418.
110. Posnick, JC, Al-Qattan, MM, Moffat, S, Armstrong, D. Cranio-orbito-zygomatic measurements from standard CT scans in unoperated Treacher Collins syndrome patients: comparison with normal controls. Cleft Palate Craniofac J. 1995; 32(1):20–24.
111. Posnick, JC, Al-Qattan, MM, Whitaker, LA. Assessment of the preferred vertical position of the ear. Plast Reconstr Surg. 1993; 91(7):1198–1203.
112. Posnick, JC, Fantuzzo, J, Orchin, J. Deliberate operative rotation of the maxillo-mandibular complex to alter the A-point to B-point relationship for enhanced facial esthetics. J Oral Maxillofac Surg. 2006; 64:1687–1695.
113. Posnick, JC, Goldstein, JA, Waitzman, A. Surgical correction of the Treacher Collins malar deficiency: quantitative CT scan analysis of long-term results. Plast Reconstr Surg. 1993; 92(1):12–22.
114. Posnick, JC, Ruiz, R. Treacher Collins syndrome: current evaluation, treatment and future directions. Cleft Palate Craniofac J. 2000; 37(5):434.
115. Posnick, JC, Ruiz, R, Tiwana, P. Treacher Collins syndrome: comprehensive evaluation and treatment. Oral Maxillofac Surg Clin North Am. 2004; 16:503–523.
116. Poswillo, DE. Otomandibular deformity: pathogenesis as a guide to reconstruction. J Maxillofac Surg. 1974; 2:64.
117. Poswillo, DE. The pathogenesis of the Treacher Collins syndrome (mandibulofacial dysostosis). Br J Oral Surg. 1975; 13:1.
118. Pron, G, Galloway, C, Armstrong, D, Posnick, JC. Ear malformation and hearing loss in patients with Treacher Collins syndrome. Cleft Palate Craniofac J. 1993; 30:97–103.
119. Pruzansky, S. Not all dwarfed mandibles are alike. Birth Defects. 1969; 1:120.
120. Rasch, DK, Browder, F, Barr, M, et al. Anaesthesia for Treacher Collins and Pierre Robin syndromes: a report of three cases. Can Anaesth Soc J. 1986; 33:364.
121. Raulo, Y. Treacher Collins syndrome: analysis, principles of treatment. In: Caronni EP, ed. Craniofacial surgery. Boston: Little, Brown, 1985.
122. Raulo, Y, Tessier, P. Mandibulofacial dysostosis. Scand J Plast Reconstr Surg. 1981; 15:251.
123. Raustia, A, Pernu, H, Pyhtinen, J, et al. Clinical and computed tomographic findings in costochondral grafts replacing the mandibular condyle. J Oral Maxillofac Surg. 1996; 54:1393.
124. Roberts, FG, Pruzansky, S, Aduss, H. An x-radiocephalometric study of mandibulofacial dysostosis in man. Arch Oral Biol. 1975; 20:265.
125. Roddi, R, Vaandrager, J, van der Meulen, J. Treacher Collins syndrome: early surgical treatment of orbitomalar malformations. J Craniofac Surg. 1995; 6:211.
126. Rogers, BO. Berry-Treacher Collins syndrome: a review of 200 cases. Br J Plast Surg. 1964; 17:107.
127. Rosen, HM. Occlusal plane rotation: aesthetic enhancement in mandibular micrognathia. Plast Reconstr Surg. 1993; 91:1231.
128. Rotten, D, Levaillant, JM, Martinez, H, et al. The fetal mandible: a 2D and 3D sonographic approach to the diagnosis of retrognathia and micrognathia. Ultrasound Obstet Gynecol. 2002; 19:122.
129. Saadeh, P, Reavey, PL, Siebert, JW. A soft-tissue approach to midfacial hypoplasia associated with Treacher Collins syndrome. Ann Plast Surg. 2006; 56:522–525.
130. Saadeh, PB, Brent, B, Mehrara, BJ, et al. Human cartilage engineering: chondrocyte extraction, proliferation, and characterization for construct development. Ann Plast Surg. 1999; 42:509.
131. Sengezer, M. Mandibular lengthening by gradual distraction [letter]. Plast Reconstr Surg. 1993; 92:372.
132. Shprintzen, RJ. Palatal and pharyngeal anomalies in craniofacial syndromes. Birth Defects. 1982; 18:53–78.
133. Shprintzen, RJ, Croft, C, Berkman, MD, Rakoff, SJ. Pharyngeal hypoplasia in Treacher Collins syndrome. Arch Otolaryngol. 1979; 105:127–131.
134. Sidman, J, Sampson, D, Templeton, B. Distraction osteogenesis of the mandible for airway obstruction in children. Laryngoscope. 2001; 111:1137.
135. Siebert, J, Anson, G, Longaker, M. Microsurgical correction of facial asymmetry in 60 consecutive cases. Plast Reconstr Surg. 1996; 97:354.
136. Silla Freitas, R, Tolazzi, ARD, Alonso, N, Cruz, GA. Evaluation of molar teeth and buds in patients submitted to mandibular distraction: Long-term results. J Plast Reconstr Surg. 2010; 121:1335–1342.
137. Singh, D, Bartlett, S. Congenital mandibular hypoplasia: analysis and classification. J Craniofac Surg. 2005; 16:291.
138. Snyder, CC. Bilateral facial agenesis (Treacher Collins syndrome). Am J Surg. 1956; 92:81.
139. Snyder, CC, Levine, GA, Swanson, HM, et al. Mandibular lengthening by gradual distraction. Plast Reconstr Surg. 1973; 51:506.
140. Somers, T, De Cubber, J, Govaerts, P, Offeciers, FE. Total auricular repair: Bone anchored prosthesis or plastic reconstruction? Acta Otorhinolaryngol Belg. 1998; 52:317.
141. Stelnicki, E, Boyd, J, Nott, R, et al. Early treatment of severe mandibular hypoplasia with distraction mesenchymogenesis and bilateral free fibula flaps. J Craniofac Surg. 2001; 12:337.
142. Stelnicki, E, Lin, W, Lee, C. Long-term outcome study of bilateral mandibular distraction: A comparison of Treacher Collins and Nager syndromes to other types of micrognathia. Plast Reconstr Surg. 2002; 109:1819.
143. Stenstrom, SJ, Sundmark, ES. Contribution to the treatment of the eyelid deformities in dysostosis mandibulofacialis. Plast Reconstr Surg. 1966; 38:567.
144. Tanna, N, Wan, DC, Kawamoto, HK, Bradley, JP. Craniofacial microsomia soft-tissue reconstruction comparison: Inframammary extended circumflex scapular flap versus serial fat grafting. Plast Reconstr Surg. 2011; 127:802.
145. Tanzer, RC. Total reconstruction of the external ear. Plast Reconstr Surg. 1959; 23:1.
146. Tanzer, RC. Total reconstruction of the auricle: the evolution of a plan of treatment. Plast Reconstr Surg. 1971; 47:523.
147. Teber, OA, Gillessen-Kaesbach, G, Fischer, S, et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. 2004; 12:879–890.
148. Tessier, P. Chirurgische behandlung der genetisch bedingten palpebralen und orbito-facialen missbildungen. Klin Monatschr Augenheilkd. 1968; 50:82.
149. Tessier, P. Autogenous bone grafts taken from the calvarium for facial and cranial application. Clin Plast Surg. 1982; 9:531.
150. Tessier, P. Facial contouring in the Treacher Collins-Franceschetti syndrome. In: Brent B, ed. The artistry of reconstructive surgery. St. Louis: CV Mosby; 1987:343.
151. Tessier, P. Complications associated with the harvesting of cranial bone grafts [discussion]. Plast Reconstr Surg. 1995; 95:14.
152. Tessier, P, Tulasne, J. Treacher Collins syndrome. In: Marchac D, ed. Craniofacial surgery. New York: Springer-Verlag, 1985.
153. Tessier, P, Tulasne, JF. Stability in the correction of hypertelorbitism and Treacher Collins syndromes. Clin Plast Surg. 1989; 16:195.
154. The Treacher Collins Syndrome Collaborative Group. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat Genet. 1996; 12:130.
155. Tollefson, TT. Advances in the treatment of microtia. Curr Opin Otolaryngol Head Neck Surg. 2006; 14:412.
156. Van der Meulen, JCH, Hauben, DJ, Vaandrager, JM, et al. The use of a temporal osteoperiosteal flap for the reconstruction of malar hypoplasia in Treacher Collins syndrome. Plast Reconstr Surg. 1984; 74:687.
157. Vento, AR, LaBrie, RA, Mulliken, JB. The OMENS classification of hemifacial microsomia. Cleft Palate Craniofac J. 1991; 28:68–76.
158. Waitzman, AA, Posnick, JC, Armstrong, D, et al. Craniofacial skeletal measurements based on computed tomography: I. Accuracy and reproducibility. Cleft Palate Craniofac J. 1992; 29:112.
159. Waitzman, AA, Posnick, JC, Armstrong, D, et al. Craniofacial skeletal measurements based on computed tomography: II. Normal values and growth trends. Cleft Palate Craniofac J. 1992; 29:118.
160. Walker, JS, Dorian, RS, Marsh, NJ. Anaesthetic management of a child with Nager syndrome. Anaesth Analg. 1994; 79:1025.
161. Walker, MB, Trainor, PA. Craniofacial malformations: Intrinsic vs extrinsic neural crest cell defects in Treacher Collins and 22q11 deletion syndromes. Clin Genet. 2006; 69:471.
162. Walton, RL, Beahm, EK. Auricular reconstruction for microtia: Part II. Surgical techniques. Plast Reconstr Surg. 2002; 110:234.
163. Wan, D, Taub, P, et al. Distraction osteogenesis of costocartilaginous rib grafts and treatment algorithm for severely hypoplastic mandibles. Plast Reconstr Surg. 2011; 127:2005–2013.
164. Wijbenga, JG, Verlinden, CR, Jansma, J, et al. Long lasting neurosensory disturbance following advancement of the retrognathic mandible: distraction osteogenesis versus bilateral sagittal split osteotomy. Int J Oral Maxillofac Surg. 2009; 38:719.
165. Winokur, ST, Shiang, R. The Treacher Collins syndrome (TCOF1) gene product, treacle, is targeted to the nucleolus by signals in its C-terminus. Hum Mol Genet. 1998; 7:1947.
166. Wittenborn, W, Panchal, J, Marsh, J, et al. Neonatal distraction surgery for micrognathia reduces obstructive sleep apnea and the need for tracheotomy. J Craniofac Surg. 2004; 15:623.
167. Wolfe, SA, Vitenas, P, Jr. Malar augmentation using autogenous materials. Clin Plast Surg. 1991; 18:39.
168. Wolford, L. Occlusal plane alteration in orthognathic surgery–Part I: Effects on function and esthetics. Am J Orthod Dentofacial Orthop. 1994; 106(3):304–316.
169. Zanini, S, Viterbo, F, Parro, F, et al. Bone graft of the zygoma in a patient with Treacher Collins syndrome. J Craniofac Surg. 1994; 5:270.
*References 6, 9, 24, 26–28, 38, 41, 42, 46, 50, 51, 56, 57, 65, 78, 79, 84, 96, 116, 117, 126, 128, 138, 157
*References 1–3, 5, 16, 21, 25, 81, 97–100, 105–107, 109, 110, 112, 114, 115, 121, 122, 127, 153, 151
*References 17, 21, 47, 48, 53, 59, 61, 67, 70, 71, 90–93, 131, 134, 136, 139, 141, 142, 160, 166
*References 1, 2, 16, 29, 33, 38, 47, 48, 53, 61, 67, 70, 71, 90–94, 108, 123, 131, 136, 139, 141, 142
*References 1, 2, 16, 29, 33, 38, 47, 48, 53, 61, 67, 70, 71, 80, 90–94, 108, 123, 131, 136, 139, 141, 142, 163
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]27
Treacher Collins Syndrome
Evaluation and Treatment
• Inheritance, Genetic Markers, and Testing
• Considerations during Infancy and into Early Childhood
• Dysmorphology with Treacher Collins Syndrome
• Facial Growth Potential with Treacher Collins Syndrome
• Classification of Temporomandibular Joint and Mandibular Malformation
• Staging of Skeletal Reconstruction: Timing and Techniques
• Indications for First-Stage Mandibular Reconstruction in the Newborn
• Indications for First-Stage Mandibular Reconstruction during the Mixed Dentition
• Facial Soft-Tissue Reconstruction
Treacher Collins syndrome (TCS), which is also known as mandibulofacial dysostosis, is an autosomal dominant condition with variable expressivity.* It is generally characterized by bilaterally symmetric abnormalities of the structures within the first and second branchial arches. Early descriptions are attributed to Berry,10 Treacher Collins,51 and Franceschetti and Klein.41 To date, the physical findings of many hundreds of individuals and families with TCS have been published in the world literature.
The adult patient with fully expressed TCS has a convex horizontally deficient facial profile with a prominent nasal dorsum above a retrusive lower jaw and chin (Fig. 27-1). The eye region is characterized by an antimongoloid slant of the palpebral fissure as a result of colobomata and hypoplasia of the lower lids and the lateral canthi, including the partial absence of the eyelid cilia and inferolateral orbital hypoplasia and dystopia. Tongue-shaped processes of hair frequently extend into the preauricular region. The external ears are absent, malformed, or malposed and hearing is impaired as a result of variable degrees of hypoplasia of the external auditory canals and the ossicles of the middle ears. A characteristic finding is hypoplasia of the malar bones, often with clefting through the arches and limited formation of the residual zygomas, including the glenoid fossa component. The maxilla and mandible bones are also characteristically hypoplastic, with variable effects on the temporomandibular joints (TMJs), the muscles of mastication, and the muscles of facial expression. Interestingly, hypoplasia of the centrally located soft tissues of the face is not seen with TCS. In general, there is an Angle Class II anterior open-bite malocclusion and a steep clockwise-rotated maxillomandibular complex. The posterior facial height is short, and the anterior lower facial height is long. The A-point–to–B-point relationship in profile is consistent with the clockwise rotation of the jaws. The presence of cleft palate (with or without cleft lip and choanal atresia of the nasal cavity) is variable. Dental anomalies are present in 60% of individuals; these include tooth agenesis (33.3%), enamel opacities (20%), and the ectopic eruption of the maxillary first molars (13.3%).
Figure 27-1 An 11-year-old boy with fully expressed Treacher Collins syndrome. His eyes are characterized by an antimongoloid slant of the palpebral fissures as a result of colobomata and hypoplasia of the lower lids and the lateral canthi, including the partial absence of the eyelid cilia and inferolateral orbital dystopia. The appearance of the malpositioned adnexal structures is a reflection of both zygomatic–orbital dystopia and hypoplasia of the soft tissues. The maxilla and the mandible are also deficient and malformed. The patient demonstrates a Type IIA mandibular malformation. The external ears are malformed and lack antihelical folds. A, Frontal facial and three-dimensional computed tomography scan views. B, Oblique facial and three-dimensional computed tomography scan views. C, Profile facial and three-dimensional computed tomography scan views.
Individuals with TCS have a reduced cranial base angle (i.e., basilar kyphosis). Craniosynostosis is not a feature of TCS, but the neurocranium may have an abnormal shape (i.e., decreased anteroposterior length and diminished bitemporal width) that is evident during childhood and remains through adulthood. 110 The degree of craniofacial malformation present at birth is believed to be relatively stable and non-progressive with age (this is discussed in more detail later in this chapter). TCS should not be confused with similar entities such as oculo–auriculo–vertebral dysplasia or Goldenhar syndrome. Distinguishing facial features of patients with TCS often include coloboma of the lower eyelids; the upper eyelids are more likely affected among patients with Goldenhar syndrome.
Inheritance, Genetic Markers, and Testing
The occurrence of TCS is in the range of 1 in 25,000 to 1 in 50,000 live births. Inheritance is autosomal dominant, and male and female individuals are equally affected.51 About 60% of probands with TCS have the disorder as the result of a de novo gene mutation. Each child of an individual with TCS has a 50% chance of inheriting the mutation (Fig. 27-2). Teber and colleagues identified TCOF1 mutations in 28 out of 36 (78%) individuals with a clinically unequivocal diagnosis of TCS.147 The most frequent clinical findings were downward-slanting palpebral fissures, hypoplasia of the zygomatic complex, hypoplasia of the mandible, conductive deafness, any degree of microtia, and atresia of the external ear canal. Although there were interfamilial and intrafamilial variations that ranged from mild to severe, there were no genotype or phenotype correlations. Four clinically unaffected parents were heterozygous for the TCOF1 mutation. The authors concluded that modifying factors are important for phenotypic expression.
Figure 27-2 This mother and daughter demonstrate the extent of variation in the expression of Treacher Collins syndrome within a family. The mother was not aware that she carried the Treacher Collins gene until after the birth of her daughter. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997.
Genetic counseling for TCS is the process of providing individuals and families with information about the nature, inheritance, and implications of the syndrome to help them make informed medical and personal decisions. Molecular testing should be considered for any individual who presents with at least two major features or three minor features of TCS.30,32,34,62,63,72,85,88,89,154,161,165 Until recently, penetrance in TCS was believed to be complete. Marres and colleagues confirmed the absence of clinical and radiographic findings in an individual with a pathogenic TCOF1 mutation. For this reason, testing is acceptable for individuals with any degree of severity of TCS features to confirm or refute the diagnosis.
Considerations during Infancy and into Early Childhood
At the time of the birth of a child with TCS, concerns will center on the adequacy of the airway, swallowing, feeding, hearing, vision (corneal protection), the presence of cleft palate (with or without cleft lip), any other associated malformations, and the psychosocial well-being of the family.8,17,27,31,77,94,95,101,103,104,120,160 The airway may be compromised as a result of multiple factors. The first is maxillary hypoplasia, which occurs with a degree of choanal stenosis or atresia with the blockage of the nasal airway. Second, mandibular micrognathia with a retropositioned tongue that at least partially obstructs the oropharyngeal and hypopharyngeal spaces will be present. In addition, tracheomalacia, vocal cord anomalies, or neuromotor involvement may also negatively affect the upper airway. Depending on the severity of the anomalies, a spectrum of upper airway compromise may necessitate, at a minimum, special infant positioning, an extended hospital stay, and pulse oximetry monitoring. Surgical considerations for the management of airway compromise in the infant or young adult may include tongue–lip adhesion, the use of a custom-made oral appliance, immediate or delayed tracheostomy, or an urgent mandibular advancement procedure.
When a cleft of the secondary palate is documented, potential additive effects on the airway before and after repair must be considered. When Nager syndrome is present, the soft palate is both clefted and severely hypoplastic to the extent that the achievement of a functioning palate is not feasible.65,96 A primary pharyngeal flap—although helpful for velopharyngeal function—is rarely indicated, because further obstruction of the airway would likely result. Evaluation by a medical geneticist is indicated to clarify the diagnosis and all presenting anomalies, to discuss family planning issues, and to analyze chromosomal studies.
Dysmorphology with Treacher Collins Syndrome
Cranio–Orbito–Zygomatic Region
In earlier published studies, 14 reproducible cranio-orbito-zygomatic measurements taken from each of 26 standard axial CT scans of individuals with symmetric forms of TCS who had not been operated on were compared with the measurements of age-matched controls.110 The interorbital measurements (i.e., medial and lateral orbital wall separations) of the patients with TCS were at the mean when compared with their cohort group (i.e., no measurable orbital hypertelorism or hypotelorism), whereas the zygomatic measurements were significantly less than normal, thereby confirming the extent of malar hypoplasia. The congenitally deficient lateral aspect of the orbits in individuals with TCS was confirmed by the greater than normal values measured for globe protrusion and medial orbital wall protrusion in conjunction with diminished lateral orbital wall lengths. These measurements use the lateral orbital rim as a reference point and confirm the limited depth of the lateral orbits as a result of hypoplasia of the lateral orbital rims. The lateral orbital rims are components of the hypoplastic zygomatic complex. The abnormal shape of the anterior cranial vault in patients with TCS was documented as a diminished intercoronal (bitemporal) distance (width) and decreased cephalic length as compared with values for normal age-matched controls. In general, the CT measurements documented in the described study agree with the clinically observed morphology of TCS. The measurements carried out in the zygomatic structures confirmed the extent of hypoplasia of the zygomas for the group as a whole and in each individual patient; they also documented the extent of globe protrusion and decreased upper face width in patients with TCS. It appears as though the bones immediately adjacent to the involved zygomatic complex also experience a degree of hypoplasia or at least distortion.4,28,39,87,110,113,125,158,159,167,169
Maxillomandibular Region
In 1975, Roberts and colleagues completed a radiographic cephalometric study of TCS. Serial cephalometric radiographs were available for eight patients who presented with the full range of features that are characteristic of this syndrome.124 The expected facial convexity in patients with TCS was substantiated by cephalometric measurements. The extent of convexity was attributed to the severity of mandibular retrognathia (i.e., a decreased sella–nasion–B-point angle). Interestingly, the horizontal projection of the maxilla to the cranial base (i.e., the sella–nasion–A-point angle) remained within normal limits in the patients that were measured. Over time and with growth, the facial convexity remained relatively constant, thus confirming that the facial profile morphology of the infant with TCS was remarkably similar to that of the adult. Anterior facial heights were measured as “upper facial height” (i.e., the nasion to the anterior nasal spine) and “total facial height” (i.e., the nasion to the menton). The upper facial height was found to be relatively normal, whereas the total facial height was often excessive. This is the result of a combination of anterior open-bite malocclusion, mandibular retrognathism, and chin dysplasia characterized by increased vertical length and horizontal retrusion. The angle between the sella–nasion plane and the mandibular plane was obtuse, which confirmed the steepness of the clockwise rotation of the mandibular plane. The angle between the sella–nasion plane and the palatal plane was also obtuse, thus confirming the steepness of the clockwise rotation of the maxillary plane. The posterior facial height was markedly shorter for patients with TCS, with values in five of the eight patients found to be a minimum of four standard deviations less than those of the normal cohort.
The effective horizontal length of the mandible was markedly reduced, with values in most of the patients falling below four standard deviations as compared with those in the normal cohort. The decreased mandible size was evident in both the ramus height and the body length. The gonial angle was confirmed to be obtuse, with values of two standard deviations from the mean as compared with control values. The extent of antigonial notching was also documented in patients with TCS and was found to be similar to that observed among patients with condylar arrest early during their lives (e.g., as a result of juvenile rheumatoid arthritis or TMJ trauma at a young age). The steepness of the clockwise rotation of the maxillary and mandibular planes combined with the anterior and posterior vertical height disproportions and severe horizontal deficiency of the mandible is also indirectly seen in the A-point–to–B-point discrepancy. All of these morphologic findings explain the observed facial dysmorphology.4, 39,49,78,79,87
Facial Soft Tissues
Soft-tissue anomalies in individuals with TCS are manifested primarily in four clinical regions: the external ears; the eyelid–adnexal structures; the preauricular–cheek skin; and the temporal fossa (e.g., temporalis muscle hypoplasia).10,11,22,35,36,64,76,86,132,133,143,148,156 The extent of deficiency within the soft-tissue envelope is variable and, to large extent, reflective of the skeletal involvement. Each patient is unique and must be evaluated individually. The extent of soft-tissue anomalies continues to represent the most difficult obstacle to a favorable reconstruction.
External Auditory Canal, Middle and Inner Ear Structures, and Audiologic Findings
Relatively symmetric (i.e., from side to side) outer and middle ear malformations are characteristic of TCS. Middle-ear abnormalities (e.g., hypoplastic or absent cavities and ossicles) are an almost universal finding.18,55,58,66,118 The majority of patients have a unilateral or bilateral moderate or greater conductive hearing loss, with a flat or reverse sloping configuration on the audiogram. Most of the hearing loss in these patients is attributable to middle-ear malformations. In 1993, Pron and colleagues reported the results of a detailed CT radiologic and audiologic investigation of a large series of pediatric patients with TCS.118 The objectives of the study were 1) to determine the degree and symmetry of the external auditory canal and middle-ear abnormalities and evaluate hearing loss; and 2) to explore the relationship between hearing loss and external auditory canal and middle-ear abnormalities in patients with TCS. Data were available from 29 children who had not yet undergone any ear interventions and who had undergone audiometric testing and focused standardized petrous temporal bone CT scans. A review of the CT scans allowed for the evaluation of the external ear canal and the middle-ear anatomy. The majority of patients (88%) had largely symmetric external auditory canal abnormalities that were either stenotic (31%), atretic (54%), or normal (15%). Middle-ear cavity ossicular deformities were generally symmetric (96%), and cavities were either hypoplastic (85%) or missing (4%). Cavities that were hypoplastic were also deformed and assumed a rectangular rather than an oval shape. Ossicle deformities were generally completely symmetric and consisted largely of hypoplastic (46%) or missing (46%) ossicles. Ossicles (both the malleus and the incus) that were hypoplastic also tended to be ankylosed (82%) to either the lateral or medial wall of the tympanic recess. The structures of the external auditory canal and the middle ear are generally related. Increasing degrees of external ear canal malformations (i.e., normal, stenotic, or atretic) are directly associated with ossicle and cavity malformations. Of the patients with atretic canals, 67% had missing ossicles, and 33% had small or ankylosed ossicles. No abnormalities were noted in the inner-ear structures (i.e., cochlea, vestibule, canals, and internal auditory meatus) in the patients with TCS who were studied. According to the study by Pron and colleagues, hearing loss for the patient with TCS ranges from mild to severe (average, 58 dB of hearing loss).118 The majority of individuals (96%) have a moderate or greater degree of hearing loss.
Relationships exist between the external auditory canal, the middle-ear ossicles, and the degree and configuration of hearing loss.18,55,58,66,118 There is an association between the degree of external ear canal malformation and the degree of hearing loss in patients with TCS. In addition, hypoplastic, ankylosed, and missing ossicles appear to be associated with increasing levels of hearing loss; on average, the hearing loss is 52 dB, 56 dB, and 60 dB of hearing loss, respectively.
Facial Growth Potential with Treacher Collins Syndrome
The degree of malformation present at the time of birth in a newborn with TCS is believed to be relatively stable and non-progressive with age (Figs. 27-3 through 27-6).53,102,109,114,115,124 Roberts and associates reviewed serial cephalometric radiographs from individuals who were known to have TCS.124 They documented the stability of the mandible, which continued to grow but not to catch up. Through independent research, Garner reported findings of the cephalometric analysis of three patients of varied ages who were confirmed to have TCS.49 He also documented the relative stability of the anomalies without the progression of deformity at various ages.
Figure 27-3 A child with a severe form of Treacher Collins syndrome was followed up longitudinally without treatment intervention. No progression of the deformity was seen during clinical or computed tomography scan examination. A, Frontal and B, profile views at 2 months and 2 years of age. C and D, Computed tomography scan views of the craniofacial region at 2 months and 2 years, respectively, demonstrating the consistent growth of the malformed condyles, the coronoid processes, and the ascending rami of the mandible. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997.
Figure 27-4 A girl who was born with Treacher Collins syndrome was followed up longitudinally at 1 and 5 years of age without treatment intervention. There is consistent growth of the craniofacial region without progression of the deformity. As a result of chronic nasal obstruction and an open-mouth posture, excess vertical facial growth can be seen. A, Frontal views at 1 and 5 years of age. B, Profile views at 1 and 5 years of age. C, Computed tomography scan views at 5 years of age. There is severe hypoplasia of the zygomatic complex with lateroinferior orbital deficiency. The mandible demonstrates a Type IIA malformation.
Figure 27-5 A child who was born with Treacher Collins syndrome was followed up longitudinally without treatment intervention. No progression of the facial deformity occurred with growth. A, Frontal views at 10 months and 6 years of age. B, Profile views at 10 months and 6 years of age. C, and D, Computed tomography scans.
Classification of Temporomandibular Joint and Mandibular Malformation
The extent of TMJ–mandibular malformation in patients with TCS will influence the timing and techniques of reconstruction.69,137 A lack of clinically relevant definitions of the presenting glenoid fossa–condyle–ascending ramus malformations has resulted in confusion and miscommunication. Kaban and colleagues revised and clarified the previously described Pruzansky classification system for the degree of TMJ–mandibular malformation observed in patients with hemifacial microsomia.69,119 This classification is also useful to define the presenting anomalies and then to direct reconstruction in the patient with TCS (Fig. 27-7).
Figure 27-7 Kaban and colleagues described a classification system to define the degree of temporomandibular joint–mandibular malformation observed in patients with hemifacial microsomia. By definition, a Type IIB condylar deficiency will require the construction of a neo-condyle. The Type III malformation will also require neo-glenoid fossa construction. The same classification system is used for Treacher Collins syndrome. Craniofacial computed tomography (CT) scans demonstrate the classification system. A, CT scan views of a Type I mandibular malformation. B, C, and D, CT scan views of a Type IIA mandibular malformation. E and F, CT scan views of a Type III glenoid fossa–mandibular malformation.
A Type I TMJ–mandibular malformation involves minimal hypoplasia of the glenoid fossa and the condyle–ascending ramus on each side. All of the skeletal components are present (see Fig 27-7, A). Mandibular retrognathia and a mild anterior open bite are likely to be present, but TMJ function is essentially normal. The muscles of mastication are intact, and the condyle and the glenoid fossa are also fully intact. There is no need or advantage to constructing a neo-condyle or neo-glenoid fossa.
A Type IIA TMJ–mandible malformation involves a moderate degree of glenoid fossa and condyle–ascending ramus hypoplasia (see Fig. 27-7, B, C, and D). The anatomic location of each joint is anterior and medial, but TMJ function remains satisfactory. One or more of the paired muscles of mastication are likely to be hypoplastic. By definition, in Type IIA malformations, the condyle and the glenoid fossa have function that is at a level deemed better than could reliably be achieved through TMJ replacement. Even in patients with more severe Type IIA malformations, when applying superior and posterior pressure to the chin, the condyles can be seated into a consistent location against the skull base–glenoid fossa. By completing ramus osteotomies, the proximal segments can be positioned into a stable location against the skull base, and the distal mandible can then be reoriented into the preferred location. This will avoid the need to construct neo-condyles (e.g., costochondral grafts). Coronoidectomies may be required to accommodate the short condyles.
The Type III glenoid fossa–mandibular malformation involves deficiency to the extent that no posterior stop of the lower jaw against the cranial base is possible (see Fig. 27-7, E). The condyles and the ascending rami are not present. The disc, the TMJ capsule, and the glenoid fossa on each side are also not developed. Severe mandibular dysmorphology is present, including horizontal retrognathia and decreased posterior ramus height. A tracheostomy is generally required shortly after birth to manage the airway. Eventual construction of both a neo-glenoid fossa and a neo-condyle ascending ramus will be required on each side. Reconstruction with bone grafts (e.g., cranial grafts for the zygomatic complex and either costochondral or a vascularized fibula flap for the mandible) will be required (see discussion to follow).
Staging of Skeletal Reconstruction: Timing and Techniques
The reconstruction and rehabilitation of the Treacher Collins malformation must address unique and specific components of the anomalies, including the following: 1) the zygomatic and orbital region; 2) the maxillomandibular region; 3) the nasal region; 4) the soft-tissue envelope; 5) the external ears; 6) the external auditory canals; and 7) the middle-ear structures (Figs. 27-8 through 27-10).*
Figure 27-8 A, A child who was born with Treacher Collins syndrome is shown during the mixed dentition. She has severe zygomatic complex and orbital deficiency as well as a Type IIA condylar malformation. B, Illustration of the location of the full-thickness cranial bone graft donor site for zygomatic complex reconstruction. The zygomatic complex graft is also shown inset at the recipient site. C, Illustrations of a maxillofacial complex seen in a teenager with Treacher Collins syndrome (Type IIA mandibular malformation). The cheekbone orbital reconstruction has previously been completed. The proposed maxillary, mandibular, and chin osteotomies are marked out and then completed. Counterclockwise rotation of the maxillomandibular complex is demonstrated as part of the reconstruction.
Figure 27-9 Additional images of the patient from Fig. 27-8 showing the stigmata of Treacher Collins syndrome. At the age of 17 years, the patient was referred to this surgeon and agreed to a staged reconstruction that included three operations. The initial preoperative orthodontic treatment included the extraction of mandibular first premolars with the retraction of the anterior dentition to remove compensations. Orthognathic surgery included a Le Fort I osteotomy (counterclockwise rotation and anterior maxillary intrusion); bilateral sagittal split ramus osteotomies (advancement and counterclockwise rotation); and an osseous genioplasty (vertical reduction and advancement). During a second operation, the patient underwent zygomatic complex reconstruction with fixed full-thickness autogenous cranial bone grafts as described in this chapter and as illustrated in Fig. 27-8. This was followed a third operation: septorhinoplasty (open approach) to in-fracture the nasal bones, reduce the dorsal hump (bone and cartilage), and reshape the tip, including septal (caudal strut) grafting (see Chapter 38). A, Right profile views before and after reconstruction. B, Left profile views before and after reconstruction. C, Lateral cephalometric radiographs before and after reconstruction. D, Articulated dental casts that indicate analytic model planning. E, Occlusal views before treatment, early after reconstruction, and 3 years after reconstruction. A degree of maxillary relapse (skeletal versus dental) has occurred but with a retained functional occlusion and favorable facial aesthetics. F, Crafted autogenous cranial grafts shown before and then after inset into the zygomatic defects. The advancement of the hypoplastic temporalis muscles is also demonstrated. The soft-tissue hypoplasia involves multiple layers (i.e., skin, subcutaneous tissue, fascia, and fat tissue) and therefore can not be fully corrected simply by advancing the hypoplastic temporalis muscle. G, Axial computed tomography scan views shown through the zygomas before and after malar reconstruction indicating the improved interzygomatic buttress width, interzygomatic arch width, and zygomatic arch length. A, From Posnick JC, Treacher Collins syndrome: Perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997. F (bottom left, bottom right), G, From Posnick JC, Goldstein JA, Waitzman A: Surgical correction of Treacher Collins malar deficiency: quantitative CT scan analysis of long-term results, Plast Reconstr Surg 92:12, 1993.
Figure 27-10 A 16-year-old boy born with Treacher Collins syndrome with a Type I mandibular malformation was followed up longitudinally to confirm no progression of the deformity. He then underwent two operations, including orbitozygomatic and orthognathic reconstruction, as described previously. In preparation for orthognathic surgery, orthodontic treatment included maxillary first bicuspid extractions with the retraction of the anterior dentition to correct the arch form. The mandibular second molars are congenitally absent. The patient’s orthognathic surgery included Le Fort I osteotomy (vertical intrusion and horizontal advancement); bilateral sagittal split ramus osteotomies (horizontal advancement); and an osseous genioplasty (horizontal advancement). He is shown before and after orbitozygomatic and orthognathic surgery. A, Frontal views before and after orthognathic surgery. B, Left profile views before and after orthognathic surgery. C, Right profile views before and after reconstruction. D, Occlusal views before and after orthognathic surgery. Note that, with the upper bicuspid extractions and the absent mandibular second molars, the maxillary second molars are no longer in occlusion. E, Palatal views before and after orthognathic surgery. F,
[/not-level-membership-for-surgery-category]
