[level-membership-for-critical-care-medicine-category]
CHAPTER 24 TRAUMATIC BRAIN INJURY: PATHOPHYSIOLOGY, CLINICAL DIAGNOSIS, AND PREHOSPITAL AND EMERGENCY CENTER CARE
Death. Long-lasting or even permanent loss of function. Those are the burdens borne by many traumatic brain injury (TBI) patients and their families. Even some patients who initially appeared to have injuries that were mild according to clinical or radiographic criteria can suffer permanent injury.
INCIDENCE
According to data reported by the Centers for Disease Control and Prevention in the 1990s, 1.5 million Americans sustain a TBI every year. Of this number, hospitalization and ultimate survival occur in approximately 230,000 patients, but 50,000 will die. Long-term disability will occur in 80,000-90,000 patients annually. It has been estimated that more than 5 million men, women, and children in the United States are living with a permanent TBI-related disability.1
MECHANISM OF INJURY
Subdural Hematoma
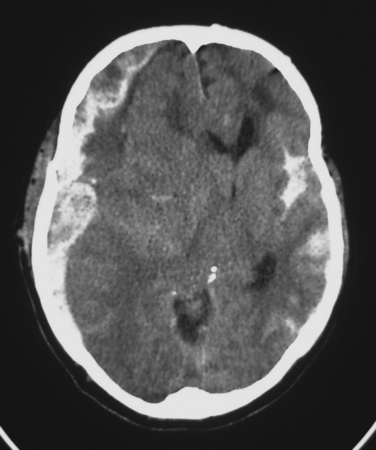
Classically, a subdural hematoma (SDH) (Figure 1) has been said to develop after tearing of a bridging vein, that is, a vein passing directly from the cortex to the overlying dura. The mechanical forces of the trauma can cause tearing of these veins. More recent evidence indicates that at least some of these hematomas actually form from splitting of inner and outer layers of the dura, that is, they may actually be “intradural hematomas.” Finally, some SDHs are caused by direct bleeding into the subdural space from parenchymal contusions or hematomas or from injured cortical arteries or veins.
Epidural Hematoma
Epidural hematomas (EDHs) classically arise after a blow to the side of the head results in a fracture of the thin temporal bone immediately overlying the middle meningeal artery. The patient may briefly lose consciousness after the initial impact, but he or she quickly awakens; thus, the brain injury was mild. Unfortunately, the fracturing of the skull lacerated the middle meningeal artery. Continued bleeding from this source produces an enlarging EDH, the presence of which may be signaled by such symptoms as severe and worsening headache, vomiting, and decreasing level of consciousness. The period between awakening from the initial concussion and subsequent lapsing into a coma has historically been described as a “lucid interval.” Importantly, loss of consciousness does not always occur after the skull is fractured, and many patients with large EDHs are awake until they begin to lapse into a terminal coma. It must also be mentioned that many EDHs are not associated with meningeal arterial bleeding. In these cases, perhaps the source of the hematoma is oozing from the overlying edges of fractured bone.
Subarachnoid Hemorrhage
The most common post-traumatic intracerebral hemorrhage is not a mass lesion, but rather diffuse subarachnoid hemorrhage (SAH) (see Figure 1). Several retrospective series report that SAH after TBI is independently associated with worse outcomes, but the mechanism that might explain such an association is unclear. In the acute setting, SAH does not seem to have much effect on patient management, which is driven instead by more immediately pressing concerns.
Ischemia
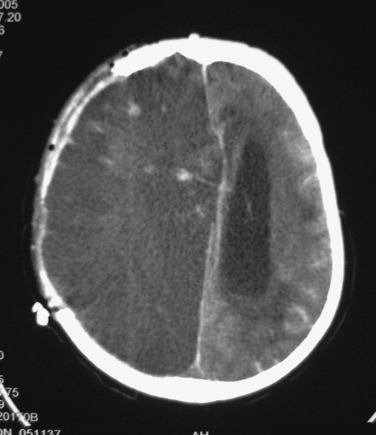
Diffuse injury may be of several types. Ischemia is a common form of diffuse injury. In some cases, mass effect from a traumatic hematoma may cause elevated intracranial pressure (ICP) and local compression of underlying tissue that can lead, respectively, to global and local reduction of cerebral blood flow (CBF). However, post-traumatic cerebral ischemia may also occur when no mass lesion is present, especially very early after injury. Although the CT scans in these cases may be relatively unimpressive, these patients may be quite vulnerable to the effects of such secondary insults as hypotension or hypoxia. Over the subsequent hours and days, CBF usually increases, but the damage from early ischemia may have already been done long before the increase in CBF (Figure 2). Some centers have used xenon CT or perfusion CT to measure CBF and identify ischemia immediately after injury.
Diffuse Axonal Injury
Another common type of diffuse injury is diffuse axonal injury (DAI). Although the axonal disconnection that characterizes DAI is commonly thought to occur at the time of injury, such immediate loss of axonal continuity probably occurs only when an injury produces severe cerebral parenchymal disruption. Instead, it appears more likely that the rotational and mechanical forces that are operant during the traumatic event produce a focal impairment of axoplasmic flow, which, in turn, culminates in axonal disconnection several hours after injury.2 This slight delay creates hope that a therapeutic window may exist for the administration of a yet-to-be-developed treatment that would prevent loss of axonal integrity and function.
CLINICAL DIAGNOSIS
Clinical Examination
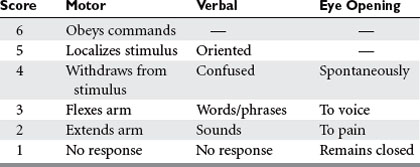
Many scales have been developed for the assessment of consciousness or neurologic status after injury, but by far the most widely used is the Glasgow Coma Scale (GCS)3 (Table 1). In conjunction with such information as the status of pupillary reactivity and the tempo or rate of change of a patient’s neurologic condition, the GCS is an extremely useful tool for assessing a patient’s baseline condition and subsequent progress.
INITIAL CLINICAL INTERVENTIONS: PREHOSPITAL AND EMERGENCY CENTER CARE
The basic principles of TBI care, which have remained unchanged for decades, are maintenance of normal homeostasis and prevention and prompt treatment of secondary insults. The acutely traumatized brain is much more vulnerable than the uninjured brain to even mild deviations from normal, such as transient episodes of hypotension or hypoxia.4 Some evidence suggests that events like febrile episodes, seizures, and hyperglycemia may also worsen outcome. Brief insults are usually tolerated well by the normal brain, but they may have a profound detrimental effect on the injured brain.
Breathing
In terms of breathing, standard recommendations advocate the lowest FiO2 capable of maintaining adequate oxygenation. Although the minimum acceptable PaO2 based on the oxygenhemoglobin dissociation curve is 60 mm Hg, most practitioners target a minimum of 80-100 mm Hg in TBI patients in order to create a bit of a cushion.
Hyperventilation is no longer recommended as a prophylactic measure to prevent intracranial hypertension.5 Hyperventilation is known to cause constriction of the cerebral vasculature, and the resultant decrease in cerebral blood volume can acutely lower ICP. However, the constriction of cerebral arteries may cause CBF to drop to critical levels. Also, within 24 hours of initiation of hyperventilation, the cerebral arteries probably dilate back to their original diameter.6 Subsequent attempts to allow the PaCO2 to increase can cause the arteries to dilate even further, possibly raising ICP.
Circulation
In years past, common practice was to dehydrate patients “to prevent the brain from swelling.” In the 1990s, the pendulum swung the other way as TBI patients were aggressively managed with intravenous fluids and pressors in order to artificially elevate their blood pressure. However, subsequent studies showed that patients treated with aggressive elevation of blood pressure through fluids and pressors did not have improved neurologic outcomes and, in fact, had a five-fold higher likelihood of developing acute respiratory distress syndrome.7
IMAGING MODALITIES: WHAT, WHEN, AND WHY
Computed Tomography Scanning
After initial assessment and stabilization of a TBI patient, the next order of business is the procurement of imaging studies. The most important radiologic study for the evaluation of acute TBI is a CT scan. This imaging modality is excellent for revealing acute hemorrhage, cerebral edema, and mass effect, which are the features of greatest interest during the initial assessment. Bone settings can also detect calvarial and skull base fractures. Another advantage is that CT scanning is a quick procedure that is widely available, at least in the United States. To overcome difficulties with prehospital intubation and the frequent use of sedation in these patients, Marshall et al.8 devised a TBI classification scheme based on CT scan findings (Table 2).
| Category | Definition |
|---|---|
| Diffuse injury I (no visible pathology) | No intracranial pathology visible on CT scan |
| Diffuse injury II | Cisterns present with midline shift |
| 0-5 mm and/or: | |
| Lesion densities present | |
| No high- or mixed-density lesion >25 cc | |
| Diffuse injury III (swelling) | Cisterns compressed or absent with midline shift 0-5 mm; no high- or mixed-density lesion >25 cc |
| Diffuse injury IV (shift) | Midline shift >5 mm, no high- or mixed-density lesion >25 cc |
| Evacuated mass lesion | Any lesion surgically evacuated |
| Nonevacuated mass lesion | High- or mixed-density lesion >25 cc, not surgically evacuated |
CT, Computed tomography.
INJURY GRADING
Glasgow Coma Scale
However, accurate determination of the GCS score is often difficult in patients who are intoxicated with alcohol or other drugs or in patients with injuries that cause such extensive periorbital edema that the eyes are swollen shut, making assessment of eye opening impossible. Other factors that complicate the determination of the GCS score reflect changes in prehospital and emergency department practices since the initial description of the GCS over a generation ago. Nowadays, many patients are endotracheally intubated in the prehospital setting, and paralytics and sedatives are often administered before an accurate and thorough neurologic assessment is performed. These problems have led to the creation of other assessment tools, such as those based on CT findings.8 Another way of getting around the problem of an inaccurate neurologic examination might be the use of serum markers to identify trauma patients who are likely to have serious central nervous system injury, analogous to the manner in which serum levels of cardiac enzymes are used in the diagnosis of acute myocardial injury.9 This is an area of rapidly growing interest.
Marshall Computed Tomography Scale
Marshall and colleagues developed a CT-based classification scheme using data from the Traumatic Coma Data Bank (see Table 2). This system, commonly referred to as the Marshall scale or the TCDB scale, classifies CT scans according to such factors as midline shift and compression of cerebrospinal fluid cisterns. Although this scale is useful in certain circumstances, it should not be considered a substitute for an accurate neurologic examination.
Abbreviated Injury Scale
The Abbreviated Injury Scale (Table 3) for the head assigns a score of 1 for minor scalp injuries such as abrasions, contusions, and lacerations.10 Longer and deeper lacerations receive a score of 2, whereas scalp injuries accompanied by significant blood loss or characterized by total scalp loss are scored as 3. Cranial nerve injuries are coded as 2.
| Score | Injury Severity | Head Injury Examples |
|---|---|---|
| 1 | Minor | Minor scalp injuries |
| 2 | Moderate | More severe scalp injuries |
| Cranial nerve injuries | ||
| Simple calvarial fractures | ||
| LOC >1 hour | ||
| Post-traumatic amnesia | ||
| 3 | Serious | Worst scalp injuries |
| Cerebral vascular injuries | ||
| Skull base fractures | ||
| Comminuted calvarial fractures | ||
| Small parenchymal contusions | ||
| Traumatic subarachnoid hemorrhage | ||
| LOC 1–6 hours | ||
| 4 | Severe | Worse cerebral vascular injuries |
| Worst skull fractures | ||
| Hematomas | ||
| LOC 6–24 hours | ||
| 5 | Critical | Worst cerebral vascular injuries |
| Larger hematomas | ||
| LOC >24 hours | ||
| 6 | Lethal | Massive destruction |
| Crush injuries |
LOC, Loss of consciousness.
Mild and Moderate Traumatic Brain Injury
Unlike the situation with severe TBI, the major concern with mild TBI is not whether a patient will live or die or whether a patient will become vegetative or severely disabled. Almost all these patients survive. Instead, the major morbidity relates to disturbances of memory, cognition, attention, emotional stability, and related areas. The lack of “hard” evidence of neurologic impairment leads many physicians to downplay the significance of these symptoms. However, these patients often go on to lose their jobs, drop out of school, divorce their spouses, or go through other major upheavals in their lives. Eventually, most of them recover, but such a process may take months. Reassurance of the patient and family may be all that is needed, and in fact, may be all that can be offered. Counseling and formal testing may be appropriate if objective documentation of injury is needed or if a physician suspects malingering or symptom magnification for secondary gain.
CONCLUSIONS AND ALGORITHM
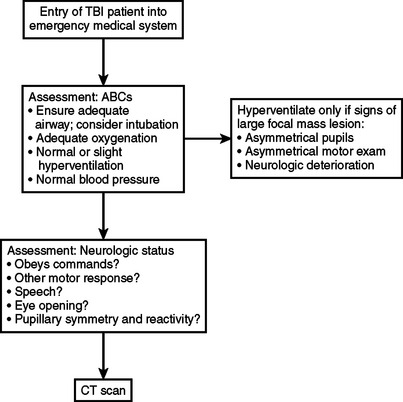
Figure 3 summarizes the priorities in assessment and initial management of TBI patients. Accurate neurologic assessment and careful attention to the ABCs are of paramount importance. These goals continue to be emphasized while patients are transported to a hospital, resuscitated, and taken to a CT scanner.
1 Centers for Disease Control and Prevention Traumatic Brain Injury in the United States: A Report to Congress. Available at http://www.cdc.gov/doc.do/id/0900f3ec8001012b
2 Stone JR, Okonkwo DO, Dialo AO, et al. Impaired axonal transport and altered axolemmal permeability occur in distinct populations of damaged axons following traumatic brain injury. Exp Neurol. 2004;190:59-69.
3 Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2(7872):81-84.
4 Miller JD, Sweet RC, Narayan R, Becker DP. Early insults to the injured brain. JAMA. 1978;240:439-442.
5 Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731-739.
6 Muizelaar JP, van der Poel HG, Li ZC, et al. Pial arteriolar vessel diameter and CO2 reactivity during prolonged hyperventilation in the rabbit. J Neurosurg. 1988;69:923-927.
7 Robertson CS, Valadka AB, Hannay HJ, et al. Prevention of secondary isch-emic insults after severe head injury. Crit Care Med. 1999;27:2086-2095.
8 Marshall LK, Marshall SB, Klauber MR, et al. A new classification of head injury based on computerized tomography. J Neurosurg. 1991;75:S14-S20.
9 Wang KK, Ottens AK, Liu MC, et al. Proteomic identification of biomarkers of traumatic brain injury. Expert Rev Proteomics. 2005;2:603-614.
10 Association for the Advancement of Automotive Medicine. The Abbreviated Injury Scale. Barrington, IL: Association for the Advancement of Automotive Medicine, 1998;2001. 1990 rev., update 1998
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
CHAPTER 24 TRAUMATIC BRAIN INJURY: PATHOPHYSIOLOGY, CLINICAL DIAGNOSIS, AND PREHOSPITAL AND EMERGENCY CENTER CARE
Death. Long-lasting or even permanent loss of function. Those are the burdens borne by many traumatic brain injury (TBI) patients and their families. Even some patients who initially appeared to have injuries that were mild according to clinical or radiographic criteria can suffer permanent injury.
INCIDENCE
According to data reported by the Centers for Disease Control and Prevention in the 1990s, 1.5 million Americans sustain a TBI every year. Of this number, hospitalization and ultimate survival occur in approximately 230,000 patients, but 50,000 will die. Long-term disability will occur in 80,000-90,000 patients annually. It has been estimated that more than 5 million men, women, and children in the United States are living with a permanent TBI-related disability.1
MECHANISM OF INJURY
Subdural Hematoma
Classically, a subdural hematoma (SDH) (Figure 1) has been said to develop after tearing of a bridging vein, that is, a vein passing directly from the cortex to the overlying dura. The mechanical forces of the trauma can cause tearing of these veins. More recent evidence indicates that at least some of these hematomas actually form from splitting of inner and outer layers of the dura, that is, they may actually be “intradural hematomas.” Finally, some SDHs are caused by direct bleeding into the subdural space from parenchymal contusions or hematomas or from injured cortical arteries or veins.
Epidural Hematoma
Epidural hematomas (EDHs) classically arise after a blow to the side of the head results in a fracture of the thin temporal bone immediately overlying the middle meningeal artery. The patient may briefly lose consciousness after the initial impact, but he or she quickly awakens; thus, the brain injury was mild. Unfortunately, the fracturing of the skull lacerated the middle meningeal artery. Continued bleeding from this source produces an enlarging EDH, the presence of which may be signaled by such symptoms as severe and worsening headache, vomiting, and decreasing level of consciousness. The period between awakening from the initial concussion and subsequent lapsing into a coma has historically been described as a “lucid interval.” Importantly, loss of consciousness does not always occur after the skull is fractured, and many patients with large EDHs are awake until they begin to lapse into a terminal coma. It must also be mentioned that many EDHs are not associated with meningeal arterial bleeding. In these cases, perhaps the source of the hematoma is oozing from the overlying edges of fractured bone.
Subarachnoid Hemorrhage
The most common post-traumatic intracerebral hemorrhage is not a mass lesion, but rather diffuse subarachnoid hemorrhage (SAH) (see Figure 1). Several retrospective series report that SAH after TBI is independently associated with worse outcomes, but the mechanism that might explain such an association is unclear. In the acute setting, SAH does not seem to have much effect on patient management, which is driven instead by more immediately pressing concerns.
Ischemia
Diffuse injury may be of several types. Ischemia is a common form of diffuse injury. In some cases, mass effect from a traumatic hematoma may cause elevated intracranial pressure (ICP) and local compression of underlying tissue that can lead, respectively, to global and local reduction of cerebral blood flow (CBF). However, post-traumatic cerebral ischemia may also occur when no mass lesion is present, especially very early after injury. Although the CT scans in these cases may be relatively unimpressive, these patients may be quite vulnerable to the effects of such secondary insults as hypotension or hypoxia. Over the subsequent hours and days, CBF usually increases, but the damage from early ischemia may have already been done long before the increase in CBF (Figure 2). Some centers have used xenon CT or perfusion CT to measure CBF and identify ischemia immediately after injury.
Diffuse Axonal Injury
Another common type of diffuse injury is diffuse axonal injury (DAI). Although the axonal disconnection that characterizes DAI is commonly thought to occur at the time of injury, such immediate loss of axonal continuity probably occurs only when an injury produces severe cerebral parenchymal disruption. Instead, it appears more likely that the rotational and mechanical forces that are operant during the traumatic event produce a focal impairment of axoplasmic flow, which, in turn, culminates in axonal disconnection several hours after injury.2 This slight delay creates hope that a therapeutic window may exist for the administration of a yet-to-be-developed treatment that would prevent loss of axonal integrity and function.
