Chapter 49 Traumatic Brain Injury
Definitions
Defining Severity of Injury
TBI is often categorized as mild, moderate, or severe. The Glasgow Coma Scale (GCS) has become the most widely used primary initial assessment tool for determining the severity of injury (Box 49-1).426 During the initial stages of diagnosis in the field or emergency facility, TBI is suspected or indicated if there has been a significant blow to the head and/or there is an alteration of or loss of consciousness at the time of injury. An initial GCS score is obtained through clinical evaluation. As indicated in Box 49-1, the score is obtained by rating the best visual, verbal, and motor responses. The total score is simply a sum of these ratings, with scores ranging from 3 to 15. A score of 3T is given if the score obtained is due to medically induced chemical paralysis associated with intubation.
Generally accepted guidelines identify three levels of severity based on GCS scores: mild (GCS = 13 to 15), moderate (GCS = 9 to 12), and severe (GCS = 3 to 8). Individuals scoring in the mild TBI range should be evaluated at an emergency facility but might require minimal or no hospitalization. For more severe injuries involving loss of consciousness, the GCS is a widely accepted measure indicating the depth of coma and is generally serially measured until emergence from coma. However, the GCS has limited applicability to young children. The Pediatric Glasgow Coma Scale (PGCS) is a modified version of the GCS for use with pediatric TBI patients.309
Epidemiology
United States and Worldwide
Approximately 1.7 million TBIs occur each year in the United States.245 Of these, approximately 52,000 result in death and 1.365 million are mild injuries (often referred to as concussions), for which individuals are treated and released from the emergency room. This leaves approximately 275,000 individuals who survive moderate to severe TBI, generally requiring significant medical care and hospital stays. Many are left with long-term disability as a result of these injuries.245 According to one study, 40% of individuals hospitalized because of TBI reported at least one ongoing issue at 1 year after injury, with improvement of memory and problem solving listed as one of the most frequently unmet needs.86 Such cognitive impairments, in addition to physical and medical issues, have significant impact on the ability of an individual to perform everyday activities. In fact, it is estimated by the Centers for Disease Control and Prevention that at least 5.3 million Americans currently have a need for long-term or lifelong assistance with activities of daily living (ADLs) as a result of TBI.435
Globally, TBI is a leading cause of death and disability. Although exact worldwide statistics are difficult to obtain, a fairly recent review estimated approximately 775,000 new TBI hospitalizations per year in Europe.422 Available information suggests that the epidemiology of TBI on a global scale is similar to trends found in the United States. Incidence of TBI by age and gender varies slightly across regions but overall appears to follow patterns similar to those found in the United States.201 For example, the distribution of mild, moderate, and severe TBI is consistent across the United States, Europe, Australia, and Asia: 80% of injuries are mild TBI, whereas moderate and severe TBI each account for 10% of injuries.422
Causes of Injury
The leading causes of TBI in the United States include falls (35.2%), traffic-related crashes (17.3%), struck by/against events (sports) (16.5%), assaults (10%), and other injuries (21%).245 Sports injuries account for 1.6 to 3.8 million TBIs a year, although most of these are mild injuries that are not treated in the hospital or emergency department.246 It should be noted that these statistics vary with age, gender, geographic location within and between countries, and with group membership as a result of multiple factors, as explained below.
What is known of the global etiology of TBI is similar to U.S. rates, with a few notable exceptions. Sixty percent of TBIs in Europe are due to road traffic injuries.167 This figure is distributed evenly across most regions, suggesting that road traffic-related injuries are not associated only with high income countries. Twenty to 30% of injuries are due to falls, 10% violence, and 10% sports or work related.167 India has the highest reported rate of TBI related to falls.201,422 Latin America, Caribbean, and sub-Saharan Africa have the highest reported TBI rates as a result of road traffic incidents and violence.201
Associated Costs
The costs of TBI including direct medical costs and lost productivity in the United States were estimated to total $60 billion dollars in 2000.127 Worldwide statistics are less easily obtained. The costs associated with TBI go well beyond just the cost of medical care. The societal costs, or indirect costs, associated with the potential loss of productive work years for those who have been injured, as well as family members or others who might need to care for them, must also be considered. TBI also results in significant alterations in personal roles and responsibilities for the injured individual and family members. Reduced participation in complex leisure or recreational activities is also often a personal cost associated with TBI.
Demographics and Risk Factors
Age and Gender
The two age-groups most at risk for sustaining a TBI are 0 to 4 year olds and 15 to 19 year olds.245 Motor vehicle accidents result in the greatest number of TBI injuries for people aged 15 to 19.245 Another group at risk for TBI is adults over the age of 65. Fall-related injuries are highest among adults over the age of 65. Patients who sustain a TBI over the age of 75 have the highest rates of TBI-related hospitalization and death.245 Men are 1. 4 times as likely as women to sustain a TBI in the civilian population.245
Socioeconomic Status
Lower socioeconomic status (SES) in U.S. cities is correlated with an increased risk for injury as a result for increased exposure to high-risk occupations, personal violence, older vehicles, and substandard housing.235 Road traffic injuries are highest in areas of low SES.405 SES disadvantages are often associated with racial characteristics and rural locations. Minority populations in urban U.S. areas are often of lower SES and have elevated risk for injury.235 African Americans have a higher rate of TBI and associated mortality than other groups.235
Rural populations display a greater incidence of unhealthy lifestyle behaviors, including high alcohol consumption and psychosocial stress. Rural populations are also less likely to participate in preventive measures such as wearing seatbelts.322,440 This increase in risk-taking behaviors is evident in poorer health outcomes, specifically with injury-related outcomes like TBI.322,440
Violence
Assault or violence-related injuries account for 10% of TBI.245 Violence-related TBIs result from firearms, cutting instruments, blunt objects, or assaults such as pushing or hitting.145 Injury as a result of firearm use is the leading cause of death from TBI.442 Sixty percent of firearm-related TBI is self-inflicted, 32% results from intentional assault, and 4% is unintentional.38 People who sustain a TBI as a result of a violence-related injury are more likely to be younger, single, male, member of a minority group, and have a history of alcohol abuse.145
Child Abuse
Intentional TBI in children is referred to by many names, including shaken baby syndrome (SBS) and inflicted childhood neurotrauma.130 Approximately 1 million children are severely abused each year.486 Child abuse is most often conducted by parents or child care providers.362 The majority of perpetrators are male, either the child’s father or a boyfriend of the child’s mother residing in the home.389 Infants are at greatest risk for sustaining a TBI as a result of violent shaking,439 although older children are still at risk.381 One third of these children survive with no consequences; one third sustain permanent injury; and one third die.486 Risk factors for the occurrence of SBS include maternal factors such as mothers less than 19 years old, education less than 12 years, single marital status, African American or Native American, limited prenatal care, and newborns less than 28 weeks old.130 Child abuse rates resulting from unintentional injury increase during periods of economic decline.50
Psychosocial Factors
Part of the reason for the higher incidence of TBI for young males could be a tendency to engage in higher risk behaviors. Substance use is also related to TBI incidence, and up to 50% of individuals injured test positive for alcohol or other substances at the time of injury.83 Previous psychiatric history is a risk factor for TBI, particularly with those who have a history of anxiety, depression, and conduct disorder.445
Military Traumatic Brain Injury Epidemiology
An emerging group at risk for sustaining TBI is members of the U.S. military. Walter Reed Army Medical Center reports that 30% of service members evacuated from the field had sustained a TBI between 2003 and 2005. Army service members are relatively young with an average age of 28 years.341 Approximately 85% of military service members are male.341 Military members who sustain a TBI are also more likely to be male because they represent a larger percentage of the military service population.
Global Demographics and Risk Factors
What is known of global statistics suggests that countries with low to middle income harbor more risk factors that contribute to TBI. Risk factors vary by country and can depend on the incidence of such factors as poor road design, substandard vehicles, higher rates of violence as a result of war, and fewer prevention measures.201 Low- to middle-income countries typically have inadequate health systems to address TBI-related care.201 Accurate information on demographics and etiology of TBI in low- to middle-income countries is especially difficult to obtain. Despite this, the burden of TBI is evident worldwide.
Pathophysiology Associated With Traumatic Brain Injury
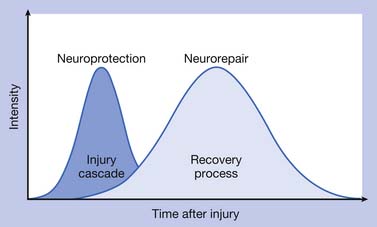
The pathophysiologic processes associated with TBI are complex and consist of (1) a primary injury that disrupts brain tissue and function at the moment of impact, (2) secondary injury through multiple biochemical cascades that propagate cellular dysfunction and lead to cell death, and (3) a chronic degeneration, repair, and regeneration process that occurs over the long term after the injury has occurred. Figure 49-1 depicts the continuation of injury and repair associated with pathophysiology after TBI.
Primary Injury
TBI occurs in conjunction with mechanical forces that cause disruption to the brain tissue. Closed head injuries can occur as a result of mechanical forces that shear axons, as well as by forces at the site of the impact or at the point opposite the impact.140 Contact forces occur when the head is prevented from moving after it is struck. Inertial forces occur when the head is set into motion and accelerates.178
Inertial forces associated with angular acceleration result in diffuse axonal injury (DAI). This is a process that causes tensile strains resulting in microscopic disruption of axons, cerebral edema, and neuronal disconnection. In angular acceleration, the brain’s center of gravity moves over a center of angulation, or fulcrum, located in the lower to middle cervical region.143 The severity of DAI depends on the duration, magnitude, direction of the angular acceleration, and associated impact.143 In severe cases of DAI, more than just superficial axons and deeper white matter structures are affected. The gray–white matter junction is also particularly vulnerable to DAI. Midline brain structures, such as the corpus callosum, are often affected by DAI, and DAI is also associated with loss of consciousness and coma. Recovery from DAI is gradual and can be linked to the duration of coma.178 Violent shaking associated with child abuse generates significant acceleration–deceleration forces, which result in shearing injury to brain tissue, disruption of blood vessels, and retinal bleeding.130
Inertial forces associated with translational acceleration result in a head movement that is in line with the brain’s center of gravity. The resulting differential movement of the brain relative to the skull causes focal injuries such as contusions. Cortical contusions often occur on gyral crests, particularly on the undersurface of the frontal and anterior temporal lobes, where bony prominences located in the basilar skull can create tissue and vascular disruption. Contusions can occur under the impact site (coup injury) and result from a rapid skull distortion during impact.143 Contusions remote from the injury site and opposite of the impact are contrecoup injuries and occur as a result of negative pressure generated from the impact, and are associated with translational acceleration.328 Patients with significant TBI can have one or more contusions, and deficits observed with cerebral contusions are linked with the lesion location and size.
DAI and focal contusions can result in neuronal disconnection, or diaschisis. The concept of diaschisis was first proposed by Constantin von Monakow in 1905 and refers to neurons remote from a site of injury, but anatomically connected to the damaged area, becoming functionally depressed. Because every structure in the brain is directly or indirectly connected to all other structures, the basic idea generated from this concept is that damage to one structure will be disruptive to other structures. Imaging studies suggest that diaschisis can result in the metabolic depression of areas both regionally associated and remote from the site of injury.121,295 Others suggest that focal cortical lesions can affect contralateral cortical functioning by way of intercallosal connections102 and can affect the function of subcortical structures, including the striatum.121 Resolution of diaschisis is one of several theories for recovery of function.121,295
Epidural hematomas (EDHs) result from local impact and subsequent laceration of underlying dural veins and arteries. The meningeal artery is commonly the source of EDHs, and damage to dural sinuses can also cause EDH. When an EDH develops from a disrupted artery, a neurologic emergency occurs because the EDH quickly expands and rapidly causes neurologic deterioration.178
Subdural hematomas (SDHs) result from inertial forces and the tearing of bridging veins.328 Bridging veins are susceptible to shear and rupture from brief high-velocity angular accelerations associated with falls.141 Traumatic subarachnoid hemorrhage (SAH) occurs when angular acceleration shears vessels located in the subarachnoid space.178
Secondary Injury
Secondary injury develops over the hours and days after the initial impact and is associated with disruption of cerebral blood flow and metabolism, massive release of neurochemicals, cerebral edema, and disruption of ion homeostasis leading to cellular injury and eventual cell death. Much of what is known about secondary injury is derived from postmortem analyses of human brain tissues, blood, cerebrospinal fluid (CSF), and parenchymal microdialysate fluid obtained early after clinical TBI. Much of the work exploring secondary injury has also used in vivo experimental models of TBI and in vitro injury methods. Experimental models of TBI produce one or more types of primary injury, which lead to the development of secondary injury cascades that closely resemble observations made in the human condition.104,276,404
Brain swelling occurs in response to the initial injury and early events involved with secondary injury and results in elevated intracranial pressure (ICP) and decreased cerebral perfusion pressure (CPP). If severe enough, brain swelling can lead to herniation, which has potentially fatal consequences. In some cases, elevated ICP is due to the development of focal extraaxial lesions like SDH and EDH described above. In other cases, elevated ICP and intracranial hypertension result from global mechanisms occurring on a cellular level that lead to brain edema. Some mechanisms that affect brain edema include vasogenic edema, increased tissue osmolarity, and vascular dysregulation resulting from increased cerebral blood volume. However, brain edema resulting from neuronal and astrocyte swelling secondary to increased glutamate metabolism after excitotoxic insult might impart the most damage.231
Excitatory amino acids (EAAs) like glutamate and aspartate are ubiquitous and important neurotransmitters for normal brain function. After TBI, excess excitatory amino acid levels are present in extracellular microdialysate fluid and CSF.53,452 Excess EAA release occurs, in part, from the mechanical stresses associated with stretch injury of axons. Elevations in extracellular glutamate levels after TBI contribute to excitotoxic injury by affecting other paths of secondary head injury through two primary mechanisms. By binding their target receptors, excess EAAs can trigger an influx of sodium and chloride that leads to acute neuronal and astrocytic swelling. Excess EAA levels can also lead to an intracellular calcium influx and the release of Ca++ from intracellular stores, leading to delayed cellular damage or death.70,415 EAA-mediated, Ca++-dependent production of nitric oxide, superoxide, and free radical damage to DNA and cellular membranes leads to cell death.265 Regional differences in the distribution of EAA-sensitive receptors contribute to selective vulnerability of some regions to excitotoxic injury like the hippocampus, a critical region for learning and memory.300
Lactate is the end product of glycolysis, and in neurons, lactate is converted to pyruvate for oxidative metabolism. Lactate/pyruvate ratios reflect the cellular energy balance and the cytosolic oxidation–reduction state.398 Increased metabolic demand after TBI is due, in part, to increased astrocytic glutamate uptake and results in increased lactate. Reduced cerebral blood flow, including pericontusional hypoperfusion, has been documented in both experimental models187 and in humans409 early after TBI and results in ischemic injury. Elevations in lactate/pyruvate have been measured in pericontusional tissue in humans448 and are associated with other aspects of secondary injury, including increased ICP and mitochondrial dysfunction.
Although extracellular lactate has classically been a marker for anaerobic metabolism with hypoxic injury, newer research indicates that lactate is also used by brain tissue during an insult as an important energy source in oxidative metabolism.356 Evidence supports a dichotomous role of lactate in which it is produced by the astrocyte via glycolytic metabolism, to be directly consumed by the neuron after uptake through lactate transporters,89,356 Physiologically337 and after TBI,356 excitatory amino acids are likely triggers to this metabolic process. After injury, estrogen facilitates lactate production for neuronal use by modulating lactate transporters.293 Experimental studies suggest that lactate is used as a neuronal energy substrate after TBI, and lactate administration after TBI is neuroprotective and decreases cognitive deficits.360
Mitochondrial dysfunction is often the result of energy failure and excitotoxic insult and results in the formation of highly reactive free radicals and subsequent oxidative damage to cell membranes and DNA. Hydroxyl radicals and other reactive oxygen species (ROS) react with many molecules found in living cells, including DNA, membrane lipids, and carbohydrates. Central nervous system (CNS) tissue is particularly vulnerable to oxidative injury because of the high rate of oxidative metabolic activity, the nonreplicating nature of neurons, high levels of transition metals, and a high membrane/cytoplasm ratio in neurons.10 Superoxide radicals are a principal mediator of the microvascular damage after TBI.242 Free-reactive iron generated from hemorrhage catalyzes the formation of free radicals.231 ROS production may also be a downstream effect of caspase-mediated cell death (apoptosis).126
Cell death in CNS tissues after brain trauma falls into two primary categories: (1) cellular necrosis and (2) apoptosis or programmed cell death. Necrotic cells display cellular and nuclear swelling as well as disruption of cellular membranes. In contrast, apoptotic cells are characterized by cellular shrinkage, nuclear condensation, and DNA fragmentation.216 Apoptosis is accompanied by a cascade of events required for cell death to occur.410 Cell death patterns in TBI often reflect a combination of both necrosis and apoptosis.
Apoptosis can be initiated through either extracellular or intracellular signaling pathways. Cysteine-aspartic acid proteases (caspases) play an essential role in cell death via both pathways. Extracellular binding of cell surface death receptors, including tumor necrosis factor (TNF)-α receptor and Fas ligand receptor, initiates apoptotic signals that include caspase-8 and caspase-3 within seconds of ligand binding. Intracellular signaling is initiated by mitochondrial dysfunction in response to disturbances in cellular energy balance, oxidative injury, and changes in ionic homeostasis. This mitochondrial stress leads to the opening of the mitochondrial transition pore and cytochrome c release. Cytosolic cytochrome c release induces multiple caspases, including caspase-9 and caspase-3, which leads to the cleavage of key cellular elements like cytoskeletal proteins and DNA repair proteins as well as the activation of endonucleases.77 In addition to this intracellular caspase-dependent process for apoptosis, intracellular apoptosis signaling can also occur via a caspase-independent pathway with apoptosis-inducing factor (AIF). AIF is released from the mitochondria and results in DNA fragmentation. The mitochondria house prosurvival and prodeath proteins that are both members of the BCL-2 protein family,2 each of which can be manipulated experimentally to effect cell survival. As an example, estrogen enhances BCL-2 expression and cell survival after CNS injury.199
Inflammation after TBI is characterized by complex cascades that largely include cytokines, or signaling molecules, that communicate with resident and infiltrating inflammatory cells. Cytokines like interleukin (IL)-1β and TNF are well studied in TBI. They are synthesized by peripheral cells and CNS neurons and glia, and are increased early after injury. IL-1β and other cytokines are also synthesized by activated microglia. IL-1β has potent leukocyte signaling properties that lead to cellular inflammatory responses after TBI. Cytokines generally can facilitate blood–brain barrier (BBB) disruption, cerebral edema, and cell death. Specifically, IL-1β and TNF can mediate synthesis of neurotoxic compounds such as arachidonic acid and are implicated in excitotoxic components of secondary injury.374
Cytokines are expressed in the brain and promote neuronal differentiation and survival under normal conditions. Cytokines play a dual role in secondary injury with TBI, and inflammatory cascades can either enhance cellular injury or provide neuroprotection. Some cytokines support neuroprotection and neurorepair through their effects on neurotrophin production, excitotoxicity, and antiinflammatory properties.223,307 Some studies suggest that cytokine effects might be dose dependent, with higher levels more likely to be associated with detrimental effects and lower levels being neuroprotective. Cytokine effects might also be time dependent, with beneficial effects more likely at delayed or chronic time points after injury.308 TNF is an example in which time-dependent functions have been demonstrated,387 with TNF expression being protective at later time points after injury.
Aside from BCL-2 and the prosurvival components of neuroinflammation, other CNS compounds serve as endogenous neuroprotectants after TBI. Adenosine is a purine ribonucleoside and is ubiquitous in the brain, where it largely functions as a major neuroinhibitory molecule. After experimental brain injury, extracellular adenosine levels increase by 50- to 100-fold compared with baseline,27 a phenomenon that is implicated as an endogenous neuroprotective mechanism. Adenosine functions as a neuroinhibitory molecule through its primary target, the A1 receptor. A1 receptor activation is a key local regulator of excitatory amino acid release during ischemia and trauma.12 Adenosine can also hyperpolarize neuronal membranes and attenuate intracellular Ca++ accumulation. Neurotrophins might also have a neuroprotective role after TBI, particularly because early increases in neurotrophin expression have been reported in experimental TBI.193,492
Concomitant Injury
TBIs (especially the more severe cases) often are not an isolated injury. High-speed vehicular crashes and active combat frequently result in multiple injuries that can affect outcome.230 In civilians, associated injuries are linked with more disability even 1 year after the injury.455 TBIs associated with multiple other injuries are also associated with longer acute care lengths of stay and need for rehabilitation resources.453 In addition to increased functional deficits, rehabilitation needs, and more disability, concomitant injuries can directly affect secondary injury and associated pathology, and these phenomena have received some attention in the TBI literature. For example, experimental models incorporating TBI and hemorrhagic shock show increased hippocampal injury compared with TBI alone, a phenomenon likely associated with increased secondary ischemia.92
Military Blast Injury
With the ongoing military conflicts in Iraq and Afghanistan, TBI is not only a common problem in the civilian population but also is a major problem for active combat military personnel. The frequent use of improvised explosive devices in these conflicts has resulted in a large number of military personnel (see previous epidemiology sections) sustaining TBI from blast injury. Blast injury can result in both blunt and penetrating trauma and follows a specific nomenclature (Box 49-2). Explosives can be categorized as high-order explosives (HE) or low-order explosives (LE), and each can cause a distinct injury pattern. HE produces a supersonic overpressurization shock wave that is associated with “primary injury.”441 In addition to middle ear damage, brain injury can occur from primary blast injury without overt evidence of injury. The rapid pressure changes generated from the overpressure wave result in shear and stress forces that lead to trauma such as concussion, SDH, and DAI.94,421 Penetrating head injury from flying debris is the result of secondary blast injury. Both HE and LE can result in tertiary blast injury from being physically thrown by a blast wind into the ground or other objects. Quaternary injury includes asphyxia or inhalation of toxic chemicals that can also contribute to brain injury. Many patients suffer from TBI induced from more than one facet of blast injury.
BOX 49-2 Immediate Effects of Blasts and Explosions in Traumatic Brain Injury
Modified from DePalma RG, Burris DG, Champion HR, et al: Blast injuries, N Engl J Med 352:1335-1342, 2008, and CDC Primer
Research is increasingly being conducted to develop relevant blast injury models of TBI to examine and characterize mechanisms of primary and secondary injury for blast TBI and to assess behavioral effects.21,60,264 The clinical difficulties after blast TBI of any severity can include headache, insomnia, decreased memory and attention, slower thinking, irritability, and depression.91 Additional research is focused on clinical symptom screening and outcomes, as well as physiologic markers and biomarkers associated with this type of TBI.7,418,436
Neurobiology of Neurotransmitter Dysfunction After Traumatic Brain Injury
The neurobiology of catecholaminergic dysfunction, particularly dopamine (DA) dysfunction, is the most characterized in the experimental literature.20 Early changes in DA levels occur after experimental TBI.278 Chronic DA receptor and synthetic protein changes after TBI also have been characterized using experimental TBI models. The DA transporter (DAT) is a key protein regulating the extracellular half-life of DA in the synaptic cleft, and the therapeutic target of neurostimulants like methylphenidate and amphetamine. Long-term changes in striatal and cortical DAT expression and intracellular trafficking have been identified451,462 that are gender specific.450 Functional changes in DAT after experimental TBI also lead to altered striatal neurotransmission.448,461 These experimental changes are corroborated by clinical neuroimaging studies demonstrating TBI-related decreases in DAT expression 1 year after injury.107,384 Other changes in postsynaptic secondary signaling mechanisms, as well as changes in presynaptic DA synthetic enzyme levels, have been identified.20
Behavioral pharmacology studies show that daily treatment with the DAT inhibitor methylphenidate (MPH) has a beneficial impact on cognition after experimental TBI.228 Recent work suggests that the cognitive benefits of MPH are more pronounced in males456 and that daily MPH therapy has a neurorestorative effect on striatal neurotransmission in males through MPH-mediated changes in DAT function.451,461 Daily treatment with the pleiotropic DA enhancer amantadine,103 as well as the D2 receptor agonist bromocriptine, also shows a benefit on learning and memory.226 Early treatment with bromocriptine also appears to decrease oxidative damage initiated by the injury.223
Evidence also suggests that norepinephrine (NE) systems are disrupted after experimental TBI but can be manipulated to improve recovery. Cortical NE levels are decreased in the first 24 hours after injury,348 and NE turnover is impaired chronically after injury.137 α1-Adrenergic receptor expression is also decreased.349 Local NE infusions early after injury enhance recovery,36 and stimulating adrenergic systems with amphetamine can decrease neuronal damage after TBI.153 Recent studies suggest that at least some of the beneficial effects of amphetamine might be due to its ability to increase neurotrophin production after injury.164 Daily treatment with low doses of the selective NE transporter (NET) inhibitor atomoxetine shows benefit on cognitive recovery after experimental TBI.355
Experimental TBI studies also demonstrate decreased hippocampal cholinergic transmission105 and alterations in cholinergic synthetic and metabolic enzyme production.67,105 Daily treatment with cytidine diphosphate (CDP)-choline, a key intermediary in the biosynthesis of phosphatidylcholine, improves cognitive recovery in experimental TBI and increases hippocampal and cortical acetylcholine levels.105 A single dose of the acetylcholinesterase inhibitor, rivastigmine, early after experimental TBI also improves cerebral edema, cognition, and motor functioning.67
Repair, Regeneration, and Recovery
One mechanism by which TBI recovery occurs is through the reversal of diaschisis. Contemporary thought on the concept of diaschisis suggests that the functional effects of diaschisis can be either reversible or permanent. The mechanisms by which diaschisis resolves, however, are poorly understood and likely multifactorial. The resolution of cerebral edema181 and blood flow regulation121 can contribute to early recovery.233 Later during the course, the alleviation of diaschisis might involve factors like synaptic plasticity,295,365 axonal sprouting,372 and cortical reorganization.238 Diaschisis after brain injury can be manipulated by both behavioral experience (rehabilitation) and pharmacologic intervention.325,392 For example, depressed thalamic metabolism after cortical infarction was reversed with administration of neurostimulants such as amphetamine.342
Experimental and clinical research suggests that DA systems are key pathways involved in attention, task salience, and cognition, and that these pathways are chronically impaired after TBI.20 Alterations in both presynaptic and postsynaptic dopaminergic proteins in the striatum and frontal cortex have been documented, including important proteins involved in DA synthesis and uptake.20,451,462,491 Decreases in striatal DAT expression, a key protein in regulating the extracellular half-life of DA, have been documented in humans.107 Further research demonstrates that striatal neurotransmission is impaired, and this might be the result of TBI-induced changes in DA protein functionality.451,462
Noradrenergic systems affect arousal, sleep–wake cycles, vigilance, and cognition, and the NET appears to affect NE neurotransmission, as well as interact with DA systems to affect DA neurotransmission.465 Noradrenergic function is impaired early after TBI. Previous reports suggest that cortical NE levels are decreased within 24 hours after brain injury, and adrenergic receptors are altered after injury.348,349 NE augmentation early after TBI is neuroprotective, however, and NE agonists aid in neurorecovery.37,108 It is important to remember that NE is critical for governing cortical plasticity and facilitating recovery post-TBI. By stimulating adrenergic systems with amphetamine and L-threo-dihydroxyphenylserine (or L-DOPS), a precursor of NE, neuronal damage after TBI can be minimized and sprouting is promoted.153,154,217,414,416
Cholinergic systems are important for memory and cognition, and evidence suggests chronic impairments can occur after TBI. Hippocampus expression of the vesicular acetylcholine transporter is increased; however, evoked release of acetylcholine in neocortex and hippocampus is reduced after experimental TBI.100,106,393 Daily local application of nerve growth factor also enhanced memory and hippocampal neuron function after experimental TBI.101
In addition to acute protection and cholinergic function, other studies support a role for neurotrophin administration in promoting long-term recovery. In contrast to acute increases in neurotrophin production, other work suggests that hippocampal brain-derived neurotrophic factor (BDNF) is decreased during chronic recovery from experimental TBI.66 An enriched environment can reverse these decreases,66 but chronic pericontusional administration of BDNF does not improve behavioral outcome or hippocampal cell survival.32 Intraparenchymal administration of both basic fibroblast growth factor and nerve growth factor reduces cognitive deficits in experimental TBI.289,402 With these positive studies, improved recovery might be secondary to stimulation of brain repair and neuroplasticity.
Evaluation and Treatment of Traumatic Brain Injury
Management of Mild Traumatic Brain Injury (Concussion)
Like more severe injuries, concussion results in significant alterations in brain physiology. After an initial increase in glucose use, brain metabolism is impaired and associated with the development of energy crisis, oxidative injury, and the initiation of biochemical cell death pathways. These derangements can occur for up to weeks after the concussion and persist even with a normal GCS score and general neurologic examination.151
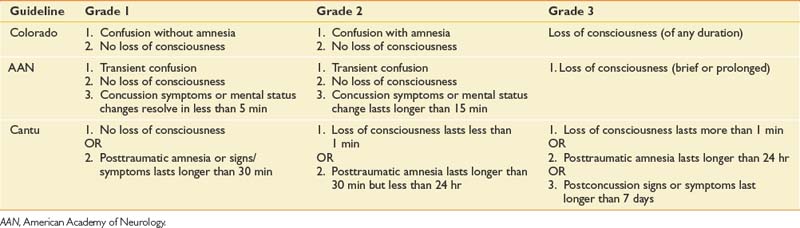
There are several criteria and scoring systems for classifying degree or severity of mild TBI. The World Health Organization Collaborating Centre Task Force on Mild TBI states that key criteria for identifying persons with a mild TBI should include (1) at least one of the following: confusion, disorientation, loss of consciousness for less than 30 minutes, PTA for less than 24 hours, or other transient focal neurologic abnormalities, and (2) GCS score of 13 to 15 after 30 minutes or presentation to a health care facility. Findings with these criteria should be made in the absence of illicit drugs, alcohol, medications with sedating side effects, or other injuries or problems.58 Patients with uncomplicated TBI typically do not have associated abnormalities on standard imaging tests like computer tomography (CT). Concussion severity nomenclature has been developed for the purposes of injury characterization and injury management. Some examples of concussion are provided in Table 49-1.57,81,215
Patients with concussion frequently complain of any number of associated symptoms, including memory loss, poor concentration, impaired emotional control, posttraumatic headaches, sleep disorders, fatigue, irritability, dizziness, changes in visual acuity, depression, anxiety, personality changes, and seizures.9,481 These problems can affect ADLs, social relationships, and abilities in the workplace. For most individuals with mild TBI, the symptoms resolve over time. A subset of patients, however, will have persistent symptoms classified as postconcussional syndrome and can require outpatient follow-up evaluation. The types of symptoms, proportion of patients who have persistent symptoms, and the duration of symptoms required to define symptoms as persistent have been widely debated. Reports of symptom duration to define persistent deficits can range between 3 and 12 months. The fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) suggests that individuals showing objective evidence of cognitive deficits, as well as three or more subjective symptoms, that represent a change in baseline function for at least 3 months can be classified with postconcussional disorder. Other classification systems, such as the International Classification of Diseases, 10th edition (ICD-10), do not include objective evidence of cognitive deficits.481 Despite variability in definitions and classification, some studies suggest that females are more likely to have postconcussional syndrome.346
Athletes comprise a large portion of the population who sustain mild TBI, and many of these athletes are children. Special issues arise with this population with regard to “on site” concussion screening tools, determining the most effective algorithm for return to play, safety equipment use, management and prevention of recurrent injuries, and academic performance. As a general rule, athletes should be free of postconcussive symptoms before returning to competition. One report shows that 30% of high school football players return to play the same day of injury.168 Many return-to-play guidelines, however, outline that athletes with a grade 1 or 2 concussion have a minimum of a 7-day period free of symptoms before returning to play.169 Same-season injuries often occur within 7 to 10 days of the initial injury, a timeframe in which there is increased vulnerability to damage with the second insult.151 Most return-to-play algorithms emphasize a graded protocol for reintroducing the athlete to physical exertion. Recent studies, however, suggest that academic and cognitive exertion can affect postconcussive symptoms and cognitive testing performance and should be considered when developing return-to-play protocols for athletes.271 Several guidelines recommend termination of sports participation for the remainder of the season if an athlete sustains a third concussion within that season.57,358 Athletes who have one concussion are at increased risk for subsequent concussion,169 and making a return to contact sports even after a single concussion is a decision that requires careful thought and consideration.
Acute Medical Management of Moderate to Severe Traumatic Brain Injury
The care of the patient with TBI begins in the field. Evidence-based guidelines put forth by the Brain Trauma Foundation and the American Association of Neurological Surgeons support a regional organized trauma care system that addresses prehospital care and triage. Direct transport of a patient with severe TBI to a Level 1 or 2 trauma center is associated with decreased mortality compared with indirect transport.183 Facilities defined by the Guidelines for Prehospital Management of Traumatic Brain Injury as being appropriate facilities for transport include those offering CT scanning, neurosurgical care, ICP monitoring, and treatment capabilities. These facilities can provide 24-hour neurosurgical care, and intensive care treatment ICP monitors are routinely used. Each state can designate trauma facilities based on the availability of such care. Although treatment guidelines for initial management are not specific, the recommendations include complete and rapid physiologic resuscitation. Correction of hypoxia, including intubation, is appropriate. Sedation and neuromuscular blockade might need to be used to optimize the transport of the head injury patient.41
ICP has been the mainstay of monitoring after severe TBI. ICP can be monitored using a ventriculostomy, which is the procedure of choice because it allows therapeutic CSF drainage. ICP monitoring is appropriate in those patients with a GCS score of 8 or less after resuscitation, head CT showing contusions or edema, or systolic blood pressure less than 90 mm Hg.316 ICP monitoring can also be considered in patients with severe TBI and a negative head CT if they are more than age 40, posturing, and/or have systolic blood pressure less than 90 mm Hg.40 Elevated ICP is defined as 20 to 25 mm Hg in clinical practice. Maneuvers to decrease ICP include elevating the head of the bed 30 degrees, treatment of hyperthermia, mannitol administration, sedation, and brief hyperventilation. Prolonged chronic hyperventilation can negatively affect cerebral blood flow.39
CPP is defined as the mean arterial pressure minus ICP and is the pressure gradient driving cerebral blood flow. Studies have documented worsened clinical outcomes in TBI patients with episodes of low CPP.69 The current therapeutic recommendation is that CPP be maintained at more than 60 mm Hg in adults.42 Measurement of cerebral oxygen tension allows the practitioner to directly measure ischemia, rather than rely on the indirect measurement of CPP or ICP. Tissue oxygenation monitors, directly placed in brain tissue via an external ventricular drain (EVD), can detect focal ischemic changes that can go undetected with global measures. Interventions are then tailored to the cause of the decrease in tissue oxygenation. Studies have shown reduction in mortality with addition of this monitoring.411
Other treatment interventions have been studied in acute care clinical trials. A multicenter trial evaluating the role of corticosteroids in management of acute TBI showed an increase in mortality 2 weeks after TBI compared with placebo.367 Elevated CSF cortisol levels in severely injured patients might contribute to this outcome.368 Progesterone is a drug with pleiotropic neuroprotective properties identified in several preclinical studies occurring over decades.408 Recent clinical trials have shown its safety, and progesterone appears to have potential as a therapeutic agent.482,487
Animal studies have evaluated the protective use of hypothermia, with several showing positive results.76,96 Clinical trials using moderate hypothermia have shown some reduction in excitotoxic insult274,452 and neurologic impairment; however, the results have not been consistent. For example, a large multicenter trial of hypothermia did not show improved outcomes in those patients with severe TBI.78 To transition this treatment to routine human use, factors such as treatment window, rewarming rates, and type of anesthesia must be defined.407 Gender effects on treatment efficacy and physiologic response to treatment should also be considered.23,417,452
Surgical treatment of TBI is indicated when intracerebral fluid collections exert a significant mass effect. Mass effect can be gauged by degree of midline shift on neuroimaging, and the decision to intervene can require serial neurologic examinations and CT scans.52 Depressed skull fractures greater than the thickness of the skull should be elevated to decrease the risk for infection, especially in the setting of a dural laceration. Surgical treatment can also be used to lower ICP. The use of decompressive craniectomy to decrease ICP is an accepted intervention, although the reduction of morbidity and mortality has not been proven.477
Secondary complications occurring in the intensive care unit (ICU) can also negatively affect TBI outcomes. Elevated glucose levels are associated with increased mortality after TBI, presumably as a result of increased lactic acid production leading to ischemia and impaired phosphorus metabolism.375 Sodium imbalance can potentiate risks for seizure, as well as worsen level of consciousness. Sodium imbalance is directly related to volume status. Hyponatremia can be exacerbated by the use of excessive volumes of intravenous fluids. This occurs even in the setting of normal saline or lactated Ringer’s solution used for fluid resuscitation, making monitoring of electrolytes and volume status important in preventing secondary complications. Prevention and aggressive treatment of infections are also important. Hyperthermia is another source of acceleration of secondary insults to brain parenchyma, increasing ischemia.273
Early initiation of nutrition is vitally important in the TBI patient. Metabolic rates can increase 40% during the early stages after injury. Guidelines recommend replacement of 140% of resting metabolism expenditure (100% of resting metabolism expenditure in paralyzed patients). The use of the gastrointestinal tract is preferred to maintain nutritional status. The goal of treatment is to maintain a positive nitrogen balance, although this is difficult to achieve. Measures such as nitrogen balance via a metabolic cart study, prealbumin levels, and liver functions should be followed in the ICU. Delaying nutrition replacement is associated with increased mortality.184
The physiatrist plays a role in evaluating patients who have sustained TBI. The early assessment includes prevention of orthopedic and immobility complications. Early consultation can result in improved mobility, improved functional outcomes, and decreased acute care length of stay. In this model, rehabilitation assessment is a portion of the acute medical care, providing education and support for patients, families, and caregivers.453
Physiologic Measurements During Acute Care
Somatosensory-evoked potentials (SSEPs) are recorded from the scalp after stimulation of a sensory or mixed nerve in the peripheral nervous system. SSEPs are often used in severe TBI cases to predict survival and evaluate posttraumatic coma and persistent vegetative states. In some cases, SSEP testing is used as an adjunct to the clinical examination in determining brain death.174 Cant et al.56 found that bilateral loss of the median SSEP was indicative of impending death, and asymmetric findings observed in the cortical response were indicative of severe residual deficits. Their study indicated that serial SSEPs often change through the hospital course, and each study does not always correlate well with the ultimate outcome.
Like other neurologic evaluations, electroencephalography (EEG) can be beneficial when assessing TBI patients to detect injury severity and depth of coma.378 Degree of unconsciousness can quickly change, and continuous EEG (cEEG) monitoring can detect electrophysiologic signs of clinical deterioration.175 Initial EEG recordings taken within 24 hours of injury, however, are generally more abnormal and of less prognostic significance than those performed after 24 to 48 hours.419 cEEG is a standard part of care at some Level 1 trauma centers, and cEEG can be used to effectively identify subclinical electrographic seizures, which are associated with other physiologic factors that contribute to poor outcome, such as ICP elevations and increases in metabolic stress.447
Pupillary reflexes have been used not only as clinical indicators of surgical emergencies and complications in TBI associated with edema and herniation but also as a tool in determining lesion location, mortality, and global outcome after injury. The presence or absence of pupillary reflexes and degrees of anisocoria have been used in models predicting the probability of intracerebral hematoma and lesion location.68
Although not yet used routinely at the bedside, biomarkers have considerable potential as adjunctive tools in TBI diagnosis, prognosis, and monitoring treatment effect. Protein biomarkers that have been studied in TBI include either (1) markers of structural damage or (2) markers reflective of the cellular and molecular cascades observed with secondary injury and repair. Protein biomarkers have also been studied in both CSF and serum. CSF biomarker studies are valuable in that measured levels are largely a reflection of CNS pathology. CSF collection is only routine, however, in severely injured patients who require an extraventricular drain for management of ICP. Serum markers are attractive because of their potential application to mild and severe cases of TBI, but serum biomarker levels are often limited by the extent of BBB permeability present after the injury. Markers of acute injury have been much more thoroughly studied compared with chronic markers of injury.
Neuroimaging for Medical Management
The head CT is a mainstay of diagnostic imaging in TBI, and findings have usefulness in identifying mass lesions that constitute a neurosurgical emergency and in correlating with outcome. Mass lesions, including SDH and EDH (Figure 49-2), can often cause shifting of the midline structures and are life threatening if not surgically evacuated (Figure 49-3). Intraparenchymal contusions can also cause shifting of midline structures (Figure 49-4). DAI is hard to appreciate on head CT unless it is severe with associated small hemorrhages forming near sheared axons (Figure 49-5). In mild TBI the head CT is often negative, but other modalities might capture subtle pathology associated with less severe injuries.
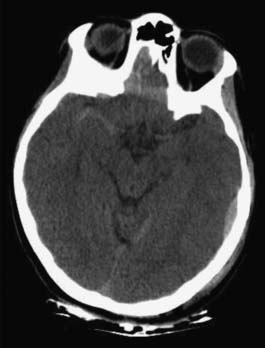
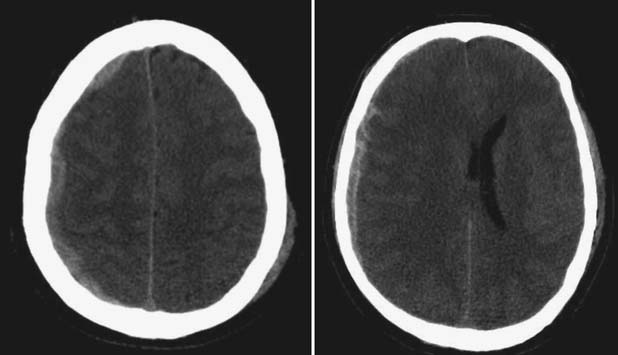
FIGURE 49-3 Computed tomography scans showing subdural hematomas, which have the potential to result in midline shift.
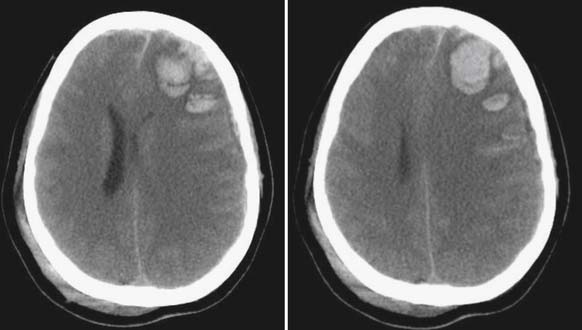
FIGURE 49-4 Hemorrhagic contusions can also cause midline shift as seen by computed tomography scan.
CT is the current standard neuroimaging modality for the initial evaluation of patients with suspected moderate to severe TBI.90,138,250 CT methods, which are based on the same principles as plain-film radiographs, are advantageous because they allow for rapid, noninvasive three-dimensional imaging. This can accurately detect facial and skull fractures, as well as acute hemorrhaging and mass effect. This allows for optimal medical management of trauma patients who might require immediate surgical intervention. Repeat CT scans can be used to evaluate recovery or to monitor for additional complications. For example, if a patient with TBI does not progress as expected or declines in neurologic status, repeat CT scans are often used to diagnose issues such as new or evolving bleeding or hydrocephalus. Because of their high clinical usefulness and relatively low cost, CT scanners have become readily available for use with TBI evaluation. As with any radiograph, there are risks associated with ionizing radiation that require clinical judgment and risk–benefit analysis when considering multiple CT evaluations over time. This is particularly an issue with certain higher risk groups, such as pregnant women and children.
Less than 10% of mild TBI cases have positive CT findings, and less than 1% require neurosurgery.206,250 These statistics have resulted in significant debate regarding the appropriate use of CT in cases of mild TBI. The New Orleans Criteria186 and the Canadian CT Head rule412 indicate that CTs should be obtained in cases of mild head trauma if specific additional criteria are met suggesting the possibility of more severe or evolving neurologic injury. Examples include age and symptoms such as seizure, vomiting, headache, and significant PTA.
Magnetic resonance imaging (MRI) is a second method of structural neuroimaging that has proven clinical usefulness in TBI.138,249,250 MRI has the capability of producing high-resolution structural images of the brain by exploiting the magnetic properties of hydrogen atoms in tissues of the body, as well as variations in the density and water content of body tissues. Compared with CT, MRI scans take longer to complete and are more susceptible to motion artifacts. This can make it more difficult to scan individuals who are agitated. MRI is also less able than CT to detect skull fracture and acute blood. Because of the magnetic environment of the MRI scanner, it also can be more complicated to scan individuals with the clinical monitoring and treatment equipment required during acute care, or with medically implanted devices or objects that might be affected by the magnet (i.e., pacemakers, aneurysm clips, plates, and rods). However, many MRI-compatible instruments and surgical implants have become available. MRI can also be contraindicated when there are causes of injury such as gunshot wounds or other accidents involving the presence of metal fragments. MRI does have resolution superior to CT and provides much higher detail in the soft tissues of the body. Therefore MRI becomes the methodology of choice for the evaluation of injury to the brain stem and frontal areas, as well as for detecting small hemorrhages and nonhemorrhagic white matter injury. As a result, MRI becomes increasingly useful with time from trauma, when patients become more medically stable and additional diagnosis of DAI or other small areas of hemorrhage might be helpful when considering treatment and prognosis.138,249,253,466
The primary sequences for conventional MRI scanning are T2-weighted images and fluid-attenuated inversion recovery (FLAIR) sequences. These sequences suppress the signal from CSF, resulting in improved visualization of cortical and periventricular lesions, as well as nonhemorrhagic shear injury associated with DAI. T2-weighted gradient recalled echo (GRE) has additional sensitivity to blood breakdown products, further adding to its sensitivity for DAI.138 Because MRIs can be obtained in sagittal, coronal, and axial planes without moving the patient, additional nonconventional MRI sequences can be helpful. For example, coronal and sagittal FLAIR images can be helpful at discerning DAI involving the fornix and corpus callosum compared with routine axial T2-weighted images.249
Management of Patients With Disorders of Consciousness
Some patients with severe TBI have prolonged periods of depressed consciousness that might never resolve. The accurate classification of these individuals is vital for appropriate prognostication. Numerous terms have been used to classify these different states. Coma is defined as a state of pathologic unconsciousness in which the eyes remain closed and there is no evidence of purposeful motor activity. Vegetative state typically follows a period of coma, where there is some evidence of wakefulness, in the form of eye opening, without any sustained or reproducible responses to the environment. Vegetative states should be described with the length of time since injury. The terms persistent and permanent should not be used to describe the length of time since injury.148 A minimally conscious state (MCS) is the condition of severely altered consciousness but in which there is definite, reproducible evidence of self-awareness or environmental awareness. The behaviors examined include command following, intelligible verbalization, recognizable yes–no responses, and movements or emotional responses triggered by environmental stimuli.146
Functional neuroimaging has been useful to understand the neuropathology of depressed consciousness, especially in defining those patients who are classified as MCS or who might have a higher likelihood of recovery of communication or functional object use. Both functional MRI and positron emission tomography (PET) imaging provide correlates of the differences between vegetative state and MCS. In PET studies, patients with clinical evidence of vegetative state typically have reductions of overall cerebral metabolism of 50% or more.377 In comparison, patients with MCS have been shown to have activation of appropriate cerebral networks in response to environmental stimuli, although the activation might not be the same as normal controls with novel or complex stimuli.388 Although the neuroimaging may eventually be used to discriminate between vegetative state and MCS, it currently does not have a role in clinical diagnosis and prognostication.
The current evaluation of patients with depressed levels of consciousness includes a thorough neurologic examination, including pupillary responses, brain stem reflexes, and ocular movements. Brain stem reflexes commonly elicited include corneal responses, gag reflex, and oculocephalic reflexes, referred to as doll’s eyes. These reflexes show integrity of brain stem pathways, with reproducible behavioral responses to specific areas of sensory stimulation. Also recorded are the direct and consensual pupillary responses, as well as a response to visual threat. Observation of spontaneous activity and responses to environmental stimuli is important. Common mistakes include attributing purposefulness to responses that are reflexive, allowing insufficient observation time for patient response to a stimulus, and underconsideration or overconsideration of family observation of behavior. To best observe behaviors, it is important to optimize environmental conditions and patient positioning and avoid sedating medications. Standardized rating scales are available to differentiate vegetative state from MCS. These allow for the detection of subtle improvements and can offer interrater and test–retest reliability when incorporated into daily clinical testing.147
A variety of interventions have been studied in these patients. The use of sensory enrichment in both naturally occurring events and in the administration of multimodal sensory stimuli has been reported in case studies and retrospective data analyses. The effectiveness of these techniques is not clear, although there is no report of harm associated with sensory stimulation.149 Pharmacologic interventions can be used to promote arousal and behavioral persistence. Medications frequently include psychostimulants, DA agonists, and tricyclic antidepressants. Bromocriptine, a DA agonist, was studied in a series of five patients in a vegetative state. Treated patients had greater physical and cognitive recovery at 12 months compared with historical controls. This study lacked a sufficient sample size, however, and did not address the possibility of spontaneous recovery.336 Recent trials have investigated the implantation of electrodes in the brain stem and thalamus to stimulate the reticular system and increase arousal. Although studies are limited, behavioral changes were observed in some patients.437
Behavioral Measures of Responding and Cognition
Emergence From Coma
Evaluation of functioning during the initial stages of emergence from coma generally involves the monitoring and serial assessment of a patient’s ability to respond to external stimuli. This is often done through the use of a standardized measure of responsiveness, such as the Coma Near Coma (CNC) scale.353 This 11-item scale was designed to measure small changes in responding in patients with severe brain injuries functioning at a low-level state. An additional scale is the JFK Coma Recovery Scale–Revised (CRS-R),147 which is a 23-item scale used to assess functioning in six areas: auditory, visual, motor, oromotor, communication, and arousal. Instruments such as the CNC and CRS-R scales are generally useful during acute care hospitalization or, at times, if an individual who is minimally responsive is transferred to the inpatient rehabilitation setting or another longer-term care facility.
Evaluation of Posttraumatic Amnesia
As the individual emerges from coma and is able to respond to verbal inquiry, instruments such as the Galveston Orientation and Amnesia Test (GOAT)257 or The Orientation Log (O-Log)203 are used to track progress through the PTA phase of recovery. Both of these instruments essentially measure and score serial responses to orientation questions and allow for the objective scoring of the responses elicited from the patient. Emergence from PTA is then defined by the consistent attainment of a score on one of these instruments indicating orientation.
Although rehabilitation scales, such as the GOAT and O-Log, generally focus on disorientation and amnesic symptoms, other elements of confusion (i.e., delirium and psychiatric types of symptoms) have also been noted during the acute phase of recovery from TBI. There is increasing awareness of the need to measure these symptoms, as well as to develop instruments to bridge gaps between scales traditionally used to measure PTA and those traditionally used to measure delirium.425 One example is the Neurobehavioral Rating Scale,255 which combines portions of the Brief Psychiatric Rating Scale337 with additional items to measure other psychiatric symptoms associated with TBI. The Confusion Assessment Protocol (CAP)395 focuses more on delirium than other scales.395
The Ranchos Levels of Cognitive Functioning Scale (LCFS),173 commonly referred to as the Ranchos Scale, is a widely accepted tool used to describe the process of cognitive recovery as an individual emerges from coma, then progresses toward emergence from PTA or delirium, and emerges to near-normal cognitive functioning (Table 49-2). This scale presents recovery as a progression through eight typical stages. It has been widely adopted as a method to assess patient functioning for purposes of rehabilitation planning and treatment, and to explain patient progress to families.
| 1 |
(Further information and rating forms for the CAP, CNC, CRS-R, GOAT, O-Log, and LCFS may be found on the “scales” page of the Center for Outcome Measurement in Brain Injury website: http://www.tbims.org/combi/index.html.)
Inpatient Rehabilitation Assessment and Management
Role of the Neuropsychologist in Inpatient Rehabilitation
The role of the neuropsychologist in inpatient rehabilitation service is multifaceted. The neuropsychologist provides ongoing assessment of recovery of cognitive function and works with therapy and nursing staff to integrate information about cognitive abilities and limitations into implementation of effective and appropriate rehabilitation goals for the patient. Ideally the neuropsychologist is available to observe and cotreat with other therapists as needed. Although neuropsychologists have specialized training in brain–behavior relationships and expertise in the testing and evaluation of cognition, they also apply that expertise to the evaluation of interactions between cognitive strengths and limitations, as well as behavior. As a result, they are generally experts in behavior management and can often assist with organization and implementation of behavior plans for patients as needed. This is often particularly relevant on TBI units, where agitation related to recovery from TBI can threaten or impede patient progress in rehabilitation. In some settings, psychologists function both as neuropsychologists and rehabilitation psychologists. Rehabilitation psychology focuses on adjustment to disability and other personal, social, and situational issues in an effort to assist individuals with all types of disabilities to work toward and maintain healthy and satisfying lifestyles.
Posttraumatic Seizures
Posttraumatic seizures (PTS) are a significant complication arising from TBI. PTS accounts for 20% of symptomatic seizures and 5% of all seizures in the general population.113 Up to 86% of patients with one seizure after TBI will have a second within 2 years.179 Moreover, PTS is associated with increased disability and limitations with ADLs.17 The neurobiology of PTS is not well understood. Neuroinhibitory molecules like adenosine and γ-aminobutyric acid (GABA), however, afford relative protection from this complication. In fact, genetic variability within the ubiquitous adenosine A1 receptor has been linked with risk for PTS.301 Conversely, excitotoxicity likely contributes to the development of PTS. PTS occurrence is related to injury severity.432 Other reported risk factors include biparietal contusions, dural penetration with bone and metal fragments, multiple intracranial operations, cortical contusions, SDH, significant midline shift, early PTS, and skull fractures.13,113 PTS has commonly been defined as occurring in the immediate period (<24 hours after injury), early period (24 hours to 7 days after injury), and late (>7 days after injury).
Phenytoin (Dilantin) is commonly used for PTS prophylaxis and treatment, and it acts by diminishing excitatory tone and augmenting inhibitory neurotransmitter systems in cortical and subcortical structures. Previous clinical trials suggest that phenytoin therapy for 1 week provides protection against early PTS.433 Based on this literature, the Association of Academic Neurologists currently recommends early intervention with phenytoin (intravenous) given as a loading dose as soon as possible, followed by a 7-day course in the asymptomatic moderate to severely brain-injured population.62 New drugs that do not require active monitoring of drug levels are commonly used clinically for PTS; however, research on their efficacy in preventing PTS specifically is limited.
Despite recommendations supporting limited treatment for seizure prophylaxis,431 people with TBI often receive PTS prophylaxis on a long-term basis with anticonvulsant medications that require regular monitoring and are often associated with unwanted side effects, including sedation. Patients who go on to develop PTS also experience similar issues with long-term treatment.98,406 Recent evidence using an experimental model of TBI demonstrates that long-term treatment with phenytoin after injury leads to increase cell death and poorer function on cognitive behavioral tasks.304 This work further supports following current guidelines for PTS prophylaxis with phenytoin, and prolonged therapy should be minimized when possible.
Heterotopic Ossification
Heterotopic ossification (HO) (Figure 49-6) is a common complication occurring after TBI and is a poorly understood process by which ectopic bone is formed outside of the skeleton. Incidence of HO after TBI ranges from 11% to 28%.188 Incidence can be higher in those sustaining military-related blast injuries, in which TBI and traumatic amputation (both of which increase the risk of HO development) often co-occur.134,347 Patients with more severe TBI and associated immobility, spasticity, and fractures seem to be at relatively greater risk for this complication. Dysautonomia is also linked with higher HO risk.188 Among acute inpatient patients undergoing rehabilitation for TBI, HO is considered a risk factor for poorer outcomes and decreased home discharge rates.207 Blood markers associated with bone metabolism, such as alkaline phosphatase and osteocalcin, are nonspecific for HO.315 Radiographs can identify HO in more advanced cases, but bone scanning is more sensitive in identifying early and asymptomatic cases.
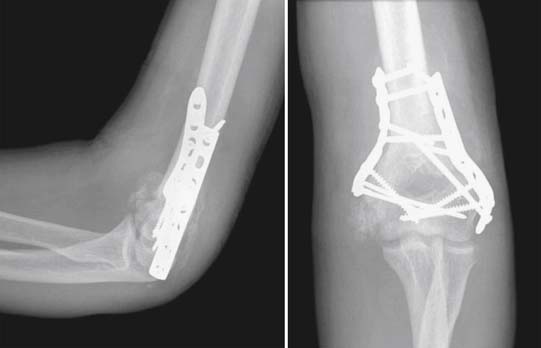
Although the pathophysiology of HO after TBI is poorly understood, evidence suggests that CNS processes facilitate HO bone formation. Bone remodeling is subject to central control through the sympathetic nervous system, which is partially regulated by the endocannabinoid system. After experimental TBI, cannabinoid-mediated down-regulation of NE at osteoblasts contributes to increased bone formation.423 Other research suggests that humeral factors in human CSF after TBI are osteoinductive, but specific factors have not been identified.141 Leptin, a metabolic protein, is also a potential mediator of HO through both central and peripheral mechanisms.63
Common prophylactic methods include antiinflammatory medications like indomethacin, irradiation, and Ca++ binding chelating agents such as etidronate (Didronel). Nonsteroidal antiinflammatory drugs (NSAIDs) are effective in preventing HO formation in total joint replacement patients,318 but less is known about the specific effects of NSAIDs on HO after TBI. At this time, a Cochrane Database review fails to provide adequate research evidence to support these therapies for acute HO treatment.182 Once HO formation occurs, it can significantly affect joint range of motion and mobility. Areas of HO formation take several months to mature. After maturation is complete, excision of the ectopic bone can often improve joint motion and mobility.305
Deep Venous Thrombosis
Deep venous thrombosis (DVT) is a significant source of morbidity and risk for mortality for patients with TBI. In patients with severe TBI, pulmonary embolus secondary to DVT is an important cause of death, and the estimated incidence of DVT is 40%.73 The effectiveness of routine screening for clot at rehabilitation admission has not been proven, and there is no standard of care for the initiation of DVT prophylaxis and treatment in patients with TBI. Increasing evidence supports the safe use of either heparin or low-molecular-weight heparin within 24 to 72 hours after severe TBI or intracranial bleed.142 This decision can be made in consultation with the neurosurgeon about the risk of rebleed in patients after review of their neuroimaging studies.
Patients at highest risk for DVT include those at advanced age, severe injury, prolonged immobilization, significant fractures, and presence of clotting disorder.369 Unfortunately the behavior of the TBI patient, including fall risk, agitation, and ability to provide a safe environment, might prevent one from taking a risk with anticoagulation or even prophylaxis. In those patients who cannot undergo pharmacologic prophylaxis of DVT because of risk of bleeding, mechanical compression devices can be used. Although they have never been completely studied as to effectiveness, it is thought that their effectiveness in clinical practice would be lower than that in controlled trials. If they are used, it is important that they are consistently used, appropriately sized, and used for the maximum hours each day. For patients with agitation or requiring restraints, tolerance of these devices can be low. The length of DVT prophylaxis is not clearly defined in the TBI population. Among general rehabilitation patients, the ability to ambulate greater than 100 feet is an important milestone to discontinue prophylaxis.
Swallowing and Nutrition
Moderate to severe TBI is associated with specific nutritional needs. Patients demonstrate increased caloric requirements because of hypermetabolism, increased energy expenditure, and increased protein loss. Early institution of enteral nutritional support might decrease morbidity and mortality, shorten hospital length of stay, and potentially improve immune function.171,494 Nutritional status monitoring can include review of laboratories and weights and, in more complex patients, metabolic cart studies. Clinical dieticians are important team members in this regard.
As the patient’s level of alertness improves, swallowing evaluation can include a bedside swallowing assessment or video fluoroscopy. Because normal gag reflexes and good cough reflex are not completely predictive of normal swallow function, video fluoroscopy often identifies silent aspiration and improves clinical decision making when relaxing dietary restrictions.476 Even after video fluoroscopy, institution of oral nutrition is often done in a step-wise manner, evaluating the effects of behavior on feeding, as well as variations in level of arousal and fatigue during a day (see also Chapter 27).
Bowel and Bladder Dysfunction
Loss of bladder and bowel control is common after TBI. Injury to cortical and subcortical structures can lead to loss of control over these functions or dyscoordination of sphincter management. The early incidence of urinary incontinence after significant TBI is approximately 62% of patients. The patterns of dysfunction include an uninhibited overactive bladder, as well as poor perception of bladder fullness and poor sphincter control. During acute care and rehabilitation, urinary tract infections are common, affecting functional outcome on discharge, skin integrity, and increasing the risk of discharge to long-term care.72 Treatment options include behavioral interventions such as timed voiding. Caution must be used in initiating anticholinergic medications because of their adverse cognitive effects (see also Chapter 28).
Bowel dysfunction after TBI includes incontinence and constipation. Constipation is common, as a result of lack of mobility, use of constipating medications, and dietary influences. Bowel programs include stool softeners, stimulant suppositories, and hydration. Bowel incontinence is often associated with more severe injury and poorer functional status. It can also be due to diarrhea, frequently seen with the use of enteral feeds or infectious causes (see also Chapter 19).135
Airway and Pulmonary Management
Pulmonary complications after TBI can be directly related to the effects of trauma, including pneumothorax, hemothorax, flail chest, and rib fractures. Neurologic level of injury might lead to respiratory failure, pulmonary edema, and airway complications. Pneumonia is the most common complication observed in acute care and rehabilitation, occurring in 60% of patients.472 The presence of respiratory failure and need for tracheostomy have a strong relationship with acute care and rehabilitation length of stay. They are also associated with functional status on the Disability Rating Scale and the Functional Independence Measure 1 year after injury.114
The majority of patients with TBI who need a tracheostomy in acute care regain sufficient pulmonary and neurologic function later to undergo decannulation. This can be considered when patients no longer require ventilation, can manage their secretions, and are at low risk for aspiration. Decannulation is usually achieved by serially decreasing the cannula diameter and then by capping the tube. Before capping the tracheostomy, a speaking valve can be used to allow phonation through the open tracheostomy. This is generally only tolerated with a smaller diameter tracheostomy, and the cuff on the tracheostomy must be deflated for safety. It is often recommended if this step is used, that the valve is only placed by trained staff, after they have verified that the cuff is deflated. Tolerance is measured by ability to maintain oxygen saturations and speak clear, long sentences without signs of breathlessness. If the patient tolerates these steps, the tracheostomy may be removed. Visualization of the airway before decannulation via laryngoscopy can assess for tracheal stenosis, subglottic stenosis, glottic stenosis, and tracheal granuloma.478 These tracheal abnormalities are associated with a high risk for respiratory difficulties requiring reintubation and possible surgical intervention.
Decision making in decannulation of patients at Rancho Levels 2 and 3 is more difficult. Poor pulmonary toilet and variable central respiratory status in this patient group leads to pneumonia and sepsis and increased morbidity and mortality with decannulation. Predictors of successful decannulation include younger age, alert cognitive status, and adequate swallowing and cough reflexes.247 In these patients one should consider whether decannulation will make care easier or more difficult in the next level of care for the patient. Timing of decannulation is also important, with it being more troublesome in the winter when respiratory illnesses are common (see also Chapter 34).
Spasticity and Contractures
Spasticity is a common problem observed with upper motor neuron damage such as that occurring with TBI. It is one portion of the upper motor neuron syndrome that contributes to motor dysfunction in these patients. Spasticity is clinically defined as a velocity-dependent increase in tonic stretch reflexes with exaggerated tendon jerk responses.243 Other components of the upper motor syndrome include loss of autonomic control, decreased dexterity, and limb weakness, which is often seen in these patients as well. Although the incidence of spasticity in the TBI population in general is unknown, among those with injuries severe enough to require inpatient rehabilitation, the incidence has been reported to be as high as 84% (see also Chapter 30).493
After significant damage to central motor pathways such as that seen in TBI, acute paralysis often occurs. This leaves the affected muscles and joint immobilized. Immobilization leads to reduction in longitudinal tension in a muscle, the basis for muscle contracture. In animal models, only 24 hours of unloading of tension in a muscle caused a 60% shortening of muscle fiber length.292 Over the next few weeks, both plastic neural and muscular reorganization leads to muscle overactivity defined as spasticity. This further aggravates the development of muscle and joint contractures.157
The early identification of contracture development and spasticity is critical. The risk factors for spasticity development include more severe injury (lower GCS), motor dysfunction (hemiplegia or tetraplegia), associated anoxic injury, spinal cord injury, and age. Development can occur as early as days after injury but is more classically observed months after injury. Patient evaluation includes a clinical and functional history, stretch reflex examination, range of motion assessment, and an active motor examination.195 Measurement of degree of passive abnormal muscle tone is often done with the Ashworth Scale or Modified Ashworth Scale (Table 49-3). The Modified Ashworth Scale assigns a 0 to 4 value based on the amount of resistance measured by an evaluator when attempting to range a joint through an available range of motion. The Tardieu scale is a true measure of spasticity, comparing the differences noted when a muscle is stretched at different velocities, and comparing the angles at which the catch is noted (Table 49-4).112
| Score | Description |
|---|---|
| 0 | No increase in muscle tone |
| 1 | Slight increase in muscle tone manifested by a catch and release at end range of motion |
| 1+ | Slight increase in muscle tone, manifested by a catch followed by minimal resistance throughout the remainder of the range of motion |
| 2 | More marked increase in tone through most of the range of motion but joint easily moved |
| 3 | Considerable increase in muscle tone; passive movement is difficult |
| 4 | Affected part is rigid in flexion or extension |
| Score | Description |
|---|---|
| 0 | No resistance throughout the course of the passive movement |
| 1 | Slight resistance throughout the course of the passive movement with no clear catch at a precise angle |
| 2 | Clear catch at a precise angle, interrupting the passive movement followed by release |
| 3 | Fatiguable clonus, less than 10 seconds when maintaining pressure, appearing at a precise angle |
| 4 | Nonfatiguable clonus, more than 10 seconds when maintaining pressure, at a precise angle |
The cornerstone of spasticity treatment is the use of static and dynamic splinting devices. When used in combination with passive range of motion exercises, splints serve to lengthen the muscles and tendons that are shortening. Although controlled trials of the use of passive range of motion are few, the pathophysiology of spasticity and clinical experience has guided use of these exercises. A stretching program can also be easily taught to family members and transitioned to a home environment. In combination with static splinting such as serial casting, measures of spasticity can decrease.400 In a setting with close monitoring for adverse effects such as pressure sore development, serial casting can be efficacious after TBI. Caution must be used in initiating it while patients are still experiencing high ICPs.496 Physical modalities, such as cryotherapy, superficial heat, and ultrasound, can be used in concert with splinting or pharmacotherapy. Multiple forms of electrical stimulation have been studied in the management of spasticity, with positive results identified in some placebo-controlled studies.158
Systemic pharmacologic agents are also used in the TBI population. Dantrolene is indicated for the treatment of spasticity of CNS origin, including TBI. It acts at the sarcoplasmic reticulum by inhibiting calcium activity. Dantrolene has been associated with hepatic toxicity, requiring regular monitoring of liver enzymes. Baclofen acts both presynaptically and postsynaptically at the GABA B receptors, effectively inhibiting spinal reflexes. Side effects include fatigue, sedation, weakness, hallucinations, and lowering of the seizure threshold. When studied in a TBI population, lower limb Ashworth scores were improved, but a similar effect was not seen in the upper limbs. Somnolence limited the maximum dose in 17% of subjects.297 Other oral agents for spasticity include benzodiazepines, tizanidine, and clonidine. Somnolence and cognitive effects often limit the usefulness of these medications in the TBI population, and some worsen recovery in experimental models.123
Focal chemodenervation can be an important tool for practitioners, without concerns for cognitive side effects. Phenol used for either motor point block or mixed nerve block can cause denaturation of the nerve, which reduces focal spasticity. Advantages include low cost and immediate effects. Complications include sensory loss, possible painful dysesthesias with mixed nerves, and vascular complications. Chemodenervation with botulinum toxin A and B is used offlabel for the treatment of focal spasticity. When used in combination with physical modalities like splinting or serial casting, improved range of motion and Ashworth scores can be achieved.488 There are common patterns of upper motor neuron system dysfunction seen in patients with TBI that can be identified and effectively treated with botulinum toxin.280
Baclofen can also be delivered intrathecally, directly to the lumbar subarachnoid space via an intrathecal pump. This delivery system overcomes the BBB and the peripheral breakdown of the orally administered medication. Although sedation often limits the oral dosing of baclofen, sedation is less frequently seen with intrathecal treatment, and the dose limit is much higher. The surgical complications of intrathecal pump placement include infection, catheter dislodgement, and CSF leak or seroma. The abrupt withdrawal of baclofen, as seen in those with catheter complications, can result in rhabdomyolysis, multiple organ failure, and death.197 When studied in TBI patients, however, after 1 year of treatment Ashworth scores were improved and maintained over the year of treatment.298
Normal Pressure Hydrocephalus
Normal pressure hydrocephalus is one of the most common treatable neurosurgical complications after severe TBI, with an estimated incidence of 45% in those with severe injury.283 Posttraumatic hydrocephalus is largely of the communicating variety; thus there is still free flow of CSF within the ventricular system. Absorption of CSF into the arachnoid granulations is limited by blood products, protein, or fibrosis, leading to dilatation of the ventricular system. Clinical presentation of acute hydrocephalus early after injury includes headache, nausea, vomiting, and lethargy. The symptoms of delayed hydrocephalus or normal pressure hydrocephalus are more subtle. The clinical triad of dementia, gait ataxia, and urinary incontinence is described, with the gait disorder being most amenable to shunting of CSF. In those patients with TBI who have neurologic impairment from their injury, however, the diagnosis should be considered in anyone who worsens or fails to progress adequately. CT scan of the head without contrast to evaluate ventricular size is necessary in evaluation. However, as many as 72% of patients with severe TBI have been reported to have ventriculomegaly.256 In patients with cerebral atrophy leading to ex vacuo dilatation of the ventricle, sulcal prominence is often seen, making it less likely they will be responsive to shunting.35 Further evaluation in questionable cases of hydrocephalus can include large-volume CSF drainage via lumbar puncture, including postprocedure neurologic evaluation, as well as MRI cine flow studies to watch spinal fluid flow through the third ventricle.
Endocrine Dysfunction Associated With Traumatic Brain Injury
Although reported frequencies and numbers vary by study, neuroendocrine disorders affect a significant portion of the population with TBI. Up to 80% of patients with acute TBI have some type of acute pituitary dysfunction, and up to 25% of long-term survivors continue to have pituitary dysfunction.26 Much of the pathology is attributed to primary and/or secondary injury to the hypothalamus and pituitary gland, structures that govern most neuroendocrine function. The hypophyseal portal vascular system that supplies the hypothalamic–pituitary region is vulnerable to traumatic injury, and frequent pituitary hemorrhage and necrosis have been noted in autopsy studies conducted in patients who die acutely from their injuries.34
Several hormones affecting reproduction and metabolism are located in the anterior pituitary gland and include follicular-stimulating hormone, luteinizing hormone, adrenocorticotropic hormone, thyroid-stimulating hormone, prolactin, and growth hormone (GH). Early after injury, several aberrations in serum gonadal hormone profiles occur with severe TBI, and are likely associated with an enhanced stress surge in response to the injury. In cases of severe TBI, serum testosterone levels decline below control levels by 48 to 72 hours after injury. Acute testosterone levels correlate with circulating gonadotropin hormone levels and injury severity. Progesterone levels rapidly decrease in females after severe TBI, calling into question the long-held hypothesis generated from experimental models that females are afforded hormone-dependent neuroprotection.368 Chronic hypogonadism can affect 10% to 17% of patients with TBI.6 In males, hypogonadism is largely secondary to pituitary dysfunction. Menstrual cycle function can be impaired for women up to several months after severe TBI and can have a negative impact on outcome.364
Hyperprolactinemia can affect more than 50% of the population with TBI.5 Trauma can lead to acute depression of the thyroid axis. Chronic hypothyroidism affects between 1% and 22% of the population with TBI and can contribute to cognitive decline.5 Thyroid function should be screened routinely, particularly if recovery is slow to progress. GH deficiency in children is associated with short stature. In adults, GH deficiency decreases muscle mass, energy levels, and cognitive performance. One report suggests that chronic GH deficiency occurs in about 16% to 18% of the TBI population, and is correlated with lower quality of life (QOL) and increased rates of depression.22 Other studies suggest impaired aerobic capacity with GH deficiency after TBI.312 GH replacement, however, can have a beneficial impact on recovery and QOL.239
Functional Evaluation and Treatment Concepts in Rehabilitation
Vestibular Dysfunction
Patients with both severe and mild TBI often present with complaints of dizziness and imbalance. The incidence of such symptoms is reported to be between 30% and 60% in various TBI populations.150 The incidence could be as high as 100% when the patient has sustained a temporal bone fracture.
There are many etiologies for these symptoms. Injury to the peripheral nervous system can result in vestibular symptoms with or without hearing loss. Benign paroxysmal positional vertigo is the most common cause for complaints of dizziness and imbalance. It is clinically characterized by brief episodes of vertigo provoked by movement of the head. Although this can resolve with time, treatments with canalith repositioning procedures can shorten the length of symptoms.189 Another peripheral vestibular injury includes labyrinthine concussion, or the sudden onset of hearing loss and vertigo after TBI in the absence of temporal bone fracture. These patients are good candidates for vestibular and balance rehabilitation. Temporal bone fractures can lead to disruption of the bony labyrinth or internal auditory canal. Trauma to the external canal clinically manifests with bloody otorrhea and severe pain and must be differentiated from a traumatic tympanic membrane perforation.
Central causes of dizziness can include direct trauma to the brain stem and cerebellum. These lesions are often associated with typical oculomotor abnormalities on neurologic examinations. These include various forms of nystagmus at rest, eye movement abnormalities resulting in diplopia, and abnormalities of pupillary response. Rehabilitation techniques to promote alternative strategies for gaze stability can be successful to induce long-term adaptation in the CNS.190 The focus of such rehabilitation programs is to decrease symptoms and allow patients to be more active. In patients with postural instability, exercises to promote static and dynamic postural stability are conducted by manipulating visual, vestibular, and somatosensory cues in the environment.394
Visual and Perceptual Dysfunction
TBI can lead to many symptoms related to vision such as diplopia, photophobia, difficulties with tracking and fixation, and visuoperceptual complaints. Injury can occur to the optic nerve, visual cortex, visual processing centers, or oculomotor nerves. Each of these areas of injury has variable recovery patterns and rehabilitation interventions.
A neuroophthalmologic evaluation can assist therapists in evaluating the impact of vision on ADLs and perceptual motor function. In patients referred for evaluation because of visual complaints after TBI, 85% have visual acuity of 20/20 and 33% have cranial nerve palsy on examination.380 Vision therapy is the clinical treatment of visual disorders with the use of nonsurgical techniques. Individually planned activities based on physical examination are repeated over a series of treatment sessions under the supervision of an optometrist or OT. Techniques include prisms, computer-based treatments, biofeedback, and stereoscopic devices.334
Exercise and Traumatic Brain Injury
In addition to the known therapeutic effects on physical condition and function, a significant amount of literature demonstrates that physical exercise and conditioning can have beneficial effects on neuroplasticity and cognition, as well as slowing neurodegenerative processes.444 Increasing evidence suggests that voluntary exercise is beneficial in TBI recovery. In experimental TBI, voluntary exercise with a running wheel has been linked with increased neurotrophin production and improved cognition compared with a sedentary control group.163 In the experimental literature, environmental stimulation, along with opportunity for social interaction and exercise, provides a loose context for a relevant rehabilitation experience. Several experimental TBI studies demonstrate the relative benefits of this type of “enriched environment” on cognitive performance after TBI compared with an impoverished environment with little environmental stimulation or social interaction.180,227,335,457 Interestingly, environmental enrichment can enhance the effectiveness of pharmacologic interventions in improving cognition.227 The beneficial effects of short-term environmental enrichment, however, appear to be more effective in males compared with females, warranting additional study on the potential gender specificity of this intervention.457
For those with concussion, physical exertion or training too early after injury can worsen cognition and associated symptoms. Exercise programs should be reintroduced in a graded fashion such that the patient remains symptom free during training266,271 This approach is supported by experimental TBI work, where voluntary exercise early after TBI resulted in worse performance on tasks of learning acquisition and memory and a reduction in plasticity-related proteins compared with nonexercising injured control subjects who underwent a sham treatment. In contrast, a delayed voluntary exercise paradigm increased neurotrophin levels and improved performance on cognitive tasks.163
Locomotion assistance systems that provide body weight support and robotic gait orthoses can be implemented for exercise in some patients with these types of deficits to provide additional support and feedback during locomotion training. Body weight–supported treadmill training has been more widely studied in stroke, and some studies suggest that treadmill training with body weight support might be more beneficial than treadmill training alone in this population.310 Although one study has shown that body weight–supported treadmill training does not necessarily improve gait mechanics in a chronic TBI population with multiple gait abnormalities,48 case reports suggest that body weight–supported treadmill training can improve cardiorespiratory status in independent walkers with severe TBI.313
Cognition After Traumatic Brain Injury
Cognitive Deficits After Traumatic Brain Injury
Arousal is one of the most basic cognitive functions required to engage in other higher levels of cognition. Arousal is governed primarily by reticular–thalamic, thalamocortical, and reticular–cortical networks. Multiple neurotransmitter systems, particularly monoamines, affect arousal. These monoaminergic nuclei originate in the brain stem at the reticular formation, and each has multiple forebrain terminal projections. Although other neurotransmitters influence arousal, NE is central to this function, which affects other cognitive domains such as attention and memory.
Attention can refer to a wide variety of different cognitive processes that are both voluntary and involuntary. In addition, attention is a widely distributed cognitive function that involves both cortical and subcortical pathways, including striatal and thalamic inputs and reticular activation. It is also generally accepted that DA plays a significant role with attention.43,478 Attention can be operationalized to include multiple processes. Sensory gating is a preattentive process that filters incoming stimuli to limit information into attention-processing networks. Selective attention involves that ability to direct cognitive resources to a salient stimulus, whereas sustained attention allows for maintenance of attentional processing on a specific stimulus. Like arousal, there is overlap of many of these attentional processes with other cognitive functions such as memory and executive functioning (EF). Further, attentional deficits can contribute to dysfunction in other cognitive domains.
The anatomy involved with memory is better characterized than that related to arousal and attention. Specific memory-processing events (e.g., retrieval, consolidation) can be ascribed to specific brain regions. For example, damage to the hippocampus has historically been associated with reproducible deficits in spatial and temporal memory processing.51 Memory involves both encoding and retrieval processes, each of which may involve different structures. Declarative memory, or memory for facts and events that can be consciously discussed, requires the hippocampus for memory consolidation and uses the frontal cortex for memory retrieval. Declarative memory retrieval is also considered associative in nature in that any area of the brain involved in encoding the information can be tapped to help initiate retrieval. In contrast, procedural memory is a hippocampal independent process that involves learning how to do something. Procedural memory has a relatively inflexible retrieval process in that it is limited specifically to the processes involved in learning the task. Procedural learning, however, is relevant for many other types of cognitive functions. Because of the central role that the hippocampus plays in memory, cholinergic systems projecting to the hippocampus are implicated in memory dysfunction. The prefrontal cortex (PFC) and corticostriatal DA signaling system, in addition to the hippocampus, are also important for memory formation.
Clinical Neuropharmacology for Traumatic Brain Injury–Mediated Cognitive Deficits
In contrast to experimental TBI models that have characterized specific alternations in neurotransmitter systems, evidence that neurotransmitter systems, including dopaminergic systems, are altered in humans after TBI is based largely on reports that DA agonists can be beneficial in attenuating cognitive deficits. Although amphetamine has been used to some degree clinically as both a DA and an NE agent, MPH is more widely accepted as an effective neurostimulant to improve cognition. According to a recent evidence-based guideline, MPH is recommended as a neurostimulant to improve cognition, and particularly to improve attention.319 Much of the evidence for this treatment guideline is derived from small but well-controlled clinical trials showing improvements in cognitive processing speed and caregiver rates of attention.470
Recent evidence-based guidelines support the use of amantadine to enhance general cognition and attention in patients with moderate and severe TBI. Previous clinical reports demonstrate improved scores on ADL scales in case reports of patients treated with amantadine.485,498 Case reports also indicate general improvements in global functioning and processing speed.61,236 PET imaging studies indicate amantadine-mediated increases in cortical glucose metabolism in TBI patients.237 In patients with DAI, amantadine appears to be effective in improving cognition independent of the timing of administration.296 Bromocriptine is less well studied in clinical research compared with MPH and amantadine. Small case series, however, show some evidence that low-dose bromocriptine might improve executive function.290 Recent work, however, suggests that daily bromocriptine is not effective for treating attentional deficits.471 Some work has been undertaken regarding noradrenergic agonists in TBI recovery. The tricyclic antidepressant and NET inhibitor desipramine has been used for years for depression, and it increased arousal and initiation in a TBI case series,357 as well as improved post-TBI depression.484 Clinical reports also suggest some potential efficacy with the selective NET inhibitor atomoxetine.363
Previous large randomized controlled trials with CDP-choline have shown improvements in memory and global function in TBI patients across the injury severity spectrum.55,253 In addition, both case series and small randomized controlled trials suggest that the cholinesterase inhibitors can improve memory and attention, as well as general cognition.399,463 Interestingly, daily treatment with the cholinesterase inhibitor donepezil results in cortical increases in metabolism, and the magnitude of this metabolic change is associated with the degree of clinical treatment response.219
Purpose of Neuropsychologic Assessment
The purpose of neuropsychologic assessment varies across the course of recovery from TBI. The first assessment might occur during the initial stages of emergence from coma. Later neuropsychologists are often responsible for evaluating a patient’s progress through PTA. Neuropsychologic testing is useful in evaluating the domains of cognitive functioning described in the previous section. The ability to complete formalized testing of any sort, however, depends on the ability of the patient to tolerate or respond to the tools used. As a result, extensive formal neuropsychologic testing is generally not completed while an individual is in PTA, but rather, brief tools and batteries are introduced as an individual is able to participate. Formalized testing of certain cognitive areas at times might not be possible for a period because of other brain injury-related issues, such as aphasia, severe limitations in motor or sensory abilities, medical issues, or behavioral issues that can impede the ability of an individual to participate. Recent studies suggest it is more feasible than previously thought to administer brief testing during acute inpatient rehabilitation211 and suggest some prognostic value to completing certain types of neuropsychologic testing within the early weeks of the recovery process.160 Full neuropsychologic test batteries are often not administered for several months after moderate to severe TBI. These detailed outpatient batteries may be used to answer questions related to return to school, work, driving, or other activities, as well as to assess ongoing recovery.
Practical Interventions for Cognitive Impairment After Traumatic Brain Injury
All the behavioral and cognitive issues discussed in this section tend to improve over the recovery period from injury. Some of the issues observed in the inpatient setting, however, can continue to be present as the patient transitions to home or other settings. In fact, according to one study, approximately 40% of individuals reported ongoing unmet needs at 1 year, with the most frequent issues cited including issues with memory and problem solving, managing stress and emotional upset, and temper control.86 Many of the routines and techniques learned in therapy, however, can be applied successfully by families in the home setting. Staff should be certain to educate families regarding the purpose and use of these techniques. Continuing to set up routines and structure to assist the individual and to increase the likelihood of success can play a significant role in decreasing depression, irritability, and difficulties with explosive anger. Treatment with medication, combined with therapy to manage adjustment to disability, anxiety, and depression, as well as to teach anger management skills, is often recommended. Box 49-3 reviews helpful behavioral and cognitive strategies for common issues after TBI.
Additional tips for working with individuals with brain injury:
Agitation
Agitation is a common factor in the acute phase of recovery from TBI. Not all patients in the acute phase of recovery from TBI become agitated, with studies suggesting prevalence rates between 11% and 42% (depending on the definition used).33,46,359 These types of behaviors occur frequently enough that they are described as a distinct phase of recovery in the Ranchos Los Amigos Scale (i.e., Rancho Level IV—Confused–Agitated), and garner attention because they pose significant challenges to rehabilitation staff and family members. Importantly, agitation can have a significant impact on a patient’s ability to productively participate in therapies. Bogner and Corrigan33 define agitation as “an excess of one or more behaviors that occurs during an altered state of consciousness.” This definition includes not only aggressive physical or verbal behaviors but also restlessness and disinhibition. “True” clinical agitation occurring during an altered state of consciousness is therefore differentiated from the description of an individual who is “irritable,” “angry,” or “aggressive” when not confused. This definition also requires observed behaviors to be “excessive,” where behaviors interfere with functional activities and are not able to be inhibited by the person either with or without cueing.33
Measures of Agitation
The behaviors associated with agitation have been most effectively described by the Agitated Behavior Scale (Box 49-4).82 This scale can be helpful for monitoring the progression of patients through recovery, as well as for evaluation of the effectiveness of interventions such as medication or behavior plans to manage agitation. In addition, valuable information can be gained regarding the timing of such as interventions and the scheduling of therapeutic activities based on scores observed. Another more recently developed agitation scale is the Overt Agitation Severity Scale,495 which limits scoring to observable behaviors, and is not specific to TBI. The Neurobehavioral Rating Scale has also been used to monitor agitation and PTA.
Treatment of Agitation
Pharmacologic treatment for agitation might be beneficial, although heavily sedating medications can prolong the period of agitation.314 Improvement in cognition (which sedating medications might impede) might be necessary for agitation to improve.84 Caution should be taken to consider the potential adverse effects of many of these psychotropic drugs on neurologic recovery. Early experimental brain injury studies have demonstrated the detrimental impact of the classic D2 receptor antagonist antipsychotic medication, haloperidol, on motor recovery.122 Later experimental TBI studies also suggest detrimental effects of daily haloperidol use on cognitive function.224,475 Atypical antipsychotic (AAP) drugs can be better choices clinically for TBI-related agitation, in part because of their more favorable side effect profile and their smaller effects as a D2 receptor antagonist. Experimental studies also show negative effects on cognitive recovery with multiple AAP medications, however, including olanzapine and risperidone.224,475 Among the AAP drugs, quetiapine is a frequently selected agent for post–TBI-related agitation for its favorable side effect profile and relatively low actions as a D2 receptor antagonist.116 A recent pilot study suggests that quetiapine is clinically effective in reducing agitation symptoms post-TBI, with associated improvements in cognition noted.218
β-Blockers are commonly used clinically to manage TBI-related agitation. Previous clinical trials suggest that propranolol can decrease agitation intensity and decrease the need for physical restraints.47 A recent Cochrane Database review suggests that β-blockers have the best evidence for effective use with minimal side effects.132 Although anticonvulsant medications such as tegretol and valproate are commonly used to manage TBI-related aggression, research on their efficacy is limited.132
Benzodiazepines are GABA-A receptor agonists that can reduce agitation symptoms post-TBI. The use of these drugs for agitation management during rehabilitation has historically been minimized because of concerns of adversely affecting cognitive recovery by the ability of these drugs to augment neuroinhibitory neurotransmitter systems. In the ICU, benzodiazepines are commonly used for sedation and behavioral management. Interestingly, limited experimental research for TBI does support the concept that benzodiazepines given early after TBI might confer a neuroprotective effect on cognitive recovery,326 presumably by limiting the degree of excitotoxic insult during this period.
Hypoarousal and Sleep Disturbance
During the acute phases of recovery from TBI, problems with arousal, attention, and fatigue are common and can be the result or combination of multiple factors. Injury to structures or connecting white matter fibers in the reticulothalamic, thalamocortical, and reticulocortical networks might cause symptoms of fatigue, hypoarousal, persistent vegetative or minimally conscious states, coma, or death, depending on the severity of injury. Neurochemical disturbance related to injury to these areas can also be a contributing factor. As an individual awakens from coma, arousal improves but can remain a problematic issue during rehabilitation. In addition, sleep disturbance, medical issues, and debilitation may all contribute to a patient’s ability to remain aroused and attentive. With hypoarousal, patients complain of fatigue or frequently ask to return to their room or bed. They might fall asleep midtask during a therapy session or require frequent cues to remain awake. When issues with arousal are problematic, medications should be reviewed for sedating side effects. The timing of medications that can have sedating side effects should also be reviewed. Discontinuation, dose reduction, changes in timing, or alternative treatments should be considered as needed. Medical treatment for arousal disorders might include the use of psychostimulants or other medications. From a behavioral standpoint, staff members can evaluate patient schedules and therapy to see whether routines can be established to improve arousal. Changes may include monitoring the time of day when arousal appears to be problematic and providing rest breaks in the schedule. It might be beneficial to schedule the most challenging tasks during periods of the day when the patient appears to be most alert. It might also be beneficial to schedule cognitive tasks after a physical activity (i.e., walking) if the patient appears to be more alert after being physically active. Often patients with arousal issues can appear to be different if they are evaluated during an alert period versus during a time when they are less alert. This can cause significant differences is scoring and ascertaining a patient’s true abilities.
Sleep disturbance is also common after TBI and can contribute to issues with arousal. Patients might have alterations in circadian rhythms, sleep patterns, and sleep quality, which can be disruptive to therapy. Acutely after TBI, sleep disturbance is largely the result of diffuse cerebral dysfunction associated with primary and secondary injury.371 Sleep patterns after TBI are characterized by decreased rapid eye movement and slow wave sleep. Total sleep time and sleep efficiency is also disturbed.80 There is also a strong link between cognitive recuperation and normalization of sleep.371 Disrupted sleep after TBI can result from pharmacologic treatments, associated neuropsychiatric conditions, agitation, drug withdrawal, pain, preexisting sleep disorders, and environmental overstimulation.263,330 Environmental factors unique to hospital units, such as noise, untimely procedures, and pharmacologic interventions, are common.19
Pharmacologic intervention is often needed to effectively treat TBI-mediated sleep disorders. The literature is limited for use of tested and clinically proven sleep-promoting drugs on the injured brain. Trazodone, a selective serotonin (5-hydroxytryptamine [5-HT]) reuptake inhibitor and 5-HT2 receptor antagonist, is frequently used with insomnia for its sedating effects in TBI.136 At lower doses its sedative properties likely result from its antagonistic effect of the 5-HT2 receptors.270 Trazodone promotes natural sleep cycles by increasing the amount of total sleep, increasing the percentage of deep sleep, and decreasing the number of intermittent awakenings.214,385,464
Cognitive–behavioral therapies (including stimulus control, sleep restriction, and sleep hygiene education) are beneficial, particularly in those with moderate TBI.329 Assisting patients with sleep hygiene might also be helpful. This may include allowing fewer naps during the day as the patient is able to tolerate it, turning off television or other sources of stimulation near bed time, helping the patient to engage in quiet activities in the evening, and dimming or turning out lights during the night-time hours, as safety permits. An evaluation of night-time medical needs may also be helpful to determine whether any medication schedules or other medical routines could be altered to allow the patient as many uninterrupted hours of sleep as possible. Sleep logs should be maintained for patients with identified sleep issues to monitor the efficacy and progress of any interventions prescribed.
Psychiatric Issues
Depression
Depression is the most common psychologic problem after TBI,116 with a prevalence between 6% and 77%.240 The variation in prevalence rates is due to different measurements used to assess depression, the time course when assessed after injury, and differences in injury severity of TBI patients. The prevalence of posttraumatic depression is highest in the first year after injury,18 but delayed depression is not uncommon.166
It is not known whether the development of posttraumatic depression is from the pathophysiologic consequences of the head injury, a premorbid predisposition, or a postinjury psychologic response to the trauma.370 Depression can result from the biochemical changes in the brain after injury. As a result, medications to treat depression are often beneficial. Clinical studies report benefit of both selective serotonin reuptake inhibitors and tricyclic antidepressants in treating post-TBI depression.117,484 A number of potential risk factors affect postinjury depression rates. Minority status, unemployment, low income, history of alcohol abuse, and low education levels are associated with posttraumatic depression.370 There is no clear consensus concerning how preinjury psychiatric history,124,209 age,152,210,391,485 gender,∗ and injury severity370 affect posttraumatic depression rates.
The overlap between symptoms of depression and symptoms of neurologic disease presents a challenge when assessing depression in TBI patients. Overlapping symptoms can include insomnia, irritability, and lack of motivation. Depression has been assessed with TBI patients using a variety of measurements, including the Zung Self-Rating Depression Scale, Hamilton Rating Scale for Depression, and Beck Depression Inventory.120,268,390 DSM-IV criteria for identifying major depression have also been used to assess depression in TBI patients.240 The Patient Health Questionnaire, which is based on DSM-IV criteria of depression, has been used to assess depression in the TBI population.115 Posttraumatic depression is associated with poor outcome after injury.116 Depression is also associated with impaired cognitive function such as psychomotor slowing, decreased information-processing speed, memory, and flexibility in problem solving,370 and impaired cognition is associated with posttraumatic depression after TBI.367
Posttraumatic Stress Disorder
There has been considerable recent debate about posttraumatic stress disorder (PTSD) after TBI, which has gained significant attention in light of combat and blast-related injuries related to the conflicts in Afghanistan and Iraq. It was previously thought that amnesia for the traumatic event was a protective mechanism, and that PTSD (which is an emotional response to a traumatic event) could not occur for an event not remembered.281 Recent reviews of the literature suggest, however, that it is possible for PTSD to occur after TBI. The severity of the injury, however, might have some protective qualities in modulating PTSD development.221 It is less likely, for example, for PTSD to occur in instances in which significant loss of consciousness and amnesia for the event occur. In comparison, it can be more likely for an individual with no loss of consciousness or brief loss of consciousness (such as with mild TBI) to develop PTSD because of the increased likelihood of recall for the injury event or events leading up to or after injury. In individuals with moderate to severe injury, however, a percentage do go on to develop symptoms of PTSD, although the exact rates are unclear.221 In those who do, few report reexperience of the event as a symptom. Some individuals, however, despite having no memory of the event, can develop an unconscious or implicit fear response for the event.248 Individuals can also develop an emotional response to information that is provided to them about the event, such as stories or pictures relayed by others185 or in response to brief periods of memory during the confusing period of PTA.220 The overlap in symptoms of PTSD and TBI is significant and complicates the diagnostic process. Some data suggest that both mild TBI and PTSD are more prevalent when injuries are a result of improvised explosive devices306; however, additional research is needed.
Behavioral Management
Pediatrics
Original outcome data in pediatrics led to the generalization that younger children have worse outcomes than older children.241 Overall, in the pediatric population, 95% survive injury. The highest mortality rates are observed in children under the age of 2.267 The mechanism of injury in up to 50% of those under the age of 7 is related to assaults or child abuse and falls. This population, compared with noninflicted TBI, had lower initial GCS scores, more prolonged impairments of consciousness, different biomarker profiles, and worse functional scores at 6 months.25,202 When functional outcome data are evaluated, memory difficulties contributed to persistent dependence after injury. Those with shorter duration of coma and younger age- groups had improved functional outcomes, postulated to be due to greater plasticity in the younger brain.110 In comparison, those patients with nonaccidental TBI early in life have more significant persistent cognitive and emotional difficulties.339
Specific practice guidelines for the acute management of severe TBI in pediatric patients exist. Evidence supports treatment in a pediatric trauma center or in an adult trauma center with added qualifications to treat pediatric patients. Trends have been seen with improved outcomes with therapeutic hypothermia, making it an option in pediatric management not proposed in the adult guidelines.3 Recent trials have shown safety of the application of moderate hypothermia in pediatric patients, but with variability in the outcomes reported.4,200
Pediatric TBI has a significant impact on the health care needs of children. During the first year after a moderate to severe TBI, one third of children had unmet or unrecognized health care needs. The largest area of need was cognitive services. The most frequent reason for this need not being met was that it was not recommended by the physician or school. Lack of insurance funding and poor family functioning were also related to unmet and unrecognized needs. When these issues are combined with a lack of concrete information on recovery after TBI, family distress is high after injury.403
When children with TBI are compared with matched peers, there is a correlation between severity of injury and performance in neurobehavioral functioning in the areas of intelligence, academic performance, memory, problem solving, and motor performance 1 year after injury. Even though the students scored within the normal range, they were substantially below their matched peers.204 These same findings persist 3 years after injury. As children with TBI age, deficits with abstract and concept learning become more apparent that were not easy to measure 1 year after injury. These findings support the need for serial neuropsychologic testing after injury in children.119 Research supports the need for increased educational support for children with TBI in an educational system that understands learning deficits after TBI.
The impact of deficits in EF and behavior can be manifested in difficulties with social interactions. The interrelationship between language, EF, and self-managing behavior is central to social success. These skills are often compromised after TBI, resulting in social difficulties especially with children injured during the teenage years.438 Social functioning is an important predictor of QOL after pediatric TBI. For children with TBI, maintaining close friendships and number of friends are more important than success in school or sports.350 This information should lead practitioners to assess this area of function when examining patients, in the same way they evaluate motor and cognitive function.
Behavioral changes are commonly reported by families postinjury. The Child Behavior Checklist can be used to define abnormal behavior in a home and academic setting. In a telephone interview study, 40% of patients were described as having behavioral problems. Preinjury behavioral issues and family function might also relate to persistent behavior issues.162 It is important to discriminate between post-TBI behavior changes and psychiatric disorders, seen in those with more severe injury, and those with premorbid or family psychiatric history.
Acute Prognostic Indicators of Outcome: A Clinical and Translational Research Perspective
Demographics
Female gender is considered to be a risk factor for worse outcome, particularly in studies that include mildly injured subjects in the analysis.24,118,459 Increases in postconcussive symptoms are often reported. In contrast, experimental TBI model studies suggest that female hormones can be neuroprotective against many biochemical mechanisms of secondary injury, including cerebral edema, excitotoxicity, oxidative injury, and inflammation.373 Recent clinical reports show that female hormones might have a beneficial effect on TBI pathophysiology, with lower lactate/pyruvate ratios, glutamate accumulation, and oxidative stress loads reported in CSF for females with severe TBI compared with males.23,449,452 Severe TBI, however, has a profound effect clinically on endogenous serum hormone profiles. Elevations in hormones like estrogen measured early after injury are linked to greater mortality and worse global outcome, regardless of gender. The pattern likely represents a stress response to the injury323,332,368 and is not innately neuroprotective. However, pharmacologic progesterone dosing early after injury is a promising neuroprotective therapy that is currently undergoing Phase 3 clinical trials in humans.482,487 Taken together, these studies suggest that gender and hormones have a complex, yet important, role in TBI physiology and outcome.
TBI in the elderly population is more commonly the result of falls than in younger age-groups. Fragile bridging veins in elderly patients contribute to higher incidence of SDH in this age-group. Underlying medical conditions such as heart arrhythmias and artificial valves that require artificial anticoagulation increase the likelihood for intracranial hemorrhage with falls. Decreased balance and syncopal episodes from a variety of causes also contribute to falls and TBI in elderly persons. Neuronal loss that occurs with increasing age likely also plays a role with incidence of falls. Age-related mitochondrial dysfunction associated with neurodegenerative diseases likely influences the secondary biochemical injury response observed in older patients with TBI.54 In vitro studies show that hippocampal vulnerability to excitotoxic injury increases as a function of age and is related to a more pronounced loss of intracellular Ca++ homeostasis and mitochondrial dysfunction.275 More oxidative stress has been noted for a given excitotoxic or ischemic insult in older adults with TBI.449 TBI in the aging population is associated with higher severity of injury, mortality, and morbidity. For older patients with TBI, there is greater acute care resource consumption per favorable outcome than in their younger counterparts.338 Rehabilitation discharge outcomes are often similar for older and younger TBI patients, but the length of inpatient rehabilitation and hospital costs needed to attain similar functional gains are greater for older patients.74
Intentional, or violent, TBI carries a higher mortality rate than other accidental mechanisms of injury.460 Among survivors, there are no differences in functional assessments after injury, but victims of intentional injury report lower levels of community integration and function than others who survive accidental injuries.460 Alcohol and illegal substances are commonly observed in the blood at the time of injury and can contribute to acute complications, longer hospitalizations, and worse outcomes from TBI.83
Clinical Variables
The GCS426 has been a mainstay in determining severity of neurologic injury after trauma and has been used in several studies with other variables to predict outcome. The revised trauma scale (RTS) score and injury severity score (ISS) are effective predictors of discharge disposition across a broad population hospitalized with TBI.454 RTS and GCS scores can also predict disability and community integration 1 year after injury in this population.455 Coma duration is linked with injury severity and outcome, and patients with coma more than 2 weeks are unlikely to have a good recovery.212 PTA duration is associated with DAI and is a strong predictor of functional outcome.213,497 In fact, the International Data Coma Bank reports that patients with less than 2 weeks of PTA often have a good recovery, whereas patients in PTA for more than 4 weeks have a much smaller likelihood of good recovery.212
Both elevated ICP and decreased CPP have been linked with poor outcome in adults and children.59,424 Although ICP is often elevated early after severe injury, late elevations are often more severe and require more intensive treatment to manage.413 Significant correlations between arterial pressure–induced decreases in CPP and ISS scores have been reported, indicating the difficulties in resuscitating TBI patients with multiple other injuries. Additionally, duration of low CPP was related to adverse outcome as late as 24 months after injury.87
Radiologic indices from CT studies have some usefulness in predicting outcome. DAI leads to cerebral atrophy99 and is associated with coma and PTA, both of which are associated with long-term outcome. Traumatic subarachnoid hemorrhage (tSAH) has been associated with worse injury and poorer outcomes compared with other types of lesions,279 and tSAH can be graded to predict outcome.161 It is unclear whether tSAH is simply a marker of more severe TBI, however, or whether it adversely affects outcome through direct mechanisms like vasospasm and ischemia.16 Midline shift reflects elevated ICP and contributes to poor outcomes, whereas patients who have EDHs evacuated often have relatively good outcomes.269
Although subject to variability, reports demonstrate that SSEPs provide some prognostic information. In severely injured patients, SSEPs have been linked to functional measures at rehabilitation discharge and 6- and 12-month functional outcome measures.284,333 Other studies show that SSEPs, including central somatosensory conduction time (CCT) and N13/N20 amplitude ratios (ARs) in the upper limb, are sensitive to global outcome. For cases of coma caused by supratentorial lesions, CCTs and ARs were normal in patients with good outcome. CCTs and ARs became more prolonged and decreased, respectively, as patient outcome worsened and patients with bilateral loss of cortical responses died or were severely disabled.379
EEG data have been classified in several ways for the purposes of prognosticating outcome. Some evidence suggests that the presence of more normal wake–sleep patterns, including the presence of sleep spindles, is indicative of better outcomes.45 EEG characteristics of coma considered favorable for survival are usually sensitive to external stimuli and include relatively normal activity, dominant and rhythmic θ activity, frontal rhythmic δ activity, and spindle coma patterns present where stage 2 sleep patterns most often appear. Patients with posttraumatic coma also exhibit burst suppression patterns when cerebral anoxia secondarily results from the injury. Additional negative prognostic characteristics involve nonreactive α-pattern coma with α-range activity predominant in the anterior regions or broadly distributed throughout the cortex.420 Impairment of the quantitative EEG parameter, percentage of α-variability, also signifies poor outcome.446 The most severe malignant factor for survival, however, is isoelectric EEG activity.420
Biomarkers
Biomarkers are a part of mainstream clinical care to quantitatively assess and define injury and disease in almost every organ system other than the brain.28 However, biomarkers have the potential to assist clinicians with TBI diagnosis, prognosis, and the development of treatment and management strategies. Although biomarkers are traditionally defined as protein measurements in biofluids, genes also represent a rich biomaterial for identifying individualized data for how a person may respond to an injury or treatment and the degree of risk for particular complication after injury.
Proteomics
Biomarkers identified in TBI often represent structural damage associated with the injury. These biomarkers are derived from the major cell types in the CNS. S100B is a calcium binding protein present in high concentrations in astrocytes, and elevated S100B levels indicate irreversible astrocyte damage.232 Myelin basic protein (MBP) is abundant in myelin. As such, elevated MBP levels represent axonal injury.191 Neuron-specific enolase (NSE) is a glycolytic enzyme localized predominantly in neuronal cytoplasm, with elevated levels reflective of neuronal injury.
In addition to its diagnostic potential in patients with mild TBI, serum S100B levels have been linked with impending mortality in severely injured patients, increased ICP, global outcomes such as Glasgow Outcome Scale (GOS), and QOL.28 NSE is linked to neuroinflammation345 as well as GCS and outcome in adults and children with reasonable sensitivity and specificity.30,167 Initial NSE levels in particular have been helpful in outcome prognostication for both adults and children.30,294 Both S100B and NSE absolute levels and duration of elevation have been linked to deficits with neuropsychologic testing.192 MBP also has been linked with both mortality and outcome.30,490
Each of these biomarkers also has some usefulness in differentiating etiology of injury. Children with inflicted TBI (i.e., SBS) had delayed peak concentrations of serum S100B, MBP, and NSE compared with children presenting with accidental TBI.29 A reduction in these markers has also been observed in children receiving therapeutic hypothermia.262 These markers might have usefulness in monitoring other promising therapeutic interventions, such as progesterone treatment. Each of these markers is currently being investigated in humans and experimental models for their usefulness in characterizing their prognostic potential in those with blast TBI.418 Some limitations should be considered as these markers move into clinical care. For example, S100B and NSE have limited reliability because of extracerebral accumulation of these markers in patients with concomitant injury or hemorrhagic shock.232 S100B is also not a useful marker in children less than 2 years of age because of high normative values in that age-group.232
The literature suggests that multiple molecular targets associated with key biochemical pathways involved with secondary injury can also have some usefulness as biomarkers in prognosticating outcome and monitoring treatment response. The majority of studies evaluating these types of markers have used CSF or extracellular fluid from human brain tissue microdialysis studies obtained during the first week after injury to examine elevations or decreases in biomarkers clinically in patients with severe TBI. In contrast, little has focused on identifying biomarker profiles obtained during the rehabilitation or chronic phases of recovery.
CNS glutamate levels are elevated early after TBI,489,499 a phenomenon associated with poor outcome.155 Gender also influences CSF glutamate levels over time, and treatments like therapeutic hypothermia can significantly reduce levels of this excitatory neurotransmitter in a gender-specific manner.452 F2-Isoprostane, a marker of oxidative injury, has similarly been associated with outcomes,449 as well as gender-specific response to hypothermia.23 Mitochondrial markers associated with apoptosis, like cytochrome c and BCL-2, have been linked with both injury type and outcome. In children, elevated CSF cytochrome c levels had a strong association with inflicted TBI. In severely injured adults, persistently elevated CSF cytochrome c levels were associated with poorer 6-month GOS scores. High CSF concentrations of the prosurvival protein BCL-2 have also been linked with improved outcomes in children with severe TBI,75 but with poor outcomes in adults.11 These specific differences in BCL-2 outcomes might be the result of inherent differences in CNS apoptosis across age-groups. Cytoskeletal proteins like αII spectrin and its breakdown products343 and both proinflammatory and antiinflammatory markers396 in the CSF have been linked with survival and outcome. Serum inflammatory markers can reflect extracranial injury but still affect outcome.396
Genomics
Apolipoprotein E (APOE) has been studied extensively with regard to its role in plasma lipoprotein transport. There are multiple isoforms of the APOE protein, with functional differences in plasma lipoprotein transport being noted between forms. Numerous studies have linked the expression of the APOE4 isoform with an increased risk of Alzheimer’s disease, and evidence suggests that TBI triggers ongoing degenerative pathology similar to that observed with Alzheimer’s dementia. Genetic association studies in TBI pathophysiology and outcome have focused on APOE. Some studies suggest that there is no association between the APOE4 allele and outcome,299 whereas several other studies link APOE4 with worse functional and cognitive outcomes.8,88,428 The largest TBI study evaluating possession of the APOE4 allele and outcome found a significant age–genotype interaction where adolescent and young adult patients carrying the APOE4 allele had worse outcomes compared with other younger patients without this allele.427 The frequency of the APOE4 allele in patients with β-amyloid deposition is 52%, which is higher than reported in the Alzheimer’s population. In those TBI subjects without β-amyloid deposition, the APOE4 frequency is 16%, however, and similar to control subjects without Alzheimer’s dementia.321 Recent work also implicates genetic variability in the promoter region of the neprolysin gene, the primary enzyme responsible for the β-amyloid degradation, and the incidence of Alzheimer’s type β-amyloid plaques in TBI.208
Although TBI pathophysiology and outcome have been linked, in part, to genetic variability with APOE, less is known about the role of other candidate genes in mediating deficits post-TBI. One study suggests that genetic variability in the IL-1β gene influences global outcome in patients with severe TBI.443 There are also several identified variants for a number of dopaminergic genes that are associated with cognitive and psychiatric conditions such as ADHD, depression, impulsivity, and Parkinson’s disease. Many symptoms associated with these diseases overlap with deficits associated with TBI. TaqIA restriction fragment length polymorphism for the D2 receptor affects D2 receptor levels and glucose metabolism in the brain. It is also linked with verbal memory performance after mild TBI.285 Additional work shows that variations in the promoter region of the DAT gene that affect gene transcription are linked with DAT receptor densities in the striatum chronically after TBI.384 This DAT variant is also associated with higher DA levels measured in CSF early after severe TBI, particularly in females, a finding that could have implications on oxidative injury load early after injury.456
Chromatin is a structure consisting of DNA plus interacting proteins called histones that help compact DNA in the nucleus and regulate gene expression. Although the human genome represents genetic diversity derived from DNA, the epigenome is a composite of chromatin plus a pattern of chromatin modification that results from interactions between the chromatin and its environment. Epigenetics includes the transmission and perpetuation of information, that is, heritable phenotypes not related to the DNA sequence, and is an important mechanism for the stable maintenance of gene expression that involves physically marking DNA or histones. Epigenetic modification significantly affects learning and cognition in animal models.252 Work in experimental TBI models shows that epigenetic modification of histones is reduced in the hippocampus after TBI, a finding that may contribute to learning and memory deficits.139 Epigenetic modification also occurs with the application of an enriched environment,128 an intervention that repeatedly has been shown to improve learning and memory in experimental TBI models.180,227,457 Although chromatin remodeling studies have not yet been linked to clinical prognosis, epigenetic biomarkers might be a promising strategy for further insight into patient prognosis, treatment efficacy, and individualized environmental response to TBI.
Pediatric Considerations
The clinical predictors often used for pediatric populations are similar to those used for adults. The GCS can be measured in children and adults, and is most predictive when applied at 6 hours after injury. The Abbreviated Injury Scale score, another severity of trauma measure, is also predictive of worse functional outcome and prolonged pediatric ICU stay.467 The severity of increased ICP is also associated with worse outcome, although the method of controlling ICP does not affect outcome.205 When stratified by age, age greater than 2 years generally portends a better functional outcome, although etiology of injury and severity of injury might be more predictive. Additional factors influencing outcome and recovery from pediatric TBI are covered elsewhere in this chapter.