[level-membership-for-neurology-category]
Chapter 22 Traumatic Brain Injury
Major Head Trauma
How Does Head Trauma Cause TBI?
Direct Force
A blow – by its direct mechanical force (a coup injury) – disrupts the underlying, delicate brain tissue (the parenchyma). As with strokes, trauma causes cell death by necrosis and its accompanying inflammatory changes, particularly monocular cell infiltration. (In contrast, neurodegenerative diseases, such as Huntington disease and amyotrophic lateral sclerosis, cause cell death by apoptosis [see Chapter 18].)
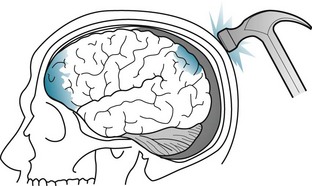
In addition to causing the coup injury, head trauma throws the brain against the opposite inner surface (table) of the skull, which causes a contrecoup injury of that surface of the brain. Damage from contrecoup injuries frequently surpasses that from the coup injury. Contrecoup injuries frequently damage the temporal and frontal lobes because their anterior surfaces abut against the sharp edges of the skull’s anterior and middle cranial fossae (Fig. 22-1). Depending on its severity, damage of the frontal and temporal lobes characteristically leads to memory impairment and personality changes.
Diffuse Axonal Shearing
Diffuse axonal shearing, which also leads to TBI, consists not only of trauma to long subcortical white-matter axons, but also damage to cytoskeletal elements and a fatal intracellular influx of calcium. Although neither computed tomography (CT) nor magnetic resonance imaging (MRI) can illustrate diffuse axonal shearing, diffusion tensor imaging readily detects it by showing traumatic disruption of white-matter tracts (see Chapter 20).
Bleeding Within the Skull, but Outside the Brain
• Pia mater, the innermost layer, is a thin, translucent, vascular membrane adherent to the cerebral gyri. It follows gyri into sulci and thus allows for a generous region, the subarachnoid space, between it and the immediately overlying layer – the arachnoid mater.
• Arachnoid mater, also thin, spans the tops of gyri, capping the sulci. The subarachnoid space, which contains the cerebrospinal fluid (CSF), envelops the brain and, continuing downward within the spinal canal to the sacrum, the spinal cord and cauda equina. Neurologists performing a spinal tap or lumbar puncture insert a special needle into the subarachnoid space below the conus medullaris (lower end of the spinal cord), which is situated between the T12 and L1 vertebrae, to sample CSF.
• Dura mater, the outermost layer of the meninges, is a thick fibrous tissue adherent to the interior surface of the skull. Two of its infoldings, the falx and tentorium, support the brain and house most of its venous drainage. Neurologists designate the space between the skull and the underlying dura as the epidural space, and between the dura and the underlying arachnoid as the subdural space. Major head trauma may cause hematomas in either or both of these areas.
Epidural hematomas, which typically result from temporal bone fractures with concomitant middle meningeal artery lacerations, are essentially rapidly expanding, high-pressure, fresh blood clots (Fig. 22-2). They compress the underlying brain and force it through the tentorial notch, i.e., they produce transtentorial herniation (see Fig. 19-3). Unless surgery can immediately arrest the bleeding, epidural hematomas are usually fatal.
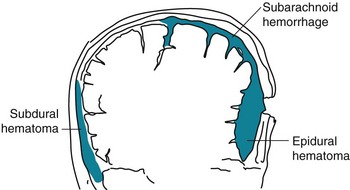
FIGURE 22-2 Meningeal arterial bleeding, which usually results from a blow forceful enough to fracture the skull, causes epidural hematomas. In contrast, venous bleeding, usually slower and under less pressure, causes subdural hematomas. Ruptured aneurysms and head trauma often cause subarachnoid hemorrhage (SAH). In SAH, blood spreads within the subarachnoid space over the convexities, between the gyri, into the interhemispheric fissure, and down into the spinal canal.
As an example, a victim of an assault with a baseball bat lost consciousness when struck. After regaining consciousness for 1 hour, the victim lapsed into coma and developed fatal decerebrate posture (see Fig. 19-3). A CT showed a temporal skull fracture and an underlying epidural hematoma (see Fig. 20-9D). Neurologists label the period when the victim transiently regained consciousness the lucid interval.
In contrast, subdural hematomas usually result from slowly bleeding bridging veins, under relatively low pressure, into the subdural space (see Fig. 20-9). Dark, venous blood generally oozes into the extensive subdural space until the expanding hematoma encounters underlying brain. The brain dampens bleeding and suppresses further expansion of the subdural hematoma. However, if the hematoma continues to expand, it may lead to cerebral transtentorial herniation or cerebellar herniation through the foramen magnum. Survivors often have permanent brain damage from the initial trauma and the pressure from the subdural hematoma.
Acute subdural hematomas, which are most apt to occur in alcoholics, individuals medicated with warfarin, or the elderly (see later), produce headache, confusion, and a deteriorating level of consciousness over several hours to 1 or 2 days. Depending on the extent of the bleeding and time until the diagnosis, patients may develop focal signs and herniation. A history of head trauma does not necessarily precede the symptoms. CTs show acute, dense blood in the subdural space (see Fig. 20-9).
Chronic subdural hematomas, ones that have developed and persisted for weeks, usually have spread extensively in the subdural space (see Fig. 20-9). They typically give rise to insidiously developing headache, change in personality, and cognitive impairment, but only subtle focal physical deficits (see Chapters 19 and 20). Although subdural hematomas may spontaneously resolve, they often require surgical evacuation. Because subdural hematoma patients’ cognitive decline is rapid, but treatment usually reverses their symptoms, neurologists often include subdural hematomas as an explanation for “rapidly developing dementia” and a “reversible cause of dementia” (see Chapter 7).
Posttraumatic Coma and Delirium
Following head trauma, as well as at other times, neurologists often classify patients’ level of consciousness as alert, lethargic, stuporous, or comatose. They may also use the Glasgow Coma Scale (GCS), which measures three readily obvious neurologic functions: eye opening, speaking, and moving (Table 22-1). In major head trauma, the GCS correlates closely with survival and neurologic sequelae; however, in minor head trauma, it correlates poorly. Moreover, physicians cannot appropriately include the GCS as part of a standard mental status examination for patients suspected of having dementia or circumscribed neuropsychologic deficits.
| Category | Score | |
|---|---|---|
| Eyes opening | Never | 1 |
| To pain | 2 | |
| To verbal stimuli | 3 | |
| Spontaneously | 4 | |
| Best verbal response | None | 1 |
| Incomprehensible sounds | 2 | |
| Inappropriate words | 3 | |
| Disoriented and converses | 4 | |
| Oriented and converses | 5 | |
| Best motor response | None | 1 |
| Extension* | 2 | |
| Flexion† | 3 | |
| Flexion withdrawal | 4 | |
| Patient localizes pain | 5 | |
| Patient obeys | 6 | |
| Total | 3–15 |
This standard scale quantitates the level of consciousness, with the lower scores indicating less neurologic function. Neurologists interpret scores of 3–8 as signifying severe traumatic brain injury (TBI) or coma; 9–12, moderate TBI; and 13–15, mild TBI. However, the GCS is not readily applicable to patients who have sustained cerebral hypoxia and, because they cannot make a verbal response, those who are intubated. Adapted, with kind permission, from Teasdale G, Jennett B. Assessment of coma and impaired consciousness: A practical scale. Lancet 1974;2:81–84.
*Decerebrate rigidity (Fig. 19-3).
†Decorticate rigidity (Fig. 11-5).
By the first day after TBI, of patients who score 3 on the GCS, which is the lowest possible score, 90% have a fatal outcome and most of the remaining never regain consciousness. By 4 weeks, almost all TBI comatose patients die, partially recover and regain consciousness, or evolve into the vegetative state (see Chapter 11). When in coma or the vegetative state, individuals cannot perceive pain and do not suffer.
Posttraumatic Epilepsy
Seizures that occur either within the first 24 hours (immediate posttraumatic seizures) or the first 7 days (early posttraumatic seizures) do not fall within the definition of PTE because neurologists label them “provoked seizures.” Only seizures that occur (and recur) more than 7 days after the traumatic injury fall into the definition of PTE. Of note, 1 week of phenytoin prophylaxis following severe TBI reduces early posttraumatic seizures, but no AED clearly reduces the prevalence of PTE. PTE remits in only 25–50% of cases. PTE usually takes the form of complex partial seizures that undergo secondary generalization (see Chapter 10). Not only does PTE cause disability and carry the risk of further head injury, AEDs may exacerbate TBI-induced cognitive and personality changes.
Cognitive Impairment
TBI-induced coma usually lasts, at most, 4 weeks. By then, most patients have either succumbed to their injuries or recovered at least some cognition. However, many patients remain in a twilight state with their eyes open, but unconscious. These patients are incapable of thinking, communicating, or deliberately moving. They cannot perceive pain and do not suffer. Most of them linger in the persistent vegetative state (see Chapter 11).
Surprisingly, the trauma’s location, with one important exception, correlates inconsistently with cognitive impairment. Left temporal lobe injuries, the exception, routinely produce vocabulary deficits similar to anomic aphasia (see Chapter 8).
In addition to TBI causing debilitating cognitive impairments, some epidemiologic studies suggest that it also constitutes a risk factor for Alzheimer disease. Studies have shown that severe head trauma causes increased levels of soluble amyloid and deposition of amyloid plaques, one of the hallmarks of Alzheimer disease. Several, but not all, studies also suggest that moderate and severe head trauma in individuals with two apolipoprotein E4 (Apo-E4) alleles correlates with a marked increased risk of developing Alzheimer disease (see Chapter 7). Surviving moderate TBI, individuals with two Apo-E4 alleles may have twice the risk of developing Alzheimer disease, and surviving severe TBI, those individuals may have four times the risk. (A confounding issue for some of these studies is that individuals with Alzheimer disease are prone to cause an accident in which they sustain TBI and come to medical attention.)
Treatment Strategies for Posttraumatic Cognitive Impairment
Neurologists and other physicians who work with TBI patients with cognitive impairment administer medications that hopefully increase their attentiveness, if not reverse their learning and memory impairments. Of the numerous medicines that purportedly help, few have undergone rigorous trials that have documented their value. For example, many trials have indicated that methylphenidate and other dopamine-enhancing medications increase patients’ attention and directly or indirectly memory. Anticholinesterases may also improve TBI patients’ memory, at least those who have suffered severe memory deficits (see Chapter 7). Treatment of comorbid PTSD, if present, may improve cognitive impairments and other posttraumatic neuropsychologic changes. At the same time, physicians should, if possible, reduce or eliminate medicines that may interfere with attention and memory, such as AEDs, antipsychotics, and minor tranquilizers. They should always bear in mind that multiple medicines are likely to lead to adverse interactions and unwanted, occasionally fatal, outcomes.
Other Mental Disturbances
Personality Changes
In addition to altering patients’ personality and possibly cognitive capacity, frontal lobe injury may disable their executive abilities. Just as with patients who sustain anterior cerebral artery infarctions, those who sustain frontal lobe trauma have impaired ability to solve abstract problems, plan sequenced actions, or execute responses. Moreover, patients who develop pseudobulbar palsy, another complication of frontal lobe trauma, may display emotional incontinence with easily precipitated fits of pathologic laughing or crying, and unexpected sudden switches between them (see Chapter 4).
Trauma in Childhood
When TBI occurs before growth spurts, affected limbs may fail to achieve their normal, expectable size. The limbs’ growth arrest resembles the spastic hemiparesis with foreshortened limbs that characterizes congenital cerebral injuries (see Fig. 13-4). If dominant hemisphere injury were to occur before age 5 years, the opposite hemisphere would usually assume control of language. For example, a left-sided cerebral injury in a 4-year-old child will probably not result in aphasia because the plasticity of the brain allows the right cerebral hemisphere to develop language centers. If TBI affects the dominant hemisphere of someone age 5 years or older, it is likely to cause language impairments, if not clear-cut aphasia.
Nonaccidental Head Injury
The shaken baby syndrome, which is another form of abusive head trauma, consists of the injuries inflicted by violent back-and-forth throws, without direct impact. In this violence, rotational (angular) deceleration produces diffuse axonal shearing, hemorrhages in the brain’s delicate parenchyma and subdural space, and retinal hemorrhages (Fig. 22-3).
Minor Head Trauma
Head Trauma in Sports
Sometimes the head trauma damages one or both trochlear (fourth) cranial nerves, causing diplopia (see Chapter 4). Head trauma may also damage the inner ear’s labyrinthine system. This injury causes vertigo, which patients may describe as “dizziness.” Patients rapidly moving their head or changing position induces this symptom. A neurologic examination of vertiginous patients may show nystagmus.
At 1 week, athletes with a headache, the most common postconcussion symptom, will usually still show impairments in memory and other neuropsychologic functions. Their other postconcussion symptoms may include irritability, mood changes, and sleep disturbances. Physicians assess injured athletes with various screening or computerized tests, but the Mini-Mental Status Examination (MMSE) (see Chapter 7) is not helpful.
Dementia Pugilistica
Boxers (pugilists) generally receive blows to the head that frequently leave them dazed. Repeated episodes of such TBI lead to cognitive impairment, often reaching the severity of dementia, and Parkinson disease-like physical deficits (see dementia pugilistica, Chapter 18). Overall, depending on the length of their career, boxers have approximately a 20% risk of dementia.
Headache
The signature of postconcussion syndrome is a dull, generalized, endless headache. Movement, bending, work, and alcohol use usually worsen it. In 50% of patients, postconcussion headache lasts longer than 1 year, and in 25%, longer than 3 years. Surprisingly, postconcussion headache occurs more frequently in mildly injured individuals than in moderately or severely injured individuals. One consideration in patients with continuous postconcussion headache is that the trauma may have exacerbated pre-existing migraine. Another is that use of analgesics, prescribed for any aspect of the injury, for as few as 15 days each month for 3 months has created chronic daily headache (see Chapter 9).
Insomnia
On the other hand, whatever the cause, physicians might consider that a pre-existing sleep disorder actually led to the TBI. For example, patients with sleep apnea, narcolepsy, or sleep deprivation might cause an MVA or be unable to avoid one (see Chapter 17).
Treatment and Recovery
For headaches, neurologists prescribe often mild, nonaddicting analgesics similar to those used for muscle contraction headaches. They typically prescribe nonsteroidal anti-inflammatory drugs (NSAIDs) and tricyclic antidepressants. These medications may alleviate insomnia and neck pain as well as headaches. Even when only a minimal migraine component exists or the patient has a history of migraine, antimigraine medicines may help (see Chapter 9).
• The patient has a history of attention deficit disorder, learning disability, or neurosis.
• Before the accident, the patient was in a low socioeconomic status, an unskilled or semiskilled worker, dissatisfied with the job, or in danger of being fired.
• An MVA caused the concussion.
• The symptoms are multiple and include bodily pains.
• Comorbidities – psychiatric conditions, medications, especially polypharmacy, substance abuse, and PTSD – impair diagnosis and treatment.
Whiplash
Mechanism of Action
The common MVA is the rear-end collision. In these MVAs, the sudden impact snaps the driver’s and passengers’ head and neck backwards and then forwards. It causes a flexion–extension neck injury (whiplash) (Fig. 22-4) because it wrenches the neck’s soft tissues, including ligaments, tendons, and the trapezius, paraspinal, and numerous, small, delicate muscles.
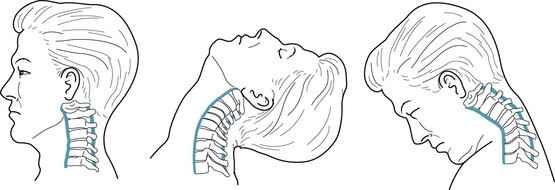
FIGURE 22-4 Cervical flexion–extension injuries, popularly called “whiplash,” may tear the anterior and posterior longitudinal ligaments (blue) and other soft tissues, herniate intervertebral disks, and exacerbate cervical spondylosis (see Chapter 5).
Whiplash may also aggravate degenerative spine disease and herniate cervical intervertebral disks (see Chapter 5). A severe rear-end MVA may fracture or dislocate cervical vertebrae, which can even transect the spinal cord.
Amen D, Newberg A, Thatcher R, et al. Impact of playing American professional football on long-term brain function. J Neuropsychiatry Clin Neurosci. 2011;23:98–106.
American Academy of Neurology Quality Standards Subcommittee. Practice parameter: The management of concussion in sports. Neurology. 1997;48:581–585.
American Academy of Neurology Sports Neurology Section, Practice Committee, and Board of Directors. Position Statement on Sports Concussion. AAN Policy. 2010.
Arciniegas DB, Harris SN, Brousseau KM, et al. Psychosis following traumatic brain injury. Int Rev Psychiatry. 2003;15:328–340.
Bombardier CH, Fann JR, Temkin NR, et al. Rates of major depressive disorder and clinical outcomes following traumatic brain injury. JAMA. 2010;303:1938–1945.
Cassidy JD, Carroll LJ, Côté P, et al. Effect of eliminating compensation for pain and suffering on the outcome of insurance claims for whiplash injury. N Engl J Med. 2000;342:1179–1186.
Chang B, Lowenstein D. Practice parameter: Antiepileptic drug prophylaxis in severe traumatic brain injury. Neurology. 2003;60:10–16.
Faul M, Xu L, Wald MM, et al. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths 2002–6. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2010.
Fleminger S, Oliver DL, Lovestone S, et al. Head injury as a risk factor for Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:857–862.
Frey L. Epidemiology of posttraumatic epilepsy: A critical review. Epilepsia. 2003;44(Suppl 10):11–17.
Fujii D, Ahmed I. Characteristics of psychotic disorder due to traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2002;14:130–140.
Green P, Rohling ML, Lees-Haley PR, et al. Effort has a greater effect on test scores than severe brain injury in compensation claimants. Brain Injury. 2001;15:1045–1060.
Hoge CW, McGurk D, Thomas JL, et al. Mild traumatic brain injury in U.S. soldiers returning from Iraq. N Engl J Med. 2008;358:453–463.
Koponen S, Taiminen T, Kairisto V, et al. APO-e4 predicts dementia but not other psychiatric disorders after traumatic brain injury. Neurology. 2004;63:749–750.
Levin H, Wilde E, Troyanskaya M, et al. Diffusion tensor imaging of mild to moderate blast-related traumatic brain injury and its sequelae. J Neurotrauma. 2010;27:683–694.
Lovell MR, Collins MW, Iverson GL, et al. Recovery from mild concussion in high school athletes. J Neurosurg. 2003;98:296–301.
McCrea M, Guskiewicz KM, Marshall SW, et al. Acute effects and recovery time following concussion in collegiate football players: The NCAA Concussion Study. JAMA. 2003;290:2556–2563.
Mckee A, Cantu R, Nowinski C, et al. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J Neuropath Experiment Neurol. 2009;68:709–735.
Mears S, Shores EA, Taylor AJ, et al. Mild traumatic brain injury does not predict acute postconcussion syndrome. J Neurol Neurol Psychiatry. 2008;79:300–306.
Omalu B, DeKosky S, Minster R, et al. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–134.
Rao V, Rosenberg P, Bertrand M, et al. Aggression after traumatic brain injury: Prevalence and correlates. J Neuropsychiatry Clin Neurosci. 2009;21:420–429.
Rohling ML, Allen LM, Green P. Who is exaggerating cognitive impairment and who is not? CNS Spectrums. 2002;7:387–395.
Ruff R. Two decades of advances in understanding of mild traumatic brain injury. J Head Trauma Rehab. 2005;1:5–18.
Schmans B, Lindboom J, Schagen S, et al. Cognitive complaints in patients after whiplash injury: The impact of malingering. J Neurol Neurosurg Psychiatry. 1998;64:339–343.
Siddall OM. Use of methylphenidate in traumatic brain injury. Ann Pharmacother. 2005;39:1309–1313.
Silver JM, McAllister RW, Yudofsky SC. Textbook of Traumatic Brain Injury, 2nd ed. Washington, DC: American Psychiatric Publishing; 2011.
Stulemeijer M, Vos PE, Bleijenberg G, et al. Cognitive complaints after mild traumatic brain injury: Things are not always what they seem. J Psychosomat Res. 2007;63:637–645.
Tateno A, Jorge RE, Robinson RG. Clinical correlates of aggressive behavior after traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2003;15:155–160.
Tateno A, Jorge RE, Robinson RG. Pathologic laughing and crying following traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2004;16:426–434.
Warden DL, Gordon B, McAllister TW. Neurobehavioral guidelines for the pharmacologic treatment of neurobehavioral sequelae of traumatic brain injury. J Neurotrauma. 2006;23:1468–1501.
Wenzel HG, Haug TT, Mykletun A, et al. A population study of anxiety and depression among persons who report whiplash traumas. J Psychosom Res. 2002;53:831–835.
Writer BW, Schillerstrom JE. Psychopharmacological treatment for cognitive impairment in survivors of traumatic brain injury: A critical review. J Neuropsychiatry Clin Neurosci. 2009;21:362–370.
Chapter 22 Questions and Answers
2. At the scene of a high-speed, single-vehicle, motor vehicle accident (MVA), emergency workers found a stuporous 24-year-old man and gave him a GCS score of 9. They dressed his head and bodily wounds and supported his circulation. During transfer to the hospital, he lapsed into coma and developed decerebrate posture and a left third cranial nerve palsy. On arrival, his GCS had fallen to 3. X-rays showed a fracture through his left temporal bone of the skull. Which is the most likely cause of his deterioration?
3. Which statement is false regarding chronic subdural hematomas?
10. After falling forward and striking his head, a 72-year-old man describes being unable to read because he sees double. To alleviate the problem, he covers one eye, avoids looking at closely held objects, or tilts his head to the left. The visual acuity in each eye is normal. Which is the most likely site of injury?
c. Neuropsychologic sequelae may not appear for several years.
28. Which statement most accurately compares the risk of developing Alzheimer disease among TBI survivors carrying two Apo-E4 alleles compared to those carrying none?
29. After the emergency room staff evaluated a 77-year-old widow who had sustained a fall but did not fracture any bones, she refused to return home to her family. Her objections were unclear, but they were so vocal and adamant that the staff solicited a psychiatry consultation. The psychiatrist found that, during the previous 3 months, the emergency room staff had evaluated her on different occasions for diabetic ketoacidosis, hypoglycemia, a wrist fracture, and rib fractures. For which condition should the staff evaluate the patient before sending her home?
30. A well-kicked soccer ball struck the head of a 16-year-old student, who was not a particularly good athlete, in the head. The blow stunned him, but did not cause a loss of consciousness. For the next week he blamed the injury on his inability to learn new material in school. Of the following deficits, which one would neuropsychologic testing most likely reveal?
32. A 32-year-old Marine, who witnessed violent death of men in his company, has flashbacks and other symptoms of posttraumatic stress disorder (PTSD). He also sustained a low back injury from a fall. His neurologic examination and MRIs of his head and low back are normal. His physicians have prescribed hypnotics, benzodiazepines, antidepressants, and, for low back pain, opioids, but he remains incapacitated. Which strategy would probably produce the greatest benefit with least risk and side effect?
35. A 37-year-old woman sustained a concussion in a rear-end MVA. She sought medical attention primarily for posttraumatic headaches which struck her three times a week. Because she carried a history of migraine since she was a teenager, the neurologist prescribed an aspirin–butalbital–caffeine compound. When that medication proved inadequate, he added an acetaminophen–codeine compound. After 4 months, she stated that she was suffering from almost daily headaches. A repeat neurologic examination, MRI of her head, and routine tests showed no abnormality. At this time, which is the most likely cause of her headaches?
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 22 Traumatic Brain Injury
Major Head Trauma
How Does Head Trauma Cause TBI?
Direct Force
A blow – by its direct mechanical force (a coup injury) – disrupts the underlying, delicate brain tissue (the parenchyma). As with strokes, trauma causes cell death by necrosis and its accompanying inflammatory changes, particularly monocular cell infiltration. (In contrast, neurodegenerative diseases, such as Huntington disease and amyotrophic lateral sclerosis, cause cell death by apoptosis [see Chapter 18].)
In addition to causing the coup injury, head trauma throws the brain against the opposite inner surface (table) of the skull, which causes a contrecoup injury of that surface of the brain. Damage from contrecoup injuries frequently surpasses that from the coup injury. Contrecoup injuries frequently damage the temporal and frontal lobes because their anterior surfaces abut against the sharp edges of the skull’s anterior and middle cranial fossae (Fig. 22-1). Depending on its severity, damage of the frontal and temporal lobes characteristically leads to memory impairment and personality changes.
Diffuse Axonal Shearing
Diffuse axonal shearing, which also leads to TBI, consists not only of trauma to long subcortical white-matter axons, but also damage to cytoskeletal elements and a fatal intracellular influx of calcium. Although neither computed tomography (CT) nor magnetic resonance imaging (MRI) can illustrate diffuse axonal shearing, diffusion tensor imaging readily detects it by showing traumatic disruption of white-matter tracts (see Chapter 20).
Bleeding Within the Skull, but Outside the Brain
• Pia mater, the innermost layer, is a thin, translucent, vascular membrane adherent to the cerebral gyri. It follows gyri into sulci and thus allows for a generous region, the subarachnoid space, between it and the immediately overlying layer – the arachnoid mater.
• Arachnoid mater, also thin, spans the tops of gyri, capping the sulci. The subarachnoid space, which contains the cerebrospinal fluid (CSF), envelops the brain and, continuing downward within the spinal canal to the sacrum, the spinal cord and cauda equina. Neurologists performing a spinal tap or lumbar puncture insert a special needle into the subarachnoid space below the conus medullaris (lower end of the spinal cord), which is situated between the T12 and L1 vertebrae, to sample CSF.
• Dura mater, the outermost layer of the meninges, is a thick fibrous tissue adherent to the interior surface of the skull. Two of its infoldings, the falx and tentorium, support the brain and house most of its venous drainage. Neurologists designate the space between the skull and the underlying dura as the epidural space, and between the dura and the underlying arachnoid as the subdural space. Major head trauma may cause hematomas in either or both of these areas.
Epidural hematomas, which typically result from temporal bone fractures with concomitant middle meningeal artery lacerations, are essentially rapidly expanding, high-pressure, fresh blood clots (Fig. 22-2). They compress the underlying brain and force it through the tentorial notch, i.e., they produce transtentorial herniation (see Fig. 19-3). Unless surgery can immediately arrest the bleeding, epidural hematomas are usually fatal.
FIGURE 22-2 Meningeal arterial bleeding, which usually results from a blow forceful enough to fracture the skull, causes epidural hematomas. In contrast, venous bleeding, usually slower and under less pressure, causes subdural hematomas. Ruptured aneurysms and head trauma often cause subarachnoid hemorrhage (SAH). In SAH, blood spreads within the subarachnoid space over the convexities, between the gyri, into the interhemispheric fissure, and down into the spinal canal.
As an example, a victim of an assault with a baseball bat lost consciousness when struck. After regaining consciousness for 1 hour, the victim lapsed into coma and developed fatal decerebrate posture (see Fig. 19-3). A CT showed a temporal skull fracture and an underlying epidural hematoma (see Fig. 20-9D). Neurologists label the period when the victim transiently regained consciousness the lucid interval.
In contrast, subdural hematomas usually result from slowly bleeding bridging veins, under relatively low pressure, into the subdural space (see Fig. 20-9). Dark, venous blood generally oozes into the extensive subdural space until the expanding hematoma encounters underlying brain. The brain dampens bleeding and suppresses further expansion of the subdural hematoma. However, if the hematoma continues to expand, it may lead to cerebral transtentorial herniation or cerebellar herniation through the foramen magnum. Survivors often have permanent brain damage from the initial trauma and the pressure from the subdural hematoma.
Acute subdural hematomas, which are most apt to occur in alcoholics, individuals medicated with warfarin, or the elderly (see later), produce headache, confusion, and a deteriorating level of consciousness over several hours to 1 or 2 days. Depending on the extent of the bleeding and time until the diagnosis, patients may develop focal signs and herniation. A history of head trauma does not necessarily precede the symptoms. CTs show acute, dense blood in the subdural space (see Fig. 20-9).
Chronic subdural hematomas, ones that have developed and persisted for weeks, usually have spread extensively in the subdural space (see Fig. 20-9). They typically give rise to insidiously developing headache, change in personality, and cognitive impairment, but only subtle focal physical deficits (see Chapters 19 and 20). Although subdural hematomas may spontaneously resolve, they often require surgical evacuation. Because subdural hematoma patients’ cognitive decline is rapid, but treatment usually reverses their symptoms, neurologists often include subdural hematomas as an explanation for “rapidly developing dementia” and a “reversible cause of dementia” (see Chapter 7).
Posttraumatic Coma and Delirium
Following head trauma, as well as at other times, neurologists often classify patients’ level of consciousness as alert, lethargic, stuporous, or comatose. They may also use the Glasgow Coma Scale (GCS), which measures three readily obvious neurologic functions: eye opening, speaking, and moving (Table 22-1). In major head trauma, the GCS correlates closely with survival and neurologic sequelae; however, in minor head trauma, it correlates poorly. Moreover, physicians cannot appropriately include the GCS as part of a standard mental status examination for patients suspected of having dementia or circumscribed neuropsychologic deficits.
| Category | Score | |
|---|---|---|
| Eyes opening | Never | 1 |
| To pain | 2 | |
| To verbal stimuli | 3 | |
| Spontaneously | 4 | |
| Best verbal response | None | 1 |
| Incomprehensible sounds | 2 | |
| Inappropriate words | 3 | |
| Disoriented and converses | 4 | |
| Oriented and converses | 5 | |
| Best motor response | None | 1 |
| Extension* | 2 | |
| Flexion† | 3 | |
| Flexion withdrawal | 4 | |
| Patient localizes pain | 5 | |
| Patient obeys | 6 | |
| Total | 3–15 |
This standard scale quantitates the level of consciousness, with the lower scores indicating less neurologic function. Neurologists interpret scores of 3–8 as signifying severe traumatic brain injury (TBI) or coma; 9–12, moderate TBI; and 13–15, mild TBI. However, the GCS is not readily applicable to patients who have sustained cerebral hypoxia and, because they cannot make a verbal response, those who are intubated. Adapted, with kind permission, from Teasdale G, Jennett B. Assessment of coma and impaired consciousness: A practical scale. Lancet 1974;2:81–84.
*Decerebrate rigidity (Fig. 19-3).
†Decorticate rigidity (Fig. 11-5).
By the first day after TBI, of patients who score 3 on the GCS, which is the lowest possible score, 90% have a fatal outcome and most of the remaining never regain consciousness. By 4 weeks, almost all TBI comatose patients die, partially recover and regain consciousness, or evolve into the vegetative state (see Chapter 11). When in coma or the vegetative state, individuals cannot perceive pain and do not suffer.
Posttraumatic Epilepsy
Seizures that occur either within the first 24 hours (immediate posttraumatic seizures) or the first 7 days (early posttraumatic seizures) do not fall within the definition of PTE because neurologists label them “provoked seizures.” Only seizures that occur (and recur) more than 7 days after the traumatic injury fall into the definition of PTE. Of note, 1 week of phenytoin prophylaxis following severe TBI reduces early posttraumatic seizures, but no AED clearly reduces the prevalence of PTE. PTE remits in only 25–50% of cases. PTE usually takes the form of complex partial seizures that undergo secondary generalization (see Chapter 10). Not only does PTE cause disability and carry the risk of further head injury, AEDs may exacerbate TBI-induced cognitive and personality changes.
Cognitive Impairment
TBI-induced coma usually lasts, at most, 4 weeks. By then, most patients have either succumbed to their injuries or recovered at least some cognition. However, many patients remain in a twilight state with their eyes open, but unconscious. These patients are incapable of thinking, communicating, or deliberately moving. They cannot perceive pain and do not suffer. Most of them linger in the persistent vegetative state (see Chapter 11).
Surprisingly, the trauma’s location, with one important exception, correlates inconsistently with cognitive impairment. Left temporal lobe injuries, the exception, routinely produce vocabulary deficits similar to anomic aphasia (see Chapter 8).
In addition to TBI causing debilitating cognitive impairments, some epidemiologic studies suggest that it also constitutes a risk factor for Alzheimer disease. Studies have shown that severe head trauma causes increased levels of soluble amyloid and deposition of amyloid plaques, one of the hallmarks of Alzheimer disease. Several, but not all, studies also suggest that moderate and severe head trauma in individuals with two apolipoprotein E4 (Apo-E4) alleles correlates with a marked increased risk of developing Alzheimer disease (see Chapter 7). Surviving moderate TBI, individuals with two Apo-E4 alleles may have twice the risk of developing Alzheimer disease, and surviving severe TBI, those individuals may have four times the risk. (A confounding issue for some of these studies is that individuals with Alzheimer disease are prone to cause an accident in which they sustain TBI and come to medical attention.)
Treatment Strategies for Posttraumatic Cognitive Impairment
Neurologists and other physicians who work with TBI patients with cognitive impairment administer medications that hopefully increase their attentiveness, if not reverse their learning and memory impairments. Of the numerous medicines that purportedly help, few have undergone rigorous trials that have documented their value. For example, many trials have indicated that methylphenidate and other dopamine-enhancing medications increase patients’ attention and directly or indirectly memory. Anticholinesterases may also improve TBI patients’ memory, at least those who have suffered severe memory deficits (see Chapter 7). Treatment of comorbid PTSD, if present, may improve cognitive impairments and other posttraumatic neuropsychologic changes. At the same time, physicians should, if possible, reduce or eliminate medicines that may interfere with attention and memory, such as AEDs, antipsychotics, and minor tranquilizers. They should always bear in mind that multiple medicines are likely to lead to adverse interactions and unwanted, occasionally fatal, outcomes.
Other Mental Disturbances
Personality Changes
In addition to altering patients’ personality and possibly cognitive capacity, frontal lobe injury may disable their executive abilities. Just as with patients who sustain anterior cerebral artery infarctions, those who sustain frontal lobe trauma have impaired ability to solve abstract problems, plan sequenced actions, or execute responses. Moreover, patients who develop pseudobulbar palsy, another complication of frontal lobe trauma, may display emotional incontinence with easily precipitated fits of pathologic laughing or crying, and unexpected sudden switches between them (see Chapter 4).
Trauma in Childhood
When TBI occurs before growth spurts, affected limbs may fail to achieve their normal, expectable size. The limbs’ growth arrest resembles the spastic hemiparesis with foreshortened limbs that characterizes congenital cerebral injuries (see Fig. 13-4). If dominant hemisphere injury were to occur before age 5 years, the opposite hemisphere would usually assume control of language. For example, a left-sided cerebral injury in a 4-year-old child will probably not result in aphasia because the plasticity of the brain allows the right cerebral hemisphere to develop language centers. If TBI affects the dominant hemisphere of someone age 5 years or older, it is likely to cause language impairments, if not clear-cut aphasia.
Nonaccidental Head Injury
The shaken baby syndrome, which is another form of abusive head trauma, consists of the injuries inflicted by violent back-and-forth throws, without direct impact. In this violence, rotational (angular) deceleration produces diffuse axonal shearing, hemorrhages in the brain’s delicate parenchyma and subdural space, and retinal hemorrhages (Fig. 22-3).
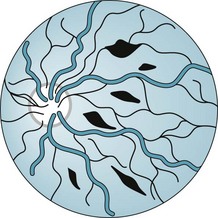
FIGURE 22-3 Head trauma causes small hemorrhages throughout the retina – not just around the disk as in papilledema.
[/not-level-membership-for-neurology-category]
