247 |
Amebiasis and Infection with Free-Living Amebas |
AMEBIASIS
DEFINITION
Amebiasis is an infection with the intestinal protozoan Entamoeba histolytica. About 90% of infections are asymptomatic, and the remaining 10% produce a spectrum of clinical syndromes ranging from dysentery to abscesses of the liver or other organs.
LIFE CYCLE AND TRANSMISSION
E. histolytica is acquired by ingestion of viable cysts from fecally contaminated water, food, or hands. Food-borne exposure is most prevalent and is particularly likely when food handlers are shedding cysts or food is being grown with feces-contaminated soil, fertilizer, or water. Besides the drinking of contaminated water, less common means of transmission include oral and anal sexual practices and—in rare instances—direct rectal inoculation through colonic irrigation devices. Motile trophozoites are released from cysts in the small intestine and, in most patients, remain as harmless commensals in the large bowel. After encystation, infectious cysts are shed in the stool and can survive for several weeks in a moist environment. In some patients, the trophozoites invade either the bowel mucosa, causing symptomatic colitis, or the bloodstream, causing distant abscesses of the liver, lungs, or brain. The trophozoites may not encyst in patients with active dysentery, and motile hematophagous trophozoites are frequently present in fresh stools. Trophozoites are rapidly killed by exposure to air or stomach acid, however, and therefore cannot transmit infection.
EPIDEMIOLOGY
![]() About 10% of the world’s population is infected with Entamoeba, the majority with noninvasive Entamoeba dispar. Amebiasis results from infection with E. histolytica and is the third most common cause of death from parasitic disease (after schistosomiasis and malaria). Invasive colitis and liver abscesses are sevenfold more common among men than among women; this difference has been attributed to a disparity in complement-mediated killing. The wide spectrum of clinical disease caused by Entamoeba is due in part to the differences between these two infecting species. E. histolytica has unique isoenzymes, surface antigens, DNA markers, and virulence properties that distinguish it from other genetically related and morphologically identical species, such as E. dispar and E. moshkovskii.
About 10% of the world’s population is infected with Entamoeba, the majority with noninvasive Entamoeba dispar. Amebiasis results from infection with E. histolytica and is the third most common cause of death from parasitic disease (after schistosomiasis and malaria). Invasive colitis and liver abscesses are sevenfold more common among men than among women; this difference has been attributed to a disparity in complement-mediated killing. The wide spectrum of clinical disease caused by Entamoeba is due in part to the differences between these two infecting species. E. histolytica has unique isoenzymes, surface antigens, DNA markers, and virulence properties that distinguish it from other genetically related and morphologically identical species, such as E. dispar and E. moshkovskii.
Most asymptomatic carriers, including men who have sex with men (MSM) and patients with AIDS, harbor E. dispar and have self-limited infections. In this respect, E. dispar is dissimilar to other enteric pathogens such as Cryptosporidium and Cystoisospora belli, which can cause self-limited illnesses in immunocompetent hosts but devastating diarrhea in patients with AIDS. These observations indicate that E. dispar is incapable of causing invasive disease. Unlike E. dispar, E. histolytica can cause invasive disease, as demonstrated in recent reports from Korea, China, and India that suggest higher prevalences of amebic seroconversion, invasive amebiasis, and amebic liver abscesses among HIV-positive than HIV-negative patients. In another study, 10% of asymptomatic patients who were colonized with E. histolytica went on to develop amebic colitis, while the rest remained asymptomatic and cleared the infection within 1 year.
The potential of E. moshkovskii to cause diarrhea, weight loss, and colitis was recently demonstrated in a mouse model of cecal infection. However, the pathogenic potential of this species is not clear. A prospective evaluation of children from the Mirpur community of Dhaka, Bangladesh, found that most children who had diarrheal diseases associated with E. moshkovskii were simultaneously infected with at least one other enteric pathogen.
Areas of highest incidence of Entamoeba infection (due to inadequate sanitation and crowding) include most developing countries in the tropics, particularly Mexico, India, and nations of Central and South America, tropical Asia, and Africa. In a 4-year follow-up study of preschool children in a highly endemic area of Bangladesh, 80% of children had at least one episode of E. histolytica infection and 53% had more than one episode. Naturally acquired immunity did develop but was usually short-lived and correlated with the presence in the stool of secretory IgA antibody to the major adherence lectin galactose N-acetylgalactosamine (Gal/GalNAc). The main groups at risk for amebiasis in developed countries are returned travelers, recent immigrants, MSM, military personnel, and inmates of institutions. Data from the GeoSentinel Surveillance Network, which come from tropical medicine clinics on six continents, showed that, among long-term travelers (trip duration, >6 months), diarrhea due to E. histolytica was among the most common diagnoses.
PATHOGENESIS AND PATHOLOGY
Both trophozoites (Fig. 247-1) and cysts (Fig. 247-2) are found in the intestinal lumen, but only trophozoites of E. histolytica invade tissue. The trophozoite is 20–60 μm in diameter and contains vacuoles and a nucleus with a characteristic central nucleolus. In animals, depletion of intestinal mucus, diffuse inflammation, and disruption of the epithelial barrier precede trophozoite contact with the colonic mucosa. Trophozoites attach to colonic mucus and epithelial cells by their Gal/GalNAc lectin. The earliest intestinal lesions are microulcerations of the mucosa of the cecum, sigmoid colon, or rectum that release erythrocytes, inflammatory cells, and epithelial cells. Proctoscopy reveals small ulcers with heaped-up margins and normal intervening mucosa (Fig. 247-3A). Submucosal extension of ulcerations under viable-appearing surface mucosa causes the classic “flask-shaped” ulcer containing trophozoites at the margins of dead and viable tissues. Although neutrophilic infiltrates may accompany the early lesions in animals, human intestinal infection is marked by a paucity of inflammatory cells, probably in part because of the killing of neutrophils by trophozoites (Fig. 247-3B). Treated ulcers characteristically heal with little or no scarring. Occasionally, however, full-thickness necrosis and perforation occur.
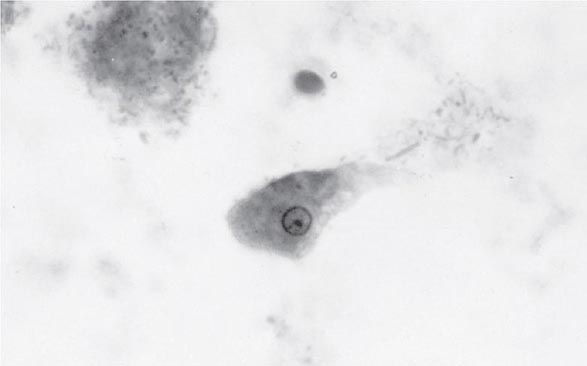
FIGURE 247-1 Trophozoite of E. histolytica. A single nucleus with a central, dot-like nucleolus is seen (trichrome stain).
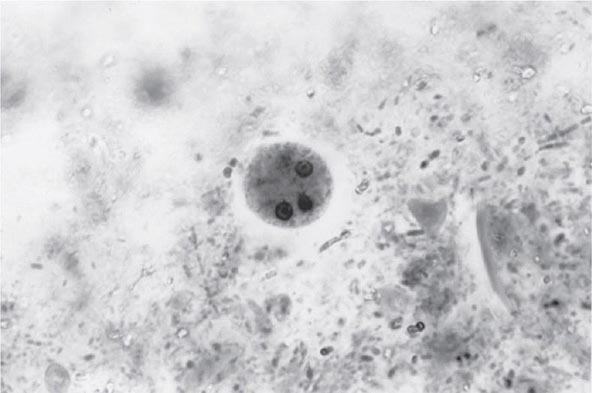
FIGURE 247-2 Cyst of E. histolytica. Three of the four nuclei are visible (trichrome stain).
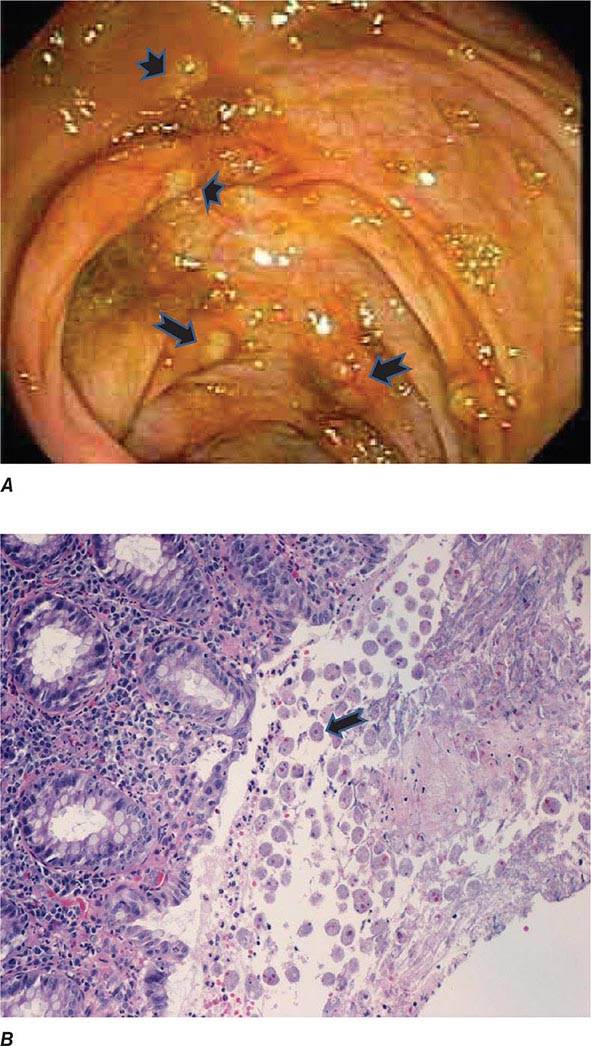
FIGURE 247-3 Endoscopic and histopathologic features of intestinal amebiasis. A. Appearance of ulcers on colonoscopy (arrows). B. Inflammatory infiltrate and E. histolytica trophozoites (arrow) in invasive amebic colitis (hematoxylin and eosin). (Courtesy of the Department of Pathology and Gastroenterology, VA San Diego Medical Center.)
Rarely, intestinal infection results in the formation of a mass lesion, or ameboma, in the bowel lumen. The overlying mucosa is usually thin and ulcerated, while other layers of the wall are thickened, edematous, and hemorrhagic; this condition results in exuberant formation of granulation tissue with little fibrous-tissue response.
A number of virulence factors have been linked to the ability of E. histolytica to invade through the interglandular epithelium. One factor consists of the extracellular cysteine proteinases that degrade collagen, elastin, IgA, IgG, and the anaphylatoxins C3a and C5a. Other enzymes may disrupt glycoprotein bonds between mucosal epithelial cells in the gut. Amebas can lyse neutrophils, monocytes, lymphocytes, and cells of colonic and hepatic lines. The cytolytic effect of amebas appears to require direct contact with target cells and may be linked to the release of phospholipase A and pore-forming peptides. E. histolytica trophozoites also cause apoptosis of human cells. Phagocytosis is a virulence factor that leads to defective parasite proliferation if inhibited. This process is potentially modulated by calmodulin-like calcium-binding protein 3, which pairs with actin and myosin during initiation and formation of phagosomes. Another virulence factor is the ability to resist reactive oxygen species, reactive nitrogen species such as nitric oxide, or S-nitrosothiols such as S-nitrosoglutathione (GSNO) and S-nitrosocysteine (CySNO). E. histolytica trophozoites are constantly exposed to reactive oxygen and nitrogen species from their own metabolism and host defenses during tissue invasion. Overexpression of hydrogen peroxide regulatory motif–binding protein appears to increase E. histolytica cytotoxicity. Since E. histolytica lacks glutathione and glutathione reductase, it relies on its thioredoxin/thioredoxin reductase system to prevent, regulate, and repair the damage caused by oxidative stress. This antioxidant system is versatile in that it can reduce reactive nitrogen species and use an alternative electron donor such as the reduced form of nicotinamide adenine dinucleotide. Metronidazole, the current standard of therapy for amebiasis, seems to exert its antiparasitic effect through the inhibition of this antioxidant system. Newer therapeutic candidates targeting this system, such as auranofin, also have demonstrated in vitro and in vivo efficacy against this parasite.
Liver abscesses are always preceded by intestinal colonization, which may be asymptomatic. Blood vessels may be compromised early by wall lysis and thrombus formation. Trophozoites invade veins to reach the liver through the portal venous system. E. histolytica is resistant to complement-mediated lysis—a property critical to survival in the bloodstream. In contrast, E. dispar is rapidly lysed by complement and is thus restricted to the bowel lumen. Inoculation of amebas into the portal system of hamsters results in an acute cellular infiltrate consisting predominantly of neutrophils. Later, the neutrophils are lysed by contact with amebas, and the release of neutrophil toxins may contribute to necrosis of hepatocytes. The liver parenchyma is replaced by necrotic material that is surrounded by a thin rim of congested liver tissue. The necrotic contents of a liver abscess are classically described as “anchovy paste,” although the fluid is variable in color and is composed of bacteriologically sterile granular debris with few or no cells. Amebas, if seen, tend to be found near the capsule of the abscess.
Host innate and adaptive immunity are important factors that determine susceptibility to invasive disease and its clinical outcome. While neutrophils were thought to contribute to tissue damage in intestinal and liver amebiasis due to their cytotoxic effects on host epithelial cells, a recent report suggests that they may exert a protective effect in susceptible mice. Neutropenia, induced with an antibody to Gr-1 (i.e., to peripheral neutrophils), led to death in C3H/HeJ mice and to severe disease in CBA mice (both of which are relatively susceptible to E. histolytica infection), while it had no effect on C57BL/6 mice, which are known for their intrinsic resistance to infection with this parasite.
Antimicrobial peptides, such as cathelicidins, are an important part of innate immunity and are induced by E. histolytica upon intestinal invasion in a mouse model. In this model, cecal cathelicidin-related antimicrobial peptide (CRAMP) mRNA increased more than fourfold by 3 days and more than 100-fold at 7 days. However, E. histolytica remained resistant to cathelicidin-mediated killing, probably because the antimicrobial peptide was digested by amebic cysteine proteinases.
![]() IgA plays a critical role in acquired immunity to E. histolytica. A study in Bangladeshi schoolchildren revealed that an intestinal IgA response to Gal/GalNAc reduced the risk of new E. histolytica infection by 64%. Serum IgG antibody is not protective; titers correlate with the duration of illness rather than with the severity of disease. Indeed, Bangladeshi children with a serum IgG response were more likely than those without such a response to develop new E. histolytica infection. In infants from this same Bangladeshi community, passive immunity conferred by maternal parasite-specific IgA via breastfeeding resulted in a 39% reduction in risk of infection and a 64% reduction in risk of diarrheal disease from E. histolytica during the first year of life.
IgA plays a critical role in acquired immunity to E. histolytica. A study in Bangladeshi schoolchildren revealed that an intestinal IgA response to Gal/GalNAc reduced the risk of new E. histolytica infection by 64%. Serum IgG antibody is not protective; titers correlate with the duration of illness rather than with the severity of disease. Indeed, Bangladeshi children with a serum IgG response were more likely than those without such a response to develop new E. histolytica infection. In infants from this same Bangladeshi community, passive immunity conferred by maternal parasite-specific IgA via breastfeeding resulted in a 39% reduction in risk of infection and a 64% reduction in risk of diarrheal disease from E. histolytica during the first year of life.
A link between nutrition and immunity is demonstrated by the elevated rate of infections due to protozoan parasites, including E. histolytica, among undernourished children in developing countries. Resistance to amebiasis is associated with a polymorphism in the receptor for the adipocytokine leptin. Children in a Bangladeshi cohort with a mutant R223 leptin receptor allele were nearly four times more likely to be infected with E. histolytica than those carrying the ancestral Q223 allele. This mutant allele is overrepresented in many geographic areas with a high prevalence of amebiasis, such as Bangladesh and India.
CLINICAL SYNDROMES
Intestinal Amebiasis The most common type of amebic infection is asymptomatic cyst passage. Even in highly endemic areas, most patients harbor E. dispar.
Symptomatic amebic colitis develops 2–6 weeks after the ingestion of infectious E. histolytica cysts. A gradual onset of lower abdominal pain and mild diarrhea is followed by malaise, weight loss, and diffuse lower abdominal or back pain. Cecal involvement may mimic acute appendicitis. Patients with full-blown dysentery may pass 10–12 stools per day. The stools contain little fecal material and consist mainly of blood and mucus. In contrast to those with bacterial diarrhea, fewer than 40% of patients with amebic dysentery are febrile. Virtually all patients have heme-positive stools.
More fulminant intestinal infection, with severe abdominal pain, high fever, and profuse diarrhea, is rare and occurs predominantly in children. Patients may develop toxic megacolon, in which there is severe bowel dilation with intramural air. Patients receiving glucocorticoids are at risk for severe amebiasis. Uncommonly, patients develop a chronic form of amebic colitis, which can be confused with inflammatory bowel disease. The association between severe amebiasis complications and glucocorticoid therapy emphasizes the importance of excluding amebiasis when inflammatory bowel disease is suspected. An occasional patient presents with only an asymptomatic or tender abdominal mass caused by an ameboma, which is easily confused with cancer on barium studies. A positive serologic test or biopsy can prevent unnecessary surgery in this setting. The syndrome of post–amebic colitis—i.e., persistent diarrhea following documented cure of amebic colitis—is controversial; no evidence of recurrent amebic infection can be found, and re-treatment usually has no effect.
Amebic Liver Abscess Extraintestinal infection by E. histolytica most often involves the liver. Of travelers who develop an amebic liver abscess after leaving an endemic area, 95% do so within 5 months. Young patients with an amebic liver abscess are more likely than older patients to present in the acute phase with prominent symptoms of <10 days’ duration. Most patients are febrile and have right-upper-quadrant pain, which may be dull or pleuritic in nature and may radiate to the shoulder. Point tenderness over the liver and right-sided pleural effusion are common. Jaundice is rare. Although the initial site of infection is the colon, fewer than one-third of patients with an amebic abscess have active diarrhea. Older patients from endemic areas are more likely to have a subacute course lasting 6 months, with weight loss and hepatomegaly. About one-third of patients with chronic presentations are febrile. Thus, the clinical diagnosis of an amebic liver abscess may be difficult to establish because the symptoms and signs are often nonspecific. Since 10–15% of patients present only with fever, amebic liver abscess must be considered in the differential diagnosis of fever of unknown origin (Chap. 26).
Complications of Amebic Liver Abscess Pleuropulmonary involvement, which is reported in 20–30% of patients, is the most frequent complication of amebic liver abscess. Manifestations include sterile effusions, contiguous spread from the liver, and rupture into the pleural space. Sterile effusions and contiguous spread usually resolve with medical therapy, but frank rupture into the pleural space requires drainage. A hepatobronchial fistula may cause cough productive of large amounts of necrotic material that may contain amebas. This dramatic complication carries a good prognosis. Abscesses that rupture into the peritoneum may present as an indolent leak or an acute abdomen and require both percutaneous catheter drainage and medical therapy. Rupture into the pericardium, usually from abscesses of the left lobe of the liver, carries the gravest prognosis; it can occur during medical therapy and requires surgical drainage.
Other Extraintestinal Sites The genitourinary tract may become involved by direct extension of amebiasis from the colon or by hematogenous spread of the infection. Painful genital ulcers, characterized by a punched-out appearance and profuse discharge, may develop secondary to extension from either the intestine or the liver. Both of these conditions respond well to medical therapy. Cerebral involvement has been reported in fewer than 0.1% of patients in large clinical series. Symptoms and prognosis depend on the size and location of the lesion.
DIAGNOSTIC TESTS
Laboratory Diagnosis Stool examinations, serologic tests, and noninvasive imaging of the liver are the most important procedures in the diagnosis of amebiasis. Fecal findings suggestive of amebic colitis include a positive test for heme, a paucity of neutrophils, and amebic cysts or trophozoites. The definitive diagnosis of amebic colitis is made by the demonstration of hematophagous trophozoites of E. histolytica (Fig. 247-1). Because trophozoites are killed rapidly by water, drying, or barium, it is important to examine at least three fresh stool specimens. Examination of a combination of wet mounts, iodine-stained concentrates, and trichrome-stained preparations of fresh stool and concentrates for cysts (Fig. 247-2) or trophozoites (Fig. 247-1) confirms the diagnosis in 75–95% of cases. Culture of amebas is more sensitive, but this diagnostic method is not routinely available. If stool examinations are negative, sigmoidoscopy with biopsy of the edge of ulcers may increase the yield, but this procedure is dangerous during fulminant colitis because of the risk of perforation. Trophozoites in a biopsy specimen from a colonic mass confirm the diagnosis of ameboma, but trophozoites are rare in liver aspirates because they are found in the abscess capsule and not in the readily aspirated necrotic center. Accurate diagnosis requires experience, since the trophozoites may be confused with neutrophils and the cysts must be differentiated morphologically from those of Entamoeba hartmanni, Entamoeba coli, and Endolimax nana, which do not cause clinical disease and do not warrant therapy. Unfortunately, the cysts of E. histolytica cannot be distinguished microscopically from those of E. dispar or E. moshkovskii. Therefore, the microscopic diagnosis of E. histolytica can be made only by the detection of Entamoeba trophozoites that have ingested erythrocytes. In terms of sensitivity, stool diagnostic tests based on the detection of the Gal/GalNAc lectin of E. histolytica compare favorably with the polymerase chain reaction and with isolation in culture followed by isoenzyme analysis.
Serology is an important addition to the methods used for parasitologic diagnosis of invasive amebiasis. Enzyme-linked immunosorbent assays and agar gel diffusion assays are positive in more than 90% of patients with colitis, amebomas, or liver abscess. Positive results in conjunction with the appropriate clinical syndrome suggest active disease because serologic findings usually revert to negative within 6–12 months. Even in highly endemic areas such as South Africa, fewer than 10% of asymptomatic individuals have a positive amebic serology. The interpretation of the indirect hemagglutination test is more difficult because titers may remain positive for as long as 10 years.
Up to 10% of patients with acute amebic liver abscess may have negative serologic findings; in suspected cases with an initially negative result, testing should be repeated in 1 week. In contrast to carriers of E. dispar, most asymptomatic carriers of E. histolytica develop antibodies. Thus, serologic tests are helpful in assessing the risk of invasive amebiasis in asymptomatic, cyst-passing individuals in nonendemic areas. Serologic tests also should be performed in patients with ulcerative colitis before the institution of glucocorticoid therapy to prevent the development of severe colitis or toxic megacolon owing to unsuspected amebiasis.
Routine hematology and chemistry tests usually are not very helpful in the diagnosis of invasive amebiasis. About three-fourths of patients with an amebic liver abscess have leukocytosis (>10,000 cells/μL); this condition is particularly likely if symptoms are acute or complications have developed. Invasive amebiasis does not elicit eosinophilia. Anemia, if present, is usually multifactorial. Even with large liver abscesses, liver enzyme levels are normal or minimally elevated. The alkaline phosphatase level is most often elevated and may remain so for months. Aminotransferase elevations suggest acute disease or a complication.
Radiographic Studies Radiographic barium studies are potentially dangerous in acute amebic colitis. Amebomas are usually identified first by a barium enema, but biopsy is necessary for differentiation from carcinoma.
Radiographic techniques such as ultrasonography, CT, and MRI are all useful for detection of the round or oval hypoechoic cyst of an amebic liver abscess. More than 80% of patients who have had symptoms for >10 days have a single abscess of the right lobe of the liver (Fig. 247-4). Approximately 50% of patients who have had symptoms for <10 days have multiple abscesses. Findings associated with complications include large abscesses (>10 cm) in the superior part of the right lobe, which may rupture into the pleural space; multiple lesions, which must be differentiated from pyogenic abscesses; and lesions of the left lobe, which may rupture into the pericardium. Because abscesses resolve slowly and may increase in size in patients who are responding clinically to therapy, frequent follow-up ultrasonography may prove confusing. Complete resolution of a liver abscess within 6 months can be anticipated in two-thirds of patients, but 10% may have persistent abnormalities for a year.
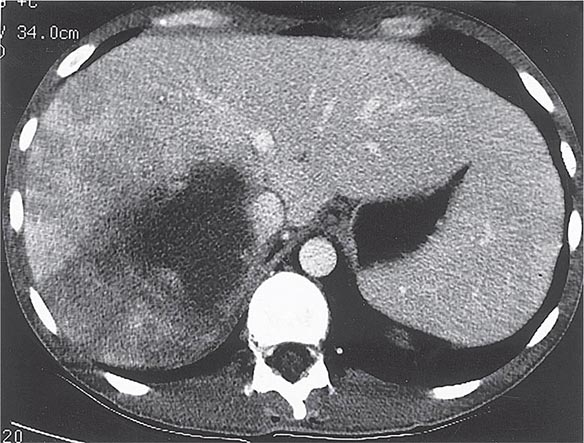
FIGURE 247-4 Abdominal CT scan of a large amebic abscess of the right lobe of the liver. (Courtesy of the Department of Radiology, UCSD Medical Center, San Diego; with permission.)
Differential Diagnosis The differential diagnosis of intestinal amebiasis includes bacterial diarrheas (Chap. 160) caused by Campylobacter (Chap. 192); enteroinvasive Escherichia coli (Chap. 186); and species of Shigella (Chap. 191), Salmonella (Chap. 190), and Vibrio (Chap. 193). Although the typical patient with amebic colitis has less prominent fever than in these other conditions as well as heme-positive stools with few neutrophils, correct diagnosis requires bacterial cultures, microscopic examination of stools, and amebic serologic testing. As has already been mentioned, amebiasis must be ruled out in any patient thought to have inflammatory bowel disease.
Because of the variety of presenting signs and symptoms, amebic liver abscess can easily be confused with pulmonary or gallbladder disease or with any febrile illness with few localizing signs, such as malaria (Chap. 248) or typhoid fever (Chap. 190). The diagnosis should be considered in members of high-risk groups who have recently traveled outside the United States (Chap. 149) and in inmates of institutions. Once radiographic studies have identified an abscess in the liver, the most important differential diagnosis is between amebic and pyogenic abscess. Patients with pyogenic abscess typically are older and have a history of underlying bowel disease or recent surgery. Amebic serology is helpful, but aspiration of the abscess, with Gram’s staining and culture of the material, may be required for differentiation of the two diseases.
|
TREATMENT |
AMEBIASIS |
INTESTINAL DISEASE
The drugs used to treat amebiasis can be classified according to their primary site of action (Table 247-1). Luminal amebicides are poorly absorbed; they reach high concentrations in the bowel, but their activity is limited to cysts and trophozoites close to the mucosa. Only two luminal drugs are available in the United States: iodoquinol and paromomycin. Indications for the use of luminal agents include eradication of cysts in patients with colitis or a liver abscess and treatment of asymptomatic carriers. The majority of asymptomatic individuals who pass cysts are colonized with E. dispar, which does not warrant specific therapy. However, it is prudent to treat asymptomatic individuals who pass cysts unless E. dispar colonization can be definitively demonstrated by specific antigen-detection tests.
|
DRUG THERAPY FOR AMEBIASIS |
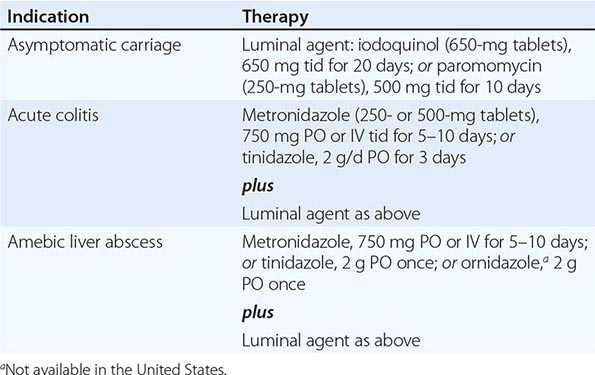
Tissue amebicides reach high concentrations in the blood and tissue after oral or parenteral administration. The development of nitroimidazole compounds, especially metronidazole, was a major advance in the treatment of invasive amebiasis. Patients with amebic colitis should be treated with IV or oral metronidazole. Side effects include nausea, vomiting, abdominal discomfort, and a disulfiram-like reaction. Another longer-acting imidazole compound, tinidazole, is also effective and available in the United States. All patients should also receive a full course of therapy with a luminal agent, since metronidazole does not eradicate cysts. Resistance to metronidazole has been selected in the laboratory but has not been found in clinical isolates. Relapses are not uncommon and probably represent reinfection or failure to eradicate amebas from the bowel because of an inadequate dosage or duration of therapy.
AMEBIC LIVER ABSCESS
Metronidazole is the drug of choice for amebic liver abscess. Longer-acting nitroimidazoles (tinidazole and ornidazole) have been effective as single-dose therapy in developing countries. With early diagnosis and therapy, mortality rates from uncomplicated amebic liver abscess are <1%. There is no evidence that combined therapy with two drugs is more effective than the single-drug regimen. Studies of South Africans with liver abscesses demonstrated that 72% of patients without intestinal symptoms had bowel infection with E. histolytica; thus, all treatment regimens should include a luminal agent to eradicate cysts and prevent further transmission. Amebic liver abscess recurs rarely.
More than 90% of patients respond dramatically to metronidazole therapy with decreases in both pain and fever within 72 h. Indications for aspiration of liver abscesses are (1) the need to rule out a pyogenic abscess, particularly in patients with multiple lesions; (2) the lack of a clinical response in 3–5 days; (3) the threat of imminent rupture; and (4) the need to prevent rupture of left-lobe abscesses into the pericardium. There is no evidence that aspiration, even of large abscesses (up to 10 cm), accelerates healing. Percutaneous drainage may be successful even if the liver abscess has already ruptured. Surgery should be reserved for instances of bowel perforation and rupture into the pericardium.
PREVENTION
Amebic infection is spread by ingestion of food or water contaminated with cysts. Since an asymptomatic carrier may excrete up to 15 million cysts per day, prevention of infection requires adequate sanitation and eradication of cyst carriage. In high-risk areas, infection can be minimized by the avoidance of unpeeled fruits and vegetables and the use of bottled water. Because cysts are resistant to readily attainable levels of chlorine, disinfection by iodination (tetraglycine hydroperiodide) is recommended. There is no effective prophylaxis.
INFECTION WITH FREE-LIVING AMEBAS
EPIDEMIOLOGY
![]() Free-living amebas of the genera Acanthamoeba and Naegleria are distributed throughout the world and have been isolated from a wide variety of fresh and brackish water, including that from lakes, taps, hot springs, swimming pools, and heating and air-conditioning units, and even from the nasal passages of healthy children. Encystation may protect the protozoa from desiccation and food deprivation. The persistence of Legionella pneumophila in water supplies may be attributable in part to chronic infection of free-living amebas, particularly Naegleria. Free-living amebas of the genus Balamuthia have been isolated from soil samples, including a sample from a flowerpot linked to a fatal infection in a child.
Free-living amebas of the genera Acanthamoeba and Naegleria are distributed throughout the world and have been isolated from a wide variety of fresh and brackish water, including that from lakes, taps, hot springs, swimming pools, and heating and air-conditioning units, and even from the nasal passages of healthy children. Encystation may protect the protozoa from desiccation and food deprivation. The persistence of Legionella pneumophila in water supplies may be attributable in part to chronic infection of free-living amebas, particularly Naegleria. Free-living amebas of the genus Balamuthia have been isolated from soil samples, including a sample from a flowerpot linked to a fatal infection in a child.
NAEGLERIA INFECTIONS
Primary amebic meningoencephalitis caused by Naegleria fowleri follows the aspiration of water contaminated with trophozoites or cysts or the inhalation of contaminated dust, leading to invasion of the olfactory neuroepithelium. Infection is most common among otherwise healthy children or young adults, who often report recent swimming in lakes or heated swimming pools. Rarely, some cases occur when contaminated water is used for nasal irrigation. After an incubation period of 2–15 days, severe headache, high fever, nausea, vomiting, and meningismus develop. Photophobia and palsies of the third, fourth, and sixth cranial nerves are common. Rapid progression to seizures and coma may follow. The prognosis is uniformly poor: most patients die within a week. Recently, two surviving children were treated with miltefosine, an investigational drug that is available through the Centers for Disease Control and Prevention (CDC) for the treatment of Naegleria infections.
The diagnosis of Naegleria infection should be considered in any patient who has purulent meningitis without evidence of bacteria on Gram’s staining, antigen detection assay, and culture. Other laboratory findings resemble those for fulminant bacterial meningitis, with elevated intracranial pressure, high white blood cell counts (up to 20,000/μL), and elevated protein concentrations and low glucose levels in cerebrospinal fluid (CSF). Diagnosis depends on the detection of motile trophozoites in wet mounts of fresh spinal fluid. Antibodies to Naegleria species have been detected in healthy adults; serologic testing is not useful in the diagnosis of acute infection.
ACANTHAMOEBA INFECTIONS
Granulomatous Amebic Encephalitis Infection with Acanthamoeba species follows a more indolent course and typically occurs in chronically ill or debilitated patients. Risk factors include lymphoproliferative disorders, chemotherapy, glucocorticoid therapy, lupus erythematosus, and AIDS. Infection usually reaches the central nervous system hematogenously from a primary focus in the sinuses, skin, or lungs. In the central nervous system, the onset is insidious, and the syndrome often mimics a space-occupying lesion. Altered mental status, headache, and stiff neck may be accompanied by focal findings such as cranial nerve palsies, ataxia, and hemiparesis. Cutaneous ulcers or hard nodules containing amebas are frequently detected in AIDS patients with disseminated Acanthamoeba infection and can be an important diagnostic site.
Examination of the CSF for trophozoites may be diagnostically helpful, but lumbar puncture may be contraindicated because of increased intracerebral pressure. CT frequently reveals cortical and subcortical lesions of decreased density consistent with embolic infarcts. In other patients, multiple enhancing lesions with edema may mimic the computed tomographic appearance of toxoplasmosis (Chap. 253). Demonstration of the trophozoites and cysts of Acanthamoeba on wet mounts or in biopsy specimens establishes the diagnosis. Culture on nonnutrient agar plates seeded with Escherichia coli also may be helpful. Fluorescein-labeled antiserum is available from the CDC for the detection of protozoa in biopsy specimens. Granulomatous amebic encephalitis in patients with AIDS may have an accelerated course (with survival for only 3–40 days) because of poor granuloma formation in these individuals. Various antimicrobial agents have been used to treat Acanthamoeba infection, but the infection is almost uniformly fatal. The CDC has now made miltefosine available because of improved survival rates when the drug is included in treatment regimens.
Keratitis The incidence of keratitis caused by Acanthamoeba has increased in the past 30 years, in part as a result of improved diagnosis. Earlier infections were associated with trauma to the eye and exposure to contaminated water. At present, most infections are linked to extended-wear contact lenses, and rare cases are associated with laser-assisted in situ keratomileusis (LASIK). Risk factors include the use of homemade saline, the wearing of lenses while swimming, and inadequate disinfection. Since contact lenses presumably cause microscopic trauma, the early corneal findings may be nonspecific. The first symptoms usually include tearing and the painful sensation of a foreign body. Once infection is established, progression is rapid; the characteristic clinical sign is an annular, paracentral corneal ring representing a corneal abscess. Deeper corneal invasion and loss of vision may follow.
The differential diagnosis includes bacterial, mycobacterial, and herpetic infection. The irregular polygonal cysts of Acanthamoeba (Fig. 247-5) may be identified in corneal scrapings or biopsy material, and trophozoites can be grown on special media. Cysts are resistant to available drugs, and the results of medical therapy have been disappointing. Some reports have suggested partial responses to propamidine isethionate eyedrops. Severe infections usually require keratoplasty.
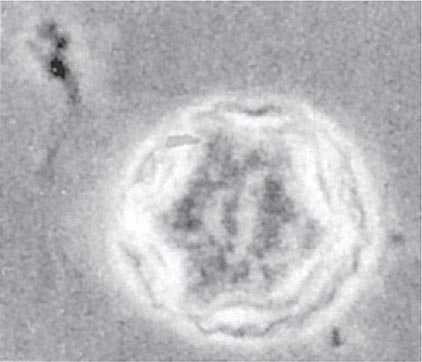
FIGURE 247-5 Double-walled cyst of Acanthamoeba castellanii, as seen by phase-contrast microscopy. (From DJ Krogstad et al, in A Balows et al [eds]: Manual of Clinical Microbiology, 5th ed. Washington, DC, American Society for Microbiology, 1991.)
BALAMUTHIA INFECTIONS
Balamuthia mandrillaris, a free-living ameba previously referred to as a leptomyxid ameba, is an important etiologic agent of amebic meningoencephalitis in immunocompetent hosts. The course is typically subacute, with focal neurologic signs, fever, seizures, and headaches leading to death within 1 week to several months after onset. Examination of CSF reveals mononuclear or neutrophilic pleocytosis, elevated protein levels, and normal to low glucose concentrations. Multiple hypodense lesions are usually detected with imaging studies (Fig. 247-6). This mixed picture of space-occupying lesions with CSF pleocytosis is suggestive of Balamuthia. Fluorescent antibody is available from the CDC for brain biopsy specimens. The variety of drugs used to treat the few surviving patients (i.e., fewer than five reported in the United States) includes pentamidine, flucytosine, sulfadiazine, and macrolides. The CDC recommends that miltefosine now be included, as for the other free-living amebas. The differential diagnosis includes tuberculomas (Chap. 202) and neurocysticercosis (Chap. 260).
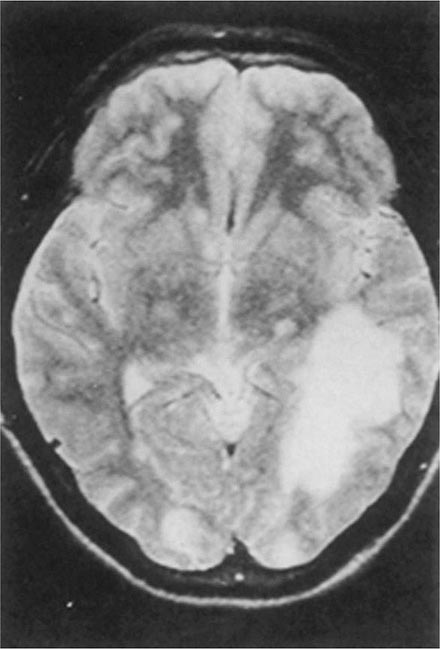
FIGURE 247-6 Brain MRI of amebic meningoencephalitis due to Balamuthia mandrillaris. A large lesion in the parieto-occipital lobe and other smaller lesions are seen. (Courtesy of the Department of Radiology, UCSD Medical Center, San Diego.)
248 |
Malaria |
Humanity has but three great enemies: Fever, famine, and war; of these by far the greatest, by far the most terrible, is fever.
—William Osler
Malaria is a protozoan disease transmitted by the bite of infected Anopheles mosquitoes. The most important of the parasitic diseases of humans, it is transmitted in 106 countries containing 3 billion people and causes approximately 2000 deaths each day; mortality rates are decreasing as a result of highly effective control programs in several countries. Malaria has been eliminated from the United States, Canada, Europe, and Russia; in the late twentieth and early twenty-first centuries, however, its prevalence rose in many parts of the tropics. Increases in the drug resistance of the parasite, the insecticide resistance of its vectors, and human travel and migration have contributed to this resurgence. Occasional local transmission after importation of malaria has occurred in several southern and eastern areas of the United States and in Europe, indicating the continual danger to nonmalarious countries. Although there are many successful new control initiatives as well as promising research initiatives, malaria remains today, as it has been for centuries, a heavy burden on tropical communities, a threat to nonendemic countries, and a danger to travelers.
ETIOLOGY AND PATHOGENESIS
Six species of the genus Plasmodium cause nearly all malarial infections in humans. These are P. falciparum, P. vivax, two morphologically identical sympatric species of P. ovale (as suggested by recent evidence), P. malariae, and—in Southeast Asia—the monkey malaria parasite P. knowlesi (Table 248-1). While almost all deaths are caused by falciparum malaria, P. knowlesi and occasionally P. vivax also can cause severe illness. Human infection begins when a female anopheline mosquito inoculates plasmodial sporozoites from its salivary gland during a blood meal (Fig. 248-1). These microscopic motile forms of the malaria parasite are carried rapidly via the bloodstream to the liver, where they invade hepatic parenchymal cells and begin a period of asexual reproduction. By this amplification process (known as intrahepatic or preerythrocytic schizogony or merogony), a single sporozoite eventually may produce from 10,000 to >30,000 daughter merozoites. The swollen infected liver cells eventually burst, discharging motile merozoites into the bloodstream. These merozoites then invade the red blood cells (RBCs) and multiply six- to twentyfold every 48 h (P. knowlesi, 24 h; P. malariae, 72 h). When the parasites reach densities of ~50/μL of blood (~100 million parasites in the blood of an adult), the symptomatic stage of the infection begins. In P. vivax and P. ovale infections, a proportion of the intrahepatic forms do not divide immediately but remain inert for a period ranging from 3 weeks to ≥1 year before reproduction begins. These dormant forms, or hypnozoites, are the cause of the relapses that characterize infection with these two species.
|
CHARACTERISTICS OF PLASMODIUM SPECIES INFECTING HUMANS |
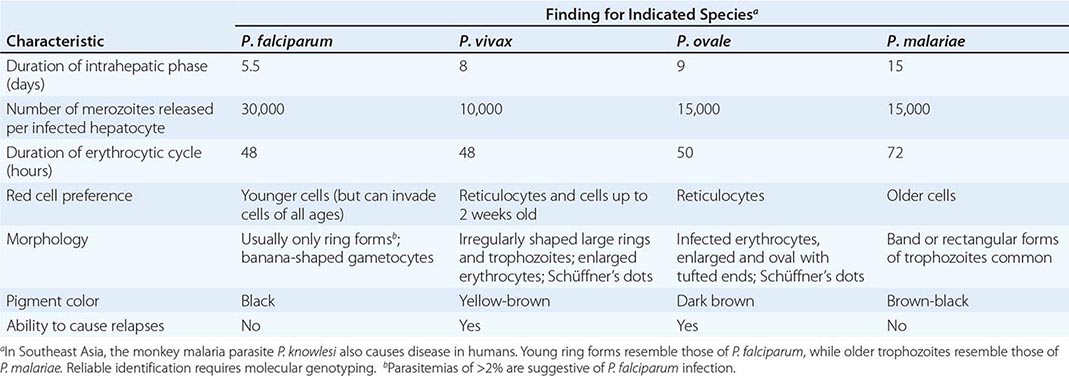
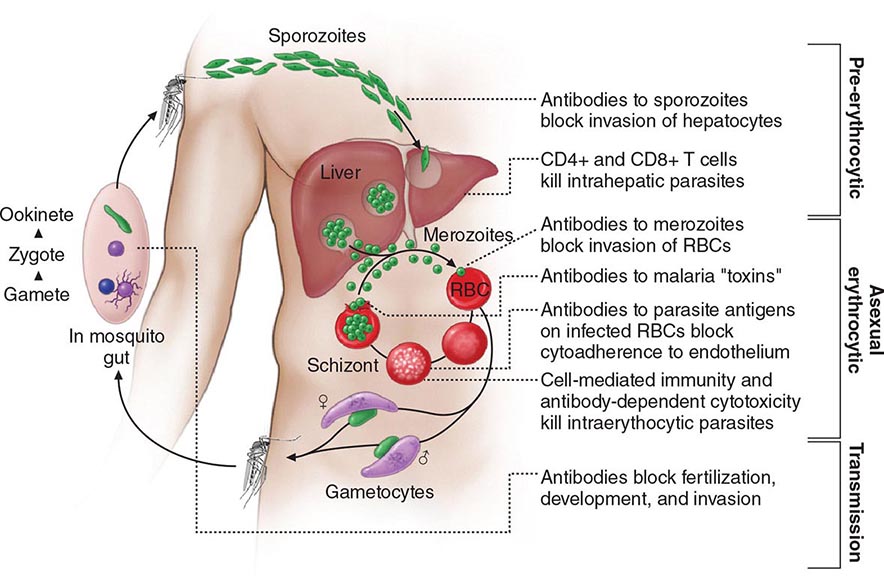
FIGURE 248-1 The malaria transmission cycle from mosquito to human and targets of immunity. RBC, red blood cell.
After entry into the bloodstream, merozoites rapidly invade erythrocytes and become trophozoites. Attachment is mediated via a specific erythrocyte surface receptor. For P. falciparum, the reticulocyte-binding protein homologue 5 (PfRh5) is indispensable for erythrocyte invasion. Basigin (CD147, EMMPRIN) is the erythrocyte receptor of PfRh5. In the case of P. vivax, this receptor is related to the Duffy blood-group antigen Fya or Fyb. Most West Africans and people with origins in that region carry the Duffy-negative FyFy phenotype and are therefore resistant to P. vivax malaria. During the early stage of intraerythrocytic development, the small “ring forms” of the different parasitic species appear similar under light microscopy. As the trophozoites enlarge, species-specific characteristics become evident, pigment becomes visible, and the parasite assumes an irregular or ameboid shape. By the end of the intraerythrocytic life cycle, the parasite has consumed two-thirds of the RBC’s hemoglobin and has grown to occupy most of the cell. It is now called a schizont. Multiple nuclear divisions have taken place (schizogony or merogony). The RBC then ruptures to release 6–30 daughter merozoites, each potentially capable of invading a new RBC and repeating the cycle. The disease in human beings is caused by the direct effects of the asexual parasite—RBC invasion and destruction—and by the host’s reaction. After release from the liver (P. vivax, P. ovale, P. malariae, P. knowlesi), some of the blood-stage parasites develop into morphologically distinct, longer-lived sexual forms (gametocytes) that can transmit malaria. In falciparum malaria, a delay of several asexual cycles precedes this switch to gametocytogenesis.
After being ingested in the blood meal of a biting female anopheline mosquito, the male and female gametocytes form a zygote in the insect’s midgut. This zygote matures into an ookinete, which penetrates and encysts in the mosquito’s gut wall. The resulting oocyst expands by asexual division until it bursts to liberate myriad motile sporozoites, which then migrate in the hemolymph to the salivary gland of the mosquito to await inoculation into another human at the next feeding.
EPIDEMIOLOGY
Malaria occurs throughout most of the tropical regions of the world (Fig. 248-2). P. falciparum predominates in Africa, New Guinea, and Hispaniola (i.e., the Dominican Republic and Haiti); P. vivax is more common in Central America. The prevalence of these two species is approximately equal in South America, the Indian subcontinent, eastern Asia, and Oceania. P. malariae is found in most endemic areas, especially throughout sub-Saharan Africa, but is much less common. P. ovale is relatively unusual outside of Africa and, where it is found, comprises <1% of isolates. Patients infected with P. knowlesi have been identified on the island of Borneo and, to a lesser extent, elsewhere in Southeast Asia, where the main hosts, long-tailed and pig-tailed macaques, are found.
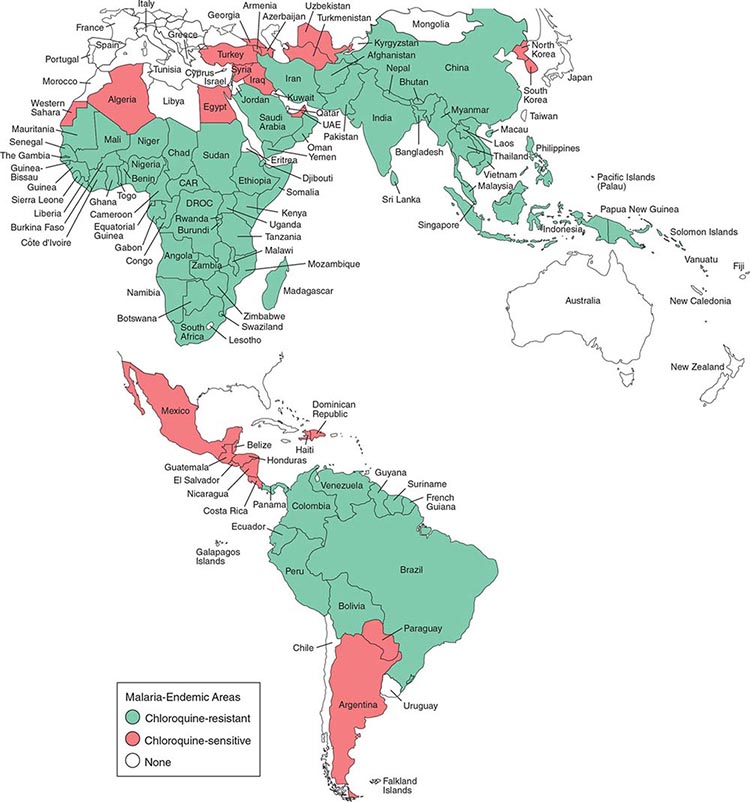
FIGURE 248-2 Malaria-endemic countries in the Americas (bottom) and in Africa, the Middle East, Asia, and the South Pacific (top), 2007. CAR, Central African Republic; DROC, Democratic Republic of the Congo; UAE, United Arab Emirates. Several countries in the Americas, the Middle East, and North Africa are close to eliminating malaria.
The epidemiology of malaria is complex and may vary considerably even within relatively small geographic areas. Endemicity traditionally has been defined in terms of parasitemia rates or palpable-spleen rates in children 2–9 years of age and classified as hypoendemic (<10%), mesoendemic (11–50%), hyperendemic (51–75%), and holoendemic (>75%). Until recently, it was uncommon to use these indices for planning control programs; however, many countries are now conducting national surveys to assess program progress. In holo- and hyperendemic areas (e.g., certain regions of tropical Africa or coastal New Guinea) where there is intense P. falciparum transmission, people may sustain more than one infectious mosquito bite per day and are infected repeatedly throughout their lives. In such settings, rates of morbidity and mortality due to malaria are considerable during early childhood. Immunity against disease is hard won in these areas, and the burden of disease in young children is high; by adulthood, however, most malarial infections are asymptomatic. As control measures progress and urbanization expands, environmental conditions become less conducive to transmission, and all age groups may lose protective immunity and become susceptible to illness. Constant, frequent, year-round infection is termed stable transmission. In areas where transmission is low, erratic, or focal, full protective immunity is not acquired, and symptomatic disease may occur at all ages. This situation usually exists in hypoendemic areas and is termed unstable transmission. Even in stable-transmission areas, there is often an increased incidence of symptomatic malaria coinciding with increased mosquito breeding and transmission during the rainy season. Malaria can behave like an epidemic disease in some areas, particularly those with unstable malaria, such as northern India (the Punjab region), the horn of Africa, Rwanda, Burundi, southern Africa, and Madagascar. An epidemic can develop when there are changes in environmental, economic, or social conditions, such as heavy rains following drought or migrations (usually of refugees or workers) from a nonmalarious region to an area of high transmission, along with failure to invest in national programs; a breakdown in malaria control and prevention services caused by war or civil disorder can intensify epidemic conditions. This situation usually results in considerable mortality among all age groups.
The principal determinants of the epidemiology of malaria are the number (density), the human-biting habits, and the longevity of the anopheline mosquito vectors. More than 100 of the >400 anopheline species can transmit malaria, but the ~40 species that do so commonly vary considerably in their efficiency as malaria vectors. More specifically, the transmission of malaria is directly proportional to the density of the vector, the square of the number of human bites per day per mosquito, and the tenth power of the probability of the mosquito’s surviving for 1 day. Mosquito longevity is particularly important because the portion of the parasite’s life cycle that takes place within the mosquito—from gametocyte ingestion to subsequent inoculation (sporogony)—lasts 8–30 days, depending on ambient temperature; thus, to transmit malaria, the mosquito must survive for >7 days. Sporogony is not completed at cooler temperatures—i.e., <16°C (60.8°F) for P. vivax and <21°C (69.8°F) for P. falciparum; thus transmission does not occur below these temperatures or at high altitudes, although malaria outbreaks and transmission have occurred in the highlands (>1500 m) of eastern Africa, which were previously free of vectors. The most effective mosquito vectors of malaria are those, such as Anopheles gambiae in Africa, that are long-lived, occur in high densities in tropical climates, breed readily, and bite humans in preference to other animals. The entomologic inoculation rate (i.e., the number of sporozoite-positive mosquito bites per person per year) is the most common measure of malaria transmission and varies from <1 in some parts of Latin America and Southeast Asia to >300 in parts of tropical Africa.
ERYTHROCYTE CHANGES IN MALARIA
After invading an erythrocyte, the growing malarial parasite progressively consumes and degrades intracellular proteins, principally hemoglobin. The potentially toxic heme is detoxified by lipid-mediated crystallization to biologically inert hemozoin (malaria pigment). The parasite also alters the RBC membrane by changing its transport properties, exposing cryptic surface antigens, and inserting new parasite-derived proteins. The RBC becomes more irregular in shape, more antigenic, and less deformable.
In P. falciparum infections, membrane protuberances appear on the erythrocyte’s surface 12–15 h after the cell’s invasion. These “knobs” extrude a high-molecular-weight, antigenically variant, strain-specific erythrocyte membrane adhesive protein (PfEMP1) that mediates attachment to receptors on venular and capillary endothelium—an event termed cytoadherence. Several vascular receptors have been identified, of which intercellular adhesion molecule 1 is probably the most important in the brain, chondroitin sulfate B in the placenta, and CD36 in most other organs. Thus, the infected erythrocytes stick inside and eventually block capillaries and venules. At the same stage, these P. falciparum–infected RBCs may also adhere to uninfected RBCs (to form rosettes) and to other parasitized erythrocytes (agglutination). The processes of cytoadherence, rosetting, and agglutination are central to the pathogenesis of falciparum malaria. They result in the sequestration of RBCs containing mature forms of the parasite in vital organs (particularly the brain), where they interfere with microcirculatory flow and metabolism. Sequestered parasites continue to develop out of reach of the principal host defense mechanism: splenic processing and filtration. As a consequence, only the younger ring forms of the asexual parasites are seen circulating in the peripheral blood in falciparum malaria, and the level of peripheral parasitemia underestimates the true number of parasites within the body. Severe malaria is also associated with reduced deformability of the uninfected erythrocytes, which compromises their passage through the partially obstructed capillaries and venules and shortens RBC survival.
In the other human malarias, sequestration does not occur, and all stages of the parasite’s development are evident on peripheral-blood smears. Whereas P. vivax, P. ovale, and P. malariae show a marked predilection for either young RBCs (P. vivax, P. ovale) or old cells (P. malariae) and produce a level of parasitemia that is seldom >2%, P. falciparum can invade erythrocytes of all ages and may be associated with very high levels of parasitemia.
HOST RESPONSE
Initially, the host responds to plasmodial infection by activating nonspecific defense mechanisms. Splenic immunologic and filtrative clearance functions are augmented in malaria, and the removal of both parasitized and uninfected erythrocytes is accelerated. The spleen is able to remove damaged ring-form parasites and return the once-infected erythrocytes to the circulation, where their survival period is shortened. The parasitized cells escaping splenic removal are destroyed when the schizont ruptures. The material released induces the activation of macrophages and the release of proinflammatory cytokines, which cause fever and exert other pathologic effects. Temperatures of ≥40°C (104°F) damage mature parasites; in untreated infections, the effect of such temperatures is to further synchronize the parasitic cycle, with eventual production of the regular fever spikes and rigors that originally served to characterize the different malarias. These regular fever patterns (quotidian, daily; tertian, every 2 days; quartan, every 3 days) are seldom seen today in patients who receive prompt and effective antimalarial treatment.
The geographic distributions of sickle cell disease, hemoglobins C and E, hereditary ovalocytosis, the thalassemias, and glucose-6-phosphate dehydrogenase (G6PD) deficiency closely resemble that of falciparum malaria before the introduction of control measures. This similarity suggests that these genetic disorders confer protection against death from falciparum malaria. For example, HbA/S heterozygotes (sickle cell trait) have a sixfold reduction in the risk of dying from severe falciparum malaria. Hemoglobin S–containing RBCs impair parasite growth at low oxygen tensions, and P. falciparum–infected RBCs containing hemoglobins S and C exhibit reduced cytoadherence because of reduced surface presentation of the adhesin PfEMP1. Parasite multiplication in HbA/E heterozygotes is reduced at high parasite densities. In Melanesia, children with α-thalassemia appear to have more frequent malaria (both vivax and falciparum) in the early years of life, and this pattern of infection appears to protect them against severe disease. In Melanesian ovalocytosis, rigid erythrocytes resist merozoite invasion, and the intraerythrocytic milieu is hostile.
Nonspecific host defense mechanisms stop the infection’s expansion, and the subsequent strain-specific immune response then controls the infection. Eventually, exposure to sufficient strains confers protection from high-level parasitemia and disease but not from infection. As a result of this state of infection without illness (premunition), asymptomatic parasitemia is common among adults and older children living in regions with stable and intense transmission (i.e., holo- or hyperendemic areas) and also in parts of low-transmission areas. Immunity is mainly specific for both the species and the strain of infecting malarial parasite. Both humoral immunity and cellular immunity are necessary for protection, but the mechanisms of each are incompletely understood (Fig. 248-1). Immune individuals have a polyclonal increase in serum levels of IgM, IgG, and IgA, although much of this antibody is unrelated to protection. Antibodies to a variety of parasitic antigens presumably act in concert to limit in vivo replication of the parasite. In the case of falciparum malaria, the most important of these antigens is the surface adhesin—the variant protein PfEMP1. Passively transferred IgG from immune adults has been shown to reduce levels of parasitemia in children. Passive transfer of maternal antibody contributes to the relative (but not complete) protection of infants from severe malaria in the first months of life. This complex immunity to disease declines when a person lives outside an endemic area for several months or longer.
Several factors retard the development of cellular immunity to malaria. These factors include the absence of major histocompatibility antigens on the surface of infected RBCs, which precludes direct T cell recognition; malaria antigen–specific immune unresponsiveness; and the enormous strain diversity of malarial parasites, along with the ability of the parasites to express variant immunodominant antigens on the erythrocyte surface that change during the course of infection. Parasites may persist in the blood for months or years (or, in the case of P. malariae, for decades) if treatment is not given. The complexity of the immune response in malaria, the sophistication of the parasites’ evasion mechanisms, and the lack of a good in vitro correlate with clinical immunity have all slowed progress toward an effective vaccine.
CLINICAL FEATURES
Malaria is a very common cause of fever in tropical countries. The first symptoms of malaria are nonspecific; the lack of a sense of well-being, headache, fatigue, abdominal discomfort, and muscle aches followed by fever are all similar to the symptoms of a minor viral illness. In some instances, a prominence of headache, chest pain, abdominal pain, cough, arthralgia, myalgia, or diarrhea may suggest another diagnosis. Although headache may be severe in malaria, the neck stiffness and photophobia seen in meningitis do not occur. While myalgia may be prominent, it is not usually as severe as in dengue fever, and the muscles are not tender as in leptospirosis or typhus. Nausea, vomiting, and orthostatic hypotension are common. The classic malarial paroxysms, in which fever spikes, chills, and rigors occur at regular intervals, are relatively unusual and suggest infection with P. vivax or P. ovale. The fever is usually irregular at first (that of falciparum malaria may never become regular); the temperature of nonimmune individuals and children often rises above 40°C (104°F) in conjunction with tachycardia and sometimes delirium. Although childhood febrile convulsions may occur with any of the malarias, generalized seizures are specifically associated with falciparum malaria and may herald the development of encephalopathy (cerebral malaria). Many clinical abnormalities have been described in acute malaria, but most patients with uncomplicated infections have few abnormal physical findings other than fever, malaise, mild anemia, and (in some cases) a palpable spleen. Anemia is common among young children living in areas with stable transmission, particularly where resistance has compromised the efficacy of antimalarial drugs. In nonimmune individuals with acute malaria, the spleen takes several days to become palpable, but splenic enlargement is found in a high proportion of otherwise healthy individuals in malaria-endemic areas and reflects repeated infections. Slight enlargement of the liver is also common, particularly among young children. Mild jaundice is common among adults; it may develop in patients with otherwise uncomplicated malaria and usually resolves over 1–3 weeks. Malaria is not associated with a rash like those seen in meningococcal septicemia, typhus, enteric fever, viral exanthems, and drug reactions. Petechial hemorrhages in the skin or mucous membranes—features of viral hemorrhagic fevers and leptospirosis—develop only very rarely in severe falciparum malaria.
SEVERE FALCIPARUM MALARIA
Appropriately and promptly treated, uncomplicated falciparum malaria (i.e., the patient can swallow medicines and food) carries a mortality rate of <0.1%. However, once vital-organ dysfunction occurs or the total proportion of erythrocytes infected increases to >2% (a level corresponding to >1012 parasites in an adult), mortality risk rises steeply. The major manifestations of severe falciparum malaria are shown in Table 248-2, and features indicating a poor prognosis are listed in Table 248-3.
|
MANIFESTATIONS OF SEVERE FALCIPARUM MALARIA |
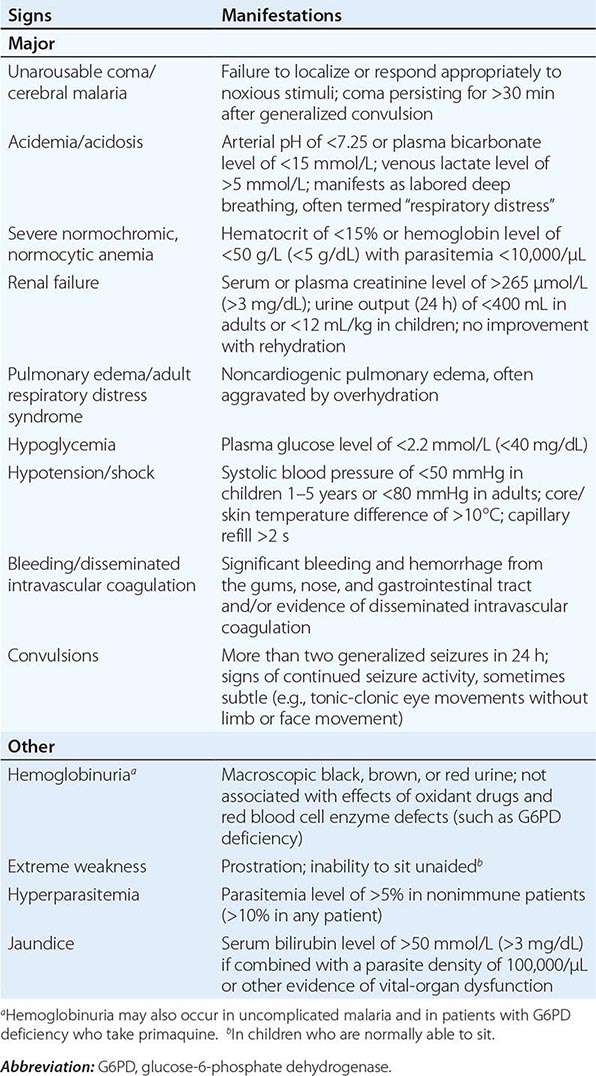
|
FEATURES INDICATING A POOR PROGNOSIS IN SEVERE FALCIPARUM MALARIA |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; PCV, packed cell volume.
Cerebral Malaria Coma is a characteristic and ominous feature of falciparum malaria and, despite treatment, is associated with death rates of ~20% among adults and 15% among children. Any obtundation, delirium, or abnormal behavior should be taken very seriously. The onset may be gradual or sudden following a convulsion.
Cerebral malaria manifests as diffuse symmetric encephalopathy; focal neurologic signs are unusual. Although some passive resistance to head flexion may be detected, signs of meningeal irritation are absent. The eyes may be divergent and a pout reflex is common, but other primitive reflexes are usually absent. The corneal reflexes are preserved, except in deep coma. Muscle tone may be either increased or decreased. The tendon reflexes are variable, and the plantar reflexes may be flexor or extensor; the abdominal and cremasteric reflexes are absent. Flexor or extensor posturing may be seen. On routine funduscopy, ~15% of patients have retinal hemorrhages; with pupillary dilation and indirect ophthalmoscopy, this figure increases to 30–40%. Other funduscopic abnormalities (Fig. 248-3) include discrete spots of retinal opacification (30–60%), papilledema (8% among children, rare among adults), cotton wool spots (<5%), and decolorization of a retinal vessel or segment of vessel (occasional cases). Convulsions, usually generalized and often repeated, occur in ~10% of adults and up to 50% of children with cerebral malaria. More covert seizure activity also is common, particularly among children, and may manifest as repetitive tonic-clonic eye movements or even hypersalivation. Whereas adults rarely (i.e., in <3% of cases) suffer neurologic sequelae, ~10% of children surviving cerebral malaria—especially those with hypoglycemia, severe anemia, repeated seizures, and deep coma—have residual neurologic deficits when they regain consciousness; hemiplegia, cerebral palsy, cortical blindness, deafness, and impaired cognition have been reported. The majority of these deficits improve markedly or resolve completely within 6 months. However, the prevalence of some other deficits increases over time; ~10% of children surviving cerebral malaria have a persistent language deficit. There may also be deficits in learning, planning and executive functions, attention, memory, and nonverbal functioning. The incidence of epilepsy is increased and life expectancy decreased among these children.
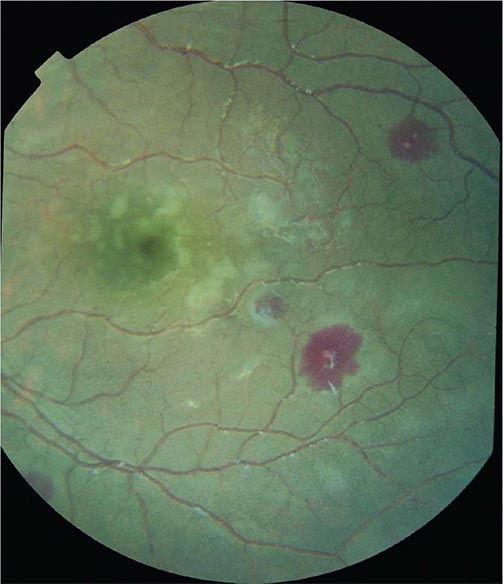
FIGURE 248-3 The eye in cerebral malaria: perimacular whitening and pale-centered retinal hemorrhages. (Courtesy of N. Beare, T. Taylor, S. Harding, S. Lewallen, and M. Molyneux; with permission.)
Hypoglycemia Hypoglycemia, an important and common complication of severe malaria, is associated with a poor prognosis and is particularly problematic in children and pregnant women. Hypoglycemia in malaria results from a failure of hepatic gluconeogenesis and an increase in the consumption of glucose by both the host and, to a much lesser extent, the malaria parasites. To compound the situation, quinine, which is still widely used for the treatment of both severe and uncomplicated falciparum malaria, is a powerful stimulant of pancreatic insulin secretion. Hyperinsulinemic hypoglycemia is especially troublesome in pregnant women receiving quinine treatment. In severe disease, the clinical diagnosis of hypoglycemia is difficult: the usual physical signs (sweating, gooseflesh, tachycardia) are absent, and the neurologic impairment caused by hypoglycemia cannot be distinguished from that caused by malaria.
Acidosis Acidosis, an important cause of death from severe malaria, results from accumulation of organic acids. Hyperlactatemia commonly coexists with hypoglycemia. In adults, coexisting renal impairment often compounds the acidosis; in children, ketoacidosis also may contribute. Other, still-unidentified organic acids are major contributors to acidosis. Acidotic breathing, sometimes called “respiratory distress,” is a sign of poor prognosis. It is followed often by circulatory failure refractory to volume expansion or inotropic drug treatment and ultimately by respiratory arrest. The plasma concentrations of bicarbonate or lactate are the best biochemical prognosticators in severe malaria. Hypovolemia is not a major contributor to acidosis. Lactic acidosis is caused by the combination of anaerobic glycolysis in tissues where sequestered parasites interfere with microcirculatory flow, lactate production by the parasites, and a failure of hepatic and renal lactate clearance. The prognosis of severe acidosis is poor.
Noncardiogenic Pulmonary Edema Adults with severe falciparum malaria may develop noncardiogenic pulmonary edema even after several days of antimalarial therapy. The pathogenesis of this variant of the adult respiratory distress syndrome is unclear. The mortality rate is >80%. This condition can be aggravated by overly vigorous administration of IV fluid. Noncardiogenic pulmonary edema can also develop in otherwise uncomplicated vivax malaria, where recovery is usual.
Renal Impairment Acute kidney injury is common in severe falciparum malaria, but oliguric renal failure is rare among children. The pathogenesis of renal failure is unclear but may be related to erythrocyte sequestration and agglutination interfering with renal microcirculatory flow and metabolism. Clinically and pathologically, this syndrome manifests as acute tubular necrosis. Renal cortical necrosis never develops. Acute renal failure may occur simultaneously with other vital-organ dysfunction (in which case the mortality risk is high) or may progress as other disease manifestations resolve. In survivors, urine flow resumes in a median of 4 days, and serum creatinine levels return to normal in a mean of 17 days (Chap. 334). Early dialysis or hemofiltration considerably enhances the likelihood of a patient’s survival, particularly in acute hypercatabolic renal failure.
Hematologic Abnormalities Anemia results from accelerated RBC removal by the spleen, obligatory RBC destruction at parasite schizogony, and ineffective erythropoiesis. In severe malaria, both infected and uninfected RBCs show reduced deformability, which correlates with prognosis and development of anemia. Splenic clearance of all RBCs is increased. In nonimmune individuals and in areas with unstable transmission, anemia can develop rapidly and transfusion is often required. As a consequence of repeated malarial infections, children in many areas of Africa and on the island of New Guinea may develop severe anemia resulting from both shortened survival of uninfected RBCs and marked dyserythropoiesis. Anemia is a common consequence of antimalarial drug resistance, which results in repeated or continued infection.
Slight coagulation abnormalities are common in falciparum malaria, and mild thrombocytopenia is usual (a normal platelet count should raise questions about the diagnosis of malaria). Of patients with severe malaria, <5% have significant bleeding with evidence of disseminated intravascular coagulation. Hematemesis from stress ulceration or acute gastric erosions also may occur rarely.
Liver Dysfunction Mild hemolytic jaundice is common in malaria. Severe jaundice is associated with P. falciparum infections; is more common among adults than among children; and results from hemolysis, hepatocyte injury, and cholestasis. When accompanied by other vital-organ dysfunction (often renal impairment), liver dysfunction carries a poor prognosis. Hepatic dysfunction contributes to hypoglycemia, lactic acidosis, and impaired drug metabolism. Occasional patients with falciparum malaria may develop deep jaundice (with hemolytic, hepatic, and cholestatic components) without evidence of other vital-organ dysfunction, in which case the prognosis is good.
Other Complications HIV/AIDS and malnutrition predispose to more severe malaria in nonimmune individuals; malaria anemia is worsened by concurrent infections with intestinal helminths, hookworm in particular. Septicemia may complicate severe malaria, particularly in children. Differentiating severe malaria from sepsis with incidental parasitemia in childhood is very difficult. In endemic areas, Salmonella bacteremia has been associated specifically with P. falciparum infections. Chest infections and catheter-induced urinary tract infections are common among patients who are unconscious for >3 days. Aspiration pneumonia may follow generalized convulsions. The frequencies of complications of severe falciparum malaria are summarized in Table 248-4.
|
RELATIVE INCIDENCE OF SEVERE COMPLICATIONS OF FALCIPARUM MALARIA |
MALARIA IN PREGNANCY
Malaria in early pregnancy causes abortion. In areas of high malaria transmission, falciparum malaria in primi- and secundigravid women is associated with low birth weight (average reduction, ~170 g) and consequently increased infant mortality rates. In general, infected mothers in areas of stable transmission remain asymptomatic despite intense accumulation of parasitized erythrocytes in the placental microcirculation. Maternal HIV infection predisposes pregnant women to more frequent and higher-density malaria infections, predisposes their newborns to congenital malarial infection, and exacerbates the reduction in birth weight associated with malaria.
In areas with unstable transmission of malaria, pregnant women are prone to severe infections and are particularly vulnerable to high parasitemias with anemia, hypoglycemia, and acute pulmonary edema. Fetal distress, premature labor, and stillbirth or low birth weight are common results. Fetal death is usual in severe malaria. Congenital malaria occurs in <5% of newborns whose mothers are infected; its frequency and the level of parasitemia are related directly to the parasite density in maternal blood and in the placenta. P. vivax malaria in pregnancy is also associated with a reduction in birth weight (average, 110 g), but, in contrast to the situation in falciparum malaria, this effect is more pronounced in multigravid than in primigravid women. About 350,000 women die in childbirth yearly, with most deaths occurring in low-income countries; maternal death from hemorrhage at childbirth is correlated with malaria-induced anemia.
MALARIA IN CHILDREN
Most of the 660,000 persons who die of falciparum malaria each year are young African children. Convulsions, coma, hypoglycemia, metabolic acidosis, and severe anemia are relatively common among children with severe malaria, whereas deep jaundice, oliguric acute kidney injury, and acute pulmonary edema are unusual. Severely anemic children may present with labored deep breathing, which in the past has been attributed incorrectly to “anemic congestive cardiac failure” but in fact is usually caused by metabolic acidosis, often compounded by hypovolemia. In general, children tolerate antimalarial drugs well and respond rapidly to treatment.
TRANSFUSION MALARIA
Malaria can be transmitted by blood transfusion, needle-stick injury, sharing of needles by infected injection drug users, or organ transplantation. The incubation period in these settings is often short because there is no preerythrocytic stage of development. The clinical features and management of these cases are the same as for naturally acquired infections. Radical chemotherapy with primaquine is unnecessary for transfusion-transmitted P. vivax and P. ovale infections.
CHRONIC COMPLICATIONS OF MALARIA
TROPICAL SPLENOMEGALY (HYPERREACTIVE MALARIAL SPLENOMEGALY)
Chronic or repeated malarial infections produce hypergammaglobulinemia; normochromic, normocytic anemia; and, in certain situations, splenomegaly. Some residents of malaria-endemic areas in tropical Africa and Asia exhibit an abnormal immunologic response to repeated infections that is characterized by massive splenomegaly, hepatomegaly, marked elevations in serum titers of IgM and malarial antibody, hepatic sinusoidal lymphocytosis, and (in Africa) peripheral B cell lymphocytosis. This syndrome has been associated with the production of cytotoxic IgM antibodies to CD8+ T lymphocytes, antibodies to CD5+ T lymphocytes, and an increase in the ratio of CD4+ to CD8+ T cells. These events may lead to uninhibited B cell production of IgM and the formation of cryoglobulins (IgM aggregates and immune complexes). This immunologic process stimulates reticuloendothelial hyperplasia and clearance activity and eventually produces splenomegaly. Patients with hyperreactive malarial splenomegaly present with an abdominal mass or a dragging sensation in the abdomen and occasional sharp abdominal pains suggesting perisplenitis. Anemia and some degree of pancytopenia are usually evident, and in some cases malarial parasites cannot be found in peripheral-blood smears. Vulnerability to respiratory and skin infections is increased; many patients die of overwhelming sepsis. Persons with hyperreactive malarial splenomegaly who are living in endemic areas should receive antimalarial chemoprophylaxis; the results are usually good. In nonendemic areas, antimalarial treatment is advised. In some cases refractory to therapy, clonal lymphoproliferation may develop and can then evolve into a malignant lymphoproliferative disorder.
QUARTAN MALARIAL NEPHROPATHY
Chronic or repeated infections with P. malariae (and possibly with other malarial species) may cause soluble immune complex injury to the renal glomeruli, resulting in the nephrotic syndrome. Other unidentified factors must contribute to this process since only a very small proportion of infected patients develop renal disease. The histologic appearance is that of focal or segmental glomerulonephritis with splitting of the capillary basement membrane. Subendothelial dense deposits are seen on electron microscopy, and immunofluorescence reveals deposits of complement and immunoglobulins; in samples of renal tissue from children, P. malariae antigens are often visible. A coarse-granular pattern of basement membrane immunofluorescent deposits (predominantly IgG3) with selective proteinuria carries a better prognosis than a fine-granular, predominantly IgG2 pattern with nonselective proteinuria. Quartan nephropathy usually responds poorly to treatment with either antimalarial agents or glucocorticoids and cytotoxic drugs.
BURKITT’S LYMPHOMA AND EPSTEIN-BARR VIRUS INFECTION
It is possible that malaria-related immune dysregulation provokes infection with lymphoma viruses. Burkitt’s lymphoma is strongly associated with Epstein-Barr virus. The prevalence of this childhood tumor is high in malarious areas of Africa.
DIAGNOSIS
DEMONSTRATION OF THE PARASITE
The diagnosis of malaria rests on the demonstration of asexual forms of the parasite in stained peripheral-blood smears. After a negative blood smear, repeat smears should be made if there is a high degree of suspicion. Of the Romanowsky stains, Giemsa at pH 7.2 is preferred; Field’s, Wright’s, or Leishman’s stain can also be used. Both thin (Figs. 248-4 and 248-5; see also Figs. 250e-3 and 250e-4) and thick (Figs. 248-6, 248-7, 248-8, and 248-9) blood smears should be examined. The thin blood smear should be rapidly air-dried, fixed in anhydrous methanol, and stained; the RBCs in the tail of the film should then be examined under oil immersion (×1000 magnification). The level of parasitemia is expressed as the number of parasitized erythrocytes per 1000 RBCs. The thick blood film should be of uneven thickness. The smear should be dried thoroughly and stained without fixing. As many layers of erythrocytes overlie one another and are lysed during the staining procedure, the thick film has the advantage of concentrating the parasites (by 40- to 100-fold compared with a thin blood film) and thus increasing diagnostic sensitivity. Both parasites and white blood cells (WBCs) are counted, and the number of parasites per unit volume is calculated from the total leukocyte count. Alternatively, a WBC count of 8000/μL is assumed. This figure is converted to the number of parasitized erythrocytes per microliter. A minimum of 200 WBCs should be counted under oil immersion. Interpretation of blood smear films requires some experience because artifacts are common. Before a thick smear is judged to be negative, 100–200 fields should be examined under oil immersion. In high-transmission areas, the presence of up to 10,000 parasites/μL of blood may be tolerated without symptoms or signs in partially immune individuals. Thus in these areas the detection of malaria parasites is sensitive but has low specificity in identifying malaria as the cause of illness. Low-density parasitemia is common in other conditions causing fever.
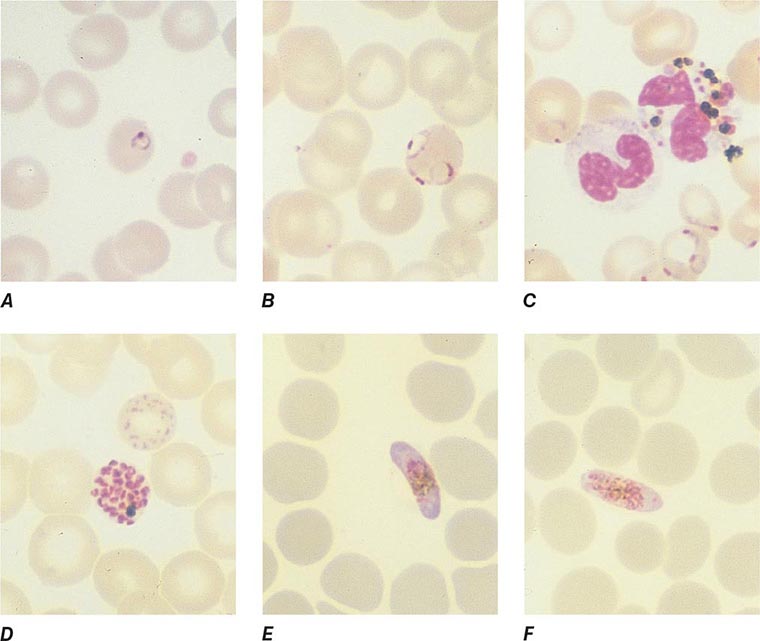
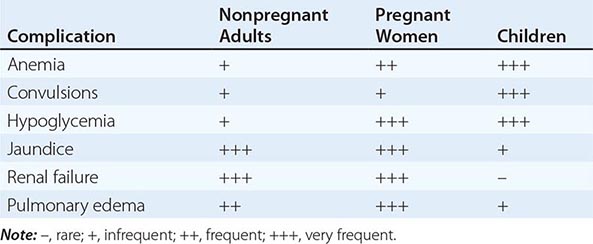
FIGURE 248-4 Thin blood films of Plasmodium falciparum. A. Young trophozoites. B. Old trophozoites. C. Pigment in polymorphonuclear cells and trophozoites. D. Mature schizonts. E. Female gametocytes. F. Male gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
FIGURE 248-5 Thin blood films of Plasmodium vivax. A. Young trophozoites. B. Old trophozoites. C. Mature schizonts. D. Female gametocytes. E. Male gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
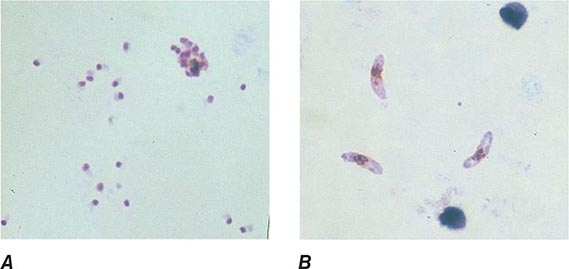
FIGURE 248-6 Thick blood films of Plasmodium falciparum. A. Trophozoites. B. Gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
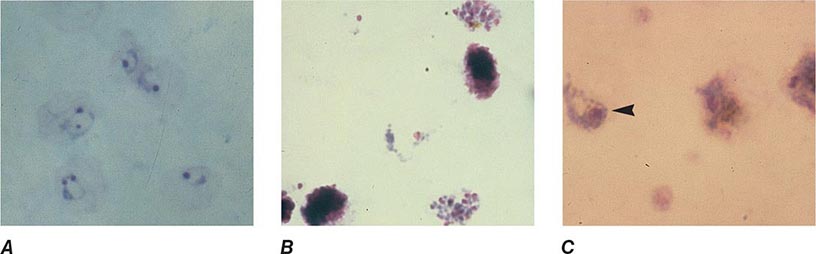
FIGURE 248-7 Thick blood films of Plasmodium vivax. A. Trophozoites. B. Schizonts. C. Gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
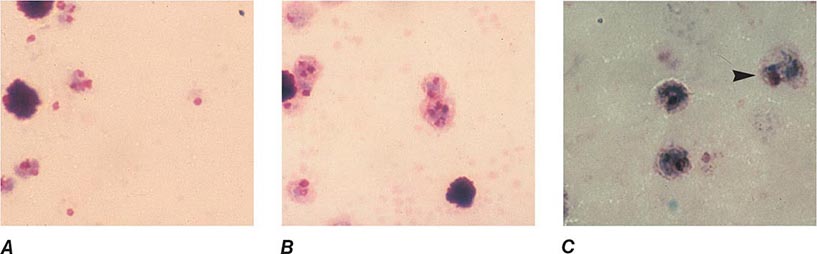
FIGURE 248-8 Thick blood films of Plasmodium ovale. A. Trophozoites. B. Schizonts. C. Gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
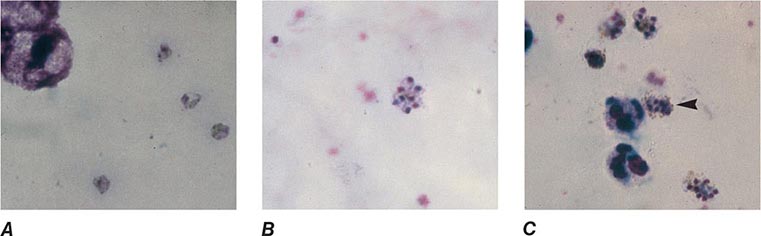
FIGURE 248-9 Thick blood films of Plasmodium malariae. A. Trophozoites. B. Schizonts. C. Gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
Rapid, simple, sensitive, and specific antibody-based diagnostic stick or card tests that detect P. falciparum–specific, histidine-rich protein 2 (PfHRP2), lactate dehydrogenase, or aldolase antigens in finger-prick blood samples are now being used widely in control programs (Table 248-5). Some of these rapid diagnostic tests carry a second antibody, which allows falciparum malaria to be distinguished from the less dangerous malarias. PfHRP2-based tests may remain positive for several weeks after acute infection. This feature is a disadvantage in high-transmission areas where infections are frequent, but it is of value in the diagnosis of severe malaria in patients who have taken antimalarial drugs and cleared peripheral parasitemia (but in whom the PfHRP2 test remains strongly positive). Rapid diagnostic tests are replacing microscopy in many areas because of their simplicity and speed. Their disadvantage is that they do not quantify parasitemia.
|
STANDARD METHODS FOR THE DIAGNOSIS OF MALARIAa |
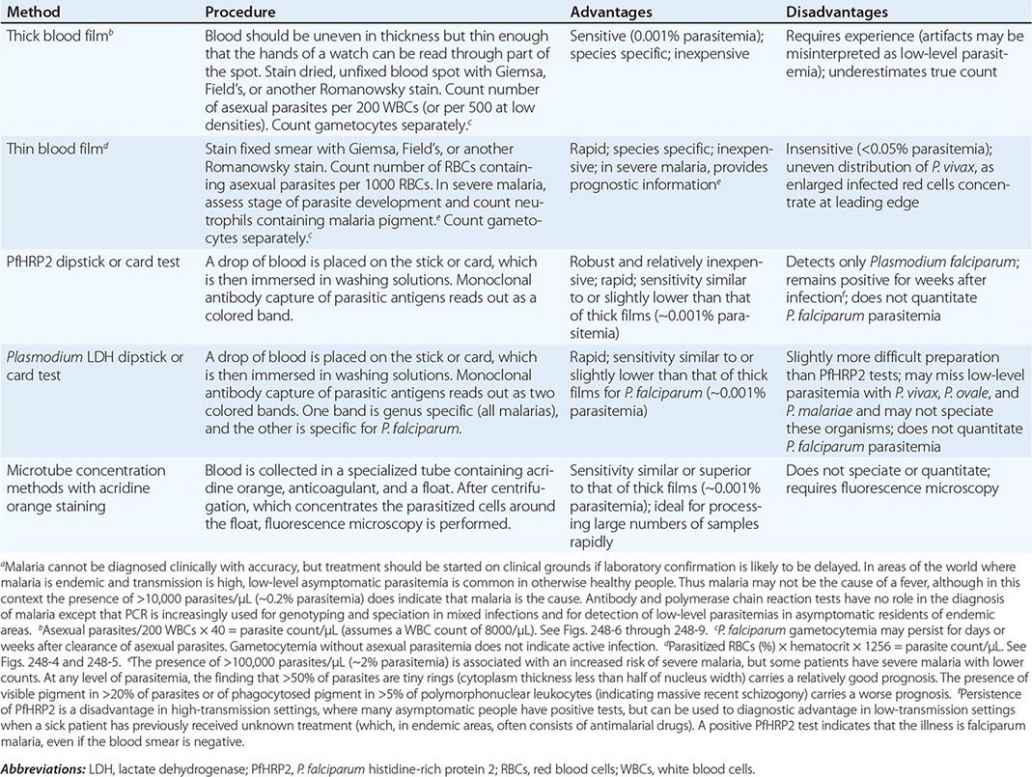
The relationship between parasitemia and prognosis is complex; in general, patients with >105 parasites/μL are at increased risk of dying, but nonimmune patients may die with much lower counts, and partially immune persons may tolerate parasitemia levels many times higher with only minor symptoms. In severe malaria, a poor prognosis is indicated by a predominance of more mature P. falciparum parasites (i.e., >20% of parasites with visible pigment) in the peripheral-blood film or by the presence of phagocytosed malarial pigment in >5% of neutrophils. In P. falciparum infections, gametocytemia peaks 1 week after the peak of asexual parasites. Because the mature gametocytes of P. falciparum (unlike those of other plasmodia) are not affected by most antimalarial drugs, their persistence does not constitute evidence of drug resistance. Phagocytosed malarial pigment is sometimes seen inside peripheral-blood monocytes or polymorphonuclear leukocytes and may provide a clue to recent infection if malaria parasites are not detectable. After the clearance of the parasites, this intraphagocytic malarial pigment is often evident for several days in the peripheral blood films or for longer in bone marrow aspirates or smears of fluid expressed after intradermal puncture. Staining of parasites with the fluorescent dye acridine orange allows more rapid diagnosis of malaria (but not speciation of the infection) in patients with low-level parasitemia.
Molecular diagnosis by polymerase chain reaction (PCR) amplification of parasite nucleic acid is more sensitive than microscopy or rapid diagnostic tests for detecting malaria parasites and defining malarial species. While currently impractical in the standard clinical setting, PCR is used in reference centers in endemic areas. In epidemiologic surveys, sensitive PCR detection may prove very useful in identifying asymptomatic infections as control and eradication programs drive parasite prevalence down to very low levels. Serologic diagnosis with either indirect fluorescent antibody or enzyme-linked immunosorbent assays may prove useful as measures of transmission intensity in future epidemiologic studies. Serology has no place in the diagnosis of acute illness.
LABORATORY FINDINGS
Normochromic, normocytic anemia is usual. The leukocyte count is generally normal, although it may be raised in very severe infections. There is slight monocytosis, lymphopenia, and eosinopenia, with reactive lymphocytosis and eosinophilia in the weeks after the acute infection. The erythrocyte sedimentation rate, plasma viscosity, and levels of C-reactive protein and other acute-phase proteins are high. The platelet count is usually reduced to ~105/μL. Severe infections may be accompanied by prolonged prothrombin and partial thromboplastin times and by more severe thrombocytopenia. Levels of antithrombin III are reduced even in mild infection. In uncomplicated malaria, plasma concentrations of electrolytes, blood urea nitrogen (BUN), and creatinine are usually normal. Findings in severe malaria may include metabolic acidosis, with low plasma concentrations of glucose, sodium, bicarbonate, calcium, phosphate, and albumin together with elevations in lactate, BUN, creatinine, urate, muscle and liver enzymes, and conjugated and unconjugated bilirubin. Hypergammaglobulinemia is usual in immune and semi-immune subjects. Urinalysis generally gives normal results. In adults and children with cerebral malaria, the mean cerebrospinal fluid (CSF) opening pressure at lumbar puncture is ~160 mm; usually the CSF content is normal or there is a slight elevation of total protein level (<1.0 g/L [<100 mg/dL]) and cell count (<20/μL).
|
TREATMENT |
MALARIA |
(Table 248-6) When a patient in or from a malarious area presents with fever, thick and thin blood smears should be prepared and examined immediately to confirm the diagnosis and identify the species of infecting parasite (Figs. 248-4 through 248-9). Repeat blood smears should be performed at least every 12–24 h for 2 days if the first smears are negative and malaria is strongly suspected. Alternatively, a rapid antigen detection card or stick test should be performed. Patients with severe malaria or those unable to take oral drugs should receive parenteral antimalarial therapy. If there is any doubt about the resistance status of the infecting organism, it should be considered resistant. Antimalarial drug susceptibility testing can be performed but is rarely available, has poor predictive value in an individual case, and yields results too slowly to influence the choice of treatment. Several drugs are available for oral treatment. The choice of drug depends on the likely sensitivity of the infecting parasites. Despite increasing evidence of chloroquine resistance in P. vivax (from parts of Indonesia, Oceania, eastern and southern Asia, and Central and South America), chloroquine remains a first-line treatment for the non-falciparum malarias (P. vivax, P. ovale, P. malariae, P. knowlesi) except in Indonesia and Papua New Guinea, where high levels of resistance in P. vivax are prevalent.
|
REGIMENS FOR THE TREATMENT OF MALARIAa |
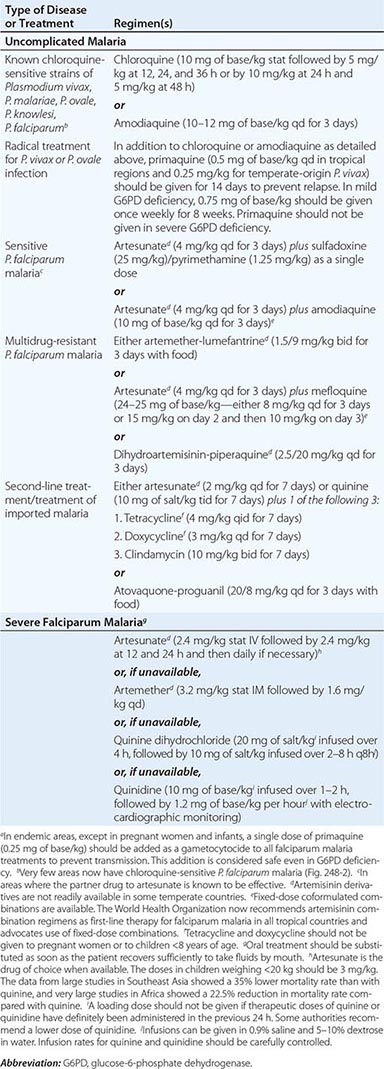
The treatment of falciparum malaria has changed radically in recent years. In all endemic areas, the World Health Organization (WHO) now recommends artemisinin-based combinations (ACTs) as first-line treatment for uncomplicated falciparum malaria. These combinations are also highly effective for the other malarias. These rapidly and reliably effective drugs are sometimes unavailable in temperate countries, where treatment recommendations are limited by the registered available drugs. Fake or substandard antimalarials are commonly sold in many Asian and African countries. Thus, careful attention is required at the time of purchase and later, especially if the patient fails to respond as expected. Characteristics of antimalarial drugs are shown in Table 248-7.
|
PROPERTIES OF ANTIMALARIAL DRUGS |
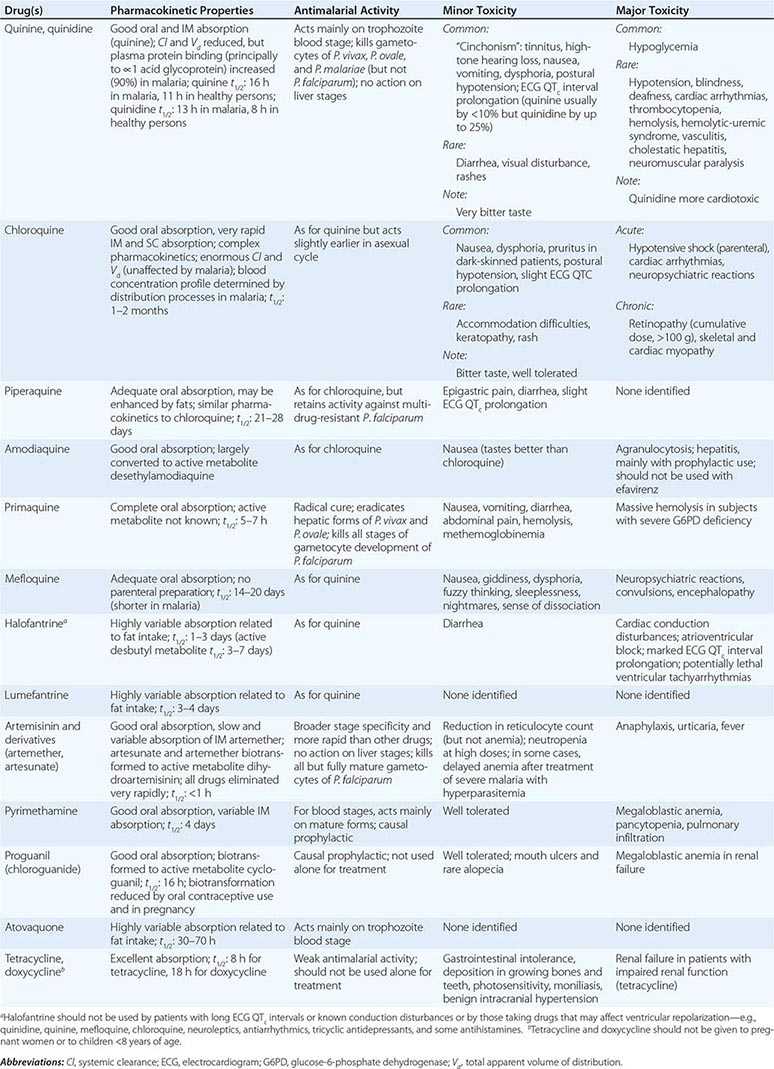
SEVERE MALARIA
In large studies, parenteral artesunate, a water-soluble artemisinin derivative, has reduced mortality rates in severe falciparum malaria among Asian adults and children by 35% and among African children by 22.5% compared with mortality rates with quinine treatment. Artesunate has therefore become the drug of choice for all patients with severe malaria everywhere. Artesunate is given by IV injection but can also be given by IM injection. Artemether and the closely related drug artemotil (arteether) are oil-based formulations given by IM injection; they are erratically absorbed and do not confer the same survival benefit as artesunate. A rectal formulation of artesunate has been developed as a community-based pre-referral treatment for patients in the rural tropics who cannot take oral medications. Pre-referral administration of rectal artesunate has been shown to decrease mortality risk among severely ill children in communities without access to immediate parenteral treatment. Although the artemisinin compounds are safer than quinine and considerably safer than quinidine, only one formulation is available in the United States. IV artesunate has been approved by the U.S. Food and Drug Administration for emergency use against severe malaria and can be obtained through the Centers for Disease Control and Prevention (CDC) Drug Service (see end of chapter for contact information). The antiarrhythmic quinidine gluconate is as effective as quinine and, as it was more readily available, replaced quinine for the treatment of malaria in the United States. The administration of quinidine must be closely monitored if dysrhythmias and hypotension are to be avoided. If total plasma levels exceed 8 μg/mL or the QTc interval exceeds 0.6 s or the QRS complex widens by more than 25% of baseline, then infusion rates should be slowed or infusion stopped temporarily. If arrhythmia or saline-unresponsive hypotension develops, treatment with this drug should be discontinued. Quinine is safer than quinidine; cardiovascular monitoring is not required except when the recipient has cardiac disease.
Severe falciparum malaria constitutes a medical emergency requiring intensive nursing care and careful management. The patient should be weighed and, if comatose, placed on his or her side. Frequent evaluation of the patient’s condition is essential. Adjunctive treatments such as high-dose glucocorticoids, urea, heparin, dextran, desferrioxamine, antibody to tumor necrosis factor α, high-dose phenobarbital (20 mg/kg), mannitol, or large-volume fluid or albumin boluses have proved either ineffective or harmful in clinical trials and should not be used. In acute renal failure or severe metabolic acidosis, hemofiltration or hemodialysis should be started as early as possible.
In severe malaria, parenteral antimalarial treatment should be started immediately. Artesunate, given by either IV or IM injection, is the agent of choice; it is simple to administer, safe, and rapidly effective. It does not require dose adjustments in liver dysfunction or renal failure, and it should be used in pregnant women with severe malaria. If artesunate is unavailable and artemether, quinine, or quinidine is used, an initial loading dose must be given so that therapeutic concentrations are reached as soon as possible. Both quinine and quinidine will cause dangerous hypotension if injected rapidly; when given IV, they must be administered carefully by rate-controlled infusion only. If this approach is not possible, quinine may be given by deep IM injections into the anterior thigh. The optimal therapeutic range for quinine and quinidine in severe malaria is not known with certainty, but total plasma concentrations of 8–15 mg/L for quinine and 3.5–8.0 mg/L for quinidine are effective and do not cause serious toxicity. The systemic clearance and apparent volume of distribution of these alkaloids are markedly reduced and plasma protein binding is increased in severe malaria, so that the blood concentrations attained with a given dose are higher. If the patient remains seriously ill or in acute renal failure for >2 days, maintenance doses of quinine or quinidine should be reduced by 30–50% to prevent toxic accumulation of the drug. The initial doses should never be reduced. If safe and feasible, exchange transfusion may be considered for patients with severe malaria, although the precise indications for this procedure have not been agreed upon and there is no clear evidence that this measure is beneficial, particularly with artesunate treatment. Convulsions should be treated promptly with IV (or rectal) benzodiazepines. The role of prophylactic anticonvulsants in children is uncertain. If respiratory support is not available, then a full loading dose of phenobarbital (20 mg/kg) to prevent convulsions should not be given as it may cause respiratory arrest.
When the patient is unconscious, the blood glucose level should be measured every 4–6 h. All patients should receive a continuous infusion of dextrose, and blood concentrations ideally should be maintained above 4 mmol/L. Hypoglycemia (<2.2 mmol/L or 40 mg/dL) should be treated immediately with bolus glucose. The parasite count and hematocrit level should be measured every 6–12 h. Anemia develops rapidly; if the hematocrit falls to <20%, then whole blood (preferably fresh) or packed cells should be transfused slowly, with careful attention to circulatory status. Renal function should be checked daily. Children presenting with severe anemia and acidotic breathing require immediate blood transfusion. Accurate assessment is vital. Management of fluid balance is difficult in severe malaria, particularly in adults, because of the thin dividing line between overhydration (leading to pulmonary edema) and underhydration (contributing to renal impairment). As soon as the patient can take fluids, oral therapy should be substituted for parenteral treatment.
UNCOMPLICATED MALARIA
Infections due to sensitive strains of P. vivax, P. knowlesi, P. malariae, and P. ovale should be treated with oral chloroquine (total dose, 25 mg of base/kg) or with an ATC known to be efficacious. In much of the tropics, drug-resistant P. falciparum has been increasing in distribution, frequency, and intensity. It is now accepted that, to prevent resistance, falciparum malaria should be treated with drug combinations and not with single drugs in endemic areas; the same rationale has been applied successfully to the treatment of tuberculosis, HIV/AIDS, and cancers. This combination strategy is based on simultaneous use of two or more drugs with different modes of action. ACT regimens are now recommended as first-line treatment for falciparum malaria throughout the malaria-affected world. These regimens are safe and effective in adults, children, and after the first trimester of pregnancy (uncertainty regarding safety currently precludes their use in the first trimester). The rapidly eliminated artemisinin component is usually an artemisinin derivative (artesunate, artemether, or dihydroartemisinin) given for 3 days, and the partner drug is usually a more slowly eliminated antimalarial to which P. falciparum is sensitive. Five ACT regimens are currently recommended by the WHO. In areas with multidrug-resistant falciparum malaria (parts of Asia and South America, including those with mefloquine-resistant parasites; Fig. 248-10), artemether-lumefantrine, artesunate-mefloquine, or dihydroartemisinin-piperaquine should be used; these regimens provide cure rates of >90%. In areas with sensitive parasites, the aforementioned combinations, artesunate-sulfadoxine-pyrimethamine, or artesunate-amodiaquine also may be used. Pyronaridine-artesunate is still under evaluation. Atovaquone-proguanil is highly effective everywhere, although it is seldom used in endemic areas because of its high cost and the propensity for rapid emergence of resistance. Of great concern is the emergence of artemisinin-resistant P. falciparum in western Cambodia and eastern Myanmar. Infections with these parasites are cleared slowly from the blood, with clearance times typically exceeding 3 days, and cure rates with ACTs are reduced.
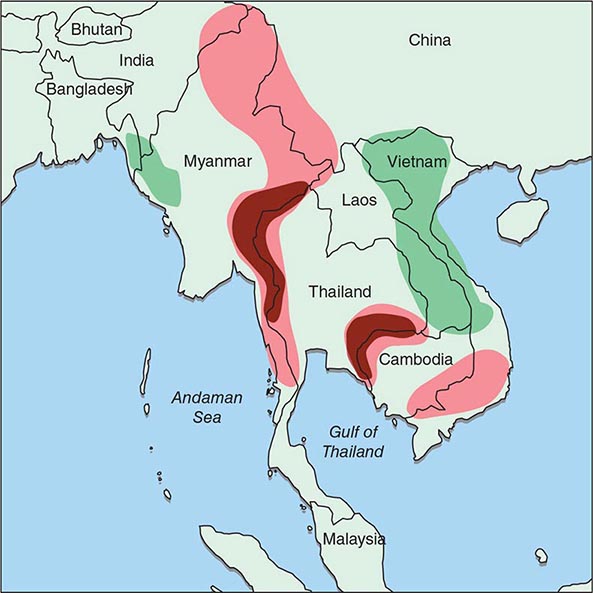
FIGURE 248-10 Mefloquine and artemisinin resistance in Plasmodium falciparum in Southeast Asia: high-level mefloquine resistance (dark red), low-level mefloquine resistance (pink), and mefloquine sensitivity (failure rate, <20%; green). There is insufficient information for other areas. Artemisinin resistance is now prevalent in areas where mefloquine resistance has been reported (pink areas).
The 3-day ACT regimens are all well tolerated, although mefloquine is associated with increased rates of vomiting and dizziness. As second-line treatments for recrudescence following first-line therapy, a different ACT regimen may be given; another alternative is a 7-day course of either artesunate or quinine plus tetracycline, doxycycline, or clindamycin. Tetracycline and doxycycline cannot be given to pregnant women or to children <8 years of age. Oral quinine is extremely bitter and regularly produces cinchonism comprising tinnitus, high-tone deafness, nausea, vomiting, and dysphoria. Adherence is poor with the required 7-day regimens of quinine.
Patients should be monitored for vomiting for 1 h after the administration of any oral antimalarial drug. If there is vomiting, the dose should be repeated. Symptom-based treatment, with tepid sponging and acetaminophen administration, lowers fever and thereby reduces the patient’s propensity to vomit these drugs. Minor central nervous system reactions (nausea, dizziness, sleep disturbances) are common. The incidence of serious adverse neuropsychiatric reactions to mefloquine treatment is ~1 in 1000 in Asia but may be as high as 1 in 200 among Africans and Caucasians. All the antimalarial quinolines (chloroquine, mefloquine, and quinine) exacerbate the orthostatic hypotension associated with malaria, and all are tolerated better by children than by adults. Pregnant women, young children, patients unable to tolerate oral therapy, and nonimmune individuals (e.g., travelers) with suspected malaria should be evaluated carefully and hospitalization considered. If there is any doubt as to the identity of the infecting malarial species, treatment for falciparum malaria should be given. A negative blood smear makes malaria unlikely but does not rule it out completely; thick blood films should be checked again 1 and 2 days later to exclude the diagnosis. Nonimmune patients receiving treatment for malaria should have daily parasite counts performed until the thick films are negative. If the level of parasitemia does not fall below 25% of the admission value in 48 h or if parasitemia has not cleared by 7 days (and adherence is assured), drug resistance is likely and the regimen should be changed.
To eradicate persistent liver stages and prevent relapse (radical treatment), primaquine (0.5 mg of base/kg or, in infections acquired in temperate areas, 0.25 mg/kg) should be given daily for 14 days to patients with P. vivax or P. ovale infections after laboratory tests for G6PD deficiency have proved negative. If the patient has a mild variant of G6PD deficiency, primaquine can be given in a dose of 0.75 mg of base/kg (45 mg maximum) once weekly for 8 weeks. Pregnant women with vivax or ovale malaria should not be given primaquine but should receive suppressive prophylaxis with chloroquine (5 mg of base/kg per week) until delivery, after which radical treatment can be given.
COMPLICATIONS
Acute Renal Failure If the plasma level of BUN or creatinine rises despite adequate rehydration, fluid administration should be restricted to prevent volume overload. As in other forms of hypercatabolic acute renal failure, renal replacement therapy is best performed early (Chap. 334). Hemofiltration and hemodialysis are more effective than peritoneal dialysis and are associated with lower mortality risk. Some patients with renal impairment pass small volumes of urine sufficient to allow control of fluid balance; these cases can be managed conservatively if other indications for dialysis do not arise. Renal function usually improves within days, but full recovery may take weeks.
Acute Pulmonary Edema (Acute Respiratory Distress Syndrome) Patients should be positioned with the head of the bed at a 45° elevation and given oxygen and IV diuretics. Pulmonary artery occlusion pressures may be normal, indicating increased pulmonary capillary permeability. Positive-pressure ventilation should be started early if the immediate measures fail (Chap. 326).
Hypoglycemia An initial slow injection of 50% dextrose (0.5 g/kg) should be followed by an infusion of 10% dextrose (0.10 g/kg per hour). The blood glucose level should be checked regularly thereafter as recurrent hypoglycemia is common, particularly among patients receiving quinine or quinidine. In severely ill patients, hypoglycemia commonly occurs together with metabolic (lactic) acidosis and carries a poor prognosis.
Other Complications Patients who develop spontaneous bleeding should be given fresh blood and IV vitamin K. Convulsions should be treated with IV or rectal benzodiazepines and, if necessary, respiratory support. Aspiration pneumonia should be suspected in any unconscious patient with convulsions, particularly with persistent hyperventilation; IV antimicrobial agents and oxygen should be administered, and pulmonary toilet should be undertaken. Hypoglycemia or gram-negative septicemia should be suspected when the condition of any patient suddenly deteriorates for no obvious reason during antimalarial treatment. In malaria-endemic areas where a high proportion of children are parasitemic, it is usually impossible to distinguish severe malaria from bacterial sepsis with confidence. These children should be treated with both antimalarials and broad-spectrum antibiotics from the outset. Because nontyphoidal Salmonella infections are particularly common, empirical antibiotics should be selected to cover these organisms. Antibiotics should be considered for severely ill patients of any age who are not responding to antimalarial treatment.
PREVENTION
In recent years, considerable progress has been made in malaria prevention, control, and research. Distribution of insecticide-treated bed-nets (ITNs) has been shown to reduce all-cause mortality in African children by 20%. New drugs have been discovered and developed, and one vaccine candidate (the RTS,S vaccine) will soon be considered for registration. Highly effective drugs, long-lasting ITNs, and insecticides for spraying dwellings are being purchased for endemic countries by the Global Fund to Fight AIDS, Tuberculosis, and Malaria; the President’s Malaria Initiative (funded by the U.S. Agency for International Development and managed by the CDC in partnership with endemic countries); UNICEF; and other organizations. Malaria research and control are being strongly supported by the National Institute of Allergy and Infectious Diseases, the CDC, the Wellcome Trust, the Bill & Melinda Gates Foundation, the Multilateral Initiative on Malaria, the Roll Back Malaria Partnership, and the WHO among others. While a laudable goal, the global eradication of malaria is not feasible in the immediate future because of the widespread distribution of Anopheles breeding sites; the great number of infected persons; the continued use of ineffective antimalarial drugs; and inadequacies in human and material resources, infrastructure, and control programs. The call for and commitment to ultimate eradication of malaria by the Gates Foundation in 2007—seconded by Margaret Chan, Director General of the WHO—added great impetus to all malaria initiatives, especially those aimed at discovery and implementation of new interventions. Malaria may be contained by judicious use of insecticides to kill the mosquito vector, rapid diagnosis, patient management, and—where effective and feasible—administration of intermittent preventive treatment, seasonal malaria chemoprevention, or chemoprophylaxis to high-risk groups such as pregnant women, young children, and travelers from nonendemic regions. Malaria researchers are intensifying their efforts to gain a better understanding of parasite-human-mosquito interactions and to develop more effective control and prevention interventions. Despite the enormous investment in efforts to develop a malaria vaccine and the 30–60% efficacy in African children of a recombinant protein sporozoite-targeted adjuvanted vaccine (RTS,S) in field trials, no safe, highly effective, long-lasting vaccine is likely to be available for general use in the near future (Chap. 148). Indeed, protection from RTS,S in the very youngest recipients dropped to 16% only 4 years after vaccination. While there is great promise for one or several malaria vaccines on the more distant horizon, prevention and control measures continue to rely on antivector and drug-use strategies. Furthermore, recent gains are threatened by increasing insecticide resistance and behavioral changes (to avoid ITN contact) in anopheline mosquito vectors and by spreading artemisinin resistance in P. falciparum.
PERSONAL PROTECTION AGAINST MALARIA
Simple measures to reduce the frequency of infected-mosquito bites in malarious areas are very important. These measures include the avoidance of exposure to mosquitoes at their peak feeding times (usually dusk to dawn) and the use of insect repellents containing 10–35% DEET (or, if DEET is unacceptable, 7% picaridin), suitable clothing, and ITNs or other insecticide-impregnated materials. Widespread use of bed nets treated with residual pyrethroids reduces the incidence of malaria in areas where vectors bite indoors at night.
CHEMOPROPHYLAXIS
(Table 248-8; wwwnc.cdc.gov/travel/yellowbook/2014/chapter-3-infectious-diseases-related-to-travel/malaria) Recommendations for prophylaxis depend on knowledge of local patterns of Plasmodium species drug sensitivity and the likelihood of acquiring malarial infection. When there is uncertainty, drugs effective against resistant P. falciparum should be used (atovaquone-proguanil [Malarone], doxycycline, or mefloquine). Chemoprophylaxis is never entirely reliable, and malaria should always be considered in the differential diagnosis of fever in patients who have traveled to endemic areas, even if they are taking prophylactic antimalarial drugs.
|
DRUGS USED IN THE PROPHYLAXIS OF MALARIA |
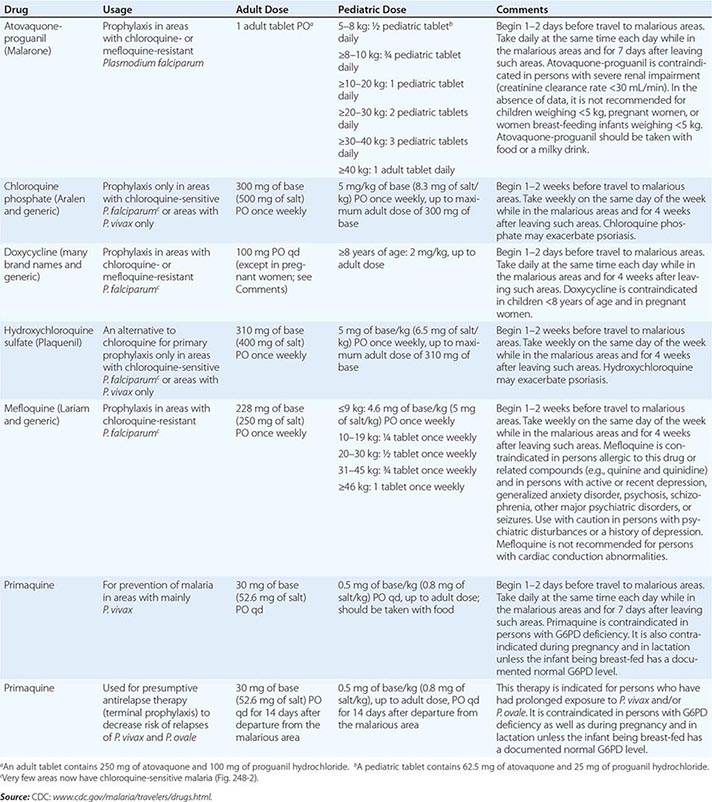
Pregnant women traveling to malarious areas should be warned about the potential risks. All pregnant women at risk in endemic areas should be encouraged to attend regular antenatal clinics. Mefloquine is the only drug advised for pregnant women traveling to areas with drug-resistant malaria; this drug is generally considered safe in the second and third trimesters of pregnancy, and the data on first-trimester exposure, although limited, are reassuring. Chloroquine and proguanil are regarded as safe. The safety of other prophylactic antimalarial agents in pregnancy has not been established. Antimalarial prophylaxis has been shown to reduce mortality rates among children between the ages of 3 months and 4 years in malaria-endemic areas; however, it is not a logistically or economically feasible option in many countries. The alternative—to give intermittent preventive treatment or seasonal malaria chemoprevention—shows promise for more widespread use in infants, young children, and pregnant women. Children born to nonimmune mothers in endemic areas (usually expatriates moving to malaria-endemic areas) should receive prophylaxis from birth.
Travelers should start taking antimalarial drugs 2 days to 2 weeks before departure so that any untoward reactions can be detected and so that therapeutic antimalarial blood concentrations will be present when needed (Table 248-8). Antimalarial prophylaxis should continue for 4 weeks after the traveler has left the endemic area, except if atovaquone-proguanil or primaquine has been taken; these drugs have significant activities against the liver stage of the infection (causal prophylaxis) and can be discontinued 1 week after departure from the endemic area. If suspected malaria develops while a traveler is abroad, obtaining a reliable diagnosis and antimalarial treatment locally is a top priority. Presumptive self-treatment for malaria with atovaquone-proguanil (for 3 consecutive days) or another drug can be considered under special circumstances; medical advice on self-treatment should be sought before departure for malarious areas and as soon as possible after illness begins. Every effort should be made to confirm the diagnosis by parasitologic studies.
Atovaquone-proguanil (Malarone; 3.75/1.5 mg/kg or 250/100 mg, daily adult dose) is a fixed-combination, once-daily prophylactic agent that is very well tolerated by adults and children, with fewer adverse gastrointestinal effects than chloroquine-proguanil and fewer adverse central nervous system effects than mefloquine. It is proguanil itself, rather than the antifolate metabolite cycloguanil, that acts synergistically with atovaquone. This combination is effective against all types of malaria, including multidrug-resistant falciparum malaria. Atovaquone-proguanil is best taken with food or a milky drink to optimize absorption. There are insufficient data on the safety of this regimen in pregnancy.
Mefloquine (250 mg of salt weekly, adult dose) has been widely used for malarial prophylaxis because it is usually effective against multidrug-resistant falciparum malaria and is reasonably well tolerated. The drug has been associated with rare episodes of psychosis and seizures at prophylactic doses; these reactions are more frequent at the higher doses used for treatment. More common side effects with prophylactic doses of mefloquine include mild nausea, dizziness, fuzzy thinking, disturbed sleep patterns, vivid dreams, and malaise. The drug is contraindicated for use by travelers with known hypersensitivity to mefloquine or related compounds (e.g., quinine, quinidine) and by persons with active or recent depression, anxiety disorder, psychosis, schizophrenia, another major psychiatric disorder, or seizures; mefloquine is not recommended for persons with cardiac conduction abnormalities although the evidence that it is cardiotoxic is very weak. Confidence is increasing with regard to the safety of mefloquine prophylaxis during pregnancy; in studies in Africa, mefloquine prophylaxis was found to be effective and safe during pregnancy. However, in one study from Thailand, treatment of malaria with mefloquine was associated with an increased risk of stillbirth; this effect was not seen subsequently.
Daily administration of doxycycline (100 mg daily, adult dose) is an effective alternative to atovaquone-proguanil or mefloquine. Doxycycline is generally well tolerated but may cause vulvovaginal thrush, diarrhea, and photosensitivity and cannot be used by children <8 years old or by pregnant women.
Chloroquine can no longer be relied upon to prevent P. falciparum infections in most areas but is used to prevent and treat malaria due to the other human Plasmodium species and for P. falciparum malaria in Central American countries west and north of the Panama Canal, Caribbean countries, and some countries in the Middle East. Chloroquine-resistant P. vivax has been reported from parts of eastern Asia, Oceania, and Central and South America. This drug is generally well tolerated, although some patients cannot take it because of malaise, headache, visual symptoms (due to reversible keratopathy), gastrointestinal intolerance, or pruritus. Chloroquine is considered safe in pregnancy. With chronic administration for >5 years, a characteristic dose-related retinopathy may develop, but this condition is rare at the doses used for antimalarial prophylaxis. Idiosyncratic or allergic reactions are also rare. Skeletal and/or cardiac myopathy is a potential problem with protracted prophylactic use; such myopathy is more likely to occur at the high doses used in the treatment of rheumatoid arthritis. Neuropsychiatric reactions and skin rashes are unusual. When used continuously, amodiaquine, a related aminoquinoline, is associated with a high risk of agranulocytosis (~1 person in 2000) and hepatotoxicity (~1 person in 16,000); thus this agent should not be used for prophylaxis.
Primaquine (daily adult dose, 0.5 mg of base/kg or 30 mg taken with food), an 8-aminoquinoline compound, has proved safe and effective in the prevention of drug-resistant falciparum and vivax malaria in adults. This drug can be considered for persons who are traveling to areas with or without drug-resistant P. falciparum and who are intolerant to other recommended drugs. Abdominal pain and oxidant hemolysis—the principal adverse effects—are not common as long as the drug is taken with food and is not given to G6PD-deficient persons, in whom it can cause serious hemolysis. Travelers must be tested for G6PD deficiency and be shown to have a level in the normal range before receiving primaquine. Primaquine should not be given to pregnant women or neonates. Primaquine, given in a single dose of 0.25 mg/kg as a gametocytocide, together with an ACT is recommended in falciparum malaria treatment regimens in malaria elimination programs.
In the past, the dihydrofolate reductase inhibitors pyrimethamine and proguanil (chloroguanide) were administered widely, but the rapid selection of resistance in both P. falciparum and P. vivax has limited their use. Whereas antimalarial quinolines such as chloroquine (a 4-aminoquinoline) act on the erythrocyte stage of parasitic development, the dihydrofolate reductase inhibitors also inhibit preerythrocytic growth in the liver (causal prophylaxis) and development in the mosquito (sporontocidal activity). Proguanil is safe and well tolerated, although mouth ulceration occurs in ~8% of persons using this drug; it is considered safe for antimalarial prophylaxis in pregnancy. The prophylactic use of the combination of pyrimethamine and sulfadoxine is not recommended because of an unacceptable incidence of severe toxicity, principally exfoliative dermatitis and other skin rashes, agranulocytosis, hepatitis, and pulmonary eosinophilia (incidence, 1:7000; fatal reactions, 1:18,000). The combination of pyrimethamine with dapsone (0.2/1.5 mg/kg weekly; 12.5/100 mg, adult dose) has been used in some countries. Dapsone may cause methemoglobinemia and allergic reactions and (at higher doses) may pose a significant risk of agranulocytosis. Proguanil and the pyrimethamine-dapsone combination are not available in the United States.
Because of the increasing spread and intensity of antimalarial drug resistance (Figs. 248-2 and 248-10), the CDC recommends that travelers and their providers consider their destination, type of travel, and current medications and health risks when choosing antimalarial chemoprophylaxis. There is an increasingly appreciated problem of counterfeit and substandard antimalarial drugs (and other medicines) on the shelves of pharmacies in Southeast Asia and sub-Saharan Africa; hence, travelers should purchase their preventive drugs from a reputable source before going to a malarious country. Consultation for the evaluation of prophylaxis failures or treatment of malaria can be obtained from state and local health departments and the CDC Malaria Hotline (770-488-7788) or the CDC Emergency Operations Center (770-488-7100).
249 |
Babesiosis |
Babesiosis is an emerging tick-borne infectious disease caused by protozoan parasites of the genus Babesia that invade and eventually lyse red blood cells (RBCs). Most cases are due to Babesia microti and occur in the United States, particularly in the Northeast and upper Midwest. The infection typically is mild in young and otherwise healthy individuals but can be severe and sometimes fatal in persons >50 years of age and in immunocompromised patients. Sporadic cases have been reported in Europe and the rest of the world.
ETIOLOGY AND EPIDEMIOLOGY
![]() Geographic Distribution More than 100 Babesia species are found in wild and domestic animals; a few of these species cause infection in humans (Fig. 249-1). B. microti, a parasite of small rodents, is the most common etiologic agent of human babesiosis and is endemic in the northeastern and upper midwestern United States. Seven states in these two regions (Connecticut, Massachusetts, Minnesota, New Jersey, New York, Rhode Island, and Wisconsin) account for >95% of the reported cases. Other etiologic agents include Babesia duncani and B. duncani–type organisms on the West Coast and Babesia divergens–like organisms in Kentucky, Missouri, and Washington State.
Geographic Distribution More than 100 Babesia species are found in wild and domestic animals; a few of these species cause infection in humans (Fig. 249-1). B. microti, a parasite of small rodents, is the most common etiologic agent of human babesiosis and is endemic in the northeastern and upper midwestern United States. Seven states in these two regions (Connecticut, Massachusetts, Minnesota, New Jersey, New York, Rhode Island, and Wisconsin) account for >95% of the reported cases. Other etiologic agents include Babesia duncani and B. duncani–type organisms on the West Coast and Babesia divergens–like organisms in Kentucky, Missouri, and Washington State.
FIGURE 249-1 Worldwide distribution of human babesiosis. Dark colors designate areas where human babesiosis is either endemic or sporadic (as defined by more than three tick-borne cases reported in a country or state). Isolated cases of babesiosis are denoted by circles. Colors designate causative Babesia species: Babesia microti and B. microti–like organisms in red, Babesia duncani and B. duncani–type organisms in orange, Babesia divergens and B. divergens–like organisms in blue, Babesia venatorum in purple, KO1 in black, and unspeciated Babesia organisms in white. Due to space constraints, the 10 cases reported from Montenegro are denoted by a single white circle, and those from Australia, Mozambique, and South Africa are not shown. Light colors denote areas that are enzootic for Ixodes tick species known to transmit one or several Babesia species but where human babesiosis has yet to be documented. (Adapted from E Vannier, PJ Krause: N Engl J Med 366:2397, 2012.)
The primary causative agent of human babesiosis in Europe is B. divergens, but Babesia venatorum and B. microti also have been reported. In Asia, cases due to B. microti–like organisms have been documented in Japan, Taiwan, and the People’s Republic of China. A case caused by B. venatorum also has been reported from the People’s Republic of China. A case of B. microti infection was described in Australia. Sporadic cases due to uncharacterized species have been reported in Colombia, Egypt, India, Mozambique, and South Africa.
Incidence More than 1100 cases were reported in the United States in 2011, the year the disease became nationally notifiable. This figure represents a fivefold increase in incidence over the past decade. The incidence of babesiosis is markedly underestimated because symptoms are nonspecific and because young healthy individuals typically experience a mild or asymptomatic infection and may not seek medical attention. Fewer than 50 cases of B. divergens, B. divergens–like, and B. venatorum infections have been reported. Babesiosis caused by B. duncani and B. duncani–type organisms has also been sporadic, with fewer than 10 reported cases.
![]() Modes of Transmission In the United States, B. microti is transmitted to humans primarily by the nymphal stage of the deer tick (Ixodes scapularis), the same tick that transmits the causative agents of Lyme disease (Chap. 210) and human granulocytotropic anaplasmosis (Chap. 211). Transmission generally occurs from May through October, with three-fourths of cases presenting in July and August. The vectors for transmission of B. duncani and B. divergens–like organisms are thought to be Ixodes pacificus and Ixodes dentatus, respectively. In Europe, Ixodes ricinus is the vector for B. divergens and B. venatorum. In Japan, B. microti–like organisms have been found in Ixodes ovatus ticks.
Modes of Transmission In the United States, B. microti is transmitted to humans primarily by the nymphal stage of the deer tick (Ixodes scapularis), the same tick that transmits the causative agents of Lyme disease (Chap. 210) and human granulocytotropic anaplasmosis (Chap. 211). Transmission generally occurs from May through October, with three-fourths of cases presenting in July and August. The vectors for transmission of B. duncani and B. divergens–like organisms are thought to be Ixodes pacificus and Ixodes dentatus, respectively. In Europe, Ixodes ricinus is the vector for B. divergens and B. venatorum. In Japan, B. microti–like organisms have been found in Ixodes ovatus ticks.
Babesiosis occasionally is acquired through transfusion of blood or blood products. B. microti is the most common transfusion-transmitted pathogen reported to the U.S. Food and Drug Administration, and more than 170 such cases have been identified. Three transfusion-transmitted cases caused by B. duncani have been documented. Transfusion-transmitted cases occur year-round, although most cases occur from June through November. More than 80% of cases occur in endemic areas. Transfusion-transmitted babesiosis occurs in nonendemic areas when unrecognized Babesia-contaminated blood products are imported from endemic areas: asymptomatically infected residents of endemic areas donate blood in nonendemic areas, or residents of nonendemic areas travel to endemic areas, become infected, and donate blood after they return home. Approximately three-quarters of the transfusion-transmitted babesiosis cases reported between 1979 and 2009 occurred in the last decade of this period, and about one-fifth of patients died.
Seven cases of probable or confirmed congenital B. microti infection have been described. Other cases of neonatal babesiosis have been acquired by transfusion or tick bite.
CLINICAL MANIFESTATIONS
Asymptomatic B. microti Infection At least 20% of adults and 40% of children do not experience symptoms following B. microti infection. Asymptomatic infection, whether treated or not, may persist for >1 year after acute babesial illness. There is no evidence of long-term complications following asymptomatic infection; however, people who are asymptomatically infected may transmit the infection when they donate blood.
Mild to Moderate B. microti Illness Symptoms typically develop following an incubation period of 1–4 weeks after tick bite and 1–9 weeks (but as long as 6 months) after transfusion of blood products. Patients experience a gradual onset of malaise, fatigue, and weakness. Fever can reach 40.9°C (105.6°F) and is accompanied by one or more of the following: chills, sweats, headache, myalgia, arthralgia, nausea, anorexia, and dry cough. Less common symptoms include sore throat, photophobia, abdominal pain, vomiting, weight loss, shortness of breath, and depression. On physical examination, fever is the salient feature. Ecchymoses and petechiae have been reported. An erythema migrans rash signifies concurrent Lyme disease (Chap. 210). Splenomegaly and hepatomegaly occasionally are noted. Lymphadenopathy is absent. Jaundice, slight pharyngeal erythema, and retinopathy with splinter hemorrhages and retinal infarcts rarely occur. Symptoms typically last 1–2 weeks, but fatigue may persist for several months.
Severe B. microti Illness Severe babesiosis requires hospital admission and typically occurs in patients with one or more of the following: age of >50 years, neonatal prematurity, male gender, asplenia, HIV/AIDS, malignancy, hemoglobinopathy, and immunosuppressive therapy. More than one-third of hospitalized patients develop complications, including acute respiratory distress syndrome, disseminated intravascular coagulation, congestive heart failure, renal failure, splenic infarcts, and splenic rupture. Patients who develop complications tend to have severe anemia (hemoglobin, ≤10 g/L). Laboratory prognostic factors for severe outcome—defined by hospitalization for >14 days, an intensive care unit stay of >2 days, or death—include an elevated alkaline phosphatase level (>125 U/L) and parasitemia of >4%. The fatality rate is 5–9% among hospitalized patients but is ~20% among immunocompromised patients and patients with transfusion-transmitted babesiosis.
![]() Other Babesia Infections Cases of B. duncani infection range in severity from asymptomatic to fatal. Clinical manifestations are similar to those reported for B. microti infection. All three patients infected with B. divergens–like organisms in the United States required hospitalization; one died. Most cases of B. divergens infection in Europe have occurred in people lacking a spleen. The incubation period is 1–3 weeks. Symptoms appear suddenly and consist of fever (>41°C or 105.8°F), shaking chills, drenching sweats, headache, myalgia, and lumbar and abdominal pain. Hemoglobinuria and jaundice are commonly noted, and mild hepatomegaly may occur. If the infection is not treated, patients often develop pulmonary edema and renal failure. All four patients infected with B. venatorum in Europe had been splenectomized; their illness ranged from mild to severe, and none died. A child in China who developed B. venatorum illness had an intact spleen and survived the infection.
Other Babesia Infections Cases of B. duncani infection range in severity from asymptomatic to fatal. Clinical manifestations are similar to those reported for B. microti infection. All three patients infected with B. divergens–like organisms in the United States required hospitalization; one died. Most cases of B. divergens infection in Europe have occurred in people lacking a spleen. The incubation period is 1–3 weeks. Symptoms appear suddenly and consist of fever (>41°C or 105.8°F), shaking chills, drenching sweats, headache, myalgia, and lumbar and abdominal pain. Hemoglobinuria and jaundice are commonly noted, and mild hepatomegaly may occur. If the infection is not treated, patients often develop pulmonary edema and renal failure. All four patients infected with B. venatorum in Europe had been splenectomized; their illness ranged from mild to severe, and none died. A child in China who developed B. venatorum illness had an intact spleen and survived the infection.
PATHOGENESIS
Anemia is a key feature of the pathogenesis of babesiosis. Hemolytic anemia caused by rupture of infected RBCs generates cell debris that may accumulate in the kidney and cause renal failure. Anemia also results from the clearance of intact RBCs as they pass through the splenic red pulp and encounter resident macrophages. Babesia antigens expressed at the RBC membrane promote opsonization and facilitate uptake by splenic macrophages. In addition, RBCs are poorly deformable as a result of oxidation generated by the parasite and the host immune response and are filtered out as they attempt to squeeze across the venous vasculature. Bone marrow suppression due to cytokine production may also contribute to anemia.
An appropriate immune response is necessary for the control and clearance of Babesia. However, several lines of evidence suggest that an excessive response contributes to pathogenesis. Studies using laboratory mice have clearly established that CD4+ T cells are critical for resistance to and resolution of B. microti infection. CD4+ T cells are a major source of interferon γ (IFN-γ), and lack of this cytokine causes resistant mice to become highly susceptible to B. microti. IFN-γ is central to host resistance in B. duncani infection, but natural killer cells are its main source. B. duncani infection is more severe than B. microti infection in rodents and is characterized by pulmonary inflammation. Tumor necrosis factor α is expressed around alveolar septa, whereas IFN-γ is detected around pulmonary vessels. Blockade of either cytokine promotes the survival of mice infected with B. duncani.
DIAGNOSIS
A diagnosis of babesiosis should be considered for any patient who lives or travels in a Babesia-endemic area and presents with a febrile illness in the late spring, summer, or early autumn or within 6 months after a blood transfusion. Co-infection with Babesia should be considered in cases of Lyme disease or human granulocytotropic anaplasmosis when symptoms are more severe or prolonged than usual.
Screening laboratory tests can help support the diagnosis of babesiosis. A complete blood count often shows anemia and thrombocytopenia. Low hematocrit, hemoglobin, and haptoglobin levels and elevated reticulocyte counts and lactate dehydrogenase levels are consistent with hemolytic anemia. Liver enzyme tests often reveal elevated levels of alkaline phosphatase, aspartate and alanine aminotransferases, and bilirubin. Urinalysis may show hemoglobinuria, excess urobilinogen, and proteinuria. Elevated levels of blood urea nitrogen and serum creatinine indicate renal compromise.
A specific diagnosis usually is established by microscopic examination of Giemsa-stained thin blood smears (Fig. 249-2). Babesia trophozoites appear round, pear-shaped, or ameboid. The ring form is most common and lacks the central brownish deposit (hemozoin) typical of Plasmodium falciparum trophozoites (see Fig. 250e-1C). Other distinguishing features are the absence of schizonts and gametocytes and the occasional presence of tetrads (“Maltese cross”). Tetrads are characteristic of B. microti, B. duncani, and B. divergens–like organisms in human erythrocytes. Because the number of parasitized RBCs may be low, particularly at the onset of symptoms, identification of the parasite may require multiple blood smears over several days. Parasitemia levels can range from 1% to 20% in immunocompetent hosts and can be as high as 85% in immunocompromised patients. If parasites cannot be identified by microscopy and the disease is still suspected, amplification of the babesial 18S rRNA gene by polymerase chain reaction (PCR) is recommended. Quantitative PCR has greatly lowered the threshold for detection of B. microti DNA.
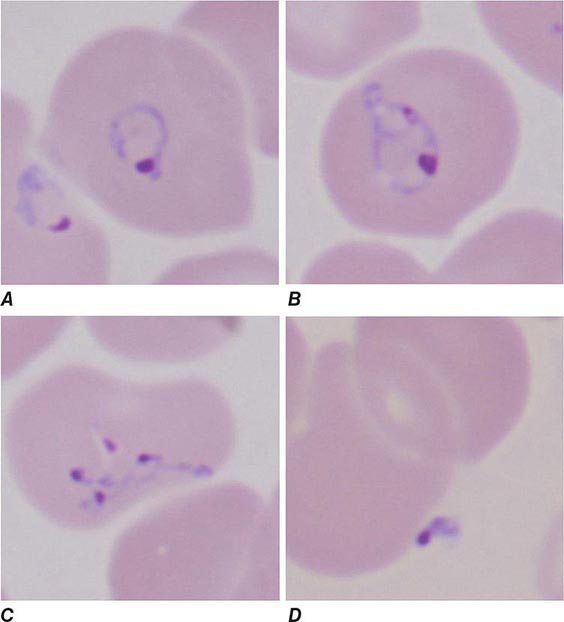
FIGURE 249-2 Giemsa-stained thin blood films showing Babesia microti parasites. B. microti are obligate parasites of erythrocytes. Trophozoites may appear as ring forms (A) or as ameboid forms (B). Merozoites can be arranged in tetrads and are pathognomonic (C). Extracellular parasites can be noted, particularly when parasitemia is high (D). (Adapted from E Vannier, PJ Krause: N Engl J Med 366:2397, 2012.)
Serology can suggest or confirm the diagnosis of babesiosis. An indirect immunofluorescent antibody test for B. microti is most commonly used. IgM titers of ≥1:64 and IgG titers of ≥1:1024 suggest active or recent infection. Titers typically decline over 6–12 months. Antibodies to B. microti do not cross-react with B. duncani or B. divergens antigen. In B. divergens infection, serology is of limited use because symptoms often appear before antibodies can be detected. Sera from patients infected with B. divergens–like organisms or B. venatorum are reactive against B. divergens antigen.
|
TREATMENT |
BABESIOSIS |
ASYMPTOMATIC B. MICROTI INFECTION
People who experience asymptomatic B. microti infection often are not diagnosed and treated. Current guidelines recommend antibiotic therapy for asymptomatic carriers only if parasitemia persists for >3 months. Laboratory-based tests are being developed for the purpose of screening the blood supply and will result in the identification of a greater number of asymptomatic B. microti carriers, raising the question of whether they should be treated.
MILD TO MODERATE B. MICROTI ILLNESS
Atovaquone plus azithromycin, given orally for 7–10 days, is the recommended antibiotic treatment combination for mild to moderate babesiosis (Table 249-1). Clindamycin plus quinine is a second choice. Symptoms usually begin to resolve within 48 h of therapy initiation, but complete resolution may take weeks to months. An atypical or poor response to therapy should raise concern about the possibility of concurrent Lyme disease (Chap. 210) or human granulocytotropic anaplasmosis (Chap. 211). In the first prospective trial of antibabesial therapy, the combination of atovaquone plus azithromycin was compared with clindamycin plus quinine in adults. These two drug combinations were equally effective in resolving symptoms and clearing parasitemia. Adverse effects were reported in 15% of trial participants who received atovaquone plus azithromycin but in 72% of those who received clindamycin plus quinine. Adverse reactions were so severe that treatment had to be stopped in about one-third of participants taking clindamycin plus quinine but in only 2% of those taking atovaquone plus azithromycin.
|
TREATMENT OF HUMAN BABESIOSIS |
SEVERE B. MICROTI ILLNESS
Clindamycin given intravenously plus quinine given orally for 7–10 days constitute the recommended treatment for severe babesiosis. Intravenous quinidine may be used instead of oral quinine but requires cardiac monitoring because of the risk of QT prolongation and polymorphic ventricular tachycardia.
Standard antimicrobial therapy is sometimes insufficient to resolve symptoms and clear parasitemia, especially in patients with marked immunosuppression due to splenectomy, HIV/AIDS, malignancy, and/or immunosuppressive therapy (including rituximab for B cell lymphomas). In such patients, antimicrobial therapy should be administered for at least 6 weeks, including 2 weeks after parasites are no longer observed on blood smear. High-dose azithromycin (600–1000 mg/d) plus atovaquone have been successfully used in immunocompromised patients. Resistance to atovaquone plus azithromycin has occurred in a few cases. In patients who are unresponsive to atovaquone plus azithromycin or who do not tolerate clindamycin plus quinine, alternative regimens have been used (Table 249-1).
Partial or complete RBC exchange transfusion is recommended in patients with high-grade parasitemia (≥10%), severe anemia (hemoglobin, <10 g/dL), or pulmonary, hepatic, or renal compromise. Parasitemia and hematocrit should be monitored daily until symptoms recede and the parasitemia level is <5%.
OTHER BABESIA INFECTIONS
The regimen for B. duncani infections typically consists of intravenous clindamycin (600 mg tid/qid or 1200 mg bid) plus oral quinine (600–650 mg tid) for 7–10 days. A regimen often used for B. divergens–like infections is intravenous clindamycin (600 mg tid/qid, 900 mg tid, or 1200 mg bid) plus oral quinine or quinidine (650 mg tid).
![]() In Europe, B. divergens infection is considered a medical emergency. The recommended treatment is immediate complete blood exchange transfusion and therapy with intravenous clindamycin plus either oral quinine or intravenous quinidine. Some cases have been cured with exchange transfusion and clindamycin monotherapy. Anemia may persist for >1 month and require additional transfusion.
In Europe, B. divergens infection is considered a medical emergency. The recommended treatment is immediate complete blood exchange transfusion and therapy with intravenous clindamycin plus either oral quinine or intravenous quinidine. Some cases have been cured with exchange transfusion and clindamycin monotherapy. Anemia may persist for >1 month and require additional transfusion.
PREVENTION
No vaccine is available for human use. There is no role for antibiotic prophylaxis. Individuals who reside in endemic areas, especially those at risk for severe babesiosis, should wear clothing that covers the lower part of the body, apply tick repellents (such as DEET) to clothing, and limit outdoor activities where ticks may abound from May through October. The skin should be thoroughly examined after outdoor activities, and ticks should be removed with tweezers. Individuals with a history of symptomatic babesiosis or with positive Babesia serology are indefinitely deferred from donating blood.
250e |
Atlas of Blood Smears of Malaria and Babesiosis |
Six species of blood protozoan parasites cause human malaria (Chap. 248): the potentially lethal and often drug-resistant Plasmodium falciparum; the relapsing parasites Plasmodium vivax and Plasmodium ovale (with what appear to be two morphologically identical sympatric species of P. ovale); Plasmodium malariae, which can persist at low densities for years; and, in infections in individuals living in or close to tropical forests in Southeast Asia, Plasmodium knowlesi, a monkey parasite that microscopically resembles P. falciparum (young forms) and P. malariae (older forms) but is identified definitively by molecular methods.
The malaria parasites are readily seen under the microscope (×1000 magnification) in thick and thin blood smears stained with supravital dyes (e.g., Giemsa’s, Field’s, Wright’s, Leishman’s). The morphologic characteristics of the parasites are summarized in Table 250e-1. In the thick film, lysis of red blood cells by water leaves the stained white cells and parasites, allowing detection of densities as low as 50 parasites/μL. This degree of sensitivity is up to 100 times greater than that of the thin film, in which the cells are fixed and the malaria parasites are seen inside the red cells. The thin film is better for speciation and provides useful prognostic information in severe falciparum malaria. Several findings are associated with increased mortality risk: high parasite counts, more mature parasites (>20% containing visible malaria pigment), and phagocytosed malaria pigment in >5% of neutrophils.
|
MORPHOLOGIC CHARACTERISTICS OF HUMAN MALARIA PARASITESa |

Babesia microti (Chap. 249) appears as a small ring form resembling P. falciparum. Unlike Plasmodium, Babesia does not cause the production of pigment in parasites, nor are schizonts or gametocytes formed.
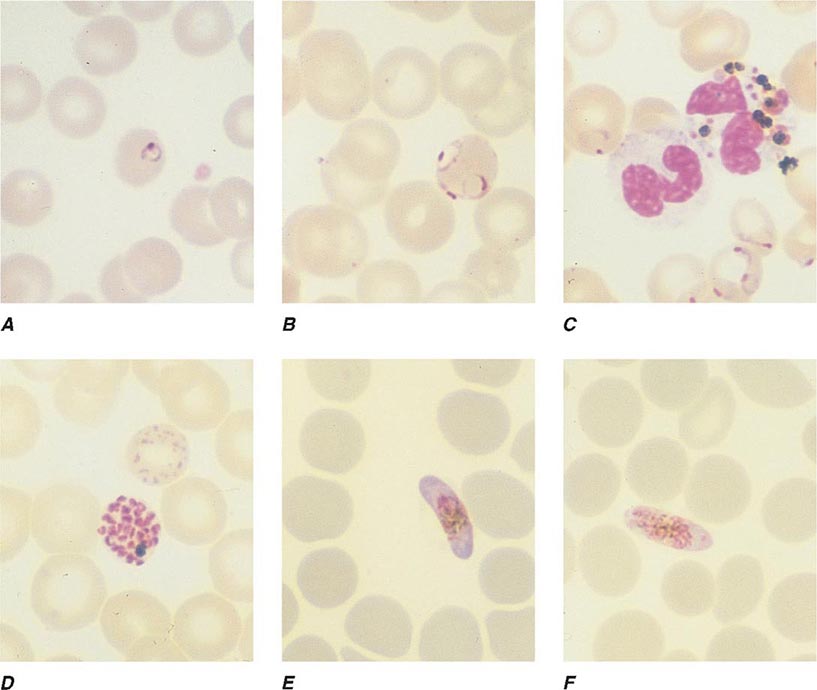
FIGURE 250e-1 Thin blood films of Plasmodium falciparum. A. Young trophozoites. B. Old trophozoites. C. Pigment in polymorphonuclear cells and trophozoites. D. Mature schizonts. E. Female gametocytes. F. Male gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
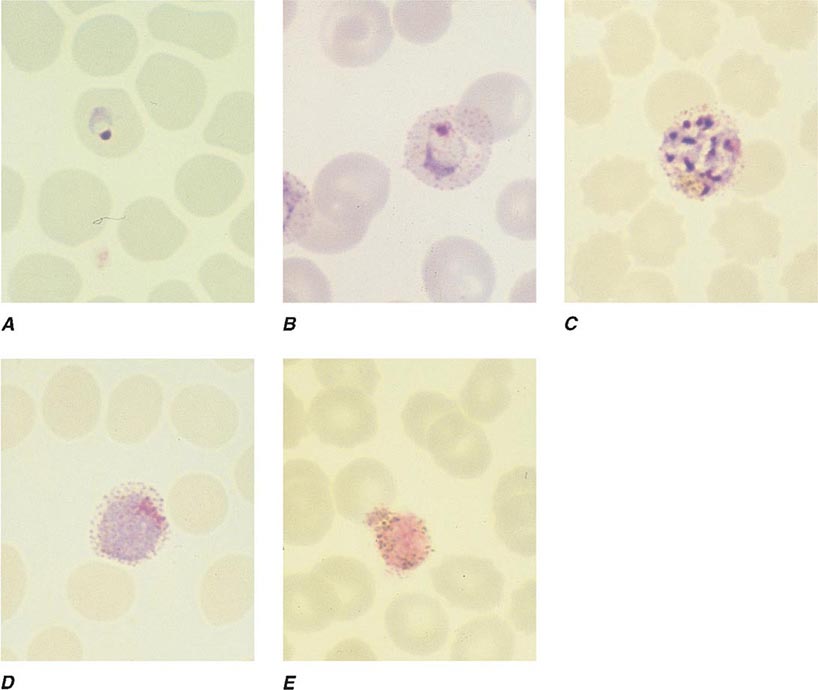
FIGURE 250e-2 Thin blood films of Plasmodium vivax. A. Young trophozoites. B. Old trophozoites. C. Mature schizonts. D. Female gametocytes. E. Male gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
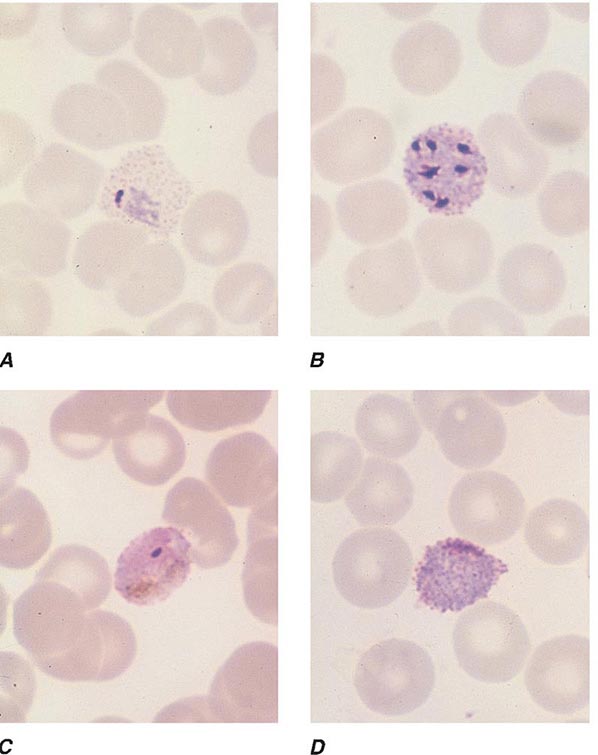
FIGURE 250e-3 Thin blood films of Plasmodium ovale. A. Old trophozoites. B. Mature schizonts. C. Male gametocytes. D. Female gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
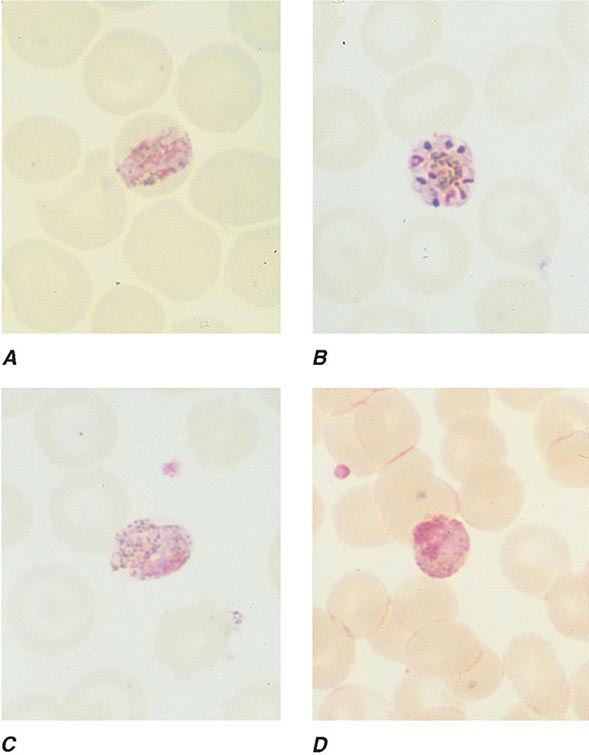
FIGURE 250e-4 Thin blood films of Plasmodium malariae. A. Old trophozoites. B. Mature schizonts. C. Male gametocytes. D. Female gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
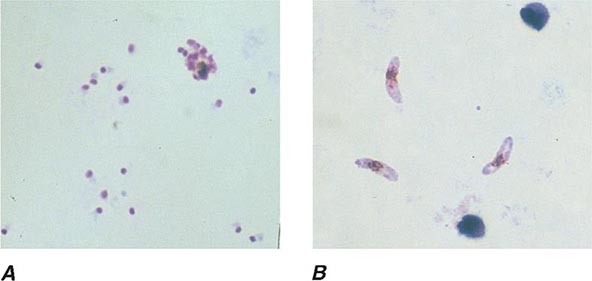
FIGURE 250e-5 Thick blood films of Plasmodium falciparum. A. Trophozoites. B. Gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
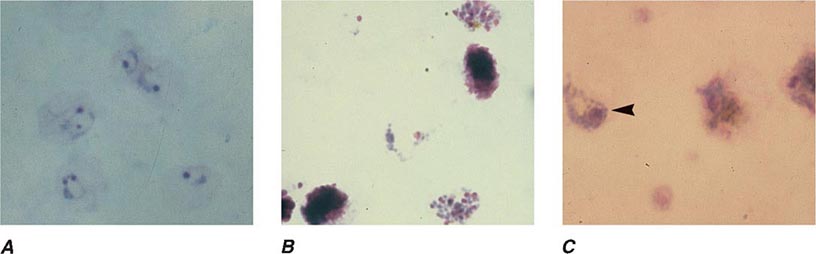
FIGURE 250e-6 Thick blood films of Plasmodium vivax. A. Trophozoites. B. Schizonts. C. Gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
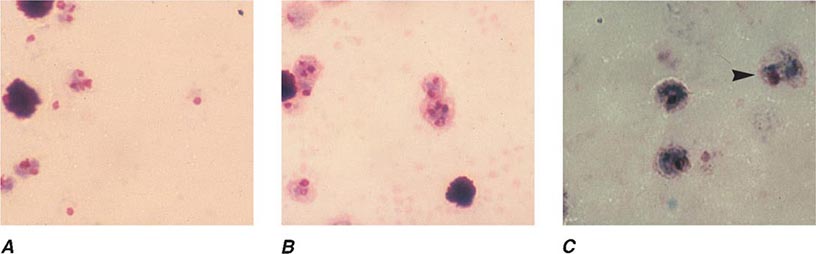
FIGURE 250e-7 Thick blood films of Plasmodium ovale. A. Trophozoites. B. Schizonts. C. Gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
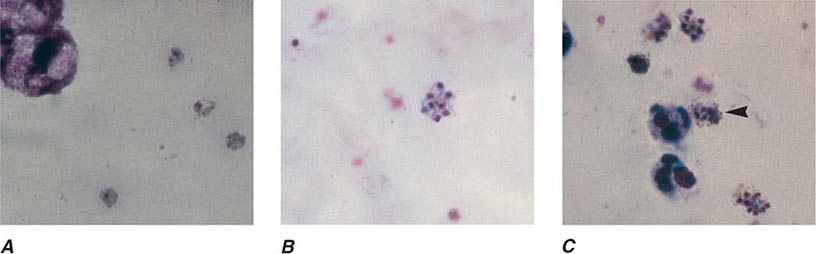
FIGURE 250e-8 Thick blood films of Plasmodium malariae. A. Trophozoites. B. Schizonts. C. Gametocytes. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
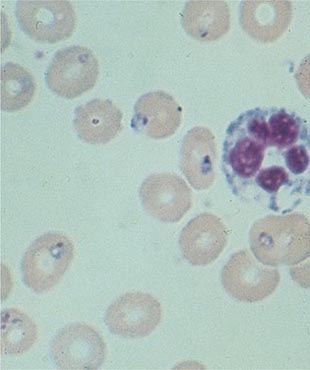
FIGURE 250e-9 Thin blood film showing trophozoites of Babesia. (Reproduced from Bench Aids for the Diagnosis of Malaria Infections, 2nd ed, with the permission of the World Health Organization.)
251 |
Leishmaniasis |
DEFINITION
Encompassing a complex group of disorders, leishmaniasis is caused by unicellular eukaryotic obligatory intracellular protozoa of the genus Leishmania and primarily affects the host’s reticuloendothelial system. Leishmania species produce widely varying clinical syndromes ranging from self-healing cutaneous ulcers to fatal visceral disease. These syndromes fall into three broad categories: visceral leishmaniasis (VL), cutaneous leishmaniasis (CL), and mucosal leishmaniasis (ML).
ETIOLOGY AND LIFE CYCLE
Leishmaniasis is caused by ~20 species of the genus Leishmania in the order Kinetoplastida and the family Trypanosomatidae (Table 251-1). Several clinically important species are of the subspecies Viannia. The organisms are transmitted by phlebotomine sandflies of the genus Phlebotomus in the “Old World” (Asia, Africa, and Europe) and the genus Lutzomyia in the “New World” (the Americas). Transmission may be anthroponotic (i.e., the vector transmits the infection from infected humans to healthy humans) or zoonotic (i.e., the vector transmits the infection from an animal reservoir to humans). Human-to-human transmission via shared infected needles has been documented in IV drug users in the Mediterranean region. In utero transmission to the fetus occurs rarely.
|
GEOGRAPHIC DISTRIBUTION AND CHARACTERISTIC EPIDEMIOLOGY OF LEISHMANIASES |
Leishmania organisms occur in two forms: extracellular, flagellate promastigotes (length, 10–20 μm) in the sandfly vector and intracellular, nonflagellate amastigotes (length, 2–4 μm; Fig. 251-1) in vertebrate hosts, including humans. Promastigotes are introduced through the proboscis of the female sandfly into the skin of the vertebrate host. Neutrophils predominate among the host cells that first encounter and take up promastigotes at the site of parasite delivery. The infected neutrophils may undergo apoptosis and release viable parasites that are taken up by macrophages, or the apoptotic cells may themselves be taken up by macrophages and dendritic cells. The parasites multiply as amastigotes inside macrophages, causing cell rupture with subsequent invasion of other macrophages. While feeding on infected hosts, sandflies pick up amastigotes, which transform into the flagellate form in the flies’ posterior midgut and multiply by binary fission; the promastigotes then migrate to the anterior midgut and can infect a new host when flies take another blood meal.
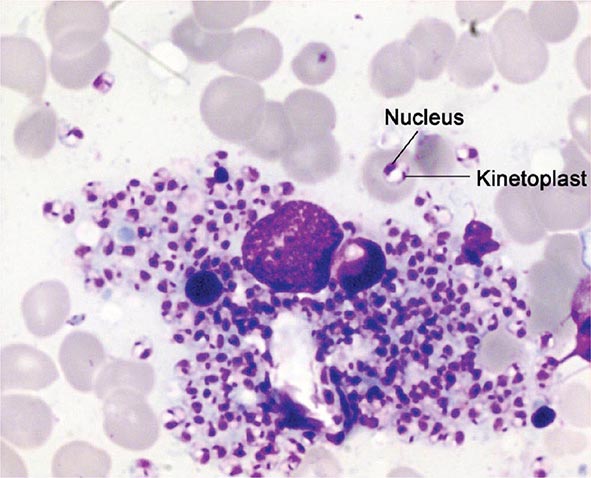
FIGURE 251-1 A macrophage with numerous intracellular amastigotes (2–4 μm) in a Giemsa-stained splenic smear from a patient with visceral leishmaniasis. Each amastigote contains a nucleus and a characteristic kinetoplast consisting of multiple copies of mitochondrial DNA. A few extracellular parasites are also visible.
EPIDEMIOLOGY
![]() Leishmaniasis occurs in 98 countries—most of them developing—in tropical and temperate regions (Fig. 251-2). More than 1.5 million cases occur annually, of which 0.7–1.2 million are CL (and its variations) and 200,000–400,000 are VL. More than 350 million people are at risk, with an overall prevalence of 12 million. Although the distribution of Leishmania is limited by the distribution of sandfly vectors, human leishmaniasis is on the increase worldwide.
Leishmaniasis occurs in 98 countries—most of them developing—in tropical and temperate regions (Fig. 251-2). More than 1.5 million cases occur annually, of which 0.7–1.2 million are CL (and its variations) and 200,000–400,000 are VL. More than 350 million people are at risk, with an overall prevalence of 12 million. Although the distribution of Leishmania is limited by the distribution of sandfly vectors, human leishmaniasis is on the increase worldwide.
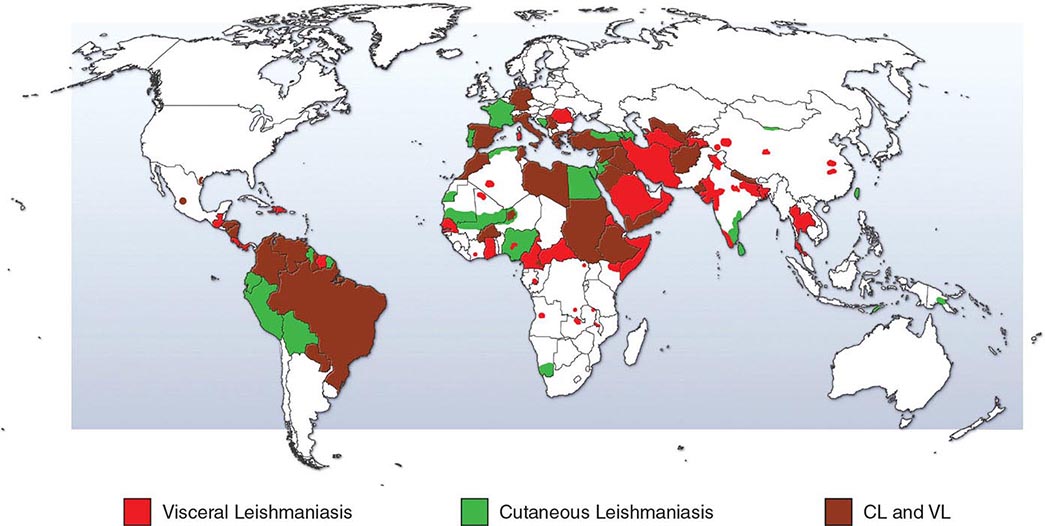
FIGURE 251-2 Worldwide distribution of human leishmaniasis. CL, cutaneous leishmaniasis; VL, visceral leishmaniasis.
VISCERAL LEISHMANIASIS
VL (also known as kala-azar, a Hindi term meaning “black fever”) is caused by the Leishmania donovani complex, which includes L. donovani and Leishmania infantum (the latter designated Leishmania chagasi in the New World); these species are responsible for anthroponotic and zoonotic transmission, respectively. India and neighboring Bangladesh, Sudan, South Sudan, Ethiopia, and Brazil are the four largest foci of VL and account for 90% of the world’s VL burden, with India being the worst affected. Zoonotic VL is reported from all countries in the Middle East, Pakistan, and other countries from western Asia to China. Endemic foci also exist in the independent states of the former Soviet Union, mainly Georgia and Azerbaijan. In the Horn of Africa, Sudan, South Sudan, Ethiopia, Kenya, Uganda, and Somalia report VL. In Sudan and South Sudan, large outbreaks are thought to be anthroponotic, although zoonotic transmission also occurs. VL is rare in West and sub-Saharan Africa.
Mediterranean VL, long an established endemic disease due to L. infantum, has a large canine reservoir and was seen primarily in infants before the advent of HIV infection. In Mediterranean Europe, 70% of adult VL cases are associated with HIV co-infection. The combination is deadly because of the impact of the two infections together on the immune system. IV drug users are at particular risk. Other forms of immunosuppression (e.g., that associated with organ transplantation) also predispose to VL. In the Americas, disease caused by L. infantum is endemic from Mexico to Argentina, but 90% of cases in the New World are reported from northeastern Brazil.
Immunopathogenesis The majority of individuals infected by L. donovani or L. infantum mount a successful immune response and control the infection, never developing symptomatic disease. Forty-eight hours after intradermal injection of killed promastigotes, these individuals exhibit delayed-type hypersensitivity (DTH) to leishmanial antigens in the leishmanin skin test (also called the Montenegro skin test). Results in mouse models indicate that the development of acquired resistance to leishmanial infection is controlled by the production of interleukin (IL) 12 by antigen-presenting cells and the subsequent secretion of interferon (IFN) γ, tumor necrosis factor (TNF) α, and other proinflammatory cytokines by the T helper 1 (TH1) subset of T lymphocytes. The immune response in patients developing active VL is complex; in addition to increased production of multiple proinflammatory cytokines and chemokines, patients with active disease have markedly elevated levels of IL-10 in serum as well as enhanced IL-10 mRNA expression in lesional tissues. The main disease-promoting activity of IL-10 in VL may be to condition host macrophages for enhanced survival and growth of the parasite. IL-10 can render macrophages unresponsive to activation signals and inhibit killing of amastigotes by downregulating the production of TNF-α and nitric oxide. Multiple antigen-presentation functions of dendritic cells and macrophages are also suppressed by IL-10. Patients with such suppression do not have positive leishmanin skin tests, nor do their peripheral-blood mononuclear cells respond to leishmanial antigens in vitro. Organs of the reticuloendothelial system are predominantly affected, with remarkable enlargement of the spleen, liver, and lymph nodes in some regions. The tonsils and intestinal submucosa are also heavily infiltrated with parasites. Bone marrow dysfunction results in pancytopenia.
Clinical Features On the Indian subcontinent and in the Horn of Africa, persons of all ages are affected by VL. In endemic areas of the Americas and the Mediterranean basin, immunocompetent infants and small children as well as immunodeficient adults are affected especially often. The most common presentation of VL is an abrupt onset of moderate- to high-grade fever associated with rigor and chills. Fever may continue for several weeks with decreasing intensity, and the patient may become afebrile for a short period before experiencing another bout of fever. The spleen may be palpable by the second week of illness and, depending on the duration of illness, may become hugely enlarged (Fig. 251-3). Hepatomegaly (usually moderate in degree) soon follows. Lymphadenopathy is common in most endemic regions of the world except the Indian subcontinent, where it is rare. Patients lose weight and feel weak, and the skin gradually develops dark discoloration due to hyperpigmentation that is most easily seen in brown-skinned individuals. In advanced illness, hypoalbuminemia may manifest as pedal edema and ascites. Anemia appears early and may become severe enough to cause congestive heart failure. Epistaxis, retinal hemorrhages, and gastrointestinal bleeding are associated with thrombocytopenia. Secondary infections such as measles, pneumonia, tuberculosis, bacillary or amebic dysentery, and gastroenteritis are common. Herpes zoster, chickenpox, boils in the skin, and scabies may also occur. Untreated, the disease is fatal in most patients, including 100% of those with HIV co-infection.
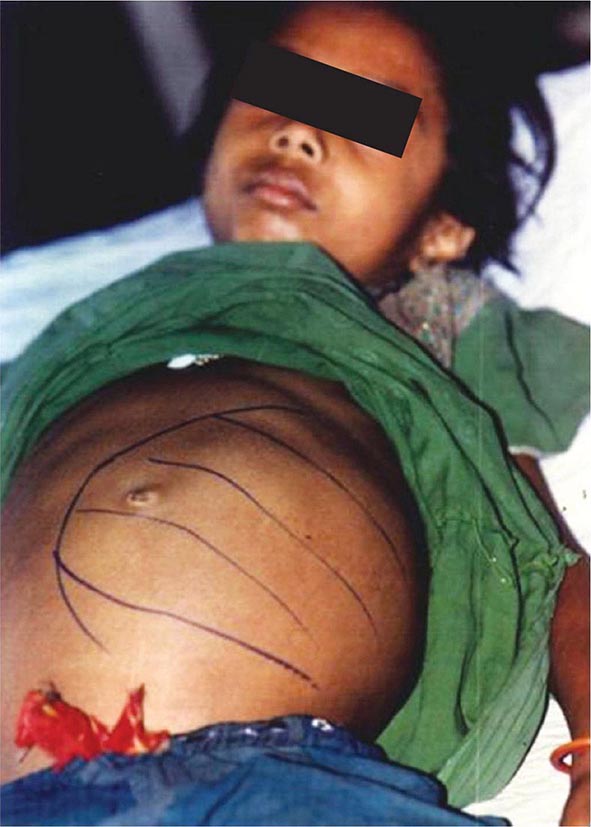
FIGURE 251-3 A patient with visceral leishmaniasis has a hugely enlarged spleen visible through the surface of the abdomen. Splenomegaly is the most important feature of visceral leishmaniasis.
Leukopenia and anemia occur early and are followed by thrombocytopenia. There is a marked polyclonal increase in serum immunoglobulins. Serum levels of hepatic aminotransferases are raised in a significant proportion of patients, and serum bilirubin levels are elevated occasionally. Renal dysfunction is uncommon.
Laboratory Diagnosis Demonstration of amastigotes in smears of tissue aspirates is the gold standard for the diagnosis of VL (Fig. 251-1). The sensitivity of splenic smears is >95%, whereas smears of bone marrow (60–85%) and lymph node aspirates (50%) are less sensitive. Culture of tissue aspirates increases sensitivity. Splenic aspiration is invasive and may be dangerous in untrained hands. Several serologic techniques are currently used to detect antibodies to Leishmania. An enzyme-linked immunosorbent assay (ELISA) and the indirect immunofluorescent antibody test (IFAT) are used in sophisticated laboratories.
![]() In the field, however, a rapid immunochromatographic test based on the detection of antibodies to a recombinant antigen (rK39) consisting of 39 amino acids conserved in the kinesin region of L. infantum is used worldwide. The test requires only a drop of fingerprick blood or serum, and the result can be read within 15 min. Except in East Africa (where both its sensitivity and its specificity are lower), the sensitivity of the rK39 rapid diagnostic test (RDT) in immunocompetent individuals is ~98% and its specificity is 90%. In Sudan, an RDT based on a new synthetic polyprotein, rK28, was more sensitive (96.8%) and specific (96.2%) than rK39-based RDTs. Qualitative detection of leishmanial nucleic acid by polymerase chain reaction (PCR) and quantitative detection by real-time PCR are confined to specialized laboratories and have yet to be used for routine diagnosis of VL in endemic areas. PCR can distinguish among the major species of Leishmania infecting humans.
In the field, however, a rapid immunochromatographic test based on the detection of antibodies to a recombinant antigen (rK39) consisting of 39 amino acids conserved in the kinesin region of L. infantum is used worldwide. The test requires only a drop of fingerprick blood or serum, and the result can be read within 15 min. Except in East Africa (where both its sensitivity and its specificity are lower), the sensitivity of the rK39 rapid diagnostic test (RDT) in immunocompetent individuals is ~98% and its specificity is 90%. In Sudan, an RDT based on a new synthetic polyprotein, rK28, was more sensitive (96.8%) and specific (96.2%) than rK39-based RDTs. Qualitative detection of leishmanial nucleic acid by polymerase chain reaction (PCR) and quantitative detection by real-time PCR are confined to specialized laboratories and have yet to be used for routine diagnosis of VL in endemic areas. PCR can distinguish among the major species of Leishmania infecting humans.
Differential Diagnosis VL is easily mistaken for malaria. Other febrile illnesses that may mimic VL include typhoid fever, tuberculosis, brucellosis, schistosomiasis, and histoplasmosis. Splenomegaly due to portal hypertension, chronic myeloid leukemia, tropical splenomegaly syndrome, and (in Africa) schistosomiasis may also be confused with VL. Fever with neutropenia or pancytopenia in patients from an endemic region strongly suggests a diagnosis of VL; hypergammaglobulinemia in patients with long-standing illness strengthens the diagnosis. In nonendemic countries, a careful travel history is essential when any patient presents with fever.
POST–KALA-AZAR DERMAL LEISHMANIASIS
On the Indian subcontinent and in Sudan and other East African countries, 2–50% of patients develop skin lesions concurrent with or after the cure of VL. Most common are hypopigmented macules, papules, and/or nodules or diffuse infiltration of the skin and sometimes of the oral mucosa. The African and Indian diseases differ in several respects; important features of post–kala-azar dermal leishmaniasis (PKDL) in these two regions are listed in Table 251-2, and disease in an Indian patient is depicted in Fig. 251-4.
|
CLINICAL, EPIDEMIOLOGIC, AND THERAPEUTIC FEATURES OF POST–KALA-AZAR DERMAL LEISHMANIASIS: EAST AFRICA AND THE INDIAN SUBCONTINENT |
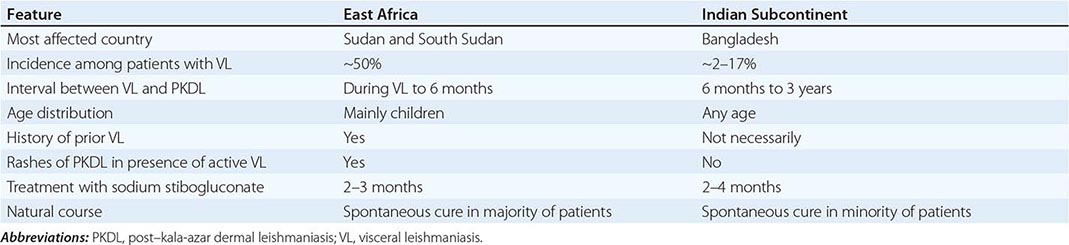
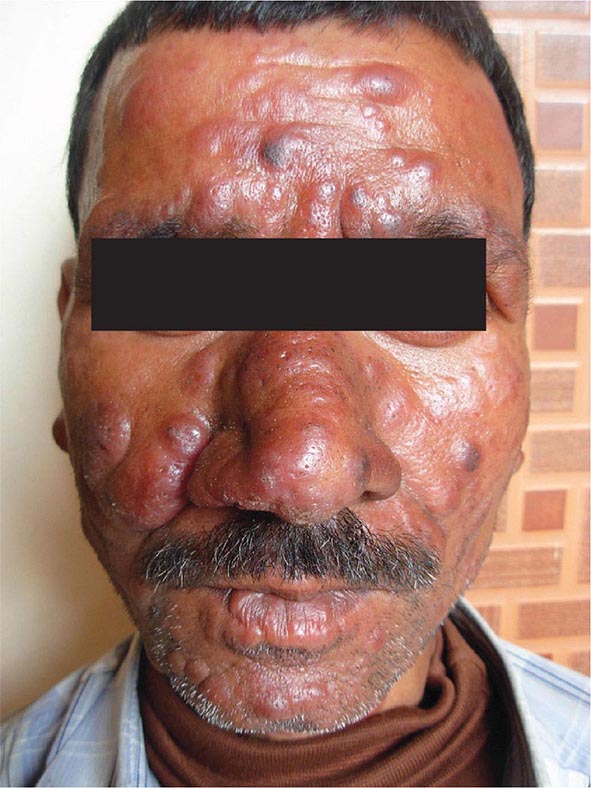
FIGURE 251-4 Post–kala-azar dermal leishmaniasis in an Indian patient. Note nodules of varying size involving the entire face. The face is erythematous, and the surface of some of the large nodules is discolored.
In PKDL, parasites are scanty in hypopigmented macules but may be seen and cultured more easily from nodular lesions. Cellular infiltrates are heavier in nodules than in macules. Lymphocytes are the dominant cells; next most common are histiocytes and plasma cells. In about half of cases, epithelioid cells—scattered individually or forming compact granulomas—are seen. The diagnosis is based on history and clinical findings, but rK39 and other serologic tests are positive in most cases. Indian PKDL is treated with pentavalent antimonials for 60–120 days. This prolonged course frequently leads to noncompliance. The alternative—several courses of AmB spread over several months—is expensive and unacceptable for most patients. Oral miltefosine for 12 weeks, in the usual daily doses, cures most patients with Indian PKDL. In East Africa, a majority of patients experience spontaneous healing. In those with persistent lesions, the response to 60 days of treatment with a pentavalent antimonial is good.
CUTANEOUS LEISHMANIASIS
CL can be broadly divided into Old World and New World forms. Old World CL caused by Leishmania tropica is anthroponotic and is confined to urban or suburban areas throughout its range. Zoonotic CL is most commonly due to Leishmania major, which naturally parasitizes several species of desert rodents that act as reservoirs over wide areas of the Middle East, Africa, and central Asia. Local outbreaks of human disease are common. Major outbreaks currently affect Afghanistan and Pakistan in association with refugees and population movement. CL is increasingly seen in tourists and military personnel on mission in CL-endemic regions of countries like Afghanistan and as a co-infection in HIV-infected patients. Leishmania aethiopica is restricted to the highlands of Ethiopia, Kenya, and Uganda, where it is a natural parasite of hyraxes. New World CL is mainly zoonotic and is most often caused by Leishmania mexicana, Leishmania (Viannia) panamensis, and Leishmania amazonensis. A wide range of forest animals act as reservoirs, and human infections with these species are predominantly rural. As a result of extensive urbanization and deforestation, Leishmania (Viannia) braziliensis has adapted to peridomestic and urban animals, and CL due to this organism is increasingly becoming an urban disease. In the United States, a few cases of CL have been acquired indigenously in Texas.
Immunopathogenesis As in VL, the proinflammatory (TH1) response in CL may result in either asymptomatic or subclinical infection. However, in some individuals, the immune response causes ulcerative skin lesions, the majority of which heal spontaneously, leaving a scar. Healing is usually followed by immunity to reinfection with that species of parasite.
Clinical Features A few days or weeks after the bite of a sandfly, a papule develops and grows into a nodule that ulcerates over some weeks or months. The base of the ulcer, which is usually painless, consists of necrotic tissue and crusted serum, but secondary bacterial infection sometimes occurs. The margins of the ulcer are raised and indurated. Lesions may be single or multiple and vary in size from 0.5 to >3 cm (Fig. 251-5). Lymphatic spread and lymph gland involvement may be palpable and may precede the appearance of the skin lesion. There may be satellite lesions, especially in L. major and L. tropica infections. The lesions usually heal spontaneously after 2–15 months. Lesions due to L. major and L. mexicana tend to heal rapidly, whereas those due to L. tropica and parasites of subspecies Viannia heal more slowly. In CL caused by L. tropica, new lesions—usually scaly, erythematous papules and nodules—develop in the center or periphery of a healed sore, a condition known as leishmaniasis recidivans. Lesions of L. mexicana and Leishmania (Viannia) peruviana closely resemble those seen in the Old World; however, lesions on the pinna of the ear are common, chronic, and destructive in the former infections. L. mexicana is responsible for chiclero’s ulcer, the so-called self-healing sore of Mexico. CL lesions on exposed body parts (e.g., the face and hands), permanent scar formation, and social stigmatization may cause anxiety and depression and may affect the quality of life of CL patients.
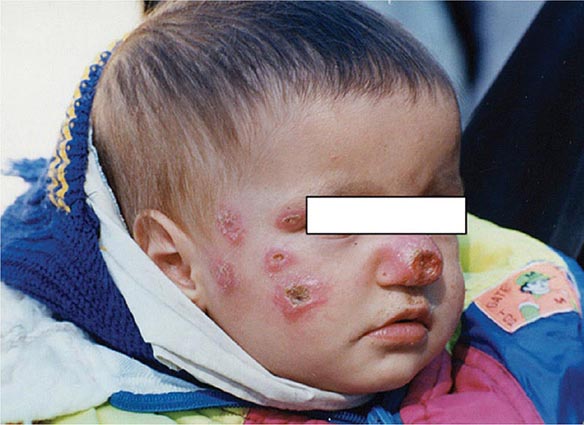
FIGURE 251-5 Cutaneous leishmaniasis in a Bolivian child. There are multiple ulcers resulting from several sandfly bites. The edges of the ulcers are raised. (Courtesy of P. Desjeux, Retired Medical Officer, World Health Organization, Geneva, Switzerland.)
Differential Diagnosis A typical history (an insect bite followed by the events leading to ulceration) in a resident of or a traveler to an endemic focus strongly suggests CL. Cutaneous tuberculosis, fungal infections, leprosy, sarcoidosis, and malignant ulcers are sometimes mistaken for CL.
Laboratory Diagnosis Demonstration of amastigotes in material obtained from a lesion remains the diagnostic gold standard. Microscopic examination of slit skin smears, aspirates, or biopsies of the lesion is used for detection of parasites. Culture of smear or biopsy material may yield Leishmania. PCR is more sensitive than microscopy and culture and allows identification of Leishmania to the species level. This information is important in decisions about therapy because responses to treatment can vary with the species. Isoenzyme profiling is used to determine species for research purposes.
Diffuse Cutaneous Leishmaniasis (DCL) DCL is a rare form of leishmaniasis caused by L. amazonensis and L. mexicana in South and Central America and by L. aethiopica in Ethiopia and Kenya. DCL is characterized by the lack of a cell-mediated immune response to the parasite, the uncontrolled multiplication of which thus continues unabated. The DTH response does not develop, and lymphocytes do not respond to leishmanial antigens in vitro. DCL patients have a polarized immune response with high levels of immunosuppressive cytokines, including IL-10, transforming growth factor (TGF) β, and IL-4, and low concentrations of IFN-γ. Profound immunosuppression leads to widespread cutaneous disease. Lesions may initially be confined to the face or a limb but spread over months or years to other areas of the skin. They may be symmetrically or asymmetrically distributed and include papules, nodules, plaques, and areas of diffuse infiltration. These lesions do not ulcerate. The overlying skin is usually erythematous in pale-skinned patients. The lesions are teeming with parasites, which are therefore easy to recover. DCL does not heal spontaneously and is difficult to treat. If relapse and drug resistance are to be prevented, treatment should be continued for some time after lesions have healed and parasites can no longer be isolated. In the New World, repeated 20-day courses of pentavalent antimonials are given, with an intervening drug-free period of 10 days. Miltefosine has been used for several months with a good initial response. Combinations should be tried. In Ethiopia, a combination of paromomycin (14 mg/kg per day) and sodium stibogluconate (10 mg/kg per day) is effective.
MUCOSAL LEISHMANIASIS
The subgenus Viannia is widespread from the Amazon basin to Paraguay and Costa Rica and is responsible for deep sores and for ML (Table 251-1). In L. (V.) braziliensis infections, cutaneous lesions may be simultaneously accompanied by mucosal spread of the disease or followed by spread years later. ML is typically caused by L. (V.) braziliensis and rarely by L. amazonensis, L. (V.) guyanensis, and L. (V.) panamensis. Young men with chronic lesions of CL are at particular risk. Overall, ~3% of infected persons develop ML. Not every patient with ML has a history of prior CL. ML is almost entirely confined to the Americas. In rare cases, ML may also be caused by Old World species like L. major, L. infantum (L. chagasi), or L. donovani.
Immunopathogenesis and Clinical Features The immune response is polarized toward a TH1 response, with marked increases of IFN-γ and TNF-α and varying levels of TH2 cytokines (IL-10 and TGF-β). Patients have a stronger DTH response with ML than with CL, and their peripheral-blood mononuclear cells respond strongly to leishmanial antigens. The parasite spreads via the lymphatics or the bloodstream to mucosal tissues of the upper respiratory tract. Intense inflammation leads to destruction, and severe disability ensues. Lesions in or around the nose or mouth (espundia; Fig. 251-6) are the typical presentation of ML. Patients usually provide a history of self-healed CL preceding ML by 1–5 years. Typically, ML presents as nasal stuffiness and bleeding followed by destruction of nasal cartilage, perforation of the nasal septum, and collapse of the nasal bridge. Subsequent involvement of the pharynx and larynx leads to difficulty in swallowing and phonation. The lips, cheeks, and soft palate may also be affected. Secondary bacterial infection is common, and aspiration pneumonia may be fatal. Despite the high degree of TH1 immunity and the strong DTH response, ML does not heal spontaneously.
FIGURE 251-6 Mucosal leishmaniasis in a Brazilian patient. There is extensive inflammation around the nose and mouth, destruction of the nasal mucosa, ulceration of the upper lip and nose, and destruction of the nasal septum. (Courtesy of R. Dietz, Universidade Federal do Espírito Santo, Vitória, Brazil.)
Laboratory Diagnosis Tissue biopsy is essential for identification of parasites, but the rate of detection is poor unless PCR techniques are used. The strongly positive DTH response fails to distinguish between past and present infection.
PREVENTION OF LEISHMANIASIS
No vaccine is available for any form of leishmaniasis. Inoculation with live L. major (“leishmanization”) is practiced in Iran. Anthroponotic leishmaniasis is controlled by case finding, treatment, and vector control with insecticide-impregnated bed nets and curtains and residual insecticide spraying. Control of zoonotic leishmaniasis is more difficult. Use of insecticide-impregnated collars for dogs, treatment of infected domestic dogs, and culling of street dogs are measures that have been used with uncertain efficacy to prevent transmission of L. infantum. In Brazil, a canine vaccine has been found to promote a decrease in the human and canine incidence of zoonotic VL. Two vaccines, Leishmune® and Leish-Tec®, are licensed in Brazil; Leishmune provides significant protection to vaccinated dogs. CaniLeish® is the first licensed canine vaccine developed in Europe. Personal prophylaxis with bed nets and repellants may reduce the risk of CL infections in the New World.
252 |
Chagas Disease and African Trypanosomiasis |
Although the genus Trypanosoma contains many species of protozoans, only T. cruzi, T. brucei gambiense, and T. brucei rhodesiense cause disease in humans. T. cruzi is the etiologic agent of Chagas disease in the Americas; T. b. gambiense and T. b. rhodesiense cause African trypanosomiasis.
CHAGAS DISEASE (AMERICAN TRYPANOSOMIASIS)
DEFINITION
Chagas disease, or American trypanosomiasis, is a zoonosis caused by the protozoan parasite T. cruzi. Acute Chagas disease is usually a mild febrile illness that results from initial infection with the organism. After spontaneous resolution of the acute illness, most infected persons remain for life in the indeterminate phase of chronic Chagas disease, which is characterized by subpatent parasitemia, easily detectable IgG antibodies to T. cruzi, and an absence of associated signs and symptoms. In 10–30% of chronically infected patients, cardiac and/or gastrointestinal symptoms develop that can lead to serious morbidity and even death.
LIFE CYCLE AND TRANSMISSION
T. cruzi is transmitted among its mammalian hosts by hematophagous triatomine insects, often called reduviid bugs. The insects become infected by sucking blood from animals or humans with circulating parasites. Ingested organisms multiply in the gut of the triatomines, and infective forms are discharged with the feces at the time of subsequent blood meals. Transmission to a second vertebrate host occurs when breaks in the skin, mucous membranes, or conjunctivae become contaminated with bug feces that contain infective parasites. T. cruzi can also be transmitted by transfusion of blood donated by infected persons, by organ transplantation, from mother to unborn child, by ingestion of contaminated food or drink, and in laboratory accidents.
PATHOLOGY
Initial infection at the site of parasite entry is characterized by local histologic changes that include the presence of parasites within leukocytes and cells of subcutaneous tissues and the development of interstitial edema, lymphocytic infiltration, and reactive hyperplasia of adjacent lymph nodes. After dissemination of the organisms through the lymphatics and the bloodstream, primarily muscles (including the myocardium) (Fig. 252-1) and ganglion cells may become heavily parasitized. The characteristic pseudocysts present in sections of infected tissues are intracellular aggregates of multiplying parasites.
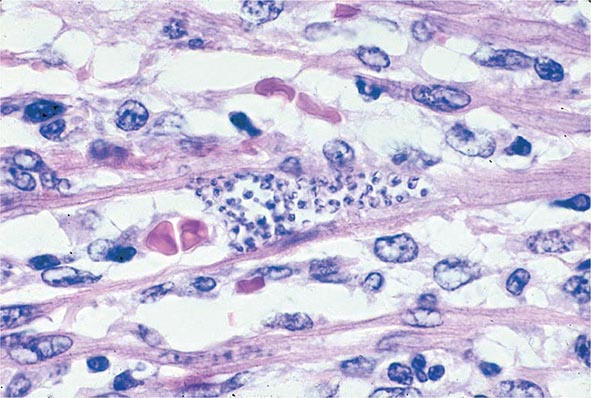
FIGURE 252-1 Trypanosoma cruzi in the heart muscle of a child who died of acute Chagas myocarditis. An infected myocyte containing several dozen T. cruzi amastigotes is in the center of the field (hematoxylin and eosin, 900 ×).
In persons with chronic T. cruzi infections who develop related clinical manifestations, the heart is the organ most commonly affected. Changes include thinning of the ventricular walls, biventricular enlargement, apical aneurysms, and mural thrombi. Widespread lymphocytic infiltration, diffuse interstitial fibrosis, and atrophy of myocardial cells are often apparent. Although parasites are difficult to find in myocardial tissue by conventional histologic methods, more sensitive techniques of parasite detection, such as immunohistochemistry and polymerase chain reaction (PCR), have more frequently demonstrated T. cruzi antigens and parasite DNA in chronic lesions. Conduction-system abnormalities often affect the right branch and the left anterior branch of the bundle of His. In chronic Chagas disease of the gastrointestinal tract (megadisease), the esophagus and colon may exhibit varying degrees of dilation. On microscopic examination, focal inflammatory lesions with lymphocytic infiltration are seen, and the number of neurons in the myenteric plexus may be markedly reduced. Accumulating evidence implicates the persistence of parasites and the accompanying chronic inflammation—rather than autoimmune mechanisms—as the basis for the pathology in patients with chronic T. cruzi infection.
EPIDEMIOLOGY
![]() T. cruzi is found only in the Americas. Wild and domestic mammals harboring T. cruzi and infected triatomines are found in spotty distributions from the southern United States to southern Argentina. Humans become involved in the cycle of transmission when infected vectors take up residence in the primitive wood, adobe, and stone houses common in much of Latin America. Thus human T. cruzi infection is a health problem primarily among the poor in rural areas of Mexico and Central and South America. Most new T. cruzi infections in rural settings occur in children, but the incidence is unknown because most cases go undiagnosed. Historically, transfusion-associated transmission of T. cruzi was a serious public health problem in many endemic countries. Transmission by this route has been largely eliminated, however, as effective programs for serologic screening of donated blood have been implemented. Several dozen patients with HIV and chronic T. cruzi infections who underwent acute recrudescence of the latter have been described. These patients generally presented with T. cruzi brain abscesses, a manifestation of the illness that does not occur in immunocompetent persons. Currently, it is estimated that 8 million people are chronically infected with T. cruzi and that 14,000 deaths due to the illness occur each year. The resulting morbidity and mortality make Chagas disease the most important parasitic disease burden in Latin America.
T. cruzi is found only in the Americas. Wild and domestic mammals harboring T. cruzi and infected triatomines are found in spotty distributions from the southern United States to southern Argentina. Humans become involved in the cycle of transmission when infected vectors take up residence in the primitive wood, adobe, and stone houses common in much of Latin America. Thus human T. cruzi infection is a health problem primarily among the poor in rural areas of Mexico and Central and South America. Most new T. cruzi infections in rural settings occur in children, but the incidence is unknown because most cases go undiagnosed. Historically, transfusion-associated transmission of T. cruzi was a serious public health problem in many endemic countries. Transmission by this route has been largely eliminated, however, as effective programs for serologic screening of donated blood have been implemented. Several dozen patients with HIV and chronic T. cruzi infections who underwent acute recrudescence of the latter have been described. These patients generally presented with T. cruzi brain abscesses, a manifestation of the illness that does not occur in immunocompetent persons. Currently, it is estimated that 8 million people are chronically infected with T. cruzi and that 14,000 deaths due to the illness occur each year. The resulting morbidity and mortality make Chagas disease the most important parasitic disease burden in Latin America.
In recent years, the rate of T. cruzi transmission has decreased markedly in several endemic countries as a result of successful programs involving vector control, screening of donated blood, and education of at-risk populations. A major program, which began in 1991 in the “southern cone” nations of South America (Uruguay, Paraguay, Bolivia, Brazil, Chile, and Argentina), has provided the framework for much of this progress. Uruguay and Chile were certified free of transmission by the main domiciliary vector species (Triatoma infestans) in the late 1990s, and Brazil was declared transmission-free in 2006. Transmission has been reduced markedly in Argentina as well. Similar control programs have been initiated in the countries of northern South America and in the Central American nations.
Acute Chagas disease is rare in the United States, where 22 cases of autochthonous transmission and seven instances of transmission by blood transfusion have been reported. Moreover, T. cruzi was transmitted to five recipients of organs from three T. cruzi–infected donors, two of whom became infected through cardiac transplants. Acute Chagas disease has been reported in only one tourist returning to the United States from Latin America, although three such instances have been reported in Europe as well as one in Canada. In contrast, the prevalence of chronic T. cruzi infections in the United States has increased considerably in recent years. An estimated 23 million immigrants from Chagas-endemic countries currently live in the United States, ~17 million of whom are Mexicans. The total number of T. cruzi–infected persons living in the United States is estimated to be 300,000. Screening of the U.S. blood supply for T. cruzi infection began in January 2007. The overall prevalence of T. cruzi infection among donors is ~1 in 13,300, and to date nearly 3000 infected donors have been identified and deferred permanently (see “Diagnosis,” below).
CLINICAL COURSE
The first signs of acute Chagas disease develop at least 1 week after invasion by the parasites. When the organisms enter through a break in the skin, an indurated area of erythema and swelling (the chagoma), accompanied by local lymphadenopathy, may appear. Romaña sign—the classic finding in acute Chagas disease, which consists of unilateral painless edema of the palpebrae and periocular tissues—can result when the conjunctiva is the portal of entry. These initial local signs may be followed by malaise, fever, anorexia, and edema of the face and lower extremities. Generalized lymphadenopathy and hepatosplenomegaly may develop. Severe myocarditis develops rarely; most deaths in acute Chagas disease are due to heart failure. Neurologic signs are not common, but meningoencephalitis occurs occasionally, especially in children <2 years old. Usually within 4–8 weeks, acute signs and symptoms resolve spontaneously in virtually all patients, with commencement of the asymptomatic or indeterminate form of chronic T. cruzi infection.
Symptomatic chronic Chagas disease becomes apparent years or even decades after the initial infection. The heart is commonly involved, and symptoms are caused by rhythm disturbances, segmental or dilated cardiomyopathy, and thromboembolism. Right bundle branch block is a common electrocardiographic abnormality, but other types of intraventricular and atrioventricular blocks, premature ventricular contractions, and tachy- and bradyarrhythmias occur frequently. Cardiomyopathy often results in biventricular heart failure, with a predominance of right-sided failure at advanced stages. Embolization of mural thrombi to the brain or other areas may take place. Sudden death is the main cause of death in Chagas heart disease; congestive heart failure and stroke are next most common. Patients with megaesophagus suffer from dysphagia, odynophagia, chest pain, and regurgitation. Aspiration can occur (especially during sleep) in patients with severe esophageal dysfunction, and repeated episodes of aspiration pneumonitis are common. Weight loss, cachexia, and pulmonary infection can result in death. Patients with megacolon are plagued by abdominal pain and chronic constipation, which predisposes to fecaloma formation. Advanced megacolon can cause obstruction, volvulus, septicemia, and death.
DIAGNOSIS
The diagnosis of acute Chagas disease requires the detection of parasites. Microscopic examination of fresh anticoagulated blood or the buffy coat is the simplest way to see the motile organisms. Parasites also can be seen in Giemsa-stained thin and thick blood smears. Microhematocrit tubes containing acridine orange as a stain can be used for the same purpose. When used repeatedly by experienced personnel, all of these methods yield positive results in a high proportion of cases of acute Chagas disease. Serologic testing does not play a major role in diagnosing acute Chagas disease. PCR assays often give positive results in infected patients in whom traditional parasitologic tests are negative, including infants with congenital Chagas disease.
Chronic Chagas disease is diagnosed by the detection of specific IgG antibodies that bind to T. cruzi antigens. Demonstration of the parasite is not of primary importance. In Latin America, ~30 assays are commercially available, including several based on recombinant antigens. Although these tests usually show good sensitivity and reasonable specificity, false-positive reactions may occur—typically with samples from patients who have other infectious and parasitic diseases or autoimmune disorders. In addition, confirmatory testing has presented a persistent challenge. For these reasons, the World Health Organization recommends that specimens be tested in at least two assays and that well-characterized positive and negative comparison samples be included in each run. The Chagas radioimmune precipitation assay (RIPA), a highly sensitive and specific confirmatory method for detecting antibodies to T. cruzi, is approved under the Clinical Laboratory Improvement Amendment and available in the laboratory of one of the authors (L.V.K.). In 2006, the U.S. Food and Drug Administration (FDA) approved a test to screen blood and organ donors for T. cruzi infection (Ortho T. cruzi ELISA Test System; Ortho-Clinical Diagnostics, Raritan, NJ). Since January 2007, the vast majority of U.S. blood donors have been screened, and positive units have undergone confirmatory testing with the Chagas RIPA. A second test for donor screening was approved by the FDA in 2010 (Abbott PRISM® Chagas Assay; Abbott Laboratories, Abbott Park, IL), as was an enzyme strip assay (Abbott ESA Chagas) in 2011. The use of PCR assays to detect T. cruzi DNA in chronically infected persons has been studied extensively; unfortunately, the sensitivity of this approach has not been shown to be reliably greater than that of serology.
|
TREATMENT |
CHAGAS DISEASE |
Therapy for Chagas disease is still unsatisfactory. For many years now, only two drugs—nifurtimox and benznidazole—have been available for this purpose. Regrettably, both drugs lack efficacy and may cause bothersome side effects.
In acute Chagas disease, nifurtimox markedly reduces the duration of symptoms and parasitemia and decreases the mortality rate. Nevertheless, limited studies have shown that only ~70% of acute infections are cured by a full course of treatment. Common adverse effects of nifurtimox include anorexia, nausea, vomiting, weight loss, and abdominal pain. Neurologic reactions to the drug may include restlessness, disorientation, insomnia, twitching, paresthesia, polyneuritis, and seizures. These symptoms usually disappear when the dosage is reduced or treatment is discontinued. The recommended daily dosage is 8–10 mg/kg for adults, 12.5–15 mg/kg for adolescents, and 15–20 mg/kg for children 1–10 years of age. The drug should be given orally in four divided doses each day, and therapy should be continued for 90–120 days. Nifurtimox is available from the Drug Service of the Centers for Disease Control and Prevention (CDC) in Atlanta (telephone number, 404-639-3670).
The efficacy of benznidazole is similar or even superior to that of nifurtimox. A cure rate of >90% among congenitally infected infants treated before their first birthday has been reported. Adverse effects include rash, peripheral neuropathy, and rare instances of granulocytopenia. The recommended oral dosage is 5 mg/kg per day for 60 days for adults and 5–10 mg/kg per day for 60 days for children, with administration of two or three divided doses. Benznidazole is generally considered the drug of choice in Latin America.
![]() The question of whether adults in the indeterminate or chronic symptomatic phase of Chagas disease should be treated with nifurtimox or benznidazole has been debated for years. The fact that cure rates in persons with long-established chronic infection are notably inferior to those in patients with acute or recent chronic infection is central to this controversy. No convincing evidence from randomized controlled trials indicates that nifurtimox or benznidazole treatment of adults in the indeterminate or chronic symptomatic phase reduces either the appearance or progression of symptoms or mortality rates. On the basis of results of some observational studies, a panel of experts convened by the CDC in 2006 recommended that adults <50 years old with presumably long-standing indeterminate T. cruzi infections—or even with mild to moderate disease—be offered treatment. A large randomized clinical trial (the BENEFIT multicenter trial) designed to assess the parasitologic and clinical efficacy of benznidazole in 2856 adults (18–75 years old) with chronic Chagas heart disease (without advanced lesions) is being performed in Brazil, Argentina, Colombia, Bolivia, and El Salvador, but results will not be available until 2015. In contrast, randomized studies have shown that treatment of children is useful, and the current consensus of Latin American authorities and the CDC panel of experts is that all T. cruzi–infected persons up to 18 years old and all adults known to have become infected recently should be given benznidazole or nifurtimox.
The question of whether adults in the indeterminate or chronic symptomatic phase of Chagas disease should be treated with nifurtimox or benznidazole has been debated for years. The fact that cure rates in persons with long-established chronic infection are notably inferior to those in patients with acute or recent chronic infection is central to this controversy. No convincing evidence from randomized controlled trials indicates that nifurtimox or benznidazole treatment of adults in the indeterminate or chronic symptomatic phase reduces either the appearance or progression of symptoms or mortality rates. On the basis of results of some observational studies, a panel of experts convened by the CDC in 2006 recommended that adults <50 years old with presumably long-standing indeterminate T. cruzi infections—or even with mild to moderate disease—be offered treatment. A large randomized clinical trial (the BENEFIT multicenter trial) designed to assess the parasitologic and clinical efficacy of benznidazole in 2856 adults (18–75 years old) with chronic Chagas heart disease (without advanced lesions) is being performed in Brazil, Argentina, Colombia, Bolivia, and El Salvador, but results will not be available until 2015. In contrast, randomized studies have shown that treatment of children is useful, and the current consensus of Latin American authorities and the CDC panel of experts is that all T. cruzi–infected persons up to 18 years old and all adults known to have become infected recently should be given benznidazole or nifurtimox.
The usefulness of antifungal azoles for the treatment of Chagas disease has been studied in laboratory animals and to a lesser extent in humans. To date, none of these drugs has exhibited a level of anti–T. cruzi activity that would justify its use in humans. Several newer drugs in this class have shown promise in animal studies and are currently being evaluated in phase 2 clinical trials.
Patients who develop cardiac and/or gastrointestinal disease in association with T. cruzi infection should be referred to appropriate subspecialists for further evaluation and treatment. Pacemakers can be useful in patients with ominous arrhythmias. The usefulness of implantable cardioverter defibrillators in persons with Chagas heart disease has not been established and currently is being studied in a prospective randomized trial. Cardiac transplantation is an option for patients with end-stage chagasic cardiomyopathy; more than 150 such transplantations have been done in Brazil and the United States. The survival rate among Chagas disease cardiac transplant recipients seems to be higher than that among persons receiving cardiac transplants for other reasons. This better outcome may be due to the fact that lesions are limited to the heart in most patients with symptomatic chronic Chagas disease.
PREVENTION
![]() Because drug therapy has limitations and vaccines are not available, the control of T. cruzi transmission in endemic countries depends on the reduction of domiciliary vector populations by spraying of insecticides, improvements in housing, and education of at-risk persons. As noted above, these measures, coupled with serologic screening of blood donors, have markedly reduced transmission of the parasite in many endemic countries. Tourists would be wise to avoid sleeping in dilapidated houses in rural areas of endemic countries. Mosquito nets and insect repellent can provide additional protection.
Because drug therapy has limitations and vaccines are not available, the control of T. cruzi transmission in endemic countries depends on the reduction of domiciliary vector populations by spraying of insecticides, improvements in housing, and education of at-risk persons. As noted above, these measures, coupled with serologic screening of blood donors, have markedly reduced transmission of the parasite in many endemic countries. Tourists would be wise to avoid sleeping in dilapidated houses in rural areas of endemic countries. Mosquito nets and insect repellent can provide additional protection.
In view of the possibly serious consequences of chronic T. cruzi infection, it would be prudent for all immigrants from endemic regions who are living in the United States to be tested for evidence of infection. Identification of persons harboring the parasite would permit periodic electrocardiographic monitoring, which is important to detect incipient heart disease and guide further diagnostic studies and treatment. The possibility of congenital transmission is yet another justification for screening. T. cruzi is classified as a Risk Group 2 agent in the United States and a Risk Group 3 agent in some European countries. Laboratory staff should work with the parasite or infected vectors and mammals at containment levels consistent with the risk group designation in their areas.
SLEEPING SICKNESS (AFRICAN TRYPANOSOMIASIS)
DEFINITION
Sleeping sickness, or human African trypanosomiasis (HAT), is caused by flagellated protozoan parasites that belong to the T. brucei complex and are transmitted to humans by tsetse flies. In untreated patients, the trypanosomes first cause a febrile illness that is followed months or years later by progressive neurologic impairment and death.
THE PARASITES AND THEIR TRANSMISSION
The East African (rhodesiense) and the West African (gambiense) forms of sleeping sickness are caused, respectively, by two trypanosome subspecies: T. b. rhodesiense and T. b. gambiense. These subspecies are morphologically indistinguishable but cause illnesses that are epidemiologically and clinically distinct (Table 252-1). The parasites are transmitted by blood-sucking tsetse flies of the genus Glossina. The insects acquire the infection when they ingest blood from infected mammalian hosts. After many cycles of multiplication in the midgut of the vector, the parasites migrate to the salivary glands. Their transmission takes place when they are inoculated into a mammalian host during a subsequent blood meal. The injected trypanosomes multiply in the blood (Fig. 252-2) and other extracellular spaces and evade immune destruction for long periods by undergoing antigenic variation, a process driven by gene switching in which the antigenic structure of the organisms’ surface coat of glycoproteins changes periodically.
|
COMPARISON OF WEST AFRICAN AND EAST AFRICAN TRYPANOSOMIASES |
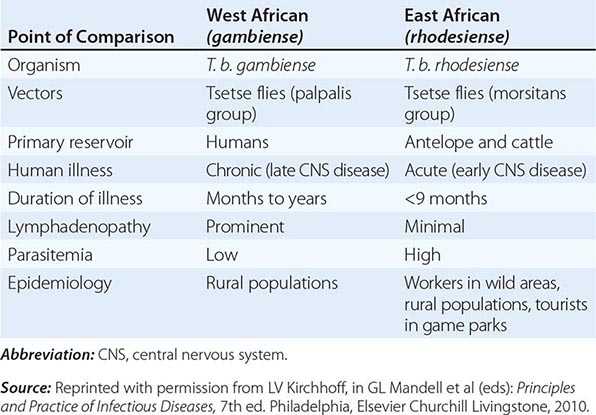
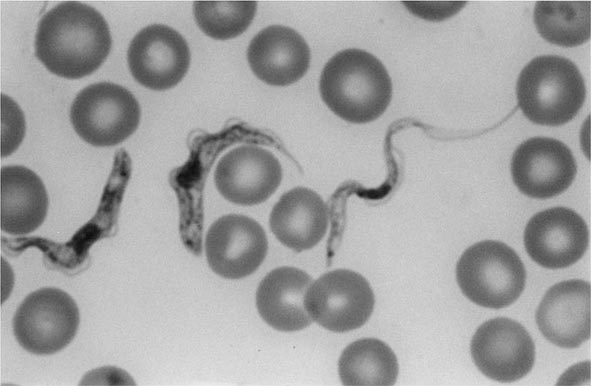
FIGURE 252-2 Trypanosoma brucei rhodesiense parasites in rat blood. The slender parasite is thought to be the form that multiplies in mammalian hosts, whereas the stumpy forms are nondividing and are capable of infecting insect vectors (Giemsa, 1200 ×). (Courtesy of Dr. G. A. Cook, Madison, WI; with permission.)
PATHOGENESIS AND PATHOLOGY
A self-limited inflammatory lesion (trypanosomal chancre) may appear a week or so after the bite of an infected tsetse fly. A systemic febrile illness then evolves as the parasites are disseminated through the lymphatics and bloodstream. Systemic HAT without central nervous system (CNS) involvement is generally referred to as stage 1 disease. In this stage, widespread lymphadenopathy and splenomegaly reflect marked lymphocytic and histiocytic proliferation and invasion of morular cells, which are plasmacytes that may be involved in the production of IgM. Endarteritis, with perivascular infiltration of both parasites and lymphocytes, may develop in lymph nodes and the spleen. Myocarditis develops frequently in patients with stage 1 disease and is especially common in T. b. rhodesiense infections.
Hematologic manifestations that accompany stage 1 HAT include moderate leukocytosis, thrombocytopenia, and anemia. High levels of immunoglobulins, consisting primarily of polyclonal IgM, are a constant feature, and heterophile antibodies, antibodies to DNA, and rheumatoid factor are often detected. High levels of antigen–antibody complexes may play a role in the tissue damage and increased vascular permeability that facilitate dissemination of the parasites.
Stage 2 disease involves invasion of the CNS. The presence of trypanosomes in perivascular areas is accompanied by intense infiltration of mononuclear cells. Abnormalities in cerebrospinal fluid (CSF) include increased pressure, elevated total protein concentration, and pleocytosis. In addition, trypanosomes are frequently found in CSF.
EPIDEMIOLOGY
![]() The trypanosomes that cause sleeping sickness are found only in sub-Saharan Africa. After its near-eradication in the mid-1960s, sleeping sickness underwent a resurgence in the 1990s, primarily in Uganda, Sudan, the Central African Republic, the Democratic Republic of the Congo, and Angola. A subsequent increase in control activities reduced the incidence in many endemic areas, however, and in 2009 fewer than 10,000 cases were reported to the World Health Organization. Although underreporting is a persistent problem, the level of control achieved to date was the basis for convening a panel of experts in 2009 to develop a vision for eradication of HAT.
The trypanosomes that cause sleeping sickness are found only in sub-Saharan Africa. After its near-eradication in the mid-1960s, sleeping sickness underwent a resurgence in the 1990s, primarily in Uganda, Sudan, the Central African Republic, the Democratic Republic of the Congo, and Angola. A subsequent increase in control activities reduced the incidence in many endemic areas, however, and in 2009 fewer than 10,000 cases were reported to the World Health Organization. Although underreporting is a persistent problem, the level of control achieved to date was the basis for convening a panel of experts in 2009 to develop a vision for eradication of HAT.
Humans are the only reservoir of T. b. gambiense, which occurs in widely distributed foci in tropical rain forests of Central and West Africa. Gambiense trypanosomiasis is primarily a problem in rural populations; tourists rarely become infected. Trypanotolerant antelope species in savanna and woodland areas of Central and East Africa are the principal reservoir of T. b. rhodesiense. Cattle can also be infected with this and other trypanosome species but generally succumb to the infection. Because risk results from contact with tsetse flies that feed on wild animals, humans acquire T. b. rhodesiense infection only incidentally, usually while visiting or working in areas where infected game and vectors are present. Roughly one or two imported cases of HAT acquired in East African parks are reported to the CDC each year.
CLINICAL COURSE
A painful trypanosomal chancre appears in some patients at the site of inoculation of the parasite. Hematogenous and lymphatic dissemination (stage 1 disease) is marked by the onset of fever. Typically, bouts of high temperatures lasting several days are separated by afebrile periods. Lymphadenopathy is prominent in T. b. gambiense trypanosomiasis. The nodes are discrete, movable, rubbery, and nontender. Cervical nodes are often visible, and enlargement of the nodes of the posterior cervical triangle, or Winterbottom’s sign, is a classic finding. Pruritus and maculopapular rashes are common. Inconstant findings include malaise, headache, arthralgias, weight loss, edema, hepatosplenomegaly, and tachycardia. The differential diagnosis of stage 1 HAT includes many diseases that are common in the tropics and are associated with fevers. HIV infection, malaria, and typhoid fever are common in populations at risk for HAT and need to be considered.
CNS invasion (stage 2 disease) is characterized by the insidious development of protean neurologic manifestations that are accompanied by progressive abnormalities in the CSF. A picture of progressive indifference and daytime somnolence develops (hence the designation “sleeping sickness”), sometimes alternating with restlessness and insomnia at night. A listless gaze accompanies a loss of spontaneity, and speech may become halting and indistinct. Extrapyramidal signs may include choreiform movements, tremors, and fasciculations. Ataxia is frequent, and the patient may appear to have Parkinson’s disease, with a shuffling gait, hypertonia, and tremors. In the final phase, progressive neurologic impairment ends in coma and death.
The most striking difference between the gambiense and rhodesiense forms of HAT is that the latter illness tends to follow a more acute course. Typically, in tourists with T. b. rhodesiense disease, systemic signs of infection, such as fever, malaise, and headache, appear before the end of the trip or shortly after the return home. Persistent tachycardia unrelated to fever is common early in the course of T. b. rhodesiense trypanosomiasis, and death may result from arrhythmias and congestive heart failure before CNS disease develops. In general, untreated T. b. rhodesiense trypanosomiasis leads to death in a matter of weeks to months, often without a clear distinction between the hemolymphatic and CNS stages. In contrast, T. b. gambiense disease can smolder for many months or even for years.
DIAGNOSIS
A definitive diagnosis of HAT requires detection of the parasite. If a chancre is present, fluid should be expressed and examined directly by light microscopy for the highly motile trypanosomes. The fluid also should be fixed and stained with Giemsa. Material obtained by needle aspiration of lymph nodes early in the illness should be examined similarly. Examination of wet preparations and Giemsa-stained thin and thick films of serial blood samples is also useful. If parasites are not seen initially in blood, efforts should be made to concentrate the organisms, which can be done in microhematocrit tubes containing acridine orange. Alternatively, the buffy coat from 10–15 mL of anticoagulated blood can be examined directly under a microscope. The likelihood of finding parasites in blood is higher in stage 1 than in stage 2 disease and in patients infected with T. b. rhodesiense rather than T. b. gambiense. Trypanosomes may also be seen in material aspirated from the bone marrow; the aspirate can be inoculated into liquid culture medium, as can blood, buffy coat, lymph node aspirates, and CSF. It is essential to examine CSF from all patients in whom HAT is suspected. Abnormalities in the CSF that may be associated with stage 2 disease include an increase in the CSF cell count as well as increases in opening pressure and in levels of total protein and IgM. Trypanosomes may be seen in the sediment of centrifuged CSF. Any CSF abnormality in a patient in whom trypanosomes have been found at other sites must be viewed as pathognomonic for CNS involvement and thus must prompt specific treatment for CNS disease. In patients with CSF pleocytosis in whom parasites are not found, tuberculous meningitis and HIV-associated CNS infections such as cryptococcosis should be considered in the differential diagnosis.
A number of serologic assays, such as the card agglutination test for trypanosomes (CATT) for T. b. gambiense, are available to aid in the diagnosis of HAT. Their ease of use makes them valuable for epidemiologic surveys, but their variable sensitivity and specificity mandate that decisions about treatment be based on demonstration of the parasite. Accurate PCR assays for detecting African trypanosomes in humans have been developed, but the lack of the necessary technical and human resources in most endemic areas stands in the way of their widespread use.
|
TREATMENT |
SLEEPING SICKNESS |
The drugs used for treatment of HAT are suramin, pentamidine, eflornithine, and the organic arsenical melarsoprol. In the United States, these drugs can be obtained from the CDC. Therapy for HAT must be individualized on the basis of the infecting subspecies, the presence or absence of CNS disease, adverse reactions, and occasionally drug resistance. The choices of drugs for the treatment of HAT are summarized in Table 252-2.
|
TREATMENT OF HUMAN AFRICAN TRYPANOSOMIASISa |
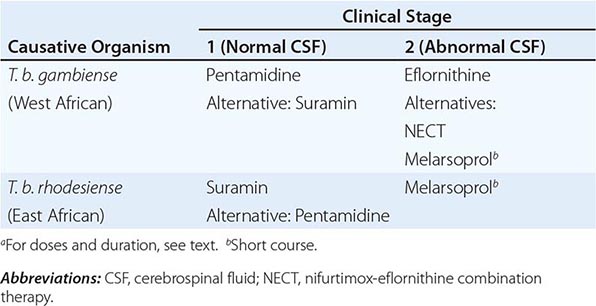
Suramin is highly effective against stage 1 rhodesiense HAT. However, it can cause serious adverse effects and must be administered under the close supervision of a physician. A 100- to 200-mg IV test dose should be given to detect hypersensitivity. The dosage for adults is 20 mg/kg on days 1, 5, 12, 18, and 26. The drug is given by slow IV infusion of a freshly prepared 10% aqueous solution. Approximately 1 patient in 20,000 has an immediate, severe, and potentially fatal reaction to the drug, developing nausea, vomiting, shock, and seizures. Less severe reactions include fever, photophobia, pruritus, arthralgias, and skin eruptions. Renal damage is the most common important adverse effect of suramin. Transient proteinuria often appears during treatment. A urinalysis should be done before each dose, and treatment should be discontinued if proteinuria increases or if casts and red cells appear in the sediment. Suramin should not be given to patients with renal insufficiency.
Pentamidine is the first-line drug for treatment of stage 1 gambiense HAT. The dose for both adults and children is 4 mg/kg per day, given IM or IV for 7–10 days. Frequent, immediate adverse reactions include nausea, vomiting, tachycardia, and hypotension. These reactions are usually transient and do not warrant cessation of therapy. Other adverse reactions include nephrotoxicity, abnormal liver function tests, neutropenia, rashes, hypoglycemia, and sterile abscesses. Suramin is an alternative agent for stage 1 T. b. gambiense disease.
Eflornithine is highly effective for treatment of both stages of gambiense sleeping sickness. In the trials on which the FDA based its approval, this agent cured >90% of 600 patients with stage 2 disease. The recommended treatment schedule is 400 mg/kg per day, given IV in four divided doses, for 2 weeks. Adverse reactions include diarrhea, anemia, thrombocytopenia, seizures, and hearing loss. The high dosage and duration of therapy required are disadvantages that make widespread use of eflornithine difficult. A randomized trial comparing the standard eflornithine regimen (400 mg/kg per day infused over 6 h for 14 days) with nifurtimox-eflornithine combination therapy (NECT; oral nifurtimox, 15 mg/kg per day in three divided doses, plus IV eflornithine, 200 mg/kg per day in two divided doses, both for 7 days) in adults with stage 2 gambiense HAT showed improved efficacy and reduced adverse effects with combination therapy, making this drug suitable for first-line use.
The arsenical melarsoprol is the drug of choice for the treatment of rhodesiense HAT with CNS involvement and is an alternative agent for stage 2 gambiense disease. The “short course” of melarsoprol that is currently recommended has been shown to be noninferior to the decades-old treatment course for T. b. rhodesiense, which was administered over several weeks and was much more toxic. The short-course regimen consists of 10 daily doses of 2.2 mg/kg IV, each given with prednisolone (1 mg/kg).
Melarsoprol is highly toxic and should be administered with great care. As noted, all patients receiving melarsoprol should be given prednisolone to reduce the likelihood of drug-induced encephalopathy. Without prednisolone prophylaxis, the incidence of reactive encephalopathy has been as high as 18% in some series. Clinical manifestations of reactive encephalopathy include high fever, headache, tremor, impaired speech, seizures, and even coma and death. Treatment with melarsoprol should be discontinued at the first sign of encephalopathy but may be restarted cautiously at lower doses a few days after signs have resolved. Extravasation of the drug results in intense local reactions. Vomiting, abdominal pain, nephrotoxicity, and myocardial damage can occur.
PREVENTION
HAT poses complex public-health and epizootic problems in Africa. Considerable progress has been made in many areas through control programs that focus on eradication of vectors and drug treatment of infected humans. People can reduce their risk of acquiring trypanosomiasis by avoiding areas known to harbor infected insects, by wearing protective clothing, and by using insect repellent. Chemoprophylaxis is not recommended, and no vaccine is available to prevent transmission of the parasites.
253 |
Toxoplasma Infections |
DEFINITION
Toxoplasmosis is caused by infection with the obligate intracellular parasite Toxoplasma gondii. Acute infection acquired after birth may be asymptomatic but is thought to result in the lifelong chronic persistence of cysts in the host’s tissues. In both acute and chronic toxoplasmosis, the parasite is responsible for clinically evident disease, including lymphadenopathy, encephalitis, myocarditis, and pneumonitis. Congenital toxoplasmosis is an infection of newborns that results from the transplacental passage of parasites from an infected mother to the fetus. These infants may be asymptomatic at birth, but most later manifest a wide range of signs and symptoms, including chorioretinitis, strabismus, epilepsy, and psychomotor retardation. In immunocompetent individuals, toxoplasmosis can also present as acute disease (typically chorioretinitis) associated with food- or waterborne sources.
ETIOLOGY
T. gondii is an intracellular coccidian that infects both birds and mammals. There are two distinct stages in the life cycle of T. gondii that yield transmissible forms of the parasite (Fig. 253-1). In the asexual stages, tissue cysts that contain bradyzoites or sporulated oocysts that contain sporozoites are ingested by an intermediate host (e.g., a human, mouse, sheep, pig, or bird). The cyst is rapidly digested by the acidic-pH gastric secretions. Bradyzoites or sporozoites are released, enter the small-intestinal epithelium, and transform into rapidly dividing tachyzoites. The tachyzoites can infect and replicate in all mammalian cells except red blood cells. The parasite actively penetrates the cell and forms a parasitophorous vacuole. Parasite replication continues within the vacuole. After the parasites reach a critical mass, intracellular signaling within the host and the parasite, including calcium fluxes, result in parasite egress from the vacuole. The host cell is destroyed, and the released tachyzoites infect adjoining cells. The tachyzoite replication cycle within an infected organ causes cytopathology. Most tachyzoites are eliminated by the host’s humoral and cell-mediated immune responses. Tissue cysts containing many bradyzoites develop 7–10 days after systemic tachyzoite infection. These tissue cysts occur in various host organs but persist principally within the central nervous system (CNS) and muscle. The development of this chronic stage completes the asexual portion of the life cycle. Active infection in the immunocompromised host is most likely to be due to the spontaneous release of encysted parasites that undergo rapid transformation into tachyzoites within the CNS and are not contained by the immune system.
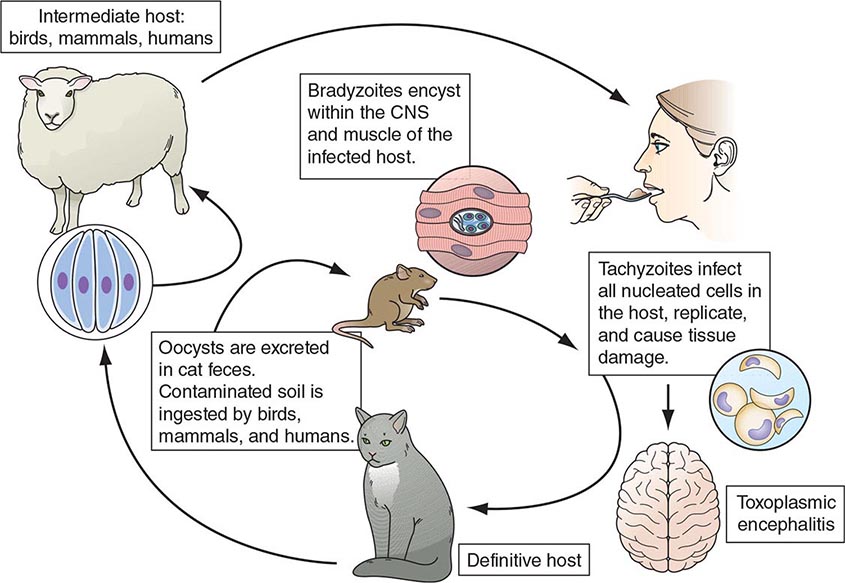
FIGURE 253-1 Life cycle of Toxoplasma gondii. The cat is the definitive host in which the sexual phase of the cycle is completed. Oocysts shed in cat feces can infect a wide range of animals, including birds, rodents, grazing domestic animals, and humans. The bradyzoites found in the muscle of food animals may infect humans who eat insufficiently cooked meat products, particularly lamb and pork. Although human disease can take many forms, congenital infection and encephalitis from reactivation of latent infection in the brains of immunosuppressed persons are the most important manifestations. CNS, central nervous system. (Courtesy of Dominique Buzoni-Gatel, Institut Pasteur, Paris; with permission.)
The sexual stage in the life cycle takes place in the cat (the definitive host). The parasite’s sexual phase is defined by the formation of oocysts within the feline host. This enteroepithelial cycle begins with the ingestion of the bradyzoite tissue cysts and, after several intermediate stages, culminates in the production of gametes. Gamete fusion produces a zygote, which envelops itself in a rigid wall and is secreted in the feces as an unsporulated oocyst. After 2–3 days of exposure to air at ambient temperature, the noninfectious oocyst sporulates to produce eight sporozoite progeny. The sporulated oocyst can be ingested by an intermediate host, such as a person emptying a cat’s litter box or a pig rummaging in a barnyard. It is in the intermediate host that T. gondii completes its life cycle.
![]() Sporulated oocysts are environmentally hardy and very infectious; they are thought to be sources of waterborne outbreaks such as those reported in Victoria (British Columbia, Canada) and in South America.
Sporulated oocysts are environmentally hardy and very infectious; they are thought to be sources of waterborne outbreaks such as those reported in Victoria (British Columbia, Canada) and in South America.
EPIDEMIOLOGY
![]() T. gondii infects a wide range of mammals and birds. Its sero-prevalence depends on the locale and the age of the population. Generally, hot arid climatic conditions are associated with a low prevalence of infection. In the United States and most European countries, the seroprevalence increases with age and exposure. For example, in the United States, 5–30% of individuals 10–19 years old and 10–67% of those >50 years old have serologic evidence of exposure. In Central America, France, Turkey, and Brazil, the seroprevalence is higher. Because of increased awareness of foodborne infections, the prevalence of seropositivity has decreased worldwide.
T. gondii infects a wide range of mammals and birds. Its sero-prevalence depends on the locale and the age of the population. Generally, hot arid climatic conditions are associated with a low prevalence of infection. In the United States and most European countries, the seroprevalence increases with age and exposure. For example, in the United States, 5–30% of individuals 10–19 years old and 10–67% of those >50 years old have serologic evidence of exposure. In Central America, France, Turkey, and Brazil, the seroprevalence is higher. Because of increased awareness of foodborne infections, the prevalence of seropositivity has decreased worldwide.
TRANSMISSION
Oral Transmission Most cases of human Toxoplasma infection are thought to be acquired by the oral route. Transmission can be attributable to ingestion of sporulated oocysts from contaminated soil, food, or water. During acute feline infection, a cat may excrete as many as 100 million parasites per day. These very stable sporozoite-containing oocysts are highly infectious and may remain viable for many years in soil or water. Humans infected during an oocyst-transmitted infection develop stage-specific antibodies to the oocyst/sporozoite.
Children and adults also can acquire infection from tissue cysts containing bradyzoites. The ingestion of a single cyst is all that is required for human infection. Undercooking or insufficient freezing of meat is an important source of infection in the developed world. In the United States, lamb products and pork products may yield evidence of cysts that contain bradyzoites, but the overall prevalence of T. gondii has been gradually decreasing. The incidence in beef is much lower—perhaps as low as 1%. Direct ingestion of bradyzoite cysts in these various meat products leads to acute infection.
Transmission via Blood or Organs In addition to being transmitted orally, T. gondii can be transmitted directly from a seropositive donor to a seronegative recipient in a transplanted heart, heart-lung, kidney, liver, or pancreas. Viable parasites can be cultured from refrigerated anticoagulated blood, which may be a source of infection in individuals receiving blood transfusions. T. gondii reactivation has been reported in bone marrow, hematopoietic stem cell, and liver transplant recipients as well as in individuals with AIDS. Although antibody titers generally are not useful in monitoring T. gondii infection, individuals with higher antibody titers may be at relatively high risk for reactivation after hematopoietic stem cell transplantation; thus routine polymerase chain reaction (PCR) screening of blood from these patients may be in order. Finally, laboratory personnel can be infected after contact with contaminated needles or glassware or with infected tissue.
Transplacental Transmission On average, about one-third of all women who acquire infection with T. gondii during pregnancy transmit the parasite to the fetus; the remainder give birth to normal, uninfected babies. Of the various factors that influence fetal outcome, gestational age at the time of infection is the most critical (see below). Few data support a role for recrudescent maternal infection as the source of congenital disease, although rare cases of transmission by immunocompromised women (e.g., those infected with HIV or those receiving high-dose glucocorticoids) have been reported. Thus, women who are seropositive before pregnancy usually are protected against acute infection and do not give birth to congenitally infected neonates.
The following general guidelines can be used to evaluate congenital infection. There is essentially no risk if the mother becomes infected ≥6 months before conception. If infection is acquired <6 months before conception, the likelihood of transplacental infection increases as the interval between infection and conception decreases. Women with documented acute toxoplasmosis should be counseled to use appropriate measures to prevent pregnancy for 6 months after infection. In pregnancy, if the mother becomes infected during the first trimester, the incidence of transplacental infection is lowest (~15%), but the disease in the neonate is most severe. If maternal infection occurs during the third trimester, the incidence of transplacental infection is greatest (65%), but the infant is usually asymptomatic at birth. Infected infants who are normal at birth may have a higher incidence of learning disabilities and chronic neurologic sequelae than uninfected children. Only a small proportion (20%) of women infected with T. gondii develop clinical signs of infection. Often the diagnosis is first appreciated when routine postconception serologic tests show evidence of specific antibody.
PATHOGENESIS
Upon the host’s ingestion of either tissue cysts containing bradyzoites or oocysts containing sporozoites, the parasites are released from the cysts by the digestive process. Bradyzoites are resistant to the effect of pepsin and invade the host’s gastrointestinal tract. Within enterocytes (or other gut-associated cells), the parasites undergo morphologic transformation, giving rise to invasive tachyzoites. These tachyzoites induce a parasite-specific secretory IgA response. From the gastrointestinal tract, parasites disseminate to a variety of organs, particularly lymphatic tissue, skeletal muscle, myocardium, retina, placenta, and the CNS. At these sites, the parasite infects host cells, replicates, and invades the adjoining cells. In this fashion, the hallmarks of the infection develop: cell death and focal necrosis surrounded by an acute inflammatory response.
In the immunocompetent host, both the humoral and the cellular immune responses control infection; parasite virulence and tissue tropism may be strain specific. Tachyzoites are sequestered by a variety of immune mechanisms, including induction of parasiticidal antibody, activation of macrophages with radical intermediates, production of interferon γ (IFN-γ), and stimulation of CD8+ cytotoxic T lymphocytes. These antigen-specific lymphocytes are capable of killing both extracellular parasites and target cells infected with parasites. As tachyzoites are cleared from the acutely infected host, tissue cysts containing bradyzoites begin to appear, usually within the CNS and the retina. Studies indicate that Toxoplasma secretes signaling molecules into infected host cells and that these molecules modulate host gene expression, host metabolism, and host immune response. While it was initially thought that cysts with bradyzoites are not eliminated by the immune system, recent studies in the murine model indicate that both CD8+ T cells and alternatively activated macrophages are able to kill cysts in vivo; some cysts persist, however, and the ability to eliminate cysts may depend on the genetic background of the infected host.




