18 Toxicology and Poisonings
Pearls
• The “ABCDE” primary assessment (Airway, Breathing, Circulation, Disability, Exposure) similar to that used to evaluate and stabilize trauma victims, is a useful tool to assess and stabilize poisoned patients.
• During stabilization of the patient with poisoning or overdose, general principles of pediatric advanced life support (PALS) apply. Focus should begin with support of airway, oxygenation, ventilation and circulation.
• Advanced life support for all poisoned patients includes meticulous attention to maintaining a patent airway and adequate oxygenation, ventilation, and circulation. Children with overdoses of some drugs may require modified resuscitation therapies or sequences.
• The critical care nurse should carefully analyze the ECG for changes that may be caused by tricyclic antidepressants (TCAs), calcium channel blockers, and beta (β)-blockers. Such changes include a widened QRS complex, prolonged corrected QT interval (QTc), bradycardia, sino-atrial (SA) and atrioventricular (AV) nodal conduction delays, ventricular tachycardia (VT), ventricular fibrillation (VF), and asystole.
• Adjustments in the bolus volume used for fluid resuscitation may be necessary for children who ingest drugs that affect myocardial contractility or drugs that may contribute to the development of noncardiogenic pulmonary edema. In these patients boluses of 5 to 10 or 10 to 15 mL/kg may be used instead of the traditional 20 mL/kg bolus. In general, fluid boluses can be administered over 5 to 20 minutes, but when myocardial contractility is compromised or pulmonary edema is present, the bolus is typically administered over about 10 to 20 minutes. Reassess the patient carefully between boluses, be prepared to support oxygenation and ventilation (with possible continuous positive airway pressure), and repeat the bolus as needed.
Scope of the problem
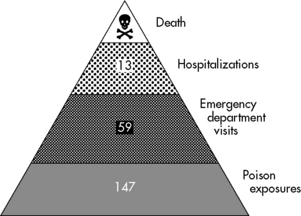
Poisonings and toxic exposures resulting in injury or death are significant problems for pediatric emergency and critical care. In 2009, approximately 1.6 million poisonings occurred in children 19 years of age or younger.26 From 1995 to 2005, poisoning accounted for 1.2 million emergency department visits.120 The average annual rate of poisoning-related visits was disproportionately higher among children under 5 years of age than among children in older age categories.107 Poisonings and drug overdoses are the most common toxicities that result in admission to pediatric critical care units.107 Figure 18-1 illustrates the burden of poisoning in the United States.
Toxic exposure can complicate resuscitation priorities and support. In unusual cases of poisoning or when life-threatening complications are anticipated, the American College of Emergency Physicians (ACEP) and American Academy of Clinical Toxicology (AACT) recommend consultation with a medical toxicologist or certified regional poison information center and transfer to a poison treatment center.2,7 Dedicated poison treatment centers can provide diagnostic and treatment services beyond those available in most hospitals. Poisoned children with life-threatening complications should ideally receive care at a children’s hospital or Emergency Department Approved for Pediatrics (EDAP) facility.
This chapter provides an overview of the general approach to the poisoned patient. It highlights the epidemiology, clinical recognition, and management of five major types of poisonings and overdose: cocaine, calcium channel blockers, β-adrenergic blockers, opioids, and TCAs. It is consistent with the detailed recommendations contained in the Toxicology chapter of the American Heart Association (AHA) 2002 Pediatric Advanced Life Support (PALS) Provider Manual,61 developed by Scalzo, Hazinski, et al. In addition, the recommendations are consistent with those in the Pediatric Advanced Life Support section of the 2010 AHA Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care.79
General approach to the poisoned patient
Life-threatening morbidity associated with poisoning may manifest as respiratory depression, seizures, depressed level of consciousness, hypotension and cardiac arrhythmias. The ten top exposures causing death are listed in Box 18-1.
Box 18-1 Ten Top Exposures Causing Deaths in 2009
• Sedatives/hypnotics/antipsychotics
• Cardiovascular drugs (i.e., calcium channel blockers and β-blockers)
• Acetaminophen in combination with narcotic analgesics (e.g., opioids)
From Bronstein AC, et al: 2009 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 27th annual report. Clin Toxicol 48:979-1178, 2010.
In addition to a thorough history and assessment of drugs and toxic chemicals in the child’s environment, physical findings beyond those detected by the primary and secondary assessments may have particular value and diagnostic significance for the patient with a toxic exposure (Box 18-2). The characteristic clinical manifestations of a specific poisoning or toxin are termed a toxidrome. Examples of toxidromes include: (1) the clinical constellation of tachycardia, mydriasis, diaphoresis, seizures, and the presence of bowel sounds suggests the sympathomimetic toxidrome (e.g., cocaine); (2) the combination of tachycardia, mydriasis, dry skin, seizures, and absent bowel sounds is consistent with the anticholinergic toxidrome (e.g., atropine).61 Recognition of common toxidromes can be vital to efficient resuscitation, supportive care, decontamination, and administration of antidotes.
Regardless of the type of toxin, protection of the airway is of fundamental importance in the management of any poisoning. Elective endotracheal intubation in a poisoned patient should be considered earlier than in a patient without a history of toxin exposure because poisoned patients are at high risk for development of sudden or progressive respiratory failure. Many toxins cause respiratory failure by depression of respiratory drive, hypoperfusion of the central nervous system (CNS), or direct toxic effects on the CNS or pulmonary systems. Toxins can affect oxygenation by causing alveolar hypoventilation (e.g., opiate intoxication)54 or direct pulmonary toxicity (e.g., TCAs).41 Table 18-1 lists potential mechanisms and toxic agents that cause decreased oxygenation in the poisoned patient.
Table 18-1 Potential Mechanisms of Decreased Oxygenation in Poisoning
| Mechanism | Examples of Toxic Etiology |
| Alveolar hypoventilation (hypercarbia with normal alveolar-arterial oxygen difference) | Opiates, tricyclic antidepressants, benzodiazepines, barbiturates, and clonidine |
| Ventilation-perfusion mismatch (includes acute respiratory distress syndrome) | Direct pulmonary toxicity (e.g., tricyclic antidepressants, calcium channel blockers, or inhaled hydrocarbons) or secondary injury resulting from decreased level of consciousness and aspiration of gastric contents, or pulmonary embolism (complication of chronic IV drug abuse) |
| Intrapulmonary shunting | Pneumothorax caused by cocaine, by internal jugular injections of heroin by drug abusers, and by iron intoxication |
| Diffusion abnormality | Chlorine, chloramine, and ammonia gas inhalation |
| Decrease in alveolar oxygen content | Simple asphyxiants: carbon dioxide, methane, inhalants (e.g., propane, butane, fluorocarbons), and nitrogen oxides |
Modified from Hazinski MF, et al: Toxicology. In PALS provider manual. Dallas, 2002, American Heart Association, p. 307.
Nondepolarizing neuromuscular blocking agents, such as vecuronium and rocuronium, are most useful for the intubation of poisoned patients. They have minimal cardiovascular side effects, rapid onset, and a relatively short duration of action (see also Chapter 5). The use of short-acting sedatives, in conjunction with short-acting neuromuscular blocking agents, enables rapid and efficient repeat assessment of a patient’s mental status. Such assessments are particularly important in the management of patients with status epilepticus.61
If the poisoned patient demonstrates effective spontaneous breathing and can maintain airway patency, consider placing the patient in a recovery position. The left lateral decubitus position reduces absorption of ingested substances154 as well as the risk for aspiration.
Gastrointestinal Decontamination
If a patent airway can be maintained in a poisoned patient, gastrointestinal decontamination may be considered. It is important to note that gastrointestinal decontamination has not been shown to improve outcome35,153 as defined by morbidity, mortality, cost, or length of hospital stay.84 In addition to achieving skill in administering gastrointestinal decontamination, critical care nurses should understand the inherent risks and benefits.
Administration of oral fluids for dilution purposes is of no proven benefit in most poisonings. In instances of acid ingestion, limited animal data suggests that oral dilution with water or milk as a demulcent may be helpful.67
Administration of syrup of ipecac is not recommended by the American Academy of Pediatrics for the treatment of poisonings.5,84 When applicable, recommended gastric decontamination basically consists of activated charcoal and rarely gastric lavage.
Activated Charcoal
Activated charcoal adsorbs many drugs as well as some other compounds.38 Although there is no evidence that administration of activated charcoal improves clinical outcome,35 it is often considered when children present to emergency care very soon after toxic ingestion. Caution is advised to limit the use of activated charcoal in children less than 6 months of age because they have a high risk of aspiration. Activated charcoal reduces the mean bioavailability of the drugs by approximately 69% when it is given within 30 minutes after drug or toxin ingestion; however, the bioavailability of drugs is only reduced by half that amount when activated charcoal is given an hour or more after ingestion.34,35
Most toxicologists and poison centers do not recommend prehospital administration of activated charcoal, although emergency department administration of activated charcoal may be useful in the treatment of some poisonings, particularly if the ingestion has occurred within 1 hour of presentation. There are insufficient data to either support or exclude the use of activated charcoal at greater than 1 hour after an ingestion.34,35
The optimal dose of activated charcoal has not been established in controlled human trials. The AACT and the European Association of Poisons Centres and Clinical Toxicologists have developed consensus oral dose recommendations for activated charcoal (Box 18-3).34,35
Repeated doses of activated charcoal can be administered to treat certain specific ingestions,3 but there is no evidence that multi-dose administration is superior to a single dose.45 Use of multiple dose activated charcoal is not recommended if the toxic agent slows gastrointestinal motility (e.g., TCAs, calcium channel blockers, and opiates), because the activated charcoal can contribute to regurgitation and aspiration or can become impacted, leading to intestinal perforation.56
Contraindications to the administration of activated charcoal include an unprotected airway, ingestion of volatile substances (e.g., hydrocarbons) and anatomic anomalies of the gastrointestinal tract. Administration of activated charcoal may lead to regurgitation and aspiration, hence placement of an endotracheal tube before administration of activated charcoal may reduce but not reliably prevent aspiration.114
Gastric Lavage
Although emergency personnel have used gastric lavage for years, there is no convincing evidence that it improves clinical outcome.153 Like activated charcoal, gastric lavage increases the risk of aspiration.78,138,152 In addition, complications of lavage tube placement include hypoxia,151 tension pneumothorax and charcoal-containing empyema,71 and esophageal9 and gastrointestinal perforation.102 For these reasons, gastric lavage is only indicated in a patient who presents soon after ingestion of some life-threatening toxins (Box 18-4).
Box 18-4 Key Issues in Gastrointestinal Decontamination
• Sudden changes in level of consciousness and respiratory depression may develop in poisoned pediatric patients. For this reason, support the ABCs and establish and maintain a patent airway before administering gastrointestinal decontamination. Alert patients may be able to maintain their airways, whereas others will require intubation.
• Syrup of ipecac is not recommended for any of the five types of poisoning discussed in this chapter.
• If activated charcoal is indicated, administer it early after ingestion because it is most effective within 1 h of ingestion.
• Gastric lavage is not a routine intervention; its effectiveness is limited in poisoned patients. Lavage may be considered for asymptomatic patients who present early (usually within 60 min or less) after ingestion of some life-threatening toxins.
• Whole bowel irrigation may be beneficial in select poisonings, but further research is needed.
Antidotes
Following the assessment and support of airway, oxygenation, ventilation, and circulation, the critical care nurse may need to administer a specific antidotal therapy. Use of true antidotes as defined by the International Programme for Chemical Safety (IPCS) is relatively infrequent in pediatric poisonings, with the exception of naloxone, which is a classic antidote that effectively reverses opiate toxicity. The IPCS classifies naloxone as an A1 agent (i.e., A: should be available within 30 min or less; 1: effectiveness is well documented).70,132 A list of other antidotes is included in Table 18-2.
| Poison | Antidote(s) |
| Acetaminophen | N-acetylcysteine (NAC) |
| Organophosphate and Carbamate Insecticides, Nerve Agents | Atropine Pralidoxime (2-PAM); (not usually required with Carbamate Insecticides) |
| Iron | Deferoxamine |
| Digoxin | Digoxin Immune Fab |
| Mercury | Dimercaprol (BAL) |
| Benzodiazepines | Flumazenil (not recommended for overdose but may be used to reverse procedural sedation) |
| Methanol, ethylene glycol | Fomepizole |
| Cyanide |
Hydroxocobalamin (preferred) Cyanide Antidote Package |
Management of specific poisonings
Cocaine
Epidemiology, Pathophysiology, and Clinical Manifestations
Cocaine has complex pharmacologic effects, and the route of administration and the form of cocaine involved can affect the onset, duration, and magnitude of the clinical signs and symptoms and potential complications.14 Cocaine is absorbed from all mucous membranes, from the gastrointestinal tract (most common route in pediatric unintentional exposure), and the genitourinary tract.55,61
Cocaine binds to the reuptake pump in presynaptic nerves, blocking the uptake of norepinephrine, dopamine, epinephrine, and serotonin from the synaptic cleft. This action leads to local accumulation of these neurotransmitters (Fig. 18-2), which produces both peripheral and CNS effects.61
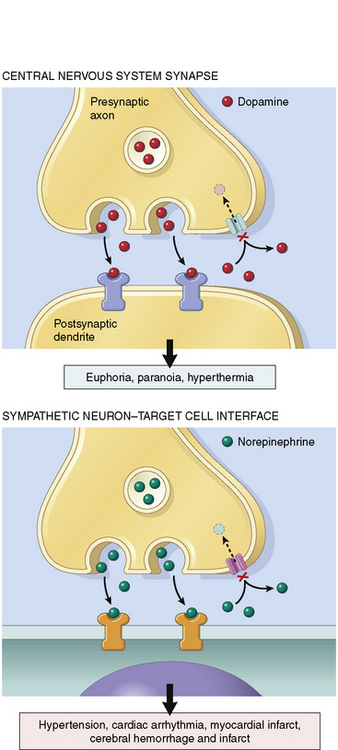
The accumulation of norepinephrine and epinephrine at β-adrenergic receptors results in tachycardia, increased myocardial contractility, tremor, diaphoresis, and mydriasis. Tachycardia increases myocardial oxygen demand while reducing the time for diastolic filling and for coronary perfusion (particularly of the left ventricle).87 Accumulation of neurotransmitters at peripheral α-adrenergic receptors results in vasoconstriction and hypertension. The peripheral endothelial nitric oxide system can also be impaired, leading to further vasoconstriction.113
Centrally mediated dopaminergic effects of cocaine include mood elevation and movement disorders. Centrally mediated stimulation of serotonin (i.e., 5-hydroxytryptamine or 5-HT) receptors results in exhilaration, hallucinations, and hyperthermia. Stimulation of peripheral 5-HT receptors also results in coronary artery vasospasm that can lead to acute coronary syndrome (ACS) and myocardial infarction. In addition, cocaine stimulates both platelet aggregation62 and increases in circulating epinephrine; these effects can lead to secondary platelet activation and coronary occlusion.73
In adults, the most frequent cause of cocaine-induced hospitalization is ACS, caused by coronary vasoconstriction and platelet aggregation with resulting myocardial ischemia, chest discomfort and possible infarction.24,65,87 Although ACS is a rare complication in children, it has been reported, particularly when ethanol and cocaine are combined.164 Concurrent use of cocaine and ethanol precipitates the formation of the cocaine metabolite, cocaethylene, which increases the cardiotoxic and neurotoxic effects of either substance alone.48 Although myocardial infarction in the neonate with a structurally normal heart and coronary arteries is rare, its association with maternal cocaine abuse has been reported.28
Cocaine-induced ACS can lead to myocardial ischemia and subsequent infarction and complications such as ventricular arrhythmias, congestive heart failure, and death.61,65 Cocaine-induced ACS is diagnosed by ECG changes characteristic of myocardial ischemia; infarction has occurred if serum troponin levels are elevated. In addition to ischemia-induced arrhythmias, cocaine also disturbs cardiac electrophysiology by altering sodium and potassium channel conduction and may induce wide-complex arrhythmias, VT, and VF, including torsades de pointes.13,14,81,127
Infants can be exposed to cocaine in breast milk,166 and infants or children may experience passive inhalation of vapors from adults smoking crack cocaine.63,116 The presence of the cocaine metabolite benzoylecgonine in the urine of children who are otherwise medically stable may reflect passive inhalation; it does not necessarily indicate poisoning or intentional cocaine administration.63 This situation raises legitimate concern for the well-being of such children, because deaths have been linked to passive inhalation of crack cocaine smoke.112
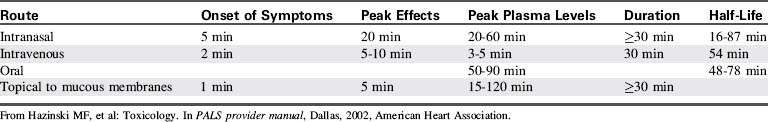
Because cocaine is rapidly metabolized, serum levels generally are of little use and often do not correlate with clinical findings.19 Table 18-3 summarizes the pharmacokinetics and pharmacodynamics of cocaine hydrochloride.
Management
General Care
Initial treatment of cocaine toxicity consists of oxygen administration, continuous ECG monitoring, and administration of a benzodiazepine (e.g., diazepam or lorazepam).42,61 This care is summarized in Box 18-5.
Box 18-5 Recognition and Management of Cocaine Toxicity
Management
Provide continuous ECG monitoring; observe for and treat ventricular arrhythmias (for arrhythmias secondary to infarction, consider lidocaine).
Treat hyperthermia aggressively.
For ACS, consider nitroglycerin, a benzodiazepine, and phentolamine.
Benzodiazepine administration is the mainstay of cocaine toxicity treatment because it offers both anticonvulsant and CNS-depressant effects, and it reduces heart rate and systemic arterial pressure.42 Benzodiazepines also appear to attenuate the toxic myocardial effects of cocaine.87 In contrast, phenothiazines and butyrophenones (e.g., haloperidol) provide no benefit and may be harmful to patients with cocaine toxidrome. β-Blockers are contraindicated in cocaine intoxication because their use has been associated with increased blood pressure, coronary vasospasm and fatal myocardial infarction.79
Because cocaine is a sodium channel blocker, sodium bicarbonate in a dose of 1 to 2 mEq/kg may be effective in the treatment of cocaine-associated ventricular arrhythmias.61,79 In an experimental model of cocaine-induced ECG changes, sodium bicarbonate significantly reduced the prolonged PR and QT intervals and reduced the QRS duration.127 Sodium bicarbonate also may be effective in treating the cocaine-associated acidemia that is thought to contribute to intraventricular conduction delays (prolonged QRS interval), arrhythmias, and depressed myocardial contractility.156
Lidocaine administration may be considered for patients with ventricular arrhythmias associated with cocaine-induced myocardial infarction that are refractory to other treatments.79 The effectiveness of lidocaine in this patient population has not been well established. Because of the fact that lidocaine inhibits fast sodium channels, it has been shown to potentiate cocaine toxicity in animals,43 although this effect has not been documented in humans. Cocaine and lidocaine together also may have additive effects that increase the likelihood of seizure activity.142,168
The effectiveness of epinephrine in the treatment of cocaine-induced circulatory failure is questionable.111 Epinephrine may exacerbate cocaine-induced arrhythmias and should not be administered for ventricular arrhythmias. If VF or pulseless VT develop, epinephrine is used to increase coronary perfusion pressure during cardiopulmonary resuscitation (CPR).61
Treatment of Hyperthermia
The CNS manifestations of cocaine intoxication often include loss of thermoregulation with resulting hyperthermia. High ambient temperature has been associated with a significant increase in mortality from cocaine overdose in humans.104,105 As a result, vigilant monitoring of body temperature is indicated for all patients with cocaine intoxication, and fever should be treated aggressively.79 External cooling is necessary for children presenting with agitation, delirium, seizures, and elevated body temperature.
Treatment of Seizures
Cocaine may produce seizures in infants and children after ingestion,37,46 and in infants when the drug is transmitted through breast milk.33 Cocaine likely causes seizures by affecting gamma aminobutyric acid (GABA) transmission; it also may stimulate the neuroexcitatory N-methyl D-aspartate (NMDA) receptor. Seizure management includes administration of a benzodiazepine. Lorazepam is often used (0.05-0.1 mg/kg, up to 2 mg/dose), with doses repeated as needed. Following administration of benzodiazepines, particularly when repeated doses are necessary (e.g., to manage prolonged cocaine-induced seizures), patients should be closely monitored for development of respiratory depression. Phenytoin and fosphenytoin may not be effective in treating cocaine-induced seizures because they lack an effect on the GABAA receptor.159 Phenobarbital is recommended for the treatment of seizures refractory to benzodiazepines. Propofol also may be of benefit to control cocaine-induced seizures because it has a short half-life, making it easy to titrate according to patient response.159
Calcium Channel Blocker Toxicity
Epidemiology, Pathophysiology, and Clinical Manifestations
The increasing use of calcium channel blockers for the treatment of hypertension and congestive heart failure makes them readily available for unintentional or intentional overdose. In 2009, a total of 10,868 exposures to calcium channel blockers were reported to the AAPCC; nearly 14% of these exposures occurred in children younger than 6 years.26
Although calcium channel blockers can be classified according to their effects on the myocardium and vascular smooth muscle, in cases of overdose these selective properties are lost and serious cardiovascular toxicity may be seen with all agents.134 All calcium channel blockers bind to calcium channels, inhibiting the influx of calcium into cells. As a result, these agents will affect impulse conduction in slow-channel-dependent tissue such as the sinoatrial (SA) and AV nodes, coupling of myocardial excitation-contraction, and vascular smooth muscle tone.
The life-threatening clinical manifestations of calcium channel toxicity include bradyarrhythmias (caused by inhibition of pacemaker cells) and hypotension (caused by vasodilation and impaired cardiac contractility).115,134 Electrocardiographic changes can include a prolonged PR interval, inverted P waves, AV dissociation, AV block,1 ST segment changes, low-amplitude T waves, sinus arrest, and asystole. Cerebral hypoperfusion can cause altered mental status (e.g., syncope, seizures, and coma).
The lung and gastrointestinal system are affected directly or indirectly by calcium channel blocker poisoning. Pulmonary complications include cardiogenic and noncardiogenic pulmonary edema,69,90 which will necessitate cautious fluid resuscitation and early support of ventilation.
Gastrointestinal complications include hypomotility, ileus,49 and constipation; these effects may be secondary to the inhibition of gastrointestinal motility hormone release.135 Patients with calcium channel blocker overdose often have absent or greatly diminished bowel sounds. Use of activated charcoal or whole-bowel irrigation may not be appropriate for these patients.
Careful serial assessment of bowel sounds should be performed if any form of gastrointestinal decontamination is being considered, particularly if the patient has ingested sustained-release products. Some experts advocate whole-bowel irrigation for patients who ingest sustained-release products to prevent further absorption,148 but controlled trials have not been performed to determine the effectiveness of whole bowel irrigation after calcium channel blocker overdose.4
Management
General Care
Although the supportive and specific therapies discussed in this section can be very effective in children, third-degree atrioventricular (AV) block with cardiac arrest161 and death have been reported.39,89 As a result, providers should monitor the patient closely and be prepared to institute resuscitation. Onset of symptoms may be immediate or delayed for up to 12 to 16 hours, especially when a sustained-release preparation has been ingested.145,157
The initial approach to therapy for calcium channel blocker overdose is to support oxygenation and ventilation, provide continuous ECG monitoring, and carefully monitor and support cardiovascular function and systemic perfusion (Box 18-6). All patients with a significant overdose require close monitoring of blood pressure because severe myocardial dysfunction and hypotension may develop. Continuous intra-arterial blood pressure monitoring should be considered for symptomatic patients.
Box 18-6 Recognition and Management of Calcium Channel Blocker Toxicity
Management
Support airway, oxygenation, and ventilation.
Establish continuous ECG monitoring; consider intra-arterial pressure monitoring.
Support blood pressure and systemic perfusion as needed (administer small fluid boluses [5-10 mL/kg], vasopressors, inotropes).
Treat bradycardia as needed with chronotropic drugs (e.g., epinephrine).
Calcium chloride (central line infusion): 20 mg/kg of 10% solution over 5-10 min, followed by infusion of 20-50 mg/kg per hour); monitor serum ionized calcium. If only peripheral IV access available, administer calcium gluconate.
• Adolescents and adults: Administer 5-10 mg over several minutes, followed by infusion of 1-5 mg/hour or higher if needed.
• Younger children: Administer 0.05-0.1 mg/kg up to 1 mg, followed by continuous infusion of 0.05-0.1 mg/kg per hour up to a maximum adult dose.
Consider mechanical support of cardiopulmonary function (see Chapter 7).
Calcium is often infused to treat calcium channel blocker overdose (to overcome the channel blockade), but the effectiveness of this therapy varies.15,68,134 Calcium chloride is generally recommended because it results in greater elevation of the ionized calcium concentration,25 but it should be administered through a central venous catheter because infiltration causes severe tissue injury. The optimal dose of calcium for treatment of calcium channel blocker overdose has not been established. Typically, a dose of 20 mg/kg (0.2 mL/kg) of 10% calcium chloride is given over 5 to 10 minutes, followed by an infusion of 20 to 50 mg/kg per hour.86 Adolescents may require additional calcium. Serum ionized calcium should be closely monitored to prevent hypercalcemia.143 If central venous access is not available, calcium gluconate should be administered by peripheral venous catheter79 (typical dose: 100 mg/kg).
High-dose norepinephrine or epinephrine has been reported to be effective for treatment of hypotension (vasopressor effects) or bradycardia (chronotropic effects) associated with severe calcium channel blocker toxicity.121,131,148 These drugs should be carefully titrated to the desired hemodynamic effect.
Two small case series21,170 suggest that hyperinsulinemia euglycemia therapy (HIET) may be beneficial in calcium channel blocker toxicity. Some clinicians advocate using HIET early in the management of severe calcium channel blocker overdose,58,93,110,123 as it enhances myocardial glucose uptake and metabolism, and has positive inotropic properties. Maximal efficacy may be obtained when HIET is administered in conjunction with IV calcium and vasopressors early in the course of serious calcium channel blocker overdose when insulin resistance is high.57 Presumably, the beneficial effects result from better myocardial glucose utilization through activation of pyruvate dehydrogenase and subsequent production of adenosine triphosphate through aerobic metabolism.
Precise dosing11,57,128 recommendations for HIET are unavailable. Using a central venous line, administer a loading dose of glucose (0.5 g/kg). The loading dose of glucose is followed by a continuous infusion at a rate of 0.5 g/kg per hour, adjusted accordingly. After the glucose bolus, an insulin bolus of 1 unit/kg of regular insulin is administered, followed by an insulin drip of 0.5-1 unit/kg per hour. Severely poisoned patients may require more than 1 unit of insulin/kg per hour. but careful attention to avoid hypoglycemia is imperative. Serum glucose concentration must be closely monitored, and it is often necessary to administer higher doses of glucose if high doses of insulin are required. The target range for glucose administration is to maintain the serum glucose concentration between 100 and 200 mg/dL by titration. Patients should be closely monitored for hypokalemia during HIET because potassium moves intracellularly with glucose. Potassium administration may be required.
Glucagon may be beneficial in the treatment of myocardial toxicity caused by calcium channel blocker overdose.1,126,171 Glucagon increases serum glucose concentration and causes a transient release of intracellular calcium. It has both positive chronotropic and inotropic effects, thereby increasing heart rate and contractility. In adults and adolescents, an initial bolus of 5 to 10 mg can be administered over several minutes, and repeated as needed. The initial bolus may be followed by a continuous glucagon infusion of 1 to 5 mg/hour or more in an adult.12,76,93 In younger children, bolus doses of 0.05 to 0.1 mg/kg up to a total dose of 1 mg or higher may be indicated. The range for continuous infusion of glucagon in children is 0.05 to 0.1 mg/kg per hour up to the typical adult dose of about 1-5 mg/hour.8,12
There is insufficient evidence to recommend for or against the use of sodium bicarbonate in the treatment of calcium channel blocker toxicity.79
Treatment of Cardiac Arrest
Cardiac arrest caused by calcium channel blocker overdose requires traditional management with high-quality CPR and epinephrine. Cardiac pacing and extracorporeal membrane oxygenation (ECMO) also may be useful.66 Mechanical cardiopulmonary support (e.g., ECMO, left ventricular assist device, intra-aortic balloon pumping) can also be effective (see Chapters 6 and 7). Aggressive resuscitation may be warranted in cases of calcium channel blocker overdose because recovery has been reported after prolonged verapamil-induced pulseless arrest.47,158 Calcium channel blockers may have some neuroprotective effects.
β-Adrenergic Blocker Toxicity
Epidemiology, Pathophysiology, and Clinical Manifestations
β-Adrenergic antagonists or β-blockers are widely prescribed and are responsible for a large number of poisonings every year. Intentional overdose by adolescents may result in severe intoxication. In 2009 a total of 22,135 ingestions of β-blockers were reported to the AAPCC, with nearly 15% of these exposures occurring in children younger than 6 years of age.26 Another retrospective review of data over an 11-year period showed more than 50,000 β-blocker exposures; overdoses of these agents accounted for 2.5% of all poison-related fatalities.101 A cohort study of 280 β-blocker exposures found that propranolol was the most commonly ingested β-blocker and the drug most frequently responsible for cardiovascular toxicity.100
β-blockade decreases intracellular cyclic adenosine monophosphate (cAMP), with resultant decrease in the metabolic, chronotropic, and inotropic activities of the heart and decreased vasoconstriction in blood vessels. A low intracellular cAMP concentration also decreases release of calcium from intracellular stores,76,163 producing bradycardia and conduction disturbances (e.g., sinus pauses, prolonged PR interval, various degrees of heart block, intraventricular conduction defects, and prolonged QRS interval). Arrhythmias including torsades de pointes,10 VF, and in rare cases, asystole149 may occur with severe poisoning. Hypotension paired with bradycardia, and various degrees of heart block are also common clinical manifestations of β-blocker toxicity.76
In addition to the cardiovascular effects, altered mental status, seizures, and coma may develop in cases of β-blocker toxicity. Altered mental status is particularly likely with agents that have high lipid solubility (e.g., propranolol) because these agents readily cross the blood-brain barrier.40,76 The CNS effects of β-blocker toxicity are direct effects separate from the effects of cerebral hypoperfusion that develop secondary to systemic hypotension. CNS toxicity may occur in the absence of clinical cardiac symptoms.98
Management
General Care
The initial approach to treatment of β-blocker overdose includes supporting adequate oxygenation and ventilation, assessing perfusion, establishing vascular access, and treating shock if present. Continuous ECG monitoring and frequent clinical reassessments are important (Box 18-7).
Box 18-7 Recognition and Management of β-Blocker Toxicity
Management
Support airway, oxygenation, and ventilation.
Establish continuous ECG monitoring; consider intra-arterial pressure monitoring.
Support blood pressure (epinephrine infusion at high doses may be required) and systemic perfusion (consider inotropes and inodilator).
Administer small fluid boluses; titrate to patient response.
• Adolescents and adults: Administer 5-10 mg over several minutes, followed by infusion of 1-5 mg/hour or higher if needed.
• Younger children: Administer 0.05-0.1 mg/kg up to 1 mg, followed by continuous infusion of 0.05-0.1 mg/kg per hour up to a maximum adult dose.
HIET (hyperinsulinemia euglycemia therapy):
• Administer glucose bolus of 0.5 g/kg.
• Administer insulin loading dose of 1 unit/kg.
• Establish continuous glucose infusion of 0.5 g/kg per hour, adjust according to serum glucose concentration (maintain approximately 100-200 mg/dL).
• Establish continuous insulin infusion of 0.5-1 unit/kg per hour (or higher as required).
Consider calcium if unresponsive to glucagon and HIET.
Consider mechanical support of cardiopulmonary function (see Chapter 7).
To overcome β-adrenergic blockade, high-dose epinephrine infusions may be effective.79,160 Other high-dose adrenergic agents (e.g., norepinephrine, dobutamine, isoproterenol, and dopamine) have been used successfully.72,92,144 Phosphodiesterase inhibitors such as inamrinone (formerly amrinone)82 or milrinone also can be used to improve myocardial contractility.
Limited experimental data76,171 and case reports92,160 suggest that glucagon may be beneficial in the treatment of β-adrenergic blocker overdose. In adults and adolescents, infusion of 5 to 10 mg of glucagon (administered over several minutes) followed by an IV infusion of 1-5 mg/hour or higher may be used.79 In younger children, bolus doses of 0.05 to 0.1 mg/kg up to 5 mg may be needed. In lieu of glucagon, HIET (hyperinsulinemia euglycemia therapy) also may be useful in the treatment of β-adrenergic blocker overdose (see doses in Box 18-7).
β-adrenergic blockade reduces cytoplasmic calcium concentration. Limited animal data99 and limited case reports23,129 suggest that calcium administration may be beneficial144 if glucagon and catecholamines are ineffective.79 Consider the administration of calcium to patients with β-blocker poisoning unresponsive to catecholamines and glucagon. In cases of intraventricular conduction delay (i.e., prolonged QRS interval), sodium bicarbonate44,141 (in addition to therapy with glucagon12,76 or HIET),110,125 catecholamines,76 and calcium122 may be given.
Nonpharmacologic Therapies
Nonpharmacologic therapies such as cardiac pacing75 and extracorporeal circulation109 may be successful in β-blocker overdose when other modalities and pharmacologic therapies fail. Children suspected of ingesting massive amounts of β-blockers or who manifest early signs of impending cardiovascular collapse may benefit from transport to a tertiary care pediatric center capable of providing these advanced therapies (see Chapters 6, 7 and 8).
Opioid Toxicity
Epidemiology, Pathophysiology, and Clinical Manifestations
Exposures related to opiates account for a large number of cases reported to poison centers and presenting to emergency departments. In 2009 a total of 88,609 exposures to opiates (either alone or in combination with other analgesics) were reported to the AAPCC.26 This number does not include heroin overdoses managed by EMS and emergency departments. According to 2009 NPDS statistics, opioids accounted for the third largest number of poison related fatalities.26 Opiate exposures and deaths have increased in many communities in the United States.31,50,53 Toxicity and overdose from opiates and opioids can occur with pediatric procedural sedation.54 Published reviews of procedural sedation and analgesia for children highlight the need for providers to be familiar with the sedative agents used and reversal agents such as naloxone.18,83,139
Narcotic overdose in children may occur from a number of different opioids (e.g., morphine, codeine, hydrocodone, oxycodone, hydromorphone, meperidine, pentazocine, and propoxyphene) and sources (i.e., intentional overdose, recreational use, and ingestion by small children). Abuse of the synthetic agent oxycodone as a recreational drug has recently increased among adolescents. Over-the-counter agents such as dextromethorphan, the d-isomer of the opiate agonist levorphanol, have been abused by adolescents, resulting in overdose deaths.119 Butorphanol nasal spray also may be available for abuse or accidental ingestion by children. A controlled-release form of oxycodone (i.e., OxyContin) is available in dosage strengths of up to 160 mg; this preparation can contribute to serious and prolonged opioid toxicity in instances of overdose and abuse.
Methadone is prescribed for chronic pain, but it is more commonly used to prevent withdrawal symptoms in patients recovering from opiate addiction. Its widespread use puts children at risk for inadvertent exposure.27,94 Because methadone has a long half-life and active metabolites, the patient with methadone overdose is usually admitted for monitoring and observation. Similar to opiates, clonidine (a centrally-acting imidazoline α2-receptor agonist) may cause respiratory depression, miosis, and coma. Clonidine has a prolonged effect, so patients require intensive monitoring.
Whenever a child is admitted with coma or respiratory depression of unknown cause, providers should consider the possibility of opiate overdose. These drugs are commonly used in both intentional abuse and Munchausen syndrome by proxy.32,108
Rapid urine immunoassays may detect opiates, but these tests can produce both false-negative and false-positive results. As an example, screening immunoassays often do not detect methadone, but more comprehensive assays will detect it.16 Whenever child maltreatment is suspected, qualitative screens such as urine immunoassays are insufficient for medicolegal purposes. In these situations, samples should be sent with documented chain of custody to a reference laboratory for analysis using advanced techniques.
Some opioids are available in transdermal patches that are formulated to release the opioid at a relatively slow rate; however, the patches themselves contain very large total amounts of the drug. Ingestion of a small amount of fentanyl from a fentanyl patch can produce severe toxicity in a child because of the potency of this opioid. The fentanyl on transdermal patches also can be inhaled. In one case severe toxicity developed within seconds after the patient heated a fentanyl patch and inhaled the vapors.103
Heroin may be inhaled, ingested, or injected. Heroin accounts for very few unintentional exposures in children, but it may be abused by adolescents.136 Heroin overdose is a major problem in most urban emergency departments.
Although children may present with respiratory failure caused by ingestion of only one opiate or opioid, adolescents often mix these agents with alcohol and other substances. Glutamate is the major excitatory neurotransmitter and GABA is the major inhibitory neurotransmitter contributing to control of respiration. Both benzodiazepines and alcohol facilitate the inhibitory effect of GABA; alcohol also decreases the excitatory effect of glutamate.162 As a result, the combination of benzodiazepines and alcohol can produce significant respiratory depression that is usually evident early after ingestion and may require support with mechanical ventilation.146
Severe opiate intoxication may cause cardiovascular symptoms such as hypotension, tachycardia or bradycardia, arrhythmias, circulatory collapse, and cardiac arrest. Noncardiogenic pulmonary edema may occur with heroin overdose.146
Decreased gastrointestinal motility is common with opioid overdose, presumably caused by peripheral opioid receptor effects.30,118 Delayed gastric emptying may cause “cyclical” coma. The first phase of drug absorption results in a decreased level of consciousness. This phase is followed by some metabolism of the drug, so the patient begins to awaken. Further delayed absorption of the drug may cause another decrease in level of consciousness. This inconsistency in level of consciousness generally precludes administration of activated charcoal unless preceded by endotracheal intubation. Seizures may occur with the opiate meperidine,59,85 further complicating management.
Management
General Care
Therapy for opiate or opioid toxicity should begin with assessment and support of the airway, oxygenation, and ventilation. If significant respiratory depression or respiratory failure is present, provide immediate bag-mask ventilation with oxygen. Endotracheal intubation with mechanical ventilation may be required.79
Naloxone is the antidote of choice for treatment of severe opiate or opioid toxicity. In the presence of the opiate toxidrome, characterized by coma, depressed respirations, and miosis (pinpoint pupils), naloxone administration should be considered once adequate ventilation has been established (Box 18-8).61
Box 18-8 Recognition and Management of Opioid Toxicity
Management
Establish patent airway, support oxygenation and ventilation.
• Less than 5 years (or 20 kg or less): 0.1 mg/kg IV/IO
• 5 years or older (or more than 20 kg): Up to 2 mg IV/IO
• In suspected narcotic addiction, use low initial doses (e.g., 0.01 mg/kg, up to 0.4 mg in a single dose; repeat as needed) to avoid withdrawal symptoms.
• For treatment of respiratory depression associated with therapeutic opioid use as in procedural sedation: 0.001-0.005 mg/kg (1-5 mcg/kg), repeat as needed.
• After initial dose, consider infusion, particularly for significant overdose or overdose with long-acting formulations.
Naloxone has been used for more than 20 years.6,74 Although patients generally tolerate naloxone well,147,169 adverse reactions, including ventricular arrhythmias, acute pulmonary edema,133 asystole, and seizures, may occur.124 The opioid and adrenergic systems are interrelated; opioid antagonists stimulate sympathetic nervous system activity.77 In addition, hypercapnia stimulates the sympathetic nervous system. Thus, administration of naloxone (an opioid antagonist) in the presence of significant hypercapnia from respiratory depression can produce substantial adrenergic stimulation with possible tachycardia, increased blood pressure, acute pulmonary edema, arrhythmias, seizures, and even cardiac arrest.79 Effective ventilation to normalize the partial pressure of CO2 (PaCO2) should be established before administration of naloxone to reduce potential adrenergic stimulation and attendant toxic effects of naloxone administration.79
For treatment of the adverse effects of opiate overdose, the recommended naloxone dose is 0.1 mg/kg by IV or intraosseous (IO) route; for children 5 years of age and older (or larger than 20 kg), administer up to 2 mg in a single dose.6 To avoid the sudden hemodynamic effects of opioid reversal, repeated doses of 0.01 to 0.03 mg/kg may be indicated. Very low doses (0.001-0.005 mg/kg [1-5 mcg/kg]) can be used to reverse respiratory depression caused by therapeutic doses of opiates.79
The half-life of naloxone is much shorter than the half-life of opiates. Following the initial doses of naloxone, a continuous infusion may be needed to reverse the toxic effects of opiate poisoning.150 Continuous infusion of naloxone also may be necessary to treat poisoning from some long-acting opioids such as methadone, continuous release oxycodone, and diphenoxylate. Naloxone may also be administered intramuscularly, subcutaneously, or through the endotracheal tube, but use of these routes may delay its onset of action, particularly if perfusion is poor.
TCAs and Other Sodium Channel Blocking Agents
Epidemiology, Pathophysiology, and Clinical Manifestations
Despite the introduction of safer treatment options for depression, tricyclic (cyclic) antidepressant toxicity continues to be a leading cause of morbidity and mortality. In 2009 a total of 102,792 ingestions involving an antidepressant and 79 total deaths caused by cyclic antidepressants were reported to the AAPCC.26 In children, TCAs are currently used to treat depression, attention deficit hyperactivity syndrome, migraine headaches, neuropathic pain, cyclic vomiting syndrome, nocturnal enuresis, and sleep disturbances, making them readily available to children for toxic ingestion.
TCAs typically are considered within the broader context of sodium channel blocking toxins, but when taken in overdose TCAs are also potassium channel blockers. The combined sodium and potassium channel blocking effects of TCAs may cause repolarization abnormalities and prolongation of the QT interval. Seizures can develop as the result of TCA blockade of the neuroinhibitory gamma aminobutyric acid (GABAA) chloride channel.36,91
Symptoms of early CNS stimulation may be the first signs of TCA toxicity. These symptoms likely result from the anticholinergic effects of the TCA, characterized by agitation, irritability, confusion, delirium, hallucinations, choreoathetosis (irregular, uncontrolled random movements, flowing from one part of the body to another), hyperactivity, seizures, and hyperpyrexia.61,155 Sinus tachycardia, hypertension, and supraventricular tachycardia may be observed early after ingestion and likely are related to excessive norepinephrine. Catecholamine depletion soon develops because norepinephrine reuptake into neurons is inhibited and the released norepinephrine is metabolized by catechol-O-methyltransferase and monoamine oxidase.61
With serious intoxication, cardiac rhythm disturbances result from prolongation of the action potential. This inhibition delays intraventricular conduction, causing QRS prolongation167 with QRS duration often exceeding 100 ms.52,60 The presence of these ECG abnormalities may be predictive of seizures as well as ventricular arrhythmias.20,52,60 Predictors of severe toxicity are an R wave in lead aVR equal to or greater than 3 mm or an R wave to S wave ratio in lead aVR equal to or greater than 0.7 (Fig. 18-3).96,97 TCA toxicity also is associated with preterminal sinus bradycardia and heart block with junctional or ventricular (wide-complex) escape rhythms.81
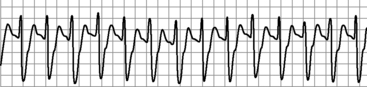
TCA overdose also may cause direct pulmonary toxicity. In addition, noncardiogenic pulmonary edema and acute lung injury in the setting of TCA overdose have been reported.137,172 The combined cardiac and respiratory manifestations of TCA overdose may precipitate cardiorespiratory arrest.
Other sodium channel blockers with toxicity similar to that of TCAs include β-adrenergic blockers (particularly propranolol and sotalol), procainamide, quinidine, local anesthetics (e.g., lidocaine), carbamazepine, type IC antiarrhythmics (e.g., flecainide and encainide), and cocaine.81 A common antihistamine, diphenhydramine, also can produce prominent sodium channel blocker effects resulting in wide-complex tachyarrhythmias with significant overdoses.117
Management
General Care
If there is no specific history of TCA exposure but an overdose is suspected based on symptoms of the TCA toxidrome (i.e., coma, convulsions, cardiac arrhythmias, and sometimes acidosis) and ECG changes, a number of bedside rapid urine immunoassays are available to screen for TCAs.140 For both suspected and known sodium channel blocker toxicity of any type, establishment of a patent airway and assessment and support of adequate oxygenation and ventilation are priorities; early endotracheal intubation should be considered.
Poison centers traditionally have recommended gastric lavage for TCA overdose, but there is no clear evidence that it is effective. If a patient ingests a potentially life-threatening amount of a TCA and presents asymptomatic within an hour of ingestion, lavage may be considered.152
Treatment of TCAs and other sodium channel blocker toxicants requires continuous ECG monitoring and treatment of arrhythmias with sodium bicarbonate (Box 18-9).22,79 Sodium bicarbonate raises the sodium concentration, which helps overcome the sodium channel blockade. The creation of alkalosis appears to contribute to the therapeutic effect by increasing protein binding of the TCA, thus reducing the amount of free drug available to cause toxicity. Administration of sodium bicarbonate should produce narrowing of the QRS complex, shortening of the QT interval, and increased myocardial contractility.61 These actions typically suppress ventricular arrhythmias, increase blood pressure, and improve systemic perfusion.64,106
Box 18-9 Recognition and Management of Tricyclic Antidepressant Toxicity
Management
General Management
Protect the airway (consider early intubation).
Maintain adequate oxygenation and ventilation.
Provide continuous ECG monitoring.
Treat arrhythmias with sodium bicarbonate: 1-2 mEq/kg IV/IO boluses until arterial pH is 7.45 or higher (or 7.50-7.55 for severe poisoning).
Follow boluses with IV/IO sodium bicarbonate infusion (150 mEq NaHCO3/L solution); titrate to maintain alkalosis.
Administer small normal saline boluses (10 mL/kg each) in addition to sodium bicarbonate as needed to support systemic perfusion; monitor for pulmonary edema.
Administer vasopressors (e.g., epinephrine, norepinephrine) to maintain adequate vascular tone and blood pressure (high doses may be required).
Consider ECMO and cardiopulmonary bypass if vasopressors are insufficient to maintain blood pressure.
Sodium bicarbonate is administered in 1 to 2 mEq/kg boluses until the arterial pH is 7.45 or higher. Sodium bicarbonate is then infused as a solution of 150 mEq NaHCO3 per liter of 5% dextrose and water, titrated to maintain alkalosis (arterial pH greater than 7.45). Additional boluses of sodium bicarbonate may be required for severe intoxications; increasing pH to a level between 7.50 and 7.55 may be warranted.79 Manipulating systemic pH to higher than this range generally is not recommended because of the risk of excessive alkalosis.81,95 Support of normal ventilation is recommended while the pH is increased with sodium bicarbonate administration. Although hyperventilation-induced alkalosis reportedly improves cardiac conduction,17 the effectiveness of respiratory alkalosis for TCA overdose has not been established.106
If ventricular arrhythmias caused by a TCA or other sodium channel blocker toxicant do not respond to sodium bicarbonate administration, consider lidocaine administration.51,61,79 Providers should be aware that many antiarrhythmics may worsen cardiovascular problems, including arrhythmias. Class IA (quinidine, procainamide) and Class IC (flecainide, propafenone) antiarrhythmics are contraindicated because they may exacerbate cardiac toxicity.95 Class III antiarrhythmics (amiodarone, sotalol) should not be administered because they prolong the QT interval.95
Treatment of Hypotension
If hypotension caused by TCA, or other sodium channel blocker toxicant is present, administer small normal saline boluses (5-10 mL/kg each), in addition to sodium bicarbonate.22,79,130 Cautious administration of intravenous fluid is essential because antidepressants and other sodium channel blocking drugs have myocardial depressant effects, and excessive or rapid intravenous fluid administration may contribute to myocardial failure and pulmonary edema.
Because TCAs block reuptake of norepinephrine at the neuromuscular junction, overdose can cause catecholamine depletion that contributes to vasodilation and hypotension. Vasopressors such as norepinephrine or epinephrine may be needed to maintain adequate vascular tone and blood pressure.61,79,80 Pure β-adrenergic agonists (e.g., dobutamine and isoproterenol) generally are not used because they may cause further vasodilation and hypotension. Furthermore, administration of dopamine may not result in sufficient vasoconstriction to correct hypotension unless given in high doses (i.e., greater than 20 mcg/kg per minute).155
If high-dose vasopressors are insufficient to maintain blood pressure, ECMO and cardiopulmonary bypass may be effective,88,165 but these therapies require rapid availability of equipment and trained personnel.79 Early identification of at-risk patients is important to enable possible referral to a facility capable of providing these therapies.
Treatment of Seizures
Benzodiazepines (e.g., lorazepam or diazepam) are indicated to treat seizures caused by TCA or other sodium channel blocker agents. In addition, patients require careful support of the airway, oxygenation, and ventilation and correction of acid-base imbalances. Phenobarbital is recommended for the treatment of seizures refractory to benzodiazepines. Phenytoin and fosphenytoin are not recommended to treat seizures, because they lack a subreceptor on the GABAA chloride channel that is partially blocked by the TCAs. As a result, these drugs will not help restore the brain equilibrium and stop the seizures. In addition, phenytoin and fosphenytoin may have pro-arrhythmic effects in patients with TCA toxicity.29
Advanced concepts
The advanced practitioner will be able to identify common toxidromes and anticipate the development of complications related to poisoning. Advanced concepts in the care of the patient with toxicologic problems are listed in Box 18-10.
Box 18-10 Advanced Practice Concepts: Toxicology
• Several toxins and drugs (e.g., tricyclic antidepressants, β-blockers, and cocaine) have sodium channel blocking properties and effects on cell membrane stability that predispose children to ventricular arrhythmias. These arrhythmias may respond to administration of sodium bicarbonate.
• Specific antidotes that are not used routinely in cardiopulmonary resuscitation may be useful in certain types of drug overdose:





1 Adams B.D., Browne W.T. Amlodipine overdose causes prolonged calcium channel blocker toxicity. Am J Emerg Med. 1998;16:527-528.
2 American Academy of Clinical Toxicology. Facility assessment guidelines for regional toxicology treatment centers. J Toxicol Clin Toxicol. 1993;31:211-217.
3 American Academy of Clinical Toxicology and European Association of Poisons Centres and Clinical Toxicologists. Position statement and practice guidelines on the use of multi-dose activated charcoal in the treatment of acute poisoning. J Toxicol Clin Toxicol. 1999;37(6):731-751.
4 American Academy of Clinical Toxicology and European Association of Poison Centres and Clinical Toxicologists. Position paper: whole bowel irrigation. J Toxicol Clin Toxicol. 2004;42(6):843-854.
5 American Academy of Clinical Toxicology and European Association of Poisons Centres and Clinical Toxicologists. Position Paper: Ipecac Syrup. J Toxicol Clin Toxicol. 2004;42(2):133-143.
6 American Academy of Pediatrics Committee on Drugs. Naloxone dosage and route of administration for infants and children: addendum to emergency drug doses for infants and children. Pediatrics. 1990;86:484-485.
7 American College of Emergency Physicians. Poison information and treatment systems. Ann Emerg Med. 2001;37:370.
8 Arroyo A.M., Kao L.W. Calcium channel blocker toxicity. Ped Emerg Care. 2009;25(8):532-538.
9 Askenasi R., et al. Esophageal perforation: an unusual complication of gastric lavage. Ann Emerg Med. 1984;13:146.
10 Assimes T.L., Malcolm I. Torsade de pointes with sotalol overdose treated successfully with lidocaine. Can J Cardiol. 1998;14:753-756.
11 Azendour H., et al. Severe amlodipine intoxication treated by hyperinsulemia-euglycemic therapy. J Emerg Med. 2010;38(1):33-35.
12 Bailey B. Glucagon in β-blocker and calcium channel blocker overdoses: a systemic review. J Toxicol Clin Toxicol. 2003;41(5):595-602.
13 Bauman J., DiDomenic R. Cocaine-induced channelopathies: emerging evidence. J Cardiovasc Pharmacol Ther. 2002;7:195-202.
14 Bauman J.L., Grawe J.J., Winecoff A.P., et al. Cocaine-related sudden cardiac death: a hypothesis correlating basic science and clinical observations. J Clin Pharmacol. 1994;34:902-911.
15 Belson M.G., Gorman S.E., Sullivan K., Geller R.J. Calcium channel blocker ingestions in children. Am J Emerg Med. 2000;18:581-586.
16 Belson M.G., Simon H.K. Utility of comprehensive toxicologic screens in children. Am J Emerg Med. 1999;17:221-224.
17 Bessen H.A., Niemann J.T. Improvement of cardiac conduction after hyperventilation in tricyclic antidepressant overdose. J Toxicol Clin Toxicol. 1985;23:537-546.
18 Bhatt M., et al. Consensus-based recommendations for standardizing terminology and reporting adverse events for emergency department procedural sedation and analgesia in children. Ann Emerg Med. 2009;53(4):426-435.
19 Blaho K., et al. Blood cocaine and metabolite concentrations, clinical findings, and outcome of patients presenting to an ED. Am J Emerg Med. 2000;18:593-598.
20 Boehnert M.T., Lovejoy F.H.Jr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985;313:474-479.
21 Boyer E.W., Shannon M. Treatment of calcium-channel-blocker intoxication with insulin infusion. N Engl J Med. 2001;344:1721-1722.
22 Bradberry S.M., et al. Management of the cardiovascular complications of tricyclic antidepressant poisoning: role of sodium bicarbonate. Toxicol Rev. 2005;24(3):195-204.
23 Brimacombe J.R., Scully M., Swainston R. Propranolol overdose—a dramatic response to calcium chloride. Med J Aust. 1991;155:267-268.
24 Brody S.L., Slovis C.M., Wrenn K.D. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325-331.
25 Broner C.W., Stidham G.L., Westenkirchner D.F., Watson D.C. A prospective, randomized, double-blind comparison of calcium chloride and calcium gluconate therapies for hypocalcemia in critically ill children. J Pediatr. 1990;117:986-989.
26 Bronstein A.C., et al. 2009 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 27th annual report. Clin Toxicol. 2010;48:979-1178.
27 Brooks D.E., Roberge R.J., Spear A. Clinical nuances of pediatric methadone intoxication. Vet Hum Toxicol. 1999;41:388-390.
28 Bulbul Z.R., Rosenthal D.N., Kleinman C.S. Myocardial infarction in the perinatal period secondary to maternal cocaine abuse: a case report and literature review. Arch Pediatr Adolesc Med. 1994;148:1092-1096.
29 Callaham M., Schumaker H., Pentel P. Phenytoin prophylaxis of cardiotoxicity in experimental amitriptyline poisoning. J Pharmacol Exp Ther. 1988;245:216-220.
30 Camilleri M. Opioid-induced constipation: challenges and therapeutic opportunities. Am J Gastroenterol. 2011;106(5):835-842.
31 Centers for Disease Control and Prevention. Unintentional opiate overdose deaths—King County, Washington, 1990-1999. MMWR. 2000;49:636-640.
32 Centers for Disease Control and Prevention. Emergency department visits involving nonmedical use of selected prescription drugs—United States, 2004-2008. MMWR. 2010;59(23):705-709.
33 Chaney N.E., Franke J., Wadlington W.B. Cocaine convulsions in a breast-feeding baby. J Pediatr. 1988;112:134-135.
34 Chyka P.A., Seger D., et alAmerican Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Position statement: single-dose activated charcoal. J Toxicol Clin Toxicol. 1997;35:721-741.
35 Chyka P.A., et al. Position paper: single-dose activated charcoal. Clin Toxicol (Phila). 2005;43(2):61-87.
36 Citak A., et al. Seizures associated with poisoning in children: tricyclic antidepressant intoxication. Pediatr Int. 2006;48(6):582-585.
37 Conway E.E.J., Mezey A.P., Powers K. Status epilepticus following the oral ingestion of cocaine in an infant. Pediatr Emerg Care. 1990;6:189-190.
38 Cooney D.O. Activated charcoal in medicinal applications. New York: Marcel Dekker; 1995.
39 Cosbey S.H., Carson D.J. A fatal case of amlodipine poisoning. J Anal Toxicol. 1997;21:221-222.
40 Cruickshank J.M., et al. (-Adrenoreceptor-blocking agents and the blood-brain barrier. Clin Sci. 1980;59(Suppl. 6):453s-455s.
41 Dahlin K.L., et al. Acute lung failure induced by tricyclic antidepressants. Toxicol Appl Pharmacol. 1997;146:309-316.
42 Derlet R.W., Albertson T.E. Diazepam in the prevention of seizures and death in cocaine-intoxicated rats. Ann Emerg Med. 1989;18:542-546.
43 Derlet R.W., Albertson T.E., Tharratt R.S. Lidocaine potentiation of cocaine toxicity. Ann Emerg Med. 1991;20:135-138.
44 Donovan K.D., Gerace R.V., Dreyer J.F. Acebutolol-induced ventricular tachycardia reversed with sodium bicarbonate. J Toxicol Clin Toxicol. 1999;37:481-484.
45 Eddleston M., et al. Multiple-dose activated charcoal in acute self-poisoning: a randomised controlled trial. Lancet. 2008;371(9612):579-587.
46 Ernst A.A., Sanders W.M. Unexpected cocaine intoxication presenting as seizures in children. Ann Emerg Med. 1989;18:774-777.
47 Evans J.S., Oram M.P. Neurological recovery after prolonged verapamil-induced cardiac arrest. Anaesth Intensive Care. 1999;27:653-655.
48 Farooq M.U., Bhatt A., Patel M. Neurotoxic and cardiotoxic effects of cocaine and ethanol. J Med Toxicol. 2009;5(3):134-138.
49 Fauville J.P., et al. Severe diltiazem poisoning with intestinal pseudo-obstruction: case report and toxicological data. J Toxicol Clin Toxicol. 1995;33:273-277.
50 Fingerhut L.A., Cox C.S.: Poisoning mortality, 1985-1995, [published correction appears in Public Health Rep 113:380, 1998], Public Health Rep 1998;113:218-233
51 Foianini A., Joseph Wiegand T., Benowitz N. What is the role of lidocaine or phenytoin in tricyclic antidepressant-induced cardiotoxicity? Clin Toxicol (Phila). 2010;48(4):325-330.
52 Foulke G.E. Identifying toxicity risk early after antidepressant overdose. Am J Emerg Med. 1995;13:123-126.
53 Friedman L.S. Real-time surveillance of illicit drug overdoses using poison center data. Clin Toxicol (Phila). 2009;47(6):573-579.
54 Gill A.M., et al. Opiate-induced respiratory depression in pediatric patients. Ann Pharmacother. 1996;30:125-129.
55 Goldfrank L.R., Hoffman R.S. The cardiovascular effects of cocaine. Ann Emerg Med. 1991;20:165-175.
56 Gomez H.F., et al. Charcoal stercolith with intestinal perforation in a patient treated for amitriptyline ingestion. J Emerg Med. 1994;12(1):57-60.
57 Greene S.L., et al. Relative safety of hyperinsulinemia/euglycemia therapy in the management of calcium channel blocker overdose: a prospective observational study. Intensive Care Med. 2007;33(11):2019-2024.
58 Hadjipavlou G., Hafeez A., Messer B., Hughes T. Management of lercanidipine overdose with hyperinsulinaemic euglycaemia therapy: case report. Scand J Trauma, Resusc Emerg Med. 2011;19:8.
59 Hagmeyer K.O., Mauro L.S., Mauro V.F. Meperidine-related seizures associated with patient-controlled analgesia pumps. Ann Pharmacother. 1993;27:29-32.
60 Harrigan R.A., Brady W.J. ECG abnormalities in tricyclic antidepressant ingestion. Am J Emerg Med. 1999;17:387-393.
61 Hazinski M.F., et al. Toxicology. In: PALS provider manual. Dallas: American Heart Association; 2002.
62 Heesch C.M., et al. Cocaine activates platelets and increases the formation of circulating platelet containing microaggregates in humans. Heart. 2000;83:688-695.
63 Heidemann S.M., Goetting M.G. Passive inhalation of cocaine by infants. Henry Ford Hosp Med J. 1990;38:252-254.
64 Hoffman J.R., et al. Effect of hypertonic sodium bicarbonate in the treatment of moderate-to-severe cyclic antidepressant overdose. Am J Emerg Med. 1993;11:336-341.
65 Hollander J.E., et al. Prospective multicenter evaluation of cocaine-associated chest pain. Cocaine Associated Chest Pain (COCHPA) Study Group. Acad Emerg Med. 1994;1:330-339.
66 Holzer M., et al. Successful resuscitation of a verapamil-intoxicated patient with percutaneous cardiopulmonary bypass. Crit Care Med. 1999;27:2818-2823.
67 Homan C.S., et al. Thermal characteristics of neutralization therapy and water dilution for strong acid ingestion: an in-vivo canine model. Acad Energ Med. 1998;5(4):286-292.
68 Horowitz B.Z., Rhee K.J. Massive verapamil ingestion: a report of two cases and a review of the literature. Am J Emerg Med. 1989;7:624-631.
69 Humbert V.H.Jr., Munn N.J., Hawkins R.F. Noncardiogenic pulmonary edema complicating massive diltiazem overdose. Chest. 1991;99:258-259.
70 Jacobsen D., Haines J.A. The relative efficacy of antidotes: the IPCS evaluation series. International Programme on Chemical Safety. Arch Toxicol Suppl. 1997;19:305-310.
71 Justiniani F.R., Hippalgaonkar R., Martinez L.O. Charcoal-containing empyema complicating treatment for overdose. Chest. 1985;87:404-405.
72 Kalman S., Berg S., Lisander B. Combined overdose with verapamil and atenolol: treatment with high doses of adrenergic agonists. Acta Anaesthesiol Scand. 1998;42:379-382.
73 Karch S.B. Cardiac arrest in cocaine users. Am J Emerg Med. 1996;14:79-81.
74 Kattwinkel J., et al. An advisory statement from the Pediatric Working Group of the International Liaison Committee on Resuscitation. Pediatrics. 1999;e56:103.
75 Kenyon C.J., et al. Successful resuscitation using external cardiac pacing in beta adrenergic antagonist-induced bradyasystolic arrest. Ann Emerg Med. 1988;17:711-713.
76 Kerns W. Management of β-adrenergic blocker and calcium channel antagonist toxicity. Emerg Med Clin N Am. 2007;25(2):309-331.
77 Kienbaum P., et al. Profound increase in epinephrine concentration in plasma and cardiovascular stimulation after mu-opioid receptor blockade in opioid-addicted patients during barbiturate-induced anesthesia for acute detoxification. Anesthesiology. 1998;88:1154-1161.
78 Klasner A.E., Luke D.A., Scalzo A.J. Pediatric orogastric and nasogastric tubes: a new formula evaluated. Ann Emerg Med. 2002;39(3):268-272.
79 Kleinman M.E., et al. Part 14: Pediatric advanced life support. 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S876-S908.
80 Knudsen K., Abrahamsson J. Effects of epinephrine, norepinephrine, magnesium sulfate, and milrinone on survival and the occurrence of arrhythmias in amitriptyline poisoning in the rat. Crit Care Med. 1994;22:1851-1855.
81 Kolecki P.F., Curry S.C. Poisoning by sodium channel blocking agents. Crit Care Clin. 1997;13:829-848.
82 Kollef M.H. Labetalol overdose successfully treated with amrinone and α-adrenergic receptor agonists. Chest. 1994;105:626-627.
83 Krauss B., Green S.M. Sedation and analgesia for procedures in children. N Engl J Med. 2000;342:938-945.
84 Krenzelok E., Vale A. Position statements: gut decontamination. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol. 1997;35:695-786.
85 Kyff J.V., Rice T.L. Meperidine-associated seizures in a child. Clin Pharm. 1990;9:337-338.
86 Lam Y.M., Tse H.F., Lau C.P. Continuous calcium chloride infusion for massive nifedipine overdose. Chest. 2001;119(4):1280-1282.
87 Lange R.A., Hillis L.D. Cardiovascular complications of cocaine use. N Engl J Med. 2001;345:351-358.
88 Larkin G.L., Graeber G.M., Hollingsed M.J. Experimental amitriptyline poisoning: treatment of severe cardiovascular toxicity with cardiopulmonary bypass. Ann Emerg Med. 1994;23:480-486.
89 Lee D.C. Fatal nifedipine ingestions in children. J Emerg Med. 2000;19:359-361.
90 Leesar M.A., et al. Noncardiogenic pulmonary edema complicating massive verapamil overdose. Chest. 1994;105:606-607.
91 Lester L., McLaughlin S. SALT: a case for sodium channel blockade toxidrome and the mnemonic SALT. Ann Emerg Med. 2008;5:214.
92 Lewis M., et al. Survival following massive overdose of adrenergic blocking agents (acebutolol and labetalol). Eur Heart J. 1983;4:328-332.
93 Lheureux P.E., et al. Bench-to-bedside review: hyperinsulinaemia/euglyceamia therapy in the management of overdose of calcium channel blockers. Crit Care. 2006;10(3):212.
94 Li L., Levine B.E., Smialek J.E. Fatal methadone poisoning in children: Maryland 1992-1996. Subst Use Misuse. 2000;35:1141-1148.
95 Liebelt E.L. Targeted management strategies for cardiovascular toxicity from tricyclic antidepressant overdose: the pivotal role for alkalinization and sodium loading. Pediatr Emerg Care. 1998;14:293-298.
96 Liebelt E.L., Francis P.D., Woolf A.D. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med. 1995;26:195-201.
97 Liebelt E.L., Ulrich A., Francis P.D., Woolf A. Serial electrocardiogram changes in acute tricyclic antidepressant overdoses. Crit Care Med. 1997;25:1721-1726.
98 Lifshitz M., Zucker N., Zalzstein E. Acute dilated cardiomyopathy and central nervous system toxicity following propranolol intoxication. Pediatr Emerg Care. 1999;15:262-263.
99 Love J.N., Hanfling D., Howell J.M. Hemodynamic effects of calcium chloride in a canine model of acute propranolol intoxication. Ann Emerg Med. 1996;28(1):1-6.
100 Love J.N., Howell J.M., Litovitz T.L., et al. Acute beta blocker overdose: factors associated with the development of cardiovascular morbidity. J Toxicol Clin Toxicol. 2000;38:275-281.
101 Love J.N., Litovitz T.L., Howell J.M., Clancy C. Characterization of fatal beta blocker ingestion: a review of the American Association of Poison Control Centers data from 1985 to 1995. J Toxicol Clin Toxicol. 1997;35:353-359.
102 Mariani P.J., Pook N. Gastrointestinal tract perforation with charcoal peritoneum complicating orogastric intubation and lavage. Ann Emerg Med. 1993;22:606-609.
103 Marquardt K.A., Tharratt R.S. Inhalation abuse of fentanyl patch. J Toxicol Clin Toxicol. 1994;32:72-75.
104 Martinez M., Devenport L., Saussy J., Martinez J. Drug-associated heat stroke. South Med J. 2002;95(8):799-802.
105 Marzuk P.M., et al. Ambient temperature and mortality from unintentional cocaine overdose. JAMA. 1998;279:1795-1800.
106 McCabe J.L., et al. Experimental tricyclic antidepressant toxicity: a randomized, controlled comparison of hypertonic saline solution, sodium bicarbonate, and hyperventilation. Ann Emerg Med. 1998;32(pt 1):329-333.
107 McCaig L.F., Burt C.W. Poisoning-related visits to emergency departments in the United States, 1993-1996. J Toxicol Clin Toxicol. 1999;37:817-826.
108 McClure R.J., et al. Epidemiology of Munchausen syndrome by proxy, non-accidental poisoning, and non-accidental suffocation. Arch Dis Child. 1996;75:57-61.
109 McVey F.K., Corke C.F. Extracorporeal circulation in the management of massive propranolol overdose. Anaesthesia. 1991;46:744-746.
110 Mégarbane B., Karyo S., Baud F.J. The role of insulin and glucose (hyperinsulinaemia/euglycaemia) therapy in acute calcium channel antagonist and beta-blocker poisoning. Toxicol Rev. 2004;23(4):215-222.
111 Mets B., Jamdar S., Landry D. The role of catecholamines in cocaine toxicity: a model for cocaine “sudden death.”. Life Sci. 1996;59(24):2021-2031.
112 Mirchandani H.G., et al. Passive inhalation of free-base cocaine (‘crack’) smoke by infants. Arch Pathol Lab Med. 1991;115:494-498.
113 Mo W., et al. Role of nitric oxide in cocaine-induced acute hypertension. Am J Hypertens. 1998;11:708-714.
114 Moll J., et al. Incidence of aspiration pneumonia in intubated patients receiving activated charcoal. J Emerg Med. 1999;17:279-283.
115 Moser L.R., Smythe M.A., Tisdale J.E. The use of calcium salts in the prevention and management of verapamil-induced hypotension. Ann Pharmacother. 2000;34:622-629.
116 Mott S.H., Packer R.J., Soldin S.J. Neurologic manifestations of cocaine exposure in childhood. Pediatrics. 1994;93:557-560.
117 Mullins M.E., Pinnick R.V., Terhes J.M. Life-threatening diphenhydramine overdose treated with charcoal hemoperfusion and hemodialysis. Ann Emerg Med. 1999;33:104-107.
118 Murphy D.B., et al. Opioid-induced delay in gastric emptying: a peripheral mechanism in humans. Anesthesiology. 1997;87:765-770.
119 Murray S., Brewerton T. Abuse of over-the-counter dextromethorphan by teenagers. South Med J. 1993;86:1151-1153.
120 Newar F.W., Niska R.W., Xu J. National Hospital Ambulatory Medical Care Survey: 2005; Emergency Department Summary. Adv Data. 2007;29(386):1-32.
121 Oe H., Taniura T., Ohgitani N. A case of severe verapamil overdose. Jpn Circ J. 1998;62:72-76.
122 O’Grady J., Anderson S., Pringle D. Successful treatment of severe atenolol overdose with calcium chloride. CJEM. 2001;3(3):224-227.
123 Ortiz-Münoz L., Rodriquez-Ospina L.F., Figueroa-Gonzalez M. Hyperinsulinemic-euglycemic therapy for intoxication with calcium channel blockers. Bol Assoc Med P R. 2005;97(3 Pt 2):182-189.
124 Osterwalder J.J. Naloxone—for intoxications with intravenous heroin and heroin mixtures—harmless or hazardous? A prospective clinical study. J Toxicol Clin Toxicol. 1996;34:409-416.
125 Page C., Hacket L.P., Isbister G.K. The use of high-dose insulin-glucose euglycemia in beta-blocker overdose: a case report. J Med Toxicol. 2009;5(3):139-143.
126 Papadopoulos J., O’Neil M.G. Utilization of a glucagon infusion in the management of a massive nifedipine overdose. J Emerg Med. 2000;18:453-455.
127 Parker R.B., et al. Comparative effects of sodium bicarbonate and sodium chloride on reversing cocaine-induced changes in the electrocardiogram. J Cardiovasc Pharmacol. 1999;34:864-869.
128 Patel N.P., Pugh M.E., Goldberg S., Eiger G. Hyperinsulinemic euglycemia therapy for verapamil poisoning: case report. Am J Crit Care. 2007;16(5):520, 518-520, 519.
129 Pertoldi F., D’Orlando L., Mercante W.P. Electromechanical dissociation 48 hours after atenolol overdose: usefulness of calcium chloride. Ann Emerg Med. 1998;31:777-781.
130 Pierog J.E., et al. Tricyclic antidepressant toxicity treated with massive sodium bicarbonate. Am J Emerg Med. 2009;27(9):1168.e3-1168.e7.
131 Proano L., Chiang W.K., Wang R.Y. Calcium channel blocker overdose. Am J Emerg Med. 1995;13:444-450.
132 Pronczuk de Garbino J., et al. Evaluation of antidotes: activities of the International Programme on Chemical Safety. J Toxicol Clin Toxicol. 1997;35:333-343.
133 Prough D.S., et al. Acute pulmonary edema in healthy teenagers following conservative doses of intravenous naloxone. Anesthesiology. 1984;60:485-486.
134 Ramoska E.A., et al. A one-year evaluation of calcium channel blocker overdoses: toxicity and treatment. Ann Emerg Med. 1993;22:196-200.
135 Ray J.M., Squires P.E., et al. L-type calcium channels regulate gastrin release from human antral G cells. Am J Physiol. 1997;273:G281-G288.
136 Remskar M., et al. Profound circulatory shock following heroin overdose. Resuscitation. 1998;38:51-53.
137 Roy T.M., et al. Pulmonary complications after tricyclic antidepressant overdose. Chest. 1989;96:852-856.
138 Scalzo A.J., Tominack R.L., Thompson M.W. Malposition of pediatric gastric lavage tubes demonstrated radiographically. J Emerg Med. 1992;10:581-586.
139 Scherrer P.D. Safe and sound: pediatric procedural sedation and analgesia. Minn Med. 2011;94(3):43-47.
140 Schwartz J.G., Hurd I.L., Carnahan J.J. Determination of tricyclic antidepressants for ED analysis. Am J Emerg Med. 1994;12:513-516.
141 Shanker U.R., Webb J., Kotze A. Sodium bicarbonate to treat massive beta blocker overdose. Emerg Med J. 2003;20(4):393.
142 Shih R.D., et al. Clinical safety of lidocaine in patients with cocaine-associated myocardial infarction. Ann Emerg Med. 1995;26:702-706.
143 Sims M.T., Stevenson F.T. A fatal case of iatrogenic hypercalcemia after calcium channel blocker overdose. J Med Toxicol. 2008;4(1):25-29.
144 Snook C.P., Sigvaldason K., Kristinsson J. Severe atenolol and diltiazem overdose. J Toxicol Clin Toxicol. 2000;38:661-665.
145 Spiller H.A., et al. Delayed onset of cardiac arrhythmias from sustained-release verapamil. Ann Emerg Med. 1991;20:201-203.
146 Sporer K.A., Dorn E. Heroin-related noncardiogenic pulmonary edema: a case series. Chest. 2001;120:1628-1632.
147 Sporer K.A., Firestone J., Isaacs S.M. Out-of-hospital treatment of opioid overdoses in an urban setting. Acad Emerg Med. 1996;3:660-667.
148 Stanek E.J., Nelson C.E., DeNofrio D. Amlodipine overdose. Ann Pharmacother. 1997;31:853-856.
149 Stinson J., Walsh M., Feely J. Ventricular asystole and overdose with atenolol. BMJ. 1992;305:693.
150 Tenenbein M. Continuous naloxone infusion for opiate poisoning in infancy. J Pediatr. 1984;105:645-648.
151 Thompson A.M., Robins J.B., Prescott L.F. Changes in cardiorespiratory function during gastric lavage for drug overdose. Hum Toxicol. 1987;6:215-218.
152 Vale J.A. Position statement: gastric lavage. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol. 1997;35:711-719.
153 Vale J.A., Kulig K., American Academy of Clinical Toxicology and European Association of Poisons Centres and Clinical Toxicologists. Position paper: gastric lavage. J Toxicol Clin Toxicol. 2004;42(7):933-943.
154 Vance M.V., Selden B.S., Clark R.F. Optimal patient position for transport and initial management of toxic ingestions. Ann Emerg Med. 1992;21:243-246.
155 Walsh D.M. Cyclic antidepressant overdose in children: a proposed treatment protocol. Pediatr Emerg Care. 1986;2:28-35.
156 Wang R.Y. pH-dependent cocaine-induced cardiotoxicity. Am J Emerg Med. 1999;17:364-369.
157 Watling S.M., et al. Verapamil overdose: case report and review of the literature. Ann Pharmacother. 1992;26:1373-1378.
158 Waxman A.B., White K.P., Trawick D.R. Electromechanical dissociation following verapamil and propranolol ingestion: a physiologic profile. Cardiology. 1997;99:478-481.
159 Weigand T. Phenytoin is not successful in preventing cocaine-induced seizures: a response to the article, “Cocaine body packing in pregnancy.”. Ann Emerg Med. 2007;49(4):543-544.
160 Weinstein R.S. Recognition and management of poisoning with β-adrenergic blocking agents. Ann Emerg Med. 1984;13:1123-1131.
161 Wells T.G., et al. Nifedipine poisoning in a child. Pediatrics. 1990;86:91-94.
162 White J.M., Irvine R.J. Mechanisms of fatal opioid overdose. Addiction. 1999;94:961-972.
163 Whitehurst V.E., et al. Reversal of propranolol blockade of adrenergic receptors and related toxicity with drugs that increase cyclic AMP. Proc Soc Exp Biol Med. 1999;221:382-385.
164 Williams J.J., Restieaux N.J., Low C.J. Myocardial infarction in young people with normal coronary arteries. Heart. 1998;79:191-194.
165 Williams J.M., et al. Extracorporeal circulation in the management of severe tricyclic antidepressant overdose. Am J Emerg Med. 1994;12:456-458.
166 Winecker R.E., et al. Detection of cocaine and its metabolites in breast milk. J Forensic Sci. 2001;46:1221-1223.
167 Wolfe T.R., Caravati E.M., Rollins D.E. Terminal 40-ms frontal plane QRS axis as a marker for tricyclic antidepressant overdose. Ann Emerg Med. 1989;18:348-351.
168 Ye J.H., et al. Cocaine and lidocaine have additive inhibitory effects on the GABA A current of acutely dissociated hippocampal pyramidal neurons. Brain Res. 1999;821:26-32.
169 Yealy D.M., et al. The safety of prehospital naloxone administration by paramedics. Ann Emerg Med. 1990;19:902-905.
170 Yuan T.H., et al. Insulin-glucose as adjunctive therapy for severe calcium channel antagonist poisoning. J Toxicol Clin Toxicol. 1999;37:463-474.
171 Zaritsky A.L., Horowitz M., Chernow B. Glucagon antagonism of calcium channel blocker-induced myocardial dysfunction. Crit Care Med. 1988;16:246-251.
172 Zuckerman G.B., Conway E.E.Jr. Pulmonary complications following tricyclic antidepressant overdose in an adolescent. Ann Pharmacother. 1993;27:572-574.