107 Toxic Megacolon and Ogilvie’s Syndrome
Acute megacolon refers to a syndrome presenting as marked colonic distension in the absence of mechanical obstruction. It results from disturbed colonic motility1,2 and may be a manifestation of Ogilvie’s syndrome or toxic megacolon. Ogilvie’s syndrome, or acute colonic pseudo-obstruction (or its precursor, critical illness–related colonic ileus [CIRCI]),3 is a disease of seriously ill hospitalized patients and associated with myriad hemodynamic, metabolic, pharmacologic, inflammatory, and postoperative conditions. In toxic megacolon, distension is caused by severe colitis and is associated with systemic toxicity. Toxic megacolon is classically described as a complication of inflammatory bowel disease (IBD), usually ulcerative colitis, but in the critically ill, toxic megacolon mostly occurs as a complication of severe infectious colitis generally caused by Clostridium difficile. Whether secondary to IBD or C. difficile colitis, progressive colonic distension can lead to gut barrier failure, sepsis, ischemia, perforation, and multiple organ dysfunction. These potentially life-threatening complications must be prevented. This chapter focuses on toxic megacolon and Ogilvie’s syndrome in critically ill patients admitted to the intensive care unit (ICU) and proposes prevention strategies.
 Clinical Features
Clinical Features
Ogilvie’s Syndrome or Acute Colonic Pseudo-Obstruction
The hallmark of Ogilvie’s syndrome is abdominal distension with or without tenderness in hospitalized patients with serious comorbid disease.4–6 Patients may present with constipation, but flatus or stools may pass as well. Bowel sounds are normal, diminished, or high, and percussion is hypertympanic. Tenderness is most pronounced over the cecum. Nausea and vomiting may occur, but gastric retention is often minimal, and enteral feeding may be tolerated. If diagnosis and treatment are delayed, progressive distension may cause peritoneal signs, respiratory compromise, nutritional depletion, sepsis, multiple organ failure, ischemia, and perforation. Perforation most commonly occurs in the cecum. The risk of perforation is unlikely when cecal diameter is less than 12 cm but increases sharply when cecal diameter is 12 cm or more. CIRCI is characterized by constipation for many days without marked colonic distension. This syndrome may herald development of Ogilvie’s syndrome.3
Toxic Megacolon
Toxic megacolon is a serious complication of colitis. Patients present with fever, abdominal tenderness, and distension or even with an acute abdomen. IBD or infectious colitis commonly present with diarrhea, but a decrease in stool frequency may herald the onset of megacolon and delay diagnosis.2,7 Altered consciousness, dehydration, hypotension, tachycardia, leukocytosis, thrombocytopenia, low albumin, and electrolyte disturbances are common. In severe cases, systemic toxicity leads to septic shock and multiple organ failure.8–10 Ascending pylephlebitis and septic emboli in the superior mesenteric vein and liver are rare complications. Patients with ulcerative colitis are at highest risk of developing toxic megacolon early in their disease.11 Factors that may trigger toxic megacolon are early discontinuation or decrease in medications, use of antidiarrheal agents such as loperamide or opioids, severe hypokalemia, barium enema, and colonoscopy.
Gastrointestinal Motility
Intestinal motility is mainly under control of the enteric nervous system, an independently functioning complex network regulated by entero-enteric reflex pathways, the so-called enteric minibrain.12
Several types of motor activity are involved in intestinal propulsion; local reflex peristalsis after feeding and the migrating motor complex (MMC) during fasting are the most important.13 Local reflex peristalsis is activated by intraluminal distension (food), which stimulates the release of the neurotransmitter, serotonin. Release of serotonin triggers afferent neurons that activate excitatory motor neurons proximal to the site of the stimulus to release acetylcholine and substance P, resulting in contraction. Distal to the site of distension, inhibitory neurons are activated to release nitric oxide (NO) and vasoactive intestinal peptide (VIP), leading to relaxation.14 This nonadrenergic, noncholinergic, intrinsic inhibitory innervation (NANCI) is more pronounced in organs with a reservoir function, explaining why the stomach and proximal colon are more susceptible to distension than the small intestine.15
The MMC or interdigestive motility pattern is initiated by the hormone, motilin. The motilin receptor is expressed on enteric neurons of the human duodenum and colon.16 Many peptides, autacoids, and hormones influence MMC activity, including insulin, cholecystokinin, serotonin, opioids, dopamine, norepinephrine, somatostatin, and NO.17–19
The enteric nervous system is modulated by the central autonomic nervous system; the parasympathetic nerves promote and the sympathetic nerves suppress motility.20,21 Parasympathetic nerves to the right and transverse colon originate from the vagal nerve and those to the distal colon from the spinal cord (S2-4); they release acetylcholine. Sympathetic innervation of the colon runs through the spinal cord and the celiac and mesenteric ganglia. Sympathetic activations suppress contractions via the release of norepinephrine, causing a presynaptic inhibition of acetylcholine release from enteric neurons and also the release of other excitatory neurotransmitters such as serotonin from enteric nerve cells.22,23 Apart from inhibiting motility, sympathetic activation contracts the sphincters by a direct effect of norepinephrine on the smooth muscle; norepinephrine released by sympathetic neurons also affects vascular tone.
 Pathogenesis of Megacolon
Pathogenesis of Megacolon
Colonic Ileus and Ogilvie’s Syndrome
The pathophysiology of Ogilvie’s syndrome is not fully understood. Increased sympathetic and suppressed or interrupted parasympathetic activity play a role. In addition, neurotransmitters, inflammatory mediators, metabolic derangement and pharmacologic interventions are directly or indirectly involved.20,21
Abdominal surgery induces hypomotility by a complex interaction of neurogenic and inflammatory mechanisms. Intestinal manipulation initiates norepinephrine release via sympathetic nerves from the spinal cord, as well as NO and VIP release via vagal nerve stimulation, causing inhibition of contractile activity and relaxation.21 Prolonged postoperative ileus involves inflammation of the intestinal muscularis, initiated by activation of peritoneal mast cells and resident macrophages. Activated mast cells release histamine and proteases, which recruit leukocytes and temporarily increase intestinal permeability with translocation of bacteria and bacterial products. Activated mast cells also stimulate resident macrophages to release cytokines such as tumor necrosis factor (TNF) and up-regulate inducible nitric oxide synthetase (iNOS) and cyclooxygenase (COX-2) expression; collectively, all of these factors inhibit motility. Local inflammation by influx of leukocytes in the muscularis mucosa and circulating cytokines subsequently activate neurogenic inhibitory adrenergic pathways, causing generalized hypomotility.21 In addition, activation of peripheral opioid receptors in the gastrointestinal (GI) tract inhibits acetylcholine release from motor neurons and promotes transmitter release from inhibitory neurons.24 Opioid receptors are stimulated by endogenous opioids, which are locally secreted upon surgical stress. Exogenous opioids used for analgesia also act on peripheral opioid receptors in the GI tract, inhibiting motility. Peritonitis and pain cause a generalized inhibition of motility via spinal afferents that connect in the spinal cord to sympathetic efferents.20
Colonic hypomotility in critically ill patients may be related to circulating bacterial products and/or proinflammatory cytokines (e.g., lipopolysaccharide or TNF), leading to increased expression of iNOS and COX-2.25,26 Colonic hypomotility also may be related to ischemia and reperfusion, causing an energy deficit, functio laesa, and oxidant-mediated tissue damage. Finally, distal colonic distension induces inhibition of proximal colonic motility, the so-called colo-colonic reflex, which passes by the paravertebral ganglia and activates inhibitory sympathetic nerves.22 In this way, colonic dilation perpetuates itself.
 Predisposing Factors
Predisposing Factors
Ogilvie’s Syndrome
Clinical factors predisposing to Ogilvie’s syndrome are summarized in Box 107-1.5,27,28 The syndrome was first described by Sir William Heneage Ogilvie (1887-1971) in two patients with malignant infiltration of the celiac plexus.29 After surgery and trauma of spine, hip, and pelvis, dysfunction of the sacral parasympathetic nerves may impair motility of the distal colon, causing atony with functional obstruction.30 In a series reporting 400 patients with Ogilvie’s syndrome, the most common underlying conditions were trauma, cardiovascular disease, and infections.5 In another series, the majority of patients had cardiovascular disease.31 Both drugs and ischemia may play a role in cardiovascular disease. Exogenous catecholamines have dose-dependent effects on intestinal motility; low doses promote and high doses suppress motility.18,32 α-Adrenergic agonists are stronger inhibitors of acetylcholine release than β-adrenergic agents. Dobutamine and dopexamine have little effect on intestinal peristalsis. Dopamine not only inhibits upper GI motility but also distal colonic motility.17,19,33 The use of dopamine is associated with late defecation.34 Clonidine and dexmedetomidine, central α2-adrenergic receptor agonists, decrease fasting colonic smooth muscle tone35 and are associated with Ogilvie’s syndrome.36 Opioids suppress the phase III migrating motor contractions.37 This inhibiting effect on gut motility is mediated by activation of mu-opioid receptors in the GI tract, reducing the release of acetylcholine from the myenteric plexus.38 Ogilvie’s syndrome with life-threatening complications is reported with the use of antipsychotic agents such as clozapine that cause generalized GI hypomotility by anticholinergic and antiserotonergic mechanisms.39,40 In patients with sepsis, bacterial products (e.g., lipopolysaccharide), proinflammatory cytokines (e.g., TNF) and NO produced by the inducible enzyme, iNOS, suppress intestinal motility.25,41,42
Toxic Megacolon
The incidence of toxic megacolon in IBD has substantially decreased with better management of severe colitis.43 The most common cause of toxic megacolon in the critically ill is pseudomembranous colitis caused by overgrowth of C. difficile.44 However, other pathogens such as enterotoxin-producing strains of Clostridium perfringens, Staphylococcus aureus, and Klebsiella oxytoca can cause colitis after antibiotic use.45 Sporadic cases of megacolon due to infections caused by Salmonella spp.,46 Shigella spp., Amoeba, herpesvirus, or cytomegalovirus (CMV) have also been described.11 In patients with human immunodeficiency virus (HIV), toxic megacolon may be a primary manifestation of the HIV infection or be related to infection with C. difficile or CMV.47 Causes of toxic megacolon are summarized in Box 107-2.
The pathogenesis of toxic megacolon is not well understood.2,11 In infectious colitis, inflammation extends into the deeper layers of the colonic wall, whereas the inflammation in ulcerative colitis is typically limited to the mucosa. Deep infiltration, microabscesses, edema, and necrosis may paralyze colonic smooth muscle and lead to dilatation. Bacterial toxins permeating through ulcerations activate the release of cytokines, with subsequent systemic toxicity. NO, locally generated in excessive amounts secondary to increased expression of iNOS in inflammatory and smooth muscle cells, is the key mediator of diminished smooth muscle function in toxic megacolon.48,49
Clostridium Difficile Infection
Clostridium difficile is a gram-positive spore-forming rod. Pathogenic strains produce two major exotoxins: A and B. Both activate cell-signaling molecules including the transcription factor, nuclear factor-κB, and mitogen-activated protein kinases in monocytes, leading to the production and release of proinflammatory cytokines. Both toxins induce colitis in humans.50 Colonic injury results from alterations of the enterocyte cytoskeleton, with disruption of tight junction function and marked inflammation in the lamina propria. Severe pseudomembranous colitis occurs in 3% to 5% of carriers. Recurrent sepsis or toxic megacolon are rare but severe complications.
Colonization with C. difficile results from alterations in the composition of the indigenous colonic microflora. Enemas containing normal human feces appear to be effective in the treatment of infected patients.51,52 Mechanisms of suppression include the production of volatile acids, hydrogen sulfide, and secondary bile acids.53 The most frequently identified clinical risk factor for C. difficile–associated diarrhea is the antecedent use of antibiotics affecting indigenous colonic microflora.54 The opportunity to acquire the organism increases with prolonged hospital stay,55 and it may spread by nosocomial transmission.56 Whether a person remains an asymptomatic carrier or develops colitis depends on the size of the C. difficile population, toxigenicity of the strain, toxin-neutralizing effects of the indigenous gut flora, and underlying disease (Box 107-3).53,57,58 Susceptibility is further increased by poor GI defense mechanisms resulting from the use of gastric acid–inhibiting drugs that facilitate intestinal transit of the bacteria,54,55 total parenteral nutrition, postpyloric enteral feeding, or recent GI surgery.59–61 A combination of factors increases the risk.
 Diagnosis and Differential Diagnosis of Acute Megacolon
Diagnosis and Differential Diagnosis of Acute Megacolon
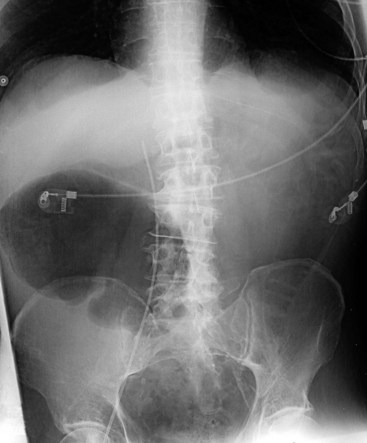
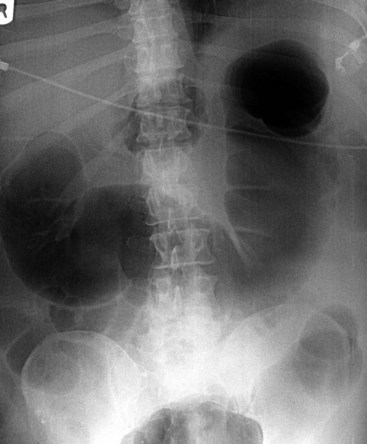
Besides history and clinical features, plain abdominal radiography is crucial for diagnosis and follow-up. Dilatation is most pronounced in the cecum, ascending, and right transverse colon. The size of the cecum may range from 6 to 20 cm. In Ogilvie’s syndrome, colonic diameter typically decreases gradually to a collapsed bowel and a normal gas and fecal pattern in the rectum (Figure 107-1). Dilation of the left colon may occur as well (Figure 107-2). Mechanical obstruction is excluded if gas is visible in all colonic segments, including the rectosigmoid. If not, an enema should be administered. The osmotic effect of water-soluble contrast medium is diagnostic and may be therapeutic in decompressing the colon.62 Some air/fluid levels and dilatation of the small bowel may be present. In Ogilvie’s syndrome, the colonic haustral and mucosal pattern is maintained, whereas the pattern is disturbed or lost in toxic megacolon. Deep ulcerations may be visible between large pseudopolypoid projections into the lumen. Pneumatosis of the bowel wall is a sign of ischemic necrosis and free peritoneal air of perforation. A diagnosis of severe colitis can be made with computed tomographic (CT) scan, but findings are nonspecific for the underlying cause.63 The scan shows a diffusely thickened or edematous colonic wall with pericolonic inflammation. CT scan may be helpful in patients presenting without diarrhea, with acute abdomen, for differential diagnosis, or to show or exclude complications.
The underlying cause of toxic megacolon, IBD, or infectious colitis must be identified (see Boxes 107-2 and 107-3). The history may reveal chronic abdominal complaints, diarrhea, bloody stools, familial occurrence of IBD, recent travel, intake of contaminated food, hospitalization, use of antibiotics, risk factors for HIV infection, or immunosuppression. Infection with CMV or C. difficile may precipitate toxic megacolon in ulcerative colitis.64 If unresponsive to therapy, both Ogilvie’s syndrome and ischemic colitis may be complicated by progressive distension with bacterial overgrowth and systemic toxicity, mimicking toxic megacolon.
For microbiological diagnosis of infectious colitis, a fresh fecal sample should immediately be submitted to the laboratory for culture on specific media. For screening of a C. difficile infection, stool can be tested for the presence of toxicogenic C. difficile. The screening assay is inexpensive, quick, and highly sensitive. Unfortunately, its specificity is low.65 To improve diagnosis, a two-stage testing strategy is recommended with an initial highly sensitive rapid screening test capable of detecting both toxin A and B, followed by a confirmatory test capable of detecting neutralizable C. difficile toxin in cell culture.65 Final results take 2 to 5 days. Although the test is positive in asymptomatic carriers, a positive test in a patient with antibiotic-associated megacolon makes infection with C. difficile highly probable. Surveillance cultures of feces are advocated to detect other pathogens such as enterotoxin-producing C. perfringens, S. aureus, and K. oxytoca. Blood cultures are warranted in all cases of toxic megacolon. They are generally positive in severe cases of typhoid fever. If stool and blood cultures remain negative, a bone marrow culture may still yield Salmonella spp. after 5 days despite antibiotic use.
Limited endoscopy with biopsy may provide useful information. Inflammatory bowel disease is characterized by diffusely abnormal crypt structure, whereas the architecture of the crypts is intact in bacterial colitis. In CMV colitis, inclusion bodies are present. Mild cases of C. difficile colitis are associated with nonspecific findings of colitis. In severe cases, focal ulcerations covered by purulent material, presenting as yellow or white plaques 2 to 4 cm in diameter, are found with normal intervening mucosa. Pseudomembranous colitis may not be detected if flexible sigmoidoscopy is performed.66 Full colonoscopy in patients with acute megacolon carries the risk of perforation, however.
 Management
Management
Ogilvie’s Syndrome
An early proactive strategy for preventing Ogilvie’s syndrome is advocated (Box 107-4). Awaiting resolution, the distended colon is at risk for life-threatening complications that need to be prevented. To exclude obstruction, a water-soluble contrast enema can be given unless the patient displays peritoneal irritation. Concomitantly, conditions that can impair colon motility must be corrected. All motility-inhibiting medications should be minimized or withdrawn. Alternatives are generally available; for example, opioids can be replaced by thoracic epidural anesthesia, an intervention that improves motility by inducing sympathetic blockade.67,68 Efforts should be made to reduce infusion rates of norepinephrine and especially dopamine.
If these measures are not effective, neostigmine is the drug of choice. In a double-blind crossover trial in a non-ICU population, an intravenous (IV) bolus of 2 mg neostigmine led to rapid colonic decompression in the majority of the patients.69 Since severe bradycardia is feared in the critically ill, a continuous infusion with neostigmine is safer. In a double-blind, placebo-controlled cross-over study in critically ill, ventilated patients with CIRCI, continuous IV administration of neostigmine at 0.4 to 0.8 mg/h resulted in defecation in 80% of the patients, whereas no defecation occurred during placebo infusion.3 If defecation does not occur, the neostigmine dose should be increased at 4-hour intervals. With this regimen, neostigmine is tolerated well, adverse events present slowly, and if necessary, the dose can be reduced. Adverse events include bradycardia, increased salivation and bronchorrhea, bronchospasm, and abdominal cramps if motility recovers. Repeat radiographs are obtained for follow-up assessment of colonic diameter.
Several case reports showed effective colonic decompression with cisapride,70 which enhances acetylcholine release in the mesenteric plexus. However, the drug was withdrawn from the U.S. market in 2000 because of its propensity to induce severe ventricular dysrhythmias. Erythromycin, a motilin agonist, also may improve colonic motility.71 Recommended dose is low (200 mg twice daily IV), since higher doses can actually inhibit motility.72
Endoscopic decompression may be indicated if decompression with neostigmine fails.73 In our experience, this intervention is seldom necessary. Colonoscopy in this setting is time consuming, difficult to perform, and not without hazards. The unprepared bowel contains copious amounts of stool. Inflation of air may increase colonic distension, impair ventilation, and lead to perforation. It is advocated to avoid the liberal use of air insufflation. Advancing the scope as far as the hepatic flexure may be sufficient to obtain adequate decompression.74 Gas should be aspirated. Colonoscopy is successful in 70% to 80% of patients, but the recurrence rate is 15% to 40%. Recurrence may be reduced if a decompressive tube is left in place, but controlled trials with this intervention are not available. Mortality rate associated with colonoscopy is between 1% and 5% in experienced hands. If signs of ischemia are encountered, the procedure should be discontinued.
Indications for surgery are failure of conservative treatment, with clinical signs of impending or actual ischemia or perforation. For surgical management, the reader is referred to the specific literature.6,28,75 The type of surgery depends on the state of the colon. If the colon is viable, some sort of venting stoma is placed. Tube cecostomy is a simple procedure and carries a lower mortality than resection.5 A large Foley catheter is left in place for 2 to 3 weeks. Stomas have relatively low immediate morbidity but relatively high late morbidity. CT-guided transperitoneal percutaneous cecostomy may be considered for patients unresponsive to medical treatment and unfit for surgery.28
Toxic Megacolon
The main initial goal of treatment is to reduce the severity of colitis and restore motility.11 Medical treatment is successful in about 50% of cases, but the patient should be assessed daily by the intensivist and the surgeon. Conditions impairing colonic motility must be corrected as far as possible (see Box 107-1). Antiperistaltic agents for diarrhea are absolutely contraindicated. Patients need general support with IV fluids, electrolyte and vitamin replacement, early optimization of circulation and, if necessary, mechanical ventilation. They are additionally treated with IV antibiotics, corticosteroids, selective decontamination of the digestive tract (SDD), and enteral nutrition. Tolerance of nutrition is monitored by gastric retention and abdominal signs. TPN offers no proven benefit.11
Systemic antibiotics are necessary to reduce septic complications and peritonitis. Systemic antibiotics should have an anaerobic and gram-negative spectrum guided by local susceptibility patterns and adjusted to fecal surveillance cultures. It is important to select antibiotics that give the least disturbance of the indigenous anaerobic flora, and in case of C. difficile, the culprit antibiotic is discontinued. SDD with the correct antibiotics (polymyxin 100 mg, tobramycin 80 mg, and amphotericin B 500 mg 4 times daily)76,77 attacks overgrowth of aerobic gram-negative bacteria and yeast species, reduces the fecal endotoxin pool, and leaves the protective flora intact.78 Systemic toxicity and associated infections are thus limited.77,79,80 In animal studies, decontamination of the bowel with oral nonabsorbable broad-spectrum antibiotics reduces iNOS expression and prevents dilatation.48 A trial with neostigmine may be useful to promote motility and defecation, allowing the oral antibiotics to reach the entire GI tract, clear bacterial overgrowth, and mitigate associated systemic toxicity.
There are two effective drugs for the treatment of C. difficile–associated diarrhea: vancomycin and metronidazole. Oral vancomycin is not absorbed, and high fecal concentrations are achieved. IV vancomycin is not effective. IV metronidazole may be secreted through an inflamed mucosa. In patients with active disease receiving oral or IV metronidazole, bactericidal fecal concentrations were achieved, but concentrations fell as the diarrhea improved, and neither substance was detectable in the feces after recovery.81 Although an older randomized controlled trial showed no difference between oral vancomycin 500 mg 4 times daily and oral metronidazole 250 mg 4 times daily,82 a recent trial comparing oral vancomycin 125 mg 4 times daily to oral metronidazole 250 mg 4 times daily for 10 days showed that metronidazole and vancomycin are equally effective for the treatment of mild C. difficile associated diarrhea, while vancomycin was superior for patients with severe diarrhea.83 Notably, treatment of asymptomatic carriers is not recommended.84 For antibiotic treatment of toxic megacolon due to C. difficile colitis, IV metronidazole (500 mg 3 times daily) may be considered in addition to vancomycin (500 mg 4 times daily, administered via the nasogastric tube).50,85 Vancomycin retention enemas might be administered as well (500 mg vancomycin in 100 mL normal saline). CMV colitis requires specific treatment with ganciclovir (5 mg/kg IV, with dose adjustment in patients with renal dysfunction).
All patients with severe colitis should be treated with corticosteroids.86 Corticosteroids are potent inhibitors of inflammation and specifically inhibit iNOS expression, preventing further colonic dilatation.48
Patients with toxic megacolon need surgery without delay if they are unresponsive to medical treatment. Surgical intervention should be considered when the patient has progressive signs of organ failure despite medical treatment, a worsening CT scan, or signs of peritonitis. Subtotal colectomy with end-ileostomy is the treatment of choice for urgent surgery.11,43,44
 Outcome
Outcome
With appropriate management, pseudo-obstruction usually resolves within a couple of days. However, hospitalization may be prolonged30 and mortality rate may be high because megacolon affects debilitated patients with other organ failure. Mortality is related to underlying disease, cecal diameter, delay in decompression, the kind of intervention, or the presence of an ischemic or perforated cecum.5,11,75 In patients with pseudo-obstruction needing surgery, mortality was 30% compared to 14% after early conservative treatment.5 In one series, all patients who died had coronary artery disease.31 In the presence of perforation, mortality rate may increase to 50%.75 Notably, none of these studies used neostigmine early after presentation. Colonic pseudo-obstruction reflects a failing organ, one that is not scored in the presently available organ failure scores.
In severe ulcerative colitis, the fatality of toxic megacolon is high, especially if surgery is delayed. The development of multiple organ failure predicts a fatal outcome.9 Mortality of toxic megacolon due to C. difficile infection rises to 80%.87 In a cohort of 59 intensive care patients with C. difficile colitis, one-fifth of the patients required surgery for progressive toxicity or peritonitis. In the surgical patients, APACHE scores at diagnosis were higher, and mortality rate was 42% compared to 15% in medical patients.44
 Strategies to Prevent Megacolon in the Critically Ill
Strategies to Prevent Megacolon in the Critically Ill
In contrast to the wide attention paid in the literature to gastric emptying, little notice is taken of defecation. Among critically ill patients, it is not unusual for the first stools to be passed after more than a week.34 Defecation removes bacteria from the gut and reduces overgrowth of pathogenic bacteria and yeasts. With respect to the potentially lethal complications of Ogilvie’s syndrome and toxic megacolon, clinical awareness and a strategy of care for the colon are crucial (see Box 107-4). This strategy includes early resuscitation of the circulation, tailored infusion therapy,88 correction of hypokalemia and hypomagnesemia, minimizing prolonged infusion of high doses of α-adrenergic drugs and dopamine,13 restrictive use of opioids, thoracic epidural anesthesia,89 avoiding antibiotics, which disrupt the growth of anaerobic fecal bacteria, early enteral feeding, avoiding routine use of proton pump inhibitors, promoting defecation, and early mobilization and ambulation. Nutrients in the gut directly stimulate proliferation of enterocytes and motility by the production of GI messengers, acting via autocrine, paracrine, and endocrine pathways. The use of antibiotics that affect the growth of indigenous protective colonic microflora should be avoided whenever possible. SDD is advocated in patients with an expected stay of more than a few days. Proper SDD76 prevents overgrowth of aerobic gram-negative bacteria and yeasts and reduces the fecal endotoxin pool and associated motility disorder, systemic toxicity,78 gram-negative infections, and mortality.77,80 With these measures, C. difficile colitis is virtually absent in the ICU. If defecation does not occur spontaneously, an enema and oral polyethylene glycol (PEG 13.125 g in 100 mL water 3 times daily) is advocated from day 3.90,91 Compared to PEG, Ogilvie’s syndrome was more often seen with lactulose. When stools do not pass and physical examination of the abdomen is without suspicion, neostigmine is started. By implementation of a protocol promoting defecation, deterioration of the patient’s condition by dilatation of the colon can be prevented.
Key Points
Saunders MD, Kimmey MB. Colonic pseudo-obstruction: the dilated colon in the ICU. Semin Gastrointest Dis. 2003;14:20-27.
Fruhwald S, Holzer P, Metzler H. Gastrointestinal motility in acute illness. Wien Klin Wochenschr. 2008;120:6-17.
Boeckxstaens GE, de Jonge WJ. Neuroimmune mechanisms in postoperative ileus. Gut. 2009;58:1300-1311.
Lomax AE, Sharkey KA, Furness JB. The participation of the sympathetic innervation of the gastrointestinal tract in disease states. Neurogastroenterol Motil. 2010;22:7-18.
Van der Spoel JI, Oudemans-van Straaten HM, Stoutenbeek CP, Bosman RJ, Zandstra DF. Neostigmine resolves critical illness-related colonic ileus in intensive care patients with multiple organ failure—a prospective, double-blind, placebo-controlled trial. Intensive Care Med. 2001;27:822-827.
van der Spoel JI, Oudemans-van Straaten HM, Kuiper MA, van Roon EN, Zandstra DF, van der Voort PH. Laxation of critically ill patients with lactulose or polyethylene glycol: a two-center randomized, double-blind, placebo-controlled trial. Crit Care Med. 2007;35:2726-2731.
Saunders MD, Cappell MS. Endoscopic management of acute colonic pseudo-obstruction. Endoscopy. 2005;37:760-763.
Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis. 2007;45:302-307.
1 Saunders MD, Kimmey MB. Colonic pseudo-obstruction: the dilated colon in the ICU. Semin Gastrointest Dis. 2003;14:20-27.
2 Bharucha AE, Phillips SF. Megacolon: Acute, Toxic, and Chronic. Curr Treat Options Gastroenterol. 1999;2:517-523.
3 van der Spoel JI, Oudemans-van Straaten HM, Stoutenbeek CP, Bosman RJ, Zandstra DF. Neostigmine resolves critical illness-related colonic ileus in intensive care patients with multiple organ failure–a prospective, double-blind, placebo-controlled trial. Intensive Care Med. 2001;27:822-827.
4 Saunders MD. Acute colonic pseudo-obstruction. Gastrointest Endosc Clin N Am. 2007;17:341-vii.
5 Vanek VW, Al Salti M. Acute pseudo-obstruction of the colon (Ogilvie’s syndrome). An analysis of 400 cases. Dis Colon Rectum. 1986;29:203-210.
6 Tack J. Acute Colonic Pseudo-Obstruction (Ogilvie’s Syndrome). Curr Treat Options Gastroenterol. 2006;9:361-368.
7 Sheikh RA, Yasmeen S, Pauly MP, Trudeau WL. Pseudomembranous colitis without diarrhea presenting clinically as acute intestinal pseudo-obstruction. J Gastroenterol. 2001;36:629-632.
8 Caprilli R, Latella G, Vernia P, Frieri G. Multiple organ dysfunction in ulcerative colitis. Am J Gastroenterol. 2000;95:1258-1262.
9 Latella G, Vernia P, Viscido A, et al. GI distension in severe ulcerative colitis. Am J Gastroenterol. 2002;97:1169-1175.
10 Dobson G, Hickey C, Trinder J. Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome. Intensive Care Med. 2003;29:1030.
11 Sheth SG, LaMont JT. Toxic megacolon. Lancet. 1998;351:509-513.
12 Goyal RK, Hirano I. The enteric nervous system. N Engl J Med. 1996;334:1106-1115.
13 Fruhwald S, Holzer P, Metzler H. Gastrointestinal motility in acute illness. Wien Klin Wochenschr. 2008;120:6-17.
14 Bult H, Boeckxstaens GE, Pelckmans PA, Jordaens FH, Van Maercke YM, Herman AG. Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature. 1990;345:346-347.
15 Takahashi T, Owyang C. Regional differences in the nitrergic innervation between the proximal and the distal colon in rats. Gastroenterology. 1998;115:1504-1512.
16 Feighner SD, Tan CP, McKee KK, et al. Receptor for motilin identified in the human gastrointestinal system. Science. 1999;284:2184-2188.
17 Marzio L, Neri M, Pieramico O, Delle DM, Peeters TL, Cuccurullo F. Dopamine interrupts gastrointestinal fed motility pattern in humans. Effect on motilin and somatostatin blood levels. Dig Dis Sci. 1990;35:327-332.
18 Fruhwald S, Scheidl S, Toller W, et al. Low potential of dobutamine and dopexamine to block intestinal peristalsis as compared with other catecholamines. Crit Care Med. 2000;28:2893-2897.
19 Fruhwald S, Herk E, Petnehazy T, et al. Sufentanil potentiates the inhibitory effect of epinephrine on intestinal motility. Intensive Care Med. 2002;28:74-80.
20 Lomax AE, Sharkey KA, Furness JB. The participation of the sympathetic innervation of the gastrointestinal tract in disease states. Neurogastroenterol Motil. 2010;22:7-18.
21 Boeckxstaens GE, de Jonge WJ. Neuroimmune mechanisms in postoperative ileus. Gut. 2009;58:1300-1311.
22 Hughes SF, Scott SM, Pilot MA, Williams NS. Adrenoceptors and colocolonic inhibitory reflex. Dig Dis Sci. 1999;44:2462-2468.
23 Wood JD. Neurotransmission at the interface of sympathetic and enteric divisions of the autonomic nervous system. Chin J Physiol. 1999;42:201-210.
24 Greenwood-Van Meerveld B, Gardner CJ, Little PJ, Hicks GA, Dehaven-Hudkins DL. Preclinical studies of opioids and opioid antagonists on gastrointestinal function. Neurogastroenterol Motil. 2004;16(Suppl 2):46-53.
25 Cullen JJ, Caropreso DK, Ephgrave KS. Effect of endotoxin on canine gastrointestinal motility and transit. J Surg Res. 1995;58:90-95.
26 Russo A, Fraser R, Adachi K, Horowitz M, Boeckxstaens G. Evidence that nitric oxide mechanisms regulate small intestinal motility in humans. Gut. 1999;44:72-76.
27 Saunders MD. Acute colonic pseudo-obstruction. Best Pract Res Clin Gastroenterol. 2007;21:671-687.
28 De Giorgio R, Knowles CH. Acute colonic pseudo-obstruction. Br J Surg. 2009;96:229-239.
29 Ogilvie WH. William Heneage Ogilvie 1887-1971. Large-intestine colic due to sympathetic deprivation. A new clinical syndrome. Dis Colon Rectum. 1987;30:984-987.
30 Petrisor BA, Petruccelli DT, Winemaker MJ, de Beer JV. Acute colonic pseudo-obstruction after elective total joint arthroplasty. J Arthroplasty. 2001;16:1043-1047.
31 Tenofsky PL, Beamer L, Smith RS. Ogilvie syndrome as a postoperative complication. Arch Surg. 2000;135:682-686.
32 MacDonald A, McLaughlin DP, Fulton J, MacDonald E, Scott PJ. Effects of catecholamines on isolated human colonic smooth muscle. J Auton Pharmacol. 1996;16:213-220.
33 Walker JK, Gainetdinov RR, Mangel AW, Caron MG, Shetzline MA. Mice lacking the dopamine transporter display altered regulation of distal colonic motility. Am J Physiol Gastrointest Liver Physiol. 2000;279:G311-G318.
34 van der Spoel JI, Schultz MJ, van der Voort PH, de Jonge E. Influence of severity of illness, medication and selective decontamination on defecation. Intensive Care Med. 2006;32:875-880.
35 Viramontes BE, Malcolm A, Camilleri M, et al. Effects of an alpha(2)-adrenergic agonist on gastrointestinal transit, colonic motility, and sensation in humans. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1468-G1476.
36 Stieger DS, Cantieni R, Frutiger A. Acute colonic pseudoobstruction (Ogilvie’s syndrome) in two patients receiving high dose clonidine for delirium tremens. Intensive Care Med. 1997;23:780-782.
37 Ferraz AA, Cowles VE, Condon RE, Schulte WJ. Opioid and nonopioid analgesic drug effects on colon contractions in monkeys. Dig Dis Sci. 1995;40:1417-1419.
38 Kurz A, Sessler DI. Opioid-induced bowel dysfunction: pathophysiology and potential new therapies. Drugs. 2003;63:649-671.
39 Palmer SE, McLean RM, Ellis PM, Harrison-Woolrych M. Life-threatening clozapine-induced gastrointestinal hypomotility: an analysis of 102 cases. J Clin Psychiatry. 2008;69:759-768.
40 Peyriere H, Roux C, Ferard C, et al. Antipsychotics-induced ischaemic colitis and gastrointestinal necrosis: a review of the French pharmacovigilance database. Pharmacoepidemiol Drug Saf. 2009;18:948-955.
41 Cullen JJ, Caropreso DK, Hemann LL, Hinkhouse M, Conklin JL, Ephgrave KS. Pathophysiology of adynamic ileus. Dig Dis Sci. 1997;42:731-737.
42 Lindberg G. Nitric oxide and the migrating motor complex. Gut. 1999;44:7.
43 Teeuwen PH, Stommel MW, Bremers AJ, van der Wilt GJ, de Jong DJ, Bleichrodt RP. Colectomy in patients with acute colitis: a systematic review. J Gastrointest Surg. 2009;13:676-686.
44 Grundfest-Broniatowski S, Quader M, Alexander F, Walsh RM, Lavery I, Milsom J. Clostridium difficile colitis in the critically ill. Dis Colon Rectum. 1996;39:619-623.
45 Gorkiewicz G. Nosocomial and antibiotic-associated diarrhoea caused by organisms other than Clostridium difficile. Int J Antimicrob Agents. 2009;33(Suppl 1):S37-S41.
46 Chaudhuri A, Bekdash BA. Toxic megacolon due to Salmonella: a case report and review of the literature. Int J Colorectal Dis. 2002;17:275-279.
47 Beaugerie L, Ngo Y, Goujard F, et al. Etiology and management of toxic megacolon in patients with human immunodeficiency virus infection. Gastroenterology. 1994;107:858-863.
48 Mourelle M, Vilaseca J, Guarner F, Salas A, Malagelada JR. Toxic dilatation of colon in a rat model of colitis is linked to an inducible form of nitric oxide synthase. Am J Physiol. 1996;270:G425-G430.
49 Mourelle M, Casellas F, Guarner F, et al. Induction of nitric oxide synthase in colonic smooth muscle from patients with toxic megacolon. Gastroenterology. 1995;109:1497-1502.
50 Kyne L, Farrell RJ, Kelly CP. Clostridium difficile. Gastroenterol Clin North Am. 2001;30:753-775.
51 Bowden TAJr, Mansberger ARJr, Lykins LE. Pseudomembraneous enterocolitis: mechanism for restoring floral homeostasis. Am Surg. 1981;47:178-183.
52 van Nood E, Speelman P, Kuijper EJ, Keller JJ. Struggling with recurrent Clostridium difficile infections: is donor faeces the solution? Euro Surveill. 14, 2009.
53 Wilson KH. The microecology of Clostridium difficile. Clin Infect Dis. 1993;16(Suppl 4):S214-S218.
54 Dupont HL, Garey K, Caeiro JP, Jiang ZD. New advances in Clostridium difficile infection: changing epidemiology, diagnosis, treatment and control. Curr Opin Infect Dis. 2008;21:500-507.
55 Southern WN, Rahmani R, Aroniadis O, et al. Postoperative Clostridium difficile-associated diarrhea. Surgery. 2010;148:24-30.
56 Gasink LB, Brennan PJ. Isolation precautions for antibiotic-resistant bacteria in healthcare settings. Curr Opin Infect Dis. 2009;22:339-344.
57 Kyne L, Sougioultzis S, McFarland LV, Kelly CP. Underlying disease severity as a major risk factor for nosocomial Clostridium difficile diarrhea. Infect Control Hosp Epidemiol. 2002;23:653-659.
58 Deneve C, Janoir C, Poilane I, Fantinato C, Collignon A. New trends in Clostridium difficile virulence and pathogenesis. Int J Antimicrob Agents. 2009;33(Suppl 1):S24-S28.
59 Riddle DJ, Dubberke ER. Clostridium difficile infection in the intensive care unit. Infect Dis Clin North Am. 2009;23:727-743.
60 Thorson MA, Bliss DZ, Savik K. Re-examination of risk factors for non-Clostridium difficile-associated diarrhoea in hospitalized patients. J Adv Nurs. 2008;62:354-364.
61 Cunningham R, Dial S. Is over-use of proton pump inhibitors fuelling the current epidemic of Clostridium difficile-associated diarrhoea? J Hosp Infect. 2008;70:1-6.
62 Schermer CR, Hanosh JJ, Davis M, Pitcher DE. Ogilvie’s syndrome in the surgical patient: a new therapeutic modality. J Gastrointest Surg. 1999;3:173-177.
63 Imbriaco M, Balthazar EJ. Toxic megacolon: role of CT in evaluation and detection of complications. Clin Imaging. 2001;25:349-354.
64 Ananthakrishnan AN, Issa M, Binion DG. Clostridium difficile and inflammatory bowel disease. Med Clin North Am. 2010;94:135-153.
65 Planche T, Aghaizu A, Holliman R, et al. Diagnosis of Clostridium difficile infection by toxin detection kits: a systematic review. Lancet Infect Dis. 2008;8:777-784.
66 Tedesco FJ, Corless JK, Brownstein RE. Rectal sparing in antibiotic-associated pseudomembranous colitis: a prospective study. Gastroenterology. 1982;83:1259-1260.
67 Lee JT, Taylor BM, Singleton BC. Epidural anesthesia for acute pseudo-obstruction of the colon (Ogilvie’s syndrome). Dis Colon Rectum. 1988;31:686-691.
68 Carli F, Trudel JL, Belliveau P. The effect of intraoperative thoracic epidural anesthesia and postoperative analgesia on bowel function after colorectal surgery: a prospective, randomized trial. Dis Colon Rectum. 2001;44:1083-1089.
69 Ponec RJ, Saunders MD, Kimmey MB. Neostigmine for the treatment of acute colonic pseudo-obstruction. N Engl J Med. 1999;341:137-141.
70 MacColl C, MacCannell KL, Baylis B, Lee SS. Treatment of acute colonic pseudoobstruction (Ogilvie’s syndrome) with cisapride. Gastroenterology. 1990;98:773-776.
71 Longo WE, Vernava AMIII. Prokinetic agents for lower gastrointestinal motility disorders. Dis Colon Rectum. 1993;36:696-708.
72 Wilmer A, Dits H, Malbrain ML, Frans E, Tack J. Gastric emptying in the critically ill–the way forward. Intensive Care Med. 1997;23:928-929.
73 Saunders MD, Cappell MS. Endoscopic management of acute colonic pseudo-obstruction. Endoscopy. 2005;37:760-763.
74 Rex DK. Colonoscopy and acute colonic pseudo-obstruction. Gastrointest Endosc Clin N Am. 1997;7:499-508.
75 Dorudi S, Berry AR, Kettlewell MG. Acute colonic pseudo-obstruction. Br J Surg. 1992;79:99-103.
76 Oudemans-van Straaten HM, van Saene HK, Zandstra DF. Selective decontamination of the digestive tract: use of the correct antibiotics is crucial. Crit Care Med. 2003;31:334-335.
77 de Smet AM, Kluytmans JA, Cooper BS, et al. Decontamination of the digestive tract and oropharynx in ICU patients. N Engl J Med. 2009;360:20-31.
78 Van Saene JJM, Stoutenbeek CP, Van Saene HKF. Reduction of the intestinal endotoxin pool by three different SDD regimens in human volunteers. J Endotoxin Research. 1996;3:337-343.
79 Conraads VM, Jorens PG, De Clerck LS, et al. Selective intestinal decontamination in advanced chronic heart failure: a pilot trial. Eur J Heart Fail. 2004;6:483-491.
80 de Jonge E, Schultz MJ, Spanjaard L, et al. Effects of selective decontamination of digestive tract on mortality and acquisition of resistant bacteria in intensive care: a randomised controlled trial. Lancet. 2003;362:1011-1016.
81 Bolton RP, Culshaw MA. Faecal metronidazole concentrations during oral and intravenous therapy for antibiotic associated colitis due to Clostridium difficile. Gut. 1986;27:1169-1172.
82 Teasley DG, Gerding DN, Olson MM, et al. Prospective randomised trial of metronidazole versus vancomycin for Clostridium difficile-associated diarrhoea and colitis. Lancet. 1983;2:1043-1046.
83 Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis. 2007;45:302-307.
84 Johnson S, Homann SR, Bettin KM, et al. Treatment of asymptomatic Clostridium difficile carriers (fecal excretors) with vancomycin or metronidazole. A randomized, placebo-controlled trial. Ann Intern Med. 1992;117:297-302.
85 Fekety R. Guidelines for the diagnosis and management of Clostridium difficile-associated diarrhea and colitis. American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 1997;92:739-750.
86 Jakobovits SL, Travis SP. Management of acute severe colitis. Br Med Bull. 2005;75-6:131-144.
87 Prendergast TM, Marini CP, D’Angelo AJ, Sher ME, Cohen JR. Surgical patients with pseudomembranous colitis: factors affecting prognosis. Surgery. 1994;116:768-774.
88 Kehlet H, Holte K. Review of postoperative ileus. Am J Surg. 2001;182:3S-10S.
89 Holte K, Kehlet H. Postoperative ileus: a preventable event. Br J Surg. 2000;87:1480-1493.
90 van der Spoel JI, Oudemans-van Straaten HM, Kuiper MA, van Roon EN, Zandstra DF, van der Voort PH. Laxation of critically ill patients with lactulose or polyethylene glycol: a two-center randomized, double-blind, placebo-controlled trial. Crit Care Med. 2007;35:2726-2731.
91 Sgouros SN, Vlachogiannakos J, Vassiliadis K, et al. Effect of polyethylene glycol electrolyte balanced solution on patients with acute colonic pseudo obstruction after resolution of colonic dilation: a prospective, randomised, placebo controlled trial. Gut. 2006;55:638-642.
92 Ross JP, Small TR, Lepage PA. Imipramine overdose complicated by toxic megacolon. Am Surg. 1998;64:242-244.
93 Brubacher JR, Levine B, Hoffman RS. Intestinal pseudo-obstruction (Ogilvie’s syndrome) in theophylline overdose. Vet Hum Toxicol. 1996;38:368-370.
