[level-membership-for-pulmolory-and-respiratory-category]
Chapter 56 Toxic Inhalational Lung Injury
A variety of chemicals, when liberated into the atmosphere as gases, fumes, or mist, can cause irritant lung injury or asphyxiation. As summarized in Table 56-1, any level of the respiratory tract can be the target for toxins, which produce a wide range of disorders, from tracheitis and bronchitis to pulmonary edema.
Table 56-1 Inhalational Toxins and Range of Toxicity
| Toxins | Effects |
|---|---|
| Ammonia | Mucous membrane irritation and sloughing Bronchiectasis, pulmonary interstitial fibrosis |
| Ammonia, phosgene, hydrogen sulfide | Laryngeal edema and obstruction |
| Carbon monoxide, cyanide, hydrogen sulfide | Asphyxiation |
| Chlorine, phosgene | Acute respiratory distress syndrome |
| Hydrofluoric acid | Systemic effects, hypocalcemia, hypomagnesemia |
| Hydrogen chloride, chlorine | Tracheobronchitis |
| Hydrogen fluoride, mustard gas | Chemical pneumonitis |
| Hydrogen sulfide | Bacterial pneumonia |
| Nitric oxide | Systemic effects, methemoglobinemia |
| Oxides of nitrogen, sulfur oxides | Obliterative bronchiolitis (bronchiolitis obliterans) |
| Sulfur dioxide, hydrogen chloride, oxides of nitrogen, ozone | Bronchoconstriction, airway edema, asthma |
Etiology and Risk Factors
Occupational injuries more often occur when workers handle chemicals, work in areas that are inadequately ventilated, or enter exposed areas with improper protective equipment. Table 56-2 lists sources of occupational exposure to major chemical causes of irritant lung injury and asphyxiation.
| Toxin | Sources of Exposure |
|---|---|
| Ammonia | Agriculture, explosives, plastics |
| Carbon monoxide | Firefighters, smoke inhalation, smelters, miners, transportation, home furnaces |
| Chlorine | Household cleaners, paper, textiles, sewage treatment, swimming pools |
| Hydrofluoric acid | Fertilizers, insecticides, glass and ceramic etching, masonry, metalworking, pharmaceuticals, chemical manufacture |
| Hydrogen chloride | Fertilizers, textiles, dyes, rubber manufacture |
| Hydrogen cyanide | Metallurgy, electroplating, plastics, polyurethane manufacture |
| Hydrogen sulfide | Metallurgy, chemical manufacture, wastewater treatment, natural gas and oil drilling, paper mills, coke ovens, rayon manufacture, rubber vulcanization |
| Mustard gas | Chemical warfare |
| Oxides of nitrogen | Air pollution, welding, hockey rinks, chemical and dye manufacture, agriculture |
| Ozone | Welding, air pollution, high altitude, chemical manufacture |
| Phosgene | Firefighters; paint strippers; chemical, pharmaceutical, and dye manufacturing; chemical warfare |
| Sulfur dioxide | Air pollution, smelting, power plants, chemical manufacture, paper manufacture, food preparation |
Factors that influence the acute effects of toxic chemicals include solubility, particle size, concentration, duration of exposure, chemical properties, and individual factors such as minute ventilation. The more water-soluble compounds dissolve in the upper respiratory tract and airways, whereas the less water-soluble agents tend to bypass the upper airway and affect peripheral airways and pulmonary parenchyma (Figure 56-1).
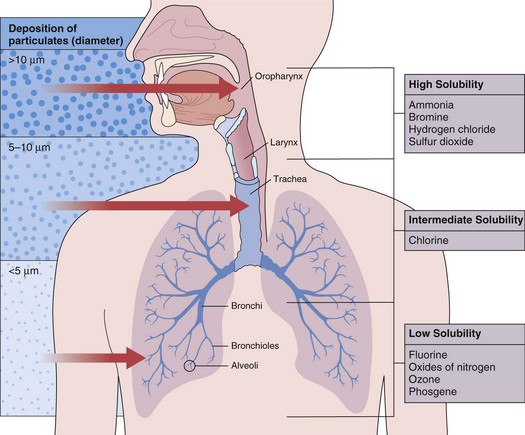
Figure 56-1 Distribution of gases and particulate matter in the respiratory tract influences the site of toxic injury.
Pathology
In general, the upper airway can be affected by most inhaled toxins, which result in edema of the nasal passage, posterior oropharynx, and larynx. In severe cases, mucous membrane ulceration and hemorrhage ensue. Toxins of low water solubility may reach the lung parenchyma without necessarily producing upper airway lesions. If breath holding, laryngospasm, and normal “scrubbing” activities of the nasopharynx fail to contain the exposure, lesions develop in the trachea and bronchi (e.g., paralysis of cilia, increased mucus production, goblet cell hyperplasia, injury to airway epithelium, epithelial denudation, exudation, submucosal hemorrhage, edema). Pseudomembranes may form along the trachea and bronchi, causing various degrees of bronchiolitis, bronchiolitis obliterans (Figure 56-2), and organizing pneumonia (Figure 56-3). Bronchiolitis has been associated with exposures to oxides of nitrogen—nitric oxide (NO), nitrogen dioxide (NO2), and nitrogen peroxide (N2O4)—as well as sulfur dioxide, ammonia, chlorine (Cl2), phosgene, fly ash that contains trichloroethylene (C2HCl3), ozone (O3), hydrogen sulfide, hydrogen fluoride (HF), metal oxide fumes, dusts (e.g., asbestos, silica, talc, grain dust), free-base cocaine, tobacco smoke, and fire smoke.
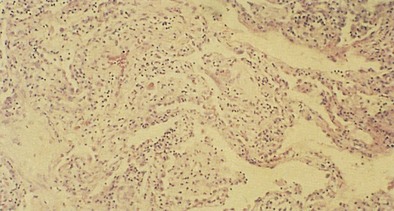
Figure 56-2 Histologic appearance of bronchiolitis obliterans caused by inhalation of oxides of nitrogen in silo filler.
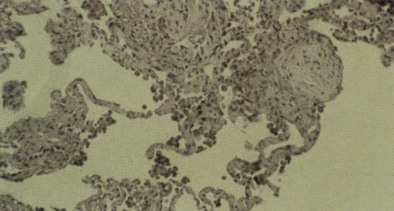
Parenchymal injury is less common than airway damage. When alveolar or interstitial injury occurs, both epithelial damage and endothelial damage are observed, resulting in alveolocapillary leak and the pathologic changes of adult respiratory distress syndrome (ARDS). Diffuse alveolar damage (DAD) is a common histologic pattern in acute interstitial lung disease caused by inhaled toxins. It is characterized by widespread, diffuse edema, epithelial necrosis and cell sloughing (with exudates that fill the alveolar spaces), and formation of hyaline membranes (Figure 56-4). Later, DAD may organize, which leads to proliferation of type II pneumonocytes, resorption of the hyaline membranes and exudates, and fibroblast proliferation. Long-term survivors of such parenchymal injury may fully recover or may have various degrees of permanent interstitial fibrosis.
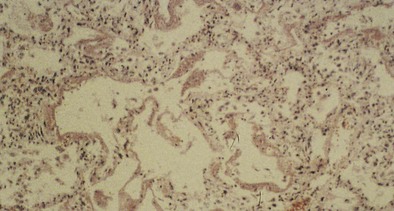
Clinical Features and Diagnosis
Diagnosis should focus on the nature of the compound inhaled, the magnitude and duration of exposure, and the water solubility of the inhaled agent. Inhalational injury is suspected in those who have facial burns or inflamed nares. Headache and dizziness, along with chest pains and emesis, suggest systemic poisons (e.g., cyanide, H2S). Unconscious victims found in confined spaces are assumed to have received longer inhalational exposures than conscious ones because of the unprotected airways and concentrated exposures. Evidence of hoarseness, upper airway stridor, wheezing or rales, cough, and sputum production is assessed. Chest radiographs may show pulmonary edema, atelectasis, or infiltrates (Figure 56-5), although they are often negative early after exposure. Flow-volume loops are the most sensitive noninvasive indicators of upper and lower airway obstruction. Hypoxemia in the face of a normal arterial partial pressure of oxygen (PaO2) suggests CO toxicity. Carboxyhemoglobin levels are obtained for all fire and explosion victims. Metabolic acidosis may indicate cyanide or H2S intoxication.
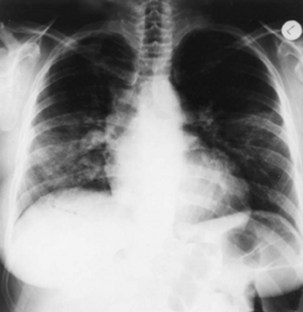
Figure 56-5 Radiographic changes produced by diffuse alveolar damage. Chest radiograph of the same patient as in Figure 56-4 shows acute, diffuse alveolar and interstitial infiltrates.
Chemical Irritants
Inflammatory and Autoimmune Agents
Biochemical Toxins and Weaponized Agents
A variety of agents have been developed and used to cause mass injury or disruption. Table 56-3 provides an overview of pulmonary effects and treatment of these toxicities. With all chemical exposures, individuals should be removed from exposure and given supportive care.
Table 56-3 Categories of Biochemical Poisons and Weapons: Specific Agents, Pulmonary Effects, and Treatment
| Toxin | Pulmonary Effect | Treatment |
|---|---|---|
| Biotoxins/Poisoning Agents | ||
| Ricin (phytotoxin) | Cough, pulmonary edema | Pulmonary toilet, oxygen, mechanical ventilation, cardiopulmonary resuscitation |
| Blister Agents/Vesicants | ||
| Lewisite | Respiratory tract irritation, coryza sneezing, hoarseness, epistaxis, dyspnea and cough, pneumonitis, pulmonary edema, respiratory failure | Dimercaprol (antilewisite, British anti-lewisite [BAL]) |
| Lung-Damaging Agents | ||
| Chloropicrin | Coughing, dyspnea, chest tightness, pulmonary edema | β2-Adrenergic agonists and corticosteroids for bronchospasm |
| Nerve Agents | ||
| Sarin, Soman, Tabun, VX | Respiratory failure | Atropine, 2-PAM (pralidoxime); benzodiazepines for convulsions |
| Riot Control/Tearing Agents | ||
| Chloroacetophenone | Rhinorrhea, cough, sneezing, chest tightness, dyspnea, pulmonary edema, bronchospasm | |
| Vomiting Agents | ||
| Adamsite (diphenylamine chlorarsine) | Rhinorrhea, cough, sneezing | Oxygen; bronchodilators for bronchospasm |
Aldrich TK, Gustave J, Hall CB, et al. Lung function in rescue workers at the World Trade Center after 7 years. N Engl J Med. 2010;362:1263–1272.
Amshel CE, Fealk MH, Phillips BJ, Caruso DM. Anhydrous ammonia burns: case report and review of the literature. Burns. 2000;26:493–497.
Banauch GL, Dhala A, Prezant DJ. Pulmonary disease in rescue workers at the World Trade Center site. Curr Opin Pulm Med. 2005;11:160–168.
Das R, Blanc PD. Chlorine gas exposure and the lung: a review. Toxicol Ind Health. 1993;9:439–455.
Hnizdo E, Sullivan PA, Moon Bang K, Wagner G. Association between chronic obstructive pulmonary disease and employment by industry and occupation in the US population: a study of data from the Third National Health and Nutrition Examination Survey. Am J Epidemiol. 2002;156:738–746.
Mapp CE, Boschetto P, Maestrelli P, Fabbri LM. Occupational asthma. Am J Respir Crit Care Med. 2005;172:280–305.
Miller K, Chang A. Acute inhalation injury. Emerg Med Clin North Am. 2003;21:533–557.
Newman LS. Current concepts: occupational illness. N Engl J Med. 1995;333:1128–1134.
Perkner JJ, Fennelly KP, Balkissoon R, et al. Irritant-associated vocal cord dysfunction. J Occup Environ Med. 1998;40:136–143.
US National Institute for Occupational Safety and Health. The emergency response safety and health database. www.cdc.gov/NIOSH/ershdb/AgentListCategory.html, 2008.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Chapter 56 Toxic Inhalational Lung Injury
A variety of chemicals, when liberated into the atmosphere as gases, fumes, or mist, can cause irritant lung injury or asphyxiation. As summarized in Table 56-1, any level of the respiratory tract can be the target for toxins, which produce a wide range of disorders, from tracheitis and bronchitis to pulmonary edema.
Table 56-1 Inhalational Toxins and Range of Toxicity
| Toxins | Effects |
|---|---|
| Ammonia | Mucous membrane irritation and sloughing Bronchiectasis, pulmonary interstitial fibrosis |
| Ammonia, phosgene, hydrogen sulfide | Laryngeal edema and obstruction |
| Carbon monoxide, cyanide, hydrogen sulfide | Asphyxiation |
| Chlorine, phosgene | Acute respiratory distress syndrome |
| Hydrofluoric acid | Systemic effects, hypocalcemia, hypomagnesemia |
| Hydrogen chloride, chlorine | Tracheobronchitis |
| Hydrogen fluoride, mustard gas | Chemical pneumonitis |
| Hydrogen sulfide | Bacterial pneumonia |
| Nitric oxide | Systemic effects, methemoglobinemia |
| Oxides of nitrogen, sulfur oxides | Obliterative bronchiolitis (bronchiolitis obliterans) |
| Sulfur dioxide, hydrogen chloride, oxides of nitrogen, ozone | Bronchoconstriction, airway edema, asthma |
Etiology and Risk Factors
Occupational injuries more often occur when workers handle chemicals, work in areas that are inadequately ventilated, or enter exposed areas with improper protective equipment. Table 56-2 lists sources of occupational exposure to major chemical causes of irritant lung injury and asphyxiation.
| Toxin | Sources of Exposure |
|---|---|
| Ammonia | Agriculture, explosives, plastics |
| Carbon monoxide | Firefighters, smoke inhalation, smelters, miners, transportation, home furnaces |
| Chlorine | Household cleaners, paper, textiles, sewage treatment, swimming pools |
| Hydrofluoric acid | Fertilizers, insecticides, glass and ceramic etching, masonry, metalworking, pharmaceuticals, chemical manufacture |
| Hydrogen chloride | Fertilizers, textiles, dyes, rubber manufacture |
| Hydrogen cyanide | Metallurgy, electroplating, plastics, polyurethane manufacture |
| Hydrogen sulfide | Metallurgy, chemical manufacture, wastewater treatment, natural gas and oil drilling, paper mills, coke ovens, rayon manufacture, rubber vulcanization |
| Mustard gas | Chemical warfare |
| Oxides of nitrogen | Air pollution, welding, hockey rinks, chemical and dye manufacture, agriculture |
| Ozone | Welding, air pollution, high altitude, chemical manufacture |
| Phosgene | Firefighters; paint strippers; chemical, pharmaceutical, and dye manufacturing; chemical warfare |
| Sulfur dioxide | Air pollution, smelting, power plants, chemical manufacture, paper manufacture, food preparation |
Factors that influence the acute effects of toxic chemicals include solubility, particle size, concentration, duration of exposure, chemical properties, and individual factors such as minute ventilation. The more water-soluble compounds dissolve in the upper respiratory tract and airways, whereas the less water-soluble agents tend to bypass the upper airway and affect peripheral airways and pulmonary parenchyma (Figure 56-1).
Figure 56-1 Distribution of gases and particulate matter in the respiratory tract influences the site of toxic injury.
Pathology
In general, the upper airway can be affected by most inhaled toxins, which result in edema of the nasal passage, posterior oropharynx, and larynx. In severe cases, mucous membrane ulceration and hemorrhage ensue. Toxins of low water solubility may reach the lung parenchyma without necessarily producing upper airway lesions. If breath holding, laryngospasm, and normal “scrubbing” activities of the nasopharynx fail to contain the exposure, lesions develop in the trachea and bronchi (e.g., paralysis of cilia, increased mucus production, goblet cell hyperplasia, injury to airway epithelium, epithelial denudation, exudation, submucosal hemorrhage, edema). Pseudomembranes may form along the trachea and bronchi, causing various degrees of bronchiolitis, bronchiolitis obliterans (Figure 56-2), and organizing pneumonia (Figure 56-3). Bronchiolitis has been associated with exposures to oxides of nitrogen—nitric oxide (NO), nitrogen dioxide (NO2), and nitrogen peroxide (N2O4)—as well as sulfur dioxide, ammonia, chlorine (Cl2), phosgene, fly ash that contains trichloroethylene (C2HCl3), ozone (O3), hydrogen sulfide, hydrogen fluoride (HF), metal oxide fumes, dusts (e.g., asbestos, silica, talc, grain dust), free-base cocaine, tobacco smoke, and fire smoke.
Figure 56-2 Histologic appearance of bronchiolitis obliterans caused by inhalation of oxides of nitrogen in silo filler.
Parenchymal injury is less common than airway damage. When alveolar or interstitial injury occurs, both epithelial damage and endothelial damage are observed, resulting in alveolocapillary leak and the pathologic changes of adult respiratory distress syndrome (ARDS). Diffuse alveolar damage (DAD) is a common histologic pattern in acute interstitial lung disease caused by inhaled toxins. It is characterized by widespread, diffuse edema, epithelial necrosis and cell sloughing (with exudates that fill the alveolar spaces), and formation of hyaline membranes (Figure 56-4). Later, DAD may organize, which leads to proliferation of type II pneumonocytes, resorption of the hyaline membranes and exudates, and fibroblast proliferation. Long-term survivors of such parenchymal injury may fully recover or may have various degrees of permanent interstitial fibrosis.
Clinical Features and Diagnosis
Diagnosis should focus on the nature of the compound inhaled, the magnitude and duration of exposure, and the water solubility of the inhaled agent. Inhalational injury is suspected in those who have facial burns or inflamed nares. Headache and dizziness, along with chest pains and emesis, suggest systemic poisons (e.g., cyanide, H2S). Unconscious victims found in confined spaces are assumed to have received longer inhalational exposures than conscious ones because of the unprotected airways and concentrated exposures. Evidence of hoarseness, upper airway stridor, wheezing or rales, cough, and sputum production is assessed. Chest radiographs may show pulmonary edema, atelectasis, or infiltrates (Figure 56-5), although they are often negative early after exposure. Flow-volume loops are the most sensitive noninvasive indicators of upper and lower airway obstruction. Hypoxemia in the face of a normal arterial partial pressure of oxygen (PaO2) suggests CO toxicity. Carboxyhemoglobin levels are obtained for all fire and explosion victims. Metabolic acidosis may indicate cyanide or H2S intoxication.
Figure 56-5 Radiographic changes produced by diffuse alveolar damage. Chest radiograph of the same patient as in Figure 56-4 shows acute, diffuse alveolar and interstitial infiltrates.
