Hepatitis B Until 1982, prevention of hepatitis B was based on passive immunoprophylaxis either with standard Ig, containing modest levels of anti-HBs, or hepatitis B immunoglobulin (HBIG), containing high-titer anti-HBs. The efficacy of standard IG has never been established and remains questionable; even the efficacy of HBIG, demonstrated in several clinical trials, has been challenged, and its contribution appears to be in reducing the frequency of clinical illness, not in preventing infection. The first vaccine for active immunization, introduced in 1982, was prepared from purified, noninfectious 22-nm spherical HBsAg particles derived from the plasma of healthy HBsAg carriers. In 1987, the plasma-derived vaccine was supplanted by a genetically engineered vaccine derived from recombinant yeast. The latter vaccine consists of HBsAg particles that are nonglycosylated but are otherwise indistinguishable from natural HBsAg; two recombinant vaccines are licensed for use in the United States. Current recommendations can be divided into those for pre-exposure and postexposure prophylaxis.
For pre-exposure prophylaxis against hepatitis B in settings of frequent exposure (health workers exposed to blood; first-responder public safety workers; hemodialysis patients and staff; residents and staff of custodial institutions for the developmentally handicapped; injection drug users; inmates of long-term correctional facilities; persons with multiple sexual partners or who have had a sexually transmitted disease; men who have sex with men; persons such as hemophiliacs who require long-term, high-volume therapy with blood derivatives; household and sexual contacts of persons with chronic HBV infection; persons living in or traveling extensively in endemic areas; unvaccinated children under the age of 18; unvaccinated children who are Alaskan natives, Pacific Islanders, or residents in households of first-generation immigrants from endemic countries; persons born in countries with a prevalence of HBV infection ≥2%; patients with chronic liver disease; persons <age 60 with diabetes mellitus [those ≥60 at the discretion of their physicians]; persons with end-stage renal disease; persons with HIV infection), three IM (deltoid, not gluteal) injections of hepatitis B vaccine are recommended at 0, 1, and 6 months (other, optional schedules are summarized in Table 360-8). Pregnancy is not a contraindication to vaccination. In areas of low HBV endemicity such as the United States, despite the availability of safe and effective hepatitis B vaccines, a strategy of vaccinating persons in high-risk groups was not effective. The incidence of new hepatitis B cases continued to increase in the United States after the introduction of vaccines; <10% of all targeted persons in high-risk groups were actually vaccinated, and ~30% of persons with sporadic acute hepatitis B did not fall into any high-risk-group category. Therefore, to have an impact on the frequency of HBV infection in an area of low endemicity such as the United States, universal hepatitis B vaccination in childhood has been recommended. For unvaccinated children born after the implementation of universal infant vaccination, vaccination during early adolescence, at age 11–12 years, was recommended, and this recommendation has been extended to include all unvaccinated children age 0–19 years. In HBV-hyperendemic areas (e.g., Asia), universal vaccination of children has resulted in a marked 10- to 15-year decline in hepatitis B and its complications, including hepatocellular carcinoma.
|
PRE-EXPOSURE HEPATITIS B VACCINATION SCHEDULES |
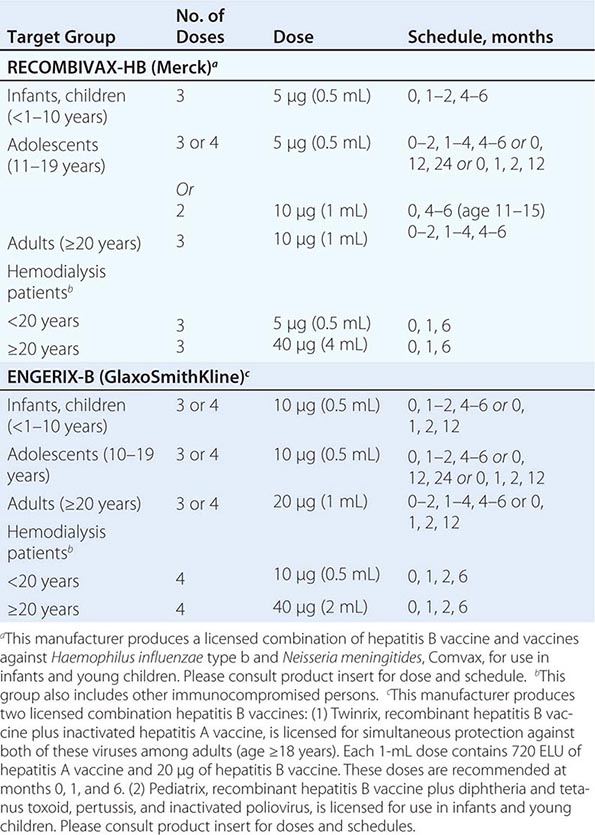
The two available recombinant hepatitis B vaccines are comparable, one containing 10 μg of HBsAg (Recombivax-HB) and the other containing 20 μg of HBsAg (Engerix-B), and recommended doses for each injection vary for the two preparations (Table 360-8). Combinations of hepatitis B vaccine with other childhood vaccines are available as well (Table 360-8).
For unvaccinated persons sustaining an exposure to HBV, post-exposure prophylaxis with a combination of HBIG (for rapid achievement of high-titer circulating anti-HBs) and hepatitis B vaccine (for achievement of long-lasting immunity as well as its apparent efficacy in attenuating clinical illness after exposure) is recommended. For perinatal exposure of infants born to HBsAg-positive mothers, a single dose of HBIG, 0.5 mL, should be administered IM in the thigh immediately after birth, followed by a complete course of three injections of recombinant hepatitis B vaccine (see doses above) to be started within the first 12 h of life. For those experiencing a direct percutaneous inoculation or transmucosal exposure to HBsAg-positive blood or body fluids (e.g., accidental needle stick, other mucosal penetration, or ingestion), a single IM dose of HBIG, 0.06 mL/kg, administered as soon after exposure as possible, is followed by a complete course of hepatitis B vaccine to begin within the first week. For those exposed by sexual contact to a patient with acute hepatitis B, a single IM dose of HBIG, 0.06 mL/kg, should be given within 14 days of exposure, to be followed by a complete course of hepatitis B vaccine. When both HBIG and hepatitis B vaccine are recommended, they may be given at the same time but at separate sites. Testing adults for anti-HBs after a course of vaccine is advisable to document the acquisition of immunity, but, because hepatitis B vaccine immunogenicity is nearly universal in infants, postvaccination anti-HBs testing of children is not recommended.
The precise duration of protection afforded by hepatitis B vaccine is unknown; however, ~80–90% of immunocompetent adult vaccinees retain protective levels of anti-HBs for at least 5 years, and 60–80% for 10 years, and protective antibody has been documented to last for at least two decades after vaccination in infancy. Thereafter and even after anti-HBs becomes undetectable, protection persists against clinical hepatitis B, hepatitis B surface antigenemia, and chronic HBV infection. Currently, booster immunizations are not recommended routinely, except in immunosuppressed persons who have lost detectable anti-HBs or immunocompetent persons who sustain percutaneous HBsAg-positive inoculations after losing detectable antibody. Specifically, for hemodialysis patients, annual anti-HBs testing is recommended after vaccination; booster doses are recommended when anti-HBs levels fall to <10 mIU/mL. As noted above, for persons at risk of both hepatitis A and B, a combined vaccine is available containing 720 enzyme-linked immunoassay units (ELUs) of inactivated HAV and 20 μg of recombinant HBsAg (at 0, 1, and 6 months).
Hepatitis D Infection with hepatitis D can be prevented by vaccinating susceptible persons with hepatitis B vaccine. No product is available for immunoprophylaxis to prevent HDV superinfection in HBsAg carriers; for them, avoidance of percutaneous exposures and limitation of intimate contact with persons who have HDV infection are recommended.
Hepatitis C IG is ineffective in preventing hepatitis C and is no longer recommended for postexposure prophylaxis in cases of perinatal, needle stick, or sexual exposure. Although prototype vaccines that induce antibodies to HCV envelope proteins have been developed, currently, hepatitis C vaccination is not feasible practically. Genotype and quasispecies viral heterogeneity as well as rapid evasion of neutralizing antibodies by this rapidly mutating virus, conspire to render HCV a difficult target for immunoprophylaxis with a vaccine. Prevention of transfusion-associated hepatitis C has been accomplished by the following successively introduced measures: exclusion of commercial blood donors and reliance on a volunteer blood supply; screening donor blood with surrogate markers such as ALT (no longer recommended) and anti-HBc, markers that identify segments of the blood donor population with an increased risk of bloodborne infections; exclusion of blood donors in high-risk groups for AIDS and the introduction of anti-HIV screening tests; and progressively sensitive serologic and virologic screening tests for HCV infection.
In the absence of active or passive immunization, prevention of hepatitis C includes behavior changes and precautions to limit exposures to infected persons. Recommendations designed to identify patients with clinically inapparent hepatitis as candidates for medical management have as a secondary benefit the identification of persons whose contacts could be at risk of becoming infected. A so-called look-back program has been recommended to identify persons who were transfused before 1992 with blood from a donor found subsequently to have hepatitis C. In addition, anti-HCV testing is recommended for persons born between 1945 and 1965, anyone who received a blood transfusion or a transplanted organ before the introduction of second-generation screening tests in 1992, those who ever used injection drugs (or took other illicit drugs by noninjection routes), chronically hemodialyzed patients, persons with clotting disorders who received clotting factors made before 1987 from pooled blood products, persons with elevated aminotransferase levels, health workers exposed to HCV-positive blood or contaminated needles, recipients of blood or organs from a donor found to be positive for hepatitis C, persons with HIV infection, health care and public safety personnel following a needle stick or other nonpercutaneous exposure to HCV-infected material, sexual partners of persons with hepatitis C, and children born to HCV-positive mothers (Table 360-4).
For stable, monogamous sexual partners, sexual transmission of hepatitis C is unlikely, and sexual barrier precautions are not recommended. For persons with multiple sexual partners or with sexually transmitted diseases, the risk of sexual transmission of hepatitis C is increased, and barrier precautions (latex condoms) are recommended. A person with hepatitis C should avoid sharing such items as razors, toothbrushes, and nail clippers with sexual partners and family members. No special precautions are recommended for babies born to mothers with hepatitis C, and breast-feeding does not have to be restricted.
Hepatitis E Whether IG prevents hepatitis E remains undetermined. Safe and effective recombinant genotype 1 vaccines, which protect against other genotypes as well, have been developed and are available in endemic areas but not in the United States.
361 |
Toxic and Drug-Induced Hepatitis |
Liver injury is a possible consequence of ingestion of any xenobiotic, including industrial toxins, pharmacologic agents, and complementary and alternative medications (CAMs). Among patients with acute liver failure, drug-induced liver injury is the most common cause, and evidence for hepatotoxicity detected during clinical trials for drug development is the most common reason for failure of compounds to reach approval status. Drug-induced liver injury requires careful history taking to identify unrecognized exposure to chemicals used in work or at home, drugs taken by prescription or bought over the counter, and herbal or dietary supplement medicines. Hepatotoxic drugs can injure the hepatocyte directly, e.g., via a free-radical or metabolic intermediate that causes peroxidation of membrane lipids and that results in liver cell injury. Alternatively, a drug or its metabolite may activate components of the innate or adaptive immune system, stimulate apoptotic pathways, or initiate damage to bile excretory pathways (Fig. 361-1). Interference with bile canalicular pumps can allow endogenous bile acids, which can injure the liver, to accumulate. Such secondary injury, in turn, may lead to necrosis of hepatocytes; injure bile ducts, producing cholestasis; or block pathways of lipid movement, inhibit protein synthesis, or impair mitochondrial oxidation of fatty acids, resulting in lactic acidosis and intracellular triglyceride accumulation (expressed histologically as microvesicular steatosis). In other instances, drug metabolites sensitize hepatocytes to toxic cytokines. The differences observed between susceptible and nonsusceptible drug recipients may be attributable to HLA haplotypes that determine binding of drug-related haptens on the cell surface as well as to polymorphisms in elaboration of competing, protective cytokines, as has been suggested for acetaminophen hepatotoxicity (see below). Immune mechanisms may include cytotoxic lymphocytes or antibody-mediated cellular cytotoxicity. In addition, a role has been shown for activation of nuclear transporters, such as the constitutive androstane receptor (CAR) or, more recently, the pregnane × receptor (PXR), in the induction of drug hepatotoxicity.
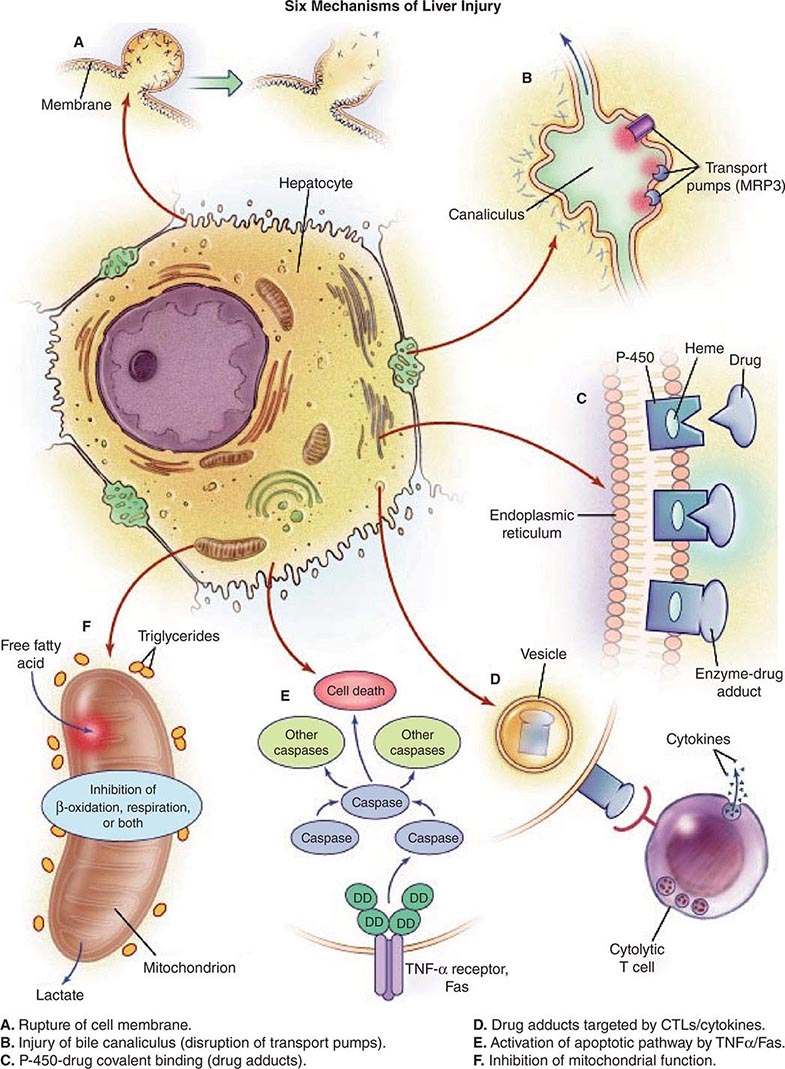
FIGURE 361-1 Potential mechanisms of drug-induced liver injury. The normal hepatocyte may be affected adversely by drugs through (A) disruption of intracellular calcium homeostasis that leads to the disassembly of actin fibrils at the surface of the hepatocyte, resulting in blebbing of the cell membrane, rupture, and cell lysis; (B) disruption of actin filaments next to the canaliculus (the specialized portion of the cell responsible for bile excretion), leading to loss of villous processes and interruption of transport pumps such as multidrug resistance–associated protein 3 (MRP3), which, in turn, prevents the excretion of bilirubin and other organic compounds; (C) covalent binding of the heme-containing cytochrome P450 enzyme to the drug, thus creating nonfunctioning adducts; (D) migration of these enzyme-drug adducts to the cell surface in vesicles to serve as target immunogens for cytolytic attack by T cells, stimulating an immune response involving cytolytic T cells and cytokines; (E) activation of apoptotic pathways by tumor necrosis factor α (TNF-α) receptor or Fas (DD denotes death domain), triggering the cascade of intercellular caspases, resulting in programmed cell death; or (F) inhibition of mitochondrial function by a dual effect on both β-oxidation and the respiratory-chain enzymes, leading to failure of free fatty acid metabolism, a lack of aerobic respiration, and accumulation of lactate and reactive oxygen species (which may disrupt mitochondrial DNA). Toxic metabolites excreted in bile may damage bile-duct epithelium (not shown). CTLs, cytolytic T lymphocytes. (Reproduced from WM Lee: Drug-induced hepatotoxicity. N Engl J Med 349:474, 2003, with permission.)
DRUG METABOLISM
Most drugs, which are water-insoluble, undergo a series of metabolic steps, culminating in a water-soluble form appropriate for renal or biliary excretion. This process begins with oxidation or methylation mediated initially by the microsomal mixed-function oxygenases, cytochrome P450 (phase I reaction), followed by glucuronidation or sulfation (phase II reaction) or inactivation by glutathione. Most drug hepatotoxicity is mediated by a phase I toxic metabolite, but glutathione depletion, precluding inactivation of harmful compounds by glutathione S-transferase, can contribute as well.
LIVER INJURY CAUSED BY DRUGS
In general, two major types of chemical hepatotoxicity have been recognized: (1) direct toxic and (2) idiosyncratic. As shown in Table 361-1, direct toxic hepatitis occurs with predictable regularity in individuals exposed to the offending agent and is dose-dependent. The latent period between exposure and liver injury is usually short (often several hours), although clinical manifestations may be delayed for 24–48 h. Agents producing toxic hepatitis are generally systemic poisons or are converted in the liver to toxic metabolites. The direct hepatotoxins result in morphologic abnormalities that are reasonably characteristic and reproducible for each toxin. For example, carbon tetrachloride and trichloroethylene characteristically produce a centrilobular zonal necrosis, whereas yellow phosphorus poisoning typically results in periportal injury. The hepatotoxic octapeptides of Amanita phalloides usually produce massive hepatic necrosis; the lethal dose of the toxin is ~10 mg, the amount found in a single deathcap mushroom. Liver injury, which is often only one facet of the toxicity produced by the direct hepatotoxins, may go unrecognized until jaundice appears.
|
SOME FEATURES OF TOXIC AND DRUG-INDUCED HEPATIC INJURY |
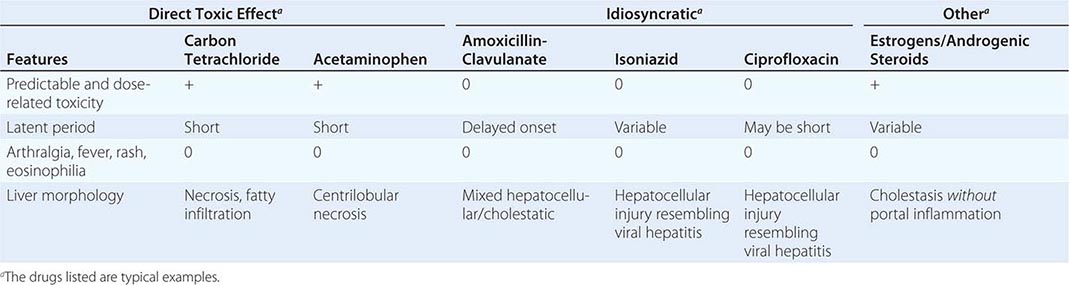
In idiosyncratic drug reactions, the occurrence of hepatitis is usually infrequent (1 in 103–105 patients) and unpredictable; the response is not as clearly dose-dependent as is injury associated with direct hepatotoxins, and liver injury may occur at any time during or shortly after exposure to the drug. That said, recent data suggest that most agents causing idiosyncratic toxicity are given at a daily dose exceeding 100 mg, suggesting a role for dose—drugs with low potency must be given in higher doses that engender greater chances for “off-target” effects. Adding to the difficulty of predicting or identifying idiosyncratic drug hepatotoxicity is the occurrence of mild, transient, nonprogressive serum aminotransferase elevations that resolve with continued drug use. Such “adaptation,” the mechanism of which is unknown, is well recognized for drugs such as isoniazid, valproate, phenytoin, and HMG-CoA reductase inhibitors (statins). Extrahepatic manifestations of hypersensitivity, such as rash, arthralgias, fever, leukocytosis, and eosinophilia, occur in about one-quarter of patients with idiosyncratic hepatotoxic drug reactions but are characteristic for certain drugs and not others. Both primary immunologic injury and direct hepatotoxicity related to idiosyncratic differences in generation of toxic metabolites have been invoked to explain idiosyncratic drug reactions. The most current data appear to implicate the adaptive immune system responding to the formation of immune stimulatory compounds resulting from phase I metabolic activation of the offending drug. Differences in host susceptibility may result from varying kinetics of toxic metabolite generation and genetic polymorphisms in downstream drug-metabolizing pathways or cytokine activation; in addition, certain HLA haplotypes have been associated with hepatotoxicity of certain drugs such as amoxicillin-clavulanate and flucloxacillin. Occasionally, however, the clinical features of an allergic reaction (prominent tissue eosinophilia, autoantibodies, etc.) are difficult to ignore and suggest activation of IgE pathways. A few instances of drug hepatotoxicity are observed to be associated with autoantibodies, including a class of antibodies to liver-kidney microsomes, anti-LKM2, directed against a cytochrome P450 enzyme.
Idiosyncratic reactions lead to a morphologic pattern that is more variable than those produced by direct toxins; a single agent is often capable of causing a variety of lesions, although certain patterns tend to predominate. Depending on the agent involved, idiosyncratic hepatitis may result in a clinical and morphologic picture indistinguishable from that of viral hepatitis (e.g., isoniazid or ciprofloxacin). So-called hepatocellular injury is the most common form, featuring spotty necrosis in the liver lobule with a predominantly lymphocytic infiltrate resembling that observed in acute hepatitis A, B, or C. Drug-induced cholestasis ranges from mild to increasingly severe: (1) bland cholestasis with limited hepatocellular injury (e.g., estrogens, 17,α-substituted androgens); (2) inflammatory cholestasis (e.g., amoxicillin-clavulanic acid [the most frequently implicated antibiotic among cases of drug-induced liver injury], oxacillin, erythromycin estolate); (3) sclerosing cholangitis (e.g., after intrahepatic infusion of the chemotherapeutic agent floxuridine for hepatic metastases from a primary colonic carcinoma); and (4) disappearance of bile ducts, “ductopenic” cholestasis, similar to that observed in chronic rejection (Chap. 368) following liver transplantation (e.g., carbamazepine, levofloxacin). Cholestasis may result from binding of drugs to canalicular membrane transporters, accumulation of toxic bile acids resulting from canalicular pump failure, or genetic defects in canalicular transporter proteins. Clinically, the distinction between a hepatocellular and a cholestatic reaction is indicated by the R value, the ratio of alanine aminotransferase (ALT) to alkaline phosphatase values, both expressed as multiples of the upper limit of normal. An R value of >5.0 is associated with hepatocellular injury, R <2.0 with cholestatic injury, and R between 2.0 and 5.0 with mixed hepatocellular-cholestatic injury.
Morphologic alterations may also include bridging hepatic necrosis (e.g., methyldopa) or, infrequently, hepatic granulomas (e.g., sulfonamides). Some drugs result in macrovesicular or microvesicular steatosis or steatohepatitis, which, in some cases, has been linked to mitochondrial dysfunction and lipid peroxidation. Severe hepatotoxicity associated with steatohepatitis, most likely a result of mitochondrial toxicity, is being recognized with increasing frequency among patients receiving antiretroviral therapy with reverse transcriptase inhibitors for HIV infection (e.g., zidovudine, didanosine), although many of these drugs have been withdrawn because of such hepatotoxicity (Chap. 226). Generally, such mitochondrial hepatotoxicity of these antiretroviral agents is reversible, but dramatic, nonreversible hepatotoxicity associated with mitochondrial injury (inhibition of DNA polymerase γ) was the cause of acute liver failure encountered during early clinical trials of now-abandoned fialuridine, a fluorinated pyrimidine analogue with potent antiviral activity against hepatitis B virus. Another potential target for idiosyncratic drug hepatotoxicity is sinusoidal lining cells; when these are injured, such as by high-dose chemotherapeutic agents (e.g., cyclophosphamide, melphalan, busulfan) administered prior to bone marrow transplantation, venoocclusive disease can result. Nodular regenerative hyperplasia, a subtle form of portal hypertension, may also result from vascular injury to portal venous endothelium following systemic chemotherapy, such as with oxaliplatin, as part of adjuvant treatment for colon cancer.
Not all adverse hepatic drug reactions can be classified as either toxic or idiosyncratic. For example, oral contraceptives, which combine estrogenic and progestational compounds, may result in impairment of hepatic tests and, occasionally, jaundice; however, they do not produce necrosis or fatty change, manifestations of hypersensitivity are generally absent, and susceptibility to the development of oral contraceptive–induced cholestasis appears to be genetically determined. Such estrogen-induced cholestasis is more common in women with cholestasis of pregnancy, a disorder linked to genetic defects in multidrug resistance–associated canalicular transporter proteins.
Any idiosyncratic reaction that occurs in <1:10,000 recipients will go unrecognized in most clinical trials, which involve only several thousand recipients. The U.S. Food and Drug Administration (FDA) and pharmaceutical companies have learned to look for even subtle indications of serious toxicity and monitor regularly the number of trial subjects in whom any aminotransferase elevations develop, as a possible surrogate for more serious toxicity. Even more valid as a predictor of severe hepatotoxicity is the occurrence of jaundice in patients enrolled in a clinical drug trial, so called “Hy’s Law,” named after Hyman Zimmerman, one of the pioneers of the field of drug hepatotoxicity. He recognized that, if jaundice occurred during a phase III trial, more serious liver injury was likely, with a 10:1 ratio between cases of jaundice and liver failure—10 patients with jaundice to 1 patient with acute liver failure. Thus, the finding of such Hy’s Law cases during drug development often portends failure of approval, particularly if any of the subjects sustains a bad outcome. Troglitazone, a peroxisome proliferator-activated receptor γ agonist, was the first in its class of thiazolidinedione insulin-sensitizing agents. Although in retrospect, Hy’s Law cases of jaundice had occurred during phase III trials, no instances of liver failure were recognized until well after the drug was introduced, underlining the importance of postmarketing surveillance in identifying toxic drugs and in leading to their withdrawal from use. Fortunately, such hepatotoxicity is not characteristic of the second-generation thiazolidinedione insulin-sensitizing agents rosiglitazone and pioglitazone; in clinical trials, the frequency of aminotransferase elevations in patients treated with these medications did not differ from that in placebo recipients, and isolated reports of liver injury among recipients are extremely rare.
Proving that an episode of liver injury is caused by a drug is difficult in many cases. Drug-induced liver injury is nearly always a presumptive diagnosis, and many other disorders produce a similar clinicopathologic picture. Thus, causality may be difficult to establish and requires several separate supportive assessment variables to lead to a high level of certainty, including temporal association (time of onset, time to resolution), clinical-biochemical features, type of injury (hepatocellular versus cholestatic), extrahepatic features, likelihood that a given agent is to blame based on its past record, and exclusion of other potential causes. Scoring systems such as the Roussel-Uclaf Causality Assessment Method (RUCAM) yield residual uncertainty and have not been adopted widely. Currently, the U.S. Drug-Induced Liver Injury Network (DILIN) relies on a structured expert opinion process requiring detailed data on each case and a comprehensive review by three experts who arrive at a consensus on a five-degree scale of likelihood (definite, highly likely, probable, possible, unlikely); however, this approach is not practical for routine clinical application.
Generally, drug hepatotoxicity is not more frequent in persons with underlying chronic liver disease, although the severity of the outcome may be amplified. Reported exceptions include hepatotoxicity of aspirin, methotrexate, isoniazid (only in certain experiences), antiretroviral therapy for HIV infection, and certain drugs such as conditioning regimens for bone marrow transplantation in the presence of hepatitis C.
In Table 361-2, several classes of chemical agents are listed together with examples of the pattern of liver injury produced by them. Certain drugs appear to be responsible for the development of chronic as well as acute hepatic injury. For example, nitrofurantoin, minocycline, hydralazine, and methyldopa have been associated with moderate to severe chronic hepatitis with autoimmune features. Methotrexate, tamoxifen, and amiodarone have been implicated in the development of cirrhosis. Portal hypertension in the absence of cirrhosis may result from alterations in hepatic architecture produced by vitamin A or arsenic intoxication, industrial exposure to vinyl chloride, or administration of thorium dioxide. The latter three agents have also been associated with angiosarcoma of the liver. Oral contraceptives have been implicated in the development of hepatic adenoma and, rarely, hepatocellular carcinoma and hepatic vein occlusion (Budd-Chiari syndrome). Another unusual lesion, peliosis hepatis (blood cysts of the liver), has been observed in some patients treated with anabolic or contraceptive steroids. The existence of these hepatic disorders expands the spectrum of liver injury induced by chemical agents and emphasizes the need for a thorough drug history in all patients with liver dysfunction. A helpful LiverTox website that contains up-to-date information on drug-induced liver injury is available through the National Institute of Diabetes and Digestive and Kidney Diseases and the National Library of Medicine (www.livertox.nih.gov).
|
PRINCIPAL ALTERATIONS OF HEPATIC MORPHOLOGY PRODUCED BY SOME COMMONLY USED DRUGS AND CHEMICALSa |
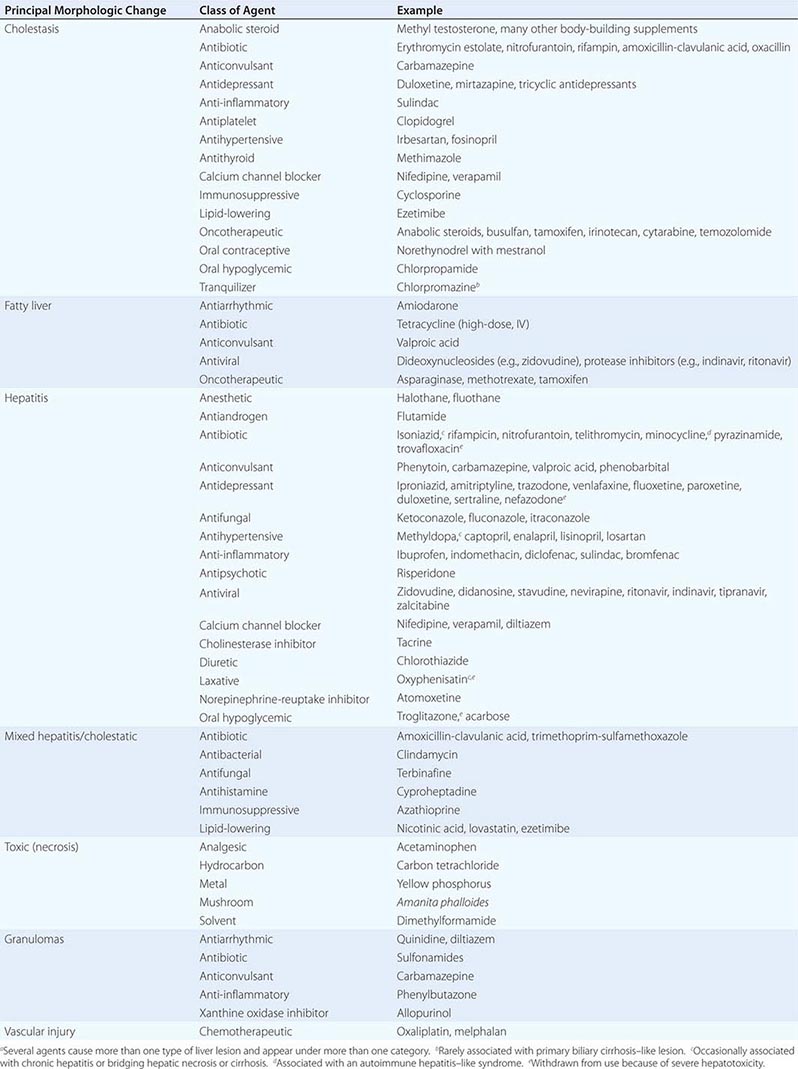
The following are patterns of adverse hepatic reactions for some prototypic agents.
ACETAMINOPHEN HEPATOTOXICITY (DIRECT TOXIN)
Acetaminophen represents the most prevalent cause of acute liver failure in the Western world; up to 72% of patients with acetaminophen hepatotoxicity in Scandinavia—somewhat lower frequencies in the United Kingdom and the United States—progress to encephalopathy and coagulopathy. Acetaminophen causes dose-related centrilobular hepatic necrosis after single-time-point ingestions, as intentional self-harm, or over extended periods, as unintentional overdoses, when multiple drug preparations or inappropriate drug amounts are used daily for several days, e.g., for relief of pain or fever. In these instances, 8 g/d, twice the daily recommended maximum dose, over several days can readily lead to liver failure. Use of opioid-acetaminophen combinations appears to be particularly harmful, because habituation to the opioid may occur with a gradual increase in opioid-acetaminophen combination dosing over days or weeks. A single dose of 10–15 g, occasionally less, may produce clinical evidence of liver injury. Fatal fulminant disease is usually (although not invariably) associated with ingestion of ≥25 g. Blood levels of acetaminophen correlate with severity of hepatic injury (levels >300 μg/mL 4 h after ingestion are predictive of the development of severe damage; levels <150 μg/mL suggest that hepatic injury is highly unlikely). Nausea, vomiting, diarrhea, abdominal pain, and shock are early manifestations occurring 4–12 h after ingestion. Then 24–48 h later, when these features are abating, hepatic injury becomes apparent. Maximal abnormalities and hepatic failure are evident 3–5 days after ingestion, and aminotransferase levels exceeding 10,000 IU/L are not uncommon (i.e., levels far exceeding those in patients with viral hepatitis). Renal failure and myocardial injury may be present. Whether or not a clear history of overdose can be elicited, clinical suspicion of acetaminophen hepatotoxicity should be raised by the presence of the extremely high aminotransferase levels in association with low bilirubin levels that are characteristic of this hyperacute injury. This biochemical signature should trigger further questioning of the subject if possible; however, denial or altered mentation may confound diagnostic efforts. In this setting, a presumptive diagnosis is reasonable, and the proven antidote, N-acetylcysteine—both safe and presumed to be effective even when injury has already begun to evolve—should be instituted.
Acetaminophen is metabolized predominantly by a phase II reaction to innocuous sulfate and glucuronide metabolites; however, a small proportion of acetaminophen is metabolized by a phase I reaction to a hepatotoxic metabolite formed from the parent compound by the cytochrome P450 CYP2E1. This metabolite, N-acetyl-p-benzoquinone-imine (NAPQI), is detoxified by binding to “hepatoprotective” glutathione to become harmless, water-soluble mercapturic acid, which undergoes renal excretion. When excessive amounts of NAPQI are formed, or when glutathione levels are low, glutathione levels are depleted and overwhelmed, permitting covalent binding to nucleophilic hepatocyte macromolecules forming acetaminophen-protein “adducts.” These adducts, which can be measured in serum by high-performance liquid chromatography, hold promise as diagnostic markers of acetaminophen hepatotoxicity, and a point-of-care assay for acetaminophen-Cys adducts is under development. The binding of acetaminophen to hepatocyte macromolecules is believed to lead to hepatocyte necrosis; the precise sequence and mechanism are unknown. Hepatic injury may be potentiated by prior administration of alcohol, phenobarbital, isoniazid, or other drugs; by conditions that stimulate the mixed-function oxidase system; or by conditions such as starvation (including inability to maintain oral intake during severe febrile illnesses) that reduce hepatic glutathione levels. Alcohol induces cytochrome P450 CYP2E1; consequently, increased levels of the toxic metabolite NAPQI may be produced in chronic alcoholics after acetaminophen ingestion, but the role of alcohol in potentiating acute acetaminophen injury is still debated. Alcohol also suppresses hepatic glutathione production. Therefore, in chronic alcoholics, the toxic dose of acetaminophen may be as low as 2 g, and alcoholic patients should be warned specifically about the dangers of even standard doses of this commonly used drug. In a 2006 study, aminotransferase elevations were identified in 31–44% of normal subjects treated for 14 days with the maximal recommended dose of acetaminophen, 4 g daily (administered alone or as part of an acetaminophen-opioid combination); because these changes were transient and never associated with bilirubin elevation, the clinical relevance of these findings remains to be determined. Although underlying hepatitis C virus (HCV) infection was found to be associated with an increased risk of acute liver injury in patients hospitalized for acetaminophen overdose, generally, in patients with nonalcoholic liver disease, acetaminophen taken in recommended doses is well tolerated. Acetaminophen use in cirrhotic patients has not been associated with hepatic decompensation. On the other hand, because of the link between acetaminophen use and liver injury, and because of the limited safety margin between safe and toxic doses, the FDA has recommended that the daily dose of acetaminophen be reduced from 4 g to 3 g (even lower for persons with chronic alcohol use), that all acetaminophen-containing products be labeled prominently as containing acetaminophen, and that the potential for liver injury be prominent in the packaging of acetaminophen and acetaminophen-containing products. Within opioid combination products, the limit for the acetaminophen component has been lowered to 325 mg per tablet.
|
TREATMENT |
ACETAMINOPHEN OVERDOSAGE |
Treatment includes gastric lavage, supportive measures, and oral administration of activated charcoal or cholestyramine to prevent absorption of residual drug. Neither charcoal nor cholestyramine appears to be effective if given >30 min after acetaminophen ingestion; if they are used, the stomach lavage should be done before other agents are administered orally. The chances of possible, probable, and high-risk hepatotoxicity can be derived from a nomogram plot (Fig. 361-2), readily available in emergency departments, as a function of measuring acetaminophen plasma levels 8 h after ingestion. In patients with high acetaminophen blood levels (>200 μg/mL measured at 4 h or >100 μg/mL at 8 h after ingestion), the administration of N-acetylcysteine reduces the severity of hepatic necrosis. This agent provides sulfhydryl donor groups to replete glutathione, which is required to render harmless toxic metabolites that would otherwise bind covalently via sulfhydryl linkages to cell proteins, resulting in the formation of drug metabolite-protein adducts. Therapy should be begun within 8 h of ingestion but may be at least partially effective when given as late as 24–36 h after overdose. Later administration of sulfhydryl compounds is of uncertain value. Routine use of N-acetylcysteine has substantially reduced the occurrence of fatal acetaminophen hepatotoxicity. N-acetylcysteine may be given orally but is more commonly used as an IV solution, with a loading dose of 140 mg/kg over 1 h, followed by 70 mg/kg every 4 h for 15–20 doses. Whenever a patient with potential acetaminophen hepatotoxicity is encountered, a local poison control center should be contacted. Treatment can be stopped when plasma acetaminophen levels indicate that the risk of liver damage is low. If signs of hepatic failure (e.g., progressive jaundice, coagulopathy, confusion) occur despite N-acetylcysteine therapy for acetaminophen hepatotoxicity, liver transplantation may be the only option. Early arterial blood lactate levels among such patients with acute liver failure may distinguish patients highly likely to require liver transplantation (lactate levels >3.5 mmol/L) from those likely to survive without liver replacement. Acute renal injury occurs in nearly 75% of patients with severe acetaminophen injury but is virtually always self-limited.
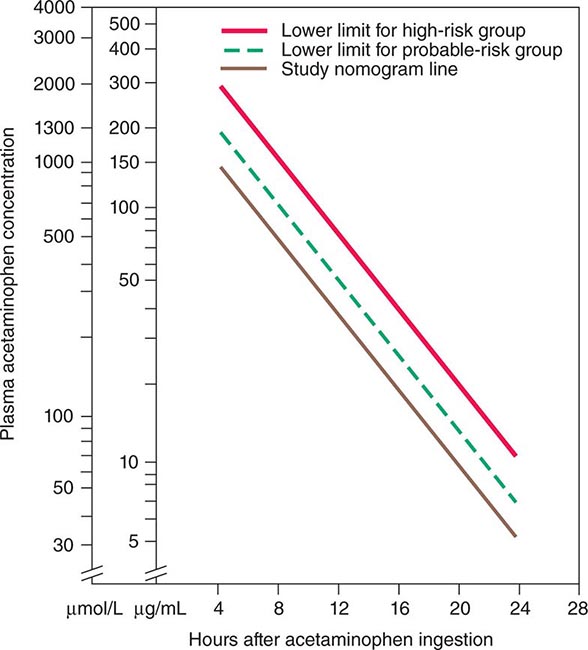
FIGURE 361-2 Nomogram to define risk of acetaminophen hepatotoxicity according to initial plasma acetaminophen concentration. (After BH Rumack, H Matthew: Pediatrics 55:871, 1975.)
Survivors of acute acetaminophen overdose rarely, if ever, have ongoing liver injury or sequelae.
ISONIAZID HEPATOTOXICITY (TOXIC AND IDIOSYNCRATIC REACTION)
Isoniazid (INH) remains central to most antituberculous prophylactic and therapeutic regimens, despite its long-standing recognition as a hepatotoxin. In 10% of patients treated with INH, elevated serum aminotransferase levels develop during the first few weeks of therapy; however, these elevations in most cases are self-limited, mild (values for ALT <200 IU/L), and resolve despite continued drug use. This adaptive response allows continuation of the agent if symptoms and progressive enzyme elevations do not follow the initial elevations. Acute hepatocellular drug-induced liver injury secondary to INH is evident with a variable latency period up to 6 months and is more frequent in alcoholics and patients taking certain other medications, such as barbiturates, rifampin, and pyrazinamide. If the clinical threshold of encephalopathy is reached, severe hepatic injury is likely to be fatal or to require liver transplantation. Liver biopsy reveals morphologic changes similar to those of viral hepatitis or bridging hepatic necrosis. Substantial liver injury appears to be age-related, increasing substantially after age 35; the highest frequency is in patients over age 50, and the lowest is in patients under the age of 20. Even for patients >50 years of age monitored carefully during therapy, hepatotoxicity occurs in only ~2%, well below the risk estimate derived from earlier experiences. Fever, rash, eosinophilia, and other manifestations of drug allergy are distinctly unusual. Recently, antibodies to INH have been detected in INH recipients, but a link to causality of liver injury remains unclear. A clinical picture resembling chronic hepatitis has been observed in a few patients. Many public health programs that require INH prophylaxis for a positive tuberculin skin test or Quantiferon test include monthly monitoring of aminotransferase levels, although this practice has been called into question. Even more effective in limiting serious outcomes may be encouraging patients to be alert for symptoms such as nausea, fatigue, or jaundice, because most fatalities occur in the setting of continued INH use despite clinically apparent illness.
SODIUM VALPROATE HEPATOTOXICITY (TOXIC AND IDIOSYNCRATIC REACTION)
Sodium valproate, an anticonvulsant useful in the treatment of petit mal and other seizure disorders, has been associated with the development of severe hepatic toxicity and, rarely, fatalities, predominantly in children but also in adults. Among children listed as candidates for liver transplantation, valproate is the most common antiepileptic drug implicated. Asymptomatic elevations of serum aminotransferase levels have been recognized in as many as 45% of treated patients. These “adaptive” changes, however, appear to have no clinical importance, because major hepatotoxicity is not seen in the majority of patients despite continuation of drug therapy. In the rare patients in whom jaundice, encephalopathy, and evidence of hepatic failure are found, examination of liver tissue reveals microvesicular fat and bridging hepatic necrosis, predominantly in the centrilobular zone. Bile duct injury may also be apparent. Most likely, sodium valproate is not directly hepatotoxic, but its metabolite, 4-pentenoic acid, may be responsible for hepatic injury. Valproate hepatotoxicity is more common in persons with mitochondrial enzyme deficiencies and may be ameliorated by IV administration of carnitine, which valproate therapy can deplete. Recently, valproate toxicity has been linked to HLA haplotypes (DR4 and B*1502) and to mutations in mitochondrial DNA polymerase gamma 1.
NITROFURANTOIN HEPATOTOXICITY (IDIOSYNCRATIC REACTION)
This commonly used antibiotic for urinary tract infections may cause an acute hepatitis leading to fatal outcome or, more frequently, chronic hepatitis of varying severity but indistinguishable from autoimmune chronic hepatitis. These two scenarios may reflect the frequent use and reuse of the drug for treatment of recurrent cystitis in women. Although most toxic agents manifest injury within 6 months of first ingestion, nitrofurantoin may have a longer latency period, in part perhaps because of its intermittent, recurrent use. Autoantibodies to nuclear components, smooth muscle, and mitochondria are seen and may subside after resolution of infection; however, glucocorticoid or other immunosuppressive medication may be necessary to resolve the autoimmune injury, and cirrhosis may be seen in cases that are not recognized quickly. Interstitial pulmonary fibrosis presenting as chronic cough and dyspnea may be present and resolve slowly with medication withdrawal. Histologic findings are identical to those of autoimmune hepatitis. A similar disease pattern can be observed with minocycline that is used repeatedly for the treatment of acne in teenagers as well as with hydralazine and alpha methyldopa.
AMOXICILLIN-CLAVULANATE HEPATOTOXICITY (IDIOSYNCRATIC MIXED REACTION)
Currently, the most common agent implicated as causing drug-induced liver injury in the United States and in Europe is amoxicillin-clavulanate (most frequent brand name: Augmentin). This medication causes a very specific syndrome of mixed or primarily cholestatic injury. Because hepatotoxicity may follow amoxicillin-clavulanate therapy after a relatively long latency period, the liver injury may begin to manifest at the time of drug withdrawal or after the drug has been withdrawn. The high prevalence of hepatotoxicity reflects in part the very frequent use of this drug for respiratory tract infections, including community-acquired pneumonia. The mechanism of hepatotoxicity is unclear, but the liver injury is thought to be caused by amoxicillin toxicity that is potentiated in some way by clavulanate, which itself appears not to be toxic. Symptoms include nausea, anorexia, fatigue, and jaundice—which may be prolonged—with pruritus. Rash is quite uncommon. On occasion, amoxicillin-clavulanate, like other cholestatic hepatotoxic drugs, causes permanent injury to small bile ducts, leading to the so-called “vanishing bile duct syndrome.” In vanishing bile duct syndrome, initially, liver injury is minimal except for severe cholestasis; however, over time, histologic evidence of bile duct abnormalities is replaced by a paucity and eventual absence of discernible ducts on subsequent biopsies.
PHENYTOIN HEPATOTOXICITY (IDIOSYNCRATIC REACTION)
Phenytoin, formerly diphenylhydantoin, a mainstay in the treatment of seizure disorders, has been associated in rare instances with the development of severe hepatitis-like liver injury leading to fulminant hepatic failure. In many patients, the hepatitis is associated with striking fever, lymphadenopathy, rash (Stevens-Johnson syndrome or exfoliative dermatitis), leukocytosis, and eosinophilia, suggesting an immunologically mediated hypersensitivity mechanism. Despite these observations, evidence suggests that metabolic idiosyncrasy may be responsible for hepatic injury. In the liver, phenytoin is converted by cytochrome P450 to metabolites, including the highly reactive electrophilic arene oxides. These metabolites are normally metabolized further by epoxide hydrolases. A defect (genetic or acquired) in epoxide hydrolase activity could permit covalent binding of arene oxides to hepatic macromolecules, thereby leading to hepatic injury. Hepatic injury is usually manifest within the first 2 months after beginning phenytoin therapy. With the exception of an abundance of eosinophils in the liver, the clinical, biochemical, and histologic picture resembles that of viral hepatitis. In rare instances, bile duct injury may be the salient feature of phenytoin hepatotoxicity, with striking features of intrahepatic cholestasis. Asymptomatic elevations of aminotransferase and alkaline phosphatase levels have been observed in a sizable proportion of patients receiving long-term phenytoin therapy. These liver changes are believed by some authorities to represent the potent hepatic enzyme-inducing properties of phenytoin and are accompanied histologically by swelling of hepatocytes in the absence of necroinflammatory activity or evidence of chronic liver disease.
AMIODARONE HEPATOTOXICITY (TOXIC AND IDIOSYNCRATIC REACTION)
Therapy with this potent antiarrhythmic drug is accompanied in 15–50% of patients by modest elevations of serum aminotransferase levels that may remain stable or diminish despite continuation of the drug. Such abnormalities may appear days to many months after beginning therapy. A proportion of those with elevated aminotransferase levels have detectable hepatomegaly, and clinically important liver disease develops in <5% of patients. Features that represent a direct effect of the drug on the liver and that are common to the majority of long-term recipients are ultrastructural phospholipidosis, unaccompanied by clinical liver disease, and interference with hepatic mixed-function oxidase metabolism of other drugs. The cationic amphiphilic drug and its major metabolite desethylamiodarone accumulate in hepatocyte lysosomes and mitochondria and in bile duct epithelium. The relatively common elevations in aminotransferase levels are also considered a predictable, dose-dependent, direct hepatotoxic effect. On the other hand, in the rare patient with clinically apparent, symptomatic liver disease, liver injury resembling that seen in alcoholic liver disease is observed. The so-called pseudoalcoholic liver injury can range from steatosis to alcoholic hepatitis–like neutrophilic infiltration and Mallory’s hyaline to cirrhosis. Electron-microscopic demonstration of phospholipid-laden lysosomal lamellar bodies can help to distinguish amiodarone hepatotoxicity from typical alcoholic hepatitis. This category of liver injury appears to be a metabolic idiosyncrasy that allows hepatotoxic metabolites to be generated. Rarely, an acute idiosyncratic hepatocellular injury resembling viral hepatitis or cholestatic hepatitis occurs. Hepatic granulomas have occasionally been observed. Because amiodarone has a long half-life, liver injury may persist for months after the drug is stopped.
ERYTHROMYCIN HEPATOTOXICITY (CHOLESTATIC IDIOSYNCRATIC REACTION)
The most important adverse effect associated with erythromycin, more common in children than adults, is the infrequent occurrence of a cholestatic reaction. Although most of these reactions have been associated with erythromycin estolate, other erythromycins may also be responsible. The reaction usually begins during the first 2 or 3 weeks of therapy and includes nausea, vomiting, fever, right upper quadrant abdominal pain, jaundice, leukocytosis, and moderately elevated aminotransferase and alkaline phosphatase levels. The clinical picture can resemble acute cholecystitis or bacterial cholangitis. Liver biopsy reveals variable cholestasis; portal inflammation comprising lymphocytes, polymorphonuclear leukocytes, and eosinophils; and scattered foci of hepatocyte necrosis. Symptoms and laboratory findings usually subside within a few days of drug withdrawal, and evidence of chronic liver disease has not been found on follow-up. The precise mechanism remains ill-defined.
ORAL CONTRACEPTIVE HEPATOTOXICITY (CHOLESTATIC REACTION)
The administration of oral contraceptive combinations of estrogenic and progestational steroids leads to intrahepatic cholestasis with pruritus and jaundice in a small number of patients weeks to months after taking these agents. Especially susceptible seem to be patients with recurrent idiopathic jaundice of pregnancy, severe pruritus of pregnancy, or a family history of these disorders. With the exception of liver biochemical tests, laboratory studies are normal, and extrahepatic manifestations of hypersensitivity are absent. Liver biopsy reveals cholestasis with bile plugs in dilated canaliculi and striking bilirubin staining of liver cells. In contrast to chlorpromazine-induced cholestasis, portal inflammation is absent. The lesion is reversible on withdrawal of the agent. The two steroid components appear to act synergistically on hepatic function, although the estrogen may be primarily responsible. Oral contraceptives are contraindicated in patients with a history of recurrent jaundice of pregnancy. Primarily benign, but rarely malignant, neoplasms of the liver, hepatic vein occlusion, and peripheral sinusoidal dilatation have also been associated with oral contraceptive therapy. Focal nodular hyperplasia of the liver is not more frequent among users of oral contraceptives.
ANABOLIC STEROIDS (CHOLESTATIC REACTION)
The most common form of liver injury caused by complementary and alternative medications is the profound cholestasis associated with anabolic steroids used by body builders. Unregulated agents sold in gyms and health food stores as diet supplements, which are taken by athletes to improve their performance, may contain anabolic steroids. Jaundice in a young male that is accompanied by a cholestatic, rather than a hepatitic, laboratory profile almost invariably will turn out to be caused by the use of one of a variety of androgen congeners. Such agents have the potential to injure bile transport pumps and to cause intense cholestasis; the time to onset is variable, and resolution, which is the rule, may require many weeks to months. Initially, anorexia, nausea, and malaise may occur, followed by pruritus in some but not all patients. Serum aminotransferase levels are usually <100 IU/L and serum alkaline phosphatase levels are generally moderately elevated with bilirubin levels frequently exceeding 342 μmol/L (20 mg/dL). Examination of liver tissue reveals cholestasis without substantial inflammation or necrosis. Anabolic steroids have also been used by prescription to treat bone marrow failure. In this setting, hepatic sinusoidal dilatation and peliosis hepatis have been reported in rare patients, as have hepatic adenomas and hepatocellular carcinoma.
TRIMETHOPRIM-SULFAMETHOXAZOLE HEPATOTOXICITY (IDIOSYNCRATIC REACTION)
This antibiotic combination is used routinely for urinary tract infections in immunocompetent persons and for prophylaxis against and therapy of Pneumocystis carinii pneumonia in immunosuppressed persons (transplant recipients, patients with AIDS). With its increasing use, its occasional hepatotoxicity is being recognized with growing frequency. Its likelihood is unpredictable, but when it occurs, trimethoprim-sulfamethoxazole hepatotoxicity follows a relatively uniform latency period of several weeks and is often accompanied by eosinophilia, rash, and other features of a hypersensitivity reaction. Biochemically and histologically, acute hepatocellular necrosis predominates, but cholestatic features are quite frequent. Occasionally, cholestasis without necrosis occurs, and, very rarely, a severe cholangiolytic pattern of liver injury is observed. In most cases, liver injury is self-limited, but rare fatalities have been recorded. The hepatotoxicity is attributable to the sulfamethoxazole component of the drug and is similar in features to that seen with other sulfonamides; tissue eosinophilia and granulomas may be seen. The risk of trimethoprim-sulfamethoxazole hepatotoxicity is increased in persons with HIV infection.
HMG-COA REDUCTASE INHIBITORS (STATINS) (IDIOSYNCRATIC MIXED HEPATOCELLULAR AND CHOLESTATIC REACTION)
Between 1 and 2% of patients taking lovastatin, simvastatin, pravastatin, fluvastatin, or one of the newer statin drugs for the treatment of hypercholesterolemia experience asymptomatic, reversible elevations (>threefold) of aminotransferase activity. Acute hepatitis-like histologic changes, centrilobular necrosis, and centrilobular cholestasis have been described in a very small number of cases. In a larger proportion, minor aminotransferase elevations appear during the first several weeks of therapy. Careful laboratory monitoring can distinguish between patients with minor, transitory changes, who may continue therapy and those with more profound and sustained abnormalities, who should discontinue therapy. Because clinically meaningful aminotransferase elevations are so rare after statin use and do not differ in meta-analyses from the frequency of such laboratory abnormalities in placebo recipients, a panel of liver experts recommended to the National Lipid Association’s Safety Task Force that liver test monitoring was not necessary in patients treated with statins and that statin therapy need not be discontinued in patients found to have asymptomatic isolated aminotransferase elevations during therapy. Statin hepatotoxicity is not increased in patients with chronic hepatitis C, hepatic steatosis, or other underlying liver diseases, and statins can be used safely in these patients.
TOTAL PARENTERAL NUTRITION (STEATOSIS, CHOLESTASIS)
Total parenteral nutrition (TPN) is often complicated by cholestatic hepatitis attributable to steatosis, cholestasis, or gallstones (or gallbladder sludge). Steatosis or steatohepatitis may result from the excess carbohydrate calories in these nutritional supplements and is the predominant form of TPN-associated liver disorder in adults. The frequency of this complication has been reduced substantially by the introduction of balanced TPN formulas that rely on lipid as an alternative caloric source. Cholestasis and cholelithiasis, caused by the absence of stimulation of bile flow and secretion resulting from the lack of oral intake, is the predominant form of TPN-associated liver disease in infants, especially in premature neonates. Often, cholestasis in such neonates is multifactorial, contributed to by other factors such as sepsis, hypoxemia, and hypotension; occasionally, TPN-induced cholestasis in neonates culminates in chronic liver disease and liver failure. When TPN-associated liver test abnormalities occur in adults, balancing the TPN formula with more lipid is the intervention of first recourse. In infants with TPN-associated cholestasis, the addition of oral feeding may ameliorate the problem. Therapeutic interventions suggested, but not shown, to be of proven benefit, include cholecystokinin, ursodeoxycholic acid, S -adenosyl methionine, and taurine.
ALTERNATIVE AND COMPLEMENTARY MEDICINES (IDIOSYNCRATIC HEPATITIS, STEATOSIS)
Herbal medications that are of scientifically unproven efficacy and that lack prospective safety oversight by regulatory agencies currently account for more than 20% of drug-induced liver injury in the United States. Besides anabolic steroids, the most common category of dietary or herbal products is weight loss agents. Included among the herbal remedies associated with toxic hepatitis are Jin Bu Huan, xiao-chai-hu-tang, germander, chaparral, senna, mistletoe, skullcap, gentian, comfrey (containing pyrrolizidine alkaloids), ma huang, bee pollen, valerian root, pennyroyal oil, kava, celandine, Impila (Callilepis laureola), LipoKinetix, Hydroxycut, herbal nutritional supplements, and herbal teas containing Camellia sinensis (green tea extract). Well characterized are the acute hepatitis-like histologic lesions following Jin Bu Huan use: focal hepatocellular necrosis, mixed mononuclear portal tract infiltration, coagulative necrosis, apoptotic hepatocyte degeneration, tissue eosinophilia, and microvesicular steatosis. Megadoses of vitamin A can injure the liver, as can pyrrolizidine alkaloids, which often contaminate Chinese herbal preparations and can cause a venoocclusive injury leading to sinusoidal hepatic vein obstruction. Because some alternative medicines induce toxicity via active metabolites, alcohol and drugs that stimulate cytochrome P450 enzymes may enhance the toxicity of some of these products. Conversely, some alternative medicines also stimulate cytochrome P450 and may result in or amplify the toxicity of recognized drug hepatotoxins. Given the widespread use of such poorly defined herbal preparations, hepatotoxicity is likely to be encountered with increasing frequency; therefore, a drug history in patients with acute and chronic liver disease should include use of “alternative medicines” and other nonprescription preparations sold in so-called health food stores.
HIGHLY ACTIVE ANTIRETROVIRAL THERAPY (HAART) FOR HIV INFECTION (MITOCHONDRIAL TOXIC, IDIOSYNCRATIC, STEATOSIS; HEPATOCELLULAR, CHOLESTATIC, AND MIXED)
The recognition of drug hepatotoxicity in persons with HIV infection is complicated in this population by the many alternative causes of liver injury (chronic viral hepatitis, fatty infiltration, infiltrative disorders, mycobacterial infection, etc.), but drug hepatotoxicity associated with HAART is an emerging and common type of liver injury in HIV-infected persons (Chap. 226). Although no one antiviral agent is recognized as a potent hepatotoxin, combination regimens including reverse transcriptase and protease inhibitors cause hepatotoxicity in ~10% of treated patients. Implicated most frequently are combinations including nucleoside analogue reverse transcriptase inhibitors zidovudine, didanosine, and, to a lesser extent, stavudine; protease inhibitors ritonavir and indinavir (and amprenavir when used together with ritonavir), as well as tipranavir; and nonnucleoside reverse transcriptase inhibitors nevirapine and, to a lesser extent, efavirenz. These drugs cause predominantly hepatocellular injury but cholestatic injury as well, and prolonged (>6 months) use of reverse transcriptase inhibitors has been associated with mitochondrial injury, steatosis, and lactic acidosis. Indirect hyperbilirubinemia, resulting from direct inhibition of bilirubin-conjugating activity by UDP-glucuronosyltransferase, usually without elevation of aminotransferase or alkaline phosphatase activities, occurs in ~10% of patients treated with the protease inhibitor indinavir. Distinguishing the impact of HAART hepatotoxicity in patients with HIV and hepatitis virus co-infection is made challenging by the following: (1) both chronic hepatitis B and hepatitis C can affect the natural history of HIV infection and the response to HAART, and (2) HAART can have an impact on chronic viral hepatitis. For example, immunologic reconstitution with HAART can result in immunologically mediated liver-cell injury in patients with chronic hepatitis B co-infection if treatment with an antiviral agent for hepatitis B (e.g., the nucleoside analogue lamivudine) is withdrawn or if nucleoside analogue resistance emerges. Infection with HIV, especially with low CD4+ T cell counts, has been reported to increase the rate of hepatic fibrosis associated with chronic hepatitis C, and HAART therapy can increase levels of serum aminotransferases and HCV RNA in patients with hepatitis C co-infection. Didanosine or stavudine should not be used with ribavirin in patients with HIV/HCV co-infection because of an increased risk of severe mitochondrial toxicity and lactic acidosis.
ACKNOWLEDGMENT
Kurt J. Isselbacher, MD, contributed to this chapter in previous editions of Harrison’s.
362 |
Chronic Hepatitis |
Chronic hepatitis represents a series of liver disorders of varying causes and severity in which hepatic inflammation and necrosis continue for at least 6 months. Milder forms are nonprogressive or only slowly progressive, while more severe forms may be associated with scarring and architectural reorganization, which, when advanced, lead ultimately to cirrhosis. Several categories of chronic hepatitis have been recognized. These include chronic viral hepatitis, drug-induced chronic hepatitis (Chap. 361), and autoimmune chronic hepatitis. In many cases, clinical and laboratory features are insufficient to allow assignment into one of these three categories; these “idiopathic” cases are also believed to represent autoimmune chronic hepatitis. Finally, clinical and laboratory features of chronic hepatitis are observed occasionally in patients with such hereditary/metabolic disorders as Wilson’s disease (copper overload), α1 antitrypsin deficiency (Chaps. 365 and 429), and nonalcoholic fatty liver disease (Chap. 367e) and even occasionally in patients with alcoholic liver injury (Chap. 363). Although all types of chronic hepatitis share certain clinical, laboratory, and histopathologic features, chronic viral and chronic autoimmune hepatitis are sufficiently distinct to merit separate discussions. For discussion of acute hepatitis, see Chap. 360.
CLASSIFICATION OF CHRONIC HEPATITIS
Common to all forms of chronic hepatitis are histopathologic distinctions based on localization and extent of liver injury. These vary from the milder forms, previously labeled chronic persistent hepatitis and chronic lobular hepatitis, to the more severe form, formerly called chronic active hepatitis. When first defined, these designations were believed to have prognostic implications, which were not corroborated by subsequent observations. Categorization of chronic hepatitis based primarily on histopathologic features has been replaced by a more informative classification based on a combination of clinical, serologic, and histologic variables. Classification of chronic hepatitis is based on (1) its cause; (2) its histologic activity, or grade; and (3) its degree of progression, or stage. Thus, neither clinical features alone nor histologic features—requiring liver biopsy—alone are sufficient to characterize and distinguish among the several categories of chronic hepatitis.
CLASSIFICATION BY CAUSE
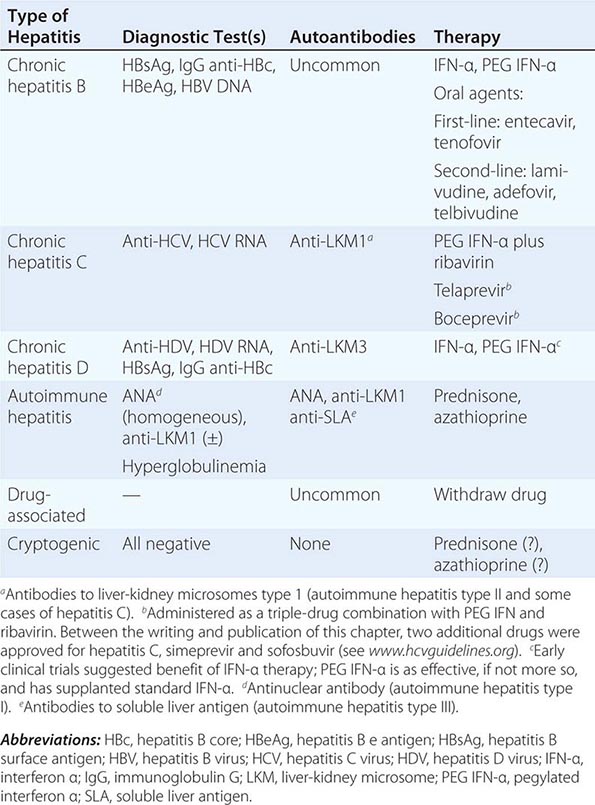
Clinical and serologic features allow the establishment of a diagnosis of chronic viral hepatitis, caused by hepatitis B, hepatitis B plus D, or hepatitis C; autoimmune hepatitis, including several subcategories, I and II (perhaps III), based on serologic distinctions; drug-associated chronic hepatitis; and a category of unknown cause, or cryptogenic chronic hepatitis (Table 362-1). These are addressed in more detail below.
|
CLINICAL AND LABORATORY FEATURES OF CHRONIC HEPATITIS |
CLASSIFICATION BY GRADE
Grade, a histologic assessment of necroinflammatory activity, is based on examination of the liver biopsy. An assessment of important histologic features includes the degree of periportal necrosis and the disruption of the limiting plate of periportal hepatocytes by inflammatory cells (so-called piecemeal necrosis or interface hepatitis); the degree of confluent necrosis that links or forms bridges between vascular structures—between portal tract and portal tract or even more important bridges between portal tract and central vein—referred to as bridging necrosis; the degree of hepatocyte degeneration and focal necrosis within the lobule; and the degree of portal inflammation. Several scoring systems that take these histologic features into account have been devised, and the most popular are the histologic activity index (HAI), used commonly in the United States, and the METAVIR score, used in Europe (Table 362-2). Based on the presence and degree of these features of histologic activity, chronic hepatitis can be graded as mild, moderate, or severe.
|
HISTOLOGIC GRADING AND STAGING OF CHRONIC HEPATITIS |
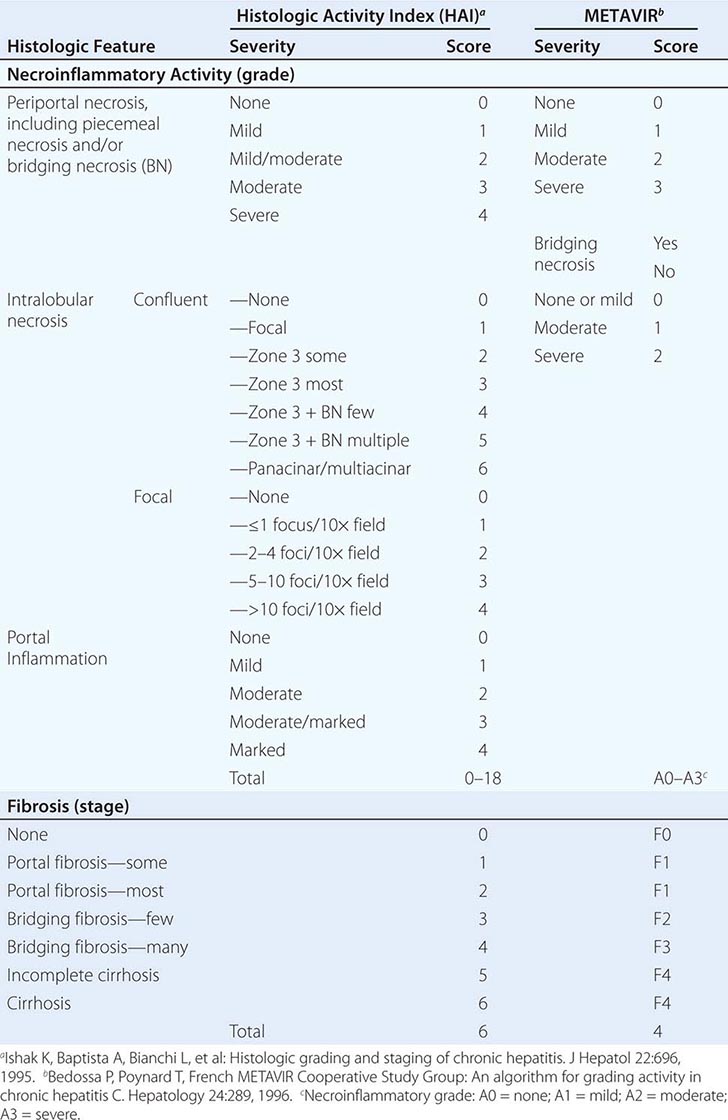
CLASSIFICATION BY STAGE
The stage of chronic hepatitis, which reflects the level of progression of the disease, is based on the degree of hepatic fibrosis. When fibrosis is so extensive that fibrous septa surround parenchymal nodules and alter the normal architecture of the liver lobule, the histologic lesion is defined as cirrhosis. Staging is based on the degree of fibrosis as categorized on a numerical scale from 0–6 (HAI) or 0–4 (METAVIR) (Table 362-2). Several noninvasive approaches have been introduced to provide approximations of hepatic histologic stage, including serum biomarkers of fibrosis and imaging determinations of liver elasticity.
CHRONIC VIRAL HEPATITIS
Both the enterically transmitted forms of viral hepatitis, hepatitis A and E, are self-limited and do not cause chronic hepatitis (rare reports notwithstanding in which acute hepatitis A serves as a trigger for the onset of autoimmune hepatitis in genetically susceptible patients or in which hepatitis E (Chap. 360) can cause chronic liver disease in immunosuppressed hosts, e.g., after liver transplantation). In contrast, the entire clinicopathologic spectrum of chronic hepatitis occurs in patients with chronic viral hepatitis B and C as well as in patients with chronic hepatitis D superimposed on chronic hepatitis B.
CHRONIC HEPATITIS B
The likelihood of chronicity after acute hepatitis B varies as a function of age. Infection at birth is associated with clinically silent acute infection but a 90% chance of chronic infection, whereas infection in young adulthood in immunocompetent persons is typically associated with clinically apparent acute hepatitis but a risk of chronicity of only approximately 1%. Most cases of chronic hepatitis B among adults, however, occur in patients who never had a recognized episode of clinically apparent acute viral hepatitis. The degree of liver injury (grade) in patients with chronic hepatitis B is variable, ranging from none in inactive carriers to mild to moderate to severe. Among adults with chronic hepatitis B, histologic features are of prognostic importance. In one long-term study of patients with chronic hepatitis B, investigators found a 5-year survival rate of 97% for patients with mild chronic hepatitis, 86% for patients with moderate to severe chronic hepatitis, and only 55% for patients with chronic hepatitis and postnecrotic cirrhosis. The 15-year survival in these cohorts was 77%, 66%, and 40%, respectively. On the other hand, more recent observations do not allow us to be so sanguine about the prognosis in patients with mild chronic hepatitis; among such patients followed for 1–13 years, progression to more severe chronic hepatitis and cirrhosis has been observed in more than a quarter of cases.
More important to consider than histology alone in patients with chronic hepatitis B is the degree of hepatitis B virus (HBV) replication. As reviewed in Chap. 360, chronic HBV infection can occur in the presence or absence of serum hepatitis B e antigen (HBeAg), and generally, for both HBeAg-reactive and HBeAg-negative chronic hepatitis B, the level of HBV DNA correlates with the level of liver injury and risk of progression. In HBeAg-reactive chronic hepatitis B, two phases have been recognized based on the relative level of HBV replication. The relatively replicative phase is characterized by the presence in the serum of HBeAg and HBV DNA levels well in excess of 103–104 IU/mL, sometimes exceeding 109 IU/mL; by the presence in the liver of detectable intrahepatocyte nucleocapsid antigens (primarily hepatitis B core antigen [HBcAg]); by high infectivity; and by accompanying liver injury. In contrast, the relatively nonreplicative phase is characterized by the absence of the conventional serum marker of HBV replication (HBeAg), the appearance of anti-HBe, levels of HBV DNA below a threshold of ~103 IU/mL, the absence of intrahepatocytic HBcAg, limited infectivity, and minimal liver injury. Patients in the replicative phase tend to have more severe chronic hepatitis, whereas those in the nonreplicative phase tend to have minimal or mild chronic hepatitis or to be inactive hepatitis B carriers. The likelihood in a patient with HBeAg-reactive chronic hepatitis B of converting spontaneously from relatively replicative to nonreplicative infection is approximately 10% per year. Distinctions in HBV replication and in histologic category, however, do not always coincide. In patients with HBeAg-reactive chronic HBV infection, especially when acquired at birth or in early childhood, as recognized commonly in Asian countries, a dichotomy is common between very high levels of HBV replication during the early decades of life (when the level of host tolerance of HBV is relatively high) and negligible levels of liver injury. Yet despite the relatively immediate, apparently benign nature of liver disease for many decades in this population, in the middle decades, activation of liver injury emerges as relative tolerance of the host to HBV declines, and these patients with childhood-acquired HBV infection are ultimately at increased risk later in life of cirrhosis, hepatocellular carcinoma (HCC) (Chap. 111), and liver-related death. A discussion of the pathogenesis of liver injury in patients with chronic hepatitis B appears in Chap. 360.
 HBeAg-negative chronic hepatitis B (i.e., chronic HBV infection with active virus replication, readily detectable HBV DNA but without HBeAg [anti-HBe-reactive]), is more common than HBeAg-reactive chronic hepatitis B in Mediterranean and European countries and in Asia (and, correspondingly, in HBV genotypes other than A). Compared to patients with HBeAg-reactive chronic hepatitis B, patients with HBeAg-negative chronic hepatitis B have levels of HBV DNA that are several orders of magnitude lower (no more than 105–106 IU/mL) than those observed in the HBeAg-reactive subset. Most such cases represent precore or core-promoter mutations acquired late in the natural history of the disease (mostly early-life onset; age range 40–55 years, older than that for HBeAg-reactive chronic hepatitis B); these mutations prevent translation of HBeAg from the precore component of the HBV genome (precore mutants) or are characterized by downregulated transcription of precore mRNA (core-promoter mutants; Chap. 360). Although their levels of HBV DNA tend to be lower than among patients with HBeAg-reactive chronic hepatitis B, patients with HBeAg-negative chronic hepatitis B can have progressive liver injury (complicated by cirrhosis and HCC) and experience episodic reactivation of liver disease reflected in fluctuating levels of aminotransferase activity (“flares”). The biochemical and histologic activity of HBeAg-negative disease tends to correlate closely with levels of HBV replication, unlike the case mentioned above of Asian patients with HBeAg-reactive chronic hepatitis B during the early decades of their HBV infection. An important point worth reiterating is the observation that the level of HBV replication is the most important risk factor for the ultimate development of cirrhosis and HCC in both HBeAg-reactive and HBeAg-negative patients. Although levels of HBV DNA are lower and more readily suppressed by therapy to undetectable levels in HBeAg-negative (compared to HBeAg-reactive) chronic hepatitis B, achieving sustained responses that permit discontinuation of antiviral therapy is less likely in HBeAg-negative patients (see below). Inactive carriers are patients with circulating hepatitis B surface antigen (HBsAg), normal serum aminotransferase levels, undetectable HBeAg, and levels of HBV DNA that are either undetectable or present at a threshold of ≤103 IU/mL. This serologic profile can occur not only in inactive carriers but also in patients with HBeAg-negative chronic hepatitis B during periods of relative inactivity; distinguishing between the two requires sequential biochemical and virologic monitoring over many months.
HBeAg-negative chronic hepatitis B (i.e., chronic HBV infection with active virus replication, readily detectable HBV DNA but without HBeAg [anti-HBe-reactive]), is more common than HBeAg-reactive chronic hepatitis B in Mediterranean and European countries and in Asia (and, correspondingly, in HBV genotypes other than A). Compared to patients with HBeAg-reactive chronic hepatitis B, patients with HBeAg-negative chronic hepatitis B have levels of HBV DNA that are several orders of magnitude lower (no more than 105–106 IU/mL) than those observed in the HBeAg-reactive subset. Most such cases represent precore or core-promoter mutations acquired late in the natural history of the disease (mostly early-life onset; age range 40–55 years, older than that for HBeAg-reactive chronic hepatitis B); these mutations prevent translation of HBeAg from the precore component of the HBV genome (precore mutants) or are characterized by downregulated transcription of precore mRNA (core-promoter mutants; Chap. 360). Although their levels of HBV DNA tend to be lower than among patients with HBeAg-reactive chronic hepatitis B, patients with HBeAg-negative chronic hepatitis B can have progressive liver injury (complicated by cirrhosis and HCC) and experience episodic reactivation of liver disease reflected in fluctuating levels of aminotransferase activity (“flares”). The biochemical and histologic activity of HBeAg-negative disease tends to correlate closely with levels of HBV replication, unlike the case mentioned above of Asian patients with HBeAg-reactive chronic hepatitis B during the early decades of their HBV infection. An important point worth reiterating is the observation that the level of HBV replication is the most important risk factor for the ultimate development of cirrhosis and HCC in both HBeAg-reactive and HBeAg-negative patients. Although levels of HBV DNA are lower and more readily suppressed by therapy to undetectable levels in HBeAg-negative (compared to HBeAg-reactive) chronic hepatitis B, achieving sustained responses that permit discontinuation of antiviral therapy is less likely in HBeAg-negative patients (see below). Inactive carriers are patients with circulating hepatitis B surface antigen (HBsAg), normal serum aminotransferase levels, undetectable HBeAg, and levels of HBV DNA that are either undetectable or present at a threshold of ≤103 IU/mL. This serologic profile can occur not only in inactive carriers but also in patients with HBeAg-negative chronic hepatitis B during periods of relative inactivity; distinguishing between the two requires sequential biochemical and virologic monitoring over many months.
The spectrum of clinical features of chronic hepatitis B is broad, ranging from asymptomatic infection to debilitating disease or even end-stage, fatal hepatic failure. As noted above, the onset of the disease tends to be insidious in most patients, with the exception of the very few in whom chronic disease follows failure of resolution of clinically apparent acute hepatitis B. The clinical and laboratory features associated with progression from acute to chronic hepatitis B are discussed in Chap. 360.
Fatigue is a common symptom, and persistent or intermittent jaundice is a common feature in severe or advanced cases. Intermittent deepening of jaundice and recurrence of malaise and anorexia, as well as worsening fatigue, are reminiscent of acute hepatitis; such exacerbations may occur spontaneously, often coinciding with evidence of virologic reactivation; may lead to progressive liver injury; and, when superimposed on well-established cirrhosis, may cause hepatic decompensation. Complications of cirrhosis occur in end-stage chronic hepatitis and include ascites, edema, bleeding gastroesophageal varices, hepatic encephalopathy, coagulopathy, or hypersplenism. Occasionally, these complications bring the patient to initial clinical attention. Extrahepatic complications of chronic hepatitis B, similar to those seen during the prodromal phase of acute hepatitis B, are associated with deposition of circulating hepatitis B antigen–antibody immune complexes. These include arthralgias and arthritis, which are common, and the more rare purpuric cutaneous lesions (leukocytoclastic vasculitis), immune-complex glomerulonephritis, and generalized vasculitis (polyarteritis nodosa) (Chaps. 360 and 385).
Laboratory features of chronic hepatitis B do not distinguish adequately between histologically mild and severe hepatitis. Aminotransferase elevations tend to be modest for chronic hepatitis B but may fluctuate in the range of 100–1000 units. As is true for acute viral hepatitis B, alanine aminotransferase (ALT) tends to be more elevated than aspartate aminotransferase (AST); however, once cirrhosis is established, AST tends to exceed ALT. Levels of alkaline phosphatase activity tend to be normal or only marginally elevated. In severe cases, moderate elevations in serum bilirubin (51.3–171 μmol/L [3–10 mg/dL]) occur. Hypoalbuminemia and prolongation of the prothrombin time occur in severe or end-stage cases. Hyperglobulinemia and detectable circulating autoantibodies are distinctly absent in chronic hepatitis B (in contrast to autoimmune hepatitis). Viral markers of chronic HBV infection are discussed in Chap. 360.
|
TREATMENT |
CHRONIC HEPATITIS B |
Although progression to cirrhosis is more likely in severe than in mild or moderate chronic hepatitis B, all forms of chronic hepatitis B can be progressive, and progression occurs primarily in patients with active HBV replication. Moreover, in populations of patients with chronic hepatitis B who are at risk for HCC (Chap. 111), the risk is highest for those with continued, high-level HBV replication and lower for persons in whom initially high-level HBV DNA falls spontaneously over time. Therefore, management of chronic hepatitis B is directed at suppressing the level of virus replication. Although clinical trials tend to focus on clinical endpoints achieved over 1–2 years (e.g., suppression of HBV DNA to undetectable levels, loss of HBeAg/HBsAg, improvement in histology, normalization of ALT), these short-term gains translate into reductions in the risk of clinical progression, hepatic decompensation, and death. To date, seven drugs have been approved for treatment of chronic hepatitis B: injectable interferon (IFN) α; pegylated interferon (long-acting IFN bound to polyethylene glycol, PEG [PEG IFN]); and the oral agents lamivudine, adefovir dipivoxil, entecavir, telbivudine, and tenofovir.
Antiviral therapy for hepatitis B has evolved rapidly since the mid-1990s, as has the sensitivity of tests for HBV DNA. When IFN and lamivudine were evaluated in clinical trials, HBV DNA was measured by insensitive hybridization assays with detection thresholds of 105–106 virions/mL; when adefovir, entecavir, telbivudine, tenofovir, and PEG IFN were studied in clinical trials, HBV DNA was measured by sensitive amplification assays (polymerase chain reaction [PCR]) with detection thresholds of 101–103 viral copies/mL or IU/mL. Recognition of these distinctions is helpful when comparing results of clinical trials that established the efficacy of these therapies (reviewed below in chronological order of publication of these efficacy trials).
INTERFERON
IFN-α was the first approved therapy for chronic hepatitis B. Although it is no longer used to treat hepatitis B, standard IFN is important historically, having provided important lessons about antiviral therapy in general. For immunocompetent adults with HBeAg-reactive chronic hepatitis B (who tend to have high-level HBV DNA [>105–106 virions/mL] and histologic evidence of chronic hepatitis on liver biopsy), a 16-week course of IFN given subcutaneously at a daily dose of 5 million units, or three times a week at a dose of 10 million units, results in a loss of HBeAg and hybridization-detectable HBV DNA (i.e., a reduction to levels below 105–106 virions/mL) in ~30% of patients, with a concomitant improvement in liver histology. Seroconversion from HBeAg to anti-HBe occurred in approximately 20%, and, in early trials, approximately 8% lost HBsAg. Successful IFN therapy and seroconversion are often accompanied by an acute hepatitis-like elevation in aminotransferase activity, which has been postulated to result from enhanced cytolytic T cell clearance of HBV-infected hepatocytes. Relapse after successful therapy is rare (1 or 2%). The likelihood of responding to IFN is higher in patients with lower levels of HBV DNA and substantial elevations of ALT. Although children can respond as well as adults, IFN therapy has not been effective in very young children infected at birth. Similarly, IFN therapy has not been effective in immunosuppressed persons, Asian patients with neonatal acquisition of infection and minimal-to-mild ALT elevations, or patients with decompensated chronic hepatitis B (in whom such therapy can actually be detrimental, sometimes precipitating decompensation, often associated with severe adverse effects). Among patients with HBeAg loss during therapy, long-term follow-up has demonstrated that 80% experience eventual loss of HBsAg (i.e., all serologic markers of infection, and normalization of ALT over a 9-year posttreatment period). In addition, improved long-term and complication-free survival as well as a reduction in the frequency of HCC have been documented among IFN responders, supporting the conclusion that successful antiviral therapy improves the natural history of chronic hepatitis B.
Initial trials of brief-duration IFN therapy in patients with HBeAg-negative chronic hepatitis B were disappointing, suppressing HBV replication transiently during therapy but almost never resulting in sustained antiviral responses. In subsequent IFN trials among patients with HBeAg-negative chronic hepatitis B, however, more protracted courses, lasting up to 1.5 years, have been reported to result in sustained remissions documented to last for several years, with suppressed HBV DNA and aminotransferase activity, in ~20%.
Complications of IFN therapy include systemic “flu-like” symptoms; marrow suppression; emotional lability (irritability, depression, anxiety); autoimmune reactions (especially autoimmune thyroiditis); and miscellaneous side effects such as alopecia, rashes, diarrhea, and numbness and tingling of the extremities. With the possible exception of autoimmune thyroiditis, all these side effects are reversible upon dose lowering or cessation of therapy.
Although no longer competitive with the newer generation of antivirals, IFN did represent the first successful antiviral approach and set a standard against which to measure subsequent drugs in the achievement of durable virologic, serologic, biochemical, and histologic responses; consolidation of virologic and biochemical benefit in the ensuing years after therapy; and improvement in the natural history of chronic hepatitis B. Standard IFN has been supplanted by long-acting PEG IFN (see below), and IFN nonresponders are now treated with one of the newer oral nucleoside analogues.
LAMIVUDINE
The first of the nucleoside analogues to be approved, the dideoxynucleoside lamivudine inhibits reverse transcriptase activity of both HIV and HBV and is a potent and effective agent for patients with chronic hepatitis B. Although generally superseded by newer, more potent agents, lamivudine is still used in regions of the world where newer agents are not yet approved are or not affordable. In clinical trials among patients with HBeAg-reactive chronic hepatitis B, lamivudine therapy at daily doses of 100 mg for 48–52 weeks suppressed HBV DNA by a median of approximately 5.5 log10 copies/mL and to undetectable levels, as measured by PCR amplification assays, in approximately 40% of patients. Therapy was associated with HBeAg loss in 32–33%; HBeAg seroconversion (i.e., conversion from HBeAg-reactive to anti-HBe-reactive) in 16–21%; normalization of ALT in 40–75%; improvement in histology in 50–60%; retardation in fibrosis in 20–30%; and prevention of progression to cirrhosis. HBeAg responses can occur even in subgroups who are resistant to IFN (e.g., those with high-level HBV DNA) or who failed in the past to respond to it. As is true for IFN therapy of chronic hepatitis B, patients with near-normal ALT activity tend not to experience HBeAg responses (despite suppression of HBV DNA), and those with ALT levels exceeding 5 × the upper limit of normal can expect 1-year HBeAg seroconversion rates of 50–60%. Generally, HBeAg seroconversions are confined to patients who achieve suppression of HBV DNA to <104 copies/mL (equivalent to ~103 IU/mL). Lamivudine-associated HBeAg responses are accompanied by a posttreatment HBsAg seroconversion rate comparable to that seen after IFN-induced HBeAg responses. Among Western patients who undergo HBeAg responses during a year-long course of therapy and in whom the response is sustained for 4–6 months after cessation of therapy, the response is durable thereafter in the vast majority (>80%); therefore, the achievement of an HBeAg response represents a viable stopping point in therapy. Reduced durability has been reported in Asian patients; therefore, to support the durability of HBeAg responses, patients should receive a period of consolidation therapy of ≥6 months in Western patients and ≥1 year in Asian patients after HBeAg seroconversion. Close posttreatment monitoring is necessary to identify HBV reactivation promptly and to resume therapy. If HBeAg is unaffected by lamivudine therapy, the current approach is to continue therapy until an HBeAg response occurs, but long-term therapy may be required to suppress HBV replication and, in turn, limit liver injury; HBeAg seroconversions can increase to a level of 50% after 5 years of therapy. Histologic improvement continues to accrue with therapy beyond the first year; after a cumulative course of 3 years of lamivudine therapy, necroinflammatory activity is reduced in the majority of patients, and even cirrhosis has been shown to regress to precirrhotic stages in as many as three-quarters of patients.
Losses of HBsAg have been few during the first year of lamivudine therapy, and this observation had been cited as an advantage of IFN-based over lamivudine therapy; however, in head-to-head comparisons between standard IFN and lamivudine monotherapy, HBsAg losses were rare in both groups. Trials in which lamivudine and IFN were administered in combination failed to show a benefit of combination therapy over lamivudine monotherapy for either treatment-naïve patients or prior IFN nonresponders.
In patients with HBeAg-negative chronic hepatitis B (i.e., in those with precore and core-promoter HBV mutations), 1 year of lamivudine therapy results in HBV DNA suppression and normalization of ALT in three-quarters of patients and in histologic improvement in approximately two-thirds. Therapy has been shown to suppress HBV DNA by approximately 4.5 log10 copies/mL (baseline HBV DNA levels are lower than in patients with HBeAg-reactive hepatitis B) and to undetectable levels in approximately 70%, as measured by sensitive PCR amplification assays. Lacking HBeAg at the outset, patients with HBeAg-negative chronic hepatitis B cannot achieve an HBeAg response—a stopping point in HBeAg-reactive patients; almost invariably, when therapy is discontinued, reactivation is the rule. Therefore, these patients require long-term therapy; with successive years, the proportion with suppressed HBV DNA and normal ALT increases.
Clinical and laboratory side effects of lamivudine are negligible and indistinguishable from those observed in placebo recipients. Still, lamivudine doses should be reduced in patients with reduced creatinine clearance. During lamivudine therapy, transient ALT elevations, resembling those seen during IFN therapy and during spontaneous HBeAg-to-anti-HBe seroconversions, occur in one-fourth of patients. These ALT elevations may result from restored cytolytic T cell activation permitted by suppression of HBV replication. Similar ALT elevations, however, occur at an identical frequency in placebo recipients, but ALT elevations associated with HBeAg seroconversion are confined to lamivudine-treated patients. When therapy is stopped after a year of therapy, two- to threefold ALT elevations occur in 20–30% of lamivudine-treated patients, representing renewed liver-cell injury as HBV replication returns. Although these posttreatment flares are almost always transient and mild, rare severe exacerbations, especially in cirrhotic patients, have been observed, mandating close and careful clinical and virologic monitoring after discontinuation of treatment. Many authorities caution against discontinuing therapy in patients with cirrhosis, in whom posttreatment flares could precipitate decompensation.
Long-term monotherapy with lamivudine is associated with methionine-to-valine (M204V) or methionine-to-isoleucine (M204I) mutations, primarily at amino acid 204 in the tyrosine-methionine-aspartate-aspartate (YMDD) motif of HBV DNA polymerase, analogous to mutations that occur in HIV-infected patients treated with this drug. During a year of therapy, YMDD mutations occur in 15–30% of patients; the frequency increases with each year of therapy, reaching 70% at year 5. Ultimately, patients with YMDD mutants experience degradation of clinical, biochemical, and histologic responses; therefore, if treatment is begun with lamivudine monotherapy, the emergence of lamivudine resistance, reflected clinically by a breakthrough from suppressed levels of HBV DNA and ALT, is managed by adding another antiviral to which YMDD variants are sensitive (e.g., adefovir, tenofovir; see below).
Currently, although lamivudine is very safe and still used widely in other parts of the world, in the United States and Europe, lamivudine has been eclipsed by more potent antivirals that have superior resistance profiles (see below); it is no longer recommended as first-line therapy. Still, as the first successful oral antiviral agent for use in hepatitis B, lamivudine has provided proof of the concept that polymerase inhibitors can achieve virologic, serologic, biochemical, and histologic benefits. In addition, lamivudine has been shown to be effective in the treatment of patients with decompensated hepatitis B (for whom IFN is contraindicated), in some of whom decompensation can be reversed. Moreover, among patients with cirrhosis or advanced fibrosis, lamivudine has been shown to be effective in reducing the risk of progression to hepatic decompensation and, marginally, the risk of HCC. In the half decade following the introduction in the United States of lamivudine therapy for hepatitis B, referral of patients with HBV-associated end-stage liver disease for liver transplantation was reduced by ~30%, supporting further the beneficial impact of oral antiviral therapy on the natural history of chronic hepatitis B.
Because lamivudine monotherapy can result universally in the rapid emergence of YMDD variants in persons with HIV infection, patients with chronic hepatitis B should be tested for anti-HIV prior to therapy; if HIV infection is identified, lamivudine monotherapy at the HBV daily dose of 100 mg is contraindicated. These patients should be treated for both HIV and HBV with an HIV drug regimen that includes or is supplemented by at least two drugs active against HBV; antiretroviral therapy (ART) often contains two drugs with antiviral activity against HBV (e.g., tenofovir and emtricitabine), but if lamivudine is part of the regimen, the daily dose should be 300 mg (Chap. 226). The safety of lamivudine during pregnancy has not been established; however, the drug is not teratogenic in rodents and has been used safely in pregnant women with HIV infection and with HBV infection. Limited data even suggest that administration of lamivudine during the last months of pregnancy to mothers with high-level hepatitis B viremia (≥108 IU/mL) can reduce the likelihood of perinatal transmission of hepatitis B.
ADEFOVIR DIPIVOXIL
At an oral daily dose of 10 mg, the acyclic nucleotide analogue adefovir dipivoxil, the prodrug of adefovir, reduces HBV DNA by approximately 3.5–4 log10 copies/mL and is equally effective in treatment-naïve patients and IFN nonresponders. In HBeAg-reactive chronic hepatitis B, a 48-week course of adefovir dipivoxil was shown to achieve histologic improvement (and reduce the progression of fibrosis) and normalization of ALT in just over one-half of patients, HBeAg seroconversion in 12%, HBeAg loss in 23%, and suppression to an undetectable level of HBV DNA in 13–21%, as measured by PCR. Similar to IFN and lamivudine, adefovir dipivoxil is more likely to achieve an HBeAg response in patients with high baseline ALT (e.g., among adefovir-treated patients with ALT level >5 × the upper limit of normal), and HBeAg seroconversions occurred in 25%. The durability of adefovir-induced HBeAg responses is high (91% in one study); therefore, HBeAg response can be relied upon as a stopping point for adefovir therapy, after a period of consolidation therapy, as outlined above. Although data on the impact of additional therapy beyond 1 year are limited, biochemical, serologic, and virologic outcomes improve progressively as therapy is continued.
In patients with HBeAg-negative chronic hepatitis B, a 48-week course of 10 mg/d of adefovir dipivoxil resulted in histologic improvement in two-thirds, normalization of ALT in three-fourths, and suppression of HBV DNA to PCR-undetectable levels in one-half to two-thirds. As was true for lamivudine, because HBeAg responses—a potential stopping point—cannot be achieved in this group, reactivation is the rule when adefovir therapy is discontinued, and indefinite, long-term therapy is required. Treatment beyond the first year consolidates the gain of the first year; after 5 years of therapy, improvement in hepatic inflammation and regression of fibrosis were observed in three-fourths of patients, ALT was normal in 70%, and HBV DNA was undetectable in almost 70%. In one study, stopping adefovir after 5 years was followed by sustained suppression of HBV DNA and ALT, but most HBeAg-negative patients are treated indefinitely unless HBsAg loss, albeit very rare, is achieved.
Adefovir contains a flexible acyclic linker instead of the L-nucleoside ring of lamivudine, avoiding steric hindrance by mutated amino acids. In addition, the molecular structure of phosphorylated adefovir is very similar to that of its natural substrate; therefore, mutations to adefovir would also affect binding of the natural substrate, dATP. Hypothetically, these are among the reasons that resistance to adefovir dipivoxil is much less likely than resistance to lamivudine; no resistance was encountered in 1 year of clinical trial therapy. In subsequent years, however, adefovir resistance begins to emerge (asparagine to threonine at amino acid 236 [N236T] and alanine to valine or threonine at amino acid 181 [A181V/T], primarily), occurring in 2.5% after 2 years, but in 29% after 5 years of therapy (reported in HBeAg-negative patients). Among patients co-infected with HBV and HIV and who have normal CD4+ T cell counts, adefovir dipivoxil is effective in suppressing HBV dramatically (by 5 logs10 in one study). Moreover, adefovir dipivoxil is effective in lamivudine-resistant, YMDD-mutant HBV and can be used when such lamivudine-induced variants emerge. When lamivudine resistance occurs, adding adefovir (i.e., maintaining lamivudine to preempt the emergence of adefovir resistance) is superior to switching to adefovir. Almost invariably, patients with adefovir-mutant HBV respond to lamivudine (or newer agents, such as entecavir, see below). When, in the past, adefovir had been evaluated as therapy for HIV infection, doses of 60–120 mg were required to suppress HIV, and, at these doses, the drug was nephrotoxic. Even at 30 mg/d, creatinine elevations of 44 μmol/L (0.5 mg/dL) occurred in 10% of patients; however, at the HBV-effective dose of 10 mg, such elevations of creatinine are rarely encountered. If any nephrotoxicity does occur, it rarely appears before 6–8 months of therapy. Although renal tubular injury is a rare potential side effect, and although creatinine monitoring is recommended during treatment, the therapeutic index of adefovir dipivoxil is high, and the nephrotoxicity observed in clinical trials at higher doses was reversible. For patients with underlying renal disease, frequency of administration of adefovir dipivoxil should be reduced to every 48 h for creatinine clearances of 30–49 mL/min; to every 72 h for creatinine clearances of 10–29 mL/min; and once a week, following dialysis, for patients undergoing hemodialysis. Adefovir dipivoxil is very well tolerated, and ALT elevations during and after withdrawal of therapy are similar to those observed and described above in clinical trials of lamivudine. An advantage of adefovir is its relatively favorable resistance profile; however, it is not as potent as the other approved oral agents, it does not suppress HBV DNA as rapidly or as uniformly as the others, it is the least likely of all agents to result in HBeAg seroconversion, and 20–50% of patients fail to suppress HBV DNA by 2 log10 (“primary nonresponders”). For these reasons, adefovir, which has been supplanted in both treatment-naïve and lamivudine-resistant patients by the more potent, less resistance-prone nucleotide analogue tenofovir (see below), is no longer recommended as first-line therapy.
PEGYLATED INTERFERON
After long-acting PEG IFN was shown to be effective in the treatment of hepatitis C (see below), this more convenient drug was evaluated in the treatment of chronic hepatitis B. Once-a-week PEG IFN is more effective than the more frequently administered, standard IFN, and several large-scale trials of PEG IFN versus oral nucleoside analogues have been conducted among patients with HBeAg-reactive and HBeAg-negative chronic hepatitis B.
In HBeAg-reactive chronic hepatitis B, two large-scale studies were done. One study evaluated PEG IFN-α 2b (100 μg weekly for 32 weeks, then 50 μg weekly for another 20 weeks for a total of 52 weeks, with a comparison arm of combination PEG IFN with oral lamivudine) in 307 subjects. The other study involved PEG IFN-α 2a (180 μg weekly for 48 weeks) in 814 primarily Asian patients, three-fourths of whom had ALT ≥2 × the upper limit of normal, with comparison arms of lamivudine monotherapy and combination PEG IFN plus lamivudine. At the end of therapy (48–52 weeks) in the PEG IFN monotherapy arms, HBeAg loss occurred in approximately 30%, HBeAg seroconversion in 22–27%, undetectable HBV DNA (<400 copies/mL by PCR) in 10–25%, normal ALT in 34–39%, and a mean reduction in HBV DNA of 2 log10 copies/mL (PEG IFN-α 2b) to 4.5 log10 copies/mL (PEG IFN-α 2a). Six months after completing PEG IFN monotherapy in these trials, HBeAg losses were present in approximately 35%, HBeAg seroconversion in approximately 30%, undetectable HBV DNA in 7–14%, normal ALT in 32–41%, and a mean reduction in HBV DNA of 2–2.4 log10 copies/mL. Although the combination of PEG IFN and lamivudine was superior at the end of therapy in one or more serologic, virologic, or biochemical outcomes, neither the combination arm (in both studies) nor the lamivudine monotherapy arm (in the PEG IFN-α 2a trial) demonstrated any benefit compared to the PEG IFN monotherapy arms 6 months after therapy. Moreover, HBsAg seroconversion occurred in 3–7% of PEG IFN recipients (with or without lamivudine); some of these seroconversions were identified by the end of therapy, but many were identified during the posttreatment follow-up period. The likelihood of HBeAg loss in PEG IFN–treated HBeAg-reactive patients is associated with HBV genotype A > B > C > D (shown for PEG IFN-α2b but not for α-2a).
Based on these results, some authorities concluded that PEG IFN monotherapy should be the first-line therapy of choice in HBeAg-reactive chronic hepatitis B; however, this conclusion has been challenged. Although a finite, 1-year course of PEG IFN results in a higher rate of sustained response (6 months after treatment) than is achieved with oral nucleoside/nucleotide analogue therapy, the comparison is confounded by the fact that oral agents are not discontinued at the end of 1 year. Instead, taken orally and free of side effects, therapy with oral agents is extended indefinitely or until after the occurrence of an HBeAg response. The rate of HBeAg responses after 2 years of oral-agent nucleoside analogue therapy is at least as high as, if not higher than, that achieved with PEG IFN after 1 year; favoring oral agents is the absence of injections, difficult-to-tolerate side effects, and laboratory monitoring as well as lower direct and indirect medical care costs and inconvenience. The association of HBsAg responses with PEG IFN therapy occurs in such a small proportion of patients that subjecting everyone to PEG IFN for the marginal gain of HBsAg responses during or immediately after therapy in such a very small minority is questionable. Moreover, HBsAg responses occur in a comparable proportion of patients treated with early-generation nucleoside/nucleotide analogues in the years after therapy, and, with the newer, more potent nucleoside analogues, the frequency of HBsAg loss during the first year of therapy equals that of PEG IFN and is exceeded during year 2 and beyond (see below). Of course, resistance is not an issue during PEG IFN therapy, but the risk of resistance is much lower with new agents (≤1% up to 3–6 years in previously treatment-naïve, entecavir-treated and tenofovir-treated patients; see below). Finally, the level of HBV DNA inhibition that can be achieved with the newer agents, and even with lamivudine, exceeds that which can be achieved with PEG IFN, in some cases by several orders of magnitude.
In HBeAg-negative chronic hepatitis B, a trial of PEG IFN-α 2a (180 μg weekly for 48 weeks versus comparison arms of lamivudine monotherapy and of combination therapy) in 564 patients showed that PEG IFN monotherapy resulted at the end of therapy in suppression of HBV DNA by a mean of 4.1 log10 copies/mL, undetectable HBV DNA (<400 copies/mL by PCR) in 63%, normal ALT in 38%, and loss of HBsAg in 4%. Although lamivudine monotherapy and combination lamivudine–PEG IFN therapy were both superior to PEG IFN at the end of therapy, no advantage of lamivudine monotherapy or combination therapy was apparent over PEG IFN monotherapy 6 months after therapy—suppression of HBV DNA by a mean of 2.3 log10 copies/mL, undetectable HBV DNA in 19%, and normal ALT in 59%. In subjects involved in this trial followed for up to 5 years, among the two-thirds followed who had been treated initially with PEG IFN, 17% maintained HBV DNA suppression to <400 copies/mL, but ALT remained normal in only 22%; HBsAg loss increased gradually to 12%. Among the half followed who had been treated initially with lamivudine monotherapy, HBV DNA remained <400 copies/mL in 7% and ALT normal in 16%; by year 5, 3.5% had lost HBsAg. As was the case for standard IFN therapy in HBeAg-negative patients, only a small proportion maintained responsiveness after completion of PEG IFN therapy, raising questions about the relative value of a finite period of PEG IFN, versus a longer course with a potent, low-resistance oral nucleoside analogue in these patients. Moreover, the value of PEG IFN for HBeAg-negative chronic hepatitis B has not been confirmed. In the only other controlled clinical trial of PEG IFN for HBeAg-negative chronic hepatitis B, the hepatitis C regimen of PEG IFN plus ribavirin was compared to PEG IFN monotherapy. In this trial, HBV DNA suppression (<400 copies/mL) occurred in only 7.5% of the two groups combined, and no study subject lost HBsAg.
In patients treated with PEG IFN, HBeAg and HBsAg responses have been associated with IL28B genotype CC, the favorable genotype identified in trials of PEG IFN for chronic hepatitis C. Also, reductions in quantitative HBsAg levels have been shown to correlate with and to be predictive of responsiveness to PEG IFN in chronic hepatitis B. If HBsAg levels fail to fall within the first 12–24 weeks or to reach <20,000 IU/mL by week 24, PEG IFN therapy is unlikely to be effective and should be discontinued.
ENTECAVIR
Entecavir, an oral cyclopentyl guanosine analogue polymerase inhibitor, appears to be the most potent of the HBV antivirals and is just as well tolerated as lamivudine. In a 709-subject clinical trial among HBeAg-reactive patients, oral entecavir, 0.5 mg daily, was compared to lamivudine, 100 mg daily. At 48 weeks, entecavir was superior to lamivudine in suppression of HBV DNA (mean 6.9 versus 5.5 log10 copies/mL), percentage with undetectable HBV DNA (<300 copies/mL by PCR; 67% versus 36%), histologic improvement (≥2-point improvement in necroinflammatory HAI score; 72% versus 62%), and normal ALT (68% versus 60%). The two treatments were indistinguishable in percentage with HBeAg loss (22% versus 20%) and seroconversion (21% versus 18%). Among patients treated with entecavir for 96 weeks, HBV DNA was undetectable cumulatively in 80% (versus 39% for lamivudine), and HBeAg seroconversions had occurred in 31% (versus 26% for lamivudine). After 3–6 years of entecavir, HBeAg seroconversions have been observed in 39–44% and HBsAg loss in 5–6%. Similarly, in a 638-subject clinical trial among HBeAg-negative patients, at week 48, oral entecavir, 0.5 mg daily, was superior to lamivudine, 100 mg daily, in suppression of HBV DNA (mean 5.0 versus 4.5 log10 copies/mL) and in percentage with undetectable HBV DNA (90% versus 72%), histologic improvement (70% versus 61%), and normal ALT (78% versus 71%). No resistance mutations were encountered in previously treatment-naïve, entecavir-treated patients during 96 weeks of therapy, and in a cohort of subjects treated for up to 6 years, resistance emerged in only 1.2%. Entecavir-induced HBeAg seroconversions are as durable as those achieved with other antivirals. Its high barrier to resistance coupled with its high potency renders entecavir a first-line drug for patients with chronic hepatitis B.
Entecavir is also effective against lamivudine-resistant HBV infection. In a trial of 286 lamivudine-resistant patients, entecavir, at a higher daily dose of 1 mg, was superior to lamivudine, as measured at week 48, in achieving suppression of HBV DNA (mean 5.1 versus 0.48 log10 copies/mL), undetectable HBV DNA (72% versus 19%), normal ALT (61% versus 15%), HBeAg loss (10% versus 3%), and HBeAg seroconversion (8% versus 3%). In this population of lamivudine-experienced patients, however, entecavir resistance emerged in 7% at 48 weeks. Although entecavir resistance requires both a YMDD mutation and a second mutation at one of several other sites (e.g., T184A, S202G/I, or M250V), resistance to entecavir in lamivudine-resistant chronic hepatitis B has been recorded to increase progressively to 43% at 4 years; therefore, entecavir is not as attractive a choice as adefovir or tenofovir for patients with lamivudine-resistant hepatitis B.
In clinical trials, entecavir has an excellent safety profile; in addition, on-treatment and posttreatment ALT flares are relatively uncommon and relatively mild in entecavir-treated patients. Doses should be reduced for patients with reduced creatinine clearance. Entecavir does have low-level antiviral activity against HIV and cannot be used as monotherapy to treat HBV infection in HIV/HBV co-infected persons.
TELBIVUDINE
Telbivudine, a cytosine analogue, is similar in efficacy to entecavir but slightly less potent in suppressing HBV DNA (a slightly less profound median 6.4 log10 reduction in HBeAg-reactive disease and a similar 5.2 log10 reduction in HBeAg-negative disease). In its registration trial, telbivudine at an oral daily dose of 600 mg suppressed HBV DNA to <300 copies/mL in 60% of HBeAg-positive and 88% of HBeAg-negative patients, reduced ALT to normal in 77% of HBeAg-positive and 74% of HBeAg-negative patients, and improved histology in 65% of HBeAg-positive and 67% of HBeAg-negative patients. Although resistance to telbivudine (M204I, not M204V, mutations) was less frequent than resistance to lamivudine at the end of 1 year, resistance mutations after 2 years of treatment occurred in up to 22%. Generally well tolerated, telbivudine has been associated with a low frequency of asymptomatic creatine kinase elevations and with a very low frequency of peripheral neuropathy; frequency of administration should be reduced for patients with impaired creatinine clearance. Its excellent potency notwithstanding, the inferior resistance profile of telbivudine has limited its appeal; telbivudine is neither recommended as first-line therapy nor widely used.
Tenofovir disoproxil fumarate, an acyclic nucleotide analogue and potent antiretroviral agent used to treat HIV infection, is similar to adefovir but more potent in suppressing HBV DNA and inducing HBeAg responses; it is highly active against both wild-type and lamivudine-resistant HBV and active in patients whose response to adefovir is slow and/or limited. At an oral once-daily dose of 300 mg for 48 weeks, tenofovir suppressed HBV DNA by 6.2 log10 (to undetectable levels [<400 copies/mL] in 76%) in HBeAg-positive patients and by 4.6 log10 (to undetectable levels in 93%) in HBeAg-negative patients; reduced ALT to normal in 68% of HBeAg-positive and 76% of HBeAg-negative patients; and improved histology in 74% of HBeAg-positive and 72% of HBeAg-negative patients. In HBeAg-positive patients, HBeAg seroconversions occurred in 21% by the end of year 1, 27% by year 2, 34% by year 3, and 40% by year 5 of tenofovir treatment; HBsAg loss occurred in 3% by the end of year 1 and 6% at year 2, and 8% by year 5. After 5 years of tenofovir therapy, 87% of patients experienced histologic improvement, including reduction in fibrosis score (51%) and regression of cirrhosis (71%). The 5-year safety (negligible renal toxicity, in 1%, and mild reduction in bone density, in ~0.5%) and resistance profiles (none recorded through 5 years) of tenofovir are very favorable as well; therefore, tenofovir has supplanted adefovir both as first-line therapy for chronic hepatitis B and as add-on therapy for lamivudine-resistant chronic hepatitis B. Frequency of tenofovir administration should be reduced for patients with impaired creatinine clearance.
A comparison of the six antiviral therapies in current use appears in Table 362-3; their relative potencies in suppressing HBV DNA are shown in Fig. 362-1.
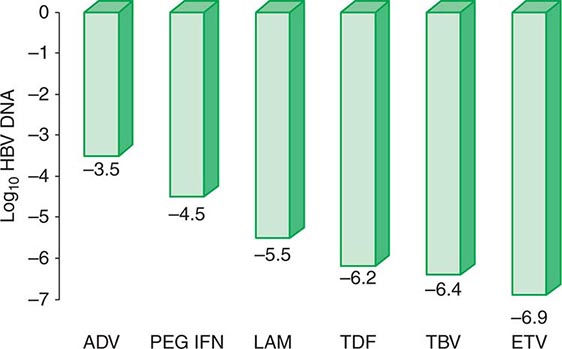
FIGURE 362-1 Relative potency of antiviral drugs for hepatitis B, as reflected by median log10 HBV DNA reduction in HBeAg-positive chronic hepatitis B. These data are from individual reports of large, randomized controlled registration trials that were the basis for approval of the drugs. In most instances, these data do not represent direct comparisons among the drugs, because study populations were different, baseline patient variables were not always uniform, and the sensitivity and dynamic range of the HBV DNA assays used in the trials varied. ADV, adefovir dipivoxil; ETV, entecavir; LAM, lamivudine; PEG IFN, pegylated interferon α2a; TBV, telbivudine; TDF, tenofovir disoproxil fumarate.
|
COMPARISON OF PEGYLATED INTERFERON (PEG IFN), LAMIVUDINE, ADEFOVIR, ENTECAVIR, TELBIVUDINE, AND TENOFOVIR THERAPY FOR CHRONIC HEPATITIS Ba |
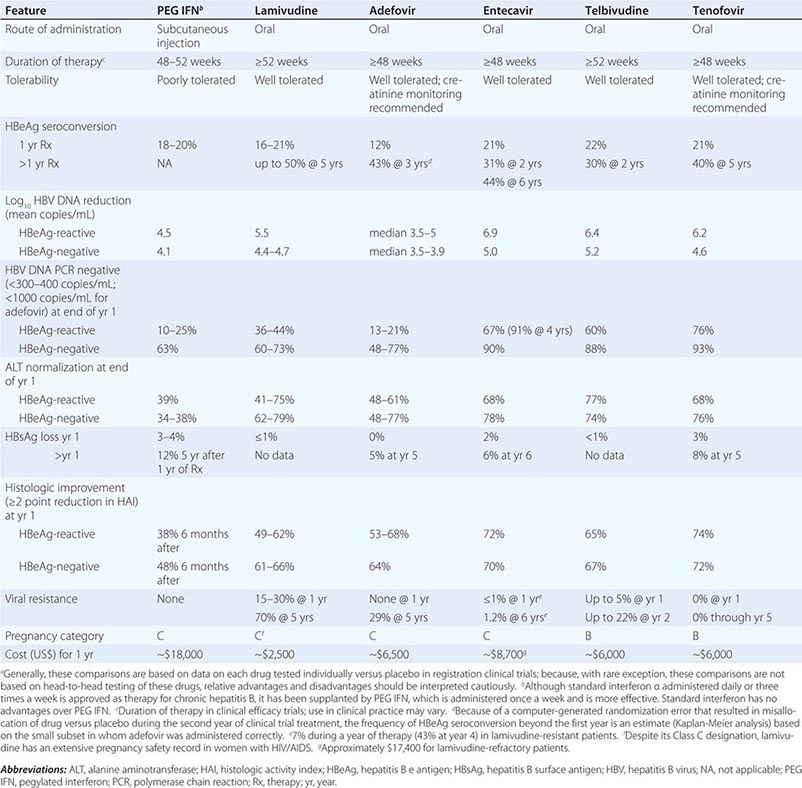
COMBINATION THERAPY
Although the combination of lamivudine and PEG IFN suppresses HBV DNA more profoundly during therapy than does monotherapy with either drug alone (and is much less likely to be associated with lamivudine resistance), this combination used for a year is no better than a year of PEG IFN in achieving sustained responses. To date, combinations of oral nucleoside/nucleotide agents have not achieved an enhancement in virologic, serologic, or biochemical efficacy over that achieved by the more potent of the combined drugs given individually. In a 2-year trial of combination entecavir and tenofovir versus entecavir monotherapy, for a small subgroup of patients with very high HBV DNA levels (≥108 IU/mL), a reduction in HBV DNA to <50 IU/mL was higher in the combination group (79% versus 62%); however, no differences in HBeAg responses or any other endpoint were observed between the combination-therapy and monotherapy groups, even in the high-HBV DNA subgroup. On the other hand, combining agents that are not cross-resistant (e.g., lamivudine and adefovir or tenofovir) has the potential to reduce the risk or perhaps even to preempt entirely the emergence of drug resistance. In the future, the treatment paradigm may shift from the current approach of sequential monotherapy to preemptive combination therapy, perhaps not for all patients but for subsets (e.g., patients with very high levels of HBV DNA, immunosuppressed patients); however, designing and executing clinical trials that demonstrate superior efficacy and resistance profile of combination therapy over monotherapy with entecavir or tenofovir will remain challenging.
NOVEL ANTIVIRALS AND STRATEGIES
In addition to the seven approved antiviral drugs for hepatitis B, emtricitabine, a fluorinated cytosine analogue very similar to lamivudine in structure, efficacy, and resistance profile, offers no advantage over lamivudine. A combination of emtricitabine and tenofovir is approved for the treatment of HIV infection and is an appealing combination therapy for hepatitis B, especially for lamivudine-resistant disease; however, neither emtricitabine nor the combination is approved yet for hepatitis B. Several initially promising antiviral agents have been abandoned because of toxicity (e.g., clevudine, which was linked to myopathy during its clinical development). Because direct-acting antivirals have been so successful in the management of chronic hepatitis B, more unconventional approaches—e.g., immunologic (e.g., toll receptor agonists) or genetic manipulation (e.g., RNA interference—gene silencing—to reduce HBV DNA transcription)—are not likely to be competitive, unless they can be shown to go beyond current antivirals in achieving recovery (HBsAg seroconversion) from HBV infection. Finally, initial emphasis in the development of antiviral therapy for hepatitis B was placed on monotherapy; whether combination regimens will yield additive or synergistic efficacy remains to be determined.
TREATMENT RECOMMENDATIONS
Several learned societies and groups of expert physicians have issued treatment recommendations for patients with chronic hepatitis B; the most authoritative and updated (and free of financial support by pharmaceutical companies) are those of the American Association for the Study of Liver Diseases (AASLD) and of the European Association for the Study of the Liver (EASL). Although the recommendations differ slightly, a consensus has emerged on most of the important points (Table 362-4). No treatment is recommended or available for inactive “nonreplicative” hepatitis B carriers (undetectable HBeAg with normal ALT and HBV DNA ≤103 IU/mL documented serially over time). In patients with detectable HBeAg and HBV DNA levels >2 × 104 IU/mL, treatment is recommended by the AASLD for those with ALT levels above 2 × the upper limit of normal. (The EASL recommends treatment in HBeAg-positive patients for HBV DNA levels >2 × 103 IU/mL and ALT above the upper limit of normal.) For HBeAg-positive patients with ALT ≤2 × the upper limit of normal, in whom sustained responses are not likely and who would require multiyear therapy, antiviral therapy is not recommended currently. This pattern is common during the early decades of life among Asian patients infected at birth; even in this group, therapy would be considered for those >40 years of age, ALT persistently at the high end of the twofold range, and/or with a family history of HCC, especially if the liver biopsy shows moderate to severe necroinflammatory activity or fibrosis. In this group, when, eventually, ALT becomes elevated later in life, antiviral therapy should be instituted. For patients with HBeAg-negative chronic hepatitis B, ALT >2 × the upper limit of normal (above the upper limit of normal according to EASL), and HBV DNA >2 × 103 IU/mL, antiviral therapy is recommended. If HBV DNA is >2 × 103 IU/mL and ALT is 1 to >2 × the upper limit of normal, liver biopsy should be considered to help in arriving at a decision to treat if substantial liver injury is present (treatment in this subset would be recommended according to EASL guidelines, because ALT is elevated).
|
RECOMMENDATIONS FOR TREATMENT OF CHRONIC HEPATITIS Ba |
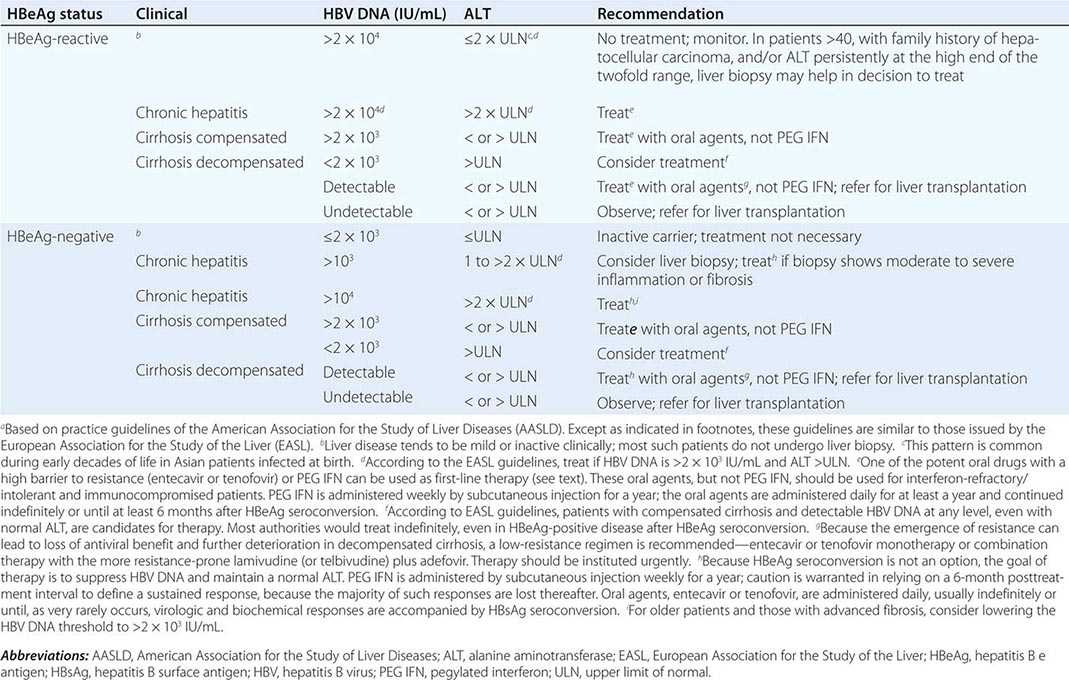
For patients with compensated cirrhosis, because antiviral therapy has been shown to retard clinical progression, treatment is recommended regardless of HBeAg status and ALT as long as HBV DNA is detectable at >2 × 103 IU/mL (detectable at any level according to the EASL); monitoring without therapy is recommended for those with HBV DNA <2 × 103 IU/mL, unless ALT is elevated. For patients with decompensated cirrhosis, treatment is recommended regardless of serologic and biochemical status, as long as HBV DNA is detectable. Patients with decompensated cirrhosis should be evaluated as candidates for liver transplantation.
Among the seven available drugs for hepatitis B, PEG IFN has supplanted standard IFN, entecavir has supplanted lamivudine, and tenofovir has supplanted adefovir. PEG IFN, entecavir, or tenofovir is recommended as first-line therapy (Table 362-3). PEG IFN requires finite-duration therapy, achieves the highest rate of HBeAg responses after a year of therapy, and does not support viral mutations, but it requires subcutaneous injections and is associated with inconvenience, more intensive clinical and laboratory monitoring, and intolerability. Oral nucleoside analogues require long-term therapy in most patients, and when used alone, lamivudine and telbivudine foster the emergence of viral mutations, adefovir somewhat less so, and entecavir (except in lamivudine-experienced patients) and tenofovir rarely at all. Oral agents do not require injections or cumbersome laboratory monitoring, are very well tolerated, lead to improved histology in 50–90% of patients, suppress HBV DNA more profoundly than PEG IFN, and are effective even in patients who fail to respond to IFN-based therapy. Although oral agents are less likely to result in HBeAg responses during the first year of therapy, as compared to PEG IFN, treatment with oral agents tends to be extended beyond the first year and, by the end of the second year, yields HBeAg responses (and even HBsAg responses) comparable in frequency to those achieved after 1 year of PEG IFN (and without the associated side effects) (Table 362-5). Although adefovir and tenofovir are safe, creatinine monitoring is recommended. Substantial experience with lamivudine during pregnancy (see above) has identified no teratogenicity. Although interferons do not appear to cause congenital anomalies, interferons have antiproliferative properties and should be avoided during pregnancy. Adefovir during pregnancy has not been associated with birth defects; however, there may be an increased risk of spontaneous abortion. Data on the safety of entecavir during pregnancy have not been published. Sufficient data in animals and limited data in humans suggest that telbivudine and tenofovir can be used safely during pregnancy. In general, except perhaps for lamivudine, and until additional data become available, the other antivirals for hepatitis B should be avoided or used with extreme caution during pregnancy.
|
PEGYLATED INTERFERON VERSUS ORAL NUCLEOSIDE ANALOGUES FOR THE TREATMENT OF CHRONIC HEPATITIS B |
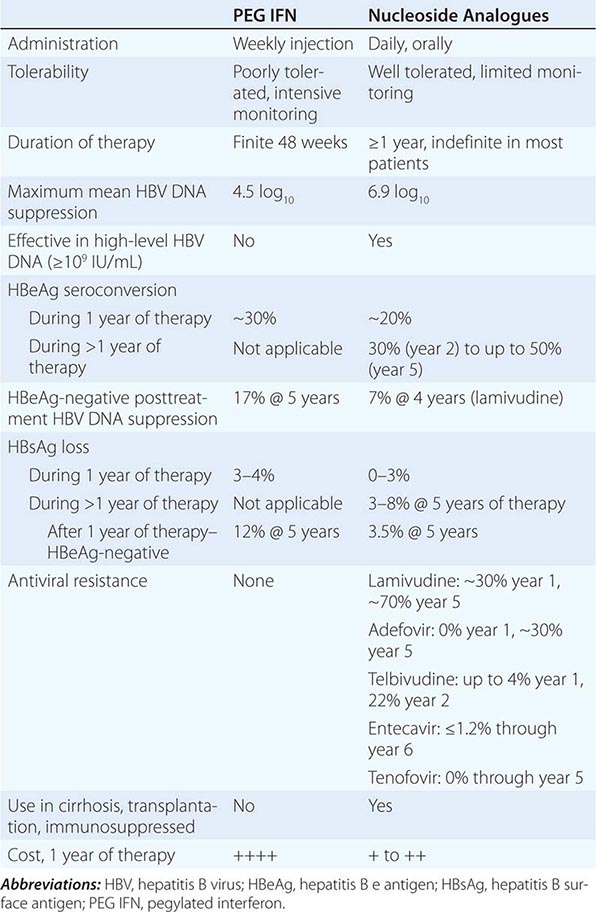
As noted above, some physicians prefer to begin with PEG IFN, while other physicians and patients prefer oral agents as first-line therapy. For patients with decompensated cirrhosis, the emergence of resistance can result in further deterioration and loss of antiviral effectiveness. Therefore, in this patient subset, the threshold for relying on therapy with a very favorable resistance profile (e.g., entecavir or tenofovir) or on combination therapy is low. PEG IFN should not be used in patients with compensated or decompensated cirrhosis.
For patients with end-stage chronic hepatitis B who undergo liver transplantation, reinfection of the new liver is almost universal in the absence of antiviral therapy. The majority of patients become high-level viremic carriers with minimal liver injury. Before the availability of antiviral therapy, an unpredictable proportion experienced severe hepatitis B–related liver injury, sometimes a fulminant-like hepatitis and sometimes a rapid recapitulation of the original severe chronic hepatitis B (Chap. 360). Currently, however, prevention of recurrent hepatitis B after liver transplantation has been achieved definitively by combining hepatitis B immune globulin with one of the oral nucleoside or nucleotide analogues (Chap. 368); preliminary data suggest that the newer, more potent, and less resistance-prone oral agents may be used instead of hepatitis B immune globulin for posttransplantation therapy.
Patients with HBV-HIV co-infection can have progressive HBV-associated liver disease and, occasionally, a severe exacerbation of hepatitis B resulting from immunologic reconstitution following ART. Lamivudine should never be used as monotherapy in patients with HBV-HIV infection because HIV resistance emerges rapidly to both viruses. Adefovir has been used successfully to treat chronic hepatitis B in HBV-HIV co-infected patients but is no longer considered a first-line agent for HBV. Entecavir has low-level activity against HIV and can result in selection of HIV resistance; therefore, it should be avoided in HBV-HIV co-infection. Tenofovir and the combination of tenofovir and emtricitabine in one pill are approved therapies for HIV and represent excellent choices for treating HBV infection in HBV-HIV co-infected patients. Generally, even for HBV-HIV co-infected patients who do not yet meet treatment criteria for HIV infection, treating for both HBV and HIV is recommended.
Patients with chronic hepatitis B who undergo cytotoxic chemotherapy for treatment of malignancies as well as patients treated with immunosuppressive, anticytokine, or antitumor necrosis factor therapies experience enhanced HBV replication and viral expression on hepatocyte membranes during chemotherapy coupled with suppression of cellular immunity. When chemotherapy is withdrawn, such patients are at risk for reactivation of hepatitis B, often severe and occasionally fatal. Such rebound reactivation represents restoration of cytolytic T cell function against a target organ enriched in HBV expression. Preemptive treatment with lamivudine prior to the initiation of chemotherapy has been shown to reduce the risk of such reactivation. The newer, more potent oral antiviral agents are even more effective in preventing hepatitis B reactivation and with a lower risk of antiviral drug resistance. The optimal duration of antiviral therapy after completion of chemotherapy is not known, but a suggested approach is 6 months for inactive hepatitis B carriers and longer-duration therapy in patients with baseline HBV DNA levels >2 × 103 IU/mL, until standard clinical endpoints are met (Table 362-4).
CHRONIC HEPATITIS D (DELTA HEPATITIS)
Chronic hepatitis D virus (HDV) may follow acute co-infection with HBV but at a rate no higher than the rate of chronicity of acute hepatitis B. That is, although HDV co-infection can increase the severity of acute hepatitis B, HDV does not increase the likelihood of progression to chronic hepatitis B. When, however, HDV superinfection occurs in a person who is already chronically infected with HBV, long-term HDV infection is the rule, and a worsening of the liver disease is the expected consequence. Except for severity, chronic hepatitis B plus D has similar clinical and laboratory features to those seen in chronic hepatitis B alone. Relatively severe and progressive chronic hepatitis, with or without cirrhosis, is the rule, and mild chronic hepatitis is the exception. Occasionally, however, mild hepatitis or even, rarely, inactive carriage occurs in patients with chronic hepatitis B plus D, and the disease may become indolent after several years of infection. A distinguishing serologic feature of chronic hepatitis D is the presence in the circulation of antibodies to liver-kidney microsomes (anti-LKM); however, the anti-LKM seen in hepatitis D, anti-LKM3, are directed against uridine diphosphate glucuronosyltransferase and are distinct from anti-LKM1 seen in patients with autoimmune hepatitis and in a subset of patients with chronic hepatitis C (see below). The clinical and laboratory features of chronic HDV infection are summarized in Chap. 360.
CHRONIC HEPATITIS C
Regardless of the epidemiologic mode of acquisition of hepatitis C virus (HCV) infection, chronic hepatitis follows acute hepatitis C in 50–70% of cases; chronic infection is common even in those with a return to normal in aminotransferase levels after acute hepatitis C, adding up to an 85% likelihood of chronic HCV infection after acute hepatitis C. Few clues had emerged to explain host differences associated with chronic infection until recently, when variation in a single nucleotide polymorphism (SNP) on chromosome 19, IL28B (which codes for IFN-λ3), was identified that distinguished between responders and nonresponders to antiviral therapy (see below). The same variants correlated with spontaneous resolution after acute infection: 53% in genotype C/C, 30% in genotype C/T, but only 23% in genotype T/T. The association with HCV clearance after acute infection is even stronger when IL28B haplotype is combined with haplotype G/G of an SNP near HLA class II DBQ1*03:01.
In patients with chronic hepatitis C followed for 20 years, progression to cirrhosis occurs in about 20–25%. Such is the case even for patients with relatively clinically mild chronic hepatitis, including those without symptoms, with only modest elevations of aminotransferase activity, and with mild chronic hepatitis on liver biopsy. Even in cohorts of well-compensated patients with chronic hepatitis C referred for clinical research trials (no complications of chronic liver disease and with normal hepatic synthetic function), the prevalence of cirrhosis may be as high as 50%. Most cases of hepatitis C are identified initially in asymptomatic patients who have no history of acute hepatitis C (e.g., those discovered while attempting to donate blood, while undergoing lab testing as part of an application for life insurance, or as a result of routine laboratory tests). The source of HCV infection in many of these cases is not defined, although a long-forgotten percutaneous exposure (e.g., injection drug use) in the remote past can be elicited in a substantial proportion and probably accounts for most infections; most of these infections were acquired in the 1960s and 1970s, coming to clinical attention decades later.
Approximately one-third of patients with chronic hepatitis C have normal or near-normal aminotransferase activity; although one-third to one-half of these patients have chronic hepatitis on liver biopsy, the grade of liver injury and stage of fibrosis tend to be mild in the vast majority. In some cases, more severe liver injury has been reported—even, rarely, cirrhosis, most likely the result of previous histologic activity. Among patients with persistent normal aminotransferase activity sustained over ≥5–10 years, histologic progression has been shown to be rare; however, approximately one-fourth of patients with normal aminotransferase activity experience subsequent aminotransferase elevations, and histologic injury can be progressive once abnormal biochemical activity resumes. Therefore, continued clinical monitoring is indicated, even for patients with normal aminotransferase activity.
Despite this substantial rate of progression of chronic hepatitis C, and despite the fact that liver failure can result from end-stage chronic hepatitis C, the long-term prognosis for chronic hepatitis C in a majority of patients is relatively benign. Mortality over 10–20 years among patients with transfusion-associated chronic hepatitis C has been shown not to differ from mortality in a matched population of transfused patients in whom hepatitis C did not develop. Although death in the hepatitis group is more likely to result from liver failure, and although hepatic decompensation may occur in ~15% of such patients over the course of a decade, the majority (almost 60%) of patients remain asymptomatic and well compensated, with no clinical sequelae of chronic liver disease. Overall, chronic hepatitis C tends to be very slowly and insidiously progressive, if at all, in the vast majority of patients, whereas in approximately one-fourth of cases, chronic hepatitis C will progress eventually to end-stage cirrhosis. In fact, because HCV infection is so prevalent, and because a proportion of patients progress inexorably to end-stage liver disease, hepatitis C is the most frequent indication for liver transplantation (Chap. 368). In the United States, hepatitis C accounts for up to 40% of all chronic liver disease, and, as of 2007, mortality caused by hepatitis C surpassed that associated with HIV/AIDS. Moreover, because the prevalence of HCV infection is so much higher in the “baby boomer” cohort borne between 1945 and 1965, three-quarters of the mortality associated with hepatitis C occurs in this age cohort. Referral bias may account for the more severe outcomes described in cohorts of patients reported from tertiary care centers (20-year progression of ≥20%) versus the more benign outcomes in cohorts of patients monitored from initial blood-product-associated acute hepatitis or identified in community settings (20-year progression of only 4–7%). Still unexplained, however, are the wide ranges in reported progression to cirrhosis, from 2% over 17 years in a population of women with hepatitis C infection acquired from contaminated anti-D immune globulin to 30% over ≤11 years in recipients of contaminated intravenous immune globulin.
Progression of liver disease in patients with chronic hepatitis C has been reported to be more likely in patients with older age, longer duration of infection, advanced histologic stage and grade, more complex quasispecies diversity, increased hepatic iron, concomitant other liver disorders (alcoholic liver disease, chronic hepatitis B, hemochromatosis, α1 antitrypsin deficiency, and steatohepatitis), HIV infection, and obesity. Among these variables, however, duration of infection appears to be one of the most important, and some of the others probably reflect disease duration to some extent (e.g., quasispecies diversity, hepatic iron accumulation). No other epidemiologic or clinical features of chronic hepatitis C (e.g., severity of acute hepatitis, level of aminotransferase activity, level of HCV RNA, presence or absence of jaundice during acute hepatitis) are predictive of eventual outcome. Despite the relatively benign nature of chronic hepatitis C over time in many patients, cirrhosis following chronic hepatitis C has been associated with the late development, after several decades, of HCC (Chap. 111); the annual rate of HCC in cirrhotic patients with hepatitis C is 1–4%, occurring primarily in patients who have had HCV infection for 30 years or more.
Perhaps the best prognostic indicator in chronic hepatitis C is liver histology; the rate of hepatic fibrosis may be slow, moderate, or rapid. Patients with mild necrosis and inflammation as well as those with limited fibrosis have an excellent prognosis and limited progression to cirrhosis. In contrast, among patients with moderate to severe necroinflammatory activity or fibrosis, including septal or bridging fibrosis, progression to cirrhosis is highly likely over the course of 10–20 years. The pace of fibrosis progression may be accelerated by such factors as concomitant HIV infection, other causes of liver disease, excessive alcohol use, and hepatic steatosis. Among patients with compensated cirrhosis associated with hepatitis C, the 10-year survival rate is close to 80%; mortality occurs at a rate of 2–6% per year; decompensation at a rate of 4–5% per year; and, as noted above, HCC at a rate of 1–4% per year. A discussion of the pathogenesis of liver injury in patients with chronic hepatitis C appears in Chap. 360.
Clinical features of chronic hepatitis C are similar to those described above for chronic hepatitis B. Generally, fatigue is the most common symptom; jaundice is rare. Immune complex–mediated extrahepatic complications of chronic hepatitis C are less common than in chronic hepatitis B (despite the fact that assays for immune complexes are often positive in patients with chronic hepatitis C), with the exception of essential mixed cryoglobulinemia (Chap. 360), which is linked to cutaneous vasculitis and membranoproliferative glomerulonephritis as well as lymphoproliferative disorders such as B-cell lymphoma and unexplained monoclonal gammopathy. In addition, chronic hepatitis C has been associated with extrahepatic complications unrelated to immune-complex injury. These include Sjögren’s syndrome, lichen planus, porphyria cutanea tarda, type 2 diabetes mellitus, and the metabolic syndrome (including insulin resistance and steatohepatitis).
Laboratory features of chronic hepatitis C are similar to those in patients with chronic hepatitis B, but aminotransferase levels tend to fluctuate more (the characteristic episodic pattern of aminotransferase activity) and to be lower, especially in patients with long-standing disease. An interesting and occasionally confusing finding in patients with chronic hepatitis C is the presence of autoantibodies. Rarely, patients with autoimmune hepatitis (see below) and hyperglobulinemia have false-positive immunoassays for anti-HCV. On the other hand, some patients with serologically confirmable chronic hepatitis C have circulating anti-LKM. These antibodies are anti-LKM1, as seen in patients with autoimmune hepatitis type 2 (see below), and are directed against a 33-amino-acid sequence of cytochrome P450 IID6. The occurrence of anti-LKM1 in some patients with chronic hepatitis C may result from the partial sequence homology between the epitope recognized by anti-LKM1 and two segments of the HCV polyprotein. In addition, the presence of this autoantibody in some patients with chronic hepatitis C suggests that autoimmunity may be playing a role in the pathogenesis of chronic hepatitis C.
Histopathologic features of chronic hepatitis C, especially those that distinguish hepatitis C from hepatitis B, are described in Chap. 360.
|
TREATMENT |
CHRONIC HEPATITIS C |
Therapy for chronic hepatitis C has evolved substantially in the two decades since IFN-α was introduced for this indication. The therapeutic armamentarium has grown to include PEG IFN with ribavirin and, in 2011, the introduction of protease inhibitors telaprevir and boceprevir used in combination with PEG IFN and ribavirin in patients with HCV genotype 1. When first approved, IFN-α was administered via subcutaneous injection three times a week for 6 months but achieved a sustained virologic response (SVR) (Fig. 362-2) (a reduction of HCV RNA to undetectable levels by PCR when measured ≥6 months after completion of therapy) below 10%. Doubling the duration of therapy—but not increasing the dose or changing IFN preparations—increased the SVR rate to ~20%, and addition to the regimen of daily ribavirin, an oral guanosine nucleoside, increased the SVR rate to 40%. When used alone, ribavirin is ineffective and does not reduce HCV RNA levels appreciably, but ribavirin enhances the efficacy of IFN by reducing the likelihood of virologic relapse after the achievement of an end-treatment response (Fig. 362-2) (response measured during, and maintained to the end of, treatment). Proposed mechanisms to explain the role of ribavirin include subtle direct reduction of HCV replication, inhibition of host inosine monophosphate dehydrogenase activity (and associated depletion of guanosine pools), immune modulation, induction of virologic mutational catastrophe, and enhancement of IFN-stimulated gene expression. IFN therapy results in activation of the JAK-STAT signal transduction pathway, which culminates in the intracellular elaboration of genes and their protein products that have antiviral properties. Hepatitis C proteins inhibit JAK-STAT signaling at several steps along the pathway, and exogenous IFN restores expression of IFN-stimulated genes and their antiviral effects.
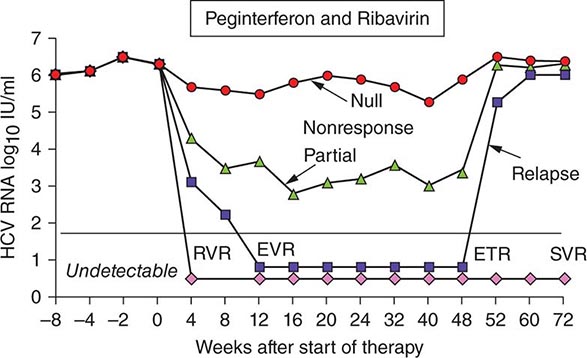
FIGURE 362-2 Classification of virologic responses based on outcomes during and after a 48-week course of pegylated interferon (PEG IFN) plus ribavirin antiviral therapy in patients with hepatitis C, genotype 1 or 4 (for genotype 2 or 3, the course would be 24 weeks). Nonresponders can be classified as null responders (hepatitis C virus [HCV] RNA reduction of <2 log10 IU/mL) or partial responders (HCV RNA reduction ≥2 log10 IU/mL but not suppressed to undetectable) by week 24 of therapy. In responders, HCV RNA can become undetectable, as shown with sensitive amplification assays, within 4 weeks (RVR, rapid virologic response); can be reduced by ≥2 log10 IU/mL within 12 weeks (early virologic response, EVR; if HCV RNA is undetectable at 12 weeks, the designation is “complete” EVR); or at the end of therapy, 48 weeks (ETR, end-treatment response). In responders, if HCV RNA remains undetectable for 24 weeks after ETR, week 72, the patient has a sustained virologic response (SVR), but if HCV RNA becomes detectable again, the patient is considered to have relapsed. In patients treated with protease inhibitor–based therapy, several additional milestones are monitored: (1) among boceprevir-treated patients, the level of HCV RNA reduction (>1 log10 or ≥1 log10 IU/mL) during the 4-week PEG IFN–ribavirin lead-in phase; (2) during boceprevir therapy, undetectable HCV RNA at week 8 (week 4 of triple-drug therapy; RVR); and (3) among telaprevir-treated patients, undetectable HCV RNA at week 4 and 12 (extended RVR). (Reproduced with permission, courtesy of Marc G. Ghany, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health and the American Association for the Study of Liver Diseases. Hepatology 49:1335, 2009.)
Treatment with the combination of PEG IFN and ribavirin increased responsiveness (frequency of SVR) to as high as 55% overall, to >40% in genotypes 1 and 4, and to >80% in genotypes 2 and 3. Still, many important lessons about antiviral therapy for chronic hepatitis C were learned from the experience with IFN monotherapy and combination IFN-ribavirin therapy. Even in the absence of biochemical and virologic responses, histologic improvement occurs in approximately three-fourths of all treated patients. In chronic hepatitis C, unlike the case in hepatitis B, responses to therapy are not accompanied by transient, acute hepatitis-like aminotransferase elevations. Instead, ALT levels fall precipitously during therapy. Up to 90% of virologic responses are achieved within the first 12 weeks of therapy; responses thereafter are rare. Most relapses occur within the first 12 weeks after treatment; therefore, an SVR at week 12 posttreatment is roughly equivalent to a 24-week SVR. SVRs are very durable; normal ALT, improved histology, and absence of HCV RNA in serum and liver have been documented a decade after successful therapy, and “relapses” 2 years after sustained responses are almost unheard of. Thus, an SVR to antiviral therapy of chronic hepatitis C is tantamount to a cure.
Patient variables that tend to correlate with sustained virologic responsiveness to IFN-based therapy include favorable genotype (genotypes 2 and 3 as opposed to genotypes 1 and 4); low baseline HCV RNA level (<2 million copies/mL, which is equivalent to <800,000 IU/mL, the current convention of quantitation); histologically mild hepatitis and minimal fibrosis; age <40; female gender; and absence of obesity, insulin resistance, and type 2 diabetes mellitus. Patients with cirrhosis can respond, but they are less likely to do so. For patients treated with combination IFN-ribavirin, therapy for those with genotype 1 should last a full 48 weeks, whereas in those with genotypes 2 and 3, a 24-week course of therapy suffices (although refined tailoring of treatment duration may be indicated based on rapidity of response or associated cofactors, see below). The response rate in African Americans is disappointingly low for reasons that are not fully understood. Potentially contributing to, but not explaining entirely, low responsiveness in African Americans are a higher proportion with genotype 1, slower early viral kinetics during therapy, impaired HCV-specific immunity, and recently recognized host genetic differences in IL28B alleles, described below. The response rate in Latino patients is also low, despite the fact that the frequency of the favorable IL28B C allele is as common in Hispanic patients as in whites. Moreover, the likelihood of a sustained response is best if adherence to the treatment regimen is high (i.e., if patients receive ≥80% of the IFN and ribavirin doses and if they continue treatment for ≥80% of the anticipated duration of therapy). Other variables reported to correlate with increased responsiveness include brief duration of infection, low HCV quasispecies diversity, immunocompetence, absence of hepatic steatosis, and low liver iron levels. High levels of HCV RNA, more histologically advanced liver disease, and high quasispecies diversity all go hand in hand with advanced duration of infection, which is one of the most important clinical variables determining IFN responsiveness. The ironic fact, then, is that patients whose disease is least likely to progress are the ones most likely to respond to IFN and vice versa.
Genetic changes in the virus may explain differences in treatment responsiveness in some patients (e.g., among patients with genotype 1b, responsiveness to IFN is enhanced in those with amino-acid-substitution mutations in the nonstructural protein 5A gene). As described above in the discussion of spontaneous recovery from acute hepatitis C, IFN gene variants discovered recently in genome-wide association studies have been shown to have a substantial impact on responsiveness of patients with genotype 1 to antiviral therapy. In studies of patients treated with PEG IFN and ribavirin, variants of the IL28B SNP that code for IFN-λ3 (a type III IFN, the receptors for which are more discretely distributed than IFN-α receptors and more concentrated in hepatocytes) correlate significantly with responsiveness. Patients homozygous for the C allele at this locus have the highest frequency of achieving an SVR (~80%), those homozygous for the T allele at this locus are least likely to achieve an SVR (~25%), and those heterozygous at this locus (C/T) have an intermediate level of responsiveness (SVRs in ~35%). The fact that C/C is common in whites of European ancestry and even more so in Japanese persons but rare in African Americans helps explain the differences in observed responsiveness among these population groups.
Side effects of IFN therapy are described above in the section on treatment of chronic hepatitis B. The most pronounced side effect of ribavirin therapy is hemolysis; a reduction in hemoglobin of up to 2–3 g or in hematocrit of 5–10% can be anticipated. A small, unpredictable proportion of patients experience profound, brisk hemolysis, resulting in symptomatic anemia; therefore, close monitoring of blood counts is crucial, and ribavirin should be avoided in patients with anemia or hemoglobinopathies and in patients with coronary artery disease or cerebrovascular disease, in whom anemia can precipitate an ischemic event. When symptomatic anemia occurs, ribavirin dose reductions or addition of erythropoietin to boost red blood cell levels may be required; erythropoietin has been shown to improve patients’ quality of life but not the likelihood of achieving an SVR. If ribavirin is stopped during therapy, SVR rates fall, but responsiveness can be maintained as long as ribavirin is not stopped and the total ribavirin dose exceeds 60% of the planned dose. In addition, ribavirin, which is excreted renally, should not be used in patients with renal insufficiency; the drug is teratogenic, precluding its use during pregnancy and mandating the scrupulous use of efficient contraception during therapy (IFNs, too, because of their antiproliferative properties, are contraindicated during pregnancy).
Ribavirin can also cause nasal and chest congestion, pruritus, and precipitation of gout. Combination IFN-ribavirin therapy is more difficult to tolerate than IFN monotherapy. In one large clinical trial of combination therapy versus monotherapy, among those in the 1-year treatment group, 21% of the combination group (but only 14% of the monotherapy group) had to discontinue treatment, whereas 26% of the combination group (but only 9% of the monotherapy group) required dose reductions.
Studies of viral kinetics have shown that despite a virion half-life in serum of only 2–3 h, the level of HCV is maintained by a high replication rate of 1012 hepatitis C virions per day. IFN-α blocks virion production or release with an efficacy that increases with increasing drug doses; moreover, the calculated death rate for infected cells during IFN therapy is inversely related to level of HCV RNA; patients with the most rapid death rate of infected hepatocytes are more likely to achieve undetectable HCV RNA at 3 months; in practice, failure to achieve an early virologic response (EVR), a ≥2-log10 reduction in HCV RNA by week 12, predicts failure to experience a subsequent SVR. Similarly, patients in whom HCV RNA becomes undetectable within 4 weeks (i.e., who achieve a rapid virologic response [RVR]) have a very high likelihood of achieving an SVR (Fig. 362-2). Therefore, to achieve rapid viral clearance from serum and the liver, high-dose induction therapy has been advocated. In practice, however, high-dose induction with IFN-based therapy has not yielded higher sustained response rates.
For the treatment of chronic hepatitis C, standard IFNs were supplanted by PEG IFNs. These have elimination times up to sevenfold longer than standard IFNs (i.e., a substantially longer half-life) and achieve prolonged concentrations, permitting administration once (rather than three times) a week. Instead of the frequent drug peaks (linked to side effects) and troughs (when drug is absent) associated with frequent administration of short-acting IFNs, administration of PEG IFNs results in drug concentrations that are more stable and sustained over time. Once-a-week PEG IFN monotherapy is twice as effective as monotherapy with its standard IFN counterpart, approaches the efficacy of combination standard IFN plus ribavirin, and is as well tolerated as standard IFNs, without more difficult-to-manage thrombocytopenia and leukopenia than standard IFNs. For most of the decade prior to 2011, when protease inhibitors were introduced for HCV genotype 1 (see below), the standard of care was a combination of PEG IFN plus ribavirin for all HCV genotypes.
Two PEG IFNs are available: PEG IFN-α2b and -α2a. PEG IFN-α2b consists of a 12-kD, linear PEG molecule bound to IFN-α2b, whereas PEG IFN-α2a consists of a larger, 40-kD, branched PEG molecule bound to IFN-α2a; because of its larger size and smaller volume of extravascular distribution, PEG IFN-α2a can be given at a uniform dose independent of weight, whereas the dose of the smaller PEG IFN-α2b, which has a much wider volume distribution, must be weight-based (Table 362-6). In the registration trial for PEG IFN-α2b plus ribavirin, the best regimen was 48 weeks of 1.5 μg/kg of PEG IFN once a week plus 800 mg of ribavirin daily. A post hoc analysis suggested that weight-based dosing of ribavirin would have been more effective than the fixed 800-mg dose used in the study (a broader dose/weight range was approved subsequently; see below). In the first registration trial for PEG IFN-α2a plus ribavirin, the best regimen was 48 weeks of 180 μg of PEG IFN plus 1000 mg (for patients <75 kg) to 1200 mg (for patients ≥75 kg) of ribavirin. SVRs of 54 and 56% were reported in these two studies, respectively. A subsequent study of PEG IFN-α2a plus ribavirin showed that, for patients with genotypes 2 and 3, a duration of 24 weeks and a ribavirin dose of 800 mg were sufficient. Among the three studies, for patients in the optimal treatment arm, SVR rates for patients with genotype 1 were 42–51%, and for patients with genotypes 2 and 3, rates were 76–82%. Between genotypes 2 and 3, genotype 3 is somewhat more refractory, and some authorities would extend therapy for a full 48 weeks in patients with genotype 3, especially if they have advanced hepatic fibrosis or cirrhosis and/or high-level HCV RNA.
|
PEGYLATED INTERFERON (PEG IFN) α2a AND α2b FOR CHRONIC HEPATITIS C |
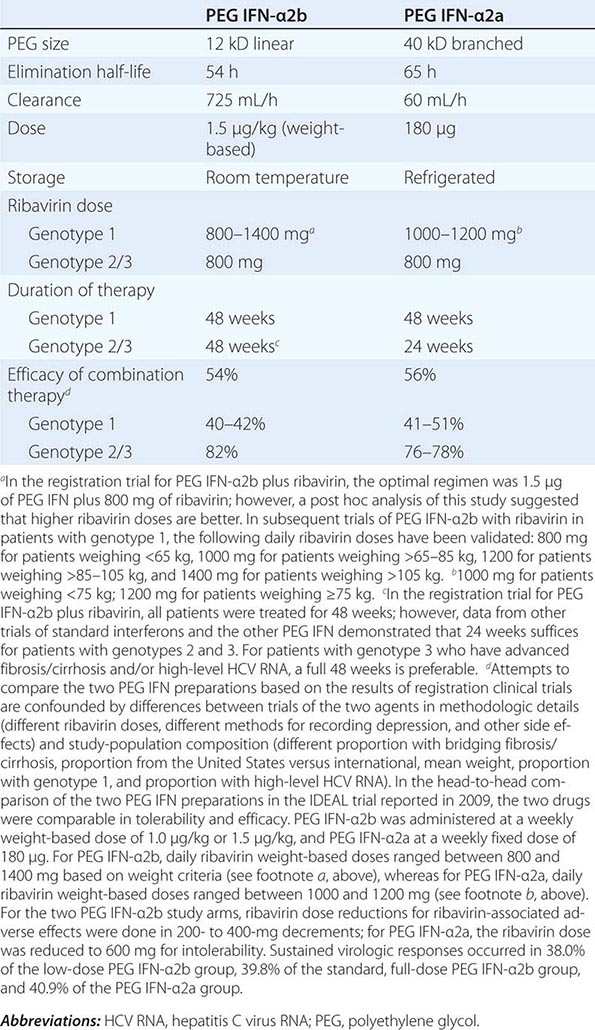
In the initial registration trials for combination PEG IFN plus ribavirin, both combination PEG IFN regimens were compared to standard IFN-α2b plus ribavirin. Side effects of the combination PEG IFN-α2b regimen were comparable to those for the combination standard IFN regimen; however, when the combination PEG IFN-α2a regimen was compared with the combination standard IFN-α2b regimen, flu-like symptoms and depression were less common in the combination PEG IFN group. Although ascertainment of side effects differed between studies of the two drugs, when each was tested against standard IFN-α2b plus ribavirin, combination PEG IFN-α2a plus ribavirin appeared to be better tolerated. In a head-to-head trial of the two PEG IFNs (the IDEAL trial), the two PEG IFNs were found to be comparable in efficacy (achievement of SVR) and tolerability, although headache, nausea, fever, myalgia, depression, and drug discontinuation for any reason were less frequent in patients treated with PEG IFN-α2a than standard-dose PEG IFN-α2b. In contrast, neutropenia and rash were more frequent in patients treated with PEG IFN-α2a than standard-dose PEG IFN-α2b. In two subsequent head-to-head trials and a systematic review of randomized trials, PEG IFN-α2a was more effective than PEG IFN-α2b (SVR in genotypes 1–4: 48–55% versus 32–40%, respectively). In trials of PEG IFN-α2b among patients with HCV genotype 1, a broader range of weight-based daily ribavirin doses has been validated: 800 mg for weight <65 kg, 1000 mg for weight 65–85 kg, 1200 mg for weight >85–105 kg, and 1400 mg for weight >105 kg. Recommended doses for the two PEG IFNs plus ribavirin and other comparisons between the two therapies are shown in Table 362-6.
Until the 2011 introduction of protease inhibitors, unless ribavirin was contraindicated (see above), combination PEG IFN plus ribavirin was the recommended course of therapy—24 weeks for genotypes 2 and 3 and 48 weeks for genotype 1. For patients with genotypes 1 and 4, the standard of care now includes protease inhibitors or other direct-acting antiviral agents (see below); however, PEG IFN–ribavirin remained the standard of care for patients with genotypes 2 and 3 until late 2013. For patients treated with combination PEG IFN–ribavirin, measurement of quantitative HCV RNA levels at 12 weeks is helpful in guiding therapy; if a 2-log10 drop in HCV RNA has not been achieved by this time, chances for an SVR are negligible, and additional therapy is futile. If the 12-week HCV RNA has fallen by 2 log10 (EVR), the chances for an SVR at the end of therapy are approximately two-thirds; if the 12-week HCV RNA is undetectable (“complete” EVR), the chances for an SVR exceed 80% (Fig. 362-2). Because absence of an EVR is such a strong predictor of the absence of an ultimate SVR, therapy is discontinued for failure to achieve a 12-week 2-log10 drop in HCV RNA (EVR).
Studies have suggested that the frequency of an SVR to PEG IFN–ribavirin therapy can be increased in patients with baseline variables weighing against a response (e.g., HCV RNA >8 × 105 IU/mL, weight >85 kg) by raising the dose of PEG IFN (e.g., to as high as 270 μg of PEG IFN-α2a) and/or the dose of ribavirin to as high as 1600 mg daily (if tolerated or supplemented by erythropoietin) or by tailoring treatment based on viral response to prolong the duration of viral clearance before discontinuing therapy, i.e., extending therapy from 48 to 72 weeks for patients with genotype 1 and a slow virologic response (i.e., those whose HCV RNA has not fallen rapidly to undetectable levels within 4 weeks [absence of RVR]). Tailoring therapy based on the kinetics of HCV RNA reduction has also been applied to abbreviating the duration of therapy in patients with genotype 1 (and 4). The results of several clinical trials suggest that, in patients with genotype 1 (and 4) who have a 4-week RVR (which occurs in ≤20%), especially in the subset with low baseline HCV RNA, 24 weeks of therapy with PEG IFN and weight-based ribavirin suffices, yielding SVR rates of ~90% and comparable to those achieved in this cohort with 48 weeks of therapy. Although initial reports suggested that, for patients with genotype 2 and (somewhat less so) genotype 3, in rapid virologic responders with undetectable HCV RNA at week 4, the total duration of therapy required to achieve an SVR could be as short as 12–16 weeks, a very sizable, definitive subsequent trial showed that relapse is increased if treatment duration is curtailed and that a full 24 weeks is superior for these genotypes (except for the minority with very low baseline levels of HCV RNA).
Persons with chronic HCV infection have been shown to suffer increased liver-related mortality. On the other hand, successful antiviral therapy of chronic hepatitis C resulting in an SVR has been shown to improve survival (and to reduce the need for liver transplantation), to lower the risk of liver failure and liver-related death and all-cause deaths, to slow the progression of chronic hepatitis C, and to reverse fibrosis and even cirrhosis. Although successful treatment reduces mortality in cirrhotic patients (and those with advanced fibrosis) and reduces the likelihood of HCC, the risk of liver-related death and HCC persists, albeit at a much reduced level, necessitating continued clinical monitoring and cancer surveillance after SVR in cirrhotics. On the other hand, in the absence of an SVR, routine-dose/duration IFN-based therapy does not reduce the risk of HCC. Similarly, for nonresponders to PEG IFN–ribavirin therapy, three trials of long-term maintenance therapy with PEG IFN have shown no benefit in reducing the risk of histologic progression or clinical decompensation, including the development of HCC. For PEG IFN–ribavirin nonresponders who have had a full, adequate course of therapy, the benefit of retreatment—with higher doses or a longer course of the original PEG IFN regimen or the alternative PEG IFN regimen or with a different type of IFN preparation (e.g., consensus IFN)—is marginal at best. Fortunately, such nonresponders can now be retreated with protease inhibitor-based therapy (see following).
FIRST-GENERATION PROTEASE INHIBITORS (2011–2013)
The HCV RNA genome encodes a single polyprotein, which is cleaved during and after translation by host and viral-encoded proteases. One protease involved in the cleavage of the viral polyprotein is an NS3-4A viral protein that has serine protease activity. Telaprevir and boceprevir are serine protease inhibitors that target NS3-4A. In 2011, telaprevir and boceprevir used in combination with PEG IFN and ribavirin were approved by the U.S. Food and Drug Administration (FDA) for the treatment of hepatitis C genotype 1 in adults with stable liver disease, both in patients who have not been treated before or who have failed previous treatment. Because the presently available HCV protease inhibitors have not been studied comprehensively in patients with genotypes other than 1, their use in these populations is not recommended.
Because resistance develops rapidly, both telaprevir and boceprevir must be used in combination with a PEG IFN and ribavirin-based regimen and should never be used alone. Ribavirin in particular appears to reduce relapse rates significantly in protease inhibitor–based regimens, such that those who cannot take or are intolerant to ribavirin are unlikely to benefit from the addition of these agents. All current telaprevir and boceprevir regimens consist of periods of triple therapy (protease inhibitor plus PEG IFN plus ribavirin) and periods of dual therapy (PEG IFN plus ribavirin). Telaprevir regimens begin with 12 weeks of triple therapy followed by dual therapy of a duration based on HCV RNA status at weeks 4 and 12 (“response-guided therapy”) and prior treatment status. Boceprevir-based regimens consist of a 4-week lead-in period of dual (PEG IFN–ribavirin) therapy followed by triple therapy and, in some instances, a further extension of dual therapy, with duration of response-guided therapy based on HCV RNA status at weeks 4, 8, and 24 and prior treatment status (Table 362-7).
|
INDICATIONS AND RECOMMENDATIONS FOR ANTIVIRAL THERAPY OF CHRONIC HEPATITIS Ca |
aAs this chapter was going to press, two additional drugs were approved for hepatitis C, simeprevir and sofosbuvir. Rapidly evolving new recommendations are supplanting the recommendations in this table; for up-to-date treatment recommendations, please see www.hcvguidelines.org. bLess influential in patients treated with protease inhibitors.
Abbreviations: ALT, alanine aminotransferase; HCV, hepatitis C virus; IFN, interferon; PEG IFN, pegylated interferon; IU, international units (1 IU/mL is equivalent to ~2.5 copies/mL).
For patients with HCV genotype 1, protease inhibitors have significantly improved the frequency of RVRs and SVRs as compared to PEG IFN plus ribavirin alone. In treatment-naïve patients treated with telaprevir, an SVR was seen in up to 79% of patients who received 12 weeks of triple therapy followed by 12–36 weeks of dual therapy, and among those with EVRs (undetectable HCV RNA at weeks 4 and 12) and response-guided therapy stopped at week 24 (12 weeks of triple therapy, then 12 weeks of dual therapy), the rate of SVRs was 83–89% (92% in a subsequent study). In studies with boceprevir in treatment-naïve patients, SVRs were seen in 59–66% of patients, and among those with undetectable HCV RNA at 8 weeks, the SVR rate increased to 86–88%.
Protease inhibitors have also been studied in patients previously treated unsuccessfully with PEG IFN plus ribavirin. In studies with telaprevir, SVRs were seen in 83–88% of patients who had a previous relapse, 54–59% of partial responders (HCV RNA reduced by ≥2 log10 IU/mL but not to undetectable levels), and 29–33% of null responders (HCV RNA reduced by <2 log10 IU/mL). With boceprevir, SVRs occurred in 75% of prior relapsers and in 40–52% of previous partial responders; response rates in null responders are similar to those achieved with telaprevir-based therapy. In a substantial proportion of protease inhibitor nonresponders, resistance-associated variants can be identified, but these variants are not archived, and wild-type HCV reemerges in almost all cases within 1.5 to 2 years. SVRs to these protease inhibitors are highest in prior relapsers and treatment-naïve patients (white > black ethnicity), lower in prior partial responders, lower still in prior null responders, and lowest in cirrhotic prior null responders (Fig. 362-3). Responses to protease inhibitor triple-drug regimens are higher in patients with IL28B C than non-C genotypes, HCV genotype 1b than genotype 1a, less advanced than more advanced fibrosis stage, whites than blacks, lower body mass index (BMI) than elevated BMI, and, for boceprevir, achievement of a >1 log10 HCV RNA reduction during 4 weeks of PEG IFN–ribavirin lead-in therapy. Age and HCV RNA level are less influential and insulin resistance is noninfluential on response to these antiviral agents.
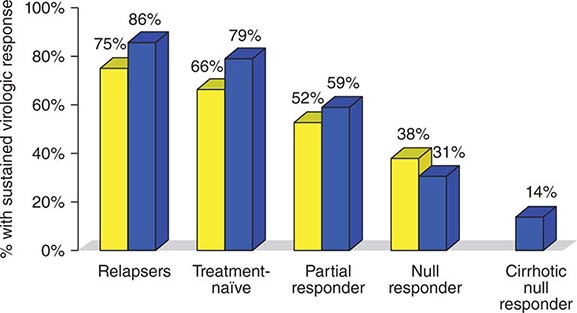
FIGURE 362-3 Maximal efficacy (sustained virologic responses, SVR) of telaprevir (blue bars) and boceprevir (yellow bars) reported in phase III clinical trials. (Figure created using data from Bacon BR et al: N Engl J Med 364:1207, 2011; Jacobson IM et al: N Engl J Med 364:2405, 2011; Poordad F et al: N Engl J Med 364:1195, 2011; Zeuzem S et al: N Engl J Med 364:2417, 2011; Vierling JM et al: Hepatology 54 [Suppl 1]:796A-797A, 2011; Ghany MG et al: Hepatology 54:1433, 2011.)
Both protease inhibitors have potential toxicities. Telaprevir is associated with a severe, generalized (trunk and extremities), often confluent, maculopapular, pruritic rash in ~6% of treated patients. Other common side effects include pruritus, rectal burning, nausea, diarrhea, fatigue, and anemia, which may be relatively refractory, occasionally requiring transfusion. Complete blood counts should be obtained at baseline and then at 2, 4, 8, and 12 weeks after starting telaprevir. Anemia can occur in half of boceprevir-treated patients, as can neutropenia in up to 30% and thrombocytopenia in 3–4%. Complete blood counts should be obtained at baseline and then at 4, 8, and 12 weeks after starting boceprevir. Other side effects of boceprevir include fatigue, nausea, headache, dysgeusia (altered or unpleasant taste), dry mouth, vomiting, and diarrhea.
Use of protease inhibitors is further complicated by numerous drug-drug interactions. As telaprevir and boceprevir are both eliminated by and inhibit CYP3A4, these agents should not be administered with other medications that induce CYP3A4 or are dependent on CYP3A4 for elimination. Care should be taken to examine for any potential interactions between protease inhibitors and other medications the patient may be taking, because serious adverse events can occur. A convenient website is available to check for such drug-drug interactions (www.hep-druginteractions.org).
TREATMENT RECOMMENDATIONS*
Prior to therapy, HCV genotype should be determined, because the genotype dictates the duration of therapy and potentially the agents to be used. PEG IFN plus ribavirin represents the foundation of treatment for all HCV genotypes; patients infected with genotype 1 should also receive a protease inhibitor (telaprevir or boceprevir) when these are available and not contraindicated (Table 362-7). For chronic HCV genotype 1 infection, the AASLD and EASL published treatment guidelines in 2011 reflecting FDA-approved indications for the new protease inhibitors, and in 2012, United Kingdom and French consensus guidelines were published. For treatment-naïve patients and prior relapsers, response-guided therapy with telaprevir or boceprevir is recommended. For telaprevir, the regimen consists of 12 weeks of triple therapy, followed by 12 or 36 weeks of PEG IFN–ribavirin consolidation, depending on whether extended RVR milestones (HCV RNA undetectable at weeks 4 and 12) are met. For boceprevir, the regimen consists of a 4-week PEG IFN–ribavirin lead-in period, followed by 24–32 weeks of triple-drug therapy, depending on whether HCV RNA milestones (undetectable at weeks 8 and 24) are met; if HCV RNA is detectable at week 8 but undetectable at week 24, after 36 weeks of therapy (4-week PEG IFN–ribavirin lead-in plus 32 weeks of triple-drug therapy), an additional 12 weeks of PEG IFN–ribavirin consolidation is recommended. For prior partial and null responders, a full 48-week course of telaprevir (no lead-in period, no response-guided therapy) is recommended; for boceprevir, a 4-week PEG IFN–ribavirin lead-in period is followed by response-guided therapy (32 weeks of triple-drug therapy if HCV RNA is undetectable at weeks 8 and 24 or, if HCV RNA is still detectable at week 8 [but undetectable at week 24], 32 weeks of triple-drug therapy followed by 12 weeks of PEG IFN–ribavirin consolidation). For cirrhotic patients (and for any boceprevir-treated patient whose HCV RNA does not fall by >1 log10 by week 4), a full 48-week course without response-guided therapy should be considered.
Monitoring of HCV plasma RNA is crucial in assessing response to therapy. The goal of treatment is to eradicate HCV RNA, which is predicted by the absence of HCV RNA by PCR 6 months after stopping treatment (SVR). When therapy relied on PEG IFN and ribavirin, failure to achieve a 2-log10 drop in HCV RNA by week 12 of therapy (EVR) rendered it unlikely that further therapy would result in an SVR. When PEG IFN and ribavirin are part of a protease inhibitor regimen, HCV RNA should be measured at baseline and at weeks 4, 8 (for boceprevir), 12, and 24 to assess response to treatment and to aid in decisions regarding treatment duration (response-guided therapy), as well as 12 and 24 weeks after therapy. Stopping rules are important to prevent the emergence of resistance; if HCV RNA is >1000 IU/mL at 4 or 12 weeks of telaprevir (or still detectable at week 24), or if HCV RNA is ≥100 IU/mL at week 12 of boceprevir (or detectable at week 24), all treatment should be stopped.
INDICATIONS FOR ANTIVIRAL THERAPY*
Patients with chronic hepatitis C who have detectable HCV RNA in serum, whether or not aminotransferase levels are increased, and chronic hepatitis of at least moderate grade and stage (portal or bridging fibrosis) are candidates for antiviral therapy with PEG IFN plus ribavirin. Most authorities recommend 800 mg of ribavirin for patients with genotypes 2 and 3 for both types of PEG IFN and weight-based 1000–1200 mg (when used with PEG IFN-α2a) or 800–1400 mg (when used with PEG IFN-α2b) ribavirin for patients with genotype 1 (and 4), unless ribavirin is contraindicated (Table 362-7). These PEG IFN and ribavirin doses are used with protease inhibitors for patients with genotype 1 (Table 362-7). Although patients with persistently normal ALT activity tend to progress histologically very slowly or not at all, they respond to antiviral therapy just as well as do patients with elevated ALT levels; therefore, although observation without therapy is an option, such patients are potential candidates for antiviral therapy. As noted above, therapy with IFN has been shown to improve survival and complication-free survival and to slow progression of (and to reverse) fibrosis.
HCV genotype determines the duration of PEG IFN and ribavirin therapy: 24 weeks for those with genotypes 2 and 3 and 48 weeks for patients with genotypes 4 and 1 (in patients for whom protease inhibitors are not available or contraindicated). For patients with genotype 4, treatment should be discontinued in patients who do not achieve an EVR at week 12. For patients with genotypes 2 and 3, a full, 24-week course is most effective, although the duration may be reduced to 12–16 weeks for patients with genotype 2, a low baseline level of viremia, and an RVR, especially to be considered for patients who tolerate therapy poorly. Also, consideration should be given to increasing the duration of therapy to 48 weeks for patients with genotype 3 who have advanced fibrosis and/or a high baseline level of viremia. As noted above, the absence of a 2-log10 drop in HCV RNA at week 12 (EVR) weighs heavily against the likelihood of an SVR; therefore, measuring HCV RNA at 12 weeks is recommended routinely (Fig. 362-2), and therapy can be discontinued if an EVR is not achieved. Among patients with genotype 4 who achieve an EVR (≥2-log10 HCV RNA reduction) but in whom HCV RNA remains detectable at week 24, an SVR is unlikely, and therapy can be discontinued. Although response rates are lower in patients with certain pretreatment variables, selection for treatment should not be based on symptoms, genotype, HCV RNA level, mode of acquisition of hepatitis C, or advanced hepatic fibrosis. Patients with cirrhosis can respond and should not be excluded as candidates for therapy. For patients being treated with telaprevir and boceprevir, treating physicians should explain the negative impact of non-C IL28B genotype and advanced fibrosis on outcome.
Patients who have relapsed after, or failed to respond to (Fig. 362-2), a course of IFN monotherapy are potential candidates for retreatment with PEG IFN plus ribavirin (i.e., a more effective treatment regimen is required), and this approach remains current for patients with genotypes 2, 3, or 4; however, for patients with genotype 1, combination protease inhibitor/PEG IFN/ribavirin therapy is indicated. For patients with genotypes 2, 3, or 4 who were nonresponders to a prior course of IFN monotherapy, retreatment with IFN monotherapy or combination IFN plus ribavirin therapy is unlikely to achieve an SVR; however, a trial of combination PEG IFN plus ribavirin may be worthwhile, although an SVR is the outcome in <15–20% of patients. SVRs to retreatment of nonresponders are more frequent in those who had never received ribavirin in the past, those with genotypes 2 and 3, those with low pretreatment HCV RNA levels, and noncirrhotics, but less frequent in African Americans, those who failed to achieve a substantial reduction in HCV RNA during their previous course of therapy (null responders, Fig. 362-2), and those who required ribavirin dose reductions. Potential approaches to improving responsiveness to PEG IFN–ribavirin in prior nonresponders include longer duration of treatment; higher doses of PEG IFN, ribavirin, or both; and switching to a different IFN preparation; however, as noted above, none of these approaches achieves more than a marginal benefit. Treatment with a protease inhibitor–based regimen should be pursued in patients with genotype 1 who have relapsed after or not responded to prior treatment with IFN monotherapy or PEG IFN plus ribavirin, unless these protease inhibitors are not available or contraindicated (Table 362-7).
Early PEG IFN treatment is indicated for persons with acute hepatitis C; ribavirin, which is used frequently in such instances, has not been shown to improve efficacy over that of PEG IFN alone, and the new protease inhibitors have not been approved for acute hepatitis C (Chap. 360). In patients with biochemically and histologically mild chronic hepatitis C, the rate of progression is slow, and monitoring without therapy is an option; however, such patients respond just as well to combination PEG IFN plus ribavirin therapy or triple-drug, protease-based therapy (for genotype 1) as those with elevated ALT and more histologically severe hepatitis. Therefore, therapy for these patients should be considered and the decision made based on such factors as patient motivation, genotype, stage of fibrosis, age, and comorbid conditions. A pretreatment liver biopsy to assess histologic grade and stage provides substantial information about progression of hepatitis C in the past, has prognostic value for future progression, and can identify such histologic factors as steatosis and stage of fibrosis, which can influence responsiveness to therapy. As therapy has improved for patients with a broad range of histologic severity, and as noninvasive laboratory markers and imaging correlates of fibrosis have gained popularity, some authorities, especially in Europe, place less value on, and do not recommend, pretreatment liver biopsies. On the other hand, serum markers of fibrosis are not considered sufficiently accurate, and histologic findings provide important prognostic information to physician and patient. Therefore, although the contemporary role of a pretreatment liver biopsy commands less of a consensus, a pretreatment liver biopsy still provides useful information and should be considered.
Patients with compensated cirrhosis can respond to therapy, although their likelihood of a sustained response is lower than in noncirrhotics; moreover, survival has been shown to improve after successful antiviral therapy in cirrhotics. Similarly, although several retrospective studies have suggested that antiviral therapy in cirrhotics with chronic hepatitis C, independent of treatment outcome per se, reduces the frequency of HCC, less advanced disease in the treated cirrhotics, not treatment itself (i.e., lead-time bias), may have accounted for the reduced frequency of HCC observed in the treated cohorts in these reports; prospective studies to address this question have failed to demonstrate benefit, unless an SVR is achieved. Patients with decompensated cirrhosis are not candidates for IFN-based antiviral therapy but should be referred for liver transplantation. Some liver transplantation centers have evaluated progressively escalated, low-dose antiviral therapy in an attempt to eradicate hepatitis C viremia prior to transplantation; however, such therapy has been shown to reduce but not to prevent the risk of HCV reinfection after transplantation. After liver transplantation for end-stage liver disease caused by hepatitis C, recurrent hepatitis C is the rule, and the pace of disease progression is more accelerated than in immunocompetent patients (Chap. 368). Current therapy with PEG IFN and ribavirin after liver transplantation is unsatisfactory in most patients, but attempts to minimize immunosuppression are beneficial. Early experience with protease inhibitor–based therapy is encouraging, but inhibition of CYP3A4 by protease inhibitors can lead to markedly increased levels of immunosuppressive calcineurin inhibitors (especially tacrolimus), which requires intensive monitoring and can be very challenging. The cutaneous and renal vasculitis of HCV-associated essential mixed cryoglobulinemia (Chap. 360) may respond to antiviral therapy, but sustained responses are rare after discontinuation of therapy; therefore, prolonged, perhaps indefinite, therapy (as reported with IFN-based therapy) is recommended in this group (no indication for prolonged protease inhibitor therapy exists currently). Anecdotal reports suggest that antiviral therapy may be effective in porphyria cutanea tarda or lichen planus associated with hepatitis C.
In patients with HCV/HIV co-infection, hepatitis C is more progressive and severe than in HCV-monoinfected patients. Although patients with HCV/HIV co-infection respond to antiviral therapy for hepatitis C, they do not respond as well as patients with HCV infection alone. Four large national and international trials of antiviral therapy among patients with HCV/HIV co-infection have shown that PEG IFN (both α2a and α2b) plus ribavirin (daily doses ranging from flat-dosed 600–800 mg to weight-based 1000/1200 mg) is superior to standard IFN regimens; however, SVR rates were lower than in HCV-monoinfected patients, ranging from 14 to 38% for patients with genotypes 1 and 4 and from 44 to 73% for patients with genotypes 2 and 3. In the three largest trials, all patients, including those with genotypes 2 and 3, were treated for a full 48 weeks. In addition, tolerability of therapy was lower than in HCV-monoinfected patients; therapy was discontinued because of side effects in 12–39% of patients in these clinical trials. Based on these trials, weekly PEG IFN plus daily ribavirin at a daily dose of at least 600–800 mg, up to full weight-based doses, at doses recommended for HCV-monoinfected patients, if tolerated, is recommended for a full 48 weeks, regardless of genotype. An alternative recommendation for ribavirin doses was issued by a European Consensus Conference and consisted of standard, weight-based 1000–1200 mg for genotypes 1 and 4, but 800 mg for genotypes 2 and 3. A head-to-head trial of combination PEG IFN–ribavirin therapy in HCV/HIV co-infection demonstrated statistically indistinguishable efficacy of the two types of PEG IFN, despite a small advantage for PEG IFN-α2a: For PEG IFN-α2b and -α2a, SVRs occurred in 28% versus 32%, respectively, of patients with genotypes 1 and 4 and in 62% versus 71%, respectively, of patients with genotypes 2 and 3.
Although data are limited and recommendations pending, protease inhibitors may be used for genotype 1; however, because of potential drug-drug interactions between HCV protease inhibitors and HIV antiretroviral drugs (especially in ritonavir-boosted HIV protease inhibitors), HCV protease inhibitors should be used cautiously in HCV-HIV co-infected patients. If protease inhibitors are used, a full 48-week course is recommended without response-guided therapy: for boceprevir, 4 weeks of PEG IFN–ribavirin lead-in, followed by 44 weeks of triple-drug therapy (PEG IFN, ribavirin, boceprevir), and for telaprevir, 12 weeks of triple-drug therapy (PEG IFN, ribavirin, telaprevir), followed by 36 weeks of PEG IFN–ribavirin therapy. In preliminary trials among HIV-HCV co-infected patients, telaprevir-based triple-drug therapy (independent of whether they were receiving antiretroviral therapy [no antiretroviral drugs, efavirenz-tenofovir-emtricitabine, or ritonavir-boosted atazanavir-tenofovir-emtricitabine or lamivudine]) resulted in an SVR in 28 of 38 patients (74%), compared with 10 of 22 control patients (45%) treated with PEG IFN–ribavirin (60 study subjects); boceprevir-based triple-drug therapy (all were also receiving antiretroviral therapy) resulted in an SVR in 40 of 64 patients (63%), compared with 10 of 34 control patients (29%) treated with PEG IFN–ribavirin (98 study subjects). Thus, for the prior standard of care, PEG IFN plus ribavirin, although the likelihood of an SVR is lower for HIV-HCV co-infected patients than for HCV-monoinfected patients, for protease inhibitor–based regimens, rates of SVR are comparable in HIV-HCV co-infected and HCV-monoinfected patients.
In HCV/HIV-infected patients, ribavirin can potentiate the toxicity of didanosine (e.g., lactic acidosis) and the lipoatrophy of stavudine, and zidovudine can exacerbate ribavirin-associated hemolytic anemia; therefore, these drug combinations should be avoided.
Patients with a history of injection drug use and alcoholism can be treated successfully for chronic hepatitis C, preferably in conjunction with drug and alcohol treatment programs. Because ribavirin is excreted renally, patients with end-stage renal disease, including those undergoing dialysis (which does not clear ribavirin), are not ideal candidates for ribavirin therapy. Rare reports suggest that reduced-dose ribavirin can be used, but the frequency of anemia is very high, and data on efficacy are limited. If patients with renal failure (glomerular filtration rate <60 mL/min) are treated, the PEG IFN-α2a dose should be reduced from 180 to 135 μg weekly and the PEG IFN-α2b dose reduced from 1.5 to 1 μg/kg weekly; similarly, the daily ribavirin dose in this population should be reduced to 200–800 mg (but not used or used cautiously at very low doses) if hemodialysis is required. Neither the optimal regimen nor the efficacy of therapy is well established in this population.
NOVEL ANTIVIRALS*
To date, attempts to develop better-tolerated ribavirin successors or improved types of IFN-α or longer acting IFNs than PEG IFN have not been successful. The demonstration that responsiveness to antiviral therapy is influenced by genetic variation in IL28B, which codes for IFN-λ (as noted above), raises the possibility that IFN-λ might be an effective or even more effective IFN for treating hepatitis C; early trials are in progress, but elevations of aminotransferase levels in treated subjects have raised concerns and delayed development. Beyond telaprevir and boceprevir, other direct antivirals that target HCV polymerase, protease, or NS5A (a membrane phosphoprotein component of the viral replication complex) are being investigated, as well as agents that can target host-encoded proteins. Among the novel antivirals are drugs with improved pharmacokinetic and resistance profiles, less treatment complexity, pangenotypic activity, fewer side effects, and fewer drug-drug interactions.* The pace of successful trials of all-oral regimens has accelerated. All-oral combinations of a second-generation protease inhibitor (asunaprevir) plus an NS5A inhibitor (daclatasvir); of a uridine nucleoside polymerase inhibitor (sofosbuvir)* plus ribavirin; of a polymerase inhibitor (sofosbuvir) plus an NS5A inhibitor (ledipasvir or daclatasvir) and ribavirin; and of combinations of a ritonavir-boosted protease inhibitor (ABT-450) plus a nonnucleoside polymerase inhibitor (ABT-333) plus an NS5A inhibitor (ABT-267) with or without ribavirin have been studied in clinical trials. Several of these drug combinations have achieved SVR rates exceeding 90%, even approaching 100%, for both treatment-naïve and treatment-experienced patients (including patients who failed to respond to first-generation protease inhibitors), across all HCV genotypes and independent of host IL28B genotype, and with treatment durations of 12–24 weeks or even shorter (8 weeks). Potentially, as early as 2014 or 2015, such combinations of direct antiviral agents will be used in drug cocktails that may replace IFN-based regimens entirely.
Less advanced is development of inhibitors of host proteins, such as oral, nonimmunosuppressive inhibitors of cyclophilin A (which interacts with NS5A during HCV replication) and subcutaneous antisense antagonists of host liver-expressed micro-RNA-122 (which promotes HCV replication). Given the accelerated progress of all-oral, short-treatment-duration, high-efficacy, direct-acting antivirals, these alternative approaches may not be practical or competitive.
AUTOIMMUNE HEPATITIS
DEFINITION
Autoimmune hepatitis is a chronic disorder characterized by continuing hepatocellular necrosis and inflammation, usually with fibrosis, which can progress to cirrhosis and liver failure. When fulfilling criteria of severity, this type of chronic hepatitis, when untreated, may have a 6-month mortality of as high as 40%. Based on contemporary estimates of the natural history of autoimmune hepatitis, the 10-year survival is 80–98% for treated and 67% for untreated patients. The prominence of extrahepatic features of autoimmunity and seroimmunologic abnormalities in this disorder supports an autoimmune process in its pathogenesis; this concept is reflected in the prior labels lupoid and plasma cell hepatitis. Autoantibodies and other typical features of autoimmunity, however, do not occur in all cases; among the broader categories of “idiopathic” or cryptogenic chronic hepatitis, many, perhaps the majority, are probably autoimmune in origin. Cases in which hepatotropic viruses, metabolic/genetic derangements (including nonalcoholic fatty liver disease), and hepatotoxic drugs have been excluded represent a spectrum of heterogeneous liver disorders of unknown cause, a proportion of which are most likely autoimmune hepatitis.
IMMUNOPATHOGENESIS
The weight of evidence suggests that the progressive liver injury in patients with autoimmune hepatitis is the result of a cell-mediated immunologic attack directed against liver cells. In all likelihood, predisposition to autoimmunity is inherited, whereas the liver specificity of this injury is triggered by environmental (e.g., chemical, drug [e.g., minocycline], or viral) factors. For example, patients have been described in whom apparently self-limited cases of acute hepatitis A, B, or C led to autoimmune hepatitis, presumably because of genetic susceptibility or predisposition. Evidence to support an autoimmune pathogenesis in this type of hepatitis includes the following: (1) In the liver, the histopathologic lesions are composed predominantly of cytotoxic T cells and plasma cells; (2) circulating autoantibodies (nuclear, smooth muscle, thyroid, etc.; see below), rheumatoid factor, and hyperglobulinemia are common; (3) other autoimmune disorders—such as thyroiditis, rheumatoid arthritis, autoimmune hemolytic anemia, ulcerative colitis, membranoproliferative glomerulonephritis, juvenile diabetes mellitus, celiac disease, and Sjögren’s syndrome—occur with increased frequency in patients and in their relatives who have autoimmune hepatitis; (4) histocompatibility haplotypes associated with autoimmune diseases, such as HLA-B1, -B8, -DR3, and -DR4 as well as extended haplotype DRB1*0301 and DRB1*0401 alleles, are common in patients with autoimmune hepatitis; and (5) this type of chronic hepatitis is responsive to glucocorticoid/immunosuppressive therapy, effective in a variety of autoimmune disorders.
Cellular immune mechanisms appear to be important in the pathogenesis of autoimmune hepatitis. In vitro studies have suggested that in patients with this disorder, CD4+ T lymphocytes are capable of becoming sensitized to hepatocyte membrane proteins and of destroying liver cells. Molecular mimicry by cross-reacting antigens that contain epitopes similar to liver antigens is postulated to activate these T cells, which infiltrate, and result in injury to, the liver. Abnormalities of immunoregulatory control over cytotoxic lymphocytes (impaired regulatory CD4+CD25+ T cell influences) may play a role as well. Studies of genetic predisposition to autoimmune hepatitis demonstrate that certain haplotypes are associated with the disorder, as enumerated above, as are polymorphisms in cytotoxic T lymphocyte antigens (CTLA-4) and tumor necrosis factor α (TNFA*2). The precise triggering factors, genetic influences, and cytotoxic and immunoregulatory mechanisms involved in this type of liver injury remain incompletely defined.
Intriguing clues into the pathogenesis of autoimmune hepatitis come from the observation that circulating autoantibodies are prevalent in patients with this disorder. Among the autoantibodies described in these patients are antibodies to nuclei (so-called antinuclear antibodies [ANAs], primarily in a homogeneous pattern) and smooth muscle (so-called anti-smooth-muscle antibodies, directed at actin, vimentin, and skeletin), antibodies to F-actin, antibodies to liver-kidney microsomes (anti-LKM, see below), antibodies to “soluble liver antigen” (directed against a uracil-guanine-adenine transfer RNA suppressor protein), antibodies to α-actinin, and antibodies to the liver-specific asialoglycoprotein receptor (or “hepatic lectin”) and other hepatocyte membrane proteins. Although some of these provide helpful diagnostic markers, their involvement in the pathogenesis of autoimmune hepatitis has not been established.
Humoral immune mechanisms have been shown to play a role in the extrahepatic manifestations of autoimmune and idiopathic hepatitis. Arthralgias, arthritis, cutaneous vasculitis, and glomerulonephritis occurring in patients with autoimmune hepatitis appear to be mediated by the deposition of circulating immune complexes in affected tissue vessels, followed by complement activation, inflammation, and tissue injury. While specific viral antigen-antibody complexes can be identified in acute and chronic viral hepatitis, the nature of the immune complexes in autoimmune hepatitis has not been defined.
CLINICAL FEATURES
Many of the clinical features of autoimmune hepatitis are similar to those described for chronic viral hepatitis. The onset of disease may be insidious or abrupt; the disease may present initially like, and be confused with, acute viral hepatitis; a history of recurrent bouts of what had been labeled acute hepatitis is not uncommon. In approximately a quarter of patients, the diagnosis is made in the absence of symptoms, based on abnormal liver laboratory tests. A subset of patients with autoimmune hepatitis has distinct features. Such patients are predominantly young to middle-aged women with marked hyperglobulinemia and high-titer circulating ANAs. This is the group with positive lupus erythematosus (LE) preparations (initially labeled “lupoid” hepatitis) in whom other autoimmune features are common. Fatigue, malaise, anorexia, amenorrhea, acne, arthralgias, and jaundice are common. Occasionally, arthritis, maculopapular eruptions (including cutaneous vasculitis), erythema nodosum, colitis, pleurisy, pericarditis, anemia, azotemia, and sicca syndrome (keratoconjunctivitis, xerostomia) occur. In some patients, complications of cirrhosis, such as ascites and edema (associated with portal hypertension and hypoalbuminemia), encephalopathy, hypersplenism, coagulopathy, or variceal bleeding may bring the patient to initial medical attention.
The course of autoimmune hepatitis may be variable. In patients with mild disease or limited histologic lesions (e.g., piecemeal necrosis without bridging), progression to cirrhosis is limited, but, even in this subset, clinical monitoring is important to identify progression; up to half left untreated can progress to cirrhosis over the course of 15 years. In North America, cirrhosis at presentation is more common in African Americans than in whites. In those with severe symptomatic autoimmune hepatitis (aminotransferase levels >10 times normal, marked hyperglobulinemia, “aggressive” histologic lesions—bridging necrosis or multilobular collapse, cirrhosis), the 6-month mortality without therapy may be as high as 40%. Such severe disease accounts for only 20% of cases; the natural history of milder disease is variable, often accentuated by spontaneous remissions and exacerbations. Especially poor prognostic signs include the presence histologically of multilobular collapse at the time of initial presentation and failure of serum bilirubin to improve after 2 weeks of therapy. Death may result from hepatic failure, hepatic coma, other complications of cirrhosis (e.g., variceal hemorrhage), and intercurrent infection. In patients with established cirrhosis, HCC may be a late complication (Chap. 111) but occurs less frequently than in cirrhosis associated with viral hepatitis.
Laboratory features of autoimmune hepatitis are similar to those seen in chronic viral hepatitis. Liver biochemical tests are invariably abnormal but may not correlate with the clinical severity or histopathologic features in individual cases. Many patients with autoimmune hepatitis have normal serum bilirubin, alkaline phosphatase, and globulin levels with only minimal aminotransferase elevations. Serum AST and ALT levels are increased and fluctuate in the range of 100–1000 units. In severe cases, the serum bilirubin level is moderately elevated (51–171 μmol/L [3–10 mg/dL]). Hypoalbuminemia occurs in patients with very active or advanced disease. Serum alkaline phosphatase levels may be moderately elevated or near normal. In a small proportion of patients, marked elevations of alkaline phosphatase activity occur; in such patients, clinical and laboratory features overlap with those of primary biliary cirrhosis (Chap. 365). The prothrombin time is often prolonged, particularly late in the disease or during active phases.
Hypergammaglobulinemia (>2.5 g/dL) is common in autoimmune hepatitis, as is the presence of rheumatoid factor. As noted above, circulating autoantibodies are also prevalent, most characteristically ANAs in a homogeneous staining pattern. Smooth-muscle antibodies are less specific, seen just as frequently in chronic viral hepatitis. Because of the high levels of globulins achieved in the circulation of some patients with autoimmune hepatitis, occasionally the globulins may bind nonspecifically in solid-phase binding immunoassays for viral antibodies. This has been recognized most commonly in tests for antibodies to hepatitis C virus, as noted above. In fact, studies of autoantibodies in autoimmune hepatitis have led to the recognition of new categories of autoimmune hepatitis. Type I autoimmune hepatitis is the classic syndrome prevalent in North America and northern Europe occurring in young women, associated with marked hyperglobulinemia, lupoid features, circulating ANAs, and HLA- DR3 or HLA-DR4 (especially B8-DRB1*03). Also associated with type I autoimmune hepatitis are autoantibodies against actin and atypical perinuclear antineutrophilic cytoplasmic antibodies (pANCA).
Type II autoimmune hepatitis, often seen in children, more common in Mediterranean populations, and linked to HLA- DRB1 and HLA-DQB1 haplotypes, is associated not with ANA but with anti-LKM. Actually, anti-LKM represent a heterogeneous group of antibodies. In type II autoimmune hepatitis, the antibody is anti-LKM1, directed against cytochrome P450 2D6. This is the same anti-LKM seen in some patients with chronic hepatitis C. Anti-LKM2 is seen in drug-induced hepatitis, and anti-LKM3 (directed against uridine diphosphate glucuronyltransferases) is seen in patients with chronic hepatitis D. Another autoantibody observed in type II autoimmune hepatitis is directed against liver cytosol formiminotransferase cyclodeaminase (anti-liver cytosol 1). More controversial is whether or not a third category of autoimmune hepatitis exists, type III autoimmune hepatitis. These patients lack ANA and anti-LKM1 but have circulating antibodies to soluble liver antigen. Most of these patients are women and have clinical features similar to, perhaps more severe than, those of patients with type I autoimmune hepatitis. Type III autoimmune hepatitis does not appear to represent a distinct category but, instead, is part of the spectrum of type I autoimmune hepatitis; this subcategory has not been adopted by a consensus of international experts.
Liver biopsy abnormalities are similar to those described for chronic viral hepatitis. Expanding portal tracts and extending beyond the plate of periportal hepatocytes into the parenchyma (designated interface hepatitis or piecemeal necrosis) is a mononuclear cell infiltrate that, in autoimmune hepatitis, may include the presence of plasma cells. Necroinflammatory activity characterizes the lobular parenchyma, and evidence of hepatocellular regeneration is reflected by “rosette” formation, the occurrence of thickened liver cell plates, and regenerative “pseudolobules.” Septal fibrosis, bridging fibrosis, and cirrhosis are frequent. In patients with early autoimmune hepatitis presenting as an acute-hepatitis-like illness, lobular and centrilobular (as opposed to the more common periportal) necrosis has been reported. Bile duct injury and granulomas are uncommon; however, a subgroup of patients with autoimmune hepatitis has histologic, biochemical, and serologic features overlapping those of primary biliary cirrhosis (Chap. 365).
DIAGNOSTIC CRITERIA
An international group has suggested a set of criteria for establishing a diagnosis of autoimmune hepatitis. Exclusion of liver disease caused by genetic disorders, viral hepatitis, drug hepatotoxicity, and alcohol are linked with such inclusive diagnostic criteria as hyperglobulinemia, autoantibodies, and characteristic histologic features. This international group has also suggested a comprehensive diagnostic scoring system that, rarely required for typical cases, may be helpful when typical features are not present. Factors that weigh in favor of the diagnosis include female gender; predominant aminotransferase elevation; presence and level of globulin elevation; presence of nuclear, smooth muscle, LKM1, and other autoantibodies; concurrent other autoimmune diseases; characteristic histologic features (interface hepatitis, plasma cells, rosettes); HLA-DR3 or -DR4 markers; and response to treatment (see below). A more simplified, more specific scoring system relies on four variables: autoantibodies, serum IgG level, typical or compatible histologic features, and absence of viral hepatitis markers. Weighing against the diagnosis are predominant alkaline phosphatase elevation, mitochondrial antibodies, markers of viral hepatitis, history of hepatotoxic drugs or excessive alcohol, histologic evidence of bile duct injury, or such atypical histologic features as fatty infiltration, iron overload, and viral inclusions.
DIFFERENTIAL DIAGNOSIS
Early during the course of chronic hepatitis, autoimmune hepatitis may resemble typical acute viral hepatitis (Chap. 360). Without histologic assessment, severe chronic hepatitis cannot be readily distinguished based on clinical or biochemical criteria from mild chronic hepatitis. In adolescence, Wilson’s disease (Chaps. 365 and 429) may present with features of chronic hepatitis long before neurologic manifestations become apparent and before the formation of Kayser-Fleischer rings (copper deposition in Descemet’s membrane in the periphery of the cornea). In this age group, serum ceruloplasmin and serum and urinary copper determinations plus measurement of liver copper levels establish the correct diagnosis. Postnecrotic or cryptogenic cirrhosis and primary biliary cirrhosis (Chap. 365) share clinical features with autoimmune hepatitis, and both alcoholic hepatitis (Chap. 363) and nonalcoholic steatohepatitis (Chap. 367e) may present with many features common to autoimmune hepatitis; historic, biochemical, serologic, and histologic assessments are usually sufficient to allow these entities to be distinguished from autoimmune hepatitis. Of course, the distinction between autoimmune and chronic viral hepatitis is not always straightforward, especially when viral antibodies occur in patients with autoimmune disease or when autoantibodies occur in patients with viral disease. Furthermore, the presence of extrahepatic features such as arthritis, cutaneous vasculitis, or pleuritis—not to mention the presence of circulating autoantibodies—may cause confusion with rheumatologic disorders such as rheumatoid arthritis and systemic lupus erythematosus. The existence of clinical and biochemical features of progressive necroinflammatory liver disease distinguishes chronic hepatitis from these other disorders, which are not associated with severe liver disease. Rarely, hepatic venous outflow obstruction (Budd-Chiari syndrome) may present with features suggestive of autoimmune hepatitis, but painful hepatomegaly, ascites, and vascular imaging provide distinguishing diagnostic clues. Other diagnostic considerations would include celiac disease and ischemic liver disease, which would be readily distinguishable by clinical and laboratory features from autoimmune hepatitis.
Finally, occasionally, features of autoimmune hepatitis overlap with features of autoimmune biliary disorders such as primary biliary cirrhosis, primary sclerosing cholangitis (Chaps. 365 and 369), or, even more rarely, mitochondrial antibody-negative autoimmune cholangitis. Such overlap syndromes are difficult to categorize, and often response to therapy may be the distinguishing factor that establishes the diagnosis.
ACKNOWLEDGMENT
Kurt J. Isselbacher, MD, contributed to this chapter in previous editions of Harrison’s.
*As this chapter was going to press, two additional antiviral drugs, a second-generation protease inhibitor simeprevir and nucleoside analogue polymerase inhibitor sofosbuvir were approved for the treatment of hepatitis C. Simerprevir, which is effective for genotype 1, must be administered, like first-generation protease inhibitors, for 12 weeks with PEG IFN and ribavirin, followed by another 12 weeks of PEG IFN and ribavirin (no response-guided therapy). Sofosbuvir, the more convenient and broadly applicable of the two new drugs, must be administered with PEG IFN and ribavirin but for only 12 weeks in patients with genotyes 1, 4-6; for patients with genotypes 2 and 3, PEG IFN is not required. Sofosbuvir plus ribavirin are administered for 12 weeks in genotype 2 and for 24 weeks in genotype 3. Antiviral therapy is evolving very rapidly; by the end of 2014, all-oral, interferon-free combinations (e.g., sofosbuvir plus the NS5a inhibitor ledipasvir) will supplant earlier treatment regimens. For updated treatment recommendations, please consult www.hcvguidelines.org.




