Tissue Engineering
In 1954, Dr Joseph Murray performed the first successful human kidney transplant, introducing the world to an entire new field of medicine.1 Organ and tissue transplant is a remarkable and life-saving option for many patients. Nevertheless, it is limited by donor scarcity, need for lifelong immunosuppression, and the risk of infection and rejection.2 For these reasons, tissue engineers are working toward developing methods of growing whole organs from a patient’s own cells or banked lines of cells that can be used to replace diseased and damaged tissues.
By definition, a tissue-engineered organ must be capable of performing the key functions of the replaced organ or tissue. Also, it should be durable, self-repairing, and match the longevity of the patient. It should not require the sacrifice or prioritization of other organs as is the case for operations such as gastric pull-up, vein harvest, or a myocutaneous flap. Tissue engineering strategies often employ overlapping techniques that can be applied to a number of organ systems. These approaches include: (1) a progenitor cell source; (2) a biological or synthetic scaffold for in vivo implantation; and (3) a method of expanding cell mass via in vitro cell culture, bioreactor, or in vivo implantation. Figure 79-1 illustrates a common approach for cell isolation, seeding of a scaffold, and implantation into the host. This general concept has been adapted to multiple organ systems and animal models.
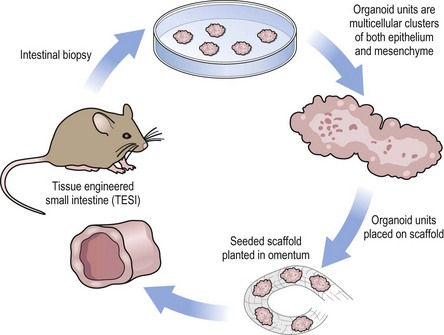
FIGURE 79-1 A representative schematic of tissue-engineered technique. In the example above, murine tissue-engineered small intestine is generated. However similar techniques have been applied to many organ systems. Tissue is obtained for the isolation of a cellular source. These cells are used to seed a scaffold which is then implanted into a host. The tissue-engineered product is harvested after a period of growth.
Cell Source
Concurrent with the expansion of regenerative medicine and tissue engineering programs in the last decade, remarkable progress has been made in understanding stem and progenitor cells. Embryonic stem cells (ESC) are harvested from the inner cell mass of the blastocyst in embryos and are considered pluripotent cells, able to develop into any cell type except for the extra-embryonic tissues. This is advantageous because they are a potential source for any tissue in the body.3 However, work performed with ESC, particularly human ESC, is subject to considerable scrutiny and ethical concerns. Moreover, ESC may possess tumorigenic potential.4 Somatic or adult stem cells can be derived from the patient who requires the tissue-engineered organ, but lack totipotency. Instead, these cells are committed toward a particular lineage, but maintain the potential for self-renewal and possess multipotency, which is the ability to differentiate into multiple, but not all cell types.3 For example, the intestinal stem cell is capable of self-renewal and producing all differentiated cells of the intestinal epithelium, but it usually does not differentiate into a hepatocyte. A central dogma is that once a cell has differentiated, it is committed to that cell line.
In 2006, a method of reprogramming somatic cells to a pluripotent state was developed.5 These cells are termed induced pluripotent stem cells (iPSC). These cells overcome many of the ethical and immunologic concerns surrounding ESC; however, one disadvantage is that they maintain tumorigenic potential.4 Many of the current techniques for reprogramming these cells require the introduction of oncogenic viral vectors, thereby limiting clinical application.6 Additional sources of pluripotent cells include mesenchymal stem cells (MSC). MSC were initially described in 1968 as a population of bone marrow derived cells capable of osteogenic differentiation.7 Since then, MSC capable of self-renewal and differentiation have also been isolated from adipose, skin, blood, synovial membrane and amniotic fluid, and other mesodermally derived tissues.8
Scaffold
Synthetic scaffolds are designed to be porous enough to allow imbibition of nutrients until vasculogenesis and angiogenesis occurs, but also rigid enough to provide structural support. Frequently used materials include polyglycolic acid (PGA) and poly(L-lactic acid) (PLLA) that are biodegradable polymers designed to be hydrolyzed and absorbed by the recipient as the engineered product grows.9 With advancements in the field of biomaterials, synthetic scaffolds have become increasingly complex. Polyhedral oliogmericsilsesquioxane bonded to poly-[carbonate-urea] urethane (POSS-PCU), for example, is biocompatible, nonreactive, nontoxic, and can be designed to fit the patient’s needs. This scaffold has already been successfully used in a tracheobronchial transplant.9 The structure of some commonly used synthetic scaffolds are demonstrated in Figure 79-2.
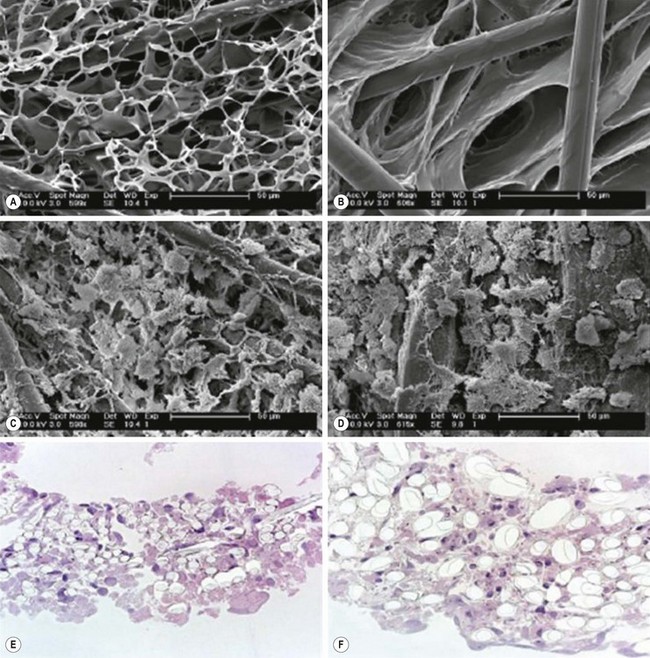
FIGURE 79-2 Common synthetic scaffolds for in vivo tissue engineering. Highly porous structures are necessary to support tissue growth as demonstrated by scanning electron microscopy of (A) PGA-P(CL/LA) scaffold and (B) PLLA-P(CL/LA) scaffold. The scaffolds can be seeded with cells as demonstrated in (C) PGA-P(CL/LA) and (D) PLLA-P(CL/LA). These cells survive by imbibition until neovascularization occurs. The close approximation of cells and scaffold can be further demonstrated by staining with hematoxylin and eosin for both (E) PGA-P(CL/LA) and (F) PLLA-P(CL/LA) scaffolds. PGA, polyglycolic acid; PLLA, poly-L-lactic acid; P(CL/LA), copolymer sealant δ-caprolactone and L-lactide. (Adapted and reproduced with permission from Roh JD, Nelson GN, Brennan MP, et al. Small-diameter biodegradable scaffolds for functional vascular tissue engineering in the mouse model. Biomaterials 2008;29:1454–63.)
Biological scaffolds composed of naturally occurring macromolecules, in particular those that formulate the ECM, have also been described. These include porous collagen lattice, chitosan, glycosaminoglycans, silk fibroin, alginate, and starch.10 More recently, in an attempt to create more complex biological scaffolds, researchers have turned toward decellularized extracellular matrices as a potential material. Decellularization is a process in which organs are treated with detergents and enzymes to remove nuclear, cytoplasmic, and antigenic material while leaving behind the ECM (Figure 79-3). This methodology preserves the macroscopic and microscopic architecture of the original tissue, including vascular inflow and outflow, facilitating future vascular anastomosis and circulation.11 Subsequent recellularization is possible using an appropriate cell source.
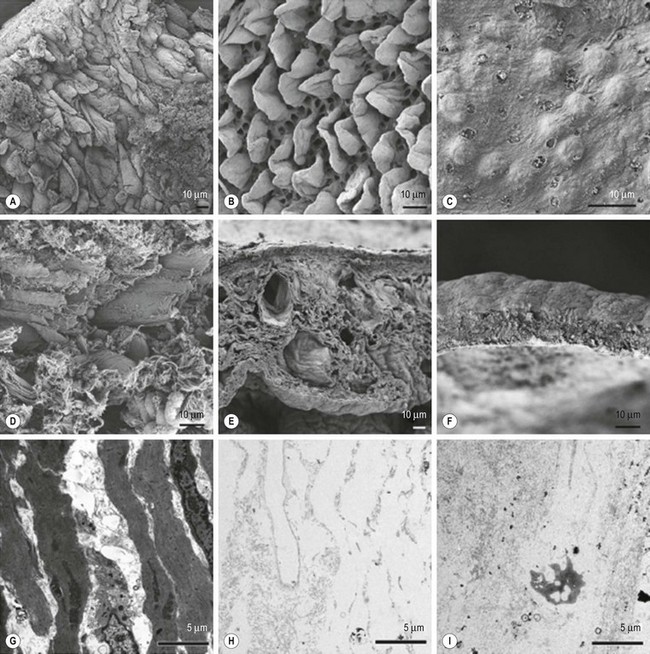
FIGURE 79-3 Biological scaffolds derived from decellularized organs have been attempted. (A) Scanning electron microscopy (SEM) of native intestine. During decellurization, crypt/villus structure of the extracellular matrix (ECM) is preserved after (B) one cycle, but is lost (C) after four cycles. The collagen fiber network as it appears on SEM for (D) native intestine remains intact after (E) one cycle of decellurization, but is lost (F) after four cycles. A transmission electron micrograph (TEM) of (G) native intestine demonstrates normal cellularity. TEM confirms acellularity of the biological scaffold following (H) one and (I) four cycles of decellularization. (Adapted and reproduced with permission from Totonelli G, Maghsoudlou P, Garriboli M, et al. A rat decellularized small bowel scaffold that preserves villus-crypt architecture for intestinal regeneration. Biomaterials 2012;33:3401–12.)
Cell Expansion and Growth
A bioengineered organ must be of sufficient size to adequately support function. Methods for culturing cells include one or more of the following techniques. Many scientists have designed artificial bioreactors that attempt to mimic many of the physiologic processes that occur in an organism. These include control of pH, oxygen tension, temperature, waste removal, and even the pulsatile flow of blood.12 However, no bioreactor can fully recreate the complexity of a living animal. An alternative approach is to expand cells in vivo in what can be thought of as a living bioreactor. This has the advantage of encouraging the growth of blood vessels that will ultimately be needed to support the growing engineered tissue and access to circulating factors that may be important for cell growth. A marked disadvantage is the unpredictable number of variables introduced by the host, which increases the challenge of deciphering the cellular and molecular mechanisms of growing the tissue-engineered organ. The greatest challenge facing most tissue engineers is expanding the volume of tissue growth to a clinically relevant size while maintaining specific tissue functions with this growth.
Respiratory System
Trachea and Bronchi
Tissue-engineered trachea is conceptualized for the replacement of long-segment tracheal defects. Primary disease of the trachea, such as tracheal agenesis, is rare with an incidence of less than 1 in 50,000 live births.13 However, iatrogenic injury or malignant invasion may necessitate segmental resection. Primary repair is possible if the defect is less than 5 cm in adults and one-third the tracheal length in children.14 Although an allograft transplant is possible, it is limited by the lack of a suitable vascular pedicle and the probable need for lifelong immunosuppression.15 Prosthetic conduits have also been attempted. However, these are frequently complicated by product migration, infection, erosion, and disruption.14
Implantation of a tissue-engineered airway has been performed successfully in humans.16 The human donor trachea was decellularized to remove all cells and major histocompatibility complex (MHC) antigens. The recipient’s own epithelial cells and chondrocytes were then cultured and used to seed this biological scaffold. The bioengineered trachea was then anastomosed to replace the left main stem bronchus in a 30-year old woman with end-stage bronchomalacia. Follow-up 4 months later confirmed the patient was breathing normally without the need for immunosuppressive medications.
The first long-term follow-up for a tissue-engineered tracheal replacement in a child was published in 2012.17 The authors reported the case of a 12-year-old boy born with long-segment tracheal stenosis and a pulmonary sling. After years of failed interventions, including multiple stainless steel expandable stents and emergent repair of an aortotracheal fistula, he was the recipient of a tissue-engineered trachea. Prior to the operation, MSC from the patient were isolated and a cadaveric donor trachea was decellularized. At the time of the operation, the decellularized scaffold was seeded with MSC and patches of tracheal epithelium, and anastomosed to replace a 7 cm segment of stenotic trachea. In addition, the scaffold was treated intraoperatively with erythropoietin, granulocyte colony-stimulating factor, and transforming growth factor beta (TGF-β). These factors serve to enhance angiogenesis, improve recruitment of MSC, and induce chondrogenesis, respectively. During the immediate postoperative recovery, the child required stent placement. However, 2 years later, the child was breathing comfortably and had returned to school without the need for tracheal stenting. The graft had revascularized, demonstrating an epithelial lining, and appeared normal on computed tomography (CT) and ventilation-perfusion scan.
A limiting factor in the technique described above is the need for donor trachea that is subsequently decellularized. To avoid this need, synthetic scaffolds have also been used. In nonhuman studies, PGA was tubularized and coated with sheets of cultured MSC. MSC treated with glucocorticoids and TGF-β will differentiate along a chondrogenic lineage. These bioengineered, cartilaginous tubes closely resemble human trachea.18 Other cell sources and scaffold materials have also been tried including sheep nasal chondrocytes on a PGA and silicon tube matrix, or human nasal chondrocytes on hydrogel and high-density polyethylene.19,20 Although these methods are successful in generating a cartilaginous conduit, they have not been applied in humans.
Tissue-engineered lung parenchyma has posed a significantly greater challenge. Development of a tissue-engineered lung with fully differentiated pneumocytes and clara cells has not been achieved to date.21 Current methods are unable deliver cells in the appropriate cellular distribution and architecture to participate in normal gas exchange.
Cardiovascular System
Heart
The Centers for Disease Control (CDC) reports that 40,000 infants are born with congenital heart defects each year in the USA. Among adults, heart disease is the leading cause of death. The CDC estimates the annual financial burden of cardiovascular disease approximates $444 billion. Tissue engineering cardiac replacements such as myocardial tissue and heart valves may help alleviate some of these costs. Murine ESC can be expanded in culture and differentiated into cardiomyocytes.22 Similar results have been shown with human cells.23 Ethical concerns have limited the transition to human experiments prompting increased interest in iPSC24 which can be driven towards a cardiac lineage after reprogramming.24,25
In 2001, implantation of autologous skeletal myoblasts into scar tissue was performed on a patient during coronary artery bypass grafting. Echocardiography performed five months after the operation demonstrated improved areas of myocardial contractility within the scar.26 However, in a randomized placebo-controlled study performed on 94 patients, these results could not demonstrate an improvement in heart function, partly due to the development of arrhythmic areas.27 Aiming to improve delivery and distribution of cells, laboratories are looking into seeding myoblasts on a polyurethane scaffold prior to implantation. In a rat model of myocardial infarction, such an approach prevented progression to heart failure more effectively than sham operation or unloaded scaffold implantation.28
Heart Valves
Native heart valves have three layers, namely: fibrosa (interstitial cells and collagen), spongiosa (proteoglycans), and ventricularis (elastin sheets).29 These valves possess remarkable mechanical durability, capable of opening and closing three billion times over a lifespan.30 Decellularized porcine valves, fibrin, collagen-rich material, and PLLA have been used as scaffolds. Cells for TEHV have generally been applied by one of two methods: either directly seeded on the scaffold prior to in situ implantation, or subsequent attachment of endothelial cells from the blood stream after implantation of an empty scaffold. Bone marrow MSC, umbilical blood progenitor cells, and circulating endothelial progenitor cells have served as possible cell sources.
Expression of the glycoprotein antigen CD133 has been used as a marker for progenitor cells, and is important for the identification and isolation of hematopoietic stem cells.31 Translation to human use has already been demonstrated. Dohmen et al produced TEHV by combining cadaveric human decellularized pulmonary heart valve scaffolds that were seeded with autologous vascular endothelial cells and were implanted into 11 patients.32 At ten-year follow-up, all patients were alive with no regurgitation or accelerated velocities on transthoracic echocardiography and no structural defects on CT. These are encouraging results for the future of TEHV, but require additional validation prior to becoming standard of care.33
Blood Vessels
Vascular bypass is frequently needed as an organ preserving and life-saving operation. Autologous venous and arterial grafts are preferred over synthetic materials, but necessitate the sacrifice of the donor vessel.34 Synthetic materials are prone to infection and maintain patency rates of only 38–49% at 5 years.35 Similar to synthetic valves, synthetic blood vessels do not increase in size with a growing child. In the mid-1980s, Weinberg and Bell ushered in the dawn of tissue-engineered vascular grafts (TEVG; Fig. 79-4) when they seeded endothelial cells onto a scaffold made from bovine smooth muscle cells, cultured fibroblasts, and Dacron mesh.36 Although these grafts failed at pressures greater than 180 mmHg, this research demonstrated that generation of a TEVG is possible. Standards for producing TEVG dictate that durable replacements should have a burst pressure greater than 2000 mmHg.37,38 Since then, multiple scaffolds have been developed, including PGA and decellularized allograft vessels, that are capable of withstanding burst pressures upwards of 2100 mmHg.34
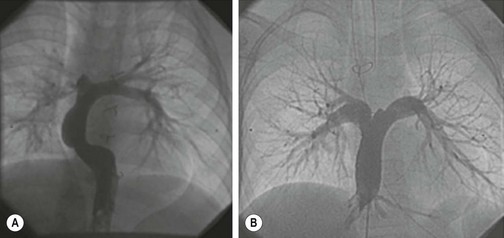
FIGURE 79-4 Treatment of human disease with TEVG. Angiography confirms that there were no stenotic, aneurysmal, or calcified lesions at (A) 5 years and (B) 4 years post implantation for these two patients. (Adapted and reproduced with permission from Hibino N, McGillicuddy E, et al. Late-term results of tissue-engineered vascular grafts in humans. The Journal of Thoracic and Cardiovasc Surg 2010;139:431–6.)
In 2001, the first TEVG was transplanted into a 4-year-old girl to replace her right intermediate pulmonary artery that had occluded following a Fontan procedure.39 A peripheral vein was harvested providing cells for culture. Once cultured to 12 × 106, they were seeded on a 2 cm long polycaprolactone-polylactic acid and PGA scaffold. This group performed a similar procedure on two additional patients, before changing the cell source to aspirated bone marrow cells. At 5.5 years of follow-up, all 25 patients were alive and there was no evidence of aneurysm, rupture, infection, or calcification. Four patients were successfully treated percutaneously for stenosis, and one patient required anticoagulation for a partial thrombus.40 Currently, another group of surgeons are conducting a phase I clinical trial for the implantation of TEVG.41 Based on these results, it is possible that TEVG will become a component in the management algorithm for the routine treatment of vascular disease in the near future.
Alimentary System
Esophagus
Esophageal lengthening via mobilization, circular myotomy, or a staged approach may close gaps up to 3 cm in babies with isolated esophageal atresia. Beyond this distance, an esophageal replacement is often needed.42 An adequate replacement conduit can be constructed via gastric pull-up or the interposition of colon or jejunum, but these approaches rely on prioritizing and sacrificing an area of the gastrointestinal tract.43–45 Tissue-engineered esophagus (TEE) has been successful in rat and dog esophageal replacement models.46,47 In dogs, an amniotic membrane was cultured with oral keratinocytes and fibroblasts. These cultured cells were fashioned into a tube on a PGA scaffold and implanted with smooth muscle tissue into the omentum of the host dog. Then, at 3 weeks, the TEE was transferred as a pedicle graft to close a 3 cm esophageal gap. More than a year later, dogs who underwent this operation were alive and healthy. Although peristalsis of the TEE segment was not evident on imaging, both solids and liquids were able to pass easily from mouth to stomach.
An earlier study in rats isolated esophageal organoid units (OU) for the production of TEE. OU are cellular constructs containing all cells necessary to generate full-thickness tissue such as progenitor cells, supportive mesenchymal cells, and differentiated epithelial cells. OUs were loaded onto a PGA/PLLA scaffold and implanted into the omentum of host rats. After 4 weeks of growth, the TEE was either harvested for histology or anastomosed as an interposition graft (n = 3) or onlay patch (n = 3). Both the interposition and patch groups had one mortality and two survivors. The surviving animals in both groups gained weight and possessed a patent TEE when evaluated with fluoroscopy. Histologic evaluation confirmed the presence of a stratified squamous epithelial lining similar to native esophagus.46
A similar technique has been demonstrated in a sheep model. The omental implantation of ovine esophageal epithelial cells seeded onto a tube of collagen produced TEE with areas of normal appearing esophageal epithelium. During the proliferative phase, the lumen was maintained by a stent that was removed at harvest. These constructs also demonstrated vascular ingrowth, a necessary feature for successful in vivo tissue engineering.48,49 A variety of cellular sources as well as synthetic and biological scaffold materials have been tested for TEE. In one study, various cell and scaffold arrangements were evaluated. It was found that human esophageal squamous cells and a porcine esophageal matrix scaffold grew TEE that most closely and reliably reproduced normal appearing esophageal morphology.50
Stomach
Conditions requiring total gastrectomy such as gastric volvulus with necrosis and gastric cancer are rare in children. However, among adults, gastric cancer is the second leading cause of cancer mortality in the world and a common indication for gastric resection.51 For both adults and children requiring gastrectomy, subsequent esophagojejunostomy lacks the reservoir capacity and digestive physiology of native stomach. Tissue-engineered stomach (TES) is a promising means of restoring normal gastric function.
TES has been grown in a rat model with OUs. OUs were seeded onto a synthetic scaffold that was implanted into the omentum of the host animal. As the TES grew, the host animal hydrolyzed the scaffold. After 30 days of growth, the TES was a well-vascularized, muscular sphere with a mucosal lining similar to native stomach.52,53
In a study designed to test the function of rat TES, the construct was sutured in situ as a gastric replacement.54 The control group underwent Roux-en-Y reconstruction. After 24 weeks, the TES and control groups survived without evidence of obstruction. On histologic evaluation, the harvested TES demonstrated smooth muscle-like layers with an epithelial lining containing parietal and G cells. Interestingly, the TES group was less anemic, which may have been secondary to increased production of intrinsic factor by the parietal cells in the TES. Of note, however, there was no difference between the two groups when comparing weight, total protein, or lipid profile. TES has been similarly generated in the mouse model. Transition to a mouse model may allow investigators to elucidate cellular and molecular mechanisms driving TES growth, and may reveal potential targets and improve tissue yield.55
Small Intestine
A leading cause of intestinal failure in children is short bowel syndrome (SBS).56 SBS occurs when intestinal length is reduced to the point where the patient is unable to absorb sufficient nutrition, resulting in profound malnutrition, dehydration, and electrolyte derangements. Common causes of SBS in children include midgut volvulus, gastroschisis, and necrotizing enterocolitis (NEC).57 Currently, SBS is treated with diet modification, total parenteral nutrition (TPN), medications to delay intestinal transit, and operation. Operative interventions include bowel elongation and intestinal transplant.58 Outcomes from intestinal transplant have improved greatly, yet they are still limited by donor scarcity, lifelong immunosuppression, infection, and rejection.59 Tissue-engineered small intestine (TESI) may be used at some point as an intestinal replacement for SBS. The success of TESI is dependent on the ability to develop normal small intestine structure and function.
The mucosa of the small intestine is arranged as finger-like projections known as villi and intervillus invaginations called crypts. The villi greatly increase the absorptive surface area of the small intestine. There are four main differentiated epithelial cells: enterocytes, goblet cells, enteroendocrine cells, and Paneth cells. The enterocyte is a simple columnar epithelial cell and is the predominant cell population in the villus. It is responsible for the absorption of water, ions, carbohydrates, peptides, lipids, and unconjugated bile salts.60 Mucus secreted by goblet cells is necessary for lubrication and barrier defense.61 Enteroendocrine cells produce over 30 hormones and peptides.62 The remaining secretory cell, the Paneth cell, is located in the crypt and is interspersed with the crypt base columnar cell (CBCC). The Paneth cell secretes antimicrobial defensins63 and has recently been shown to support the stem cell niche. Improved proliferation and differentiation were found when cultured intestinal stem cells (ISC) were paired with Paneth cells in comparison to ISC cultured in isolation. This appears related to the production of growth hormones acting via paracrine signaling between the adjacent Paneth cells and ISC.64
A population of stem cells, first described in the 1970s, is characteristically quiescent and located, on average, four cell positions from the crypt base and thought to be necessary in response to injury and healing.65,66 Interestingly, the CBCC has recently been identified as a second population of ISC residing within the crypt base. These cells, marked by their expression of a G-protein, Lgr5, are rapidly cycling and capable of self-renewal and differentiation into all cell types.67 The theory of two stem cell populations, a traditional quiescent population necessary for response to injury and a second population important for the rapid cellular turnover of intestinal homeostasis, is a fascinating new paradigm in the study of intestinal regeneration.68
All of the above cell types, as well as smooth muscle, vessels, and nerves are necessary for the normal structure and function of the small intestine. Within the field of intestinal regeneration, some investigators have focused on mechanisms of epithelial development and regeneration while others are committed to therapeutic translation, and have focused on the generation of full-thickness intestine. In an effort to refine studies in intestinal epithelial physiology, one group of investigators implanted collagen I matrix scaffolds with cultured human colonic fibroblasts and umblical vein endothelial cells grafted with human intestinal epithelial cells inside a rotating bioreactor.69 In ten to 15 days, the system grew villus-like structures containing differentiated epithelial cells and components of intestinal microanatomy such as brush borders, tight junctions, and glucose transporters. Others have created a three-dimensional culture system for the study of intestinal drug transport across the gut-blood barrier.70 Recently, iPSC that had been cultured in conditions mirroring embyronic development were driven to differentiate along intestinal lineages. This generated three-dimensional structures with crypt and villus-like architecture similar to intestinal morphology.71 These in vitro systems are superior to traditional two-dimensional culture methods as they more closely approximate native intestine. For complete organ replacement, however, full-thickness TESI is needed.
Experiments designed to grow full-thickness intestine began in the late 1980s. The Vacanti group adapted a technique from Evans et al. for the isolation of intestinal OU, seeding a biodegradable polymer scaffold and implanting these constructs in host rats.72,73 The resulting TESI demonstrated fully differentiated secretory and absorptive cells, as well as supportive mesenchyme and muscle. In an experiment aiming to investigate function, two groups were generated. The rat control group underwent a massive small bowel resection resembling SBS. The experimental group also underwent massive small bowel resection, with the TESI anastomosed in-line with the severely truncated intestine. Both groups initially lost weight. However, at 40 days, the TESI group had regained significantly more weight, reaching 98% of preoperative values. The TESI group also had normal B12 levels compared to low levels in the control group.
A current limitation in the transition of TESI to human therapy is the amount of intestine that can be generated. The addition of growth factors to the PGA scaffold has resulted in a modest ability to increase TESI yield in comparison to TESI generated without growth factors. Holo-transferrin increased TESI surface area (9.11 ± 0.66 mm vs 3.01 ± 0.22 mm, P < 0.01) and glucagon-like peptide 2 increased the total number of TESI cysts (8.88 ± 0.46 vs 4.18 ± 0.25, P < 0.01).74 To increase the amount of TESI that can be grown to a clinically relevant volume, investigators are looking at the cellular mechanisms responsible for TESI growth. The goal is to identify targets in the form of growth factors or improved donor populations that may be added to augment TESI growth. Studying TESI growth in the mouse, in which transgenic and molecular tools are available, may lead to identification of these targets.
TESI in a mouse model demonstrates normal appearing crypt and villus architecture (Fig. 79-5). In addition, it contains a fully differentiated epithelium with enterocytes, goblet cells, enteroendocrine cells, Paneth cells, a smooth muscle layer as well as intestinal subepithelial myofibroblasts (ISEMF).75 ISEMF are located immediately beneath the crypt and are thought to support the stem cell niche with an active role in injury and immune response.76 Nerve components and blood vessels are also present, verifying the presence of all necessary cells required for normal structure and function.
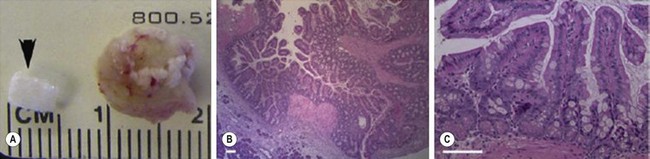
FIGURE 79-5 Murine TESI. (A) An unseeded synthetic PGA scaffold tube sealed with PLLA and coated with collagen (arrowhead) adjacent to TESI explanted four weeks after omental implantation. (B) A 20 × magnification of TESI stained with H&E reveals an epithelium toward the lumen. (C) At 40 × magnification, TESI stained with H&E reveals a simple columnar epithelium arranged in crypts and villi. Additionally, goblet and Paneth cells appear in their normal position and distribution. Scale bar: 40 µM. TESI, tissue-engineered small intestine; PGA, polyglycolic acid; PLLA, poly-L-lactic acid; H&E, hematoxylin and eosin stain. (Adapted and reproduced with permission from Sala F, Matthews J, Speer A, et al. A multicellular approach forms a significant amount of tissue-engineered small intestine in the mouse. Tissue Eng 2011;17:1841–9.)
Prior to human trials, it is necessary to establish the feasibility of generating autologous donor cells as would be expected in a sick neonate with NEC. These cells need to be harvested, prepared, loaded on a scaffold, and then implanted in a single operation. This has been performed in a Yorkshire swine model.77
Large Intestine and Anus
Infants and children with NEC, Hirschsprung disease, familial adenomatosis polyposis, or inflammatory bowel disease face specific morbidities associated with colon resection. Ileoanal anastomosis may be associated with diminished quality of life secondary to day or nighttime fecal incontinence, depression and anxiety, and dietary restrictions and daily requirement of antidiarrheal medication.78,79 End ileostomy is complicated by skin irritation, poor adherence of the ostomy appliance, with leaking as well as a decreased quality of life.80,81 Severe diarrhea causing dehydration is a frequent cause of readmission following creation of an ileostomy.82 In addition, complete colectomy leads to malabsorption of bile acids secondary to loss of intestinal microbiota.83
Tissue-engineered colon (TEC) has been grown in a rat model and found to have normal appearing architecture. To assess function in a replacement model of TEC, two groups of rats were compared after undergoing colectomy with end ileostomy. The first group underwent an ileostomy to TEC anastomosis while the control group had the ileostomy. The rats with TEC demonstrated greater weight gain, increased stool transit time, less diarrhea, and improved bile acid absorption.84
Restoration of colonic absorption and normalization of the enterohepatic circulation and stool microbiota are key points required for translation of TEC to humans. Restitution of fecal continence requires a competent anal sphincter. Interventions designed to treat conditions such as imperforate anus or anal fissures may predispose patients to develop an incompetent anal sphincter. The anal sphincter may also be damaged or excised secondary to trauma or with abdominoperineal resection. In mice, smooth muscle cells have been harvested from the internal anal sphincter (IAS) and seeded onto a fibrin mold. When cultured, these cells naturally formed a ring that could be implanted subcutaneously into a host mouse.85 Further testing revealed the bioengineered IAS could generate resting tonicity and was able to relax and contract with stimulation.85,86 Additionally, fetal mouse enteric neurons have been cultured with human IAS smooth muscle and implanted into a murine host with preserved muscle and nerve function.87 In the future, it may be possible to restore bowel continence and enhance the options for lower intestinal reconstruction with a bioengineered IAS. This has the potential to greatly improve the lives of many patients.
Integumentary and Musculoskeletal System
Skin
Complex wound closure and treatment of severe burns pose significant challenges for plastic and reconstructive surgery. Adequate skin replacement has been sought since Reverdin first described the use of skin grafts in the late 19th century.88 Skin grafts can provide adequate coverage, but they may also result in function limiting wound contracture. Also, cosmetic results are frequently suboptimal. Furthermore, the site for autologous donor skin is painful, has the potential to heal poorly, and may not be of sufficient surface area. These limitations may be overcome with tissue-engineered skin. Potential regenerative strategies can be divided into two groups. The first are biological substitutes that offer temporary wound coverage until patient skin appendages re-epithelialize the wound surface or a skin graft is placed. These include cadaveric skin, porcine xenograft, porcine small intestine submucosa, and cryopreserved amniotic membrane.
A second group involves tissue-engineered skin products that substitute epidermal, dermal, or both layers. There are a large number of commercially available products for the treatment of full and partial thickness burns and ulcers. Many of these are variable constructs of sheets of human fibroblasts and keratinocytes.89 However, current Food and Drug Administration (FDA) approved bioengineered skin replacements are inadequate for treatment of many large wounds. Although these products may facilitate wound healing, they do not recreate normal skin architecture and frequently fail to revascularlize. Skin is deceptively complex and more than a simple watertight barrier of stratified epithelium. Hair follicles, sweat and sebaceous glands, dendritic cells, blood vessels, nerve, and stem cells all contribute to the normal function of skin. Over the last decade, scientists have sought to expand stem cells for skin regeneration.90
The addition of MSC to tissue-engineered skin has resulted in better keratinization, less wound contracture, and improved vascularization when grafted onto pigs.91 Generating full-thickness tissue-engineered skin remains challenging. Preliminary success in a mouse model was achieved using adipose stem cells to regenerate the dermal layer upon which an epidermal layer can be reconstructed. This technique also permits manipulation of the adipogenic potential of these stromal cells, allowing for development of the hypodermis as well.92 Skin substitutes, composed of cultured keratinocytes and fibroblasts, self-assemble into a stratified and cornified epithelium.93 These self-assembled skin substitutes are the subject of a recently completed phase II trial evaluating their success in closing venous ulcers. The results of this trial have yet to be published.94
Bone
Large bone defects can result from trauma, cancer resections, and congenital defects. Infusion of aspirated bone marrow containing MSC has been used for the treatment of bony nonunion since the early 1990s.95 It has recently been demonstrated that transplant of culture-expanded bone marrow cells (directed toward osteogenic lineage with dexamethasone treatment) and platelet-rich plasma can accelerate callus formation.96 However, generation of a vascularized bone replacement remains a challenge. For repair of larger defects, cellular-seeded scaffolds may be needed. Quarto et al. reported the successful ex vivo expansion of osteoprogenitor cells, seeding of a hydroxyapatite scaffold, and in vivo implantation for the treatment of large bone defects. They reported three patients (defects ranging from 4–7 cm) who regained limb function up to 15 months later.97 Over the last 10 years, a large variety of autologous cell sources and scaffold materials have been explored along with the use of bioreactors to improve in vitro growth.98
Tendons and Ligaments
Normal tendons and ligaments are composed of collagen I, fibroblasts and fibrocytes (ligament), or tenoblasts and tenocytes (tendon). Injury is common among athletes, the elderly, and working members of society. One’s natural ability to regenerate these tissues is poor. Current management is stratified into either conservative or invasive approaches. Usual conservative therapy includes rest, ice packs, and anti-inflammatory medications or injections. Operative intervention may involve primary repair of an acute rupture, autograft from a patellar tendon, semitendinosus tendon or anterior cruciate ligament (ACL), allograft, or artificial prostheses.99 Delayed healing, joint stiffness or instability, allograft rejection, and long-term failure frequently limit the therapeutic outcomes from these approaches.
Tissue engineering cell sources have included autologous tenocytes harvested from patellar or calcaneal tendon, cells removed from the tendon sheath, and dermis fibroblasts obtained from skin biopsy.99 ACL-derived stem cells (LSC) can be induced to express type I collagen, type III collagen, fibronectin, and alpha-smooth muscle actin following exposure to TGF-β. This has promising applications as a future cell source for a bioengineered ligament.100
Urinary System
Kidney
End-stage renal disease (ESRD) affects approximately 1200 children each year in the USA. Common etiologies include renal dysplasia, obstructive uropathy, and focal sclerosing glomerular nephritis.101 ESRD treatment components include dialysis and transplantation.102 Complications from dialysis as well as the shortcomings of allograft transplants could be avoided with a functioning tissue-engineered kidney. Bioengineered renal grafts have been created using decellularized rat kidney scaffolds that were injected with ESC ex vivo from the arterial and ureteral routes. These tissue-engineered kidneys were incubated in a perfusion bioreactor for 3 days prior to immunohistologic evaluation. Interestingly, despite not adding growth factors, the seeded cells expressed markers of renal differentiation such as pancytokeratin, Pax-2, and Ksp-Cadherin. These observations have implications not only for tissue engineering, but also in understanding the dynamic contributions the ECM may have on cellular differentiation.103
Similar experiments have been performed on a larger scale. Nakayama et al. layered Rhesus monkey fetal kidney tissue onto a decellularized kidney and demonstrated the feasibility of this technique to support the growth of renal tissue.104 Further experiments will be required to verify this technique with postnatal tissue to avoid the ethical concerns and clinical limitations of a therapy requiring fetal tissue.
Urethra
For the repair of a long urethral stricture or large urethral defect, urethroplasty has been performed in rabbits using a tissue-engineered urethra to replace a 1 cm defect in the rabbit’s native urethra.105 Initially, bladder biopsies were performed to obtain cells for expansion. Following culture, the cells were seeded onto tubularized collagen. After one month, there was no evidence of urethral stricture. Histologic evaluation demonstrated smooth muscle and epithelial cells approximating normal architecture. Urethral repair with collagen matrix as well as acellular bladder matrix and demineralized bone matrix have both been successful in humans.106,107
In 2011, the results of urethral reconstructions with tissue-engineered urethra in five boys was reported.108 These boys with urethral defects underwent muscle and urothelial cell biopsy. The cells were expanded in culture for three to six weeks, then seeded on a tubularized PGA scaffold, followed by implantation for urethral reconstruction. Two of the five boys required a second procedure to improve voluntary voiding. Median follow-up at 71 months showed all patients to have a patent urethra without radiographic or endoscopic evidence of stricture. These are encouraging results, but will require further validation in greater numbers prior to widespread acceptance. In addition, some researchers have expressed concern over the ability of urethra-like tissue to fully replace the hydrolyzed scaffold and for adequate neovascularization to occur.109 However, if these limitations can be overcome, these techniques may become the preferred approach for treatment of large urethral defects.
Bladder
Replacement of the urinary bladder with intestinal segments can be problematic and synthetic materials frequently fail. As with other organ systems, cellular seeding onto scaffolds has been attempted and urothelial cells have often been chosen as the donor cells.110,111 A human trial studied autologous urothelial and muscle cells expanded in culture and seeded onto a collagen and PGA sphere for patients with myelomeningocele and poor bladder function. The neobladder took 2 months to prepare prior to implantation and was transplanted into seven children (mean age 11 years). At 3.5 years follow-up, the patients had improved urinary continence with a normal architecture on biopsy.112 There was no evidence of renal failure, calculi, or abnormal mucous production. Long-term follow-up will be needed to validate the durability of these repairs.
Hepatobiliary System
Liver
Biliary atresia is the most common indication for liver transplantation in children.113 Other causes of liver failure include viral hepatitis, hepatotoxic medications, and metabolic disorders. Common regenerative strategies have been applied to tissue engineering of the liver, including hepatocyte growth on synthetic and biological scaffolds.114 The in vitro metabolic function of human hepatocytes cultured in a spherical polyurethane foam was found to be improved in comparison to a monolayer culture. Ammonia removal and amino acid and bile acid metabolism was increased when the hepatocytes were cultured in the three-dimensional synthetic scaffold as opposed to routine monolayer culture. 115
Unfortunately, the organized structure of the portal triad has not been created by means of tissue engineering as yet. Despite the fact that a tissue-engineered whole liver has not yet been developed, cultured hepatocytes have been used for developing bioartificial liver support systems. The blood of patients with liver failure is circulated through a device embedded with hepatocytes. This artificial hepatic support may be sufficient to bridge the gap to liver transplant or hepatic recovery. Currently, there are several clinical trials evaluating the safety and utility of these devices.116
Additionally, advances in liver stem cell research are likely to yield important advances in liver bioengineering. Hepatocyte-like cells have already been generated from growth factor treated ESC.117 Ex vivo portal vein infusion of adult rat hepatocytes into a decellularized liver matrix generated liver grafts with cells capable of in vitro albumin secretion, urea synthesis, and cytochrome P450 expression.118 Moreover, hepatocyte morphology and enzymatic function was maintained following in vivo transplantation of these grafts into recipient rats. Grafts were anastomosed to the renal vein and artery, and perfused for 8 hours before harvest and immunohistochemical analysis. This marked a critical first step toward establishing a three-dimensional, vascularized bioengineered liver graft for the treatment of hepatic failure.
Pancreas
Tissue-engineered pancreas would be an optimal treatment for type 1 diabetics. Regarding cell-based therapies, islet cell transplantation is possible and temporarily restores normoglycemia.119 Unfortunately, this approach frequently fails over time and requires donor islet cells.120 The generation of differentiated beta cells from progenitor cells may be possible in the future.121 Kodama et al. dissociated rat pancreatic biopsies into single cells and seeded them onto PGA scaffolds, which were cultured in growth factor enriched media for 40 days prior to implantation into diabetic mice.122 All mice that received transplanted tissue-engineered pancreas attained normoglycemia without insulin administration. Histologic staining demonstrated differentiated cells that could secrete insulin, somatostatin, and glucagon. These are encouraging results for the future of pancreatic tissue engineering.
References
1. Merrill, JP, Harrison, JH, Murray, J, et al. Successful homotransplantation of the kidney in an identical twin. Trans Am Clin Climatol Assoc. 1956; 67:166–173.
2. Ueno, T, Fukuzawa, M. Current status of intestinal transplantation. Surg Today. 2010; 40:1112–1122.
3. Sylvester, KG, Longaker, MT. Stem cells: Review and update. Arch Surg. 2004; 139:93–99.
4. Ben-David, U, Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer. 2011; 11:268–277.
5. Takahashi, K, Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663–676.
6. Li, M, Chen, M, Han, W, et al. How far are induced pluripotent stem cells from the clinic? Ageing Res Rev. 2010; 9:257–264.
7. Friedenstein, AJ, Petrakova, KV, Kurolesova, AI, et al. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968; 6:230–247.
8. Keating, A. Mesenchymal stromal cells: New directions. Cell Stem Cell. 2012; 10:709–716.
9. Jungebluth, P, Alici, E, Baiguera, S, et al. Tracheobronchial transplantation with a stem-cell-seeded bioartificial nanocomposite: A proof-of-concept study. Lancet. 2011; 378:1997–2004.
10. Carletti, E, Motta, A, Migliaresi, C. Scaffolds for tissue engineering and 3D cell culture. Methods Mol Biol. 2011; 695:17–39.
11. Badylak, SF, Weiss, DJ, Caplan, A, et al. Engineered whole organs and complex tissues. Lancet. 2012; 379:943–952.
12. Mabvuure, N, Hindocha, S, Khan, WS. The role of bioreactors in cartilage tissue engineering. Curr Stem Cell Res Ther. 2012; 7:287–292.
13. Ergun, S, Tewfik, T, Daniel, S. Tracheal agenesis: A rare but fatal congenital anomaly. Mcgill J Med. 2011; 13:10.
14. Grillo, H. Tracheal replacement: A critical review. Ann Thorac Surg. 2002; 73:1995–2004.
15. Delaere, P, Vranckx, J, Verleden, G, et al. Tracheal allotransplantation after withdrawal of immunosuppressive therapy. N Engl J Med. 2010; 362:138–145.
16. Macchiarini, P, Jungebluth, P, Go, T, et al. Clinical transplantation of tissue-engineered airway. The Lancet. 2008; 372:2023–2030.
17. Elliott, MJ, De Coppi, P, Speggiorin, S, et al. Stem-cell-based, tissue engineered tracheal replacement in a child: A 2-year follow-up study. Lancet. 2012; 380:994–1000.
18. Liu, L, Wu, W, Tuo, X, et al. Novel strategy to engineer trachea cartilage graft with marrow mesenchymal stem cell macroaggregate and hydrolyzable scaffold. Artificial Organs. 2010; 34:426–433.
19. Ruszymah, B, Chua, K, Latif, M, et al. Formation of in vivo tissue engineered human hyaline cartilage in the shape of a trachea with internal support. Int J Pediatr Otorhinolaryngol. 2005; 69:1489–1495.
20. Kojima, K, Bonassar, L, Ignotz, R, et al. Comparison of tracheal and nasal chondrocytes for tissue engineering of the trachea. Ann Thorac Surg. 2003; 76:1884–1888.
21. Song, JJ, Ott, HC. Bioartificial lung engineering. Am J Transplant. 2012; 12:283–288.
22. Yuasa, S, Itabashi, Y, Koshimizu, U, et al. Transient inhibition of BMP signaling by Noggin induces cardiomyocyte differentiation of mouse embryonic stem cells. Nat Biotechnol. 2005; 23:607–611.
23. Mummery, C, Ward-van Oostwaard, D, Doevendans, P, et al. Differentiation of human embryonic stem cells to cardiomyocytes: Role of coculture with visceral endoderm-like cells. Circulation. 2003; 107:2733–2740.
24. Liau, B, Zhang, D, Bursac, N. Functional cardiac tissue engineering. Regen Med. 2012; 7:187–206.
25. Mummery, CL, Zhang, J, Ng, ES, et al. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: A methods overview. Circ Res. 2012; 111:344–358.
26. Menasche, P, Hagege, AA, Scorsin, M, et al. Myobast transplantation for heart failure. The Lancet. 2001; 357:279–280.
27. Menasché, P, Alfieri, O, Janssens, S, et al. The myoblast autologous grafting in ischemic cardiomyopathy (MAGIC) trial: First randomized placebo-controlled study of myoblast transplantation. Circulation. 2008; 117:1189–1200.
28. Siepe, M, Giraud, M-N, Pavlovic, M, et al. Myoblast-seeded biodegradable scaffolds to prevent post-myocardial infarction evolution toward heart failure. J Thorac Cardiovasc Surg. 2006; 132:124–131.
29. Rippel, RA, Ghanbari, H, Seifalian, AM. Tissue-Engineered heart valve: Future of cardiac surgery. World J Surg. 2012; 36:1581–1591.
30. Schoen, FJ. Heart valve tissue engineering: Quo vadis? Curr Opin Biotechnol. 2011; 22:698–705.
31. Yin, AH, Miraglia, S, Zanjani, ED, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997; 90:5002–5012.
32. Dohmen, PM, Lembcke, A, Holinski, S, et al. Ten years of clinical results with a tissue-engineered pulmonary valve. Ann Thorac Surg. 2011; 92:1308–1314.
33. Mendelson, K, Schoen, FJ. Heart valve tissue engineering: Concepts, approaches, progress, and challenges. Ann Biomed Eng. 2006; 34:1799–1819.
34. Orlando, G, Wood, KJ, De Coppi, P, et al. Regenerative medicine as applied to general surgery. Ann Surg. 2012; 225:867–880.
35. Takagi, H, Goto, SN, Matsui, M, et al. A contemporary meta-analysis of Dacron versus polytetrafluoroethylene grafts for femoropopliteal bypass grafting. J Vasc Surg. 2010; 52:232–236.
36. Weinberg, C, Bell, E. A blood vessel model constructed from collagen and cultured vascular cells. Science. 1986; 231:397–400.
37. Drilling, S, Gaumer, J, Lannutti, J. Fabrication of burst pressure competent vascular grafts via electrospinning: Effects of microstructure. J Biomed Mater Res A. 2009; 88:923–934.
38. Niklason, LE, Gao, J, Abbott, WM, et al. Functional arteries grown in vitro. Science. 1999; 284:489–493.
39. Shin’oka, T, Imai, Y, Ikada, Y. Transplantation of tissue-engineered pulmonary artery. N Engl J Med. 2001; 344:532–533.
40. Hibino, N, McGillicuddy, E, Matsumura, G, et al. Late-term results of tissue-engineered vascular grafts in humans. J Thorac Cardiovasc Surg. 2010; 139:431–436.
41. Breuer C. A pilot study investigating the clinical use of tissue engineered vascular grafts in congenital heart surgery. ClinicalTrialsgov. 2010: Phase 1; Identifier: NCT01034007.
42. Brown, AK, Tam, PK. Measurement of gap length in esophageal atresia: A simple predictor of outcome. J Am Coll Surg. 1996; 182:41–45.
43. Spitz, L, Ruangtrakool, R. Esophageal substitution. Semin Pediatr Surg. 1998; 7:130–133.
44. Cusick, EL, Batchelor, AA, Spicer, RD. Development of a technique for jejunal interposition in long-gap esophageal atresia. J Pediatr Surg. 1993; 28:990–994.
45. Raffensperger, JG, Luck, SR, Reynolds, M, et al. Intestinal bypass of the esophagus. J Pediatr Surg. 1996; 31:38–47.
46. Grikscheit, T, Ochoa, ER, Srinivasan, A, et al. Tissue-engineered esophagus: Experimental substitution by onlay patch or interposition. J Thorac Cardiovasc Surg. 2003; 126:537–544.
47. Nakase, Y, Nakamura, T, Kin, S, et al. Intrathoracic esophageal replacement by in situ tissue-engineered esophagus. J Thorac Cardiovasc Surg. 2008; 136:850–859.
48. Saxena, A, Ainoedhofer, H, Hollwarth, M. Culture of Ovine Esohageal Eithelial Cells and In Vitro Esophagus Tissue Engineering. Tissue Eng: Part C. 2010; 16:109–114.
49. Saxena, A, Baumgart, H, Komann, C, et al. Esophagus tissue engineering: In situ generation of rudimentary tubular vascularized esophageal conduit using the ovine model. J Pediatr Surg. 2010; 45:859–864.
50. Green, N, Huang, Q, Kahn, L, et al. The development and characterization of an organotypic tissue-engineered human esophageal mucosal model. Tissue Eng: Part A. 2010; 16:1053–1064.
51. Parkin, D, Bray, F, Ferlay, J, et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005; 55:74–108.
52. Maemura, T, Shin, M, Sato, M, et al. A tissue-engineered stomach as a replacement of the native stomach. Transplantation. 2003; 76:61–65.
53. Grikscheit, T, Srinivasan, A, Vacanti, JP. Tissue-engineered stomach: A preliminary report of a versatile in vivo model with therapeutic potential. J Pediatr Surg. 2003; 38:1305–1309.
54. Maemura, T, Shin, M, Kinoshita, M, et al. A tissue-engineered stomach shows presence of proton pump and G-cells in a rat model, resulting in improved anemia following total gastrectomy. Artif Organs. 2008; 32:234–239.
55. Speer, AL, Sala, FG, Matthews, JA, et al. Murine tissue-engineered stomach demonstrates epithelial differentiation. J Surg Res. 2011; 156:205–212.
56. Barclay, AR, Beattie, LM, Weaver, LT, et al. Systematic review: Medical and nutritional interventions for the management of intestinal failure and its resultant complications in children. Aliment Pharmacol Ther. 2011; 33:175–184.
57. Miyasaka, EA, Brown, PI, Kadoura, S, et al. The adolescent child with short bowel syndrome: New onset of failure to thrive and need for increased nutritional supplementation. J Pediatr Surg. 2010; 45:1280–1286.
58. Misiakos, E, Macheras, A, Kapetanakis, T, et al. Short bowel syndrome: Current medical and surgical trends. J Clin Gastroenterol. 2007; 41:5–18.
59. Kato, T, Tzakis, A, Selvaggi, G, et al. Intestinal and multivisceral transplantation in children. Ann Surg. 2006; 243:764–766.
60. Thomson, AB, Keelan, M, Thiesen, A, et al. Small bowel review: Normal physiology part 1. Dig Dis Sci. 2001; 46:2567–2587.
61. Dharmani, P, Srivastava, V, Kissoon-Singh, V, et al. Role of intestinal mucins in innate host defense mechanisms against pathogens. J Innate Immun. 2009; 1:123–135.
62. Ahlman, H, Nilsson. The gut as the largest endocrine organ in the body. Ann Oncol. 2001; 12(Suppl 2):S63–S68.
63. Sato, T, van Es, JH, Snippert, H, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011; 469:415–419.
64. Bevins, CL. The Paneth cell and the innate immune response. Curr Opin Gastroenterol. 2004; 20:572–580.
65. Cheng, H, Leblond, C. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian theory of the origin of the four epithelial cell types. Am J Anat. 1974; 141:537–562.
66. Potten, C, Gandara, R, Mahida, Y, et al. The stem cells of the small intestinal crypts: Where are they? Cell Prolif. 2009; 42:731–750.
67. Barker, N, van Es, JH, Kuipers, J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007; 449:1003–1008.
68. Yan, KS, Chia, LA, Li, X, et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci U S A. 2012; 109:466–471.
69. Salerno-Goncalves, R, Fasano, A, Sztein, M. Engineering of a multicellular organotypic model of the human intestinal mucosa. Gastroenterology. 2011; 141:18–21.
70. Pusch, J, Votteler, M, Göhler, S, et al. The physiological performance of a three-dimensional model that mimics the microenvironment of the small intestine. Biomaterials. 2011; 32:7469–7478.
71. Spence, JR, Mayhew, CN, Rankin, SA, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011; 470:105–109.
72. Evans, G, Flint, N, Somers, A, et al. The development of a method for the preparation of rat intestinal epithelial cell primary cultures. J Cell Sci. 1992; 101:219–231.
73. Choi, RS, Vacanti, JP. Preliminary studies of tissue-engineered intestine using isolated epithelial organoid units on tubular synthetic biodegradable scaffolds. Transplant Proc. 1997; 29:848–851.
74. Wulkersdorfer, B, Kao, KK, Agopian, VG, et al. Growth factors adsorbed on polyglycolic acid mesh augment growth of bioengineered intestinal neomucosa. J Surg Res. 2011; 169:169–178.
75. Sala, FG, Matthews, JA, Speer, AL, et al. A multicellular approach forms a significant amount of tissue-engineered small intestine in the mouse. Tissue Eng Part A. 2011; 17:1841–1850.
76. Lahar, N, Lei, NY, Wang, J, et al. Intestinal subepithelial myofibroblasts support in vitro and in vivo growth of human small intestinal epithelium. PLoS One. 2011; 6:e26898.
77. Sala, FG, Kunisaki, SM, Ochoa, ER, et al. Tissue-engineered small intestine and stomach form from autologous tissue in a preclinical large animal model. J Surg Res. 2009; 156:205–212.
78. Durno, CA, Wong, J, Berk, T, et al. Quality of life and functional outcome for individuals who underwent very early colectomy for familial adenomatous polyposis. Dis Colon Rectum. 2012; 55:436–443.
79. You, YN, Chua, HK, Nelson, H, et al. Segmental vs. extended colectomy: Measurable differences in morbidity, function, and quality of life. Dis Colon Rectum. 2008; 51:1036–1043.
80. Formijne Jonkers, HA, Draaisma, WA, Roskott, AM, et al. Early complications after stoma formation: A prospective cohort study in 100 patients with 1-year follow-up. Int J Colorectal Dis. 2012; 27:1095–1099.
81. Karada , A, Mente
, A, Mente , BB, Uner, A, et al. Impact of stomatherapy on quality of life in patients with permanent colostomies or ileostomies. Int J Colorectal Dis. 2003; 18:234–238.
, BB, Uner, A, et al. Impact of stomatherapy on quality of life in patients with permanent colostomies or ileostomies. Int J Colorectal Dis. 2003; 18:234–238.
82. Messaris, E, Sehgal, R, Deiling, S, et al. Dehydration is the most common indication for readmission after diverting ileostomy creation. Dis Colon Rectum. 2012; 55:175–180.
83. Nissinen, MJ, Gylling, H, Järvinen, HJ, et al. Ileal pouch-anal anastomosis, conventional ileostomy and ileorectal anastomosis modify cholesterol metabolism. Dig Dis Sci. 2004; 49:1444–1453.
84. Grikscheit, TC, Ochoa, ER, Ramsanahie, A, et al. Tissue-engineered large intestine resembles native colon with appropriate in vitro physiology and architecture. Ann Surg. 2003; 238:35–41.
85. Raghavan, S, Miyasaka, EA, Hashish, M, et al. Successful implantation of physiologically functional bioengineered mouse internal anal sphincter. Am J Physiol Gastrointest Liver Physiol. 2010; 299:G430–G439.
86. Hashish, M, Raghavan, S, Somara, S, et al. Surgical implantation of a bioengineered internal anal sphincter. J Pediatr Surg. 2010; 45:52–58.
87. Raghavan, S, Gilmont, RR, Miyasaka, EA, et al. Successful implantation of bioengineered, intrinsically innervated, human internal anal sphincter. Gastroenterol. 2011; 141:310–319.
88. Priya, SG, Jungvid, H, Kumar, A. Skin tissue engineering for tissue repair and regeneration. Tissue Eng: Part B. 2008; 14:105–118.
89. Auger, F, Lacroix, D, Germain, L. Skin substitutes and wound healing. Skin Pharmacol Physiol. 2009; 22:94–102.
90. Cerqueira, MT, Marques, AP, Reis, RL. Using stem cells in skin regeneration: Possibilities and reality. Stem Cells Dev. 2012; 21:1201–1214.
91. Liu, P, Deng, Z, Han, S, et al. Tissue-engineered skin containing mesenchymal stem cells improves burn wounds. Artificial Organs. 2008; 32:925–931.
92. Trotteir, V, Marceau-Fortier, G, Germain, L, et al. IFATS collection: Using human adipose-derived stem/stromal cells for the production of new skin substitutes. Stem Cells. 2008; 26:2713–2723.
93. Cvetkovska, B, Islam, N, Goulet, F, et al. Identification of functional markers in a self-assembled skin substitute in vitro. In Vitro Cell Dev Biol Anim. 2008; 44:444–450.
94. Auger FA. Treatment of cutaneous ulcers with a novel biological dressing. ClinicalTrialsgov. 2012:ultrasound National Institutes of Health; Identifier NCT00207818.
95. Connolly, J, Guse, R, Tiedeman, J, et al. Autologous marrow injection as a substitute for operative grafting of tibial nonunions. Clin Orthop Rel Res. 1991; 266:259–270.
96. Kitoh, H, Kawasumi, M, Kaneko, H, et al. Differential effects of culture-expanded bone marrow cells on the regeneration of bone between the femoral and the tibial lengthenings. J Pediatr Orthop. 2009; 29:643–649.
97. Quarto, R, Mastrogiacomo, M, Cancedda, R, et al. Repair of large bone defects with the use of autologous bone marrow stromal cells. N Eng J Med. 2001; 344:385–386.
98. Salter, E, Goh, B, Hung, B, et al. Bone tissue engineering bioreactors: A role in the clinic? Tissue Eng: Part B. 2012; 18:62–75.
99. Rodrigues, MT, Reis, RL, Gomes, ME. Engineering tendon and ligament tissues: Present developments towards successful clinical products. J Tissue Eng Regen Med. 2012.
100. Cheng M-T, Liu, C-L, Chen, T-H, et al. Comparison of potentials between stem cells isolated from human anterior cruciate ligament and bone marrow for ligament tissue engineering. Tissue Eng: Part A. 2010; 16:2237–2253.
101. Rilke, RM, Elegies, D. Pediatric end-stage renal disease; 2010 Atlas of End-Stage Renal Disease in the United States. http://www.usrds.org, 2010. [[cited 2012 June 1]].
102. Kanzelmeyer, NK, Pape, L. State of pediatric kidney transplantation in 2011. Minerva Pediatr. 2012; 64:205–211.
103. Ross, EA, Williams, MJ, Hamazaki, T, et al. Embryonic stem cells proliferate and differentiate when seeded into kidney scaffolds. J Am Soc Nephrol. 2009; 20:2338–2347.
104. Nakayama, KH, Batchelder, CA, Lee, CI, et al. Decellularized rhesus monkey kidney as a three-dimensional scaffold for renal tissue engineering. Tissue Eng: Part A. 2010; 16:2207–2216.
105. De Filippo, R, Yoo, J, Atala, A. Urethral replacement using cel seeded tubularized collagen matrices. J Urol. 2002; 168:1789–1792.
106. El-Kassaby, A, Retik, A, Yoo, J. Urethral stricture matrix with an off-the shelf collagen matrix. J Urol. 2003; 169:170–173.
107. El-Kassaby, A, AbouShwareb, T, Atala, A. Randomized comparative study between buccal mucosal and acellular bladder matrix grafts in comples anterior urethral strictures. J Urol. 2008; 179:1432–1436.
108. Raya-Rivera, A, Esquiliano, DR, Yoo, JJ, et al. Tissue-engineered autologous urethras for patients who need reconstruction: An observational study. Lancet. 2011; 377:1175–1182.
109. Yang, B, Peng, B, Zheng, J. Cell-based tissue-engineered urethras. Lancet. 2011; 378:568–569.
110. Atala, A, Freeman, M, Vacanti, J, et al. Formation of urothelial structures consisting of rabbit and human urothelium and human bladder muscle. J Urol. 1993; 150:608–612.
111. Pariente, J, Kim, B, Atala, A. In vitro biocompatability assessment of naturally derived and synthetic biomaterials using normal human urothelial cells. J Biomed Mat Res. 2001; 55:33–39.
112. Atala, A, Bauer, SB, Soker, S, et al. Tissue-engineered autologous bladders for patients needing cystoplasty. The Lancet. 2006; 367:1241–1245.
113. Santos, JL, Choquette, M, Bezerra, JA. Cholestatic liver disease in children. Curr Gastroenterol Rep. 2010; 12:30–39.
114. Chistiakov, DA. Liver regenerative medicine: Advances and challenges. Cells Tissues Organs. 2012; 1–22.
115. Yamashita, Y, Shimada, M, Tsujita, E, et al. High metabolic function of primary human and porcine hepatocytes in a polyurethane foam/spheroid culture system in plasma from patients with fulminant hepatic failure. Cell Transplant. 2002; 11:379–384.
116. Rozga, J, Malkowski, P. Artificial liver support: Quo vadis? Ann Transplant. 2010; 15:92–101.
117. Imamura, T, Cui, L, Teng, R, et al. Embryonic stem cell-derived embryoid bodies in three-dimensional culture systems form hepatocyte-like cells in vitro and in vivo. Tissue Eng. 2004; 10:1716–1724.
118. Uygun, BE, Soto-Gutierrez, A, Yagi, H, et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med. 2010; 16:814–821.
119. Shapiro, AM, Lakey, JR, Ryan, EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000; 343:230–238.
120. Shapiro, AM, Ricordi, C, Hering, BJ, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med. 2006; 355:1318–1330.
121. Hebrok, M. Generating beta cells from stem cells-the story so far. Cold Spring Harb Perspect Med. 2012; 2:1–12.
122. Kodama, S, Kojima, K, Furuta, S, et al. Engineering functional islets from cultured cells. Tissue Eng Part A. 2009; 15:3321–3329.