[level-membership-for-neurology-category]
Chapter 11 TIAs and Strokes
Transient Ischemic Attacks
Carotid Artery TIAs
Platelet emboli that form on plaques at the common carotid artery bifurcation (Fig. 11-1) can lead to cerebral-hemisphere TIAs. Contralateral hemiparesis, hemisensory loss, paresthesias, or homonymous hemianopsia characterize carotid artery TIAs. Depending on whether the dominant or nondominant carotid artery gives rise to the problem, a TIA may induce neuropsychologic aberrations accompanied or unaccompanied by hemiparesis and other physical deficits. For example, dominant-hemisphere TIAs may suddenly, but briefly, cause aphasia with or without hemiparesis or homonymous hemianopsia. Similarly, nondominant-hemisphere TIAs may cause brief neglect or hemi-inattention with or without homonymous hemiparesis.
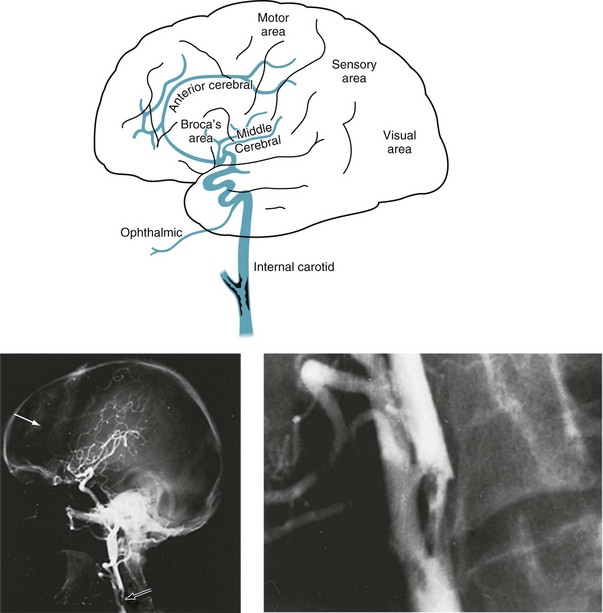
FIGURE 11-1 Top, At its bifurcation in the neck, the common carotid artery divides to form the external and internal carotid arteries. Giving off no branches until it is within the skull, the internal carotid artery immediately sends off the ophthalmic artery. The internal carotid artery then divides into the anterior and middle cerebral arteries. It also gives rise to the posterior communicating artery – not the posterior cerebral artery. Thus, each internal carotid artery perfuses the ipsilateral eye and anterior and middle portions of the ipsilateral cerebral hemisphere. The middle cerebral artery, a major branch of the internal carotid artery, supplies the deep and mid-section of the hemisphere. This region contains most of the motor cortex, sensory cortex, and, in the dominant hemisphere, the perisylvian language arc (see Fig. 8-1). Each anterior cerebral artery, another branch of the internal carotid artery, supplies the frontal lobe, including the medial surface of the motor cortex, which contains the motor innervation for the leg. The posterior cerebral artery (see Fig. 11-2), terminal branches of the basilar artery, supplies the occipital lobe, which houses the visual cortex, and most of the temporal lobe. Bottom left, An arteriogram of the carotid artery and its branches prominently displays the bifurcation (black arrow), the typical “candelabra” of branches that comprise the middle cerebral artery, and a faint anterior cerebral artery that sweeps from anterior to posterior (white arrow). Bottom right, A magnification of the bifurcation reveals an extensive circumferential plaque constricting the internal carotid artery. The remaining blood flow appears as an “apple core.” The rough interior surface of the artery gives rise to retinal and cerebral emboli.
Sometimes a TIA causes only brief visual loss in one eye, i.e., monocular blindness. Neurologists call this distinctive symptom amaurosis fugax (Greek, fleeting darkness). The underlying mechanism consists of emboli from the internal carotid artery flying into the ophthalmic artery, the carotid artery’s first branch, to induce several minutes of ischemia in the retina and optic nerve (Box 11-1). Unlike migraines, which can also cause transient visual loss, TIAs rarely cause headache or scintillations.
In patients with mild cognitive impairment, TIAs may produce a brief but marked confusional state. A similar disturbance may occur after atherosclerosis slowly occludes one carotid artery, forcing the other carotid artery to supply both cerebral hemispheres through the circle of Willis (Fig. 11-2, top right). In this case, the cerebral blood supply is tenuous and emboli from the patent artery produce bilateral cerebral ischemia.
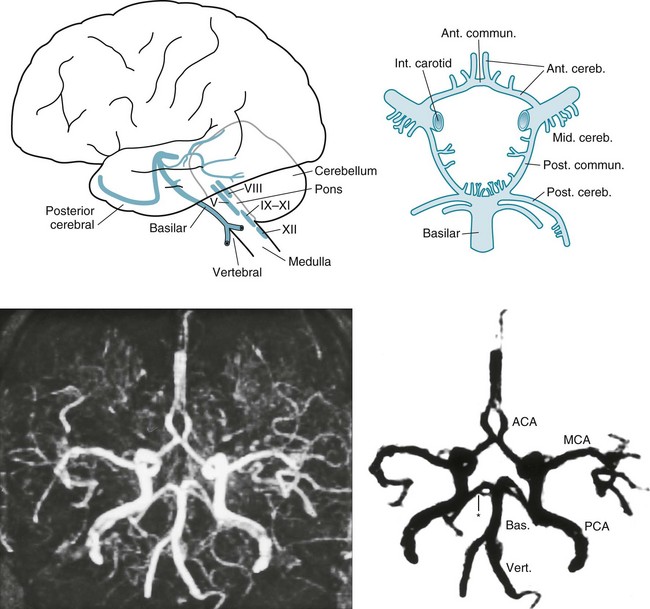
FIGURE 11-2 Top left, After ascending, encased in the cervical vertebrae, the two vertebral arteries enter the skull. They join to form the basilar artery at the base of the brain. Small, delicate branches of the basilar artery supply the brainstem and its contents. (The Roman numerals refer to cranial nerve nuclei.) Large branches, as if wrapping their arms around the brainstem, supply the cerebellum and posterior portion of the cerebrum (i.e., the occipital lobes and inferomedial portions of the temporal lobes). The posterior cerebral arteries are, for practical purposes, the terminal branches of the basilar artery. They supply the occipital cortex and the posterior, inferior aspect of the temporal lobes. Top right, Belying its name, the circle of Willis, the “great anastomoses,” is completely patent in only about 20% of people. Connections between the basilar and internal carotid arteries ideally form the circle, which should give off the anterior, middle, and posterior cerebral arteries. The circle should provide anastomoses between anterior–posterior and right–left cerebral circulations. Despite the advantages that the circle confers, junctions of the arteries are weak spots. Defects at the junctions may balloon outward to form berry aneurysms that rupture and produce subarachnoid hemorrhages. Bottom left, This axial magnetic resonance angiogram (MRA) shows the major cerebral arteries that form the circle of Willis and anterior and posterior circulations. Bottom right, This MRA highlights the major cerebral arteries and shows the anterior and posterior circulations. The anterior circulation consists of the middle cerebral arteries (MCA), anterior cerebral arteries (ACA), and the anterior communicating artery (∧), which joins the ACAs. The posterior circulation consists of the basilar (Bas.) artery and two vertebral (Vert.), posterior cerebral (PCA), and posterior communicating arteries (*), which connect the PCAs and the basilar artery.
Preventive Measures
For patients who cannot undergo carotid endarterectomy, an alternative is intravascular insertion of stents. These devices are essentially expandable tubes that neuroradiologists insert intravascularly into the carotid artery to widen atherosclerotic stenoses (see Fig. 20-28). They also trap underlying atheromatous debris against the inner surface of the arterial wall to reduce the likelihood of emboli. In contrast to the benefits of inserting stents into the extracranial carotid arteries, inserting stents into intracerebral arteries, such as the basilar or middle cerebral artery, carries too much risk to justify its use.
Basilar Artery TIAs
The vertebrobasilar system, basilar artery system, or simply the posterior circulation supplies the brainstem, cerebellum, and the posterior inferior portion of the cerebrum (the occipital and medial inferior portion of the temporal lobes [see Fig. 11-2]). Emboli-generating plaques tend to develop at both the origin of the vertebral arteries (in the chest) and their junction at the base of the brain.
Symptoms and signs of basilar artery TIAs, which usually result from patchy brainstem ischemia, differ greatly from those of carotid artery TIAs (Box 11-2). Typical basilar artery TIA symptoms include tingling around the mouth (circumoral paresthesias), dysarthria, nystagmus, diplopia, ataxia, and vertigo. On rare occasions, when all blood flow through the basilar artery momentarily stops, almost the entire brainstem suffers from ischemia. The brainstem ischemia interrupts consciousness and body tone, which causes patients to collapse. This TIA, termed a drop attack, strikes suddenly and unexpectedly. (It appears similar to cataplexy [see Chapter 17].)
Transient Global Amnesia
TIAs sometimes impair the circulation of the basilar artery’s terminal branches, the posterior cerebral arteries, which supply the temporal lobes (see Fig. 11-2). Because the temporal lobes contain portions of the limbic system, particularly the hippocampi (see Fig. 16-5), posterior circulation TIAs may induce episodes of temporary amnesia and personality change – called transient global amnesia (TGA).
Even in the absence of a confirmatory laboratory test for TGA, its clinical features differentiate it from other neurologic conditions. TGA patients’ preserved intellect and general knowledge, as well as their remaining fully conscious, distinguishes them from patients in delirium. TGA patients, despite their amnesia, also do not confabulate in the manner of Wernicke–Korsakoff patients. Complex partial seizures differ by producing dulling of the sensorium, simple repetitive actions, paroxysmal or other epileptiform EEG changes, and a high rate of recurrence (see Chapter 10).
Strokes
Risk Factors
Reflecting the necessity of preventing strokes, epidemiologic studies constantly search for stroke risk factors (Box 11-3). Although physicians customarily list stroke risk factors as individual threats, the factors actually tend to cluster. For example, obesity, elevated low-density lipoprotein cholesterol, and hypertension frequently occur together in the metabolic syndrome. Some factors, by themselves, have a weak correlation with stroke, but pose a synergistic risk when paired with others. For example, cigarette smoking correlates with stroke; however, with concomitant use of oral contraceptives or in sufferers of migraine with aura, it becomes a moderately powerful risk factor. More strikingly, smokers who are hypertensive have a 20-fold greater risk. Curiously, total serum cholesterol carries a strong risk for myocardial infarctions but a weak risk for stroke.
Elevated serum concentration of homocysteine serves as a marker for an increased risk. In the autosomal recessive genetic condition, homocystinuria, children have a Marfan-like habitus, ocular lens displacement, and other anatomic abnormalities. More importantly, they routinely suffer strokes in childhood. Elevated homocysteine levels in adults are associated with the use of antiepileptic drugs and coronary artery and peripheral vascular disease, as well as stroke. Folic acid reduces elevated serum homocysteine concentrations (see Fig. 5-8), as well as reducing the incidence of fetal neural tube defects (see Chapter 13), but surprisingly, it does not reduce the incidence of strokes.
Thrombosis and Embolus
In addition to creating permanent clinical deficits, stroke scars are potentially epileptogenic. Approximately 50% of seizures that develop in adults older than 65 years originate from these lesions (see Chapter 10). Thus, patients with a history of having sustained a stroke who develop confusion, unresponsiveness, or abnormal behavior may have suffered a complex partial seizure as well as another stroke or TIA.
Necrosis
Infarctions in the Carotid Artery Distribution
Cerebral artery thrombosis and embolism produce an infarction in the distribution of the occluded artery. These strokes result in well-known clinical deficits (see Fig. 11-1 and Box 11-4):
• Anterior cerebral artery infarction damages the anterior and medial aspects of the frontal lobe. It typically causes paresis and apraxia of the contralateral leg. With bilateral anterior cerebral artery infarctions, the resulting frontal lobe damage, which is extensive, typically causes pseudobulbar palsy, apathy, mutism, personality changes, and urinary incontinence (see Chapter 7), along with both legs having paresis and apraxia.
• Middle cerebral artery infarction, which is the most common stroke, usually results in contralateral hemiparesis, hemisensory loss, and either aphasia with dominant hemisphere lesions or hemi-inattention, including neglect, with nondominant hemisphere strokes.
Infarctions in the Basilar Artery Distribution
Infarctions in the basilar artery distribution cause brainstem, cerebellar, or posterior cerebral injuries. Small brainstem infarctions usually cause constellations of cranial nerve injuries and hemiparesis, and large ones usually cause coma, if not immediate death. In contrast to cerebral-hemisphere infarctions, brainstem infarctions generally spare language or intellectual function and do not cause seizures (see Box 11-4).
The posterior cerebral arteries, as previously noted, are actually terminal branches of the basilar artery. Infarction of a posterior cerebral artery causes a contralateral homonymous hemianopsia and occasionally alexia without agraphia (see Chapter 8). Bilateral posterior cerebral artery strokes can cause (cortical) blindness, which sometimes leads to Anton syndrome (see Chapter 12).
Neurologists love to localize small brainstem infarctions for both clinical and academic reasons. Lesions of the midbrain cause ipsilateral oculomotor nerve and contralateral paresis (see Fig. 4-9). Those in the pons cause ipsilateral abducens nerve and contralateral paresis (see Fig. 4-11). Infarctions involving the midline of the midbrain or pons cause the medial longitudinal fasciculus syndrome (see Chapters 12 and 15). Finally, lateral medullary infarctions, which are the most common brainstem infarction, cause a complex, apparently disparate, combination of ipsilateral limb ataxia, palatal paresis, Horner syndrome, and alternating hypalgesia (see Wallenberg syndrome, Fig. 2-10).
Hemorrhages
Hemorrhages, which are most often the result of hypertension, usually erupt in the basal ganglia, thalamus, pons, or cerebellum (see Figs 20-13 and 20-14). Trauma and use of cocaine also cause hemorrhages, but their location is not as predictable as with hypertension-induced ones. In a classic problem, patients who take a monamine oxidase inhibitor (MAOI) antidepressant and then, inadvertently or in a suicide attempt, consume certain foodstuffs, notoriously aged cheese or red wine, or receive meperidine (Demerol), develop cerebral hemorrhages. These combinations, which are sympathomimetic, lead to a burst of hypertension sufficient to cause the hemorrhage. (Notably, MAOI-A antidepressants may cause this problem, but MAOI-B medications, such as those used to treat Parkinson disease, generally do not [see Chapter 9].)
Subarachnoid hemorrhage (SAH), although usually traumatic in origin, sometimes results from a ruptured berry aneurysm. SAH most often produces a prostrating headache that patients classically describe as the “worst headache” of their life. SAH also produces nuchal rigidity and lethargy, but usually no lateralized signs. Exercising, straining at stool, and other usually benign activities can rupture an aneurysm and precipitate an SAH. A diagnostic dilemma occurs when a sudden, profound headache interrupts sex. In this case, the exertion may have led to an SAH, but more often coital cephalalgia is responsible (see Chapter 9). Thus, TGA, SAH, and a migraine variant are hazards of sexual activity.
Neuropsychologic Sequalae
Several large or many small (lacunar) strokes cause vascular cognitive impairment, previously labeled vascular dementia, and before that multi-infarct dementia. The preliminary version of the DSM-5 labels it Vascular Neurocognitive Disorder. Ironically, despite the implication of these terms, the dementia stems from the neuropathologic changes of Alzheimer disease that accompany those of cerebral vascular disease (see Chapter 7).
Occlusion of a cerebral artery typically destroys a discrete critical area of the cerebral cortex. Commonly occurring strokes produce well-known neuropsychologic syndromes, such as aphasia, Gerstmann syndrome, and hemi-inattention (see Chapter 8). Strokes of both frontal lobes, which may occur either simultaneously or in succession, cause pseudobulbar palsy and frontal lobe dysfunction (see Chapters 4 and 8).
In another mechanism, hypoxia, hypotension, carbon monoxide poisoning, or similar catastrophes cause generalized cerebral anoxia. This insult leads to dementia, cortical blindness, a persistent vegetative state (PVS: see later), and postanoxic myoclonus (see Chapter 18). A variation of generalized anoxia occurs when cerebral anoxia affects only the fine, terminal branches of cerebral arteries that perfuse the extensive circumferential regions of the cerebral cortex, which neurologists call the watershed or borderzone area. An episode of cerebral anoxia that reduces the oxygen supply below a critical point in the watershed area produces a watershed infarction. These infarctions often leave patients with cognitive motor impairment and, if the relatively well-perfused perisylvian language arc survives, isolation aphasia (see Fig. 8-3D).
Post-Stroke Depression
Before leaping to the diagnosis of post-stroke depression, physicians should keep in mind that some stroke symptoms mimic depression. Failing to appreciate them may lead to misdiagnosis and unwarranted prescriptions. For example, patients with a nondominant-hemisphere stroke may deny having a hemiparesis, not because of depression, but because of anosognosia. Also, their conversations may lose their emotional tone because of aprosody (see Chapter 8). In another situation, strokes – individually or in succession – that damage both frontal lobes frequently result in apathy, pseudobulbar palsy with unprovoked crying and emotional incontinence (see Chapter 4), or a paucity of verbal output because of abulia (see Chapter 7). Finally, following several strokes, patients often develop sleep disturbances and vegetative symptoms.
Altered Levels of Consciousness
Coma, which neurologists often grade using the Glasgow Coma Scale (see Chapter 22), is the most profound depression of consciousness. Comatose patients have closed eyes, make little or no verbal response, and move their limbs only as a reflex.
Locked-In Syndrome
The locked-in syndrome usually results from a stroke that damages the base (ventral portion) of the pons (basis pontis, see Fig. 2-9) and medulla (bulb). The exact cause is usually a thrombosis or embolus that occludes a branch of the basilar artery (see Fig. 11-2). The stroke causes bulbar palsy and bilateral interruption of the corticospinal tracts. It renders patients mute, quadriplegic, apneic, and dysphagic. Patients almost always require tracheostomy, ventilator support, and feeding tubes. Although locked-in patients have lost almost all their motor ability, their higher functions, like language comprehension and cognitive ability, remain intact.
The locked-in syndrome may not only result from brainstem strokes. Several peripheral nervous system (PNS) diseases, such as myasthenia gravis, ALS, and Guillain–Barré syndrome (see Chapters 5 and 6), may cause extensive enough paresis of cranial and limb muscles to cause it and require mechanical supports.
Despite the devastating damage in the locked-in syndrome, the upper brainstem, reticular activating system, and cerebral cortex remain intact. Moreover, the physiologic circuits between the cerebral cortex and upper brainstem, including the thalamus, continue to reverberate (Fig. 11-3). Thus, locked-in patients retain cognition, affective capacity, and, given sufficient clues, a sleep–wake cycle. Patients who are otherwise completely paralyzed can still purposefully move their eyes and eyelids. By closing their eyelids in a “yes” or “no” pattern, they can communicate by answering questions. The preserved circuits between the thalamus and cerebral cortex generate a relatively normal EEG.
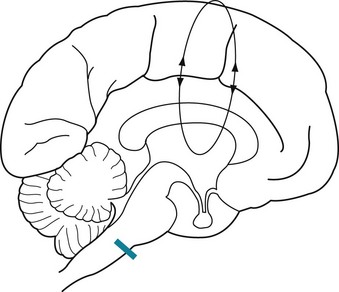
FIGURE 11-3 The locked-in syndrome usually results from an infarction of the ventral or basilar portion of the lower brainstem, typically at the basis pontis (see Fig. 2-9). A lesion in this area (indicated by the bar) would sever the corticospinal tracts and directly injure cranial nerves IX through XII. However, it would not damage several vital systems: (1) the reticular activating system of the brainstem; (2) the cerebral hemispheres, particularly the cerebral cortex; and (3) the cerebral and brainstem system that governs ocular movement (see Fig. 12-12). This lesion, which is nowhere near the cortex, would not affect the brain’s cognitive, language, or visual centers. The EEG is relatively normal because the lesion also spares the circuits reverberating between the thalamus and the cerebral cortex (indicated by the loop), which generate the organized, relatively regular background EEG activity.
Persistent Vegetative State
Extensive cerebral cortex damage – without brainstem damage – may cause PVS. Like locked-in patients, PVS patients remain bedridden with quadriparesis and incontinence, but with their eyelids open and eyes moving about. However, in stark contrast, because of the cortex damage, PVS patients lack awareness of themselves and their surroundings, have no cognitive function, and cannot communicate in any manner. With their lack of consciousness, patients in PVS can neither perceive pain nor suffer. Neurologists sometimes describe this state as “wakefulness without awareness” (Figs 11-4 and 11-5). Nevertheless, the undamaged brainstem continues to regulate the body’s vegetative functions, e.g., metabolism, breathing, temperature regulation, and digestion. Also, unlike comatose patients, PVS patients continue their sleep–wake cycle.
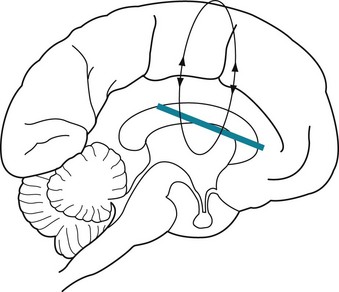
FIGURE 11-4 The persistent vegetative state, which can be caused by cerebral anoxia or numerous other conditions, results from extensive damage to the cerebral cortex or the tissue immediately underlying it (indicated by the bar). These injuries impair all cerebral functions, including cognitive ability, purposeful motor activity, vision, and speech.
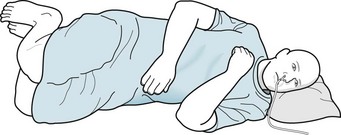
FIGURE 11-5 Patients in the persistent vegetative state tend to assume a decorticate (flexed or fetal) posture because of extensive cerebral damage. Appearing to be awake, they remain mute, unable to respond to visitors or examiners, and, except for breathing and roving eye movements, motionless. Patients almost always require combinations of nasogastric tubes, intravenous lines, urinary catheters, and tracheostomies. Being immobile and requiring mechanical devices, they are vulnerable to aspiration pneumonia, urinary tract infections, and decubitus ulcers.
Managing Stroke
Laboratory Tests
In most cases, CT or MRI confirms a diagnosis of stroke or reveals common alternatives, such as brain tumor, abscess, or subdural hematoma (see Chapter 20). CT indicates the presence and location of most strokes, except those that are very acute, small, or located in the brainstem or other regions of the brain located in the posterior fossa (where bone artifacts may obscure them). Compared to CT, MRI provides better resolution of small strokes, visualizes ones in areas of the brain surrounded by bone, and, by using special software subprograms, shows acute infarctions. MRA can visualize extra- and intracranial cerebral arteries and most regions of stenosis and occlusion (see Chapter 20). As diagnostic tests in this situation, skull X-rays and EEGs are superfluous.
Examination of the CSF through an LP may reveal signs of an SAH or infection, such as meningitis and encephalitis, that mimic a stroke. However, LP is unnecessary for a routine stroke and physicians should avoid performing the procedure in the presence of an intracranial mass lesion (see transtentorial herniation, Chapter 19).
Barnes DE, Haight TJ, Mehta KM, et al. Second hand smoke, vascular disease, and dementia incidence: Findings from the cardiovascular health cognition study. Am J Epidemiol. 2010;171:292–302.
Brott TG, Hobson RW, Howard G, et al. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med. 2010;363:11–23.
Chimowitz MI, Lynn MJ, Derdeyn CO, et al. Stenting versus aggressive medical therapy for intracranial arterial stenosis. N Engl J Med. 2011;365:993–1003.
Cruse D, Chennu S, Chatelle C, et al. Bedside detection of awareness in the vegetative state. Lancet. 2011;378:2088–2094.
Dafer RM, Rao M, Shareef A, et al. Poststroke depression. Top Stroke Rehabil. 2008;15:13–21.
Dong JY, Zang YH, Tong J, et al. Depression and risk of stroke: A meta-analysis of prospective studies. Stroke. 2012;42:32–37.
Giacino JT, Ashwal A, Childs N, et al. The minimally conscious state: Definition and diagnostic criteria. Neurology. 2002;58:349–353.
Goldstein LB, Adams R, Alberts MJ, et al. Primary prevention of ischemic stroke: A guideline from the American Heart Association/American Stroke Association Stroke Council. Stroke. 2006;37:1583–1633.
O’Donnell MJ, Xavier D, Liu L, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries. Lancet. 2010;376:112–123.
Pan A, Sun Q, Okereke OI, et al. Depression and risk of stroke morbidity and mortality: A meta-analysis and systemic review. JAMA. 2011;306:1241–1249.
Quinette P, Guillery-Girard B, Dayan J, et al. What does transient global amnesia really mean? Review of the literature and thorough study of 142 cases. Brain. 2006;129:1640–1658.
Schmid AA, Kroenke K, Hendrie HC, et al. Poststroke depression and treatment effects on functional outcomes. Neurology. 2011;76:1000–1005.
Solfrizzi V, Scafato E, Capurso C, et al. Metabolic syndrome and the risk of vascular dementia: The Italian Longitudinal Study on Ageing. J Neurol Neurosurg Psychiatry. 2010;81:433–440.
Surtees PG, Wainwright NWJ, Luben RN, et al. Psychological distress, major depressive disorder, and risk of stroke. Neurology. 2008;70:788–794.
Wechsler LR. Intravenous thrombolytic therapy for acute ischemic stroke. N Engl J Med. 2011;364:2138–2146.
Willey JZ, Disla N, Moon YP, et al. Early depressed mood after stroke predicts long-term disability. Stroke. 2010;41:1896–1900.
Chapter 11 Questions and Answers
1. Left hemiparesis with relative sparing of the leg
2. Left lower extremity monoparesis
3. Monocular blindness from optic nerve ischemia
4. Left homonymous hemianopsia
5. Left palate paresis, left limb ataxia
6. Right third cranial nerve palsy with left hemiparesis
7. Right hemiparesis with aphasia
8. Quadriplegia and mutism with intact mentation
9. Left sixth and seventh cranial nerve palsy with right hemiparesis
21. No. Cerebral hemorrhages are usually catastrophic processes, heralded by a severe headache, that develop between several minutes and a few hours.
22. No. The headaches of a subarachnoid hemorrhage are usually cataclysmic and incapacitating. The examination would show nuchal rigidity and a depressed level of consciousness. Also, because subarachnoid hemorrhage occurs outside the brain substance, patients usually do not have lateralized signs, such as aphasia and homonymous hemianopsia. An exception would be a posterior communicating artery aneurysm rupture, which compresses the adjacent third cranial nerve.
23. Yes. A brain tumor is a valid diagnostic choice in this case. Tumors, such as the glioblastoma, produce symptoms over a period of weeks. Also, they may cause multiple signs because they infiltrate or, as in the case of metastases, are multicentric.
24. Unlikely. Although the headache and hemiparesis are consistent, subdural hematomas and other masses outside the brain substance (i.e., extra-axial lesions) rarely cause aphasia or hemianopsia.
25. No. A basilar artery occlusion would interrupt the reticular activating system. It would cause coma, apnea, and quadriplegia.
26. Yes. During a week, carotid artery stenosis can lead to complete occlusion and infarction of an entire cerebral hemisphere. Carotid artery stenosis may also cause ipsilateral headache.
27. Yes. As with a brain tumor, an abscess and its surrounding edema can expand rapidly enough to impair an entire cerebral hemisphere. Surprisingly, traditional signs of infection, such as fever and leukocytosis, are often minimal or entirely absent in cases of brain abscess.
28. No. Cerebral toxoplasmosis develops almost only as a complication of immunosuppression. Moreover, because toxoplasmosis typically produces multiple sites of infection, it produces multiple symptoms and signs.
29. No. Although the findings are compatible, the time course is inconsistent. Emboli occur acutely.
30. No. The single focus and older age are inconsistent with MS. Moreover, it usually does not cause a headache.
31. No. He does not have aphasia in any form because he can understand spoken language and respond appropriately. He cannot speak or write because of motor impairment.
32. No. His vision is not impaired because he can read written questions.
33. No. He has no indication of cerebral cortex damage. His ability to communicate indicates that cortical functions remain intact. Instead, the palate weakness and other motor pareses reflect brainstem damage.
34. The EEG should appear normal because his cortical functions are intact.
35. His being fully alert, cognizant, and communicative, but almost completely paralyzed, indicates that he suffers from the locked-in syndrome.
36. The lesion is in the ventral surface of the lower brainstem, i.e., the base of the pons or basis pontis.
37. The new lesion is in the left (dominant) hemisphere. With the history of a prior right cerebral infarction, he now has bilateral cerebral infarctions.
38. The sudden, painless onset suggests a thrombotic or embolic stroke. The rapid onset and absence of signs of increased intracranial pressure exclude a mass lesion, such as a tumor or abscess.
39. The EEG will be abnormal because of extensive cerebral damage.
40. No. He has global aphasia with impairment of all avenues of communication. Moreover, as the result of extensive cerebral cortex strokes, he probably has vascular cognitive impairment (VCI), previously labeled “vascular dementia,” which the preliminary version of Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) designates Vascular Neurocognitive Disorder.
41. Although proper clinical assessment requires more time, he will probably have pseudobulbar palsy because he has sustained bilateral cerebral rather than brainstem infarctions.
42. Cerebral thrombosis associated with high-dose estrogen oral contraceptives
43. Cerebral vasculitis from lupus or drug abuse
44. Cerebral embolus from mitral stenosis
46. Septic cerebral embolus from bacterial endocarditis
47. Cerebral embolus from an atrial myxoma
49. Stroke from sickle cell disease
50. Migraine-induced transient paresis, i.e., hemiplegic migraine
53. A patient who seems to have global aphasia and a right hemiparesis would usually have a left cerebral hemisphere lesion.
55. A psychogenic disturbance is the most likely cause.
56. A normal EEG, computed tomography (CT), and magnetic resonance imaging (MRI) would support the diagnosis of a psychogenic disturbance.
57. Because he has ataxia and dysmetria the lesion is in the cerebellum.
58. When unilateral, abnormal cerebellar findings are referable to the ipsilateral hemisphere, which, in this case, is his right side.
59. In view of the patient’s age and the sudden onset of the cerebellar deficits, a stroke is the most likely cause. Because he has a severe headache, physicians must first consider cerebellar hemorrhage. Hypertension, including cocaine-induced hypertension, is the most common cause of cerebellar hemorrhage. Occasionally, hemorrhage into tumors results in cerebellar hematomas.
60. If the hemorrhage were to expand, it would compress the fourth ventricle and cause obstructive hydrocephalus. With further expansion, the hemorrhage would compress the brainstem. At that point, unless neurosurgeons were to evacuate the cerebellar hematoma, it would probably lead to coma and death.
a, b. In the absence of B12, which acts as a coenzyme, homocysteine and methylmalonic acid will accumulate. Administration of folate, which also acts as a coenzyme, will alleviate some features of B12 deficiency, such as the megaloblastic anemia. However, without concomitant administration of B12, folate will not prevent combined system disease (see Fig. 5-8).
64. A 35-year-old woman sought chiropractic treatment of neck muscle spams. In the middle of vigorous manipulation, she suddenly developed vertigo, nausea, and vomiting. On examination, a neurologist found dysarthria, nystagmus, right-sided ptosis and miosis, numbness of the left arm and leg, and ataxia on right finger-to-nose movement. What is the most likely etiology of her condition?
67. An internist asked a psychiatry consultant to evaluate a 60-year-old stroke patient who, for no apparent reason, began to scream at the nurses, complained loudly about the food, and demanded that the physical therapists leave his room. The internist had admitted the patient the previous day after he awoke with left hemiplegia. Although the patient had obviously sustained a stroke, he was not eligible for tissue plasminogen activator (tPA) because more than 3 hours probably elapsed since its onset. A CT and MRI showed a right middle cerebral artery distribution infarction. After a cardiac evaluation disclosed atrial fibrillation and mitral stenosis, the cardiologist planned to institute anticoagulation. The psychiatrist found that the patient, who was fully alert, attentive, and lucid, had been functioning very well both at work and at home. He had no preceding psychiatric history and had been using no psychotropics, illicit drugs, or excessive alcohol. The patient insisted that he did not know why he was in the hospital or that he had any cardiac disease. He also said that he was “fine” until the internist, apparently confusing him with the other patient in the room, gave him a “preposterous” diagnosis of a stroke. He added that he wanted to leave the hospital that evening, but the nurses and the “idiot doctor” refused to discharge him. Which is the most likely explanation for the patient’s irrational and potentially self-injurious behavior?
68. Following her third miscarriage, a 29-year-old woman sought psychiatric consultation for depression. In reviewing her history, the psychiatrists found that her migraines had flared up and, because of a persistent recent calf cramp, she was no longer able to exercise. Which of the following tests should her physician order?
69. A 10-year-old boy, according to his parents, has episodes of being unresponsive and having weakness on one side or the other of his body. The episodes began about 1 year before the visit, occur approximately once a month, and last several hours. He agrees with his parents’ description and adds that, during the episodes, his head hurts and he is nauseous. Between episodes, he is normal in every respect. Of the following, which is the most likely diagnosis?
74. A 28-year-old man with mild generalized headache has had a 3-day history of increasing left arm weakness and clumsiness. Examination reveals only mild left arm weakness and hyperactive DTRs. Routine medical evaluation reveals no abnormalities. Both CTs and MRIs show five large ring-enhancing cerebral lesions. Of the following, which is the most likely cause of his neurologic difficulties?
75–86. Match the sign (75–86) with the following:
78. Reflexive movement to light and sound
79. Sleep–wake cycles may be preserved
80. Capacity to suffer conscious pain
83. Due to lesion in base of pons that spares reticular activating system
84. Due to lesions that damage virtually all of the cerebral cortex
85. Guillain–Barré syndrome or myasthenia gravis may be responsible
86. Anoxia or insulin injections, as in attempted murder, may be responsible
75-c; 76-c; 77-a; 78-c; 79-c; 80-a; 81-a; 82-b; 83-a; 84-b; 85-a; 86-b.
93. Three months after surviving a drug overdose, a 30-year-old man lies in his hospital bed, quadriparetic and almost mute. However, he has regained enough strength to reach for his food and, with some assistance, feed himself with a spoon. He looks directly at examiners and appears to watch televised sports events. He usually says, “no, no” when someone begins to change the channel. His EEG when he is alert shows slow background activity and PET shows markedly decreased cerebral cortical metabolism. Which of the following diagnoses most accurately describes his condition?
96. The parents of a 4-year-old boy bring him to the emergency room after he suddenly developed aphasia and right-sided hemiparesis. There is no indication of trauma. CT confirms the clinical impression of a left middle cerebral artery stroke. Blood tests reveal marked lactic acidosis. Results of sickle cell testing and homocysteine levels are normal. Which of the following would be the most appropriate diagnostic test?
97. Which of the following statements most closely describes the course of post-stroke depression in the majority of cases?
c. Although it is effective, ECT may cause more confusion than usual in stroke victims.
102. A 40-year-old man reported suddenly developing weakness in his left arm and leg after slipping on ice and striking his neck and back. When the neurologist examined him, she found that the degree of weakness was inconsistent, the DTRs were normal, and plantar reflexes were flexor. CTs of his head, neck, and lower back showed no abnormalities. When he still seemed to have left hemiplegia, she re-examined him. When asked to raise his right leg, he pushed down (extended) his left. His strength was so great in left leg extension that, using it as a level, she raised his lower trunk from the bed. Similarly, when asked to push downward with the right leg, he reflexively raised his left leg. What is the best description of his left leg movements?
a. The neurologist has demonstrated the Hoover sign, which is an indication of psychogenic hemiparesis. Another sign of psychogenic hemiparesis is the abductor sign (see Chapter 3). Other suggestions of psychogenic hemiparesis included the intermittent nature of his weakness, normal DTRs, and absence of a Babinski sign.
103. In the recovery room following coronary artery bypass surgery, an 85-year-old man complained that he was unable to move his legs. Neurologists found that he had a clear sensorium, good memory and judgment, intact cranial nerves, and, in his arm and hands, good strength and normal DTRs. They also found that he had flaccid areflexic paraplegia, hypalgesia to pin below the umbilicus, and urinary retention that he did not appreciate. In contrast, position and vibration sensations were preserved in his legs and feet. Where is the lesion?
c. The patient sustained an infarction of the anterior spinal artery where it perfuses the thoracic spinal cord, i.e., a stroke of the spinal cord. The anterior spinal artery is thin, delicate, and vulnerable to atheromatous debris dislodged during any surgery involving the aorta. The anterior spinal artery supplies the anterior two-thirds of the spinal cord (see Chapter 2). Infarctions of this artery cause paraplegia and anesthesia because of damage to the corticospinal and lateral spinothalamic tracts. Immediately after acute spinal cord injuries, patients lose DTRs and muscle tone because of “spinal shock,” but several days to weeks later they develop hyperactive DTRs, Babinski signs, and spasticity. Because multiple small intercostal arteries supply the posterior portion of the spinal cord, infarction of the anterior spinal artery spares the posterior columns. In this infarction, position and vibration sensations are spared (see figure).
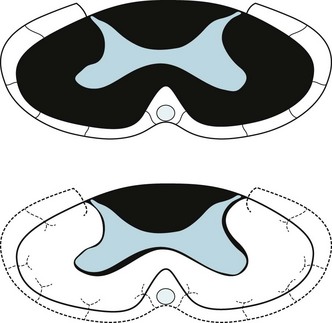
Top: A cross-section of a myelin stain of the normal thoracic spinal cord shows that the anterior spinal artery perfuses the anterior two-thirds of the cord. This region contains the corticospinal and lateral spinothalamic tracts (see Fig. 2-15). Bottom: If atheromatous material were to occlude the anterior spinal artery, this region of the spinal cord would undergo infarction. The patient would lose strength in the legs and sensation below the site of the infarction. However, because this infarction spares the posterior columns, the patient would still perceive position and vibration sensation.
105. If he survives the acute stroke superimposed on his pre-existing stroke and Alzheimer disease, which will most likely be his status?
b. CT requires only a few minutes and detects even small hemorrhages. Patients with metal devices, including pacemakers, and those with claustrophobia may undergo the test with impunity. In consulting on patients who have just sustained a thrombotic stroke, neurologists require the ease and rapidity of head CT because it must show no sign of intracranial hemorrhage before they administer tPA within the limited time frame. MRI requires 20–40 minutes, precludes patients with metal devices, and requires sedation, which confuses the clinical situation, for those with claustrophobia. Lumbar punctures will not provide useful information in cases of intracerebral hemorrhage. Moreover, in the case of a large cerebral hemorrhage, the procedure may lead to transtentorial herniation (see Fig. 19-3).
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 11 TIAs and Strokes
Transient Ischemic Attacks
Carotid Artery TIAs
Platelet emboli that form on plaques at the common carotid artery bifurcation (Fig. 11-1) can lead to cerebral-hemisphere TIAs. Contralateral hemiparesis, hemisensory loss, paresthesias, or homonymous hemianopsia characterize carotid artery TIAs. Depending on whether the dominant or nondominant carotid artery gives rise to the problem, a TIA may induce neuropsychologic aberrations accompanied or unaccompanied by hemiparesis and other physical deficits. For example, dominant-hemisphere TIAs may suddenly, but briefly, cause aphasia with or without hemiparesis or homonymous hemianopsia. Similarly, nondominant-hemisphere TIAs may cause brief neglect or hemi-inattention with or without homonymous hemiparesis.
FIGURE 11-1 Top, At its bifurcation in the neck, the common carotid artery divides to form the external and internal carotid arteries. Giving off no branches until it is within the skull, the internal carotid artery immediately sends off the ophthalmic artery. The internal carotid artery then divides into the anterior and middle cerebral arteries. It also gives rise to the posterior communicating artery – not the posterior cerebral artery. Thus, each internal carotid artery perfuses the ipsilateral eye and anterior and middle portions of the ipsilateral cerebral hemisphere. The middle cerebral artery, a major branch of the internal carotid artery, supplies the deep and mid-section of the hemisphere. This region contains most of the motor cortex, sensory cortex, and, in the dominant hemisphere, the perisylvian language arc (see Fig. 8-1). Each anterior cerebral artery, another branch of the internal carotid artery, supplies the frontal lobe, including the medial surface of the motor cortex, which contains the motor innervation for the leg. The posterior cerebral artery (see Fig. 11-2), terminal branches of the basilar artery, supplies the occipital lobe, which houses the visual cortex, and most of the temporal lobe. Bottom left, An arteriogram of the carotid artery and its branches prominently displays the bifurcation (black arrow), the typical “candelabra” of branches that comprise the middle cerebral artery, and a faint anterior cerebral artery that sweeps from anterior to posterior (white arrow). Bottom right, A magnification of the bifurcation reveals an extensive circumferential plaque constricting the internal carotid artery. The remaining blood flow appears as an “apple core.” The rough interior surface of the artery gives rise to retinal and cerebral emboli.
Sometimes a TIA causes only brief visual loss in one eye, i.e., monocular blindness. Neurologists call this distinctive symptom amaurosis fugax (Greek, fleeting darkness). The underlying mechanism consists of emboli from the internal carotid artery flying into the ophthalmic artery, the carotid artery’s first branch, to induce several minutes of ischemia in the retina and optic nerve (Box 11-1). Unlike migraines, which can also cause transient visual loss, TIAs rarely cause headache or scintillations.
In patients with mild cognitive impairment, TIAs may produce a brief but marked confusional state. A similar disturbance may occur after atherosclerosis slowly occludes one carotid artery, forcing the other carotid artery to supply both cerebral hemispheres through the circle of Willis (Fig. 11-2, top right). In this case, the cerebral blood supply is tenuous and emboli from the patent artery produce bilateral cerebral ischemia.
FIGURE 11-2 Top left, After ascending, encased in the cervical vertebrae, the two vertebral arteries enter the skull. They join to form the basilar artery at the base of the brain. Small, delicate branches of the basilar artery supply the brainstem and its contents. (The Roman numerals refer to cranial nerve nuclei.) Large branches, as if wrapping their arms around the brainstem, supply the cerebellum and posterior portion of the cerebrum (i.e., the occipital lobes and inferomedial portions of the temporal lobes). The posterior cerebral arteries are, for practical purposes, the terminal branches of the basilar artery. They supply the occipital cortex and the posterior, inferior aspect of the temporal lobes. Top right, Belying its name, the circle of Willis, the “great anastomoses,” is completely patent in only about 20% of people. Connections between the basilar and internal carotid arteries ideally form the circle, which should give off the anterior, middle, and posterior cerebral arteries. The circle should provide anastomoses between anterior–posterior and right–left cerebral circulations. Despite the advantages that the circle confers, junctions of the arteries are weak spots. Defects at the junctions may balloon outward to form berry aneurysms that rupture and produce subarachnoid hemorrhages. Bottom left, This axial magnetic resonance angiogram (MRA) shows the major cerebral arteries that form the circle of Willis and anterior and posterior circulations. Bottom right, This MRA highlights the major cerebral arteries and shows the anterior and posterior circulations. The anterior circulation consists of the middle cerebral arteries (MCA), anterior cerebral arteries (ACA), and the anterior communicating artery (∧), which joins the ACAs. The posterior circulation consists of the basilar (Bas.) artery and two vertebral (Vert.), posterior cerebral (PCA), and posterior communicating arteries (*), which connect the PCAs and the basilar artery.
Preventive Measures
For patients who cannot undergo carotid endarterectomy, an alternative is intravascular insertion of stents. These devices are essentially expandable tubes that neuroradiologists insert intravascularly into the carotid artery to widen atherosclerotic stenoses (see Fig. 20-28). They also trap underlying atheromatous debris against the inner surface of the arterial wall to reduce the likelihood of emboli. In contrast to the benefits of inserting stents into the extracranial carotid arteries, inserting stents into intracerebral arteries, such as the basilar or middle cerebral artery, carries too much risk to justify its use.
Basilar Artery TIAs
The vertebrobasilar system, basilar artery system, or simply the posterior circulation supplies the brainstem, cerebellum, and the posterior inferior portion of the cerebrum (the occipital and medial inferior portion of the temporal lobes [see Fig. 11-2]). Emboli-generating plaques tend to develop at both the origin of the vertebral arteries (in the chest) and their junction at the base of the brain.
Symptoms and signs of basilar artery TIAs, which usually result from patchy brainstem ischemia, differ greatly from those of carotid artery TIAs (Box 11-2). Typical basilar artery TIA symptoms include tingling around the mouth (circumoral paresthesias), dysarthria, nystagmus, diplopia, ataxia, and vertigo. On rare occasions, when all blood flow through the basilar artery momentarily stops, almost the entire brainstem suffers from ischemia. The brainstem ischemia interrupts consciousness and body tone, which causes patients to collapse. This TIA, termed a drop attack, strikes suddenly and unexpectedly. (It appears similar to cataplexy [see Chapter 17].)
Transient Global Amnesia
TIAs sometimes impair the circulation of the basilar artery’s terminal branches, the posterior cerebral arteries, which supply the temporal lobes (see Fig. 11-2). Because the temporal lobes contain portions of the limbic system, particularly the hippocampi (see Fig. 16-5), posterior circulation TIAs may induce episodes of temporary amnesia and personality change – called transient global amnesia (TGA).
Even in the absence of a confirmatory laboratory test for TGA, its clinical features differentiate it from other neurologic conditions. TGA patients’ preserved intellect and general knowledge, as well as their remaining fully conscious, distinguishes them from patients in delirium. TGA patients, despite their amnesia, also do not confabulate in the manner of Wernicke–Korsakoff patients. Complex partial seizures differ by producing dulling of the sensorium, simple repetitive actions, paroxysmal or other epileptiform EEG changes, and a high rate of recurrence (see Chapter 10).
Strokes
Risk Factors
Reflecting the necessity of preventing strokes, epidemiologic studies constantly search for stroke risk factors (Box 11-3). Although physicians customarily list stroke risk factors as individual threats, the factors actually tend to cluster. For example, obesity, elevated low-density lipoprotein cholesterol, and hypertension frequently occur together in the metabolic syndrome. Some factors, by themselves, have a weak correlation with stroke, but pose a synergistic risk when paired with others. For example, cigarette smoking correlates with stroke; however, with concomitant use of oral contraceptives or in sufferers of migraine with aura, it becomes a moderately powerful risk factor. More strikingly, smokers who are hypertensive have a 20-fold greater risk. Curiously, total serum cholesterol carries a strong risk for myocardial infarctions but a weak risk for stroke.
Elevated serum concentration of homocysteine serves as a marker for an increased risk. In the autosomal recessive genetic condition, homocystinuria, children have a Marfan-like habitus, ocular lens displacement, and other anatomic abnormalities. More importantly, they routinely suffer strokes in childhood. Elevated homocysteine levels in adults are associated with the use of antiepileptic drugs and coronary artery and peripheral vascular disease, as well as stroke. Folic acid reduces elevated serum homocysteine concentrations (see Fig. 5-8), as well as reducing the incidence of fetal neural tube defects (see Chapter 13), but surprisingly, it does not reduce the incidence of strokes.
Thrombosis and Embolus
In addition to creating permanent clinical deficits, stroke scars are potentially epileptogenic. Approximately 50% of seizures that develop in adults older than 65 years originate from these lesions (see Chapter 10). Thus, patients with a history of having sustained a stroke who develop confusion, unresponsiveness, or abnormal behavior may have suffered a complex partial seizure as well as another stroke or TIA.
Necrosis
Infarctions in the Carotid Artery Distribution
Cerebral artery thrombosis and embolism produce an infarction in the distribution of the occluded artery. These strokes result in well-known clinical deficits (see Fig. 11-1 and Box 11-4):
• Anterior cerebral artery infarction damages the anterior and medial aspects of the frontal lobe. It typically causes paresis and apraxia of the contralateral leg. With bilateral anterior cerebral artery infarctions, the resulting frontal lobe damage, which is extensive, typically causes pseudobulbar palsy, apathy, mutism, personality changes, and urinary incontinence (see Chapter 7), along with both legs having paresis and apraxia.
• Middle cerebral artery infarction, which is the most common stroke, usually results in contralateral hemiparesis, hemisensory loss, and either aphasia with dominant hemisphere lesions or hemi-inattention, including neglect, with nondominant hemisphere strokes.
Infarctions in the Basilar Artery Distribution
Infarctions in the basilar artery distribution cause brainstem, cerebellar, or posterior cerebral injuries. Small brainstem infarctions usually cause constellations of cranial nerve injuries and hemiparesis, and large ones usually cause coma, if not immediate death. In contrast to cerebral-hemisphere infarctions, brainstem infarctions generally spare language or intellectual function and do not cause seizures (see Box 11-4).
The posterior cerebral arteries, as previously noted, are actually terminal branches of the basilar artery. Infarction of a posterior cerebral artery causes a contralateral homonymous hemianopsia and occasionally alexia without agraphia (see Chapter 8). Bilateral posterior cerebral artery strokes can cause (cortical) blindness, which sometimes leads to Anton syndrome (see Chapter 12).
Neurologists love to localize small brainstem infarctions for both clinical and academic reasons. Lesions of the midbrain cause ipsilateral oculomotor nerve and contralateral paresis (see Fig. 4-9). Those in the pons cause ipsilateral abducens nerve and contralateral paresis (see Fig. 4-11). Infarctions involving the midline of the midbrain or pons cause the medial longitudinal fasciculus syndrome (see Chapters 12 and 15). Finally, lateral medullary infarctions, which are the most common brainstem infarction, cause a complex, apparently disparate, combination of ipsilateral limb ataxia, palatal paresis, Horner syndrome, and alternating hypalgesia (see Wallenberg syndrome, Fig. 2-10).
Hemorrhages
Hemorrhages, which are most often the result of hypertension, usually erupt in the basal ganglia, thalamus, pons, or cerebellum (see Figs 20-13 and 20-14). Trauma and use of cocaine also cause hemorrhages, but their location is not as predictable as with hypertension-induced ones. In a classic problem, patients who take a monamine oxidase inhibitor (MAOI) antidepressant and then, inadvertently or in a suicide attempt, consume certain foodstuffs, notoriously aged cheese or red wine, or receive meperidine (Demerol), develop cerebral hemorrhages. These combinations, which are sympathomimetic, lead to a burst of hypertension sufficient to cause the hemorrhage. (Notably, MAOI-A antidepressants may cause this problem, but MAOI-B medications, such as those used to treat Parkinson disease, generally do not [see Chapter 9].)
Subarachnoid hemorrhage (SAH), although usually traumatic in origin, sometimes results from a ruptured berry aneurysm. SAH most often produces a prostrating headache that patients classically describe as the “worst headache” of their life. SAH also produces nuchal rigidity and lethargy, but usually no lateralized signs. Exercising, straining at stool, and other usually benign activities can rupture an aneurysm and precipitate an SAH. A diagnostic dilemma occurs when a sudden, profound headache interrupts sex. In this case, the exertion may have led to an SAH, but more often coital cephalalgia is responsible (see Chapter 9). Thus, TGA, SAH, and a migraine variant are hazards of sexual activity.
Neuropsychologic Sequalae
Several large or many small (lacunar) strokes cause vascular cognitive impairment, previously labeled vascular dementia, and before that multi-infarct dementia. The preliminary version of the DSM-5 labels it Vascular Neurocognitive Disorder. Ironically, despite the implication of these terms, the dementia stems from the neuropathologic changes of Alzheimer disease that accompany those of cerebral vascular disease (see Chapter 7).
Occlusion of a cerebral artery typically destroys a discrete critical area of the cerebral cortex. Commonly occurring strokes produce well-known neuropsychologic syndromes, such as aphasia, Gerstmann syndrome, and hemi-inattention (see Chapter 8). Strokes of both frontal lobes, which may occur either simultaneously or in succession, cause pseudobulbar palsy and frontal lobe dysfunction (see Chapters 4 and 8).
In another mechanism, hypoxia, hypotension, carbon monoxide poisoning, or similar catastrophes cause generalized cerebral anoxia. This insult leads to dementia, cortical blindness, a persistent vegetative state (PVS: see later), and postanoxic myoclonus (see Chapter 18). A variation of generalized anoxia occurs when cerebral anoxia affects only the fine, terminal branches of cerebral arteries that perfuse the extensive circumferential regions of the cerebral cortex, which neurologists call the watershed or borderzone area. An episode of cerebral anoxia that reduces the oxygen supply below a critical point in the watershed area produces a watershed infarction
[/not-level-membership-for-neurology-category]
