Thrombocytopenia
Thrombocytopenia can be simply defined as a blood platelet count of below 150 × 109/L. With the routine measurement of platelet number by automated cell counters it is a relatively common laboratory finding. Before initiating further investigations it is important to confirm that a low platelet count is genuine by careful inspection of the blood sample and film. Either a small clot in the sample or platelet clumping (Fig 34.1) can cause artefactual thrombocytopenia.
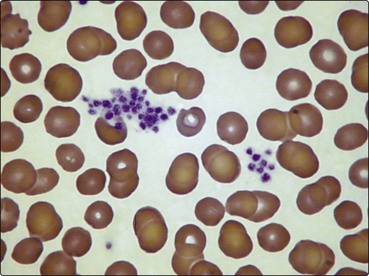
Fig 34.1 Blood film showing clumping of platelets.
This phenomenon causes an artefactual thrombocytopenia in the automated blood count.
Causes
Major causes of thrombocytopenia are listed in Table 34.1. Many of the diseases and syndromes are discussed elsewhere.
Table 34.1
| Pathogenesis | Disease examples |
| Failure of production | Leukaemia, myelodysplasia, aplastic anaemia, megaloblastic anaemia, myelofibrosis, malignant infiltration, infection, drugs1 |
| Shortened lifespan | |
| Immune | ITP, drugs1, connective tissue disorders, antiphospholipid antibody syndrome, infection, post-transfusion purpura, neonatal alloimmune thrombocytopenia |
| Non-immune | DIC, thrombotic thrombocytopenic purpura |
| Sequestration | Hypersplenism, cardiopulmonary bypass surgery |
| Dilution | Massive blood transfusion |
ITP, immune thrombocytopenia; DIC, disseminated intravascular coagulation.
In general terms there are four possible processes leading to thrombocytopenia:
 Failure of marrow production. The bone marrow failure of haematological disease (e.g. aplastic anaemia, leukaemia) usually causes pancytopenia. However, thrombocytopenia may be the only sign of intrinsic marrow disease or marrow suppression associated with infection or chemotherapy.
Failure of marrow production. The bone marrow failure of haematological disease (e.g. aplastic anaemia, leukaemia) usually causes pancytopenia. However, thrombocytopenia may be the only sign of intrinsic marrow disease or marrow suppression associated with infection or chemotherapy.


 Dilution. Normal platelets are diluted by massive blood transfusion.
Dilution. Normal platelets are diluted by massive blood transfusion.
Clinical presentation
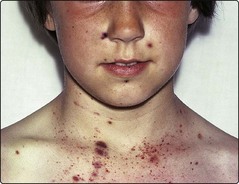
Patients with thrombocytopenia are particularly prone to bleeding from mucous membranes. It should be emphasised that spontaneous bleeding is usually only seen with platelet counts of less than 10–20 × 109/L, although patients with associated platelet dysfunction may bleed at higher counts. Conjunctival haemorrhage, nose and gum bleeding and menorrhagia are all relatively common, with haematuria and melaena less frequent. Intracranial bleeding is of serious import but, thankfully, is rare. Possible examination findings include purpura and more extensive petechial haemorrhages involving the skin and mucous membranes (Fig 34.2). The retina should be routinely inspected for haemorrhages.
Clinical syndromes
ITP is a disease characterised by immune thrombocytopenia mediated by platelet antibodies that accelerate platelet destruction and inhibit their production. It is a heterogeneous disorder but it is conventional to divide it into two discrete entities: acute ITP and chronic ITP (Table 34.2). This division is convenient for discussion of pathogenesis and apt for most patients, but in ‘real life’ there is overlap between the two syndromes.
Table 34.2
Comparison of classic acute and chronic ITP
| Characteristic | Acute ITP | Chronic ITP |
| Age | Childhood | Adult life |
| Previous viral infection | Frequent | Unusual |
| Platelet count (µ 109/L) | Often <20 | Variable |
| Onset | Sudden | Insidious |
| Duration | Few weeks | Years/lifelong |
| Spontaneous remission | Around 90% | Rare |
Chronic ITP
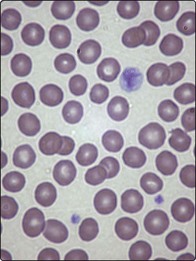
The blood film confirms thrombocytopenia; often the platelets are increased in size (Fig 34.3). There is no single specific test for ITP. A bone marrow aspirate and trephine biopsy will show increased megakaryocytes but it is often not necessary if other features are typical. Further investigations are designed to exclude other causes of isolated thrombocytopenia such as connective tissue disorders and antiphospholipid antibody syndrome. Apparent ‘primary’ ITP may be secondary to subclinical viral infections such as hepatitis C, cytomegalovirus, HIV and Helicobacter pylori. In younger patients congenital thrombocytopenias may be confused with ITP. A thorough drug history is essential.
Drug-induced thrombocytopenia
Many drugs have been linked with isolated thrombocytopenia (Table 34.3). The mechanism is usually the formation of antiplatelet antibodies. General management is withdrawal of the offending drug and platelet transfusion for significant bleeding.
Table 34.3
Some drugs associated with thrombocytopenia
| Heparin | Penicillin |
| Quinine/quinidine | Diazepam |
| Gold salts | Tolbutamide |
| Sulphonamides | Aspirin |
| Thiazides | Cephalosporins |
| Rifampicin | Ranitidine |




