The thalassaemias
Classification
The classification illustrated in Table 16.1 is based on the mode of inheritance of thalassaemia.
Table 16.1
Classification of thalassaemia
| Type of thalassaemia | Heterozygote | Homozygote |
| α-Thalassaemia1 | ||
| α0 (– –/) | Thal. minor | Hydrops fetalis |
| α+ (–α/) | Thal. minor | Thal. minor |
| β-Thalassaemia | ||
| β0 | Thal. minor | Thal. major |
| β+ | Thal. minor | Thal. major or intermedia |
Clinical syndromes
β-Thalassaemias
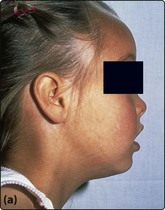
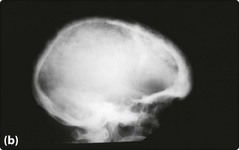
The characteristic severe anaemia (Hb less than 70 g/L) is caused by α-chain excess leading to ineffective erythropoiesis and haemolysis. Anaemia first becomes apparent at 3–6 months when production of HbF declines. The child fails to thrive and develops hepatosplenomegaly. Compensatory expansion of the marrow space causes the typical facies with skull bossing and maxillary enlargement (Fig 16.1a). The ‘hair-on-end’ radiological appearance of the skull (Fig 16.1b) is due to expansion of bone marrow into cortical bone. If left untreated further complications can include repeated infections, bone fractures and leg ulcers. Red cell membrane abnormalities contribute to hypercoagulability.
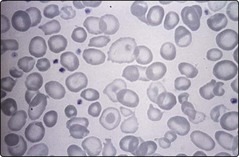
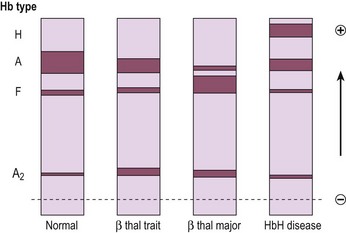
Laboratory testing should precede blood transfusion. There is a severe hypochromic microcytic anaemia with a characteristic blood film (Fig 16.2) and Hb electrophoresis demonstrates absence or near absence of HbA with small amounts of HbA2 and the remainder HbF (Fig 16.3).
Thalassaemia intermedia
Thalassaemia intermedia is a clinical syndrome which may result from a variety of genetic abnormalities (Table 16.2). The clinical features are less severe than in β-thalassaemia major as the α/β-globin chain imbalance is less pronounced. Patients usually present later than is the case for β-thalassaemia major (often at 2–4 years), and have relatively high haemoglobin levels (80–100 g/L), moderate bone changes and normal growth. Transfusion may be required but requirements are less than in β-thalassaemia major.





Prenatal diagnosis
The thalassaemias





