Chapter 11 The nervous system
The neurological history
The neurological history begins in detail with the presenting problem or problems (Table 11.1). The patient should be allowed to describe the symptoms in his or her own words to begin with, and then the clinician needs to ask questions to clarify information and obtain more detail. It is particularly important to ascertain the temporal course of the illness, as this may give important information about the underlying aetiology.
| Presenting symptoms* |
* Note particularly the temporal course of the illness, whether symptoms suggest focal or diffuse disease, and the likely level of involvement of the nervous system.
A stroke or cerebrovascular accident usually causes symptoms which appear over minutes or are present when the patient wakes from sleep. There is a focal problem with function of the brain. Patients may be unable to move one side of the body (hemiplegia) or have difficulty with speech or swallowing, There may have been previous episodes. When there is resolution of the symptoms within 24 hours the episode is called a transient ischaemic attack—TIA). The rapid onset of focal symptoms almost always has a vascular cause—embolism, infarction or haemorrhage. If the patient can answer questions it is important to ask about the onset of the symptoms and about risk factors for stroke (Questions box 11.1).
Questions box 11.1
The sudden onset of weakness on one side of the body followed by resolution and a severe headache is characteristic of hemiplegic migraine. Sudden resolution without headache suggests a transient ischaemic episode. The very gradual onset of muscle weakness suggests a muscle abnormality such as myopathy rather than a vascular event.
A subacute onset (hours to days) occurs with inflammatory disorders (e.g. meningitis, cerebral abscess or the Guillain-Barréa syndrome—acute inflammatory polyradiculoneuropathy).
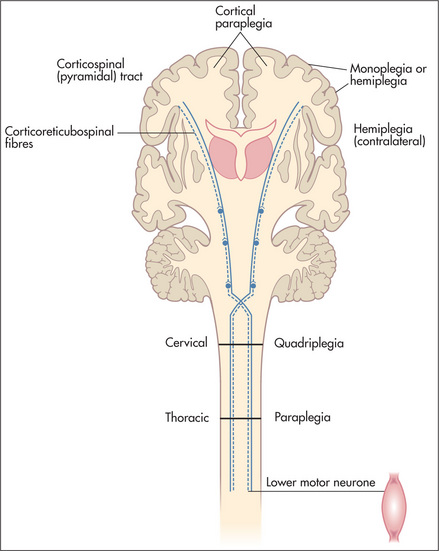
Based on the history (and physical examination), a judgment is made as to whether the disease process is localised or diffuse, and which levels of the nervous system are involved (the nervous system may be thought of as having four different levels: the peripheral nervous system, the spinal cord, the posterior fossa, and the cerebral hemispheres). Consideration of the time course and the levels of involvement will usually lead to a logical differential diagnosis of the patient’s symptoms. After detailed questions about the presenting problem, ask about previous neurological symptoms and about previous neurological diagnoses or investigations. The patient may know the results of CT or magnetic resonance imaging brain scans performed in the past. A thorough neurological history will include routine questions about possible neurological symptoms (Questions box 11.2). If the patient answers ‘yes’ to any of these, more-detailed questions about the nature of the problem and its time course are indicated.
Headache and facial pain
Headache is a very common symptom (Questions box 11.3). It is important, as with any type of pain, to determine the character, severity, site, duration, frequency, radiation, aggravating and relieving factors and associated symptoms.1,2 Unilateral headache that is preceded by flashing lights or zigzag lines and is associated with light hurting the eyes (photophobia) is likely to be a migraine with an aura (‘classical migraine’); common migraine has no aura. Pain over one eye (or over the temple) lasting for minutes to hours, associated with lacrimation, rhinorrhoea and flushing of the forehead, and occurring in bouts that last several weeks a few times a year or less, is suggestive of cluster headache. This occurs predominantly in males and patients can’t stay still. Headache over the occiput and associated with neck stiffness may be from cervical spondylosis. Coital headache occurs during intercourse close to orgasm.
Questions box 11.3
A generalised headache that is worse in the morning and is associated with drowsiness or vomiting may reflect raised intracranial pressure, while generalised headache associated with photophobia and fever as well as with a stiff neck of more gradual onset may be due to meningitis. A persistent unilateral headache over the temporal area associated with tenderness over the temporal artery and blurring of vision suggests temporal arteritis.3,4 This condition (Table 11.2) is often associated with jaw claudication, or jaw pain during eating, which can lead to considerable loss of weight. Headache with pain or fullness behind the eyes or over the cheeks or forehead occurs in acute sinusitis. The dramatic and usually instantaneous onset of severe headache that is initially localised but becomes generalised and is associated with neck stiffness may be due to a subarachnoid haemorrhage. Morning headaches worse with coughing, especially in an obese patient, may be due to idiopathic intracranial hypertension; visual loss may occur.
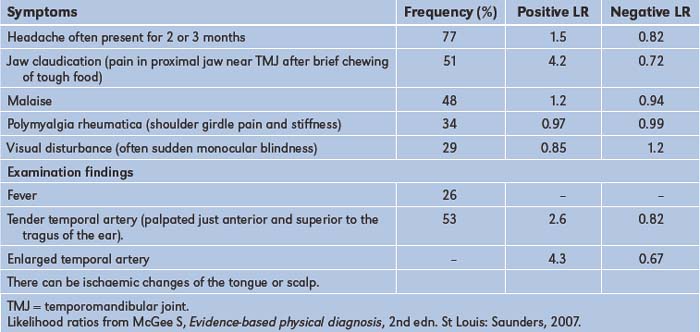
Finally, the most frequent type of headache is episodic or chronic tension-type headache; this is commonly bilateral, occurs over the frontal, occipital or temporal areas, and may be described as a sensation of tightness that lasts for hours and recurs often. There are usually no associated symptoms such as nausea, vomiting, weakness or paraesthesiae (tingling in the limbs), and the headache does not usually wake the patient at night from sleep.
Faints and fits (see also page 41)
It is important to try to differentiate syncope (transient loss of consciousness) from epilepsy (Questions box 11.4). However, primary syncopal events can cause a few clonic jerks in a significant number of cases. Generalised tonic-clonic seizures (grand mal epilepsy) cause abrupt loss of consciousness, which may be preceded by an aura. Often the patient is incontinent of urine and faeces, and the tongue may be bitten. A witness may be able to describe the type of attack that occurred. It is important to try to determine whether any seizure is generalised or localised to one side of the body: a seizure affecting part of the body may indicate a focal lesion in the central nervous system, such as a tumour or abscess. If consciousness is impaired, these partial seizures are described as ‘complex’; if consciousness is unimpaired they are termed ‘simple’. Idiopathic absence seizures (‘petit mal’) occur in children. These are frequent brief episodes of loss of awareness often associated with staring. Major motor movements do not occur with this type of epilepsy.
Questions box 11.4
Questions to ask the patient with syncope or dizziness
Transient ischaemic attacks (TIAs) affecting the brainstem can occasionally cause blackouts. Use of the term ‘drop attacks’ means the patient falls but there is no loss of consciousness. In either case the patient falls to the ground without premonition and the attacks are of brief duration. Hypoglycaemia can lead to episodes of loss of consciousness. Patients with hypoglycaemia may also report sweating, weakness and confusion before losing consciousness. Bizarre attacks of loss of consciousness occur with hysteria.b During such attacks the patient may slump to the ground without sustaining any injury and there may be apparent fluctuations in the level of consciousness for a prolonged period.
Dizziness
If a patient complains of dizziness, it is important to determine what is meant by this term. In true vertigo, there is actually a sense of motion, usually of the surroundings but also of the head itself (page 41).5 When vertigo is severe it may not be possible for the patient to stand or walk, and associated symptoms of nausea, vomiting, pallor, sweating and headache may be present. Causes of vertigo include the ‘peripheral vestibular lesions’:
Other causes of vertigo include:
Visual disturbances and deafness
Problems with vision can include double vision (diplopia), blurred vision (amblyopia), light intolerance (photophobia) and visual loss. The causes of deafness are summarised on page 348.
Disturbances of gait
Many neurological conditions can make walking difficult. These are described on page 376. Walking may also be abnormal when orthopaedic disease affects the lower limbs or spine. A bizarrely abnormal gait can sometimes be a sign of a hysterical reaction.
Disturbed sensation or weakness in the limbs
Pins and needles in the hands or feet may indicate nerve entrapment or a peripheral neuropathy (page 386) but can result from sensory pathway involvement at any level. The carpal tunnel syndrome is common; here there is median nerve entrapment, and patients experience pain and paraesthesiae in the hand and wrist. Sometimes pain may extend to the arm and even to the shoulder, but paraesthesiae are felt only in the fingers. These symptoms are usually worse at night and may be relieved by dangling the arm over the side of the bed or shaking the hand.
Tremor and involuntary movements
Tremor is a rhythmical movement (Table 11.3). A slow tremor has, by definition, a rate between 3 Hz and 5 Hz. Rapid tremors are faster than 10 Hz. Resting tremors are present mostly during relaxation of the muscles, while intention tremors occur with deliberate movement and become more pronounced towards the end of the action. Tremors become worse with fatigue or anxiety. Shivering is a type of tremor brought on by cold. It is normal for there to be a fine tremor associated with holding a posture or performing a movement slowly. This is called a physiological tremor. It becomes more obvious with fright and fatigue. It is often increased by the beta-agonist drugs used to treat asthma or by caffeine. Thyrotoxicosis is a cause of exaggeration of physiological tremor. These movements are very fine and may be difficult to see unless looked for specifically. Benign essential (familial) tremor is an inherited disorder which causes tremor, but no other signs. The tremor is most easily seen when the patient’s arms are stretched out; it can become worse during voluntary movements. It usually disappears when the muscles are at complete rest. Parkinson’s diseased may present with a resting tremor (page 397). Intention (or target-seeking) tremor is due to cerebellar disease (page 398). Chorea involves involuntary jerky movements (page 399). Definitions of the terms used to describe movement disorders are shown in Table 11.4.
| Parkinson’s disease | 3 to 5 Hz |
| Essential/familial | 4 to 7 Hz |
| Physiological | 8 to 13 Hz |
TABLE 11.4 Definitions of terms used to describe movement disorders
| Akithesia | Motor restlessness; constant semi-purposeful movements of the arms and legs |
| Asterixis | Sudden loss of muscle tone during sustained contraction of an outstretched limb |
| Athetosis | Writhing, slow sinuous movements, especially of the hands and wrists |
| Chorea | Jerky small rapid movements, often disguised by the patient with a purposeful final movement: e.g. the jerky upward arm movement is transformed into a voluntary movement to scratch the head |
| Dyskinesia | Purposeless and continuous movements, often of the face and mouth; often a result of treatment with major tranquillisers for psychotic illness |
| Dystonia | Sustained contractions of groups of agonist and antagonist muscles, usually in flexion or extremes of extension; it results in bizarre postures |
| Hemiballismus | An exaggerated form of chorea involving one side of the body: there are wild flinging movements which can injure the patient (or bystanders) |
| Myoclonic jerk | A brief muscle contraction which causes a sudden purposeless jerking of a limb |
| Myokymia | A repeated contraction of a small muscle group; often involves the orbicularis oculi muscles |
| Tic | A repetitive irresistible movement which is purposeful or semi-purposeful |
| Tremor | A rhythmical alternating movement |
Speech and mental status
Speech may be disturbed by many different neurological diseases and is discussed on page 377. A number of different diseases can also result in delirium or dementia, as described on page 377 and in Chapter 12.
Past health
Inquire about a past history of meningitis or encephalitis, head or spinal injuries, a history of epilepsy or convulsions and any previous operations. Any past history of sexually transmitted disease (e.g. risk factors for HIV infection or syphilis) should be obtained. Ask about risk factors that may predispose to the development of cerebrovascular disease (Table 11.1). A previous diagnosis of peripheral vascular disease or of coronary artery disease indicates an increased risk of cerebrovascular disease. Chronic or paroxysmal atrial fibrillation is associated with a greatly increased risk of embolic stroke, especially for people over the age of 70.
Medication history
Previous and current medications may be the cause of certain neurological or apparently neurological syndromes (Table 11.5).
| 1 Anti-hypertensives |
Social history
As smoking predisposes to cerebrovascular disease, the smoking history is relevant. It is useful to ask about occupation and exposure to toxins (e.g. heavy metals). Alcohol can also result in a number of neurological diseases (see Table 1.3, page 7).
Family history
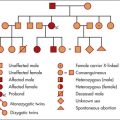
Any history of neurological or mental disease should be documented. A number of important neurological conditions are inherited (Table 11.6).
| X-linked | Colour blindness, Duchenne’s and Becker’s muscular dystrophy, Leber’s* optic atrophy |
| Autosomal dominant | Huntington’s chorea, tuberose sclerosis, dystrophia myotonica |
| Autosomal recessive | Wilson’s disease, Refsum’s† disease, Freiderich’s ataxia, Tay-Sachs’‡ disease |
| Increased incidence in families | Alzheimer’s§ disease |
* Theodor Karl von Leber (1840–1917). German ophthalmologist, professor of ophthalmology at Heidelberg. He began studying chemistry but was advised by Bunsen that there were too many chemists and so changed to studying medicine.
† Siguald Refsum (1907–91), Norwegian neurologist. He described this in 1945, calling it heredotaxia hemerlopica polyneuritiformis. This may be an argument for the use of eponymous names.
‡ Warren Tay (1843–1927), English ophthalmologist, described the ophthalmological abnormalities of the condition. Bernard Sachs (1858–1944), American neurologist and psychiatrist, described the neurological features in 1187. He studied in Germany and was a pupil of von Recklinghausen. The condition was originally called amaurotic family idiocy. This may be another reason to use eponymous names.
§ Alois Alzheimer (1864–1915), Bavarian neuropathologist, described the condition in 1906. His doctoral thesis was on the wax-producing glands of the ear.
The neurological examination
Examination anatomy
More than for any other system of the body, neurological diagnosis depends on localising the anatomical site of the lesion—in the brain, spinal cord or peripheral nerve. Figure 11.1 shows the gross anatomy of the brain and the major functional areas.
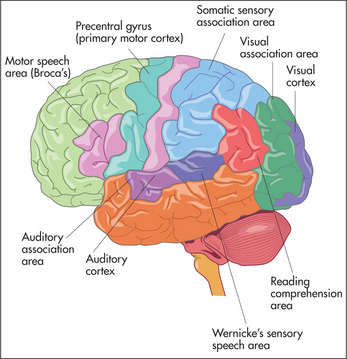
In brief, the following aspects of the examination must be attended to:
General signs
Consciousness
Note the level of consciousness. If the patient is unconscious look for responses to various stimuli (page 401).
Neck stiffness
Any patient with an acute neurological illness, or who is febrile or has altered mental status must be assessed for signs of meningism.6
With the patient lying flat in bed, the examiner slips a hand under the occiput and gently flexes the neck passively (i.e. without assistance from the patient). The chin is brought up to approach the chest wall. Meningism may be caused by pyogenic or other infection of the meninges, or by blood in the subarachnoid space secondary to subarachnoid haemorrhage. There is resistance to neck flexion due to painful spasm of the extensor muscles of the neck. Other causes of resistance to neck flexion are characterised by an equal resistance to head rotation. They include: (i) cervical spondylosis; (ii) after cervical fusion; (iii) Parkinson’s disease; and (iv) raised intracranial pressure, especially if there is impending tonsillar herniation. The Brudzinski signe is spontaneous flexion of the hips during flexion of the neck by the examiner and indicates meningism.
Kernig’s signf should also be elicited if meningitis is suspected. Flex each hip in turn, then attempt to straighten the knee while keeping the hip flexed. This is greatly limited by spasm of the hamstrings (which in turn causes pain) when there is meningism due to an inflammatory exudate around the lumbar spinal roots.
Although the diagnostic value has been questioned (combined meningeal signs had a positive LR of 0.92 and a negative LR of 0.88),6 we have found these signs useful clinically (and they have excellent specificity).
Orientation
Test orientation in person, place and time by asking the patient his or her name, present location and the date (normal patients who have been in hospital for long periods often get the day wrong as one day seems very much like another in hospital). Disorientation is not a specific localising sign and may be acute and reversible (delirium) or chronic and irreversible (dementia). The mini-mental state examination (Table 12.7, page 420) is a useful way to document the progress of a confusional state or dementia over time.
The cranial nervesg
Examination anatomy
The cranial nerves (Figure 11.2) arise as direct extensions of the brain (I and II) or from the brainstem (midbrain, pons and medulla)—Figures 11.10 (page 337), 11.13 (page 340) and 11.14 (page 341).
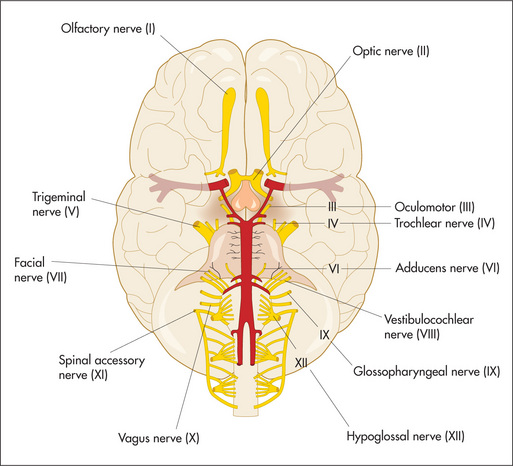
If possible, position the patient so that he or she is sitting over the edge of the bed. Look at the head, face and neck. If hydrocephalus has occurred in infancy—before closure of the cranial sutures—the head and face may resemble an inverted triangle. Acromegaly (page 307), Paget’s disease (page 320) or basilar invagination (page 321) may be obvious. A careful general inspection may reveal signs easily missed when each cranial nerve is examined separately. This is particularly true of ptosis (page 337), proptosis (page 303), pupillary inequality (page 336), skew deviation of the eyes and facial asymmetry. Inspect the whole scalp for craniotomy scars and the skin for neurofibromas (Figure 11.3). Look for skin lesions: for example, a capillary or cavernous haemangioma is seen on the face in the distribution of the trigeminal (V) nerve in the Sturge-Weber syndrome.h It is associated with an intracranial venous haemangioma of the leptomeninges and with seizures.
The cranial nerves are usually tested in approximately the order of their number.7
The first (olfactory) nervei
Examination of the nose and sense of smell
The first nerve is not tested routinely. If the patient complains of loss of smell (anosmia) or there are other signs suggesting a frontal or temporal lobe lesion, then it should be examined. Anosmic patients sometimes complain of loss of taste rather than of smell because the sense of smell plays a large part in the appreciation of taste. Test each nostril separately with a series of bottles containing essences of familiar smells, such as coffee, vanilla and peppermint (this is traditional, but not very reliable). Pungent substances such as ammonia should not be used, first because they upset the patient and second because noxious stimuli of this sort are detected by sensory fibres of the fifth (trigeminal) nerve. An easy way to test smell is to use the isopropyl alcohol wipes present in most hospital clinics. These have a distinctive and non-pungent smell.
Causes of anosmia
Most cases of anosmia are bilateral. Causes include: (i) upper respiratory tract infection (commonest); (ii) smoking and increasing age; (iii) ethmoid tumours; (iv) basal skull fracture or frontal fracture, or after pituitary surgery; (v) congenital—for example, Kallmann’s syndrome (hypogonadotrophic hypogonadism); (vi) meningioma of the olfactory groove; and (vii) following meningitis. The main unilateral causes are head trauma without a fracture, or an early meningioma of the olfactory groove.j
The second (optic) nerve
Examination anatomy
The optic nerve is not really a nerve but an extension of fibres of the central nervous system that unites the retinas with the brain. It is purely sensory, contains about a million fibres and extends for about 5 cm (Figure 11.4), passing through the optic foramen close to the ophthalmic artery and joining the nerve from the other side at the base of the brain to form the optic chiasm. The spatial orientation of fibres from different parts of the fundus is preserved so that fibres from the lower part of the retina are found in the inferior part of the chiasm, and vice versa. Fibres from the temporal visual fields (the nasal halves of the retinas) cross in the chiasm, whereas those from the nasal visual fields do not. Fibres for the light reflex from the optic chiasm finish in the superior colliculus, whence connections occur with both third nerve nuclei. The remainder of the fibres leaving the chiasm are concerned with vision, and travel in the optic tract to the lateral geniculate body. From here the fibres form the optic radiation and pass through the posterior part of the internal capsule, finishing in the visual cortex of the occipital lobe. In their course they splay out so that fibres serving the lower quadrants course through the parietal lobe, while those for the upper quadrants traverse the temporal lobe. The result of the decussation of fibres in the optic chiasm is that fibres from the left visual field terminate in the right occipital lobe, and vice versa.
Examination
Assess visual acuity, visual fields and the fundi.
Visual acuity is tested with the patient wearing his or her spectacles, if used for reading or driving, as refractive errors are not considered to be cranial nerve abnormalities. Use a hand-held eye chart or a Snellen’s chartk on the wall. Each eye is tested separately, while the other is covered by a small card.
Formal testing with a standard Snellen’s chart requires the patient to be 6 metres from the chart. Unless a very large room is available, this is done using a mirror. Normal visual acuity is present when the line marked 6 can be read correctly with each eye (6/6 acuity). If poor visual acuity improves when the patient is asked to read the chart through a pin-hole, refractive error is likely to be the cause. A patient who is unable to read even the largest letter of the chart should be asked to count fingers held up in front of each eye in turn, and if this is not possible, then perception of hand movement is tested. Failing this, light perception only may be present.
Visual fields are examined by confrontation (Figure 11.5). Always remove a patient’s spectacles first. The examiner’s head should be level with the patient’s head. Use a white- or red-tipped hat pin or pen. Test each eye separately. The examiner holds the pin at arm’s length with the coloured head upwards. It should be positioned halfway between the patient and the examiner, and brought in from just outside the examiner’s peripheral vision until the patient can see it. Make sure the patient is staring directly at the examiner’s eye and explain that he or she is looking for the first sight of the pin out of the corner of the eye. When the right eye is being tested the patient should look straight into the examiner’s left eye. The patient’s head should be at arm’s length and the eye not being tested should be covered. The pin should be brought into the visual field from the four main directions, diagonally towards the centre of the field of vision.

If a patient has such poor acuity that a pin is difficult to see, the fields should be mapped with the fingers. The examiner’s fingers can also be used to perform a quick screening test of the visual fields. Usually two fingers are held up and brought into the centre of vision in the four quadrants. The examiner wriggles the fingers and asks the patient to say ‘yes’ when movement of the fingers is first seen. The following patterns of visual field loss may be detected (Figures 11.6 and 11.7):
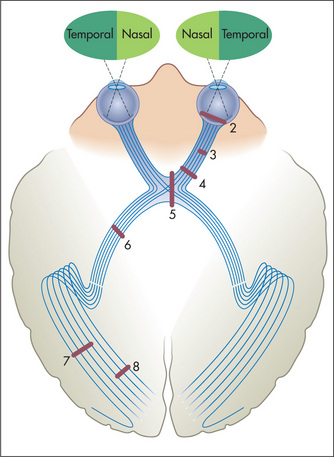
Figure 11.6 The visual fields and optic pathways Numbers indicate sites of lesions producing field defects shown in Figure 11.7.
Adapted from Snell RS, Westmoreland BF, Clinical neuroanatomy for medical students, 4th edn. Boston: Little Brown, 1997.

Figure 11.7 Visual field defects with lesions at various levels along the optic pathway, at sites indicated in Figure 11.6
Adapted from Bickerstaff ER, Spillane JA. Neurological examination in clinical practice, 5th edn. Oxford: Blackwell, 1989.
The presence of an abnormality has diagnostic value (positive LRs 4.2 to 6.8),8 but absence is largely unhelpful.
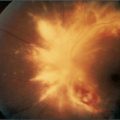
The margins of the disc must be examined with care. The disc itself is usually a shallow cup with a clearly outlined rim. Loss of the normal depression of the optic disc will cause blurring at the margins and is called papilloedema (Figure 11.8a). It indicates raised intracranial pressure. If papilloedema is suspected, the retinal veins should be examined for spontaneous pulsations. When these are present raised intracranial pressure is excluded, but their absence does not prove the pressure is raised.9 If the appearance of papilloedema is associated with demyelination in the anterior part of the optic nerve, it is called papillitis (Table 13.3, page 428). These two can be distinguished because papillitis causes visual loss but papilloedema does not.
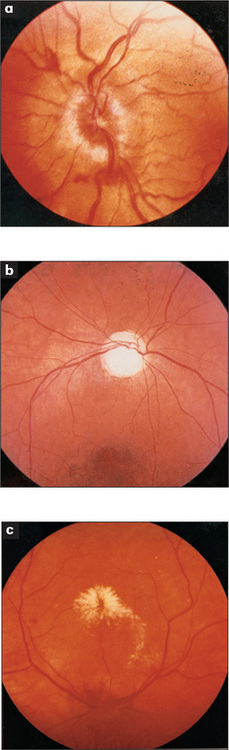
Next note the colour of the optic disc. Normally it is a rich yellow colour in contrast to the rest of the fundus which is a rich red colour. The fundus may be pigmented in some diseases and in patients with pigmented skin. When the optic disc has a pale insipid white colour, optic atrophy is usually present (Figure 11.8b).
Each of the four quadrants of the retina should be examined systematically for abnormalities. Look especially for diabetic and hypertensive changes (Figure 11.8c). Note haemorrhages or exudates.
The third (oculomotor), fourth (trochlear) and sixth (abducens) nerves—the ocular nerves
Examination anatomy
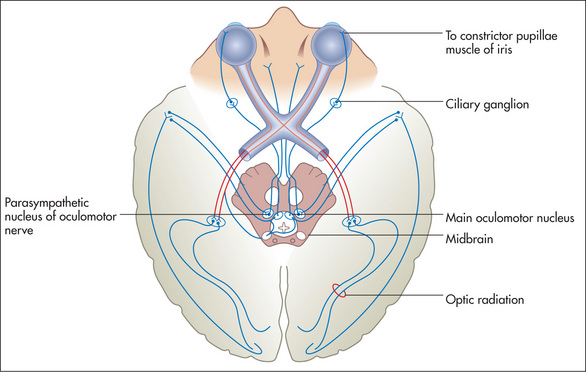
The size of the pupils depends on a balance of parasympathetic and sympathetic innervation. The parasympathetic innervation to the eyes is supplied by the Edinger-Westphal nucleusl of the third nerve (stimulation of these fibres causes pupillary constriction: miosis). The sympathetic innervation to the eye (stimulation causes pupillary dilatation: mydriasis) is as follows: fibres from the hypothalamus go to the ciliospinal centre in the spinal cord at C8, T1 and T2, synapse, and second-order neurones exit via the anterior ramus in the thoracic trunk and synapse in the superior cervical ganglion in the neck. Third-order neurones travel from here with the internal carotid artery to the eye. In addition, the pupillary reflexes (Figure 11.9) depend for their afferent limb on the optic nerve (Figure 11.4). Constriction of the pupil in response to light is relayed by the optic nerve and tract to the superior colliculus and then to the Edinger-Westphal nucleus of the third nerve in the midbrain. Efferent motor fibres from the oculomotor nucleus (Figure 11.10) travel in the wall of the cavernous sinus, where they are in association with the fourth, ophthalmic division of the fifth, and the sixth cranial nerves (see Figure 10.10, page 308). These nerves leave the skull together through the superior orbital fissure. The iridoconstrictor fibres terminate in the ciliary ganglion, whence postganglionic fibres arise to innervate the iris. The rest of the third nerve supplies all the ocular muscles except the superior oblique (fourth nerve) and the lateral rectus (sixth nerve) muscles. The third nerve also supplies the levator palpebrae superioris, which elevates the eyelid (Figure 11.11).
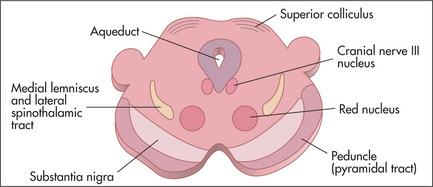
The pupils
With the patient looking at an object at an intermediate distance, examine the pupils for size, shape, equality and regularity. Slight differences in pupil size (up to 20%) may be normal.m
Move the torch in an arc from pupil to pupil. If an eye has optic atrophy or severely reduced visual acuity from another cause, the affected pupil will dilate paradoxically after a short time when the torch is moved from the normal eye to the abnormal eye. This is called an afferent pupillary defect (or the Marcus Gunn pupillary signn). It occurs because an eye with severely reduced acuity has reduced afferent impulses so that the light reflex is markedly decreased. When the light is shone from the normal eye to the abnormal one the pupil dilates, as reflex pupillary constriction in the abnormal eye is so reduced that relaxation after the consensual response dominates.
Now test accommodation. Ask the patient to look into the distance and then to focus his or her eyes on an object such as a finger or a white-tipped hat pin brought to a point about 30 cm in front of the nose. There is normally constriction of both pupils—the accommodation response. It depends on a pathway from the visual association cortex descending to the third nerve nucleus. Causes of an absent light reflex with an intact accommodation reflex include a midbrain lesion (e.g. the Argyll Robertson pupil of syphilis), a ciliary ganglion lesion (e.g. Adie’s pupilo) or Parinaud’s syndromep (page 340). Failure of accommodation alone may occur occasionally with a midbrain lesion or with cortical blindness.
Eye movements
Here failure of eye movement, double vision (diplopia) and nystagmus are assessed.
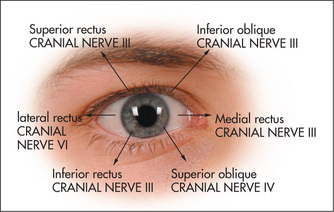
Ask the patient to look at the invaluable hat pin. (The presence of these pins in the lapel of a well-cut white coat or expensive suit often indicates that the wearer is a neurologist.) Assess voluntary eye movements in both eyes first. Ask the patient to look laterally right and left, then up and down (Figure 11.12). Remember the lateral rectus (sixth nerve) only moves the eyes horizontally outwards, while the medial rectus (third nerve) only moves the eyes horizontally inwards. The remainder of the muscle movements are a little more complicated. When the eye is abducted, the elevator is the superior rectus (third nerve), while the depressor is the inferior rectus (third nerve). When the eye is adducted, the elevator is the inferior oblique (third nerve) while the depressor is the superior oblique (fourth nerve) (Figure 11.11). The practical upshot of all this is that the testing of pure movement (that is, one muscle only) for elevation and depression is performed first with the eye adducted and then with it abducted. Therefore, ask the patient to follow the moving hat pin, held by the examiner 30–40 cm from the patient and moved in an H pattern, with both eyes and to say if double images are seen in any direction.
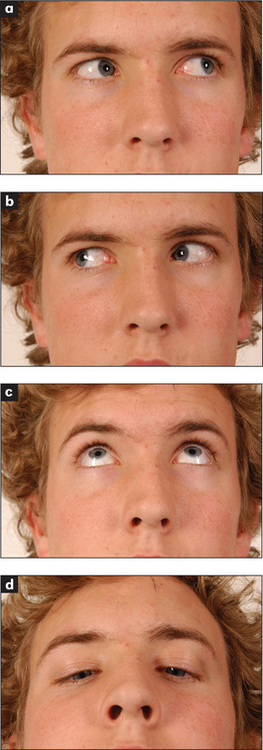
Abnormalities of conjugate gaze
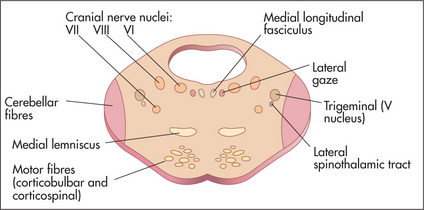
Normal eye movements occur in an organised fashion so that the visual axes remain in the same plane throughout. There are centres for conjugate gaze in the frontal lobe for saccadic movements and in the occipital lobe for pursuit movements. Conjugate movement to the right is controlled from the left side of the brain. From these centres fibres travel to the region of the sixth nerve nucleus, from which area the medial longitudinal fasciculus coordinates movement with the contralateral third nerve (medial rectus) nucleus (Figure 11.13). A brainstem lesion causes ipsilateral paralysis of horizontal conjugate gaze, and a frontal lobe lesion causes contralateral paralysis of horizontal conjugate gaze.
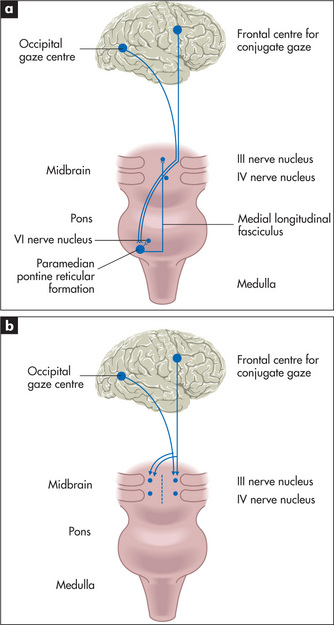
Figure 11.13 Horizontal (a) and vertical (b) eye movements
Adapted from Lance JW, McLeod JG. A physiological approach to clinical neurology, 3rd edn. London: Butterworths, 1981.
There are a number of possible causes for deviation of the eyes to one side. For example, deviation of the eyes to the left can result from: (i) a destructive lesion (usually vascular or neoplastic), which involves the pathways between the left frontal lobes and the oculomotor nuclei; (ii) a destructive lesion of the right side of the brainstem; or (iii) an irritative lesion, such as an epileptic focus, of the right frontal lobe, which stimulates deviation of the eyes to the left.
Supranuclear palsy is loss of vertical or horizontal gaze or both (Figure 11.13). The clinical features that distinguish this from third, fourth and sixth nerve palsies include: (i) both eyes are affected; (ii) pupils may be fixed and are often unequal; (iii) there is usually no diplopia; and (iv) the reflex eye movements—for example, on flexing and extending the neck—are usually intact.
Nystagmus
The eyes are normally maintained at rest in the midline by the balance of tone between opposing ocular muscles. Disturbance of this tone, which depends on impulses from the retina, the muscles of the eyes themselves and various vestibular and central connections, allows the eyes to drift in one direction. This drift is corrected by a quick movement (saccadic) back to the original position. When these movements occur repeatedly nystagmus is said to be present. The direction of the nystagmus is defined as that of the fast (correcting) movement, although it is the slow drift that is abnormal. Nystagmus from any cause tends to be accentuated by gaze in a direction away from the midline. In many instances nystagmus is not present when the eyes are at rest, and is only detected when the eyes are deviated (gaze-evoked nystagmus). At the extremes of gaze, fine nystagmus is normal (physiological). Therefore test for nystagmus by asking the patient to follow your pin out to 30 degrees from the central gaze position.
Nystagmus may be jerky or pendular.
A summary of how to approach the medical eye examination is provided on in Chapter 13.
The fifth (trigeminal) nerve
Examination anatomy
This nerve contains both sensory and motor fibres. Its motor nucleus and its sensory nucleus for touch lie in the pons (Figure 11.14), its proprioceptive nucleus lies in the midbrain, while its nucleus serving pain and temperature sensation descends through the medulla to reach the upper cervical cord. It is the largest of the cranial nerves.
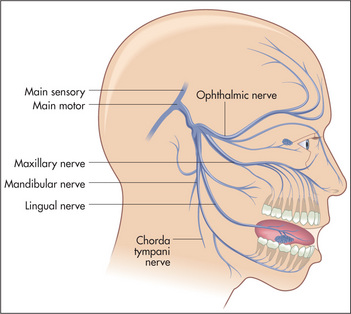
The nerve itself leaves the pons from the cerebellopontine angle and runs over the temporal lobe in the middle cranial fossa. At the petrous temporal bone the nerve forms the trigeminal (Gasserianr) ganglion and from here the three sensory divisions arise. The first (ophthalmic) division runs in the cavernous sinus with the third nerve and emerges from the superior orbital fissure to supply the skin of the forehead, the cornea and conjunctiva. The second (maxillary) division emerges from the infraorbital foramen and supplies skin in the middle of the face and the mucous membranes of the upper part of the mouth, palate and nasopharynx. The third and largest (mandibular) division runs with the motor part of the nerve, leaving the skull through the foramen ovale to supply the skin of the lower jaw and mucous membranes of the lower part of the mouth (Figures 11.15 and 11.16).
Pain and temperature fibres from the face run from the pons through the medulla as low as the upper cervical cord, terminating in the spinal tract nucleus as they descend. The second-order neurones arise in this nucleus and ascend again as the ventral trigeminothalamic tract. Touch and proprioceptive fibres terminate in the pontine or main sensory and mesencephalic nuclei, respectively, to form the dorsal and ventral mesencephalic tracts. Because of this segregation in the brainstem, lesions of the medulla or upper spinal cord can cause a dissociated sensory loss of the face—loss of pain and temperature sensation, but retention of touch and proprioception.
The motor part of the nerve supplies the muscles of mastication.
Examination
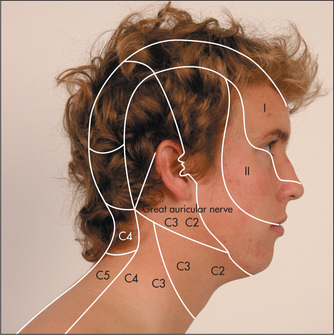
Test facial sensation in the three divisions of the nerve, comparing each side with the other (Figure 11.17). Test first with the sharp end of a new neurological pin for pain sensation (never use an old pin in these days of hepatitis B, HIV etc).10 The pin is applied lightly to the skin and the patient is asked whether it feels sharp or dull. Some examiners ask patients to shut their eyes. Loss of pain sensation will result in the pinprick feeling dull. An area of dull sensation should be mapped by testing pinprick sensation progressively: testing should go from the dull to the sharp area. Test also above the forehead progressively back over the top of the head. If the ophthalmic division is affected sensation will return when the C2 dermatome is reached (Figure 11.16). It is important to exercise caution: too sharp a pin will leave a little trail of bloody spots, which is embarrassing. Temperature is not tested routinely unless syringobulbia is suspected, as temperature loss usually accompanies loss of pain sensation.
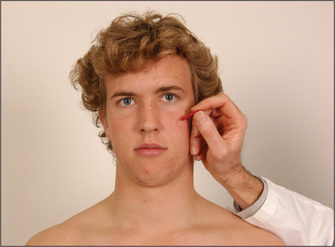
Figure 11.17 Facial sensation V, maxillary division: ‘Does this feel sharp or blunt?’—test all three divisions on each side
Now examine the motor division of the nerve. Begin by inspecting for wasting of the temporal and masseter muscles. Ask the patient then to clench the teeth and palpate for contraction of the masseter above the mandible (Figure 11.18). The strength of these muscles can be tested by asking the patient to bite forcefully onto a wooden tongue depressor with the molar teeth. The depth of the teeth marks on each side give an indication of the relative strengths of the muscles. The examiner can attempt to withdraw the tongue depressor as the patient bites it. A bite of normal strength will prevent this. Then get the patient to open the mouth (pterygoid muscles) and hold it open while the examiner attempts to force it shut. A unilateral lesion of the motor division causes the jaw to deviate towards the weak (affected) side.
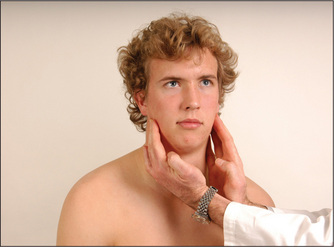
Test the jaw jerk or masseter reflex. The patient lets the mouth fall open slightly and the examiner’s finger is placed on the tip of the jaw and tapped lightly with a tendon hammer (Figure 11.19). Normally there is a slight closure of the mouth or no reaction at all. In an upper motor neurone lesion above the pons the jaw jerk is greatly exaggerated. This is commonly seen in pseudobulbar palsy).s

Causes of a fifth nerve palsy
Remember, if there is total loss of sensation in all three divisions of the nerve, this suggests that the level of the lesion is at the ganglion or the sensory root—for example, an acoustic neuroma (Figure 11.20). If there is total sensory loss in one division only, this suggests a postganglionic lesion. The ophthalmic division is most commonly affected because it runs in the cavernous sinus and through the orbital fissure, where it is vulnerable to a number of different insults.
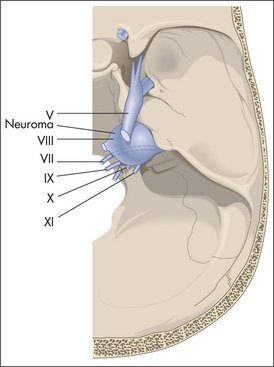
Figure 11.20 Cerebellopontine angle tumour
Adapted from Simon RP, Aminoff MJ, Greenberg DA, Clinical neurology 1989. Appleton & Lange, 1989.
If there is dissociated sensory loss (loss of pain, but preservation of touch sensation) this suggests a brainstem or upper cord lesion, such as syringobulbia, foramen magnum tumour, or infarction in the territory of the posterior inferior cerebellar artery. If touch sensation is lost but pain sensation is preserved, this is usually due to an abnormality of the pontine nuclei, such as a vascular lesion or tumour. Motor loss can also be central or peripheral.
The seventh (facial) nerve
Examination anatomy
The seventh nerve nucleus lies in the pons next to the sixth cranial nerve nucleus (Figure 11.14). The nerve (Figure 11.21) leaves the pons with the eighth nerve through the cerebellopontine angle. After entering the facial canal it enlarges to become the geniculate ganglion. The branch that supplies the stapedius muscle is given off from within the facial canal. The chorda tympani (containing taste fibres from the anterior two-thirds of the tongue) joins the nerve in the facial canal. The seventh nerve leaves the skull via the stylomastoid foramen. It then passes through the middle of the parotid gland and supplies the muscles of facial expression. The frontalis muscle receives upper motor innervation bilaterally, the other muscles receive innervation from the contralateral cortex.
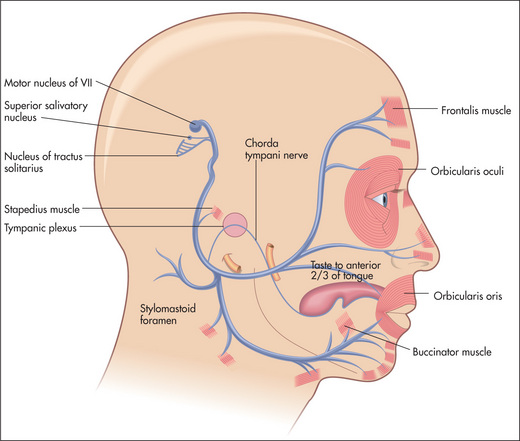
Examination
Inspect for facial asymmetry, as a seventh nerve palsy can cause unilateral drooping of the corner of the mouth, and smoothing of the wrinkled forehead and the nasolabial fold (Figure 11.22). However, with bilateral facial nerve palsies symmetry can be maintained.
Test the muscle power. Ask the patient to look up so as to wrinkle the forehead (Figure 11.23). Look for loss of wrinkling and feel the muscle strength by pushing down against the corrugation on each side. This movement is relatively preserved on the side of an upper motor neurone lesion (a lesion that occurs above the level of the brainstem nucleus) because of bilateral cortical representation of these muscles. The remaining muscles of facial expression are usually affected on the side of an upper motor neurone lesion, although occasionally the orbicularis oculi muscles are preserved. Ask the patient to puff out the cheeks (Figure 11.24). Look for asymmetry.
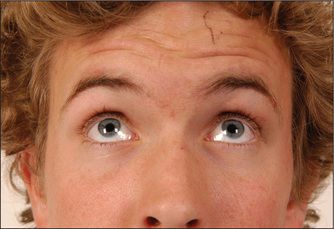

Next ask the patient to shut the eyes tightly (Figure 11.25). Compare how deeply the eyelashes are buried on the two sides and then try to force open each eye. Check whether Bell’s phenomenont is evident. Bell’s phenomenon is present in everyone, although not usually visible unless a person has a VII nerve palsy. In this case, when the patient attempts to shut the eye on the side of a lower motor neurone VII nerve palsy, there is upward movement of the eyeball and incomplete closure of the eyelid. Next ask the patient to grin (Figure 11.26) and compare the nasolabial grooves, which are smooth on the weak side.


Figure 11.26 Cranial nerve VII: ‘Show me your teeth’
(Before asking this question, make sure the patient’s teeth are not in a container beside the bed.)
If a lower motor neurone lesion is detected, check quickly for the ear and palatal vesicles of herpes zoster of the geniculate ganglion—the Ramsay Huntu syndrome.
Causes of a seventh (facial) nerve palsy
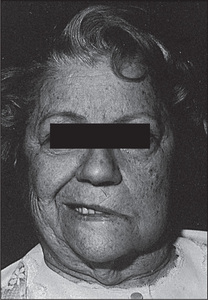
In lower motor neurone lesions, pontine causes (often associated with V and VI lesions) include vascular lesions, tumours, syringobulbia or multiple sclerosis. Posterior fossa lesions include an acoustic neuroma, a meningioma or chronic meningitis. At the level of the petrous temporal bone, Bell’s palsy (an idiopathic acute paralysis of the nerve; Figure 11.27), a fracture, the Ramsay Hunt syndrome or otitis media may occur, while the parotid gland may be affected by a tumour or sarcoidosis. Remember, Bell’s palsy is the most common cause (up to 80%) of a facial nerve palsy.v
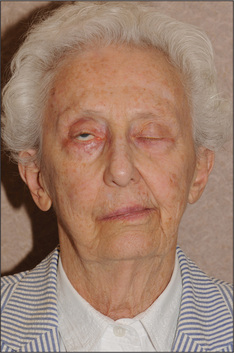
Figure 11.27 Bell’s palsy
From Mayo Clinic Images, with permission. © Mayo Clinic Scientific Press and CRC Press.
Unilateral loss of taste, without other abnormalities, can occur with middle-ear lesions involving the chorda tympani or lingual nerve, but these are very rare.
Irritative changes
Tonic and clonic movements of the facial muscles can occur in seizures. Various abnormal movements of the facial muscles can occur as a result of basal ganglia or extrapyramidal abnormalities (page 396). These include athetoid and dystonic (page 399) movements. Irritative lesions in the brainstem can cause increased secretion of saliva (sialorrhoea). This can also occur in Parkinson’s disease or accompany attacks of nausea.
The eighth (acoustic) nerve
Examination anatomy
The eighth (acoustic) nerve has two components: the cochlear, with afferent fibres subserving hearing; and the vestibular, containing afferent fibres subserving balance. Fibres for hearing originate in the organ of Cortiw and run to the cochlear nuclei in the pons. From here there is bilateral transmission to the medial geniculate bodies and thence to the superior gyrus of the temporal lobes. Fibres for balance begin in the utricle and semicircular canals, and join auditory fibres in the facial canal. They then enter the brainstem at the cerebellopontine angle. After entering the pons, vestibular fibres run widely throughout the brainstem and cerebellum.
Examination of the ear and hearing
Inspect the patient’s external auditory meatus. The adult canal angulates, so in order to see the eardrum it is necessary to pull up and backwards on the auricle before inserting the otoscope. The normal eardrum (tympanic membrane) is pearly grey and concave. Look for wax or other obstructions, and inspect the eardrum for inflammation or perforation (see Chapter 13).
Examination of vestibular function
If a patient complains of vertigo, the Hallpikezmanoeuvre should be performed. The patient sits up; having warned him or her what is about to occur, the examiner grasps the patient’s head between the hands and gets him or her to lie back quickly so that the head lies 30 degrees below the horizontal. At the same time, the head is rotated 30 degrees towards the examiner. Ask the patient to keep the eyes open. If the test is positive, after a short latent period vertigo and nystagmus (rotatory) towards the affected (lowermost) ear occur for several seconds and then abate and are not reproducible for 10 to 15 minutes. This result is seen in the condition called benign paroxysmal positioning vertigo (BPPV). It occurs with repositioning of the head and then abates so that the old name, benign positional vertigo, is not really appropriate. It is due to a disorder in the utricle and occurs, for example, following infection, trauma or vascular disease. It is caused by the presence of concretions in the semicircular canals. Inertia of these concretions following movement of the head causes the illusion of movement and nystagmus. If there is no latent period, no fatiguability or the nystagmus persists or is variable, this suggests that there is a lesion of the brainstem (e.g. multiple sclerosis) or cerebellum (e.g. metastatic carcinoma).
Causes of vestibular abnormalities
Vestibular causes include vestibular neuronitis as well as many of the causes of nerve deafness.
In the brainstem, vascular lesions, tumours of the cerebellum or fourth ventricle, demyelination or vasospastic conditions such as migraine may involve the central connections of the vestibular system.
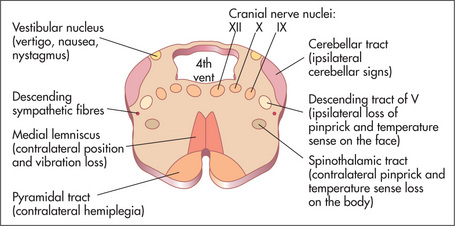
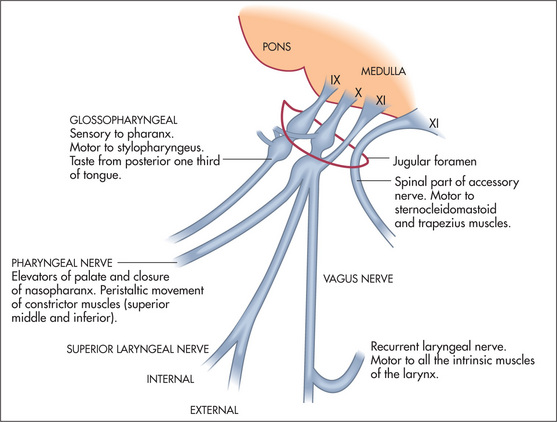
The ninth (glossopharyngeal) and tenth (vagus) nerves
Examination anatomy
These nerves have motor, sensory and autonomic functions. Nerve fibres from nuclei in the medulla (Figure 11.30) form multiple nerve rootlets as they exit the medulla. These join to form the ninth and tenth nerves, and also contribute to the eleventh nerve. The nerves emerge from the skull through the jugular foramen (Figure 11.31). The ninth nerve receives sensory fibres from the nasopharynx, pharynx, middle and inner ear and from the posterior third of the tongue (including taste fibres). It also carries secretory fibres to the parotid gland. The tenth nerve receives sensory fibres from the pharynx and larynx, and innervates muscles of the pharynx, larynx and palate.
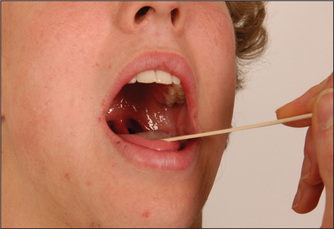
Examination
Ask the patient to open the mouth and inspect the palate with a torch. Note any displacement of the uvula. Then ask the patient to say ‘Ah!’ (Figure 11.32). Normally the posterior edge of the soft palate—the velumaa—rises symmetrically. If the uvula is drawn to one side this indicates a unilateral tenth nerve palsy. Note that the uvula is drawn towards the normal side.
Testing for the gag reflex (ninth is the sensory component and tenth the motor component) is traditional but not necessary.11 A better alternative is to touch the back of the pharynx on each side with a spatula (rather than the soft palate). The patient is asked if the touch of the spatula (ninth) is felt each time. Normally, there is reflex contraction of the soft palate. If contraction is absent and sensation is intact this suggests a tenth nerve palsy. The most common cause of a reduced gag reflex is old age. Of more concern to the examiner is the patient with an exaggerated but still normal reflex. This can lead to vomiting onto the examining clinician.
The eleventh (accessory) nerve
Examination anatomy
The central portion of this nerve arises in the medulla close to the nuclei of the ninth, tenth and twelfth nerves and its spinal portion arises from the upper five cervical segments. It leaves the skull with the ninth and tenth nerves through the jugular foramen (Figure 11.31). Its central division provides motor fibres to the vagus and the spinal division innervates the trapezius and sternocleidomastoid muscles. The motor fibres that supply the sternocleidomastoid muscle are thought to cross twice so that cortical control of the muscle is ipsilateral. This makes sense when one remembers that the muscle turns the head to the opposite side. This means that the hemisphere which receives information from and controls one side of the body also turns the head to face that side.
Examination
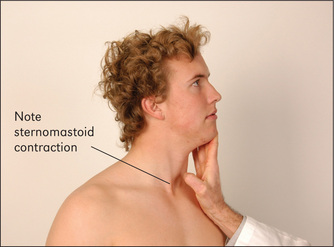
Ask the patient to shrug the shoulders (Figure 11.33). Feel the bulk of the trapezius muscles and attempt to push the shoulders down. Then instruct the patient to turn the head to the side against resistance (the examiner’s hand) (Figure 11.34). Remember that the right sternocleidomastoid turns the head to the left. Feel the muscle bulk of the sternocleidomastoids.

Weakness of these muscles is less common than torticollis, which is due to overactivity of multiple neck muscles. It is a complex movement disorder. The head appears turned to one side either permanently or in spasms. Ask the patient to turn the head to face forwards. This is usually possible at least briefly, but look to see whether he or she needs to use the hands to push the head straight.
The twelfth (hypoglossal) nerve
Examination
Inspect the tongue at rest on the floor of the mouth. The normal tongue may move a little, especially when protruded, but is not wasted. Look for wasting and fasciculations (fine, irregular, non-rhythmical muscle fibre contractions). These signs indicate a lower motor neurone lesion. Fasciculations may be unilateral or bilateral (Figure 11.35).
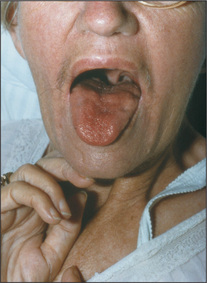
Ask the patient to poke out the tongue (Figure 11.36), which may deviate towards the weaker (affected) side if there is a unilateral lower motor neurone lesion (Figure 11.37). The tongue, like the face and palate, has a bilateral upper motor neurone innervation in most people, so a unilateral upper motor neurone lesion often causes no deviation.

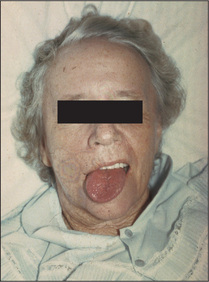
Movement disorders may affect the tongue. In Parkinson’s disease there may be a coarse tremor of the tongue, made worse by speaking or protruding the tongue. Athetoid, choreiform and tardive dyskinesia can all involve the tongue (page 399).
Causes of twelfth nerve palsy
Unilateral lower motor neurone lesions with a central cause include vascular lesions, such as thrombosis of the vertebral artery; motor neurone disease; and syringobulbia. Peripheral causes include: in the posterior fossa, aneurysms or tumours, chronic meningitis and trauma; in the upper neck, tumours or lymphadenopathy; and the Arnold-Chiari malformation.bb The Arnold-Chiari malformation is a congenital malformation of the base of the skull with herniation of a tongue of cerebellum and medulla into the spinal canal, causing lower cranial nerve palsies, cerebellar limb signs (due to tonsillar compression) and upper motor neurone signs in the legs.
Multiple cranial nerve lesions
TABLE 11.7 Clinical features of pseudobulbar and bulbar palsies
| Feature | Pseudobulbar (bilateral UMN lesions of IX, X and XII) | Bulbar (bilateral LMN lesions of IX, X and XII) |
| Gag reflex | Increased or normal | Absent |
| Tongue | Spastic | Wasted, fasciculations |
| Jaw jerk | Increased | Absent or normal |
| Speech | Spastic dysarthria | Nasal |
| Other | Bilateral limb UMN (long tract) signs | Signs of the underlying cause—e.g. limb fasciculations |
| Labile emotions | Normal emotions | |
| Causes | Bilateral cerebrovascular disease (e.g. both internal capsules) | Motor neurone disease |
| Guillain-Barré syndrome | ||
| Multiple sclerosis | Poliomyelitis | |
| Motor neurone disease | Brainstem infarction |
UMN = upper motor neurone; LMN = lower motor neurone.
TABLE 11.8 Causes of multiple cranial nerve palsies
| Nasopharyngeal carcinoma |
| Chronic meningitis—e.g. carcinoma, haematological malignancy, tuberculosis, sarcoidosis |
| Guillain-Barré syndrome (spares sensory nerves) |
| Brainstem lesions. These are usually due to vascular disease causing crossed sensory or motor paralysis (i.e. cranial nerve signs on one side and contralateral long tract signs). Patients with a brainstem tumour (e.g. in the cerebellopontine angle) may also have similar signs |
| Arnold-Chiari malformation |
| Trauma |
| Paget’s disease |
| Mononeuritis multiplex (rarely), e.g. diabetes mellitus |
The limbs and trunk
History
A variety of symptoms suggest that a patient may have a neurological problem involving the limbs and trunk and that these need to be examined. The exact nature of the symptoms will often suggest the correct diagnosis and where the examination should be directed. A thorough examination is still essential, however, if unexpected findings are not to be missed.
The patient may present with symptoms that are purely or predominately sensory or motor (Questions box 11.5), or related to disorders of movement such as tremor. Sensory symptoms include pain, numbness and paraesthesiae (tingling or pins and needles). It is important to find out if there is involvement of more than one modality, something the patient may not have noticed. The distribution, time of onset and duration may give clues to the aetiology of the symptoms or at least as to where the sensory examination should be concentrated.
Questions box 11.5
Questions to ask the patient with muscle weakness
Examination anatomy
Muscle weakness has four major causes:
General inspection
The upper limbs
The motor systemcc
General
Shake hands with the patient and introduce yourself. A patient who cannot relax his or her hand grip has myotonia (an inability to relax the muscles after voluntary contraction). The commonest cause of this is the muscle disease dystrophia myotonica (page 392). Once your hand has been extracted from the patient’s, and after pausing briefly for the vitally important general inspection, ask the patient to undress so that the arms and shoulder girdles are completely exposed.
Sit the patient over the edge of the bed if this is possible. Next ask the patient to hold out both hands, palms upward, with the arms extended and the eyes closed (Figure 11.39). Watch the arms for evidence of drifting (movement of one or both arms from the initial neutral position). There are only three causes for drift of the arms:
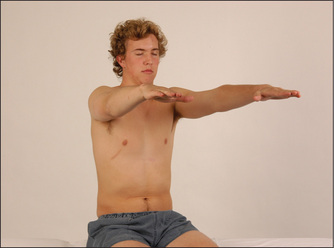
Figure 11.39 Testing for arm drift: ‘Shut your eyes and hold your arms out straight. Now turn your palms upwards.’
Ask the patient to relax the arms and rest them on his or her lap. Inspect the large muscle groups for fasciculations (Table 11.9). These are irregular contractions of small areas of muscle which have no rhythmical pattern. Fasciculation may be coarse or fine and is present at rest, but not during voluntary movement.dd If present with weakness and wasting, fasciculation indicates degeneration of the lower motor neurone. It is usually benign if unassociated with other signs of a motor lesion.
TABLE 11.9 Causes (differential diagnosis) of fasciculations
| Motor neurone disease |
| Motor root compression |
| Peripheral neuropathy—e.g. diabetic |
| Primary myopathy |
| Thyrotoxicosis |
Note: Myokymia resembles coarse fasciculation of the same muscle group, and is particularly common in the orbicularis oculi muscles, where it is usually benign. Focal myokymia, however, often represents brainstem disease, e.g. multiple sclerosis or glioma.
Fibrillation is seen only on the electromyogram.
Tone
Tone is tested at both the wrists and elbows. Rotation of the wrists with supination and pronation of the elbow joints (supporting the patient’s elbow with one hand and holding the hand with the other) is performed passively, and the patient should be told to relax to allow the examiner to move the joints freely.
Power
If power is reduced, decide whether this is symmetrical or asymmetrical, whether it involves only particular muscle groups, or whether it is proximal, distal or general. It is also important to consider whether any painful joint or muscle disease is interfering with the assessment (see Chapter 9). Asymmetrical muscle weakness is most often the result of a peripheral nerve, brachial plexus or root lesion, or an upper motor neurone lesion. As each movement is tested the important muscles involved should be observed or palpated.
Shoulder
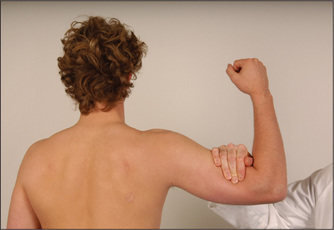
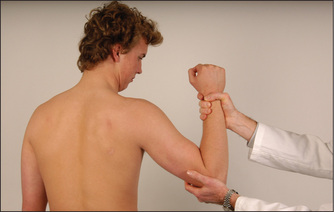
Elbow
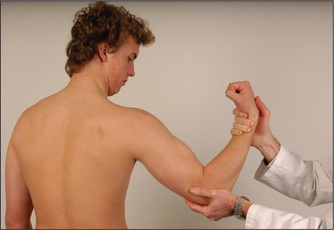
Wrist
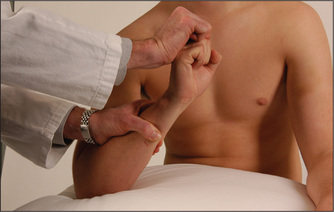
Fingers

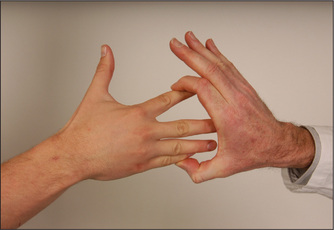
Reflexes
Tendon hammers are available in a number of designs. Sir William Gowersff used the ulnar side of his hand or part of his stethoscope. In Australia and Britain, the Queen Square hammergg is in common use (Figure 11.46). The Taylor hammer is popular in America; it is shaped like a tomahawk and has a broad rubber edge for most tendons and a more pointed side for the cutaneous reflexes.
Reflexes are graded from absent to greatly increased (Table 11.10).
| 0 | = absent |
| + | = present but reduced |
| ++ | = normal |
| +++ | = increased, possibly normal |
| ++++ | = greatly increased, often associated with clonus (page 368) |
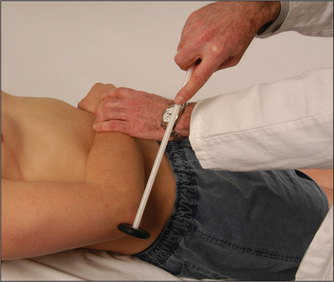
To test the biceps jerk (C5, C6), place one forefinger on the biceps tendon and tap this with the tendon hammer (Figure 11.47). The hammer should be held near its end and the head allowed to fall with gravity onto the positioned forefinger. The examiner soon learns not to hit too hard. Normally, if the reflex arc is intact, there is a brisk contraction of the biceps muscle with flexion of the forearm at the elbow, followed by prompt relaxation. Practice will help the examiner decide whether the response is within the normal range. When a reflex is greatly exaggerated, it can be elicited away from the usual zone.
An increased jerk occurs with an upper motor neurone lesion (page 354). A decreased or absent reflex occurs with a breach in any part of the reflex motor arc—the muscle itself (e.g. myopathy), the motor nerve (e.g. neuropathy), the anterior spinal cord root (e.g. spondylosis), the anterior horn cell (e.g. poliomyelitis) or the sensory arc (sensory root or sensory nerve).
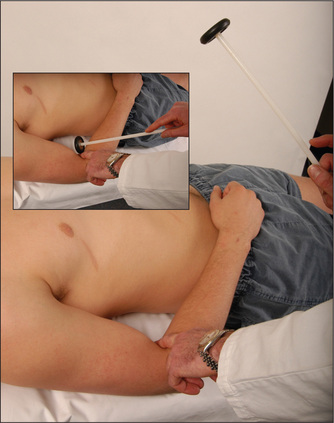
To test the triceps jerk (C7, C8), support the elbow with one hand and tap over the triceps tendon (Figure 11.48). Normally, triceps contraction results in forearm extension.
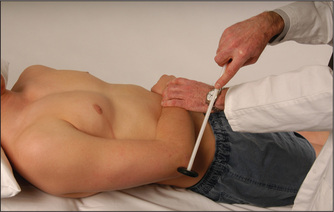
To test the brachioradialis (supinator) jerk (C5, C6), strike the lower end of the radius just above the wrist (Figure 11.49). To avoid hurting the patient by striking the radial nerve directly, place your own first two fingers over this spot and then strike the fingers, as with the biceps jerk. Normally, contraction of the brachioradialis causes flexion of the elbow.
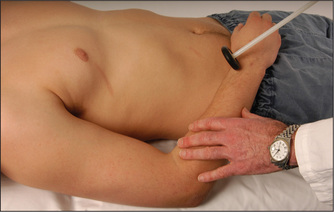
To test finger jerks (C8), the patient rests the hand palm upward, with the fingers slightly flexed. The examiner’s hand is placed over the patient’s and the hammer struck over the examiner’s fingers (Figure 11.50). Normally, slight flexion of all the fingers occurs.
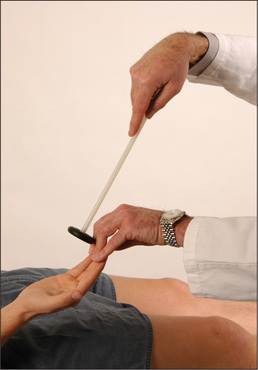
Finger–nose test
Ask the patient to touch his or her nose with the index finger and then turn the finger around and touch the examiner’s outstretched forefinger at nearly full extension of the shoulder and elbow (Figure 11.51). The test should be done both briskly and slowly, and repeated a number of times with the patient’s eyes open and later closed. Slight resistance to the patient’s movements by the examiner pushing on his or her forearm during the test may unmask less-severe abnormalities.
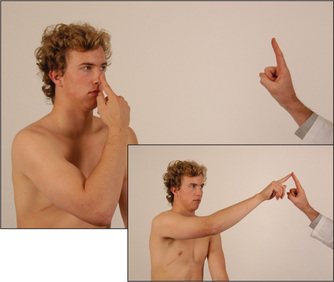
Rapidly alternating movements
Ask the patient to pronate and supinate his or her hand on the dorsum of the other hand as rapidly as possible (Figure 11.52). This movement is slow and clumsy in cerebellar disease and is called dysdiadochokinesis.hh
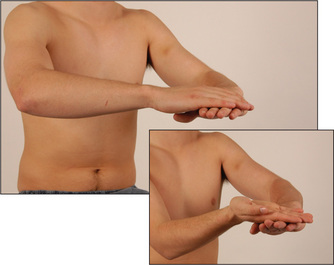
Rebound
Ask the patient to lift the arms rapidly from the sides and then stop. Hypotonia due to cerebellar disease causes delay in stopping the arms. This method of demonstrating rebound is preferable to the more often used one where the patient flexes the arm at the elbow against the examiner’s resistance. When the examiner suddenly lets go, violent flexion of the arm may occur and, unless prevented, the patient can strike himself or herself in the face. Therefore, only medical students trained in self-defence should use this method.ii
The sensory system
To test sensation, which can be a difficult assessment and frustratingly time-consuming, use the following routine.12
Spinothalamic pathway (pain and temperature)
Pain and temperature fibres enter the spinal cord and cross, a few segments higher, to the opposite spinothalamic tract (Figure 11.53). This tract ascends to the brainstem.
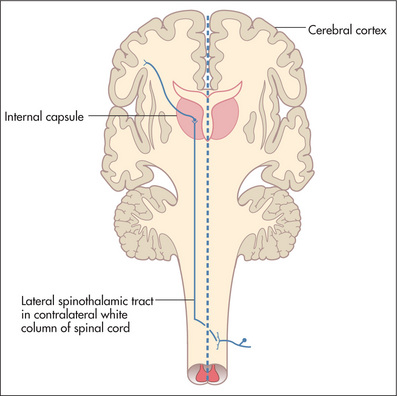
Pain (pinprick) testing
Using a new pin,10 demonstrate to the patient that this induces a relatively sharp sensation by touching lightly a normal area, such as the anterior chest wall. Then ask the patient to say whether the pinprick feels sharp or dull. Begin proximally on the upper arm and test in each dermatome—the area of skin supplied by a vertebral spinal segment (Figure 11.54). Also compare right with left in the same dermatome. Map out the extent of any area of dullness. Always do this by going from the area of dullness to the area of normal sensation.
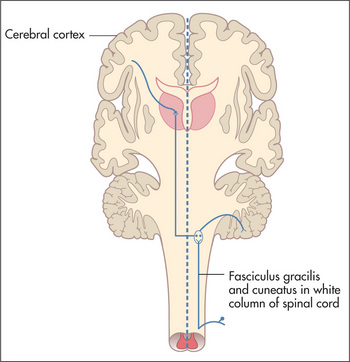
Posterior columns (vibration and proprioceptionjj)
These fibres enter and ascend ipsilaterally in the posterior columns of the spinal cord to the nucleus gracilis and nucleus cuneatus in the medulla, where they decussate (Figure 11.55).
Vibration testing
Use a 128 Hz tuning fork (not a 256 Hz fork). Ask the patient to close the eyes, and then place the vibrating tuning fork on one of the distal interphalangeal joints. The patient should be able to describe a feeling of vibration. The examiner then deadens the tuning fork with the hand, and the patient should be able to say exactly when this occurs. Compare one side with the other. If vibration sense is reduced or absent, test over the ulnar head at the wrist, then the elbows (over the olecranon) and then the shoulders to determine the level of abnormality. Although the tuning fork is traditionally placed only over bony prominences, vibration sense is just as good over soft tissues.
Light-touch testing
Test light touch by touching the skin with a wisp of cottonwool. Ask the patient to shut the eyes and say ‘yes’ when the touch is felt. Do not stroke the skin because this moves hair fibres. Test each dermatome,kk comparing left and right sides.
Interpretation of sensory abnormalities
Try to fit the distribution of any sensory loss into a dermatome (due to a spinal cord or nerve root lesion), a single peripheral nerve territory, a peripheral neuropathy pattern (glove distribution, page 386), or a hemisensory loss (due to spinal cord or upper brainstem or thalamic lesion).
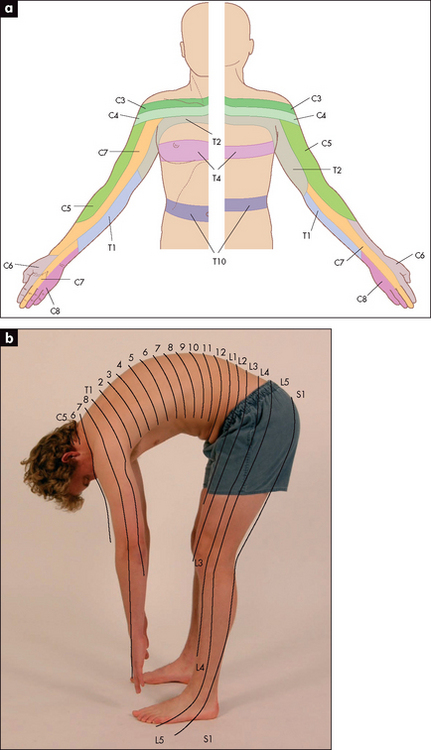
Sensory dermatomes of the upper limb (Figure 11.56) can be recognised by memorising the following rough guides: C5 supplies the shoulder tip and outer part of the upper arm; C6 supplies the lateral aspect of the forearm and thumb; C7 supplies the middle finger; C8 supplies the little finger; T1 supplies the medial aspect of the upper arm and elbow.
Examination of the peripheral nerves of the upper limb
A lesion of a peripheral nerve causes a characteristic motor and sensory loss.13 Peripheral nerve lesions may have local causes, such as trauma or compression, or may be part of a mononeuritis multiplex, where more than one nerve is affected by systemic disease.
The radial nerve (C5–C8)
Test sensation using a pin over the area of the anatomical snuff box. Sensation here is lost with a radial nerve lesion before the bifurcation into posterior interosseous and superficial radial nerves at the elbow (Figure 11.57).
Lesion at the wrist (carpal tunnel).14,15
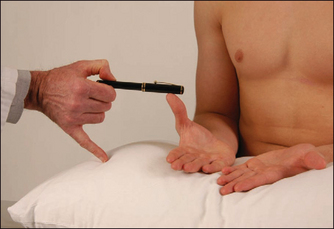
Use the pen-touching test to assess for weakness of the abductor pollicis brevis. Ask the patient to lay the hand flat, palm upwards on the table, and attempt to abduct the thumb vertically to touch the examiner’s pen held above it (Figure 11.58). This may be impossible if there is a median nerve palsy at the wrist or above. Remember, however, that most patients with the carpal tunnel syndrome have normal power and may indeed have symptoms but no signs at all.
Lesion in the cubital fossa
Ochsner’s clasping testll (for loss of flexor digitorum sublimis). Ask the patient to clasp the hands firmly together (Figure 11.59a)—the index finger on the affected side fails to flex with a lesion in the cubital fossa or higher (Figure 11.59b).
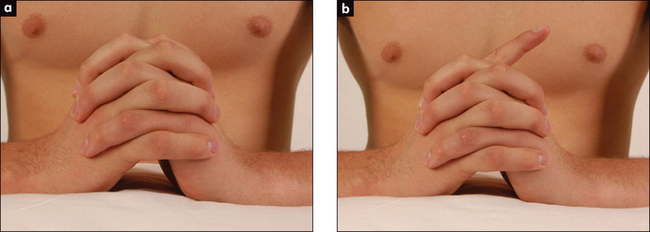
Figure 11.59 Ochsner’s clasping test
(a) Normal. (b) Abnormal due to loss of function of the flexor digitorum (simulated demonstration).
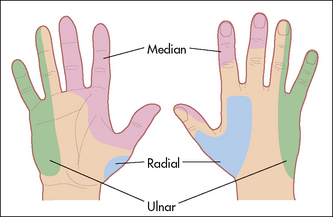
For the sensory component of the median nerve, test pinprick sensation over the hand. The constant area of loss includes the palmar aspect of the thumb, index, middle and lateral half of the ring fingers (Figure 11.57). The palm is spared in median nerve lesions in the carpal tunnel.
Froment’s signmm
Causes of a true claw hand are shown in Table 11.11, while causes of wasting of the small muscles of the hand are shown in Table 11.12; see also Figure 11.60.
TABLE 11.11 Causes (differential diagnosis) of a true claw hand (all fingers clawed)
| Ulnar and median nerve lesion (ulnar nerve palsy alone causes a claw-like hand) |
| Brachial plexus lesion (C8–T1) |
| Other neurological disease—e.g. syringomyelia, polio |
| Ischaemic contracture (late and severe) |
| Rheumatoid arthritis (advanced, untreated disease) |
TABLE 11.12 Causes (differential diagnosis) of wasting of the small muscles of the hand
| Spinal cord lesions |
Note: Distinguishing an ulnar nerve lesion from a C8 root/lower trunk brachial plexus lesion depends on remembering that sensory loss with a C8 lesion extends proximal to the wrist, and the thenar muscles are involved with a C8 root or lower trunk brachial plexus lesion. Distinguishing a C8 root from a lower trunk brachial plexus lesion is difficult clinically, but the presence of a Horner’s syndrome or an axillary mass suggests the brachial plexus is affected.
* Eric Klas Kugelberg (1913–83), professor of clinical neurophysiology at the Karolinska Institute in Stockholm, and Lisa Welander (1909–2001) described this in 1956. Lisa Welander was Sweden’s first woman professor of neurology.
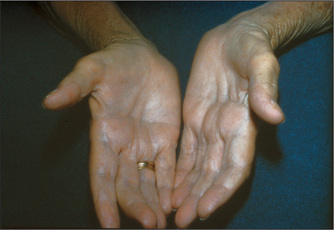
For the sensory component of the ulnar nerve, test for pinprick loss over the palmar and dorsal aspects of the little finger and the medial half of the ring finger (Figure 11.57).
The brachial plexus
Brachial plexus lesions vary from mild to complete; motor and/or sensory fibres may be involved. Nerve roots form trunks, which divide into cords and then form peripheral nerves (see Tables 11.13 and 11.14). The anatomy is shown in Figure 11.61.
| Nerve roots | Trunks | Muscles supplied |
| C5 and 6 | Upper | Shoulder (especially biceps and deltoid) |
| C7 | Middle | Triceps and some forearm muscles |
| C8 and T1 | Lower | Hand and some forearm muscles |
TABLE 11.14 Brachial plexus cords, nerves and their supplied muscles
| Cords | Nerves formed | Muscles supplied |
| Lateral | Musculocutaneous, median | Biceps, pronator teres, flexor carpi radialis |
| Medial | Median and ulnar | Hand muscles |
| Posterior | Axillary and radial | Deltoid, triceps and forearm extensors |
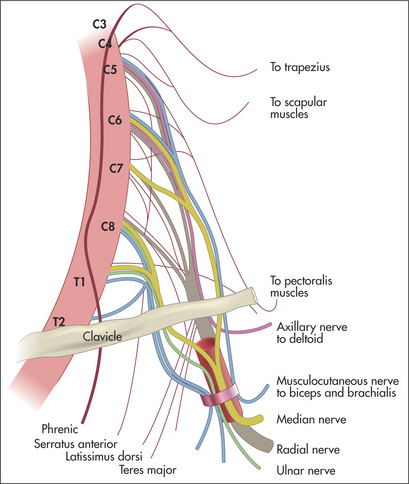
Figure 11.61 The brachial plexus
Adapted from Chusid JG, Correlative neuroanatomy and functional neurology, 19th edn. Los Altos: Lange Medical, 1985.
Examine the arms and shoulder girdle (Table 11.15). Remember that the dorsal scapular nerve (which supplies the rhomboid muscles) comes from the C5 nerve root proximal to the upper trunk, and so rhomboid function is usually spared in upper trunk lesions. Typical lesions of the brachial plexus are described in Table 11.16. The cervical rib syndrome may cause a lower brachial plexus lesion (Table 11.17). Table 11.18 suggests a scheme for distinguishing plexus and nerve root lesions.
TABLE 11.15 Shoulder girdle examination
| Method |
| Abnormalities are likely to be due to a muscular dystrophy, single nerve or a root lesion. Inspect each muscle, palpate its bulk and test function as follows: |
| 1 Trapezius (XI, C3, C4): ask the patient to elevate the shoulders against resistance and look for winging of the upper scapula. |
| 2 Serratus anterior (C5–C7): ask the patient to push the hands against the wall and look for winging of the lower scapula. |
| 3 Rhomboids (C4, C5): ask the patient to pull both shoulder blades together with the hands on the hips. |
| 4 Supraspinatus (C5, C6): ask the patient to abduct the arms from the sides against resistance. |
| 5 Infraspinatus (C5, C6): ask the patient to rotate the upper arms externally against resistance with the elbows flexed at the sides. |
| 6 Teres major (C5–C7): ask the patient to rotate the upper arms internally against resistance. |
| 7 Latissimus dorsi (C7, C8): ask the patient to pull the elbows into the sides against resistance. |
| 8 Pectoralis major, clavicular head (C5–C8): ask the patient to lift the upper arms above the horizontal and push them forward against resistance. |
| 9 Pectoralis major, sternocostal part (C6–T1) and pectoralis minor (C7): ask the patient to adduct the upper arms against resistance. |
| 10 Deltoid (C5, C6) (and axillary nerve): ask the patient to abduct the arms against resistance. |
TABLE 11.16 Brachial plexus lesions
| Complete lesion (rare) |
* Wilhelm Heinrich Erb (1840–1921), Germany’s greatest neurologist.
† Auguste Déjérine-Klumpke (1859–1927), French neurologist, described this lesion as a student. She was an American, but was educated in Switzerland. As a final-year student she married the great French neurologist Jules Déjérine.
TABLE 11.17 Cervical rib syndrome
| Clinical features |
| 1 Weakness and wasting of the small muscles of the hand (claw hand) |
| 2 C8 and T1 sensory loss |
| 3 Unequal radial pulses and blood pressure |
| 4 Subclavian bruits on arm manoeuvring (may be present in normal people) |
| 5 Palpable cervical rib in the neck (uncommon) |
TABLE 11.18 Distinguishing brachial plexus lesions and nerve root compression
| Root | Plexus | |
| Previous trauma | Occasionally | Some types |
| Insidious onset | Usually | Some types |
| Neck pain | Yes | No |
| Unilateral interscapular pain | Yes | No |
| Weakness | Mild–moderate | Often severe |
| Pattern of weakness | Most commonly triceps (C7 lesions, the most commonly affected root) | Usually shoulder and biceps or hand |
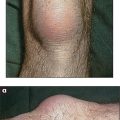
Ask the patient to stand facing away from you with the arms and hands stretched out to touch and push against the wall. Winging of the scapulae is seen typically in fascio-scapular-humeral dystrophy (Figure 11.62).
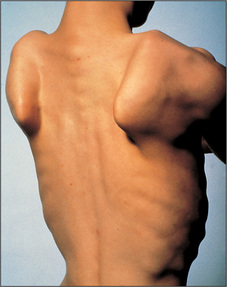
Figure 11.62 Winging of the scapulae, often a result of muscular dystrophy
From Mir MA, Atlas of Clinical Diagnosis, 2nd edn. Edinburgh: Saunders, 2003, with permission.
Causes of brachial plexus lesions include:
The lower limbs
Begin by testing gait, if this is possible (see page 376).
Inspect the legs with the patient lying in bed with the legs and thighs entirely exposed (place a towel over the groin). Note whether there is a urinary catheter present, which may indicate that there is spinal cord compression or other spinal cord disease, particularly multiple sclerosis.
The motor system
Tone
Test tone at the knees and ankles. Place one hand under a chosen knee and then abruptly pull the knee upwards, causing flexion. When the patient is relaxed this should occur without resistance. Then, supporting the thigh, flex and extend the knee at increasing velocity, feeling for resistance to muscle stretch (tone). Tone in the legs may also be tested by sitting the patient with legs hanging freely over the edge of the bed. A leg (of the patient) is raised by the examiner to the horizontal and suddenly let go. The leg will oscillate up to half a dozen times in a normal person who is completely relaxed. If hypotonia is present, as occurs in cerebellar disease, the oscillations will be wider and more prolonged. If increased tone or spasticity is present, the movements will be irregular and jerky.
Hip
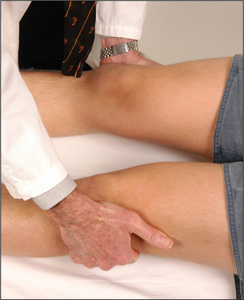
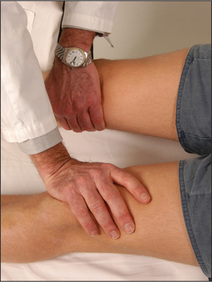
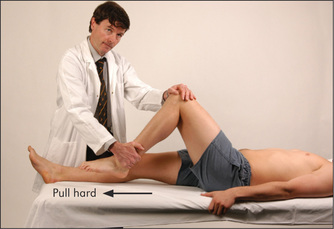
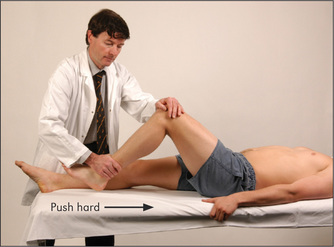
Knee
Ankle
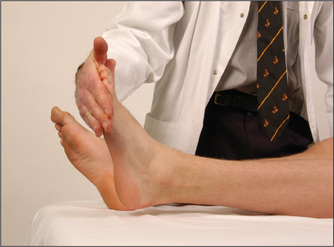
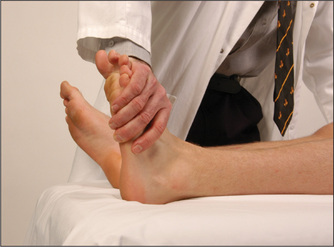
Tarsal joint
Non-organic or hysterical unilateral limb weakness may be detected by Hoover’s test. Normally when a patient attempts to resist movement, the contralateral limb works to support the effort. For example when a patient attempts to extend the leg against resistance, the other leg pushes down into the bed. If this movement is absent Hoover’snn sign is positive.
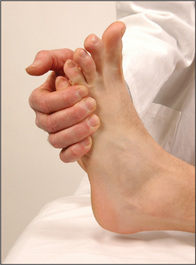
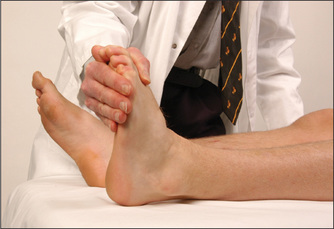
Quick test of lower limb power
The clinician in a hurry can test lower limb power quickly by asking the patient to:
This tests ankle, knee and hip power. Inability to perform any of the tests suggests a need to test more formally.
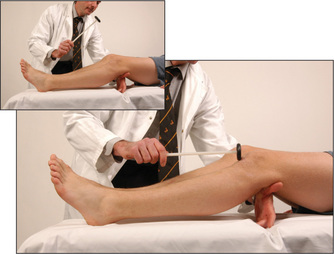
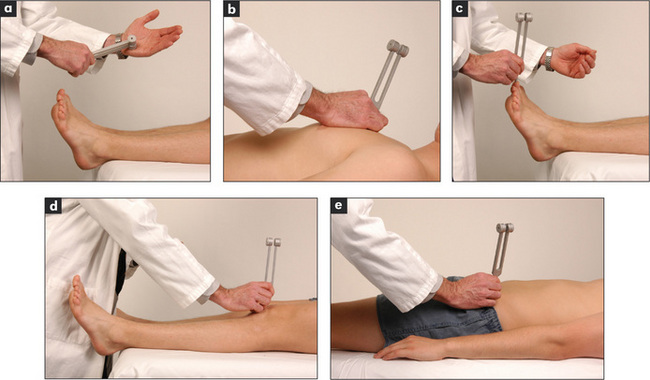
Knee jerk (L3, L4)
Slide one arm under the knees so that they are slightly bent and supported. The tendon hammer is allowed to fall onto the infrapatellar tendon (Figure 11.74). Normally, contraction of the quadriceps causes extension of the knee. Compare the two sides. If the knee jerk appears to be absent on one or both sides, it should be tested again following a reinforcement manoeuvre. Ask the patient to interlock the fingers and then pull apart hard at the moment before the hammer strikes the tendon (Jendrassik’s manoeuvreoo) (Figure 11.75). This manoeuvre has been shown to restore an apparently absent ankle jerk in 70% of elderly people. A reinforcement manoeuvre such as this, or teeth-clenching or grasping an object, should be used if there is difficulty eliciting any of the muscle stretch reflexes.
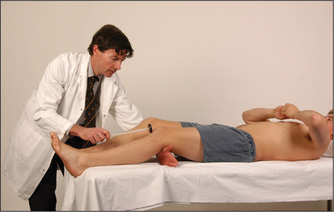
Ankle jerk (S1, S2)
Have the foot in the mid-position at the ankle with the knee bent, the thigh externally rotated on the bed, and the foot held in dorsiflexion by the examiner. The hammer is allowed to fall on the Achilles tendon (Figure 11.76). The normal response is plantar flexion of the foot with contraction of the gastrocnemius muscle. Again, test with reinforcement if appropriate. This reflex can also be tested with the patient kneeling (page 308) or by tapping the sole of the foot.16
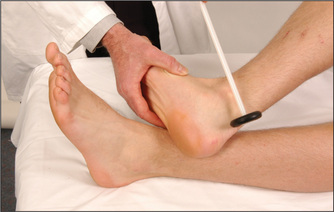
Plantar reflex (L5, S1, S2)
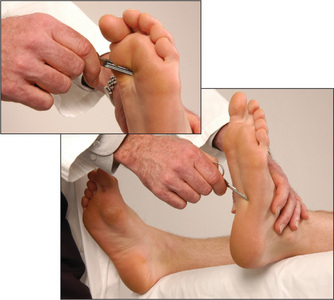
After telling the patient what is going to happen, use a blunt object (such as the key to an expensive car) to stroke up the lateral aspect of the sole, and curve inwards before it reaches the toes, moving towards the middle metatarsophalangeal (MTP) joint (Figure 11.77). The patient’s foot should be in the same position as for testing the ankle jerk. The normal response is flexion of the big toe at the MTP joint in patients over one year of age. The extensor (Babinskipp)17 response is abnormal and is characterised by extension of the big toe at the MTP joint (the upgoing toe) and fanning of the other toes. This indicates an upper motor neurone (pyramidal) lesion, although the test’s reliability can be relatively poor. Bilateral upgoing toes may also be found after a generalised seizure, or in a patient in coma.
Heel–shin test
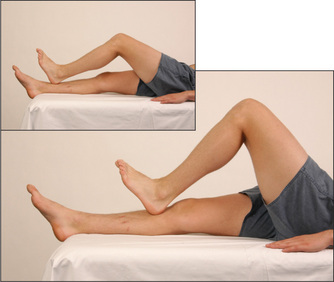
Ask the patient to run the heel of one foot up and down the opposite shin at a moderate pace and as accurately as possible (Figure 11.78). In cerebellar disease the heel wobbles all over the place, with oscillations from side to side and overshooting. Closing the eyes makes little difference to this in cerebellar disease, but if there is posterior column loss the movements are made worse when the eyes are shut—that is, there is ‘sensory ataxia’.
The sensory system
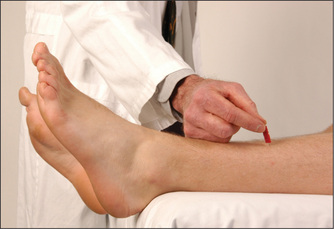
As for the upper limb, test for pain sensation first in each dermatome, comparing the right with the left side (Figure 11.79). Map out any abnormality and decide on the pattern of loss.
Then test vibration sense over the ankles and, if necessary, on the knees and the anterior superior iliac spines (Figure 11.80). Next test proprioception, using the big toes (Figure 11.81) and, if necessary, the knees and hips.
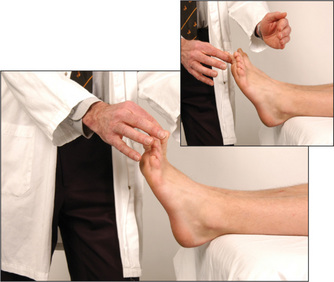
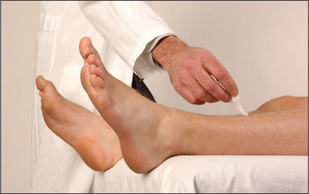
Finally, test light touch (Figure 11.82).
Dermatomes
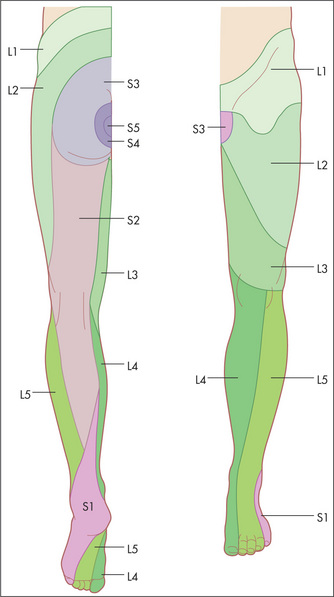
Memorise the following rough guide (Figure 11.83): L2 supplies the upper anterior thigh; L3 supplies the area around the front of the knee; L4 supplies the medial aspect of the leg; L5 supplies the lateral aspect of the leg and the medial side of the dorsum of the foot; S1 supplies the heel and most of the sole; S2 supplies the posterior aspect of the thigh; S3, S4 and S5 supply concentric rings around the anus.
The superficial or cutaneous reflexes
These reflexes occur in response to light touch or scratching of the skin or mucous membranes. The stimulus is more superficial than the tendon (muscle stretch) reflexes. As a rule these reflexes occur more slowly after the stimulus, are less constantly present and fatigue more easily.
The abdominal reflexes (epigastric T6–T9, mid-abdominal T9–T11, lower abdominal T11–L1)
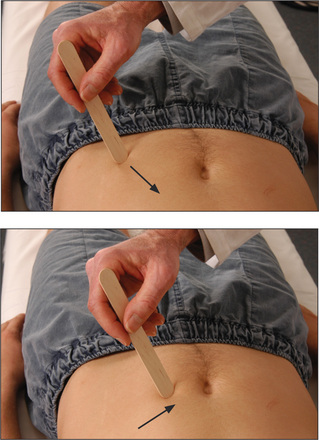
Test these by lightly stroking the abdominal wall diagonally towards the umbilicus in each of the four quadrants of the abdomen (Figure 11.84). Reflex contractions of the abdominal wall are absent in upper motor neurone lesions above the segmental level and also in patients who have had surgical operations interrupting the nerves. They disappear in coma and deep sleep, and during anaesthesia. They are usually difficult to elicit in obese patients and can also be absent in some normal people (20%). Their absence in the presence of increased tendon reflexes is suggestive of corticospinal tract abnormality.
Saddle sensation and anal reflex
Test now for saddle sensation if a cauda equina lesion is suspected (e.g. because of urinary or faecal incontinence). The only sensory loss may be on the buttocks or around the anus (S3–S5). In this case also test the anal reflex (S2, S3, S4): normal contraction of the external sphincter in response to pinprick of the perianal skin is abolished in patients with a lesion of the sacral segments of the cauda equina. If, however, the lowest sacral segments are spared but the higher ones are involved, this suggests that there is an intrinsic cord lesion.
Examination of the peripheral nerves of the lower limb
Lateral cutaneous nerve of the thigh
Test for sensory loss (Figure 11.85). A lesion of this nerve usually occurs because of entrapment between the inguinal ligament and the anterior superior iliac spine. It causes a sensory loss over the lateral aspect of the thigh with no motor loss detectable. If painful, it is called meralgia paraesthetica.
Femoral nerve (L2, L3, L4)
Test for weakness of knee extension (quadriceps paralysis). Hip flexion weakness is only slight, and adductor strength is preserved. The knee jerk is absent. The sensory loss involves the inner aspect of the thigh and leg (Figure 11.86).
Sciatic nerve (L4, L5, S1, S2)
This nerve supplies all the muscles below the knee and the hamstrings. Test for loss of power below the knee resulting in a footdrop (plantar flexed foot) and for weakness of knee flexion. Test the reflexes: with a sciatic nerve lesion the knee jerk is intact but the ankle jerk and plantar response are absent. Test sensation on the posterior thigh, lateral and posterior calf, and on the foot (lost with a proximal nerve lesion)(Figure 11.87).

Common peroneal nerve (L4, L5, S1)
This is a major terminal branch of the sciatic nerve. It supplies the anterior and lateral compartment muscles of the leg (Figure 11.88). On inspection one may notice a footdrop (Table 11.19). Test for weakness of dorsiflexion and eversion. Test the reflexes, which will all be intact. Test for sensory loss. There may be only minimal sensory loss over the lateral aspect of the dorsum of the foot. Note that these findings can be confused with an L5 root lesion, but the latter includes weakness of knee flexion and loss of foot inversion as well as sensory loss in the L5 distribution.
| Common peroneal nerve palsy |
| Sciatic nerve palsy |
| Lumbosacral plexus lesion |
| L4, L5 root lesion |
| Peripheral motor neuropathy |
| Distal myopathy |
| Motor neurone disease |
| Stroke—anterior cerebral artery or lacunar syndrome (‘ataxic hemiparesis’) |
Gait
Method
Make sure the patient’s legs are clearly visible. Now ask the patient to walk normally for a few metres and then turn around quickly and walk back. Then ask the patient to walk heel-to-toe to exclude a midline cerebellar lesion (Figure 11.89). Ask the patient to then walk on the toes (an S1 lesion will make this difficult or impossible) and then on the heels (an L4 or L5 lesion causing footdrop will make this difficult or impossible).
To test station (Rombergqq test), ask the patient to stand erect with the feet together and the eyes open (Figure 11.90a). Once the patient is stable ask him or her to close the eyes (Figure 11.90b). Compare the steadiness shown with the eyes open, then closed for up to 1 minute. Even in the absence of neurological disease a person may be slightly unsteady with the eyes closed, but the sign is positive if marked unsteadiness occurs to the point where the patient looks likely to fall. Normal people can maintain this position easily for 60 seconds. The Romberg test is positive when unsteadiness increases with eye closure. This is usually seen with the loss of proprioceptive sensation.
Marked unsteadiness with the eyes open is seen with cerebellar or vestibular dysfunction.
| Hemiplegia: the foot is plantar flexed and the leg is swung in a lateral arc |
| Spastic paraparesis: scissors gait |
| Parkinson’s disease: |
Speech and higher centres
At this stage of the examination dysarthria (difficulty with articulation), dysphonia (altered quality of the voice with reduction in volume as a result of vocal cord disease) or dysphasia (dominant higher centre disorder in the use of symbols for communication—language) may be obvious. If not, before going on to compartmentalised tests, the clinician should get the patient to talk freely—propositional or free speech. In a normal clinical encounter this comes from history taking. In viva voce or observed standardised clinical examinations (OSCEs), ask the patient to describe the room, his or her clothes, or job or daily activities, in order to promote flowing speech. Then test comprehension. This is done first without eliciting language—for example, ‘Touch your chin, then your nose and then your ear’; and then with yes and no questions—for example, ‘Do you put your shoes on before your socks?’ Then test repetition—for example, ‘Repeat the phrase “no ifs, ands or buts”.’
To complete the screening, ask the patient to name two objects pointed at, and to say a phrase such as ‘British Constitution’ or ‘West Register St’ (page 396).
There is no need to examine further if no abnormality of speech is detected in this way.
If there is an abnormality, proceed as outlined in Table 11.21.
| Fluent speech (receptive, conductive or nominal aphasia, usually) |
Dysphasia
There are four main types of dysphasia:18 receptive, expressive, nominal and conductive. The expressive aphasias are forms of motor apraxia. This means the inability to perform deliberate actions in the absence of paralysis.
To examine for dysphasia in more detail refer to Table 11.21. If the speech is fluent, but conveys information with paraphasic errors, such as ‘treen’ for ‘train’ (i.e. a word of similar sound or spelling to the one intended is used),tt the main possibilities are nominal, receptive and conductive dysphasia. Test for these by asking the patient to name an object, repeat a statement after you, and then follow commands. Then, if the above are abnormal, ask the patient to read and write, but remember that the occasional patient may be illiterate.
Dysarthria
Pseudobulbar palsy is an upper motor neurone weakness that causes a spastic dysarthria (it sounds as if the patient is trying to squeeze out words from tight lips), paralysis of the facial muscles and difficulty chewing and swallowing. The cause is infarction in both internal capsules. This causes interruption of the descending pyramidal tracts to the brainstem motor nuclei. The jaw jerk is usually increased. These patients tend to be very emotional and laugh and cry inappropriately. Their facial expressions become very animated at these times in contrast to their inability to control their facial expressions voluntarily.uu This phenomenon occurs because the nuclei that control motor responses to emotion do not reside in the motor cortex.
Patients who have bilateral lesions of the ninth and tenth cranial nerves are at risk of aspirating fluids or solids into their lungs if they try to eat or drink. Certain bedside tests can be performed to see if it is safe for them to eat or drink. These traditionally include the level of consciousness, gag reflex, pharyngeal sensation and testing swallowing water. Good signs guide 11.1 shows the likelihood ratios for these and other tests. The water swallowing test involves asking patient to sip repeatedly 5–10 mL of water. Coughing, choking or a fall in blood arterial oxygen saturation makes the test positive.
| Likelihood ratio if: | ||
| Present | Absent | |
| Voice and cough | ||
| Abnormal voluntary cough | 1.9 | 0.6 |
| Dysphonia | 1.3 | 0.4 |
| Examination | ||
| Drowsiness | 3.4 | 0.5 |
| Abnormal sensation—face and tongue | NS | NS |
| Absent pharyngeal sensation | 2.4 | 0.03 |
| Abnormal gag reflex | 1.5 | 0.6 |
| Tests | ||
| Water swallow test | 3.2 | 0.4 |
| Oxygen desaturation 2 min after swallowing | 3.1 | 0.3 |
Adapted with permission from McGee S, Evidence-based physical diagnosis, 2nd edn. St Louis: Saunders, 2007.
The cerebral hemispheres
Parietal, temporal and frontal lobe functions are tested if the patient is disoriented or has dysphasia, or if dementia is suspected. If the patient has a receptive aphasia, however, these tests cannot be performed. Their examination is otherwise not routine (Table 11.22). Students should already be familiar with the method of examining the cranial nerves and limbs.
| Parietal lobe |
* Gerstmann’s syndrome: dominant hemisphere parietal lobe only.
† Non-dominant parietal lobe signs.
‡ Non-localising parietal lobe signs.
Parietal lobe function
The parietal lobe is concerned with the reception and analysis of sensory information.
Dominant lobe signs
A mnemonic for these four dominant parietal lobe signs is AALF. Remember that Gerstmann’s syndromevv can be diagnosed only if the higher centres are intact. A demented patient would not be able to perform many of these tests.
Non-dominant and non-localising parietal lobe signs (cortical sensation)
Now test for general signs of parietal lobe dysfunction.
Temporal lobe function
An alert patient with a severe memory disturbance may make up stories to fill any gaps in his or her memory. This is called confabulation, and is typical of the syndrome of Korsakoff’s psychosisww (amnesic dementia). Confabulation can be tested by asking the patient whether he or she has met you before. However, be prepared for the very long, detailed and completely false story that may follow.
Frontal lobe function
Frontal lobe damage as a result of tumours or surgery (or both), or diffuse disease such as dementia or HIV infection, may cause changes in emotion, memory, judgment, carelessness about personal habits, and disinhibition. There may be persistent or alternating irritability and euphoria.xx These features may be clear when the history is taken but may need to be reinforced by interviewing relatives or friends. Changes of this sort in a previously reserved personality may be obvious and very distressing to relatives.
Assess first the primitive reflexes. There is controversy concerning their significance; they are not normally present in adults but may reappear in normal old age.19 The presence of an isolated primitive reflex may not be abnormal, but multiple primitive reflexes are usually associated with diffuse cerebral disease involving the frontal lobes and frontal association areas more than other parts of the brain. Dementia, encephalopathy and neoplasms are all possible causes.
Next ask the patient to interpret a proverb, such as ‘A rolling stone gathers no moss’. Patients with frontal lobe disease give concrete explanations of proverbs. Test for loss of smell (anosmia) and for gait apraxia, where there is marked unsteadiness in walking, which can be bizarre—the feet typically behave as if glued to the floor, causing a strange shuffling gait. Look in the fundi; you may rarely see optic atrophy on the side of a frontal lobe space-occupying lesion caused by compression of the optic nerve, and papilloedema on the opposite side due to secondarily raised intracranial pressure (Foster Kennedyyy syndrome).