[level-membership-for-cardiovascular-category]22
The Intercalated Disc
A Molecular Network That Integrates Electrical Coupling, Intercellular Adhesion, and Cell Excitability
Intercalated Disc Proteins in Inherited and Acquired Diseases
Structural Features of the Intercalated Disc
Ion Channel Complexes That Reside at the Intercalated Disc
Noncanonical Functions of Intercalated Disc Molecules
The Perinexus as a Site of Functional Integration
Subcellular Localization of Ion Channels and Cell-Cell Propagation: Gap Junction–Independent Electrical Coupling and the Importance of the Intercellular Cleft
Historical Perspective
This chapter focuses on the intercalated disc, a region of specialization formed at the end-end site of contact between cardiac myocytes. When first observed through light microscopy (in 1866), the intercalated disc was considered “a cementing material” at cardiac cell boundaries. However, the scientific community at the time was divided on whether cardiac cells were separate from each other or fused into a single syncytium. The latter hypothesis was in fact favored by most during the early twentieth century. The advent of electron microscopy eventually settled this debate. The studies of Sjostrand and Andersson1 and others showed that the intercalated disc consisted of a double membrane, flanked by the termination of myofibrils in dense material. Their observations led Muir2 to conclude that “the discs represent the junctions between neighboring cardiac muscle cells.” He wrote that “there is no valid evidence to contest the statement that the intercalated discs are specialized regions of cellular adhesion.” Since then, and as a result of the pioneering electrophysiology experiments of Weidmann3 and other ultrastructural observations,4 the intercalated disc has been recognized as an area of specialization that provides a physical continuum between cardiac cells through mechanical junctions (desmosomes, adherens junctions) and intercellular channels (gap junctions).
The availability of immunofluorescence microscopy allowed the demonstration that other molecular complexes, not detectable by electron microscopy, are also present in the intercalated disc. Of particular relevance to this chapter is the fact that channel protein complexes involved in both depolarization and repolarization localize preferentially to the intercalated disc. This physical proximity allows for a key functional consequence; molecules traditionally defined as junctional, such as connexin43 (Cx43) and plakophilin-2 (PKP2), actually regulate the function of ion channels responsible for the action potential. In turn, molecule accessories to ion channels are also relevant for cell adhesion and gap junction function.5–7 These data support the notion that the intercalated disc is not just the site of residence of independent junctional and nonjunctional complexes that are oblivious to the presence and function of the others. It is, rather, the home of a protein interacting network (an interactome) where molecules multitask to achieve jointly, intimately related functions: the entry and exit of charge into the cell, the transfer of charge between cells, and the anchoring of cells to each other, which provides a mechanically stable environment critical to ion channel function.
Intercalated Disc Proteins in Inherited and Acquired Diseases
Initial studies on the relation between desmosome integrity and cardiac electrophysiology were propelled by the finding that most familial cases of arrhythmogenic right ventricular cardiomyopathy (ARVC) in which a genetic link has been found associate with mutations in genes coding for desmosomal molecules.8 The latter brought forth the question of how a molecule considered purely relevant to cell adhesion altered the electrical behavior of the heart. The associations between desmosomal proteins and ion channel function (particularly the sodium current INa) are extensively reviewed in this chapter. (Recent publications refer to this disease as “arrhythmogenic cardiomyopathy,” to note the occurrence of left ventricular involvement.8)
A number of studies have demonstrated remodeling of gap junction proteins (in particular Cx43) in a number of inherited and acquired arrhythmia-related diseases.9–13 Recently, the Fishman laboratory provided evidence that aberrant posttranslational phosphorylation of Cx43 could be the common pathway leading to pathologic gap junction remodeling and arrhythmias.10,14 Although there is a relationship between Cx43 remodeling and arrhythmias, it is unclear whether these arrhythmias are exclusively consequent to changes in gap junction formation.15 Recent data show that reduced Cx43 expression alters sodium and potassium current function, and these changes could become part of the arrhythmogenic substrate.16–18
Disruption of the voltage-gated sodium channel complex is considered an important molecular substrate for arrhythmogenesis. Extensive reviews on the relation between mutations in proteins of the voltage-gated sodium channel (VGSC) complex and arrhythmias are available.19 Of particular interest in the context of this review is the observation that haploinsufficiency of a desmosomal protein, or overexpression of a mutant desmosomal protein, leads to INa deficit and increased arrhythmia susceptibility.20,21 The latter recalls the previously formulated concept that ARVC and Brugada syndrome (a channelopathy caused by mutations on genes of the VGSC complex) share common features22 and that, as Corrado et al.23 stated, there is the possibility of “an overlap in clinical manifestation and mechanisms of ventricular arrhythmias between patients with ARVC and Brugada syndrome.”
Structural Features of the Intercalated Disc
In its classic definition, the intercalated disc is composed of three electron-dense structures: adherens junctions, desmosomes, and gap junctions (Figure 22-1). Additional reviews on the characteristics of these structures can be found elsewhere.24,25 These structures are described briefly in the following sections, and the structural and molecular definitions of the intercalated disc are expanded to include the area composita, the intercellular space, and the nonjunctional ion channel complexes.
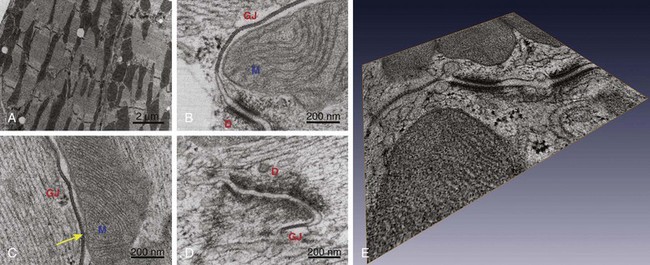
Figure 22-1 Transmission electron micrographs of intercalated disc in adult murine heart. Tissue prepared by high-pressure freezing and freeze substitution, thus greatly improving structural preservation. A, Low magnification to demonstrate the entire length of the intercalated disc as it meanders between two cells. B and C, Proximity (and contact in C, yellow arrow) between mitochondria, gap junctions, and desmosomes. D, Example of close proximity between a gap junction and a desmosome. E, Compiled three-dimensional tomographic electron microscopy reconstruction of the intercalated disc. Notice the extensive vesicular activity between the two cells. D, Desmosomes; GJ, gap junction; M, mitochondria. (Reproduced from Delmar M, Liang FX: Connexin43 and the regulation of intercalated disc function. Heart Rhythm 9:835–838, 2012.)
Adherens Junctions
Adherens junctions are specialized structures essential for the mechanical coupling between neighboring cells. The three morphologically different forms of adherens junctions are puncta adherentia, zonula adherens, and fascia adherens, with the last name corresponding to the morphology found in the cardiac intercalated disc.26 Cell-cell mechanical anchoring occurs at two crucial points: the extracellular space, within which cadherins tightly bind to each other, and the intracellular space, within which the cytoplasmic end of cadherin is indirectly attached to the actin cytoskeleton. The association between cadherin and the cytoskeleton involves at least two molecular “hinges”; cadherin binds to β-catenin and plakoglobin, and both molecules in turn bind to α-catenin (among others), the latter being in direct contact with actin. This is only a simplified description, because other interactions are likely to occur.27 This string of intermolecular interactions provides mechanical continuity between cells, allowing for the mechanical work of individual myocytes to integrate into the pumping function of the heart.
Desmosomes
The desmosome (macula adherens) appears as two parallel tripartite plaques containing an intercellular gap of approximately 30 nm bisected by a distinct line, parallel to the apposed cell membranes (see Figure 22-1).28 Desmosomes contribute to mechanical continuity between cardiac cells. Whereas adherens junctions link the actin cytoskeleton of adjacent cells, desmosomes provide continuity to the intermediate filament network (mainly desmin, in the case of heart).28,29 In the extracellular space, desmosomal cadherins (desmocollins and desmogleins) bind tightly to each other. In the intracellular space, the intermediate filaments bind to desmoplakin. The interaction between desmoplakin and the desmosomal cadherin can be in some cases direct, but it mostly occurs through their association with plakophilin and plakoglobin.28,29 The topologic organization of desmosomal molecules was studied by North et al.30 using quantitative immunogold electron microscopy. More recently, Al-Amoudi et al.31 solved the three-dimensional molecular structure of the desmosomal plaque. Overall, structural and biochemical evidence combined show that desmoplakin binds to plakophilin through their N-terminal domains,28,32 whereas desmoplakin binds to the intermediate filament by way of its C-terminal domain,28,31 yielding a highly organized structure.
Hatsell and Cowin29 once described the desmosome as “a system as staid and solid as the queen’s corsets.”29 With that analogy, it is easy to imagine the loss of containment that would follow in its absence. Mice deficient in plakoglobin, desmoplakin, or plakophilin-2 (PKP2), die during embryonic development as a result of severe myocardial rupture.33–35 More relevant from the point of view of clinical cardiology, ARVC in humans has been linked to mutations in desmosomal proteins.36 The relation between various complexes of the intercalated disc and ARVC is discussed later in this chapter and in other review articles.24,37
The Area Composita
Recent immunoelectron microscopy studies revealed the presence of a structure with mixed features of desmosomes and adherens junctions, dubbed the area composita.38 This structure is found only in the heart of higher vertebrate species including mouse and humans.39,40 The combination of components of the desmosomes and the adherens junctions allows anchorage of actin and desmin filaments to the same point, perhaps providing additional strength and flexibility to the muscle cell. Knockdown of PKP2 in neonatal cardiomyocytes leads to remodeling of the area composita.41 Additional studies suggest a role for α-catenin in the maintenance of these hybrid junctions.42 Interestingly, knockdown of α-catenin leads to PKP2 decrease only at the area composita and not at the desmosomes, suggesting that the molecular composition of the area composita, and its regulation, can be independent from that of other junctions.42 The area composita may represent a physical space where mechanical junction proteins interact with ion channel complexes.
Transcriptional Regulation by Mechanical Junction Proteins
Catenins are a part of both adherens junctions and desmosomes, thus participating in cell adhesion; however, these proteins also act as transcriptional activators. A prominent example is the participation of β-catenin in canonical Wnt signaling.43 Binding of Wnt to its Frizzled receptor leads to an increase in levels of cytosolic β-catenin and a consequent translocation of the protein to the nucleus, where it binds to Tcf/Lef complex and promotes the expression of various genes such as c-Myc or c-Fos. In the heart, the Wnt signaling and the activation of genes by β-catenin/Tcf/Lef has been associated with the regulation of physiologic and pathologic growth of the cardiomyocytes.44 Plakoglobin, another protein of the armadillo family, shows high homology with β-catenin and has been associated with the Wnt signaling. Different studies have shown that plakoglobin interacts and competes with β-catenin at multiple levels, acting as an antagonist of the Wnt/β-catenin signaling.45–47 The fact that desmosomal proteins are involved in the regulation of the Tcf/Lef complex has been invoked as a possible mechanism for the fibrofatty infiltration common in hearts affected with ARVC.48–50
Gap Junctions
In 1958, Sjostrand et al.4 described an area of specialization in the cardiac intercalated disc composed of “three dark lines with two intervening less dense lines”. This structure, which was similar to the one previously identified in the giant axon of the crayfish, was named the “longitudinal connexion” by these investigators. Years later, Revel coined the term gap junctions, thus emphasizing two key features: a gap between the cells and a junction between them.
The importance of Cx43 in the propagation of the cardiac action potential is well established. If Cx43 channels are not present, normal propagation is disrupted and lethal arrhythmias can ensue.51,52 Gap junction remodeling has been studied for various inherited and acquired diseases.10,11,14,53–55 The implications of connexin remodeling to electrophysiology are discussed later in this chapter.
Intercellular Space
The size of the space separating two cardiac cells at the intercalated disc changes depending on the proximity to the various structures, as well as the vesicular activity between the two cells (see Figure 22-1). It is a common view that the intercellular space is not relevant for electrophysiology. This view, however, is changing. Mathematical modeling studies56,57 and experimental evidence58 support the idea that the intercellular space is critical to propagation via an electric field mechanism.59 This model will be discussed later. Of note, increased size of the intercellular space has been reported in animal models of ARVC.20,21,60,61
Ion Channel Complexes That Reside at the Intercalated Disc
Voltage-Gated Sodium Channel Complex
In 1996, Cohen62 showed that cardiac sodium channel proteins were preferentially localized at the intercalated disc,62 although they are also present over the cell surface, following a striated pattern.63 Recent data emphasize how the VGSC interacts also with scaffolding, anchoring, and adhesions proteins, which regulate its function.
α-Subunit NaV1.5
The cardiac, pore-forming, α-subunit NaV1.5 contains four homologous transmembrane domains DI-DIV, linked by intracellular loops (IDI-II, IDII-III, IDIII-IV). Each domain is formed by the six transmembrane segments S1 to S6, and is involved in the voltage-dependent activation of the channel. The channel pore conducting Na+ is lined by the S6 segment and the S5-S6 pore loops in each domain.19 The inactivation gate is a complex formed by the DIII-IV loop and the C-terminus.64 This channel is defined as TTX-resistant, in contrast with other Nav channels. Of note, TTX-sensitive channel proteins, and currents, have also been found in cardiac myocytes, with the current localized primarily to the midsection of the cell.63,65
The SCN5A gene, on chromosome 3p21, codes for the NaV1.5 subunit. Mutations resulting in increased INa are associated with long QT syndrome type 3. Mutations that cause a reduction in INa are responsible for a series of different diseases, such as Brugada syndrome, progressive cardiac conduction defect, sick sinus syndrome, and a form of inherited atrial fibrillation. Some mutations are associated with a clinical spectrum encompassing more than one of those phenotypes and can manifest differently among carriers, even within the same family.19 Interestingly, some SCN5A mutations have been linked to a form of dilated cardiomyopathy with a high incidence of atrial and ventricular arrhythmias.66 It is not yet clear how mutations in the sodium channel could lead to structural damage of the myocardium. Based on the crosstalk between intercalated disc structures described in this chapter, we are tempted to speculate that the integrity of the sodium channel complex is also relevant to intercellular adhesion strength.20,21,67
β-Subunits of the Sodium Channel
The NaV β-subunit family consists of four proteins: β1-4, coded by genes SCN1B to SCN4B, respectively. These are single-span transmembrane proteins oriented with the amino terminus facing the extracellular space. The extracellular domain presents a conserved immunoglobulin domain, homologous to the one in cell adhesion molecules.68 The carboxyl terminus associates with cytoskeletal and scaffolding proteins. β1 and β2 are localized at the T-tubules–Z lines and at the intercalated discs in rat cardiac myocytes. β3 colocalizes with β1 at the T-tubules and β4 colocalizes with β2 at the intercalated disc. β1 and β2 associate with ankyrin-G and ankyrin-B in both brain and heart, and their interaction is critical for channel surface expression and modulates the channel function in vivo. β1 and β2 associate with N-cadherin and with Cx43 at the intercalated disc.69 Altogether, β-subunits have a key role in the interactions between the VGSC multiple proteins at the intercalated disc, including those relevant for cell adhesion and for electrical coupling between cells.69 Furthermore, work from the Isom lab has demonstrated that null mice for the β1 subunit show a significant increase in SCN5A mRNA in cardiac myocytes.70 Further research is needed to elucidate the role of β-subunits in transcription regulation in the heart.70 Overall, the data show that, as in the case of catenin and plakoglobin, β-subunits can have distal, contact-dependent effects, including regulation of gene transcription, with consequences to the function and structure of the heart.
Subcellular Heterogeneity of Voltage-Gated Sodium Channel
Recent studies have shown that not all sodium channels on the surface of the cardiac myocyte are equal. Instead, the molecular composition and the function of a sodium channel are different depending on whether NaV1.5 is localized to the intercalated disc or to the midsection of the cell. Petiprez et al.71 described two separate pools of VGSC in ventricular cardiomyocytes. One of these two subpopulations localizes at the lateral membrane of the myocytes, where NaV1.5 interacts with dystrophin and syntrophin; the second subpopulation of VGSC localizes at the ID, where NaV1.5 interacts with the MAGUK proteins SAP97 and ZO1, as well as with PKP2.72 Lin et al. recently demonstrated that this subcellular distribution correlates with differences in function.65 Using cell-attached macropatch recordings, these authors showed that the magnitude of INa is larger at the intercalated disc than in the midsection of the cell. Furthermore, TTX-resistant channels in the midsection showed a significant negative shift in the steady-state inactivation curve, suggesting that these channels are mostly in an inactivated state at the normal resting potential, and that the burden of excitation is on the channels at the intercalated disc. These authors also demonstrated that the amplitude of current is larger if cells remain paired, strongly suggesting that cell adhesion preserves sodium channel function. The interaction between sodium channels and proteins of the intercalated disc is discussed extensively later.
Potassium Channels at the Intercalated Disc
In 1995, Mays et al.73 described for the first time that the potassium channel protein KV1.5 localizes to the intercalated disc of adult myocardial cells. Later studies showed that KV1.5 associates with SAP97.74 Interestingly, SAP-97 also associates with NaV1.5,71 making this protein a candidate for mutual regulation of both depolarization and repolarization channels, as is the case of NaV1.5 and Kir2.1.75 Additional studies have shown that the function of KV1.5 depends on the expression of N-cadherin,76 an interesting parallelism to the interaction between PKP2 (a desmosomal molecule), and NaV1.5.72 Cheng et al.76 also showed that cortactin is required for the N-cadherin–dependent regulation of KV1.5. Of note, mice deficient in kcne2, an ancillary potassium channel subunit, display an impaired ventricular repolarization because of inhibition in the trafficking to the membrane of KV1.5.77
Another voltage-gated potassium channel that preferentially (though not exclusively) localizes to the intercalated disc is KV4.2; it is responsible for the rapid repolarization phase of the cardiac action potential.78,79 Finally, regarding Kir channels, two different subunits localize at the transverse tubules and at the intercalated disc in canine myocytes: Kir2.1 and Kir2.3.80
Noncanonical Functions of Intercalated Disc Molecules
Desmosomal Proteins Are Necessary for the Formation of Functional Gap Junctions
Studies on the relation between desmosome integrity and cardiac electrophysiology were motivated by the finding that most familial cases of ARVC where a genetic link has been found, result from mutations in genes coding for desmosomal molecules. Dr. Saffitz and his colleagues were first to propose a link between mechanical and electrical junctions.54,81 In a series of seminal studies, these investigators first provided evidence that the molecular phenotype of the ARVC-afflicted heart involves not only desmosomes, but other molecules of the intercalated disc as well.53,54 In particular, the authors showed a significant decrease in the abundance of Cx43 immunoreactive protein in the intercalated disc region of heart tissue obtained from affected patients. The hypothesis of a desmosome–gap junction crosstalk was confirmed in vitro by Oxford et al.,82 who used RNA silencing technology to reduce PKP2 expression in cardiac ventricular myocytes, as well as in epicardium-derived cells obtained from neonatal rat hearts. Their data showed that the loss of PKP2 expression led to a decrease in total Cx43 content, a significant redistribution of Cx43 to the intracellular space, and a decrease in dye coupling between cells. Separately, they demonstrated that Cx43 and PKP2 coexist in the same macromolecular complex. Follow up studies confirmed this observation,41,83,84 giving support to the notion that two complexes previously considered independent are in fact, functionally and molecularly interactive. Recent studies on samples obtained from hearts affected with ARVC have confirmed the notion that in most (though not all) cases, there is a significant loss of Cx43 immunoreactive signal associated with the loss of desmosomal integrity in the cardiac intercalated disc.85,86
Quantitative analysis in experimental models indicated that complete loss of PKP2 expression led to an approximate 50% decrease in gap junction–mediated cell-cell coupling.82 Previous studies had shown, however, that a 50% reduction in electrical coupling does not lead to significant changes in conduction velocity.87–89 We therefore speculated that in addition to interacting with gap junctions, desmosomal molecules can also interact with ion channel complexes that reside at the intercalated disc.
Desmosomal Molecules Are Necessary for Sodium Channel Function
The observations described earlier led to the question of whether Cx43 is the only molecule relevant to electrophysiology that interacts with PKP2. Given the preferential localization of the VGSC complex at the intercalated disc, we explored its possible cross-talk with desmosomal molecules. Single, adult cardiac myocytes were treated with small interfering RNA (siRNA) to prevent expression of PKP2. Control cells were treated with a nonsilencing construct. As shown in Figure 22-2, A, the amplitude of INa was significantly reduced in cells lacking PKP2 expression. This reduction in amplitude was observed across the voltage range (see Figure 22-2, B). In addition, loss of PKP2 expression caused a negative shift in the voltage dependence of steady-state inactivation (see Figure 22-2, C), and a slowing of recovery from inactivation (see Figure 22-2, D). These changes in the major excitatory current were reflected in a decrease in the velocity of action potential propagation in monolayers of neonatal rat ventricular myocytes (Figure 22-3). Interestingly, computer simulations showed that the changes in amplitude and kinetics of the INa represented the key substrate for the generation of reentrant activity in a two-dimensional model of cardiac cells.90 Overall, these data led us to propose that there is a functional crosstalk between a protein defined in the context of intercellular junctions (PKP2), and another complex primarily involved in supporting cell excitability (the VGSC complex). The results supported the hypothesis that arrhythmias that occur in patients with ARVC may have as a substrate not only changes in the macroscopic architecture of the tissue (as it would be expected once the fibrofatty infiltrations populate the heart), or in the integrity of intercellular coupling, but also changes in the electrical properties of the cardiac myocytes. Of relevance, these results revealed that a property “of the single cell” (excitability; inward sodium current [INa]) is in fact subject to modulation by proteins classically defined as belonging to the group of intercellular junction molecules.
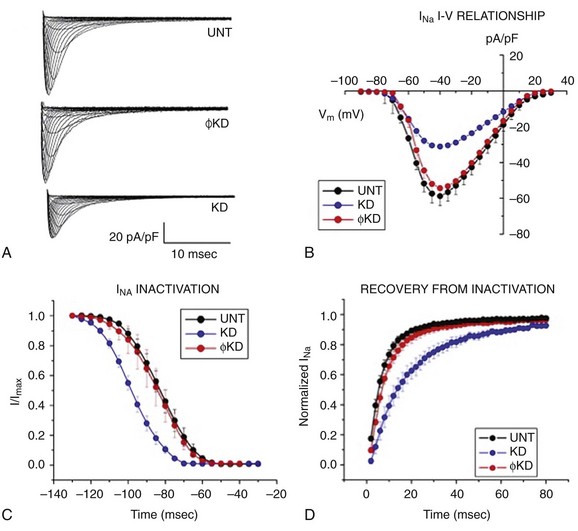
Figure 22-2 Voltage clamp data obtained from adult cardiomyocytes after knockdown of PKP2. Cells were untreated (UNT) or treated with a nonsilencing construct (ϕKD) or with oligonucleotides that prevent PKP2 expression (KD). A, Examples of sodium currents recorded under the three different conditions. B, Complete voltage dependence of sodium current density. C, Voltage dependence of steady-state inactivation. D, Recovery from inactivation. Notice that the loss of PKP2 expression decreased sodium current density, caused a negative shift in steady-state inactivation, and prolonged recovery from inactivation. Further details in.72
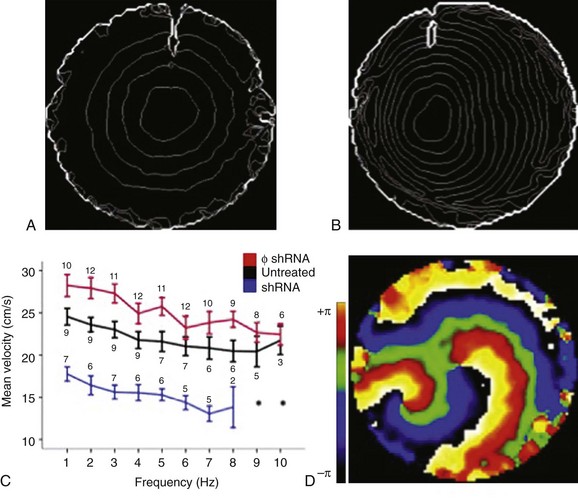
Figure 22-3 Optical mapping of action potential propagation in monolayers of neonatal rat ventricular myocytes. Isochrome maps from monolayers were treated with a nonsilencing construct (A) or silenced for PKP2 (B). C, Conduction velocity as a function of pacing frequency. D, A phase map of spontaneous reentrant activity in a PKP2-deficient monolayer. (Reproduced from Sato PY, Musa H, Coombs W, et al: Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes, Circ Res 105:523–526, 2009.)
PKP2 mutations associated with ARVC have all been found in only one allele. We therefore characterized the relation between PKP2 abundance and sodium current function in mice that were haploinsufficient for the pkp2 gene (PKP2-Hz).20 Of note, one of the most common mutations in PKP2 is the presence of a stop codon at amino acid position 79 (R79x).91 This early truncation is functionally equivalent to haploinsufficiency.92 Patch clamp experiments showed a decreased amplitude and a shift in gating and kinetics of INa in PKP2-Hz myocytes, compared with control myocytes. To further unmask INa deficiency, we exposed myocytes, Langendorff-perfused hearts and anesthetized animals to a pharmacologic challenge (flecainide). In PKP2-Hz hearts, the extent of flecainide-induced INa block, impaired ventricular conduction, and altered electrocardiographic parameters were larger than controls. As shown in Figure 22-4 and described by Cerrone et al.,20 flecainide provoked ventricular arrhythmias and death in PKP2-Hz animals, but not in wild type. These results showed that PKP2 haploinsufficiency leads to INa deficit in murine hearts, thus documenting for the first time the relation between the “desmosomal molecule” PKP2 and the VGSC complex in a living heart. Our results supported the contention that INa dysfunction contributes to generation and maintenance of arrhythmias in patients with desmosomal deficiency. It remains unclear whether pharmacologic challenges could help to unveil arrhythmia risk in patients with mutations or variants in PKP2.
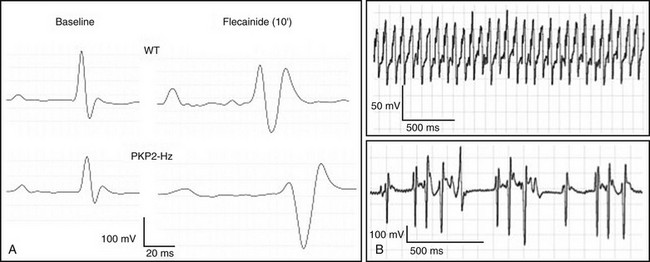
Figure 22-4 Electrocardiographic (ECG) features of PKP2-Hz mice at baseline, and in response to flecainide. A, Examples of ECG traces from wild type (top) and PKP2-Hz mice (bottom). Recordings obtained at baseline (left) and 10 minutes after flecainide (40 mg/kg intraperitoneally, right). B, ventricular tachycardia (VT) in PKP2-Hz mice. Overall, the flecainide caused a prolongation of the P and QRS durations, and of the PR and QTc intervals that were significantly more pronounced in PKP2-Hz animals than in control. Six of 12 PKP2-Hz showed ventricular arrhythmias. None of the 11 wild type mice tested presented ventricular arrhythmias. Arrhythmic death occurred in three PKP2-Hz animals and in none of the control animals. (Reproduced from Cerrone M, Noorman M, Lin X, et al: Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res 95:460–468, 2012.)
The relation between desmosomal molecules and the VGSC has also been demonstrated in mice overexpressing a desmoglein-2 (DSG-2) mutation.21 This model is interesting given that severe arrhythmias and sudden death in these mice often occur before structural damage; the latter mimics a phenomenon often observed in humans affected with ARVC, where arrhythmias and electrocardiographic changes have been described early in the history of the disease, before overt structural changes in the myocardium.24,54,93 Electrophysiological analysis of these DSG-2 transgenic mutant mice revealed prolonged ventricular activation time, decreased conduction velocity in both longitudinal and transverse directions, and increased arrhythmia susceptibility, prior to signs of fibrosis or necrosis in the myocardium. The authors also observed a decrease in maximal upstroke velocity and decreased INa amplitude. Taken together, the data demonstrate that a decrease in PKP2 abundance,20 as well as the overexpression of a DSG-2 mutation,21 lead to impaired INa function in hearts with no histologic features of ARVC. These data provide a demonstration in vivo of the interaction between desmosomal molecules and the VGSC, and they suggest impaired sodium current as a substrate for lethal arrhythmias in the concealed phase of ARVC, as proposed earlier.72
The question remains as to whether other desmosomal proteins also interact with NaV1.5, and whether these interactions occur in the human heart. Recently, Gomes et al.94 reported that mice that are haploinsufficient for desmoplakin present average peak INa density similar to control; however, careful analysis of their results suggests the possibility of technical limitations in their voltage clamp recordings, which could have masked small differences between the groups.20 However, in the same study, the authors showed that patients with heterozygous mutations in desmoplakin and without overt structural disease had significant regional conduction delays and heterogeneous NaV1.5 distribution.94 The possibility of changes in the abundance or colocalization of NaV1.5 in the intercalated disc area of the hearts of patients with ARVC is a matter of current investigation.
Connexin43 Regulates Sodium and Potassium Currents
The first indication of an interaction between Cx43 and the VGSC came from the work of Malhotra et al., showing coprecipitation of NaV1.5 with Cx43.95 The physical proximity of these molecules was recently confirmed by Rhett et al.96 Not only are Cx43 and NaV1.5 in close proximity, but these two molecules are also functionally intertwined. Indeed, as shown in Figure 22-5, Jansen et al.17 recently reported that siRNA-mediated loss of Cx43 expression in adult ventricular myocytes leads to a decrease in the amplitude of the INa. The functional effect coincided with decreased colocalization of Cx43 and NaV1.5 at the intercalated disc. A similar decrease in INa was later reported by Desplantez et al.18 in fetal atrial myocytes of Cx43-deficient mice.18 Overall, the data demonstrate that Cx43 expression is necessary for proper sodium current function, and for the accumulation of NaV1.5 at the cardiac intercalated disc. Interestingly, the regulation of INa by Cx43 is reciprocated by the fact that ankyrin-G, a molecule that is well characterized as a component of the VGSC complex, is necessary to preserve gap junction–mediated coupling between neonatal myocytes.67
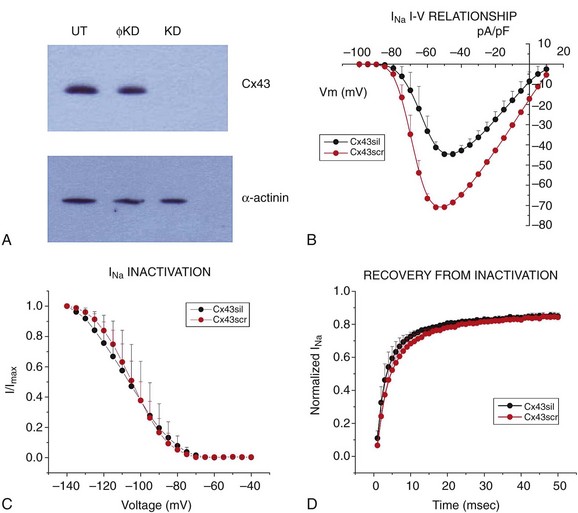
Figure 22-5 Decreased connexin43 (Cx43) expression leads to reduced INa in isolated adult rat ventricular myocytes. A, Western blot for Cx43 in cells untreated (UT), treated with an oligonucleotide that prevented Cx43 expression (KD) or a non-targeting construct (ϕKD). B, Peak sodium current density was lower in cells lacking Cx43. C and D, Loss of Cx43 expression did not affect steady-state inactivation or recovery from inactivation kinetics. (Reproduced from Jansen JA, Noorman M, Musa H, et al: Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 9:600–607, 2012.)
The VGSC is not the only electrically functional complex of the intercalated disc that is disrupted consequent to loss of Cx43 expression. In fact, the first report correlating Cx43 expression to nonjunctional currents was by Danik et al.16 These authors noted that the action potential duration recorded from the ventricle of Cx43 conditional knockout animals were significantly shorter than control. This shortening associated with higher levels of sustained repolarizing current and higher levels of inward rectifier current in myocytes from the right ventricle. Overall, the data show that Cx43 is not only a gap junction–forming molecule in the heart but also, a component of a molecular network that regulates excitability and repolarization.
AnkG and Cx43 Are Necessary to Maintain Intercellular Adhesion
The previous sections have described that molecules of the desmosome, relevant to intercellular adhesion (namely PKP2 and DSG-2), are actually important to preserve INa amplitude and electrical coupling between cardiac cells. We then speculate that, if the system works as a unit, decreased protein levels of either Cx43 or a component of the VGSC would affect intercellular adhesion strength. Consistent with this hypothesis, we have shown that intercellular adhesion strength is decreased in monolayers of neonatal rat ventricular myocytes treated with siRNA to prevent expression of AnkG.67 Similarly, loss of Cx43 expression in cultured cells significantly impaired intercellular adhesion strength,7 a result consistent with previous observations.97
The “α Personality” of the β-Subunit: Intercellular Adhesion and the Sodium Current
The finding that mutations in desmosomal molecules associate with familial cases of ARVC has highlighted the important link between cell adhesion and sodium channel function. In retrospect, the link between these two seemingly unrelated functions was first established several years ago by the Isom lab. In 1981, Hartshorne and Caterall98 purified “the saxitoxin receptor of the sodium channel from rat brain” and identified two polypeptides, which they referred to as “α” and “β.”98 They proposed that these two subunits conformed the functional sodium channel. In 1992, Isom et al.99 isolated the cDNA, sequenced and functionally expressed the β-1 subunit, concluding that this protein is crucial to the overall function of the sodium channel. In this manner, this 22,581-d protein was labeled as a “β” for its “α,” a subunit merely accessory to sodium channel function. It was 8 years later (in 2000) that the Isom lab demonstrated that “sodium channel beta subunits” also mediate cell adhesion,100 an important fact in the formation of the sodium channel complex100,101 and in sodium channel–independent functions such as cell migration, cell aggregation, and interaction with the cytoskeleton.102 Studies that followed demonstrated their critical role in cancer.103 Interestingly, the fact that β-subunits are actually independent adhesion molecules has, for the most part, missed the attention of cardiac electrophysiologists. Another case of a molecule’s identity being boxed into the function related to its original discovery. If that molecule had been found through cell adhesion assays and called adherin, perhaps its role as a modulator of INa could have gone unnoticed for years. The fact is that the sodium channel β-subunits are, together with PKP2, DSG-2, AnkG, and Cx43, members of a growing family of molecules that regulate sodium channel function and intercellular adhesion strength. Whether β-subunits regulate gap junction–mediated coupling (as the other molecules listed earlier) is a matter of future investigation.
Other Intercellular Adhesion Molecules That Cross-Talk with Cardiac Ion Channels
Two other adhesion molecules have been associated with cardiac electrical function: N-cadherin and the coxsackievirus and adenovirus receptor (CAR). N-Cadherin is well recognized as critical to the mechanical coupling between cells. However, Li et al.104 showed that restricted cardiac deletion of N-cadherin also leads to severe arrhythmias, and a loss of Cx43 from the intercalated disc. More recently, the Radice lab reported that in addition to effects on coupling, loss of N-cadherin leads to decreased density of the repolarizing current IK.slow, concurrent with decreased expression of KV1.5 and its accessory protein Kcne2.76 The properties of the INa in cadherin-deficient hearts remain undefined.
CAR is another case of a molecule whose identity is burdened by its birth name. Undoubtedly a receptor for both coxsackievirus and adenovirus, studies have demonstrated that the immunoglobulin extracellular domains of CAR are capable of homophilic binding and participate in intercellular adhesion in epithelial cells. The role of this molecule on cell adhesion in the heart is less defined. Interestingly, cardiac-restricted deletion of CAR causes significant slowing of A-V propagation and disruption of gap junctions at the intercalated disc.105,106 Research is in progress to determine whether, as other molecules with immunoglobulin extracellular domains (such as the sodium channel β-subunits), CAR expression modulates sodium current function.
The Perinexus as a Site of Functional Integration
The electron micrographic images shown in Figure 22-1 demonstrate the proximity between electron-dense structures (e.g., gap junctions to desmosomes). Distances could be even shorter for molecules in the perimeter of either a gap junction or a desmosome plaque. In that context, it is important to mention that Cx43 at the intercalated disc is not exclusively circumscribed to the gap junctions. Rhett et al.107 demonstrated that the area surrounding a gap junction plaque is populated by non–gap junction–forming Cx43 connexons. Their studies showed that Cx43 in this area, which they dubbed the perinexus, closely associates with the scaffolding protein zonula occludens-1 (ZO-1); more importantly, they demonstrated that the Cx43–ZO-1 association in the perinexus limits the abundance of Cx43 within the gap junction plaque (Figure 22-6). As such, loss of the Cx43–ZO-1 interaction increases the proportion of Cx43 involved in gap junction channel formation at the expense of a non–channel-forming pool.
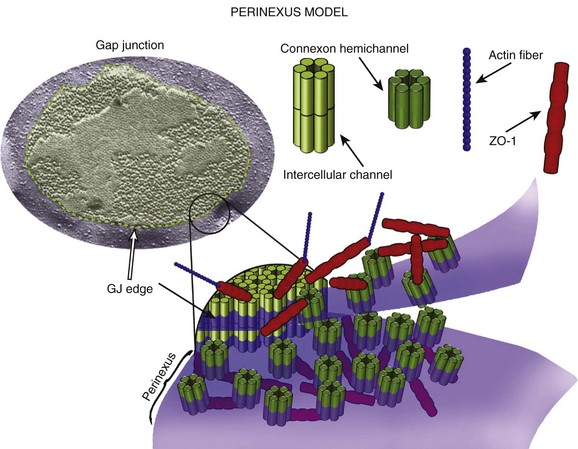
Figure 22-6 Diagram of the perinexus, described by Rhett et al.107 According to the model, connexons gather in the periphery of the gap junction. The interaction with ZO-1 prevents the transfer of the connexon into the gap junction plaque. Importantly, the connexon in the perinexus is not forming gap junctions. Instead, the perinexus may be the region where Cx43 interacts with other intercalated disc proteins, such as NaV1.5, or PKP2. (Reproduced from Rhett JM, Gourdie RG: The perinexus: a new feature of Cx43 gap junction organization. Heart Rhythm 9:619–623, 2012.)
The finding of a separate pool of Cx43 in the intercalated disc, invites speculation as to its possible function. It is unlikely that the Cx43 molecules in the perinexus are simply in standby mode, waiting idle for the signal to move into the gap junction plaque. It is also unlikely that Cx43 and ZO-1 are the only inhabitants of the perinexal space. Rather, this pool of Cx43 is likely to be exposed to a variety of other molecular complexes that approach, but are not components of, the gap junction plaque. In this space, Cx43 can act as accessory to the function of other molecules, such as NaV1.5. Biochemical evidence of a physical proximity between Cx43 and NaV1.5 was first reported by Malhotra et al.95 Recently, Rhett et al.96 used a proximity ligation assay (PLA; also known by the proprietary name of Duolink) to demonstrate, in neonatal rat ventricular myocytes, that Cx43 and NaV1.5 co-inhabit the perinexus. Loss of Cx43 in this space can lead to reduction in INa.17,18,65 Overall, although still speculative, there is reason to believe that the perinexus represents the physical space where Cx43-dependent, gap junction–independent functions take place. The data draw a portrait of Cx43 as the highly regulatable molecule that has been extensively described and as a regulator of the function of other molecular complexes.
Subcellular Localization of Ion Channels and Cell-Cell Propagation: Gap Junction–Independent Electrical Coupling and the Importance of the Intercellular Cleft
Several mathematical models of cardiac action potential propagation assume that gap junctions are the only path for transfer of charge between cells. Accordingly, they predict that decreases in junctional conductance bring about decreases in conduction velocity. This notion contrasts sharply with actual data showing that only extreme reductions in Cx43 abundance (and electrical coupling) lead to significant changes in conduction velocity.51,88,108 These results have given new impetus to the notion that, under poor gap junction–mediated coupling, propagation can be maintained via a separate “electric field mechanism.”56,57,59,109 This alternative postulates that the large INa in the proximal side of an intercellular cleft generates a negative extracellular potential within the cleft, which depolarizes the distal membrane and activates its sodium channels. Thus, propagation can continue downstream in the absence of gap junctions if there is a large INa at the intercalated disc and a narrow intercellular cleft separating the two opposing cells.
The model described here indicates the importance of the intercellular space as a critical element of cell-cell propagation. Indeed, if the width of the intercellular cleft increases, the model predicts that propagation would fail. Interestingly, separation of the intercellular space is one of the features observed in murine models of desmosomal protein deficiency, even when overt structural damage is not apparent.20,21 Figure 22-7 shows an example of tomographic electron microscopy of adult heart tissue obtained from a mouse heterozygous for PKP2. The image is reproduced from Cerrone et al.20 These images can be compared with those shown in Figure 22-1, obtained from a control animal. The results in Figure 22-7 show an expanded intercellular space that coincides with the presence of membrane invaginations in one side of the intercalated disc. Planes of the same section (see Figure 22-3, A–F) reveal that the invaginations extended several nanometers into the intracellular space; in some planes, the invaginations seemed to “pinch off,” leaving a healed membrane continuum facing the intercellular cleft. This observation was confirmed in three separate samples analyzed by tomographic electron microscopy (T-EM), and not found in controls. Clearly, this membrane separation would represent a barrier for transfer of charge mediated via an electrical field mechanism. The importance of the intercellular cleft has also been highlighted by the recent work of Veeraraghavan et al.58 and his collaborators, showing that changes in intercellular volume affect action potential propagation in the heart.
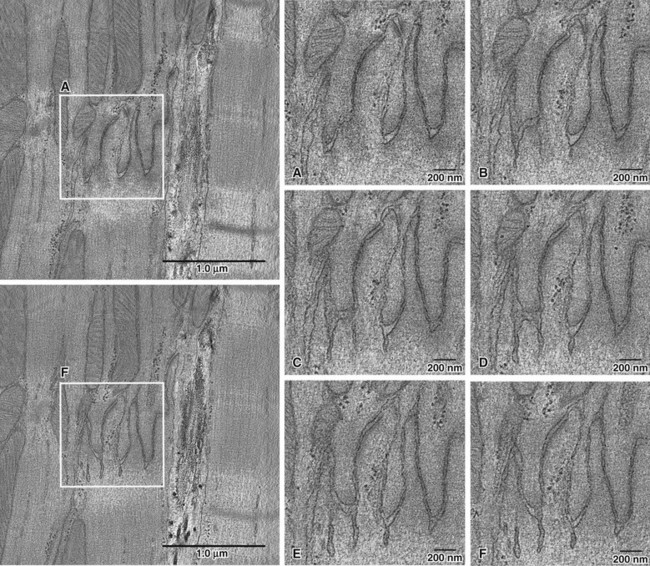
Figure 22-7 Tomographic electron microscopy images of intercalated disc in a PKP2-Hz adult heart. Outlined regions in the left panels are expanded for insets a-f. Notice the enlarged intercellular space that bulges into the cell, leaving large clefts between cells in some planes. Whether this increased gap between cells impairs electric field-mediated propagation, remains to be defined. (Reproduced from Cerrone M, Noorman M, Lin X, et al: Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res 95:460–468, 2012.)
Conclusions
This chapter has presented evidence showing that molecules classically defined as belonging to the desmosome, the VGSC, and the gap junctions, interact with each other. We have also described how, through these interactions, the function of one complex (e.g., the VGSC) is altered by changes in the expression of molecules of a different group (e.g., Cx43). The evidence suggests that intercalated disc molecules are not necessarily constrained to a single function (e.g., Cx43 is not only limited to making gap junctions). We propose that molecules of the intercalated disc multitask within a protein interacting network, working in concert toward one common function: the propagation of excitatory current from one cell to the next. From this perspective, Cx43 is a molecule that is relevant to cell excitability (by modulating INa),17,18 sodium channels can support cell-cell electrical coupling,56,57 and “adhesion molecules” are actually required for proper function of electrical complexes20 and for propagation across an intercellular cleft. In the balance, multitasking canprovide functional overlap, thus enhancing the safety factor for propagation. Future research will discern the spatial relations between these molecules. The novel concept of the perinexus110 can be extended to include the neighboring areas of mechanical junctions, or clusters of NaV1.5 molecules. Just as a mitochondrion is not just a conglomerate of independent molecules, but is an organelle sharing common functions, the intercalated disc is a single functional unit composed of molecules that interact with each other. The implication of these interactions to the understanding of arrhythmia mechanisms and arrhythmia treatment emerges as an exciting area of future investigation.
References
1. Sjostrand, FS, Andersson, E. Electron microscopy of the intercalated discs of cardiac muscle tissue. Experientia. 1954; 10(9):369–370.
2. Muir, AR. An electron microscope study of the embryology of the intercalated disc in the heart of the rabbit. J Biophys Biochem Cytol. 1957; 3(2):193–202.
3. Weidmann, S. The electrical constants of Purkinje fibres. J Physiol. 1952; 118(3):348–360.
4. Sjostrand, FS, Andersson-Cedergren, E, Dewey, MM. The ultrastructure of the intercalated discs of frog, mouse and guinea pig cardiac muscle. J Ultrastruct Res. 1958; 1(3):271–287.
5. Delmar, M. Connexin43 regulates sodium current; ankyrin-G modulates gap junctions: the intercalated disc exchanger. Cardiovasc Res. 2012; 93(2):220–222.
6. Delmar, M, Liang, FX. Connexin43 and the regulation of intercalated disc function. Heart Rhythm. 2012; 9(5):835–838.
7. Agullo-Pascual, E, Delmar, M. The Noncanonical Functions of Cx43 in the Heart. J Membr Biol. 2012; 245(3):477–482.
8. Basso, C, Bauce, B, Corrado, D, et al. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2011; 9(4):223–233.
9. Fontes, MS, van Veen, TA, de Bakker, JM, et al. Functional consequences of abnormal Cx43 expression in the heart. Biochim Biophys Acta. 2011.
10. Qu, J, Volpicelli, FM, Garcia, LI, et al. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res. 2009; 104(3):365–371.
11. Akar, FG, Nass, RD, Hahn, S, et al. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007; 293(2):H1223–H1230.
12. Zhang, Y, Wang, H, Kovacs, A, et al. Reduced expression of Cx43 attenuates ventricular remodeling after myocardial infarction via impaired TGF-beta signaling. Am J Physiol Heart Circ Physiol. 2010; 298(2):H477–H487.
13. Smyth, JW, Hong, TT, Gao, D, et al. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest. 2010; 120(1):266–279.
14. Remo, BF, Qu, J, Volpicelli, FM, et al. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res. 2011; 108(12):1459–1466.
15. Chkourko, HS, Guerrero-Serna, G, Lin, X, et al. Remodeling of mechanical junctions and of microtubule-associated proteins accompany cardiac connexin43 lateralization. Heart Rhythm. 2012; 9(7):1133–1140.
16. Danik, SB, Rosner, G, Lader, J, et al. Electrical remodeling contributes to complex tachyarrhythmias in connexin43-deficient mouse hearts. FASEB J. 2008; 22(4):1204–1212.
17. Jansen, JA, Noorman, M, Musa, H, et al. Reduced heterogeneous expression of Cx43 results in decreased Nav1. 5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart rhythm. 2012; 9(4):600–607.
18. Desplantez, T, McCain, ML, Beauchamp, P, et al. Connexin43 ablation in foetal atrial myocytes decreases electrical coupling, partner connexins, and sodium current. Cardiovasc Res. 2012; 94(1):58–65.
19. Wilde, AA, Brugada, R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011; 108(7):884–897.
20. Cerrone, M, Noorman, M, Lin, X, et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 2012; 95:460–468.
21. Rizzo, S, Lodder, EM, Verkerk, AO, et al. Intercalated disc abnormalities, reduced Na+ current density and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012.
22. Corrado, D, Buja, G, Basso, C, et al. What is the Brugada syndrome? Cardiol Rev. 1999; 7(4):191–195.
23. Corrado, D, Basso, C, Buja, G, et al. Right bundle branch block, right precordial st-segment elevation, and sudden death in young people. Circulation. 2001; 103(5):710–717.
24. Delmar, M, McKenna, WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010; 107(6):700–714.
25. Bass-Zubek, AE, Godsel, LM, Delmar, M, et al. Plakophilins: multifunctional scaffolds for adhesion and signaling. Curr Opin Cell Biol. 2009; 21(5):708–716.
26. Gottardi, CJ, Niessen, CM, Gumbiner, BM. The adherens junction. In: Beckerle MC, ed. Cell Adhesion. ed 1. Oxford: Oxford University Press; 2001:259–287.
27. Gates, J, Peifer, M. Can 1000 reviews be wrong? Actin, alpha-Catenin, and adherens junctions. Cell. 2005; 123(5):769–772.
28. Bannon, LJ, Goldfinger, LE, Jones, JCR, et al. Desmosomes and hemidesmosomes. In: Beckerle MC, ed. Cell Adhesion. ed 1. Oxford: Oxford University Press; 2001:324–368.
29. Hatsell, S, Cowin, P. Deconstructing desmoplakin. Nat Cell Biol. 2001; 3(12):E270–272.
30. North, AJ, Bardsley, WG, Hyam, J, et al. Molecular map of the desmosomal plaque. J Cell Sci. 1999; 112(Pt 23):4325–4336.
31. Al-Amoudi, A, Castano-Diez, D, Devos, DP, et al. The three-dimensional molecular structure of the desmosomal plaque. Proc Natl Acad Sci U S A. 2011; 108(16):6480–6485.
32. Chen, X, Bonne, S, Hatzfeld, M, et al. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem. 2002; 277(12):10512–10522.
33. Gallicano, GI, Kouklis, P, Bauer, C, et al. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol. 1998; 143(7):2009–2022.
34. Grossmann, KS, Grund, C, Huelsken, J, et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 2004; 167(1):149–160.
35. Ruiz, P, Brinkmann, V, Ledermann, B, et al. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 1996; 135(1):215–225.
36. Sen-Chowdhry, S, Syrris, P, McKenna, WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005; 16(8):927–935.
37. Delmar, M. Desmosome-ion channel interactions and their possible role in arrhythmogenic cardiomyopathy. Pediatr Cardiol. 2012.
38. Franke, WW, Borrmann, CM, Grund, C, et al. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 2006; 85(2):69–82.
39. Borrmann, CM, Grund, C, Kuhn, C, et al. The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Eur J Cell Biol. 2006; 85(6):469–485.
40. Pieperhoff, S, Franke, WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. VI. Different precursor structures in non-mammalian species. Eur J Cell Biol. 2008; 87(7):413–430.
41. Pieperhoff, S, Schumacher, H, Franke, WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. V. The importance of plakophilin-2 demonstrated by small interference RNA-mediated knockdown in cultured rat cardiomyocytes. Eur J Cell Biol. 2008; 87(7):399–411.
42. Li, J, Goossens, S, van Hengel, J, Gao, E, et al. Loss of alphaT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J Cell Sci. 2012; 125(Pt 4):1058–1067.
43. Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127(3):469–480.
44. Chen, X, Shevtsov, SP, Hsich, E, et al. The beta-catenin/T-cell factor/lymphocyte enhancer factor signaling pathway is required for normal and stress-induced cardiac hypertrophy. Mol Cell Biol. 2006; 26(12):4462–4473.
45. Chen, S, Guttridge, DC, You, Z, et al. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J Cell Biol. 2001; 152(1):87–96.
46. Longo, KA, Kennell, JA, Ochocinska, MJ, et al. Wnt signaling protects 3T3-L1 preadipocytes from apoptosis through induction of insulin-like growth factors. J Biol Chem. 2002; 277(41):38239–38244.
47. Ross, SE, Hemati, N, Longo, KA, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000; 289(5481):950–953.
48. Garcia-Gras, E, Lombardi, R, Giocondo, MJ, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006; 116(7):2012–2021.
49. Lombardi, R, Dong, J, Rodriguez, G, et al. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 2009; 104(9):1076–1084.
50. Lombardi, R, da Graca Cabreira-Hansen, M, Bell, A, et al. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 2011; 109(12):1342–1353.
51. Danik, SB, Liu, F, Zhang, J, et al. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004; 95(10):1035–1041.
52. Yao, JA, Gutstein, DE, Liu, F, et al. Cell coupling between ventricular myocyte pairs from connexin43-deficient murine hearts. Circ Res. 2003; 93(8):736–743.
53. Kaplan, SR, Gard, JJ, Carvajal-Huerta, L, et al. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 2004; 13(1):26–32.
54. Kaplan, SR, Gard, JJ, Protonotarios, N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 2004; 1(1):3–11.
55. Oxford, EM, Everitt, M, Coombs, W, et al. Molecular composition of the intercalated disc in a spontaneous canine animal model of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2007; 4(9):1196–1205.
56. Mori, Y, Fishman, GI, Peskin, CS. Ephaptic conduction in a cardiac strand model with 3D electrodiffusion. Proc Natl Acad Sci U S A. 2008; 105(17):6463–6468.
57. Tsumoto, K, Ashihara, T, Haraguchi, R, et al. Roles of subcellular Na+ channel distributions in the mechanism of cardiac conduction. Biophys J. 2011; 100(3):554–563.
58. Veeraraghavan, R, Salama, ME, Poelzing, S. Interstitial volume modulates the conduction velocity-gap junction relationship. Am J Physiol Heart Circ Physiol. 2012; 302(1):H278–H286.
59. Sperelakis, N. An electric field mechanism for transmission of excitation between myocardial cells. Circ Res. 2002; 91(11):985–987.
60. Kant, S, Krull, P, Eisner, S, et al. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res. 2012; 348(2):249–259.
61. Kostetskii, I, Li, J, Xiong, Y, et al. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005; 96(3):346–354.
62. Cohen, SA. Immunocytochemical localization of rH1 sodium channel in adult rat heart atria and ventricle. Presence in terminal intercalated disks. Circulation. 1996; 94(12):3083–3086.
63. Maier, SK, Westenbroek, RE, Schenkman, KA, et al. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002; 99(6):4073–4078.
64. Motoike, HK, Liu, H, Glaaser, IW, et al. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004; 123(2):155–165.
65. Lin, X, Liu, N, Lu, J, et al. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011; 8(12):1923–1930.
66. McNair, WP, Sinagra, G, Taylor, MR, et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. 2011; 57(21):2160–2168.
67. Sato, PY, Coombs, W, Lin, X, et al. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res. 2011; 109(2):193–201.
68. Patino, GA, Isom, LL. Electrophysiol and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci Lett. 2010; 486(2):53–59.
69. Meadows, LS, Isom, LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005; 67(3):448–458.
70. Lopez-Santiago, LF, Meadows, LS, Ernst, SJ, et al. Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J Mol Cell Cardiol. 2007; 43(5):636–647.
71. Petitprez, S, Zmoos, AF, Ogrodnik, J, et al. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1. 5 in cardiomyocytes. Circ Res. 2011; 108(3):294–304.
72. Sato, PY, Musa, H, Coombs, W, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009; 105(6):523–526.
73. Mays, DJ, Foose, JM, Philipson, LH, et al. Localization of the Kv1. 5 K+ channel protein in explanted cardiac tissue. J Clin Invest. 1995; 96(1):282–292.
74. Murata, M, Buckett, PD, Zhou, J, et al. SAP97 interacts with Kv1. 5 in heterologous expression systems. Am J Physiol Heart Circ Physiol. 2001; 281(6):H2575–H2584.
75. Milstein, ML, Musa, H, Balbuena, DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 2012; 109(31):E2134–2143.
76. Cheng, L, Yung, A, Covarrubias, M, et al. Cortactin is required for N-cadherin regulation of Kv1. 5 channel function. J Biol Chem. 2011; 286(23):20478–20489.
77. Roepke, TK, Kontogeorgis, A, Ovanez, C, et al. Targeted deletion of kcne2 impairs ventricular repolarization via disruption of I(K,slow1) and I(to,f). FASEB Journal. 2008; 22(10):3648–3660.
78. Barry, DM, Trimmer, JS, Merlie, JP, et al. Differential expression of voltage-gated K+ channel subunits in adult rat heart. Relation to functional K+ channels? Circ Res. 1995; 77(2):361–369.
79. Takeuchi, S, Takagishi, Y, Yasui, K, et al. Voltage-gated K(+)Channel, Kv4. 2, localizes predominantly to the transverse-axial tubular system of the rat myocyte. J Mol Cell Cardiol. 2000; 32(7):1361–1369.
80. Melnyk, P, Zhang, L, Shrier, A, et al. Differential distribution of Kir2. 1 and Kir2. 3 subunits in canine atrium and ventricle. Am J Physiol Heart Circ Physiol. 2002; 283(3):H1123–H1133.
81. Saffitz, JE. Dependence of electrical coupling on mechanical coupling in cardiac myocytes: insights gained from cardiomyopathies caused by defects in cell-cell connections. Annals of the New York Academy of Sciences. 2005; 1047:336–344.
82. Oxford, EM, Musa, H, Maass, K, et al. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 2007; 101(7):703–711.
83. Fidler, LM, Wilson, GJ, Liu, F, et al. Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J Cell Mol Med. 2009; 13(10):4219–4228.
84. Li, MW, Mruk, DD, Lee, WM, et al. Connexin 43 and plakophilin-2 as a protein complex that regulates blood-testis barrier dynamics. Proc Natl Acad Sci U S A. 2009; 106(25):10213–10218.
85. Asimaki, A, Tandri, H, Huang, H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009; 360(11):1075–1084.
86. Asimaki, A, Tandri, H, Duffy, ER, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2011; 4(5):743–752.
87. Eckardt, D, Theis, M, Degen, J, et al. Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J Mol Cell Cardiol. 2004; 36(1):101–110.
88. Morley, GE, Vaidya, D, Jalife, J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 2000; 11(3):375–377.
89. Thomas, SP, Kucera, JP, Bircher-Lehmann, L, et al. Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res. 2003; 92(11):1209–1216.
90. Deo, M, Sato, PY, Musa, H, et al. Relative contribution of changes in sodium current versus intercellular coupling on reentry initiation in 2-dimensional preparations of plakophilin-2-deficient cardiac cells. Heart Rhythm. 2011; 8(11):1740–1748.
91. van der Zwaag, PA, Cox, MG, van der Werf, C, et al. Recurrent and founder mutations in the Netherlands: Plakophilin-2 p. Arg79X mutation causing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Neth Heart J. 2010; 18(12):583–591.
92. Joshi-Mukherjee, R, Coombs, W, Musa, H, et al. Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (ARVC)-related plakophilin-2 (PKP2) mutations. Heart Rhythm. 2008; 5(12):1715–1723.
93. Bauce, B, Basso, C, Rampazzo, A, et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005; 26(16):1666–1675.
94. Gomes, J, Finlay, M, Ahmed, AK, et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J. 2012.
95. Malhotra, JD, Thyagarajan, V, Chen, C, et al. Tyrosine-phosphorylated and nonphosphorylated sodium channel beta1 subunits are differentially localized in cardiac myocytes. J Biol Chem. 2004; 279(39):40748–40754.
96. Rhett, JM, Ongstad, EL, Jourdan, J, et al. Cx43 Associates with Na(v)1. 5 in the Cardiomyocyte Perinexus. J Membr Biol. 2012; 245(7):411–422.
97. Meyer, RA, Laird, DW, Revel, JP, et al. Inhibition of gap junction and adherens junction assembly by connexin and A-CAM antibodies. J Cell Biol. 1992; 119(1):179–189.
98. Hartshorne, RP, Catterall, WA. Purification of the saxitoxin receptor of the sodium channel from rat brain. Proc Natl Acad Sci U S A. 1981; 78(7):4620–4624.
99. Isom, LL, De Jongh, KS, Patton, DE, et al. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992; 256(5058):839–842.
100. Malhotra, JD, Kazen-Gillespie, K, Hortsch, M, et al. Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem. 2000; 275(15):11383–11388.
101. Dhar Malhotra, J, Chen, C, Rivolta, I, et al. Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation. 2001; 103(9):1303–1310.
102. Isom, LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001; 7(1):42–54.
103. Brackenbury, WJ, Isom, LL. Na channel beta subunits: overachievers of the ion. channel family. Front Pharmacol. 2011; 2:53.
104. Li, J, Patel, VV, Kostetskii, I, et al. Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005; 97(5):474–481.
105. Lim, BK, Xiong, D, Dorner, A, et al. Coxsackievirus and adenovirus receptor (CAR) mediates atrioventricular-node function and connexin 45 localization in the murine heart. J Clin Invest. 2008; 118(8):2758–2770.
106. Lisewski, U, Shi, Y, Wrackmeyer, U, et al. The tight junction protein CAR regulates cardiac conduction and cell-cell communication. J Exp Med. 2008; 205(10):2369–2379.
107. Rhett, JM, Jourdan, J, Gourdie, RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011; 22(9):1516–1528.
108. Kirchhoff, S, Kim, JS, Hagendorff, A, et al. Abnormal cardiac conduction and morphogenesis in connexin40 and connexin43 double-deficient mice. Circ Res. 2000; 87(5):399–405.
109. Kucera, JP, Rohr, S, Rudy, Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res. 2002; 91(12):1176–1182.
110. Rhett, JM, Gourdie, RG. The perinexus: a new feature of Cx43 gap junction organization. Heart Rhythm. 2012; 9(4):619–623.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]22
The Intercalated Disc
A Molecular Network That Integrates Electrical Coupling, Intercellular Adhesion, and Cell Excitability
Intercalated Disc Proteins in Inherited and Acquired Diseases
Structural Features of the Intercalated Disc
Ion Channel Complexes That Reside at the Intercalated Disc
Noncanonical Functions of Intercalated Disc Molecules
The Perinexus as a Site of Functional Integration
Subcellular Localization of Ion Channels and Cell-Cell Propagation: Gap Junction–Independent Electrical Coupling and the Importance of the Intercellular Cleft
Historical Perspective
This chapter focuses on the intercalated disc, a region of specialization formed at the end-end site of contact between cardiac myocytes. When first observed through light microscopy (in 1866), the intercalated disc was considered “a cementing material” at cardiac cell boundaries. However, the scientific community at the time was divided on whether cardiac cells were separate from each other or fused into a single syncytium. The latter hypothesis was in fact favored by most during the early twentieth century. The advent of electron microscopy eventually settled this debate. The studies of Sjostrand and Andersson1 and others showed that the intercalated disc consisted of a double membrane, flanked by the termination of myofibrils in dense material. Their observations led Muir2 to conclude that “the discs represent the junctions between neighboring cardiac muscle cells.” He wrote that “there is no valid evidence to contest the statement that the intercalated discs are specialized regions of cellular adhesion.” Since then, and as a result of the pioneering electrophysiology experiments of Weidmann3 and other ultrastructural observations,4 the intercalated disc has been recognized as an area of specialization that provides a physical continuum between cardiac cells through mechanical junctions (desmosomes, adherens junctions) and intercellular channels (gap junctions).
The availability of immunofluorescence microscopy allowed the demonstration that other molecular complexes, not detectable by electron microscopy, are also present in the intercalated disc. Of particular relevance to this chapter is the fact that channel protein complexes involved in both depolarization and repolarization localize preferentially to the intercalated disc. This physical proximity allows for a key functional consequence; molecules traditionally defined as junctional, such as connexin43 (Cx43) and plakophilin-2 (PKP2), actually regulate the function of ion channels responsible for the action potential. In turn, molecule accessories to ion channels are also relevant for cell adhesion and gap junction function.5–7 These data support the notion that the intercalated disc is not just the site of residence of independent junctional and nonjunctional complexes that are oblivious to the presence and function of the others. It is, rather, the home of a protein interacting network (an interactome) where molecules multitask to achieve jointly, intimately related functions: the entry and exit of charge into the cell, the transfer of charge between cells, and the anchoring of cells to each other, which provides a mechanically stable environment critical to ion channel function.
Intercalated Disc Proteins in Inherited and Acquired Diseases
Initial studies on the relation between desmosome integrity and cardiac electrophysiology were propelled by the finding that most familial cases of arrhythmogenic right ventricular cardiomyopathy (ARVC) in which a genetic link has been found associate with mutations in genes coding for desmosomal molecules.8 The latter brought forth the question of how a molecule considered purely relevant to cell adhesion altered the electrical behavior of the heart. The associations between desmosomal proteins and ion channel function (particularly the sodium current INa) are extensively reviewed in this chapter. (Recent publications refer to this disease as “arrhythmogenic cardiomyopathy,” to note the occurrence of left ventricular involvement.8)
A number of studies have demonstrated remodeling of gap junction proteins (in particular Cx43) in a number of inherited and acquired arrhythmia-related diseases.9–13 Recently, the Fishman laboratory provided evidence that aberrant posttranslational phosphorylation of Cx43 could be the common pathway leading to pathologic gap junction remodeling and arrhythmias.10,14 Although there is a relationship between Cx43 remodeling and arrhythmias, it is unclear whether these arrhythmias are exclusively consequent to changes in gap junction formation.15 Recent data show that reduced Cx43 expression alters sodium and potassium current function, and these changes could become part of the arrhythmogenic substrate.16–18
Disruption of the voltage-gated sodium channel complex is considered an important molecular substrate for arrhythmogenesis. Extensive reviews on the relation between mutations in proteins of the voltage-gated sodium channel (VGSC) complex and arrhythmias are available.19 Of particular interest in the context of this review is the observation that haploinsufficiency of a desmosomal protein, or overexpression of a mutant desmosomal protein, leads to INa deficit and increased arrhythmia susceptibility.20,21 The latter recalls the previously formulated concept that ARVC and Brugada syndrome (a channelopathy caused by mutations on genes of the VGSC complex) share common features22 and that, as Corrado et al.23 stated, there is the possibility of “an overlap in clinical manifestation and mechanisms of ventricular arrhythmias between patients with ARVC and Brugada syndrome.”
Structural Features of the Intercalated Disc
In its classic definition, the intercalated disc is composed of three electron-dense structures: adherens junctions, desmosomes, and gap junctions (Figure 22-1). Additional reviews on the characteristics of these structures can be found elsewhere.24,25 These structures are described briefly in the following sections, and the structural and molecular definitions of the intercalated disc are expanded to include the area composita, the intercellular space, and the nonjunctional ion channel complexes.
Figure 22-1 Transmission electron micrographs of intercalated disc in adult murine heart. Tissue prepared by high-pressure freezing and freeze substitution, thus greatly improving structural preservation. A, Low magnification to demonstrate the entire length of the intercalated disc as it meanders between two cells. B and C, Proximity (and contact in C, yellow arrow) between mitochondria, gap junctions, and desmosomes. D, Example of close proximity between a gap junction and a desmosome. E, Compiled three-dimensional tomographic electron microscopy reconstruction of the intercalated disc. Notice the extensive vesicular activity between the two cells. D, Desmosomes; GJ, gap junction; M, mitochondria. (Reproduced from Delmar M, Liang FX: Connexin43 and the regulation of intercalated disc function. Heart Rhythm 9:835–838, 2012.)
Adherens Junctions
Adherens junctions are specialized structures essential for the mechanical coupling between neighboring cells. The three morphologically different forms of adherens junctions are puncta adherentia, zonula adherens, and fascia adherens, with the last name corresponding to the morphology found in the cardiac intercalated disc.26 Cell-cell mechanical anchoring occurs at two crucial points: the extracellular space, within which cadherins tightly bind to each other, and the intracellular space, within which the cytoplasmic end of cadherin is indirectly attached to the actin cytoskeleton. The association between cadherin and the cytoskeleton involves at least two molecular “hinges”; cadherin binds to β-catenin and plakoglobin, and both molecules in turn bind to α-catenin (among others), the latter being in direct contact with actin. This is only a simplified description, because other interactions are likely to occur.27 This string of intermolecular interactions provides mechanical continuity between cells, allowing for the mechanical work of individual myocytes to integrate into the pumping function of the heart.
Desmosomes
The desmosome (macula adherens) appears as two parallel tripartite plaques containing an intercellular gap of approximately 30 nm bisected by a distinct line, parallel to the apposed cell membranes (see Figure 22-1).28 Desmosomes contribute to mechanical continuity between cardiac cells. Whereas adherens junctions link the actin cytoskeleton of adjacent cells, desmosomes provide continuity to the intermediate filament network (mainly desmin, in the case of heart).28,29 In the extracellular space, desmosomal cadherins (desmocollins and desmogleins) bind tightly to each other. In the intracellular space, the intermediate filaments bind to desmoplakin. The interaction between desmoplakin and the desmosomal cadherin can be in some cases direct, but it mostly occurs through their association with plakophilin and plakoglobin.28,29 The topologic organization of desmosomal molecules was studied by North et al.30 using quantitative immunogold electron microscopy. More recently, Al-Amoudi et al.31 solved the three-dimensional molecular structure of the desmosomal plaque. Overall, structural and biochemical evidence combined show that desmoplakin binds to plakophilin through their N-terminal domains,28,32 whereas desmoplakin binds to the intermediate filament by way of its C-terminal domain,28,31 yielding a highly organized structure.
Hatsell and Cowin29 once described the desmosome as “a system as staid and solid as the queen’s corsets.”29 With that analogy, it is easy to imagine the loss of containment that would follow in its absence. Mice deficient in plakoglobin, desmoplakin, or plakophilin-2 (PKP2), die during embryonic development as a result of severe myocardial rupture.33–35 More relevant from the point of view of clinical cardiology, ARVC in humans has been linked to mutations in desmosomal proteins.36 The relation between various complexes of the intercalated disc and ARVC is discussed later in this chapter and in other review articles.24,37
The Area Composita
Recent immunoelectron microscopy studies revealed the presence of a structure with mixed features of desmosomes and adherens junctions, dubbed the area composita.38 This structure is found only in the heart of higher vertebrate species including mouse and humans.39,40 The combination of components of the desmosomes and the adherens junctions allows anchorage of actin and desmin filaments to the same point, perhaps providing additional strength and flexibility to the muscle cell. Knockdown of PKP2 in neonatal cardiomyocytes leads to remodeling of the area composita.41 Additional studies suggest a role for α-catenin in the maintenance of these hybrid junctions.42 Interestingly, knockdown of α-catenin leads to PKP2 decrease only at the area composita and not at the desmosomes, suggesting that the molecular composition of the area composita, and its regulation, can be independent from that of other junctions.42 The area composita may represent a physical space where mechanical junction proteins interact with ion channel complexes.
Transcriptional Regulation by Mechanical Junction Proteins
Catenins are a part of both adherens junctions and desmosomes, thus participating in cell adhesion; however, these proteins also act as transcriptional activators. A prominent example is the participation of β-catenin in canonical Wnt signaling.43 Binding of Wnt to its Frizzled receptor leads to an increase in levels of cytosolic β-catenin and a consequent translocation of the protein to the nucleus, where it binds to Tcf/Lef complex and promotes the expression of various genes such as c-Myc or c-Fos. In the heart, the Wnt signaling and the activation of genes by β-catenin/Tcf/Lef has been associated with the regulation of physiologic and pathologic growth of the cardiomyocytes.44 Plakoglobin, another protein of the armadillo family, shows high homology with β-catenin and has been associated with the Wnt signaling. Different studies have shown that plakoglobin interacts and competes with β-catenin at multiple levels, acting as an antagonist of the Wnt/β-catenin signaling.45–47 The fact that desmosomal proteins are involved in the regulation of the Tcf/Lef complex has been invoked as a possible mechanism for the fibrofatty infiltration common in hearts affected with ARVC.48–50
Gap Junctions
In 1958, Sjostrand et al.4 described an area of specialization in the cardiac intercalated disc composed of “three dark lines with two intervening less dense lines”. This structure, which was similar to the one previously identified in the giant axon of the crayfish, was named the “longitudinal connexion” by these investigators. Years later, Revel coined the term gap junctions, thus emphasizing two key features: a gap between the cells and a junction between them.
The importance of Cx43 in the propagation of the cardiac action potential is well established. If Cx43 channels are not present, normal propagation is disrupted and lethal arrhythmias can ensue.51,52 Gap junction remodeling has been studied for various inherited and acquired diseases.10,11,14,53–55 The implications of connexin remodeling to electrophysiology are discussed later in this chapter.
Intercellular Space
The size of the space separating two cardiac cells at the intercalated disc changes depending on the proximity to the various structures, as well as the vesicular activity between the two cells (see Figure 22-1). It is a common view that the intercellular space is not relevant for electrophysiology. This view, however, is changing. Mathematical modeling studies56,57 and experimental evidence58 support the idea that the intercellular space is critical to propagation via an electric field mechanism.59 This model will be discussed later. Of note, increased size of the intercellular space has been reported in animal models of ARVC.20,21,60,61
Ion Channel Complexes That Reside at the Intercalated Disc
Voltage-Gated Sodium Channel Complex
In 1996, Cohen62 showed that cardiac sodium channel proteins were preferentially localized at the intercalated disc,62 although they are also present over the cell surface, following a striated pattern.63 Recent data emphasize how the VGSC interacts also with scaffolding, anchoring, and adhesions proteins, which regulate its function.
α-Subunit NaV1.5
The cardiac, pore-forming, α-subunit NaV1.5 contains four homologous transmembrane domains DI-DIV, linked by intracellular loops (IDI-II, IDII-III, IDIII-IV). Each domain is formed by the six transmembrane segments S1 to S6, and is involved in the voltage-dependent activation of the channel. The channel pore conducting Na+ is lined by the S6 segment and the S5-S6 pore loops in each domain.19 The inactivation gate is a complex formed by the DIII-IV loop and the C-terminus.64 This channel is defined as TTX-resistant, in contrast with other Nav channels. Of note, TTX-sensitive channel proteins, and currents, have also been found in cardiac myocytes, with the current localized primarily to the midsection of the cell.63,65
The SCN5A gene, on chromosome 3p21, codes for the NaV1.5 subunit. Mutations resulting in increased INa are associated with long QT syndrome type 3. Mutations that cause a reduction in INa are responsible for a series of different diseases, such as Brugada syndrome, progressive cardiac conduction defect, sick sinus syndrome, and a form of inherited atrial fibrillation. Some mutations are associated with a clinical spectrum encompassing more than one of those phenotypes and can manifest differently among carriers, even within the same family.19 Interestingly, some SCN5A mutations have been linked to a form of dilated cardiomyopathy with a high incidence of atrial and ventricular arrhythmias.66 It is not yet clear how mutations in the sodium channel could lead to structural damage of the myocardium. Based on the crosstalk between intercalated disc structures described in this chapter, we are tempted to speculate that the integrity of the sodium channel complex is also relevant to intercellular adhesion strength.20,21,67
β-Subunits of the Sodium Channel
The NaV β-subunit family consists of four proteins: β1-4, coded by genes SCN1B to SCN4B, respectively. These are single-span transmembrane proteins oriented with the amino terminus facing the extracellular space. The extracellular domain presents a conserved immunoglobulin domain, homologous to the one in cell adhesion molecules.68 The carboxyl terminus associates with cytoskeletal and scaffolding proteins. β1 and β2 are localized at the T-tubules–Z lines and at the intercalated discs in rat cardiac myocytes. β3 colocalizes with β1 at the T-tubules and β4 colocalizes with β2 at the intercalated disc. β1 and β2 associate with ankyrin-G and ankyrin-B in both brain and heart, and their interaction is critical for channel surface expression and modulates the channel function in vivo. β1 and β2 associate with N-cadherin and with Cx43 at the intercalated disc.69 Altogether, β-subunits have a key role in the interactions between the VGSC multiple proteins at the intercalated disc, including those relevant for cell adhesion and for electrical coupling between cells.69 Furthermore, work from the Isom lab has demonstrated that null mice for the β1 subunit show a significant increase in SCN5A mRNA in cardiac myocytes.70 Further research is needed to elucidate the role of β-subunits in transcription regulation in the heart.70 Overall, the data show that, as in the case of catenin and plakoglobin, β-subunits can have distal, contact-dependent effects, including regulation of gene transcription, with consequences to the function and structure of the heart.
Subcellular Heterogeneity of Voltage-Gated Sodium Channel
Recent studies have shown that not all sodium channels on the surface of the cardiac myocyte are equal. Instead, the molecular composition and the function of a sodium channel are different depending on whether NaV1.5 is localized to the intercalated disc or to the midsection of the cell. Petiprez et al.71 described two separate pools of VGSC in ventricular cardiomyocytes. One of these two subpopulations localizes at the lateral membrane of the myocytes, where NaV1.5 interacts with dystrophin and syntrophin; the second subpopulation of VGSC localizes at the ID, where NaV1.5 interacts with the MAGUK proteins SAP97 and ZO1, as well as with PKP2.72 Lin et al. recently demonstrated that this subcellular distribution correlates with differences in function.65 Using cell-attached macropatch recordings, these authors showed that the magnitude of INa is larger at the intercalated disc than in the midsection of the cell. Furthermore, TTX-resistant channels in the midsection showed a significant negative shift in the steady-state inactivation curve, suggesting that these channels are mostly in an inactivated state at the normal resting potential, and that the burden of excitation is on the channels at the intercalated disc. These authors also demonstrated that the amplitude of current is larger if cells remain paired, strongly suggesting that cell adhesion preserves sodium channel function. The interaction between sodium channels and proteins of the intercalated disc is discussed extensively later.
Potassium Channels at the Intercalated Disc
In 1995, Mays et al.73 described for the first time that the potassium channel protein KV1.5 localizes to the intercalated disc of adult myocardial cells. Later studies showed that KV1.5 associates with SAP97.74 Interestingly, SAP-97 also associates with NaV1.5,71 making this protein a candidate for mutual regulation of both depolarization and repolarization channels, as is the case of NaV1.5 and Kir2.1.75 Additional studies have shown that the function of KV1.5 depends on the expression of N-cadherin,76 an interesting parallelism to the interaction between PKP2 (a desmosomal molecule), and NaV1.5.72 Cheng et al.76 also showed that cortactin is required for the N-cadherin–dependent regulation of KV1.5. Of note, mice deficient in kcne2, an ancillary potassium channel subunit, display an impaired ventricular repolarization because of inhibition in the trafficking to the membrane of KV1.5.77
Another voltage-gated potassium channel that preferentially (though not exclusively) localizes to the intercalated disc is KV4.2; it is responsible for the rapid repolarization phase of the cardiac action potential.78,79 Finally, regarding Kir channels, two different subunits localize at the transverse tubules and at the intercalated disc in canine myocytes: Kir2.1 and Kir2.3.80
