[level-membership-for-internal-medicine-category]
Chapter 3 The immune system and disease
Anatomy and principles of the immune system
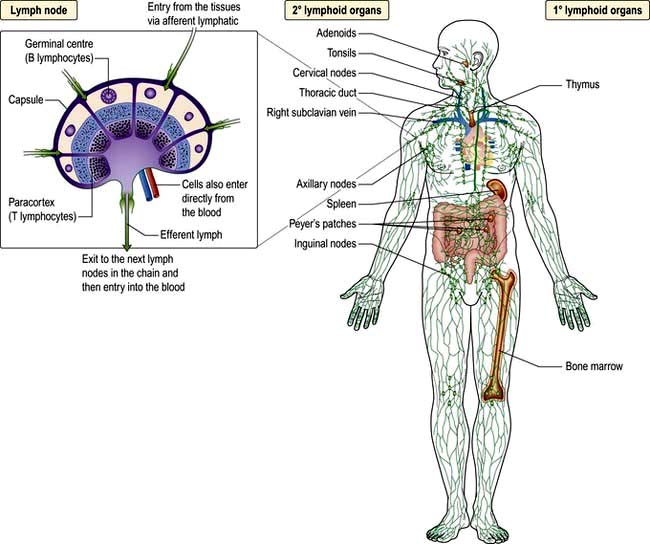
Immunity can be defined as protection from infection, whether it be due to bacteria, viruses, fungi or multicellular parasites. Like other organs involved in human physiology, the immune system is composed of cells and molecules organized into specialized tissues (Fig. 3.1).
Cells involved in immune responses: origin and function
All immune cells have a common source in the pluripotent stem cells generated in the bone marrow (Fig. 3.1). They have diverse functions (Table 3.1). T lymphocytes undergo ‘education’ in the thymus to avoid self-recognition, and populate the peripheral lymphoid tissue, where B lymphocytes also reside. Both sets of lymphocytes undergo activation in the peripheral tissue, to become mature effector cells. B lymphocytes may further differentiate into antibody-secreting plasma cells. Lymphoid tissue is frequently found at mucosal surfaces in non-encapsulated patches, termed mucosa-associated lymphoid tissue (MALT).
The immune system
Cells and molecules involved in immune responses are classified into innate and adaptive systems:
 The innate immune system is inborn and operates throughout life (pp. 51–55)
The innate immune system is inborn and operates throughout life (pp. 51–55)
 The adaptive immune system changes in response to the pathogens it encounters (pp. 58–62).
The adaptive immune system changes in response to the pathogens it encounters (pp. 58–62).
There are also non-immunological barriers that are involved in host protection, and very often it is the lowering of these that allows a pathogen to take a foothold (Table 3.2).
|
Normal barriers |
Events that may compromise barrier function |
|
Physical barriers |
|
|
Skin and mucous membranes |
Trauma, burns, i.v. cannulae |
|
Cough reflex |
Suppression, e.g. by opiates, neurological disease |
|
Mucosal function |
Ciliary paralysis (e.g. smoking) |
|
Increased mucus production (e.g. asthma) |
|
|
Abnormally viscid secretions (e.g. cystic fibrosis) |
|
|
Decreased secretions (e.g. sicca syndrome) |
|
|
Urine flow |
Stasis (e.g. prostatic hypertrophy) |
|
Chemical barriers |
Low gastric pH (gastric acid secretion inhibitors) |
|
Resistance to pathogens provided by commensal skin and gut organisms |
Changes in flora (e.g. broad-spectrum antibiotics) |
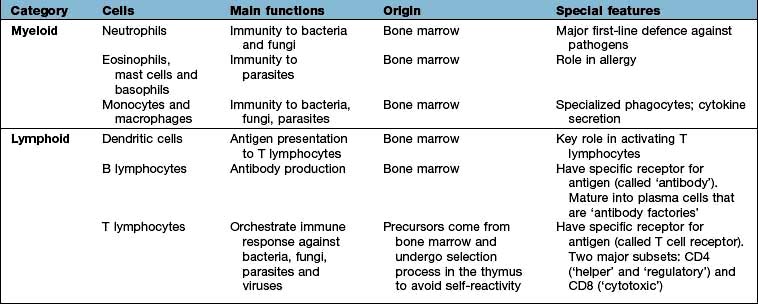
A major feature of the immune system is the complexity of the surface-bound, intracellular and soluble structures that mediate its functions. In particular, it is necessary to be aware of the CD (clusters of differentiation) classification (Box 3.1) and the functions of cytokines and chemokines.
 Box 3.1
Box 3.1
The CD classification
This is the ultimate way of defining a cell
 Immune cells are distinguished by the surface receptors and proteins that they express in order to mediate their particular range of immunological functions, e.g. cell–cell signalling, cell activation
Immune cells are distinguished by the surface receptors and proteins that they express in order to mediate their particular range of immunological functions, e.g. cell–cell signalling, cell activation


 A clinical example is the number of peripheral blood lymphocytes expressing CD4 (’the CD4 count’), which is used to monitor HIV infection (see p. 178)
A clinical example is the number of peripheral blood lymphocytes expressing CD4 (’the CD4 count’), which is used to monitor HIV infection (see p. 178)
An updated listing is available at: http://www.hcdm.org/
Cytokines
These are small polypeptides released by a cell in order to change the function of the same or another cell. These chemical messengers are found in many organ systems, but especially the immune system. Cytokines have become markers in the investigation of disease pathogenesis; therapeutic agents in their own right; and the targets of therapeutic agents (see p. 72). The key features of a cytokine are:









Cells and molecules of the innate immune system
Innate immunity provides immediate, first-line, host defence. The key features of this system are shown in Table 3.3. It is present at birth and remains operative at comparable intensity into old age. Innate immunity is mediated by a variety of cells and molecules (Table 3.4). Activation of innate immune responses is mediated through interaction between the:
 pathogen side comprising a relatively limited array of molecules (pathogen-associated molecular patterns, PAMPs)
pathogen side comprising a relatively limited array of molecules (pathogen-associated molecular patterns, PAMPs)
 host side – a limited portfolio of receptors (pattern recognition receptors, PRRs).
host side – a limited portfolio of receptors (pattern recognition receptors, PRRs).
Table 3.3 Features of the innate and adaptive immune responses
| Innate | Adaptive |
|---|---|
|
No memory: quality and intensity of response invariant |
Memory: response adapts with each exposure |
|
Recognizes limited number of non-varying, generic molecular patterns on, or made by, pathogens |
Recognizes vast array of specific antigensa on, or made by, pathogens |
|
Pattern recognition mediated by a limited array of receptors |
Antigen recognition mediated by a vast array of antigen-specific receptors |
|
Response immediate on first encounter |
Response on first encounter takes 1–2 weeks; on second encounter 3–7 days |
a Antigen is a molecular structure (protein, peptide, lipid, carbohydrate) that generates an immune response.
Activation of certain cells in the innate immune system leads to activation of the adaptive immune response (see p. 58).
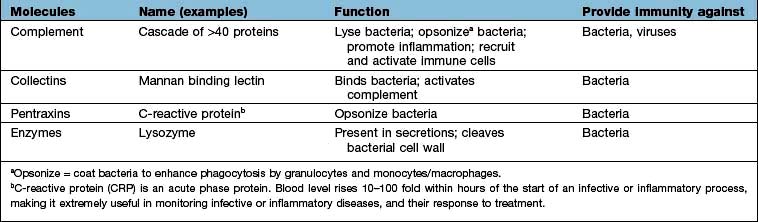
Complement
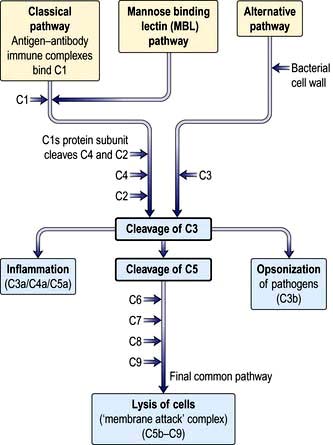
Complement proteins are produced in the liver. Each complement circulates in an inactive form until triggered to become enzymatically active, when it then activates several molecules of the next stage in a series. This complement cascade is initiated via three distinct pathways: alternative, classical and mannan-binding lectin (Fig. 3.2). These pathways are composed of three distinct enzyme cascades that culminate in the cleavage of C3 and C5. Cleavage of C3 has a number of biological consequences; breakdown of C5 achieves the same and, in addition, provides the triggering stimulus to the final common (‘membrane attack’) pathway, which provides most of the biological activity (Fig. 3.2).




Neutrophils
Neutrophils (see p. 413) phagocytose and kill microorganisms. They are derived from the bone marrow, which can produce between 1011 (healthy state) and 1012 (during infection) new cells per day. In health, neutrophils are rarely seen in the tissues.
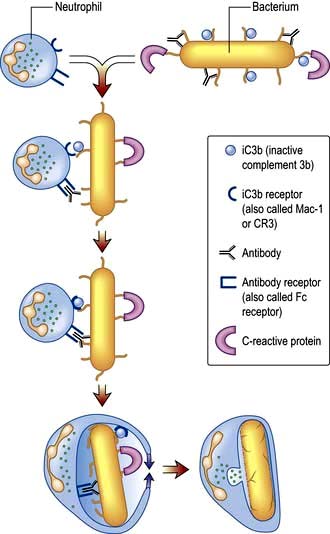
Neutrophil phagocytosis is activated by interaction with bacteria, either directly or after bacteria have been coated (opsonized) to make them more ingestible (Fig. 3.3). The contents of neutrophil granules are released both intracellularly (predominantly azurophilic granules) and extracellularly (specific granules) following fusion with the plasma membrane. Approximately 100 different molecules in neutrophil granules (Table 3.5) kill and digest microorganisms, for example:
 Myeloperoxidase and cytochrome b558 are key components of major oxygen-dependent bactericidal systems.
Myeloperoxidase and cytochrome b558 are key components of major oxygen-dependent bactericidal systems.



| Function | Primary or azurophilic granules | Secondary or specific granules |
|---|---|---|
|
Antibacterial |
Lysozyme |
Respiratory burst components (e.g. cytochrome b558) producing reactive oxygen metabolites, such as hydrogen peroxide, hydroxyl radicals and singlet oxygen |
|
Cathepsins |
Lysozyme |
|
|
Bactericidal/permeability increasing protein (BPI) |
Lactoferrin |
|
|
Cell movement |
|
Collagenase |
|
|
CD11b/CD18 (adhesion molecule) |
|
|
|
N-formyl-methionyl-leucylphenylalanine receptor (FMLP-R) |
Eosinophils
In contrast to neutrophils, several hundred times more eosinophils are present in the tissues than in the blood, particularly at epithelial surfaces where they survive for several weeks. The main role of eosinophils is protection against multicellular parasites such as worms (helminths). This is achieved by the release of pro-inflammatory mediators, which are toxic, cationic proteins. In populations and societies in which such parasites are rare, eosinophils contribute mainly to allergic disease, particularly asthma (see p. 827). Eosinophils have two types of granules:
 Specific granules (95%) contain the cationic proteins, of which there are four main types: major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil neurotoxin, which are all potently and exquisitely toxic to helminths (ECP also has some bactericidal properties), and eosinophil peroxidase, which has similar activity to neutrophil myeloperoxidase.
Specific granules (95%) contain the cationic proteins, of which there are four main types: major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil neurotoxin, which are all potently and exquisitely toxic to helminths (ECP also has some bactericidal properties), and eosinophil peroxidase, which has similar activity to neutrophil myeloperoxidase.
 Primary granules (5%) synthesize and release leukotrienes C4 and D4 and platelet-activating factor (PAF) which alter airway smooth muscle and vasculature (see p. 828).
Primary granules (5%) synthesize and release leukotrienes C4 and D4 and platelet-activating factor (PAF) which alter airway smooth muscle and vasculature (see p. 828).
Mast cells and basophils
Mast cells and basophils share features in common, especially in containing:

 high-affinity receptors for immunoglobulin E (IgE; an antibody type that is involved in allergic disease, see p. 68, 69).
high-affinity receptors for immunoglobulin E (IgE; an antibody type that is involved in allergic disease, see p. 68, 69).
Mast cells are found in tissues (especially skin and mucosae) and basophils in the blood. Both mast cells and basophils release pro-inflammatory mediators which are either pre-formed or synthesized de novo (Table 3.6).
| Mediators | Effects |
|---|---|
|
Pre-formed: |
|
|
Histamine |
Vasodilatation |
|
Vascular permeability ↑ |
|
|
Smooth muscle contraction in airways |
|
|
Proteases |
Digestion of basement membrane causes ↑vascular permeability and aids migration |
|
Proteoglycans (e.g. heparan) |
Anticoagulant activity |
|
Synthesized de novo: |
|
|
Platelet-activating factor (PAF) |
Vasodilator |
|
LTB4, LTC4, LTD4 |
Neutrophil and eosinophil activators and chemoattractants |
|
Vascular permeability ↑ |
|
|
Bronchoconstrictors |
|
|
Prostaglandins (mainly PGD2) |
Vascular permeability ↑ |
|
|
Bronchoconstrictors |
|
|
Vasodilators |
Monocytes and macrophages
Tissue macrophages
Macrophages have pro-inflammatory and microbicidal capabilities similar to those of neutrophils. Under activation conditions, antigen presentation (see p. 57, 58) is enhanced and a range of cytokines secreted, notably TNF-α, IL-1 and IFN-γ. These are necessary for the removal of certain pathogens that live within mononuclear phagocytes (e.g. mycobacteria). Macrophages and related cells may also undergo a process termed autophagy (p. 32, Ch. 2). This self-cannibalization is a critical property of many cell types under starvation conditions, but is used by the immune system to destroy intracellular pathogens such as Mycobacterium tuberculosis, which otherwise persist within cells and block normal antibacterial processes. Autophagy is also a means of enhancing antigen presentation pathways.
Tissue macrophages involved in chronic inflammatory foci may undergo terminal differentiation into multinucleated giant cells, typically found at the site of the granulomata characteristic of tuberculosis and sarcoidosis (see p. 845).
Dendritic cells
 dendritic morphology (Fig. 3.4)
dendritic morphology (Fig. 3.4)
 machinery for sensing pathogens
machinery for sensing pathogens

 the ability to dictate the T lymphocyte’s future function and differentiation.
the ability to dictate the T lymphocyte’s future function and differentiation.
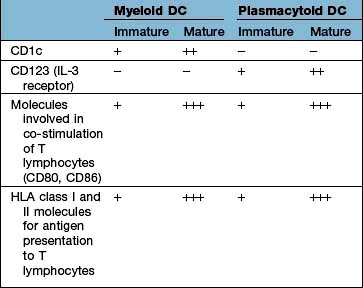
Types of dendritic cell
The major types are the myeloid DC (mDC), the plasmacytoid DC (pDC) and a variety of specialized DCs found in tissues that resemble mDCs (e.g. the Langerhans cell in the skin, see Fig. 24.1). DCs have several distinctive cell surface molecules, some of which have pathogen-sensing activity (e.g. the antigen uptake receptor DEC205 on mDCs) whilst others are involved in interaction with T lymphocytes (Table 3.7). Immature mDCs and pDCs are present in the blood, but at very low levels (<0.5% of lymphocyte/monocyte cells).
 Mannan-binding lectin, which initiates complement activity inducing opsonization (p. 51, 52).
Mannan-binding lectin, which initiates complement activity inducing opsonization (p. 51, 52).
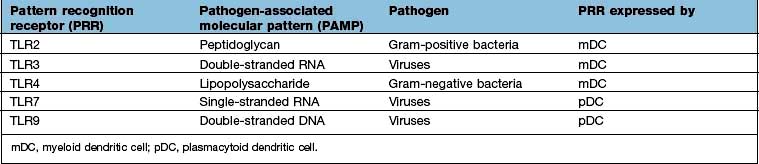
 Signalling receptors such as the PRR known as TLR4 (for toll-like receptor 4), which binds lipopolysaccharide, a molecular pattern found in the cell walls of many Gram-negative bacteria (Table 3.8), whilst others bind double-stranded and single-stranded RNA from viruses. Innate immunity critically depends on toll-like receptor signalling. These receptors act through a critical adaptor molecule, myeloid differentiation factor 88 (MyD88), to regulate the activity of nuclear factor kappa B (NF-κB) pathways.
Signalling receptors such as the PRR known as TLR4 (for toll-like receptor 4), which binds lipopolysaccharide, a molecular pattern found in the cell walls of many Gram-negative bacteria (Table 3.8), whilst others bind double-stranded and single-stranded RNA from viruses. Innate immunity critically depends on toll-like receptor signalling. These receptors act through a critical adaptor molecule, myeloid differentiation factor 88 (MyD88), to regulate the activity of nuclear factor kappa B (NF-κB) pathways.


DCs and T cell activation
In a sequence of events that spans 1–2 days, immature DCs are activated by PAMPs or DAMPs in the tissues binding to a PRR on DCs. The immature pDC is a small rounded cell that develops dendrites upon activation and secretes enormous quantities of IFN-α, a potent antiviral and pro-inflammatory cytokine. On activation, the DC migrates to the local lymph node with the engulfed pathogen. During migration the DC matures, changing its shape, gene and molecular profile and function within a matter of hours to take on a mature form, with altered functions (Table 3.9, Fig. 3.5), in particular upregulating machinery required to activate T lymphocytes. Once in the lymph node, the mature DC interacts with naive T lymphocytes (antigen presentation), resulting in two key outcomes:
| Immature mDC | Mature mDC |
|---|---|
|
Highly pinocytotic |
Ceases pinocytosis |
|
Low level expression of molecules required for T lymphocyte activation |
Upregulates CD80, CD86 and HLA molecules |
|
Low level expression of machinery required to process and present microbial antigens |
Begins to process microbial antigens (break down into small peptides) in readiness to present them to T lymphocytes (using HLA molecules) |
|
Generally localized and sedentary |
Begins active migration to local lymph node |
|
Minimal secretion of cytokines |
Active secretion of cytokines in readiness to stimulate T lymphocytes; in particular IL-12 |
mDC, myeloid dendritic cell.
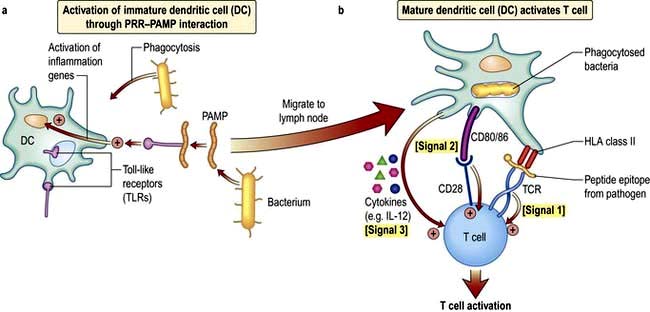
The mature DC provides three major signals to naive T cells Fig. 3.5):



Hla molecules and antigen presentation
On the short arm of chromosome 6 is a collection of genes termed the major histocompatibility complex (MHC; known as the human leucocyte antigens, or HLA in man), which plays a critical role in immune function. MHC genes code for proteins expressed on the surface of a variety of cell types that are involved in antigen recognition by T lymphocytes. The T lymphocyte receptor for antigen recognizes its ligand as a short antigenic peptide embedded within a physical groove at the extremity of the HLA molecule (Fig. 3.6).
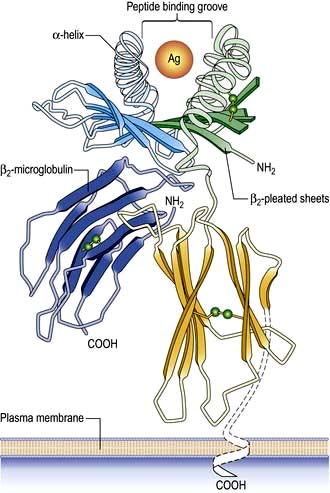
The HLA genes are particularly interesting for clinicians and biologists. First, differences in HLA molecules between individuals are responsible for tissue and organ graft rejection (hence the name ‘histo’(tissue)-compatibility). Second, possession of certain HLA genes is linked to susceptibility to particular diseases (Table 3.10).
Table 3.10 HLA associations with immune-mediated and infectious diseases
| Disease process | Disease | HLA type |
|---|---|---|
|
Autoimmunity |
Type 1 diabetes |
Class II: |
|
|
DQA1*0301/DQB1*0302 (susceptibility) |
|
|
DQA1*501/DQB1*201 (susceptibility) |
||
|
DQA1*0102/DQB1*0602 (protection) |
||
|
|
Class I: |
|
|
|
HLA-A24; HLA-B*18; HLA-B*39 |
|
|
Multiple sclerosis |
DRB1*1501 (susceptibility) |
|
|
Rheumatoid arthritis |
DRB1*0404 (susceptibility) |
|
|
Autoimmune hepatitis |
DRB1*03 and DRB1*04 (susceptibility) |
|
|
Goodpasture’s syndrome (anti-glomerular basement membrane disease) |
HLA-DRB1*1501 (susceptibility) |
|
|
Pemphigus vulgaris |
DRB1*0402; DQB1*0503 (susceptibility) |
|
|
Inflammatory |
Coeliac disease |
DQA1*0501/DQB1*0201 (susceptibility) |
|
Ankylosing spondylitis |
HLA-B27 (susceptibility) |
|
|
Psoriasis |
HLA-Cw*0602 (susceptibility) |
|
|
Infectious |
Human immunodeficiency virus infection |
HA-B27; HLA-B*51; HLA-B*57 (associated with slow progression of disease) |
|
HLA-B*35 (associated with rapid progression) |
The human major histocompatibility complex
The human MHC comprises three major classes (I, II and III) of genes involved in the immune response (Fig. 3.7).
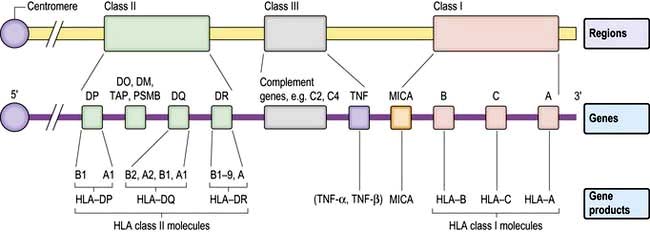
HLA classes
Classical class I HLA genes (also termed Ia), are designated HLA-A, HLA-B and HLA-C. Each encodes a class I α chain, which combines with a β chain to form the class I HLA molecule (Fig. 3.6). While there are several types of α chains, there is only one type of β chain, β2 microglobulin. The HLA class I molecule has the role of presenting short (8–10 amino acids) antigenic peptides to the T cell receptor on the subset of T lymphocytes that bear the co-receptor CD8. As an example of HLA polymorphism, there are nearly 200 allelic forms at the A gene locus. Class I HLA molecules are expressed on all nucleated cells.
Non-classical HLA class I genes are less polymorphic, have a more restricted expression on specialized cell types, and present a restricted type of peptide or none at all. These are the HLA-E, F and G (Ib genes) and MHC class I-related (MIC, or class Ic) genes, A and B. The products of these genes are predominantly found on epithelial cells, signal cellular stress and interact with lymphoid cells, especially natural killer cells (see p. 61, 62).
HLA genotypes and the range of their protein products
Some general principles apply to the HLA genes and their protein products:
 The presence of multiple genes on each chromosome, and the fact that both maternal and paternal genes are co-dominantly expressed, allows considerable breadth in the number of HLA molecules that an individual expresses.
The presence of multiple genes on each chromosome, and the fact that both maternal and paternal genes are co-dominantly expressed, allows considerable breadth in the number of HLA molecules that an individual expresses.
 The existence of polymorphisms at each locus provides great breadth in the number of HLA molecules expressed at a population level. The polymorphic forms of HLA molecules differ predominantly in the peptide-binding groove (Fig. 3.6).
The existence of polymorphisms at each locus provides great breadth in the number of HLA molecules expressed at a population level. The polymorphic forms of HLA molecules differ predominantly in the peptide-binding groove (Fig. 3.6).
Antigen presentation
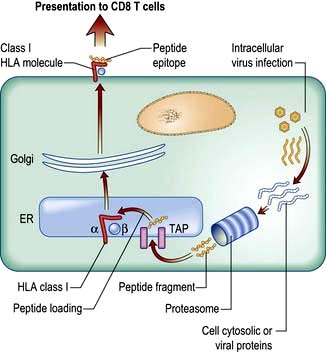
 The endogenous route (Fig. 3.8) is a property of all nucleated cells: the internal milieu is sampled to generate peptide–HLA class I complexes for display (‘presentation’) on the cell surface. In a healthy cell, the peptides are derived from self-proteins in the cytoplasm (Fig. 3.8) and are ignored by the immune system. In a virus-infected cell, viral proteins are processed and presented. The resulting viral peptide–HLA class I complex is presented to CD8 T lymphocytes that have cytotoxic (killer) function. In an immune response against a virus infection, CD8 T lymphocytes recognizing viral peptide–HLA complexes on the surface of an infected cell will kill it as a means to limit and eradicate infection.
The endogenous route (Fig. 3.8) is a property of all nucleated cells: the internal milieu is sampled to generate peptide–HLA class I complexes for display (‘presentation’) on the cell surface. In a healthy cell, the peptides are derived from self-proteins in the cytoplasm (Fig. 3.8) and are ignored by the immune system. In a virus-infected cell, viral proteins are processed and presented. The resulting viral peptide–HLA class I complex is presented to CD8 T lymphocytes that have cytotoxic (killer) function. In an immune response against a virus infection, CD8 T lymphocytes recognizing viral peptide–HLA complexes on the surface of an infected cell will kill it as a means to limit and eradicate infection.
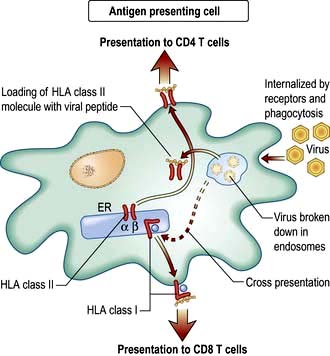
 The exogenous route (Fig. 3.9) is a property of APCs: the external milieu is sampled. Antigens are internalized, either in the process of phagocytosis of a pathogen, through pinocytosis, or through specialized surface receptors (e.g. for antigen/antibody/complement complexes). The antigen is broken down by a combination of low pH and proteolytic enzymes for ‘loading’ into HLA class II molecules. At the APC surface the pathogen peptide–HLA class II complex is presented to and able to interact with CD4 T lymphocytes. Presentation by DCs can initiate an adaptive immune response by activating a naive, pathogen-specific CD4 T lymphocyte. Presentation by monocyte/macrophages and B lymphocytes can maintain and enhance this response by activating effector and memory pathogen-specific CD4 T lymphocytes.
The exogenous route (Fig. 3.9) is a property of APCs: the external milieu is sampled. Antigens are internalized, either in the process of phagocytosis of a pathogen, through pinocytosis, or through specialized surface receptors (e.g. for antigen/antibody/complement complexes). The antigen is broken down by a combination of low pH and proteolytic enzymes for ‘loading’ into HLA class II molecules. At the APC surface the pathogen peptide–HLA class II complex is presented to and able to interact with CD4 T lymphocytes. Presentation by DCs can initiate an adaptive immune response by activating a naive, pathogen-specific CD4 T lymphocyte. Presentation by monocyte/macrophages and B lymphocytes can maintain and enhance this response by activating effector and memory pathogen-specific CD4 T lymphocytes.
 Cross-presentation refers to the ability of some APCs (mainly DCs) to internalize exogenous antigens and process them through the endogenous route (Fig. 3.8). This is an essential component in the activation of CD8 cytotoxic T cell responses against a virus.
Cross-presentation refers to the ability of some APCs (mainly DCs) to internalize exogenous antigens and process them through the endogenous route (Fig. 3.8). This is an essential component in the activation of CD8 cytotoxic T cell responses against a virus.
Cells and molecules of the adaptive immune system
The information gained by DCs that interact with a pathogen is passed on, in the form of signals 1–3 (Fig. 3.5b). These activate T lymphocytes in the adaptive immune system, which recognize the same pathogen. T lymphocytes may be involved in pathogen removal directly (e.g. by killing) or indirectly (e.g. by recruiting B lymphocytes to make specific antibody).
Antigen receptors on T and B lymphocytes
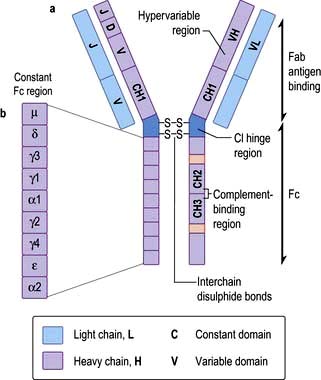
Immunoglobulins
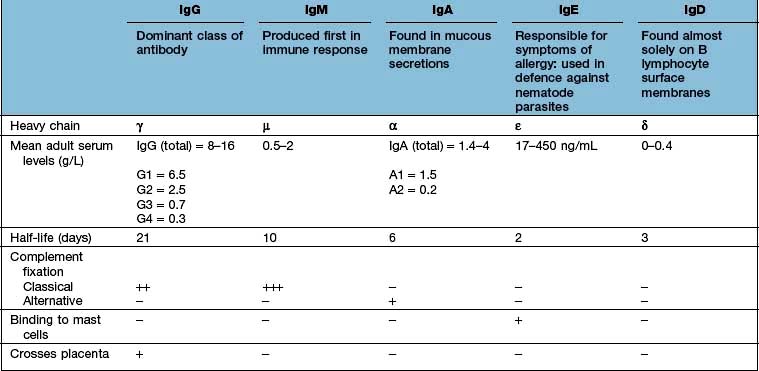
In structural terms, antibodies have four chains; two identical heavy and two identical light chains (Fig. 3.10). Each chain contains both highly variable and essentially constant regions. The variable parts of the heavy and light chains pair to form the potentially diverse part of the antibody molecule that binds antigen. The constant region of the heavy chain dictates the function of the antibody, and belongs to one of the classes M, G1–4, A1–2, D and E, giving rise to antibodies called IgM, IgG1–4, IgA1–2, IgD and IgE. The characteristics of these different isotypes are shown in Table 3.11.
Antibody production
Essential points about antibody production are:
 IgM is the first isotype to be made in a primary immune response and thus measurement of pathogen specific IgM is a useful diagnostic test for recent infection.
IgM is the first isotype to be made in a primary immune response and thus measurement of pathogen specific IgM is a useful diagnostic test for recent infection.
 IgG dominates in the second exposure to antigen.
IgG dominates in the second exposure to antigen.
 IgG and IgM are the most efficient complement activators when bound to antigen in an immune complex.
IgG and IgM are the most efficient complement activators when bound to antigen in an immune complex.
 IgG antibodies cross the placenta, and can carry both immunity and disease to the unborn fetus.
IgG antibodies cross the placenta, and can carry both immunity and disease to the unborn fetus.



Antibody function





T lymphocyte development and activation
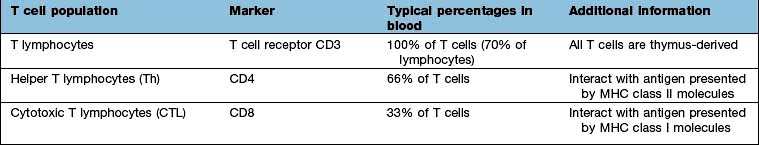
T lymphocytes are generated from precursors in the bone marrow, which migrate to the thymus (Fig. 3.11). Only 1% of the cells that enter the thymus will leave it as naive T lymphocytes to populate the lymph nodes. This process (termed thymic selection) leads to a cohort of cells (Table 3.12) with:
 functionally rearranged genes allowing surface expression of a receptor for antigen (the T cell receptor, TCR) alongside the CD3 accessory molecule involved in transducing the antigen-specific signal
functionally rearranged genes allowing surface expression of a receptor for antigen (the T cell receptor, TCR) alongside the CD3 accessory molecule involved in transducing the antigen-specific signal


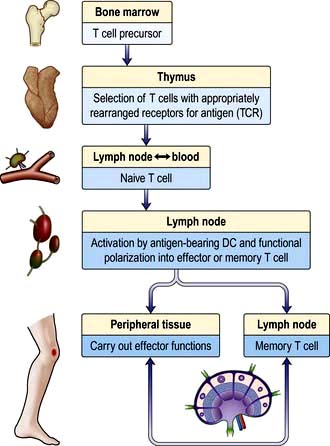
Most naive T lymphocytes are resident in the lymph nodes or spleen, whilst 2% are present in the blood, representing a recirculating pool. Naive T lymphocytes are activated for the first time in the lymph node by antigens presented to their TCRs as short peptides bound to MHC molecules on the surface of DCs (Fig. 3.5). Provision of signals 1–3 (see p. 55) sets off an intracellular cascade of signalling molecule activation, leading to induction of gene transcription in T lymphocytes.
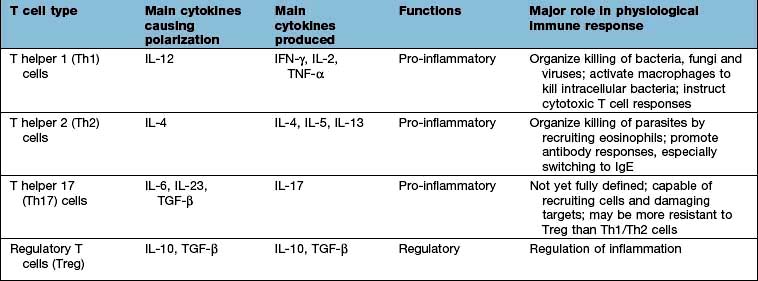
Nuclear factor kappa B (NF-κB) is a pivotal transcription factor in chronic inflammatory diseases and malignancy. It is found in the cytoplasm bound to an inhibitor IκB which prevents it from entering the nucleus (see Fig. 2.9). It is released from IκB on stimulation of the cell and passes into the nucleus, where it binds to promoter regions of target genes involved in inflammation. It is stimulated by, for example, cytokines, protein C activators and viruses. The outcome is T lymphocyte activation, cell division and functional polarization, which is the acquired ability to promote a selected type of adaptive immune response. These processes take several days to achieve. The best described polarities of T cell responses (Table 3.13) are:
 CD4+ pro-inflammatory T lymphocytes; T helper 1 (Th1), Th2 and Th17
CD4+ pro-inflammatory T lymphocytes; T helper 1 (Th1), Th2 and Th17


FURTHER READING
Bousso P. T cell activation by dendritic cells in the lymph node: lessons from the movies. Nat Rev Immunol 2008; 8:675–684.
Breart B, Bousso P. Cellular orchestration of T cell priming in lymph nodes. Curr Opin Immunol 2006; 18:483–490.
Corthay A. A three-cell model for activation of native T helper cells. Scand J Immunol 2006; 64:93–96.
CD4 T lymphocyte functions




Regulatory T lymphocytes
 CD4+ CD25hi Tregs; express high levels of CD25, the receptor for IL-2; low levels of the IL-7 receptor (CD127); regulate other T lymphocytes by cell-cell contact and also secrete the immune suppressive cytokines IL-10 and TGF-β; can be generated in the thymus or post-thymically in the periphery; have high expression of the transcription factor Foxp3.
CD4+ CD25hi Tregs; express high levels of CD25, the receptor for IL-2; low levels of the IL-7 receptor (CD127); regulate other T lymphocytes by cell-cell contact and also secrete the immune suppressive cytokines IL-10 and TGF-β; can be generated in the thymus or post-thymically in the periphery; have high expression of the transcription factor Foxp3.




Natural killer (NK) cells
The role of NK cells is to kill ‘abnormal’ host cells, typically cells that are virus-infected, or tumour cells. Killing is achieved in similar ways to CTLs. NK cells also secrete copious amounts of IFN-γ and TNF-α, through which they can mediate cytotoxic effects and activate other components of the innate and adaptive immune system. To become activated, NK cells integrate the signal from a potential target cell through a series of receptor-ligand pairings (Table 3.14). These pairings provide activating and inhibitory signals, and it is the overall balance of these that determines the outcome for the NK cell. The balance can be abnormal on a virus-infected or tumour cell, which might have altered expression of HLA molecules, for example, that mark them out for NK cytotoxicity.
| Receptors on NK cells | Ligand |
|---|---|
|
Inhibitory: |
|
|
KIR2DL1 |
HLA-C molecules |
|
KIR3DL1 |
HLA-B molecules |
|
KIRDL2 |
HLA-A molecules |
|
NKG2A |
HLA-E molecules |
|
Activating: |
|
|
CD16 (low-affinity receptor for IgG) |
IgG |
|
NKG2D |
MHC class I chain related gene A (MICA) |
|
KIR2DS1 |
HLA-C |
KIR, killer cell immunoglobulin-like receptor.
One of the targets of NK cell activating receptors, MICA (non-classical HLA class Ic molecule), is induced under conditions of cellular ‘stress’, such as might arise during infection or neoplastic transformation. Many viruses have developed immunoevasion strategies that avoid presentation of viral proteins to CTLs by interfering with the MHC class I presentation pathway, or circumvent target cell recognition by reducing MICA expression. NK cells detect the reduced levels of MHC class I molecules, which renders infected cells susceptible to killing by NK cells. Likewise, tumours that escape immune surveillance by CTLs through the outgrowth of daughter cells that have low MHC expression also become NK cell targets. That NK cells are vital to antiviral immunity, is indicated by the associations between KIR and susceptibility to chronic infection with viruses, notably the human immunodeficiency virus (see p. 173).
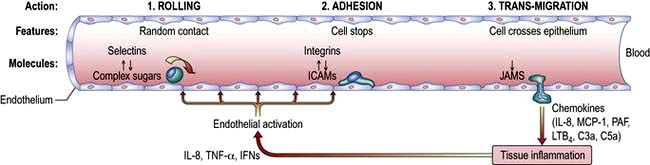
Cell migration
Taking the example of a DC migrating into the lymph node from the tissues via the lymphatics (Fig. 3.12), this is highly dependent upon expression of the chemokine receptor CCR7 by the migrating cell and of its ligand (secondary lymphoid tissue chemokine, SLC or CCL21) by the target tissue. Likewise, circulating naive lymphocytes are CCR7+ and migrate with ease into the lymph nodes from the blood or via tissue recirculation. In addition, naive lymphocytes express L-selectin which binds a glycoprotein cell adhesion molecule (GlyCAM-1) found on the high endothelial venules of lymph nodes. This system can be upregulated in an inflamed lymph node, leading to an influx of naive lymphocytes and the typical symptom of a swollen node.
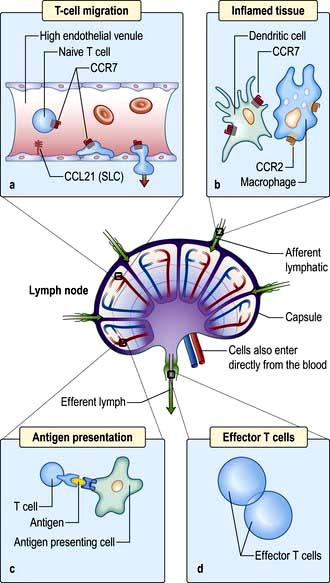
Figure 3.12 Migration of an immune cell through a lymph node. (a) T lymphocytes migrate through endothelial venules, which express the chemokine CCL21 (secondary lymphoid tissue chemokine or SLC). (b) Dendritic cells (expressing CCR7), macrophages (expressing CCR2) enter lymph nodes via afferent lymphatics and localize in the paracortex. (c) Antigen presentation (see also Figs 3.8 and 3.9) results in activation of T lymphocytes. (d) Effector T lymphocytes leave lymph node via efferent lymphatic.
Migration into inflamed tissue requires:
(a) that an affected organ or tissue signals that there is a focus of injury/infection and
(b) that responding immune cells bind and adhere specifically to that tissue.
This process is highly organized and has a similar basis for all immune cells, involving three basic steps: rolling, adhesion and trans-migration. Each of these is dependent on specialized adhesion molecules, as shown in Figure 3.13.
The immune system in concert
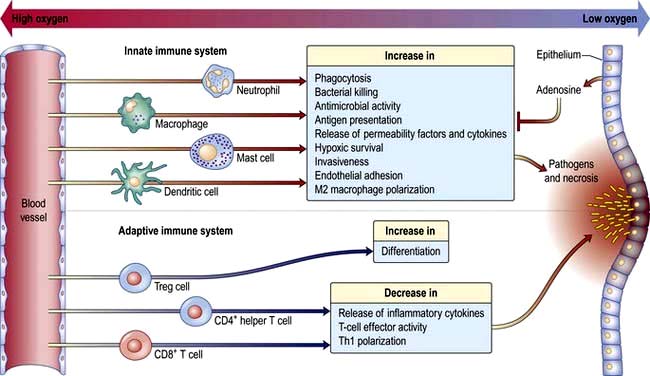
Acute inflammation: events and symptoms
 Local expansion of pathogen numbers leads to direct activation of complement in the tissues, with ensuing degranulation of mast cells.
Local expansion of pathogen numbers leads to direct activation of complement in the tissues, with ensuing degranulation of mast cells.




 At the site of pathogen entry there is often relative tissue hypoxia. The low oxygen tension has the effect of amplifying the responses of innate immune cells and suppressing the response of adaptive immune cell. This is probably an effective means of preventing excessive immune activation, which can result in collateral damage (Fig. 3.14).
At the site of pathogen entry there is often relative tissue hypoxia. The low oxygen tension has the effect of amplifying the responses of innate immune cells and suppressing the response of adaptive immune cell. This is probably an effective means of preventing excessive immune activation, which can result in collateral damage (Fig. 3.14).



 Resolution of the infection is aided by the scavenging activity of tissue macrophages.
Resolution of the infection is aided by the scavenging activity of tissue macrophages.
Chronic inflammation: events and symptoms
When the inflammation is sufficiently chronic it may take on the appearance of organized lymphoid tissue resembling a lymph node germinal centre (e.g. in the joints in rheumatoid arthritis, p. 517). There is massive cytokine production by T lymphocytes and APCs, which contributes to local tissue damage. Granulomata, which ‘wall off’ the inciting stimulus, may also arise and result in fibrosis and calcification. Symptoms typically relate to the site of the inflammation and the type of pathology, but there may also be systemic effects such as fever and weight loss.
Laboratory investigations of the immune system
Examples of the commoner tests and their interpretation are shown in Table 3.15.
Table 3.15 Examining the immune system in the clinical immunology laboratory
| Measurement | Interpretation | |
|---|---|---|
|
Proteins |
C-reactive protein |
Raised levels indicate infection or inflammation |
|
Immunoglobulins |
Low levels indicate antibody deficiency, usually a result of underlying disease or primary immunodeficiency. High levels, e.g. ↑ IgM seen in acute viral infection (hepatitis A) |
|
|
Complement C3 and C4 |
Low levels indicate consumption of complement in immune complex disease |
|
|
IgE |
Raised levels in allergy; allergen-specific IgE useful to pinpoint the inciting stimulus (e.g. pollen, grass) |
|
|
Cells |
Neutrophils |
High levels in bacterial infection; low levels in secondary immune deficiency |
|
Eosinophils |
High levels in allergic or parasitic disease |
|
|
CD4 T lymphocytes |
Low levels in HIV infection |
|
|
Function |
Neutrophil respiratory burst |
Absent in the immune deficiency chronic granulomatous disease |
|
T lymphocyte proliferation |
Abnormally low in primary T cell immune deficiency disease |
|
|
Autoantibodies (see also Table 11.3 and Box 11.16) |
Rheumatoid factor, anti-cyclic citrullinated peptide antibodies (ACPA) |
Rheumatoid arthritis |
|
Double-stranded DNA autoantibodies |
Systemic lupus erythematosus |
|
|
Acetylcholine receptor antibodies |
Myasthenia gravis |
|
|
Anti-neutrophil cytoplasmic antibodies (ANCA) |
Vasculitis |
|
|
Mitochondrial |
Primary biliary cirrhosis |
Clinical immunodeficiency
Secondary (acquired) versus primary immunodeficiency
 Acquired neutropenias, which are common (e.g. due to myelosuppression by disease or drugs or the increased rate of destruction in hypersplenism or autoimmune neutropenia) and carry a high risk of infection once the neutrophil count falls below 0.5 × 109/L.
Acquired neutropenias, which are common (e.g. due to myelosuppression by disease or drugs or the increased rate of destruction in hypersplenism or autoimmune neutropenia) and carry a high risk of infection once the neutrophil count falls below 0.5 × 109/L.

 Impairment of defence against capsulated bacteria, especially pneumococcus, following splenectomy; such patients should receive pneumococcal, meningococcal and Hib vaccinations as a matter of course (see p. 406).
Impairment of defence against capsulated bacteria, especially pneumococcus, following splenectomy; such patients should receive pneumococcal, meningococcal and Hib vaccinations as a matter of course (see p. 406).
Primary immunodeficiency is rare and arises at birth as the congenital effect of a developmental defect or as a result of genetic abnormalities (Table 3.16). Gene defects may not become manifest until later in infancy or childhood, and some forms of immunodeficiency typically present in adolescence or adulthood.
Table 3.16 Classification of immunodeficiencies and the main diseases in each category: apart from AIDS, all diseases shown here are primary immunodeficiencies
| Immune component | Examples of diseases |
|---|---|
|
T lymphocyte deficiency |
DiGeorge’s syndrome |
|
Acquired immunodeficiency syndrome/HIV infection |
|
|
T cell activation defects (e.g. CD3γ chain mutation) |
|
|
X-linked hyper-IgM syndrome (XHIM; CD40L deficiency) |
|
|
B lymphocyte deficiency |
X-linked agammaglobulinaemia (XLA) |
|
Common variable immunodeficiency (CVID) |
|
|
Selective IgA deficiency (IgAD) |
|
|
Combined T and B cell defects |
Severe combined immunodeficiency (SCID) (e.g. due to defects in common γ chain receptor for IL-2, -4, -7, -9, -15) |
|
T cell–APC interactions |
IFN-γ receptor deficiency |
|
IL-12 and IL-12 receptor deficiency |
|
|
Neutrophil defects |
Chronic granulomatous disease (CGD) |
|
Leucocyte adhesion deficiency (LAD) |
|
|
Deficiency of complement components |
Classical pathway |
|
Alternative pathway |
|
|
Common pathway |
|
|
Regulatory proteins |
|
|
Mannan binding lectin |
Clinical features of immunodeficiency
The infections associated with immunodeficiency have several typical features:
 They are often chronic, severe or recurrent
They are often chronic, severe or recurrent
 They resolve only partially with antibiotic therapy or return soon after cessation of therapy
They resolve only partially with antibiotic therapy or return soon after cessation of therapy
 The organisms involved are often unusual (‘opportunistic’ or ‘atypical’).
The organisms involved are often unusual (‘opportunistic’ or ‘atypical’).
The pattern of infection, in terms of the type of organism involved, is indicative (Table 3.17):
 ‘Opportunistic’ organisms are of low virulence but become invasive in immunodeficient states, e.g. atypical mycobacteria, Pneumocystis jiroveci, Staphylococcus epidermis.
‘Opportunistic’ organisms are of low virulence but become invasive in immunodeficient states, e.g. atypical mycobacteria, Pneumocystis jiroveci, Staphylococcus epidermis.
 Phagocyte defects cause, e.g. deep skin infections, abscesses, osteomyelitis.
Phagocyte defects cause, e.g. deep skin infections, abscesses, osteomyelitis.
 Defective antibody producers experience infections with pyogenic (‘pus-forming’) bacteria.
Defective antibody producers experience infections with pyogenic (‘pus-forming’) bacteria.
 T lymphocyte deficiency causes infection with fungi, protozoa and intracellular microorganisms.
T lymphocyte deficiency causes infection with fungi, protozoa and intracellular microorganisms.

Primary immunodeficiency
T and B lymphocyte deficiency
Neutrophil defects
Chronic granulomatous disease (CGD)
This is a rare (1/250 000) immunodeficiency due to a defect in neutrophil killing and characterized by deep-seated infections. The functional defect is an inability to generate antibacterial metabolites through the respiratory burst (Table 3.5). Typical onset is at toddler age. Neutrophil numbers are normal or increased. A simple respiratory burst test of neutrophil function is diagnostic.
Leucocyte adhesion deficiency (LAD)
This results from defects in integrins (see p. 23). Numerous underlying defects in the gene encoding one of the component chains, CD18, have been described in LAD-I. LAD-I has an autosomal recessive inheritance presenting almost immediately after birth, with delayed umbilical cord separation. Recurrent infections similar to those in chronic granulomatous disease appear during the first decade of life. Blood neutrophil levels are high but cells are absent from the sites of infection, which require aggressive antimicrobial and antifungal treatment, and SCT for cure.
Complement deficiency
The consequences of deficiency of complement proteins can be predicted from their functions (Fig. 3.2).
 Failures in innate response components (e.g. the alternative pathway) lead to impaired non-specific immunity with an increase in bacterial infections.
Failures in innate response components (e.g. the alternative pathway) lead to impaired non-specific immunity with an increase in bacterial infections.


Complement regulatory proteins
Deficiency of C1 inhibitor (C1 esterase) deficiency (see also p. 1211) is relatively rare. Since this enzyme is involved in regulation of several plasma enzyme systems (e.g. the kinin system) and is continuously consumed, a single parental chromosome defect resulting in 50% of normal production barely copes with the demand and fails under stress (hence an autosomal dominant effect). As a result, uncontrolled activation of complement and the kinins may occur, leading to oedema of the deep tissues affecting the face, trunk, viscera and the airway, hence the alternative name of hereditary angio-oedema (HAE).
Type I hypersensitivity and allergic disease
In Type I (immediate) hypersensitivity, the binding of an antigen to specific IgE bound to its high-affinity receptor on a mast cell surface results in massive and rapid cell degranulation and the inflammatory response outlined on page 53. The antigens involved are typically inert molecules present in the environment (termed allergens; see p. 824).




|
|
Treatment
Avoidance is the first-line of therapy.
 Antihistamines are effective for many immediate hypersensitivity reactions (but have no role in the treatment of asthma). A monoclonal antibody that mops up serum IgE (omalizumab) is also used (see p. 831).
Antihistamines are effective for many immediate hypersensitivity reactions (but have no role in the treatment of asthma). A monoclonal antibody that mops up serum IgE (omalizumab) is also used (see p. 831).



 Desensitization (allergen immunotherapy) can be used. The principle is that allergy can be prevented by inoculation by giving the allergen in a controlled way. It is indicated for disorders in which the hypersensitivity is IgE mediated, e.g. life-threatening allergy to insect stings, drug allergy and allergic rhinitis. An induction course of subcutaneous injections of increasing doses of the allergen extract, given once every 1–2 weeks, is followed by maintenance injections monthly for 2–3 years. A systematic review of 51 published randomized placebo-controlled clinical trials, enrolling a total of nearly 3000 participants, showed a low risk of adverse events with consistent clinical benefit. From an immunological viewpoint, desensitization seems capable of modifying the allergic response at several levels (Table 3.19). Sublingual allergen immunotherapy (using grass pollen extract tablets of Phl p 5 from Timothy grass; see p. 810) is used in hay fever that has not responded to anti-allergic drugs (the first dose is given under medical supervision).
Desensitization (allergen immunotherapy) can be used. The principle is that allergy can be prevented by inoculation by giving the allergen in a controlled way. It is indicated for disorders in which the hypersensitivity is IgE mediated, e.g. life-threatening allergy to insect stings, drug allergy and allergic rhinitis. An induction course of subcutaneous injections of increasing doses of the allergen extract, given once every 1–2 weeks, is followed by maintenance injections monthly for 2–3 years. A systematic review of 51 published randomized placebo-controlled clinical trials, enrolling a total of nearly 3000 participants, showed a low risk of adverse events with consistent clinical benefit. From an immunological viewpoint, desensitization seems capable of modifying the allergic response at several levels (Table 3.19). Sublingual allergen immunotherapy (using grass pollen extract tablets of Phl p 5 from Timothy grass; see p. 810) is used in hay fever that has not responded to anti-allergic drugs (the first dose is given under medical supervision).
Table 3.19 Mechanism of action of desensitization for allergy
| Probable mechanism | Effect |
|---|---|
|
IgG blocking antibodies |
During repeated exposure to desensitizing allergen, IgG class antibodies develop (especially IgG4 subclass); these compete with the pathogenic IgE for allergen binding, and/or prevent IgE-allergen complexes binding to mast cell high-affinity IgE receptors |
|
Regulation |
Exposure to repeated desensitizing allergen induces Treg cells which recognize allergen but invoke regulatory immune responses, dampening down migration, infiltration and inflammation |
|
Immune deviation |
A shift away from Th2- to Th1-producing CD4 cells results in the generation of cytokines (e.g. IFN-γ) which are inhibitory to IgE production |
Anaphylaxis
Anaphylaxis is a serious allergic reaction that is rapid in onset and may cause death. It arises as an acute, generalized IgE-mediated immune reaction involving specific antigen, mast cells and basophils. The reaction requires priming by the allergen, followed by re-exposure. To provoke anaphylaxis, the allergen must be systemically absorbed, either after ingestion or parenteral injection. A range of allergens that provoke anaphylaxis has been identified (Table 3.20).
Table 3.20 Sources of allergens known to provoke anaphylaxis
|
Foods |
Nuts: peanuts (protein-arachis hypogaea Ara h 1–3), Brazil, cashew |
|
Shellfish: shrimp (allergen Met e 1), lobster |
|
|
Dairy products |
|
|
Egg |
|
|
More rarely: citrus fruits, mango, strawberry, tomato |
|
|
Venoms |
Wasps, bees, yellow-jackets, hornets |
|
Medications |
Antisera (tetanus, diphtheria), dextran, latex, some antibiotics |


Treatment
Early recognition and treatment are essential (Emergency Box 3.1).
 Emergency Box 3.1
Emergency Box 3.1
Treatment of acute anaphylaxis




Management
 ABCDE (Airway Breathing Circulation Disability Exposure)
ABCDE (Airway Breathing Circulation Disability Exposure)
 Position the patient lying flat with feet raised
Position the patient lying flat with feet raised




 Administer 0.5 mg intramuscular adrenaline (epinephrine) and repeat every 5 min if shock persists.
Administer 0.5 mg intramuscular adrenaline (epinephrine) and repeat every 5 min if shock persists.
 Administer intravenous antihistamine (e.g. 10–20 mg chlorphenamine) slowly
Administer intravenous antihistamine (e.g. 10–20 mg chlorphenamine) slowly

If hypotension persists, give 1–2 L of intravenous fluid.
Autoimmune disease
1. Removal of TCRs with very strong affinity for ‘self’ in the thymus
2. The presence of naturally arising regulatory T lymphocytes (Tregs)
3. The requirement for a danger signal to license dendritic cells to activate CD4 T lymphocytes.
Autoimmune diseases affect 5% of the population at some stage of their life.
Failure of Checkpoint 1, thymic education
During thymic education, TCRs with dangerously high affinity for self are deleted. It has become apparent that this relies upon the thymic expression of self antigens. Situations which compromise the expression of a self protein would be expected to favour the development of autoimmunity. Indeed, in a rare group of patients who develop multiple autoimmune disorders affecting the adrenal and parathyroid glands (autoimmune polyglandular syndrome type 1; see p. 997), there is a defect in the AIRE gene (for autoimmune regulator), which controls thymic expression of a host of self genes. When the gene malfunctions, there is reduced expression of self proteins in the thymus and autoimmune disease is a consequence.
Failure of Checkpoint 2, regulatory T lymphocytes
An example of Treg failure is the defect in the gene encoding Foxp3, a critical transcription factor in Tregs (see p. 61) that leads to IPEX (see p. 61). IPEX is very rare, but it serves to indicate how Treg defects can lead to autoimmune disease. Laboratory studies in this area are revealing subtle Treg defects in several autoimmune diseases (e.g. type 1 diabetes, multiple sclerosis, rheumatoid arthritis).
Failure of Checkpoint 3, CD4 T lymphocyte activation against an autoantigen (or its mimic)
 Tissue damage due to infection leads to both the release of hidden self antigens and the provision of sufficient danger signals to activate DCs, which in turn activate autoreactive CD4 T lymphocytes as well as the pathogen-specific ones. This is often termed ‘bystander activation’.
Tissue damage due to infection leads to both the release of hidden self antigens and the provision of sufficient danger signals to activate DCs, which in turn activate autoreactive CD4 T lymphocytes as well as the pathogen-specific ones. This is often termed ‘bystander activation’.

It is unlikely that for the common autoimmune diseases (Table 3.21) there is a ‘single checkpoint’ explanation. Rather, it is likely that multiple subtle defects, at various checkpoints, are at play.
| Disease | Antigens |
|---|---|
|
Addison disease |
21 α-hydroxylase |
|
Goodpasture’s syndrome |
Type IV collagen (located in GBM) |
|
Graves’ thyroiditis |
Thyroid-stimulating hormone receptor |
|
Hashimoto’s thyroiditis |
Thyroid peroxidase, thyroglobulin |
|
Multiple sclerosis |
Myelin basic protein |
|
Myelin oligodendrocyte glycoprotein |
|
|
Myasthenia gravis |
Acetylcholine receptor |
|
Pemphigus vulgaris |
Desmoglein-3 |
|
Pernicious anaemia |
H+/K+-ATPase, intrinsic factor |
|
Polymyositis/dermatomyositis |
tRNA synthases |
|
Primary biliary cirrhosis |
Pyruvate dehydrogenase complex |
|
Rheumatoid arthritis |
Citrullinated cyclic peptide, IgM |
|
Scleroderma |
Topoisomerase |
|
Sjögren’s syndrome |
Ro/La ribonuclear proteins |
|
Systemic lupus erythematosus |
Sm/RNP, Ro/La (SS-A/SS-B), histone and native DNA |
|
Type 1 diabetes |
Proinsulin, glutamic acid decarboxylase, IA-2, ZNT8 |
|
Vitiligo |
Pigment cell antigens |
|
Wegener’s granulomatosis (Granulomatosis with polyangiitis). |
Neutrophil proteinase 3 |
GBM, glomerular basement membrane.
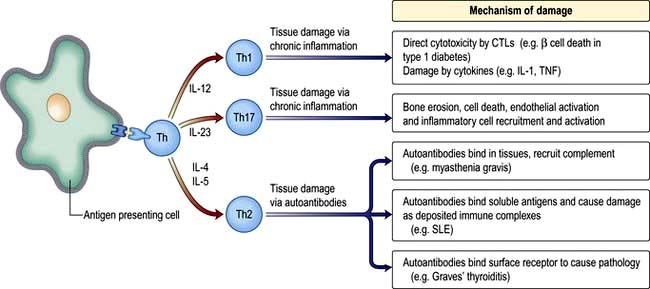
Mechanisms of tissue damage in autoimmune disease
Figure 3.15 illustrates potential mechanisms of immune damage in autoimmune disease.



Common autoimmune diseases
Major criteria






Organ rejection in clinical transplantation
The outcome of an allograft (i.e. a graft between genetically non-identical members of the same species) in the absence of adequate immunosuppressive therapy is immunological rejection. Histological analysis of rejected organs shows a range of immunological processes in action (Table 3.22). With modern tissue-matching approaches, hyperacute rejection is rare and acute rejection can usually be prevented or treated with immunosuppression. The process of chronic rejection typically takes place over several years and is the main reason for organ graft failure.
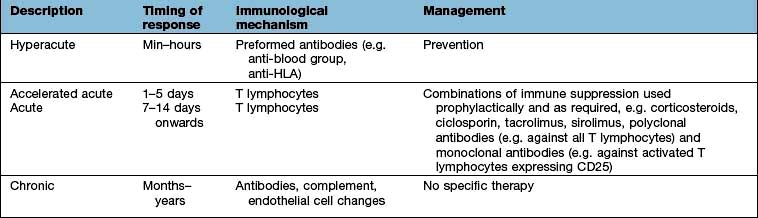
The antigens recognized in acute and chronic graft responses are donor HLA molecules (alloantigens):


Immune-based therapies
Manipulating the immune response in a therapeutic setting has seen many successes, as evidenced by the control of organ rejection in clinical transplantation through targeted immunosuppression (Table 3.22). Monoclonal antibodies offer the opportunity to neutralize the unwanted effects of cytokines, or to direct immune responses, drugs, toxins or irradiation against a specific target, whether it be a tumour cell or an immune cell involved in a damaging autoimmune response. Natural antiviral mediators, such as the interferons, are already in the clinic as therapies for, amongst other things, chronic viral infection.
Monoclonal antibody therapy (targeted therapy)
Immunosuppressive drugs
 potent effects on monocyte production of the pro-inflammatory cytokines IL-1 and TNF-α
potent effects on monocyte production of the pro-inflammatory cytokines IL-1 and TNF-α
 blockade of T lymphocyte production of IL-2 and IFN-γ
blockade of T lymphocyte production of IL-2 and IFN-γ
 reduced activation and migration of a range of innate and adaptive immune cells.
reduced activation and migration of a range of innate and adaptive immune cells.




[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 3 The immune system and disease
Anatomy and principles of the immune system
Immunity can be defined as protection from infection, whether it be due to bacteria, viruses, fungi or multicellular parasites. Like other organs involved in human physiology, the immune system is composed of cells and molecules organized into specialized tissues (Fig. 3.1).
Cells involved in immune responses: origin and function
All immune cells have a common source in the pluripotent stem cells generated in the bone marrow (Fig. 3.1). They have diverse functions (Table 3.1). T lymphocytes undergo ‘education’ in the thymus to avoid self-recognition, and populate the peripheral lymphoid tissue, where B lymphocytes also reside. Both sets of lymphocytes undergo activation in the peripheral tissue, to become mature effector cells. B lymphocytes may further differentiate into antibody-secreting plasma cells. Lymphoid tissue is frequently found at mucosal surfaces in non-encapsulated patches, termed mucosa-associated lymphoid tissue (MALT).
The immune system
Cells and molecules involved in immune responses are classified into innate and adaptive systems:
The innate immune system is inborn and operates throughout life (pp. 51–55)
The adaptive immune system changes in response to the pathogens it encounters (pp. 58–62).
There are also non-immunological barriers that are involved in host protection, and very often it is the lowering of these that allows a pathogen to take a foothold (Table 3.2).
|
Normal barriers |
Events that may compromise barrier function |
|
Physical barriers |
|
|
Skin and mucous membranes |
Trauma, burns, i.v. cannulae |
|
Cough reflex |
Suppression, e.g. by opiates, neurological disease |
|
Mucosal function |
Ciliary paralysis (e.g. smoking) |
|
Increased mucus production (e.g. asthma) |
|
|
Abnormally viscid secretions (e.g. cystic fibrosis) |
|
|
Decreased secretions (e.g. sicca syndrome) |
|
|
Urine flow |
Stasis (e.g. prostatic hypertrophy) |
|
Chemical barriers |
Low gastric pH (gastric acid secretion inhibitors) |
|
Resistance to pathogens provided by commensal skin and gut organisms |
Changes in flora (e.g. broad-spectrum antibiotics) |
A major feature of the immune system is the complexity of the surface-bound, intracellular and soluble structures that mediate its functions. In particular, it is necessary to be aware of the CD (clusters of differentiation) classification (Box 3.1) and the functions of cytokines and chemokines.
Box 3.1
The CD classification
This is the ultimate way of defining a cell
Immune cells are distinguished by the surface receptors and proteins that they express in order to mediate their particular range of immunological functions, e.g. cell–cell signalling, cell activation
A clinical example is the number of peripheral blood lymphocytes expressing CD4 (’the CD4 count’), which is used to monitor HIV infection (see p. 178)
An updated listing is available at: http://www.hcdm.org/
Cytokines
These are small polypeptides released by a cell in order to change the function of the same or another cell. These chemical messengers are found in many organ systems, but especially the immune system. Cytokines have become markers in the investigation of disease pathogenesis; therapeutic agents in their own right; and the targets of therapeutic agents (see p. 72). The key features of a cytokine are:
Cells and molecules of the innate immune system
Innate immunity provides immediate, first-line, host defence. The key features of this system are shown in Table 3.3. It is present at birth and remains operative at comparable intensity into old age. Innate immunity is mediated by a variety of cells and molecules (Table 3.4). Activation of innate immune responses is mediated through interaction between the:
pathogen side comprising a relatively limited array of molecules (pathogen-associated molecular patterns, PAMPs)
host side – a limited portfolio of receptors (pattern recognition receptors, PRRs).
Table 3.3 Features of the innate and adaptive immune responses
| Innate | Adaptive |
|---|---|
|
No memory: quality and intensity of response invariant |
Memory: response adapts with each exposure |
|
Recognizes limited number of non-varying, generic molecular patterns on, or made by, pathogens |
Recognizes vast array of specific antigensa on, or made by, pathogens |
|
Pattern recognition mediated by a limited array of receptors |
Antigen recognition mediated by a vast array of antigen-specific receptors |
|
Response immediate on first encounter |
Response on first encounter takes 1–2 weeks; on second encounter 3–7 days |
a Antigen is a molecular structure (protein, peptide, lipid, carbohydrate) that generates an immune response.
Activation of certain cells in the innate immune system leads to activation of the adaptive immune response (see p. 58).
Complement
Complement proteins are produced in the liver. Each complement circulates in an inactive form until triggered to become enzymatically active, when it then activates several molecules of the next stage in a series. This complement cascade is initiated via three distinct pathways: alternative, classical and mannan-binding lectin (Fig. 3.2). These pathways are composed of three distinct enzyme cascades that culminate in the cleavage of C3 and C5. Cleavage of C3 has a number of biological consequences; breakdown of C5 achieves the same and, in addition, provides the triggering stimulus to the final common (‘membrane attack’) pathway, which provides most of the biological activity (Fig. 3.2).
Neutrophils
Neutrophils (see p. 413) phagocytose and kill microorganisms. They are derived from the bone marrow, which can produce between 1011 (healthy state) and 1012 (during infection) new cells per day. In health, neutrophils are rarely seen in the tissues.
Neutrophil phagocytosis is activated by interaction with bacteria, either directly or after bacteria have been coated (opsonized) to make them more ingestible (Fig. 3.3). The contents of neutrophil granules are released both intracellularly (predominantly azurophilic granules) and extracellularly (specific granules) following fusion with the plasma membrane. Approximately 100 different molecules in neutrophil granules (Table 3.5) kill and digest microorganisms, for example:
Myeloperoxidase and cytochrome b558 are key components of major oxygen-dependent bactericidal systems.
| Function | Primary or azurophilic granules | Secondary or specific granules |
|---|---|---|
|
Antibacterial |
Lysozyme |
Respiratory burst components (e.g. cytochrome b558) producing reactive oxygen metabolites, such as hydrogen peroxide, hydroxyl radicals and singlet oxygen |
|
Cathepsins |
Lysozyme |
|
|
Bactericidal/permeability increasing protein (BPI) |
Lactoferrin |
|
|
Cell movement |
|
Collagenase |
|
|
CD11b/CD18 (adhesion molecule) |
|
|
|
N-formyl-methionyl-leucylphenylalanine receptor (FMLP-R) |
Eosinophils
In contrast to neutrophils, several hundred times more eosinophils are present in the tissues than in the blood, particularly at epithelial surfaces where they survive for several weeks. The main role of eosinophils is protection against multicellular parasites such as worms (helminths). This is achieved by the release of pro-inflammatory mediators, which are toxic, cationic proteins. In populations and societies in which such parasites are rare, eosinophils contribute mainly to allergic disease, particularly asthma (see p. 827). Eosinophils have two types of granules:
Specific granules (95%) contain the cationic proteins, of which there are four main types: major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil neurotoxin, which are all potently and exquisitely toxic to helminths (ECP also has some bactericidal properties), and eosinophil peroxidase, which has similar activity to neutrophil myeloperoxidase.
Primary granules (5%) synthesize and release leukotrienes C4 and D4 and platelet-activating factor (PAF) which alter airway smooth muscle and vasculature (see p. 828).
Mast cells and basophils
Mast cells and basophils share features in common, especially in containing:
high-affinity receptors for immunoglobulin E (IgE; an antibody type that is involved in allergic disease, see p. 68, 69).
Mast cells are found in tissues (especially skin and mucosae) and basophils in the blood. Both mast cells and basophils release pro-inflammatory mediators which are either pre-formed or synthesized de novo (Table 3.6).
| Mediators | Effects |
|---|---|
|
Pre-formed: |
|
|
Histamine |
Vasodilatation |
|
Vascular permeability ↑ |
|
|
Smooth muscle contraction in airways |
|
|
Proteases |
Digestion of basement membrane causes ↑vascular permeability and aids migration |
|
Proteoglycans (e.g. heparan) |
Anticoagulant activity |
|
Synthesized de novo: |
|
|
Platelet-activating factor (PAF) |
Vasodilator |
|
LTB4, LTC4, LTD4 |
Neutrophil and eosinophil activators and chemoattractants |
|
Vascular permeability ↑ |
|
|
Bronchoconstrictors |
|
|
Prostaglandins (mainly PGD2) |
Vascular permeability ↑ |
|
|
Bronchoconstrictors |
|
|
Vasodilators |
Monocytes and macrophages
Tissue macrophages
Macrophages have pro-inflammatory and microbicidal capabilities similar to those of neutrophils. Under activation conditions, antigen presentation (see p. 57, 58) is enhanced and a range of cytokines secreted, notably TNF-α, IL-1 and IFN-γ. These are necessary for the removal of certain pathogens that live within mononuclear phagocytes (e.g. mycobacteria). Macrophages and related cells may also undergo a process termed autophagy (p. 32, Ch. 2). This self-cannibalization is a critical property of many cell types under starvation conditions, but is used by the immune system to destroy intracellular pathogens such as Mycobacterium tuberculosis, which otherwise persist within cells and block normal antibacterial processes. Autophagy is also a means of enhancing antigen presentation pathways.
Tissue macrophages involved in chronic inflammatory foci may undergo terminal differentiation into multinucleated giant cells, typically found at the site of the granulomata characteristic of tuberculosis and sarcoidosis (see p. 845).
Dendritic cells
dendritic morphology (Fig. 3.4)
machinery for sensing pathogens
the ability to dictate the T lymphocyte’s future function and differentiation.
Types of dendritic cell
The major types are the myeloid DC (mDC), the plasmacytoid DC (pDC) and a variety of specialized DCs found in tissues that resemble mDCs (e.g. the Langerhans cell in the skin, see Fig. 24.1). DCs have several distinctive cell surface molecules, some of which have pathogen-sensing activity (e.g. the antigen uptake receptor DEC205 on mDCs) whilst others are involved in interaction with T lymphocytes (Table 3.7). Immature mDCs and pDCs are present in the blood, but at very low levels (<0.5% of lymphocyte/monocyte cells).
Mannan-binding lectin, which initiates complement activity inducing opsonization (p. 51, 52).
Signalling receptors such as the PRR known as TLR4 (for toll-like receptor 4), which binds lipopolysaccharide, a molecular pattern found in the cell walls of many Gram-negative bacteria (Table 3.8), whilst others bind double-stranded and single-stranded RNA from viruses. Innate immunity critically depends on toll-like receptor signalling. These receptors act through a critical adaptor molecule, myeloid differentiation factor 88 (MyD88), to regulate the activity of nuclear factor kappa B (NF-κB) pathways.
DCs and T cell activation
In a sequence of events that spans 1–2 days, immature DCs are activated by PAMPs or DAMPs in the tissues binding to a PRR on DCs. The immature pDC is a small rounded cell that develops dendrites upon activation and secretes enormous quantities of IFN-α, a potent antiviral and pro-inflammatory cytokine. On activation, the DC migrates to the local lymph node with the engulfed pathogen. During migration the DC matures, changing its shape, gene and molecular profile and function within a matter of hours to take on a mature form, with altered functions (Table 3.9, Fig. 3.5), in particular upregulating machinery required to activate T lymphocytes. Once in the lymph node, the mature DC interacts with naive T lymphocytes (antigen presentation), resulting in two key outcomes:
| Immature mDC | Mature mDC |
|---|---|
|
Highly pinocytotic |
Ceases pinocytosis |
|
Low level expression of molecules required for T lymphocyte activation |
Upregulates CD80, CD86 and HLA molecules |
|
Low level expression of machinery required to process and present microbial antigens |
Begins to process microbial antigens (break down into small peptides) in readiness to present them to T lymphocytes (using HLA molecules) |
|
Generally localized and sedentary |
Begins active migration to local lymph node |
|
Minimal secretion of cytokines |
Active secretion of cytokines in readiness to stimulate T lymphocytes; in particular IL-12 |
mDC, myeloid dendritic cell.
The mature DC provides three major signals to naive T cells Fig. 3.5):
Hla molecules and antigen presentation
On the short arm of chromosome 6 is a collection of genes termed the major histocompatibility complex (MHC; known as the human leucocyte antigens, or HLA in man), which plays a critical role in immune function. MHC genes code for proteins expressed on the surface of a variety of cell types that are involved in antigen recognition by T lymphocytes. The T lymphocyte receptor for antigen recognizes its ligand as a short antigenic peptide embedded within a physical groove at the extremity of the HLA molecule (Fig. 3.6).
The HLA genes are particularly interesting for clinicians and biologists. First, differences in HLA molecules between individuals are responsible for tissue and organ graft rejection (hence the name ‘histo’(tissue)-compatibility). Second, possession of certain HLA genes is linked to susceptibility to particular diseases (Table 3.10).
Table 3.10 HLA associations with immune-mediated and infectious diseases
| Disease process | Disease | HLA type |
|---|---|---|
|
Autoimmunity |
Type 1 diabetes |
Class II: |
|
|
DQA1*0301/DQB1*0302 (susceptibility) |
|
|
DQA1*501/DQB1*201 (susceptibility) |
||
|
DQA1*0102/DQB1*0602 (protection) |
||
|
|
Class I: |
|
|
|
HLA-A24; HLA-B*18; HLA-B*39 |
|
|
Multiple sclerosis |
DRB1*1501 (susceptibility) |
|
|
Rheumatoid arthritis |
DRB1*0404 (susceptibility) |
|
|
Autoimmune hepatitis |
DRB1*03 and DRB1*04 (susceptibility) |
|
|
Goodpasture’s syndrome (anti-glomerular basement membrane disease) |
HLA-DRB1*1501 (susceptibility) |
|
|
Pemphigus vulgaris |
DRB1*0402; DQB1*0503 (susceptibility) |
|
|
Inflammatory |
Coeliac disease |
DQA1*0501/DQB1*0201 (susceptibility) |
|
Ankylosing spondylitis |
HLA-B27 (susceptibility) |
|
|
Psoriasis |
HLA-Cw*0602 (susceptibility) |
|
|
Infectious |
Human immunodeficiency virus infection |
HA-B27; HLA-B*51; HLA-B*57 (associated with slow progression of disease) |
|
HLA-B*35 (associated with rapid progression) |
The human major histocompatibility complex
The human MHC comprises three major classes (I, II and III) of genes involved in the immune response (Fig. 3.7).
HLA classes
Classical class I HLA genes (also termed Ia), are designated HLA-A, HLA-B and HLA-C. Each encodes a class I α chain, which combines with a β chain to form the class I HLA molecule (Fig. 3.6). While there are several types of α chains, there is only one type of β chain, β2 microglobulin. The HLA class I molecule has the role of presenting short (8–10 amino acids) antigenic peptides to the T cell receptor on the subset of T lymphocytes that bear the co-receptor CD8. As an example of HLA polymorphism, there are nearly 200 allelic forms at the A gene locus. Class I HLA molecules are expressed on all nucleated cells.
Non-classical HLA class I genes are less polymorphic, have a more restricted expression on specialized cell types, and present a restricted type of peptide or none at all. These are the HLA-E, F and G (Ib genes) and MHC class I-related (MIC, or class Ic) genes, A and B. The products of these genes are predominantly found on epithelial cells, signal cellular stress and interact with lymphoid cells, especially natural killer cells (see p. 61, 62).
HLA genotypes and the range of their protein products
Some general principles apply to the HLA genes and their protein products:
The presence of multiple genes on each chromosome, and the fact that both maternal and paternal genes are co-dominantly expressed, allows considerable breadth in the number of HLA molecules that an individual expresses.
The existence of polymorphisms at each locus provides great breadth in the number of HLA molecules expressed at a population level. The polymorphic forms of HLA molecules differ predominantly in the peptide-binding groove (Fig. 3.6).
Antigen presentation
The endogenous route (Fig. 3.8) is a property of all nucleated cells: the internal milieu is sampled to generate peptide–HLA class I complexes for display (‘presentation’) on the cell surface. In a healthy cell, the peptides are derived from self-proteins in the cytoplasm (Fig. 3.8
[/not-level-membership-for-internal-medicine-category]
