8
The genetics of breast cancer, risk-reducing surgery and prevention
Genetic predisposition
The presence of a significant family history is the strongest risk factor for the development of breast cancer. Even at extremes of age, the presence of a BRCA1 mutation confers significant risks. A 25-year-old woman who carries a mutation in BRCA1 has a greater risk of developing breast cancer in the following decade than a woman aged 70 years in the general population. About 4–5% of breast cancer is thought to be due to inheritance of a high-penetrance, autosomal-dominant, cancer-predisposing gene.1,2

Inheritance of a germline mutation or deletion in a predisposing gene predisposes to early-onset, and frequently bilateral, breast cancer. Certain mutations also confer an increased susceptibility to other malignancies, such as ovary (BRCA1/2) and sarcomas (TP53)3–5 (Table 8.1).
Multiple primary cancers in one individual or related early-onset cancers in a family pedigree are highly suggestive of a predisposing gene. It is thought that over 25% of breast cancers in women under 30 years of age are due to mutation in a dominant gene, compared with less than 1% in women who develop the disease over 70 years.2 It has recently been found that at least 27% of breast cancers under 30 years of age are due to mutations in the known high-risk genes BRCA1, BRCA2 and TP53. Nonetheless, risk is still largely based on family history and the detection rate for mutations in isolated breast cancer cases even at very young ages is considerably less than 10%,6 although sporadic grade 3 triple-negative cancers have a little over a 10% chance of a BRCA1 mutation in women under 40 years of age.7
There are few families where it is possible to be certain of a dominantly inherited susceptibility. However, the Breast Cancer Linkage Consortium (BCLC) data suggest that in families with four or more cases of early-onset or bilateral breast cancer, the risk of an unaffected woman inheriting a mutation in a predisposing gene is close to 50%. These studies have estimated that the majority of such families harbour mutations in BRCA1 or BRCA2, especially when male breast cancer or ovarian cancer is present. In breast-only families, the frequency of BRCA1/2 involvement falls to below 50% in four-case families.8 Family and epidemiological studies have demonstrated that approximately 70–85% of BRCA1 and BRCA2 mutation carriers develop breast cancer in their lifetime, although the risk is a little lower for BRCA2.8–11 The very low figures published on small numbers of families from population studies have now been addressed by a meta-analysis,11 which gives risks to 70 years of age of around 70% for BRCA1 and 55% for BRCA2.
The chances that a family with a history of breast and/or ovarian cancer harbours mutations in BRCA1 or BRCA2 can be assessed from computer models.12–14 We have recently validated these models using a dataset of 258 patients and their samples tested for BRCA1/2 mutations. We found that at the lower levels of likelihood for mutations, the computer models substantially overpredict the presence of mutations, particularly for BRCA1.15 The Manchester manual model was much better at predicting a mutation in both genes and indeed was better than other manual models (Table 8.2). Further indicators for the presence of a BRCA1 mutation within a family are grade and oestrogen receptor status. BRCA1 tumours are more frequently grade 3 and oestrogen receptor negative or triple negative, and often have a medullary-like histology.16 Further studies incorporating pathology information into risk models such as the BOADICEA have improved their prediction accuracy.17 Ovarian cancers that develop in BRCA1/2 families are nearly always non-mucinous epithelial cancers.18
Table 8.2
Scoring system for identification of a pathogenic BRCA1/2 mutation
| BRCA1 | BRCA2 | |
| Female breast cancer < 30 years | 6 | 5 |
| Female breast cancer 30–39 years | 4 | 4 |
| Female breast cancer 40–49 years | 3 | 3 |
| Female breast cancer 50–59 years | 2 | 2 |
| Female breast cancer > 59 years | 1 | 1 |
| Male breast cancer < 60 years | 5 (if BRCA2 tested) | 8 |
| Male breast cancer > 59 years | 5 (if BRCA2 tested) | 5 |
| Ovarian cancer < 60 years | 8 | 5 (if BRCA1 tested) |
| Ovarian cancer > 59 years | 5 | 5 (if BRCA1 tested) |
| Pancreatic cancer | 0 | 1 |
| Prostate cancer < 60 years | 0 | 2 |
| Prostate cancer > 59 years | 0 | 1 |
The likelihood of identifying a BRCA1/2 mutation should not be confused with the ability to detect a mutation if one is present in the family. No single technique is able to detect all mutations. Even by sequencing the entire gene (exons and intron/exon boundaries), the detection rate only equates to about 75%. If a strategy is added to detect large deletions or duplications in BRCA1, this can boost detection to around 95%.19

The Ashkenazi Jewish population has three founder mutations: 185delAG and 5382insC in BRCA1, and 6174delT in BRCA2.16 The three mutations are found in over 2% of the Ashkenazi Jewish population. One study showed that one of the three mutations was present in 59% of high-risk families.20
Populations that are more outbred, such as the UK, have larger numbers of mutations and founder mutations occur at lower frequencies. In the past, some laboratories concentrated on the large exons (exon 11 in both genes and exon 10 in BRCA2) and the smaller exons commonly reported to be involved, such as exons 2 and 20 in BRCA1. This cuts down the number of polymerase chain reactions using the protein truncation test (PTT) to as little as five for BRCA1 and four for BRCA2. However, this strategy reduces the sensitivity of identifying mutations down to as little as 50%.19 With the great strides made in reducing cost and time of mutation searching using next generation sequencing these older strategies are now outdated.
Genetic testing
Once a mutation in a predisposing gene like BRCA1 or BRCA2 has been identified in a family, definitive genetic testing is possible. This can then more accurately inform women of their risks and give them an informed choice of different options, including risk-reducing surgery. Undertaking mutation analysis on an unaffected individual (without checking an affected relative), particularly in a breast cancer-only family, is problematic. Whilst identifying a pathogenic mutation will confirm a high risk, the absence of a mutation will not exclude the possibility that other genes or even a mutation refractory to the mutation screening techniques used are present. Although other genes are now being identified (see Table 8.1), many more remain to be found and screening for mutations is not clinically useful outside of BRCA1/2 and in certain circumstances TP53. Nevertheless, the outcomes of genome-wide association studies indicate that all breast cancer cases will carry at least one risk allele.21,22 Once all the lower risk alleles have been found a definitive genetic test can then be developed.
Breast cancer risk estimation
Where there is not a dominant family history or it is not possible to identify a mutation in BRCA1/2, risk estimation is based on large epidemiological studies, which give a 1.5- to 3-fold relative risk with a family history of a single affected relative.1,2 Clinicians must be careful to differentiate between lifetime and age-specific risks. Some studies quote a ninefold or greater risk associated either with bilateral disease in a mother or with severe atypical hyperplasia. However, these risks are time limited and if these at-risk individuals are observed for many years, their relative risk is reduced over time.23 Clearly, if one uses these risks and multiplies them against the lifetime incidence of 1 in 8–12 then some women will apparently have a greater than 100% chance of having the disease. Risks importantly do not multiply and may not even be additive. The best way to assess risk is to take the strongest risk factor, which in most cases is nearly always the family history. If risk is assessed on this alone, minor adjustments can then be made for other factors. It is arguable whether these other factors have any major effect on an 80% penetrant gene other than to speed up or delay the onset of breast cancer. Therefore, we can only really assume an effect on non-hereditary elements of risk. Although studies do point to an increase in risk in family history cases associated with certain factors, these may just represent an earlier age expression of the gene. Generally, therefore, non-mutant gene carriers will have risks somewhere between 40–45% and 8–10%, although lower risks are occasionally given. Higher risks are only applicable when a woman at 40–45% genetic risk is shown to have a germline mutation and to have inherited a high-risk allele or to have proliferative breast disease.

Within Europe, risk estimation in the family history setting is based mainly on the Claus dataset.2,24,25 However, within the USA, the Gail model of risk estimation is widely used.26 As well as these datasets allowing estimation of risk, there are specific computer programs available, including Tyrer–Cuzick,27 Cyrillic, BOADICEA and BRCAPRO.14
These programs take into consideration varying permutations of age of onset of diagnosis, number of affected and unaffected women, and hormonal factors; as a consequence, different programs result in different risk estimations. The Gail model does not take into account age of relatives or second-degree relatives. A newer model, the Tyrer–Cuzick,27 incorporates the majority of the currently known risk factors.
A major deficiency of current genetic models is the assumption that all inherited breast cancer is due to a single high-risk dominant gene or two genes (BRCA1 and BRCA2). The problem this causes in a program like BRCAPRO is that in order to obtain an accurate assessment for identifying a BRCA1 or BRCA2 mutation in a family, all other potential genetic factors are overlooked. Therefore, while BRCAPRO provides reasonably accurate estimates for the presence of BRCA1/2 mutations in high-risk families,12 its ability to predict breast cancer incidence is substantially hampered in smaller aggregations of breast cancer. Variation in their value in different ethnicities has also been found.28–30 We have recently found that BRCAPRO underestimates the risk of breast cancer in moderate/high-risk families by about 50%.31 The most accurate computer model was the Tyrer–Cuzick model, although a manual model incorporating the Claus tables and data from the BCLC and adjustment for hormonal and reproductive factors was similarly accurate. A fuller explanation of the manual model25,31 and a detailed review32 are available elsewhere.32
Management options
Management options available to reduce the risks of developing breast cancer for women at high lifetime risk due to their family history or for those women known to be carrying a mutation in BRCA1/2 are limited. Screening with mammography or magnetic resonance imaging (MRI) is one option but this detects cancer, it does not prevent it. Therefore, imaging surveillance can be combined with chemoprevention. However, many women consider or undergo risk-reducing mastectomy (RRM) if found to be at high risk, e.g. mutation carriers for BRCA1 or BRCA2. The efficacy of surgical procedures for reducing the risk of breast cancer is controversial but of proven benefit,33,34 although it would appear that the residual risk of breast cancer depends on the amount of residual breast tissue following the surgical procedure. It would be ideal to perform a prospective randomised clinical trial where women with the same risk were randomised to either intensive surveillance or prophylactic surgery, but it would be difficult to recruit women for this type of trial and so it is unlikely to happen. Recent work suggests that more women are considering RRM,35,36 although uptake rates even for BRCA mutation carriers vary enormously, with a much lower uptake in Israel and southern Europe, almost certainly due to different cultural beliefs, protocols and availability.36 Protocols should be in place to deal with requests for RRM at all cancer genetics and oncoplastic clinics. It has been suggested that surgery does increase life expectancy in BRCA1 or BRCA2 mutation carriers.37

The first study to demonstrate that women with a high risk of breast cancer can significantly reduce their subsequent incidence of the disease with RRM was published in 1999.38 This was followed by a Dutch study that confirmed risk reduction in those at highest risk (BRCA1/BRCA2 carriers).39 Current evidence would suggest that RRM is associated with an approximately 90–95% reduction in risk.40,41
Genetic counselling and the family history clinic
Breast cancer family history clinics started to be established in the UK in 1987,42,43 and these clinics are now established across Europe, North America and are now also emerging in Asia.44,45 Genetic counselling sessions and family history clinics are important not only to provide a means to select suitable individuals for genetic testing, but also to aid decision-making in individual women on surveillance and preventative measures. They are generally administered by consultants in medical oncology, clinical genetics and breast surgery, often with a multidisciplinary approach and with close involvement of radiologists and a psychiatrist/psychologist. At these clinics unaffected women at increased risk of breast cancer are assessed as to their lifetime and shorter-term risks of breast cancer development. After assessing risk, women are presented with a number of choices including regular surveillance, usually with a combination of clinical examination, mammography, ultrasonography and in some circumstances breast MRI46 that commences between 20 and 40 years depending on the age of onset of cancers in the family and the overall risk. Women are generally divided into three risk groups: average, moderate and high risk. It is only in the high-risk group that RRM should be considered. This usually equates to a lifetime risk of 1 in 4 (25%) or greater. As a rough guide, this equates to having a heterozygote risk of 1 in 4 with two relatives, including one first-degree relative, with breast cancer diagnosed below 50 years of age or three affected relatives under 60 years. All affected relatives should be first-degree relatives or related through a male.
In a survey of 10 European centres,42 only three (Manchester, Edinburgh, Heidelberg/Dusseldorf) routinely mentioned the possibility of RRM to women with a lifetime risk of 1 in 4 or greater. This information is often only given as a statement of the availability of the procedure as an option for prevention of breast cancer. This allows women to extend the discussion if they wish to do so, or to state that they are not interested in surgery. Many centres only mention risk-reducing surgery to potential mutation carriers undertaking a genetic test. Indeed, there is a cultural shift across Europe from north to south, with RRM being less acceptable to both physician and patient as one moves southward.47,48 In the USA, in centres where mastectomies for benign breast disease were commonplace in the 1970s and 1980s,38 there appears to be less enthusiasm for mastectomy now even among gene mutation carriers.49 What is absolutely clear is that adequate preparation of a woman contemplating RRM is essential.
The RRM protocol
Women who wish to consider RRM should be given time to consider the procedure thoroughly prior to making the decision. In Manchester, a protocol is in place for women who are assessed as at high risk who would be suitable for RRM despite not having certain knowledge of mutation status. For example, an individual’s calculated lifetime risk may be 40%, but a recognised gene mutation may not have been identified in the family. This exact same protocol may not be offered in other centres but the general principles of counselling apply. In general, most centres will offer multiple sessions of discussion of available surgical procedures and counselling. Most women would be offered a further appointment at least 1 month later. This not only gives the women time to consider more fully all options but also allows time for them to discuss it with appropriate members of their family. The mean time interval from a personal genetic diagnosis to the date of surgery of RRM is approximately 9 months.50 Usually the partner or a family member selected by the patient is encouraged to attend the clinic session to help decision-making. At the second appointment, with a geneticist or oncologist, a basic description of the surgery is provided, including the potential residual risk of different procedures. It is emphasised that a residual breast cancer risk and complication rates from surgery are still present after RRM and may be higher if the surgery preserves the nipple–areola complex (NAC). Options of simultaneous breast reconstruction can also be discussed. It is important to prepare the patient for the likely consequences of mastectomy including pain and any possible complications that, if they occur after breast reconstruction, may result in a poor cosmetic result, as well as considering the impact upon her personal life and family dynamics. Because of potential pitfalls such operations should only be undertaken by surgeons highly skilled in RRM and its possible complications, to optimise both oncological and aesthetic outcomes.
When possible, a living affected member should be the first to undertake genetic testing as this provides more information on the underlying genetic structure of the family.25,51,52 A time scale for genetic testing is discussed, and the woman is asked to consider the potential impact of proceeding with surgery, particularly if she then undergoes genetic testing and finds that she does not in fact carry the causative mutation. It is also emphasised that the genetic risk of breast cancer decreases with age and that the remaining risk of breast cancer if the woman is older (> 40 years) and has not developed breast cancer becomes lower than the lifetime risk.25 Indeed, a mutation carrier for BRCA1 may have no more than a 50% risk of breast cancer in her remaining lifetime if she has reached 50 years, but this is still a substantial personal risk. Psychological assessement and counselling should be arranged for women who are considering proceeding.

Confirmation of breast cancers in the family must be sought proactively if this has not already been done. This ensures that risk assessment is as accurate as possible. The Manchester group has previously reported the presence of factitious histories within some families where women have fabricated their family history in order to obtain surgery, or are implicated innocently as being at risk by another family member who has promoted an inaccurate family history.53
The whole process, from first consultation to the surgical procedure itself, usually takes between 6 and 12 months. This time delay is deliberate; in most centres the greatest delay is at the beginning of the protocol in order to allow women time for the decision-making process. If the protocol is run concurrently with a decision for predictive genetic testing, then the wait will generally be shorter. The full protocol of two sessions at the family history clinic, a session with a psychiatrist and sessions with the surgeon was established in 1993 in Manchester. The major difference between this protocol and that offered by some other centres both locally and internationally is that RRM in other centres is offered only when raised by the patient, and it is generally only offered in these centres when a woman is proven to be a BRCA1/2 mutation carrier. Even with a proven mutation carrier no centre actively recommends surgery, but offers this as part of a range of choices. The incorporation of counselling during decision-making and treatment has been shown to improve psychological and social outcome.54,55
Surgical consultation
The aim of risk-reducing surgery is not only to reduce cancer risk and breast cancer mortality, but also to reduce the psychological distress and anxiety of an individual.56 For unaffected mutation carriers RRM reduces lifetime risk of breast cancer to less than 5%.57,58 However, any surgical procedure, particularly in a preventative setting, warrants a balanced discussion of its benefits and risks. Due to lack of level I evidence from randomised controlled clinical trials, recommendations are usually based on cohort studies of prophylactic surgery.59,60 Moreover, to date, although risk of breast cancer is reduced there are still no data that show statistically significant improvement in survival after RRM. It is also important that when a women embarks on such a decision, a full understanding of the surgery involved is achieved and the woman understands that risk reduction of cancer is never 100%. Risk reduction surgery is not a minor cosmetic procedure but major surgery, and hence the nature and extent of the surgery and possible complications need to be considered. The expected cosmetic result, changes in the patient’s breast sensation and also the impact of RRM on a patient’s social life should be addressed. The whole range of evolving surgical techniques that can be offered should be discussed thoroughly before a decision is made for surgery.
In order to properly select suitable individuals for RRM, at least two detailed surgical consultations are needed to discuss the types of mastectomy and breast reconstruction procedures that are available. The techniques, limitations, outcomes and potential complications, and photographs of a range of aesthetic outcomes should be shown. It must be emphasised that there is still a small chance of getting cancer after RRM and that subjectively the breasts may look and feel different from the original breasts even if a successful surgical outcome is achieved. Time should be given for the women to digest this information. Consultations should involve a breast oncoplastic surgeon, plastic surgeon if appropriate and also a breast nurse specialist. More intimate issues including quality-of-life concerns, changes of body image and impact on sexual relationship with her partner may result in further questions following surgical consultation.61 Before discussing details of options for surgery, during the consultation session it is important to identify any women where RRM may not be advisable. There are relative contraindications to RRM taking place (see Box 8.1).
The decision of which surgical option to choose will vary between individuals (see Box 8.2).62 For some women a traditional total simple mastectomy without reconstruction may offer what she wants as it reduces risk and can be cosmetically acceptable if performed appropriately (see Chapter 5), whereas for others NAC-sparing mastectomy with reconstruction is the only acceptable aesthetic outcome. Skin-sparing mastectomy (SSM) has already been shown to be as oncologically safe as total simple mastectomy on selected patients, but the data available are mainly from patients with existing breast cancer. There are no specific data for prophylactic surgery using SSM, but it does appear to be an acceptable approach oncologically.63,64 Non-SSM, also known as subcutaneous mastectomy, evolved due to psychological studies that found that the feeling of mutilation is increased after removal of the NAC when performing mastectomy.65 It is unsafe to compare directly data from women who have been treated for breast cancer with those who have not; however, there have been controversies behind the safety of saving the NAC, as occult tumour involvement behind the NAC in mastectomy specimens in cancer patients has been reported to occur in 0–50% of cases.66–68 However, many of these studies were performed more than two decades ago when significant amounts of breast tissue were left behind the nipple and there are new data indicating that for selected individuals nipple-sparing mastectomy (NSM) is oncologically safe. Moreover, it is likely that, formerly, case selection was less well planned and with improvement of case selection of patients who are deemed more suitable for NSM, either by clinical presentation or after MRI with use of intraoperative frozen section to exclude patients with nipple involvement, it is likely that long-term results would be favourable.69–71 There is a risk of nipple–areola complex necrosis of about 5–15% when performing NSM, which can be partial or complete, and this is more common in smokers (see Chapter 5). In general, partial necrosis usually heals without further surgical intervention. There is a proportion of women who suffer nipple sensation loss of 25%.72–74 Loss of nipple projection and depigmentation of the areola skin are also common problems with NSM. The improvement of psychological outcome from NSM65 makes this the option of choice for most women considering RRM.
Various techniques have been suggested as incisional options for NSM.74
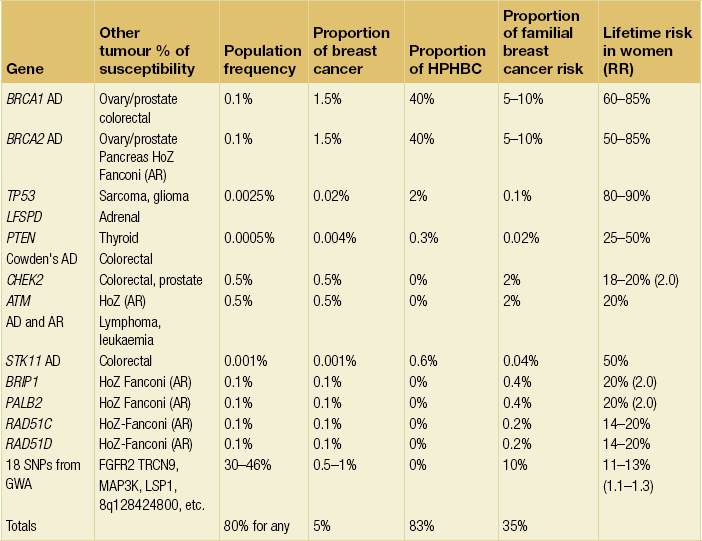
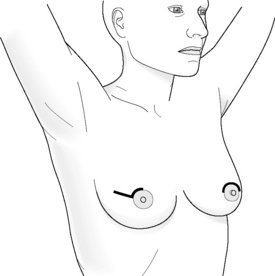
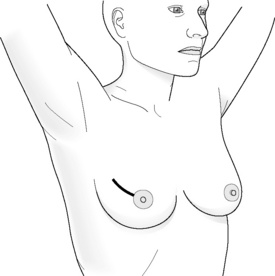
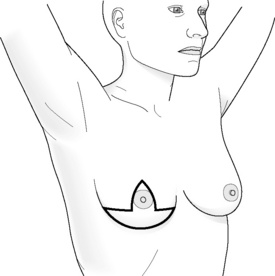
Incisions for nipple-sparing mastectomy (Figs 8.1–8.6)
In a study by Esserman and colleagues, it was found that NAC necrosis was higher when an incision crossing the NAC was used (18%).74 If the incision spanned less than one-third of the circumference of the areola the results were good. Those that cover more than one-third of the circumference are likely to compromise the blood supply of the nipple, which can result in loss of nipple–areola skin. Inframammary incisions were found to be more suitable for women with small breasts, as this incision allows only limited exposure of the upper part of the breast, being particularly problematic if performed in women who have larger breasts. A radial incision in this study provided the best access for breast tissue removal and was most likely to maintain NAC viability, but can result in nipple deviation in the direction of the incision. If reconstruction is planned, then the choice of incision is even more important and should be based on the size of the breast and the degree of ptosis.
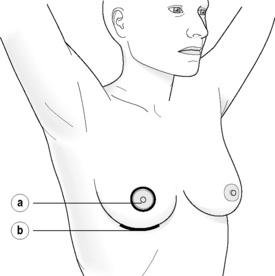
Figure 8.1 (a) Skin-sparing dissection with removal of the nipple and areola skin and replacement of the nipple alone or the nipple–areola complex as a free graft. (b) Submammary/inframammary incision.
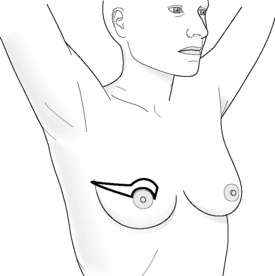
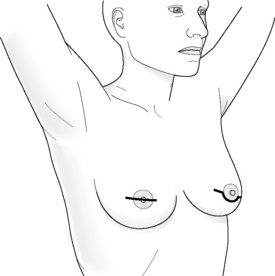
Areola-sparing mastectomy (ASM) has also been proposed as an option to NSM, as in theory it is the ducts that transverse the nipple that can harbour occult metastatis and not the areola disc, which is more similar to ‘skin’.34 Hence, over recent years this has also become an option for women who choose to have a mastectomy.
Risk-reducing surgery and breast reconstruction
Mastectomy can be a mutilating surgery for many women and so during the last century there has been increased use of reconstruction for women who opt for mastectomy. This is even more relevant for the group of high-risk women who opt for RRM, who have not themselves had breast cancer, and who need long-term reconstruction of functional and aesthetic excellence. The need for more and better breast reconstruction options after mastectomy has resulted in an increase in the number and types of different reconstructive techniques to achieve the best aesthetic outcomes with a more personalised approach based on a woman’s breast size, desired outome and technical feasibility (see Box 8.3).
Reconstructions after RRM can be performed immediately or in a staged fashion. In general, unless contraindicated, immediate reconstruction should be performed as it avoids the woman having a period of time without a ‘breast’ and it allows skin to be spared, which improves the final cosmetic result (Fig. 8.7). Although most reconstructions produce satisfactory results, it is not uncommon for a patient to have expectations that there will be no visible scars. Moreover, providing patients with photographs of possible complications is an important part of informing the patient, as is providing a specialist nurse to liaise with the woman and to address further questions that the woman may have.Sharing experience with others who have undergone the procedure may also help to better prepare an individual when deciding on RRM and breast reconstruction.
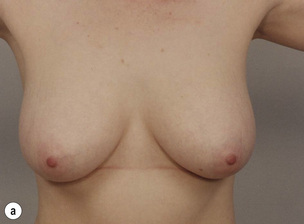
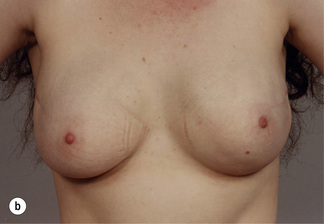
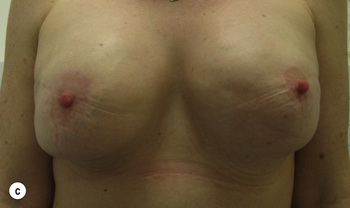
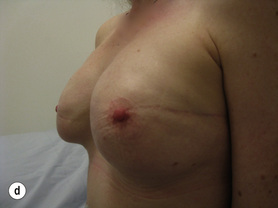
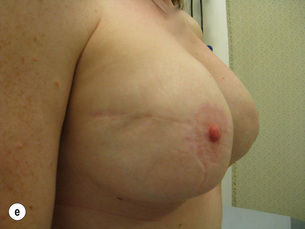
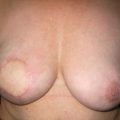
Figure 8.7 Horizontal/oblique risk-reducing mastectomy (RRM) before (a) and after (b) operation.(c–e) The same patient 15 years after RRM and implant reconstruction.
In women with large breasts, the skin envelope may need to be reduced when performing breast reconstruction (Figs 8.8 and 8.9). The placement of an acellular dermal matrix can also be used in addition to fat injection to achieve a better aesthetic result.71 Autologous myocutaneous flaps use natural tissue, and give women the feeling that they are using living tissue and have a warm soft consistency that can be more realistic. Expertise in oncoplastic and plastic surgery is required to decrease complication rates when microvascular anastomoses are warranted.
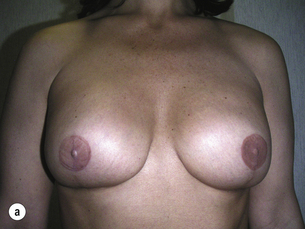
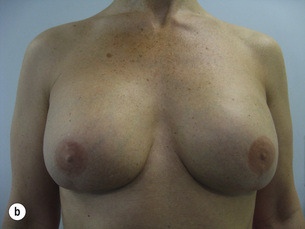
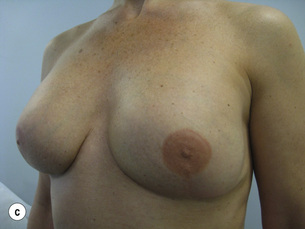
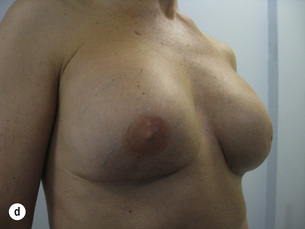
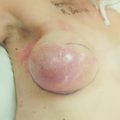
Figure 8.8 (a) Wise pattern risk-reducing mastectomies with nipple reconstructions. (b–d) The same patient 10 years later, with left lateral view (c) and right lateral view (d).
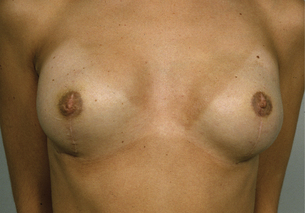
The use of implant devices after RRM seems a simpler option compared with the transfer of myocutaneous flaps but complications are common and considerable skill and expertise is required to get good, consistent results. There are different types of prosthesis in relation to shape and contour. Prostheses can be permanent or allow expansion, with saline being injected gradually into the expandable chamber of the prosthesis until the desired volume is achieved. These help in providing a natural ‘breast’ projection and may result in better contour in the upper breast.75 Anatomical expanders permit more rapid expansion and result in lower pressures within the implant, appear less likely to migrate and cause less chest deformity. Silicon gel implants are available in a range of specifications and gel types, to allow a more natural look and feel. Implants are usually placed under the chest wall muscle and, combined with well planned and carried out surgery, can give a good aesthetic outcome. Complications from such surgery includeimplant loss, haematoma, wound infection and skin flap necrosis, and the rate has been quoted as greater than 10%. For women who choose to have NSM, the use of expanders or Becker expander/prostheses allows gradual expansion to minimise skin tension and is associated with lower infection and implant loss compared with fixed-volume implants, particularly if the implants are of a volume greater than the original breast size.74 Use of permanent expander prostheses with detachable valves is associated with low infection rates and good recovery in women who have RRM and reconstruction.76 Alternative incisions have been proposed when performing RRM with reconstruction for high-risk women and these include the inverted drip incision.77 Implants can also be combined with a myocutaneous flap and acellular dermal matrix. The overall complication rate using a direct-to-implant immediate breast reconstruction using human acellular dermal matrix (ADM) or ADM from porcine or bovine skin or bovine pericardium has been reported to be low, at 3.9% in some series (implant loss 1.3%, skin necrosis 1.1%, haematoma 1.1%, human acellular dermal matrix exposure 0.6 %, capsular contracture 0.4%, and infection 0.2%).78 More recent reports using non-human acellular dermal matrices report an implant loss rate closer to 10%. There is a higher rate of seroma, infection and implant loss compared to when ADM is not used, but the aesthetic results are superior with ADM (see Chapter 9). RRM with breast reconstruction is considered a reasonably safe and acceptable procedure for risk reduction until other risk reduction methods become available.
If the NAC is removed during RRM, the NAC can be reconstructed with the areola tattoed. There has also been a description of nipple grafting, in particular if the areola is kept and the nipple is retransplanted to the reconstructed breast.76

Uptake
Data from Manchester, UK79 show that 6% of women who are at a 1 in 4 (31/902) lifetime risk or above seek further advice about RRM and 1.8% (16/902) have undergone surgery; this rises to 6% (49/798) in those at 40–45% lifetime risk. Although the uptake in BRCA1/2 carriers tended to be early, there was a continued increase in uptake so that by 7 years the actuarial uptake for RRM for BRCA1 carriers was 60% and for BRCA2 carriers was 43%. Results from the Netherlands show a similarly high uptake (52%).39 Thus far, 35 mutation carriers in our series and several in the Dutch series that initially opted not to have surgery have developed breast cancer, whereas only two of the operated cases have. Newer research including non-European/American cohorts from Asia have found that RRM is generally considered only in women who already have breast cancer and decide to opt for completion mastectomy and contralateral prophylactic mastectomy, although the uptake of prophylactic salpingo-oopherectomy is slightly higher. There is also a proportion of women who consider risk-reducing surgery after a period of intensive surveillance.44
Role of sentinel lymph node biopsy in RRM
It has been suggested that as occult cancers are found in 5% of prophylactic mastectomy specimens, to avoid a second operation sentinel lymph node biopsy (SLNB) should be performed when performing RRM. However, the latest research has found that although occult malignancy can be present in prophylactic mastectomy specimens, it is rare for the occult carcinoma to spread to the lymph nodes. A meta-analysis of available research studies found that the rate of occult invasive cancer was 1.7% and the rate of positive SLNB was 1.9%. Hence, it is concluded that SLNB should not be performed during RRM of women without cancer but may be considered if cancer is present (see chapter 7).80
Psychosocial consequences of RRM

The majority of studies exploring the psychological impact of RRM have found that surgery is associated overall with fairly high levels of satisfaction, reduced anxiety and psychological morbidity amongst women who undergo this procedure.81–88 This is particularly so when there is support from family and friends.83 A number of studies suggest that provision of presurgical multidisciplinary support appears to have a bearing on outcome, particularly in those who have initial anxiety and negative psychological reactions. These negative effects are usually related to surgical complications and reduce with longer follow-up. A minority of women do express regrets and experience adverse psychosocial events following surgery, including adverse emotional stability, self-esteem and problems in sexual relationships.87
Surgery for high-risk women with established breast cancer
For women who are diagnosed with unilateral breast cancer, there is a 2–5% risk of having cancer in the contralateral breast. For BRCA mutation carriers, studies have found that if breast-conserving surgery is performed, the 10-year cumulative incidence of ipsilateral breast cancer recurrences is 27% for mutation carriers versus 4% for sporadic controls. The 10-year cumulative incidence of contralateral breast cancer is 25% in mutation carriers compared with 1% in sporadic controls.89 Hence, it is most likely that women who are BRCA mutation carriers should consider mastectomy on the diseased breast and also consider contralateral prophylactic mastectomy (CPM). CPM can reduce contralateral cancer development by 90%, but its effectiveness in preventing breast cancer mortality is far from clear. Although most studies have found good psychological outcomes and also satisfaction in women who undergo CPM, the operation is not risk free and complications related to the surgical procedures, including breast reconstruction, do occur. These factors all need to be addressed when considering a decision on CPM. Any contralateral procedure can be delayed until treatment for the primary cancer is completed. The prognosis depends on the primary cancer tumour biology. A woman who is found to have substantial lymph node involvement is unlikely to benefit from CPM. Hence the management decision of this group of women should be decided at a multidisciplinary meeting and the aim must be to prioritise treatment of the primary malignancy.90

These women are often young, and if their family history is attributable to a gene mutation, identified or not, their personal risk of local recurrence or development of a second primary cancer in the same breast is higher than in women with sporadic breast cancer; their risk of contralateral new primary malignancy is also high.81–83
Individual circumstances vary widely; the decision on how to proceed is individual to each patient. Rates of contralateral mastectomy are high in gene mutation carriers where the efficacy of contralateral mastectomy is now well established. Proof that this may save lives is now emerging.91–98
Bilateral risk-reducing salpingo-oophorectomy

In addition to reducing the risk of ovarian cancer, prophylactic salpingo-oophorectomy has also been shown to decrease the risk of breast cancer in women with a BRCA1/2 mutation by 46–56%.99 This can be seen from the risk of breast cancer in unaffected women and in the contralateral breast for those with breast cancer. Moreover, a recent study has shown that prophylactic salpingo-oophorectomy not only reduced the risk of breast and ovarian cancer but also lowered all-cause mortality, as well as breast cancer and ovarian cancer specific mortality.57
If salpingo-oophorectomy is performed at a premenopausal age, usually at about 40 years old, particularly in BRCA1/2 mutation carriers, and if hysterectomy is performed at the same setting, the use of unopposed oestrogen as hormone replacement therapy (HRT) can ameliorate symptoms of surgical menopause and does not seem to increase the risk of breast cancer.100,101 However, as long-term effects are still not known, the effects of an early menopause and doubts over long-term HRT use have to be considered if the primary purpose is breast cancer prevention.
Use of chemopreventive agents
Aromatase inhibitors

Tamoxifen reduces breast cancer incidence by 40–50%93,102–105 and this is maintained in long-term follow-up after cessation of treatment.105 However, this reduction is almost exclusively a reduction in oestrogen receptor-positive disease and whether this translates into a reduction in cancers related to BRCA1 mutations remains unclear. The NSABP Study of Tamoxifen and Raloxifene (STAR) Trial has shown that raloxifene is nearly as effective as tamoxifen in reducing invasive breast cancer risk and has fewer adverse side-effects.106 However, there are no data available on the efficacy of raloxifene for breast cancer risk reduction among BRCA gene mutation carriers.
Acknowledgement
Thanks to Dr Louis Kwong, Jr for supplying the original line drawings in this chapter.
References
1. Newman, B., Austin, M.A., Lee, M., et al, Inheritance of human breast cancer: evidence for autosomal dominant transmission in high-risk families. Proc Natl Acad Sci U S A. 1988;85(9):3044–3048. 3362861
2. Claus, E.B., Risch, N., Thompson, W.D., Autosomal dominant inheritance of early-onset breast cancer. Implications for risk prediction. Cancer. 1994;73(3):643–651. 8299086
3. Miki, Y., Swensen, J., Shattuck-Eidens, D., et al, A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. 7545954
4. Wooster, R., Bignell, G., Lancaster, J., et al, Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789–792. 8524414
5. Malkin, D., Li, F.P., Strong, L.C., et al, Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–1238. 1978757
6. Evans, D.G., Moran, A., Hartley, R., et al, Long-term outcomes of breast cancer in women aged 30 years or younger, based on family history, pathology and BRCA1/BRCA2/TP53 status. Br J Cancer. 2010;102(7):1091–1098. 20234365
7. Evans, D.G., Howell, A., Ward, D., et al, Prevalence of BRCA1 and BRCA2 mutations in triple negative breast cancer. J Med Genet. 2011;48(8):520–522. 21653198
8. Ford, D., Easton, D.F., Stratton, M., et al, Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998;62(3):676–689. 9497246
9. Ford, D., Easton, D.F., Bishop, D.T., et al, Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994;343(8899):692–695. 7907678
10. Evans, D.G., Shenton, A., Woodward, E., et al, Penetrance estimates for BRCA1 and BRCA2 based on genetic testing in a Clinical Cancer Genetics service setting: risks of breast/ovarian cancer quoted should reflect the cancer burden in the family. BMC Cancer 2008; 8:155. 18513387
11. Antoniou, A., Pharoah, P.D., Narod, S., et al, Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–1130. 12677558
12. Parmigiani, G., Berry, D.A., Aguilar, O., Determining carrier probabilities for breast cancer-susceptibility genes BRCA1 and BRCA2. Am J Hum Genet. 1998;62(1):145–158. 9443863
13. Berry, D.A., Iversen, E.S., Gudbjartsson, D.F., et al, BRCAPRO validation, sensitivity of genetic testing of BRCA1/BRCA2, and prevalence of other breast cancer susceptibility genes. J Clin Oncol. 2002;20(11):2701–2712. 12039933
14. Antoniou, A.C., Cunningham, A.P., Peto, J., et al, The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br J Cancer. 2008;98(8):1457–1466. 18349832
15. Evans, D.G., Eccles, D.M., Rahman, N., et al, A new scoring system for the chances of identifying a BRCA1/2 mutation outperforms existing models including BRCAPRO. J Med Genet. 2004;41(6):474–480. 15173236
16. Lakhani, S.R., Van De Vijver, M.J., Jacquemier, J., et al, The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol. 2002;20(9):2310–2318. 11981002
17. Mavaddat, N., Rebbeck, T.R., Lakhani, S.R., et al, Incorporating tumour pathology information into breast cancer risk prediction algorithms. Breast Cancer Res. 2010;12(3):R28. 20482762
18. Evans, D.G., Young, K., Bulman, M., et al, Probability of BRCA1/2 mutation varies with ovarian histology: results from screening 442 ovarian cancer families. Clin Genet. 2008;73(4):338–345. 18312450
19. Evans, D.G., Bulman, M., Young, K., et al, Sensitivity of BRCA1/2 mutation testing in 466 breast/ovarian cancer families. J Med Genet. 2003;40(9):e107. 12960223
20. Struewing, J.P., Hartge, P., Wacholder, S., et al, The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336(20):1401–1408. 9145676
21. Easton, D.F., Pooley, K.A., Dunning, A.M., et al, Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447(7148):1087–1093. 17529967
22. Turnbull, C., Ahmed, S., Morrison, J., et al, Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42(6):504–507. 20453838
23. Dupont, W.D., Page, D.L., Relative risk of breast cancer varies with time since diagnosis of atypical hyperplasia. Hum Pathol. 1989;20(8):723–725. 2744746
24. Vasen, H.F., Haites, N.E., Evans, D.G., et al, Current policies for surveillance and management in women at risk of breast and ovarian cancer: a survey among 16 European family cancer clinics. European Familial Breast Cancer Collaborative Group. Eur J Cancer. 1998;34(12):1922–1926. 10023316
25. Evans, D.G., Lalloo, F., Risk assessment and management of high risk familial breast cancer. J Med Genet. 2002;39(12):865–871. 12471197
26. Gail, M.H., Brinton, L.A., Byar, D.P., et al, Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst. 1989;81(24):1879–1886. 2593165
27. Tyrer, J., Duffy, S.W., Cuzick, J., A breast cancer prediction model incorporating familial and personal risk factors. Stat Med. 2004;23(7):1111–1130. 15057881 This model incorporates the majority of currently known breast cancer risk factors.
28. Kwong, A., Wong, C.H., Suen, D.T., et al, Accuracy of BRCA1/2 mutation prediction models for different ethnicities and genders: experience in a Southern Chinese cohort. World J Surg. 2012;36(4):702–713. 22290208
29. Kurian, A.W., Gong, G.D., Chun, N.M., et al, Performance of BRCA1/2 mutation prediction models in Asian Americans. J Clin Oncol. 2008;26(29):4752–4758. 18779604
30. Kurian, A.W., Gong, G.D., John, E.M., et al, Performance of prediction models for BRCA mutation carriage in three racial/ethnic groups: findings from the Northern California Breast Cancer Family Registry. Cancer Epidemiol Biomarkers Prev. 2009;18(4):1084–1091. 19336551
31. Amir, E., Evans, D.G., Shenton, A., et al, Evaluation of breast cancer risk assessment packages in the family history evaluation and screening programme. J Med Genet. 2003;40(11):807–814. 14627668
32. Amir, E., Freedman, O.C., Seruga, B., et al, Assessing women at high risk of breast cancer: a review of risk assessment models. J Natl Cancer Inst. 2010;102(10):680–691. 20427433
33. Goodnight, J.E., Quagliana, J.M., Morton, D.L., Failure of subcutaneous mastectomy to prevent the development of breast cancer. J Surg Oncol. 1984;26(3):198–201. 6330460
34. Ziegler, L.D., Kroll, S.S., Primary breast cancer after prophylactic mastectomy. Am J Clin Oncol. 1991;14(5):451–454. 1951182
35. Evans, D., Lalloo, F., Shenton, A., et al, Uptake of screening and prevention in women at very high risk of breast cancer. Lancet. 2001;358(9285):889–890. 11567707
36. Metcalfe, K.A., Birenbaum-Carmeli, D., Lubinski, J., et al, International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int J Cancer. 2008;122(9):2017–2022. 18196574
37. Schrag, D., Kuntz, K.M., Garber, J.E., et al, Decision analysis – effects of prophylactic mastectomy and oophorectomy on life expectancy among women with BRCA1 or BRCA2 mutations. N Engl J Med. 1997;336(20):1465–1471. 9148160
38. Hartmann, L.C., Schaid, D.J., Woods, J.E., et al, Efficacy of bilateral prophylactic mastectomy in women with a family history of breast cancer. N Engl J Med. 1999;340(2):77–84. 9887158 This retrospective US cohort study examined the incidence of, and risk of death from, breast cancer after a median follow-up of 14 years among 639 women who had a family history of breast cancer and who had undergone bilateral subcutaneous or total prophylactic mastectomy. In the mastectomy group, women were divided into high-risk (n = 214) or moderate-risk (n = 425) subgroups, with most women in each subgroup having undergone subcutaneous mastectomy (89% and 90%, respectively). The study showed a reduction in the risk of breast cancer of 89.5% (P < 0.001) in moderate-risk women who had undergone prophylactic mastectomy, and a reduction in risk of 90–94% in the high-risk women.
39. Meijers-Heijboer, H., van Geel, B., van Putten, W.L.J., et al, Breast cancer after prophylactic bilateral mastectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med. 2001;345(3):159–164. 11463009
40. Evans, D.G., Baildam, A.D., Anderson, E., et al, Risk reducing mastectomy: outcomes in 10 European centres. J Med Genet. 2009;46(4):254–258. 18996907
41. Friebel, T.M., Domchek, S.M., Neuhausen, S.L., et al, Bilateral prophylactic oophorectomy and bilateral prophylactic mastectomy in a prospective cohort of unaffected BRCA1 and BRCA2 mutation carriers. Clin Breast Cancer. 2007;7(11):875–882. 18269778
42. Evans, D.G., Cuzick, J., Howell, A., Cancer genetics clinics. Eur J Cancer. 1996;32A(3):391–392. 8814678
43. Evans, D.G., Anderson, E., Lalloo, F., et al, Utilisation of prophylactic mastectomy in 10 European centres. Dis Markers. 1999;15(1–3):148–151. 10595270
44. Kwong, A., Wong, C.H., Shea, C., et al, Choice of management of southern Chinese BRCA mutation carriers. World J Surg. 2010;34(7):1416–1426. 20182723
45. Han, S.A., Park, S.K., Ahn, S.H., et al, The Korean Hereditary Breast Cancer (KOHBRA) study: protocols and interim report. Clin Oncol (R Coll Radiol). 2011;23(7):434–441. 21497495
46. Pruthi, S., Gostout, B.S., Lindor, N.M., Identification and management of women with BRCA mutations or hereditary predisposition for breast and ovarian cancer. Mayo Clin Proc. 2010;85(12):1111–1120. 21123638
47. Julian-Reynier, C., Eisinger, F., Moatti, J.P., et al, Physicians’ attitudes towards mammography and prophylactic surgery for hereditary breast/ovarian cancer risk and subsequently published guidelines. Eur J Hum Genet. 2000;8(3):204–208. 10780786
48. Julian-Reynier, C.M., Bouchard, L.J., Evans, D.G., et al, Women’s attitudes toward preventive strategies for hereditary breast or ovarian carcinoma differ from one country to another: differences among English, French, and Canadian women. Cancer. 2001;92(4):959–968. 11550171
49. Evans, D.G., Howell, A., Baildam, A., et al, Re: risk-reduction mastectomy: clinical issues and research needs. J Natl Cancer Inst. 2002;94(4):307–308. 11854394
50. Meijers-Heijboer, H., Brekelmans, C.T., Menke-Pluymers, M., et al, Use of genetic testing and prophylactic mastectomy and oophorectomy in women with breast or ovarian cancer from families with a BRCA1 or BRCA2 mutation. J Clin Oncol. 2003;21(9):1675–1681. 12721241
51. Eccles, D.M., Evans, D.G., Mackay, J., Guidelines for a genetic risk based approach to advising women with a family history of breast cancer. UK Cancer Family Study Group (UKCFSG). J Med Genet. 2000;37(3):203–209. 10699057
52. Eeles, R., Testing for the breast cancer predisposition gene, BRCA1. Br Med J. 1996;313(7057):572–573. 8806241
53. Evans, D.G., Kerr, B., Cade, D., et al, Fictitious breast cancer family history. Lancet. 1996;348(9033):1034. 8855883
54. Wasteson, E., Sandelin, K., Brandberg, Y., et al, High satisfaction rate ten years after bilateral prophylactic mastectomy – a longitudinal study. Eur J Cancer Care (Engl). 2011;20(4):508–513. 20597955
55. Brandberg, Y., Sandelin, K., Erikson, S., et al, Psychological reactions, quality of life, and body image after bilateral prophylactic mastectomy in women at high risk for breast cancer: a prospective 1-year follow-up study. J Clin Oncol. 2008;26(24):3943–3949. 18711183
56. Roukos, D.H., Briasoulis, E., Individualized preventive and therapeutic management of hereditary breast ovarian cancer syndrome. Nat Clin Pract Oncol. 2007;4(10):578–590. 17898808
57. Domchek, S.M., Friebel, T.M., Singer, C.F., et al, Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA. 2010;304(9):967–975. 20810374
58. Rebbeck, T.R., Friebel, T., Lynch, H.T., et al, Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol. 2004;22(6):1055–1062. 14981104
59. Fatouros, M., Baltoyiannis, G., Roukos, D.H., The predominant role of surgery in the prevention and new trends in the surgical treatment of women with BRCA1/2 mutations. Ann Surg Oncol. 2008;15(1):21–33. 17940826
60. Klaren, H.M., van’t Veer, L.J., van Leeuwen, F.E., et al, Potential for bias in studies on efficacy of prophylactic surgery for BRCA1 and BRCA2 mutation. J Natl Cancer Inst. 2003;95(13):941–947. 12837830
61. Tercyak, K.P., Peshkin, B.N., Brogan, B.M., et al, Quality of life after contralateral prophylactic mastectomy in newly diagnosed high-risk breast cancer patients who underwent BRCA1/2 gene testing. J Clin Oncol. 2007;25(3):285–291. 17159191
62. Guillem, J.G., Wood, W.C., Moley, J.F., et al, ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol. 2006;24(28):4642–4660. 17008706
63. Carlson, G.W., Bostwick, J., Styblo, T.M., et al, Skin-sparing mastectomy. Oncologic and reconstructive considerations. Ann Surg. 1997;225(5):570–578. 9193184
64. Newman, B., Mu, H., Butler, L.M., et al, Frequency of breast cancer attributable to BRCA1 in a population-based series of American women. JAMA. 1998;279(12):915–921. 9544765
65. Didier, F., Radice, D., Gandini, S., et al, Does nipple preservation in mastectomy improve satisfaction with cosmetic results, psychological adjustment, body image and sexuality? Breast Cancer Res Treat. 2009;118(3):623–633. 19003526
66. Laronga, C., Kemp, B., Johnston, D., et al, The incidence of occult nipple–areola complex involvement in breast cancer patients receiving a skin-sparing mastectomy. Ann Surg Oncol. 1999;6(6):609–613. 10493632
67. Lakhani, S.R., Reis-Filho, J.S., Fulford, L., et al, Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin Cancer Res. 2005;11(14):5175–5180. 16033833
68. Wertheim, U., Ozzello, L., Neoplastic involvement of nipple and skin flap in carcinoma of the breast. Am J Surg Pathol. 1980;4(6):543–549. 7212147
69. Petit, J.Y., Veronesi, U., Orecchia, R., et al, Nipple sparing mastectomy with nipple areola intraoperative radiotherapy: one thousand and one cases of a five years experience at the European institute of oncology of Milan (EIO). Breast Cancer Res Treat. 2009;117(2):333–338. 19152026
70. Sacchini, V., Pinotti, J.A., Barros, A.C., et al, Nipple-sparing mastectomy for breast cancer and risk reduction: oncologic or technical problem? J Am Coll Surg. 2006;203(5):704–714. 17084333
71. Maxwell, G.P., Storm-Dickerson, T., Whitworth, P., et al, Advances in nipple-sparing mastectomy: oncological safety and incision selection. Aesthet Surg J. 2011;31(3):310–319. 21385742
72. Gerber, B., Krause, A., Dieterich, M., et al, The oncological safety of skin sparing mastectomy with conservation of the nipple–areola complex and autologous reconstruction: an extended follow-up study. Ann Surg. 2009;249(3):461–468. 19247035
73. Crowe, J.P., Patrick, R.J., Yetman, R.J., et al, Nipple-sparing mastectomy update: one hundred forty-nine procedures and clinical outcomes. Arch Surg. 2008;143(11):1106–1110. 19015470
74. Wijayanayagam, A., Kumar, A.S., Foster, R.D., et al, Optimizing the total skin-sparing mastectomy. Arch Surg. 2008;143(1):38–45. 18209151
75. May, J.W., Attwood, J., Bartlett, S., Staged use of soft-tissue expansion and lower thoracic advancement flap in breast reconstruction. Plast Reconstr Surg. 1987;79(2):272–277. 3809275
76. Wickman, M., Sandelin, K., Arver, B., Technical aspects and outcome after prophylactic mastectomy and immediate breast reconstruction in 30 consecutive high-risk patients. Plast Reconstr Surg. 2003;111(3):1069–1077. 12621176
77. Van Geel, A.N., Contant, C.M., Wai, R.T., et al, Mastectomy by inverted drip incision and immediate reconstruction: data from 510 cases. Ann Surg Oncol. 2003;10(4):389–395. 12734087
78. Salzberg, C.A., Ashikari, A.Y., Koch, R.M., et al, An 8-year experience of direct-to-implant immediate breast reconstruction using human acellular dermal matrix (AlloDerm). Plast Reconstr Surg. 2011;127(2):514–524. 21285756
79. Evans, D.G., Lalloo, F., Ashcroft, L., et al, Uptake of risk-reducing surgery in unaffected women at high risk of breast and ovarian cancer is risk, age, and time dependent. Cancer Epidemiol Biomarkers Prev. 2009;18(8):2318–2324. 19661091
80. Zhou, W.B., Liu, X.A., Dai, J.C., et al, Meta-analysis of sentinel lymph node biopsy at the time of prophylactic mastectomy of the breast. Can J Surg. 2011;54(5):300–306. 21651834
81. Hatcher, M.B., Fallowfield, L., A’Hern, R., The psychosocial impact of bilateral prophylactic mastectomy: prospective study using questionnaires and semistructured interviews. Br Med J. 2001;322(7278):76. 11154619
82. Hopwood, P., Lee, A., Shenton, A., et al, Clinical follow-up after bilateral risk reducing (‘prophylactic’) mastectomy: mental health and body image outcomes. Psychooncology. 2000;9(6):462–472. 11180581
83. Stefanek, M.E., Helzlsouer, K.J., Wilcox, P.M., et al, Predictors of and satisfaction with bilateral prophylactic mastectomy. Prev Med. 1995;24(4):412–419. 7479633
84. Borgen, P.I., Hill, A.D., Tran, K.N., et al, Patient regrets after bilateral prophylactic mastectomy. Ann Surg Oncol. 1998;5(7):603–606. 9831108
85. Lloyd, S.M., Watson, M., Oaker, G., et al, Understanding the experience of prophylactic bilateral mastectomy: a qualitative study of ten women. Psychooncology. 2000;9(6):473–485. 11180582
86. Josephson, U., Wickman, M., Sandelin, K., Initial experiences of women from hereditary breast cancer families after bilateral prophylactic mastectomy: a retrospective study. Eur J Surg Oncol. 2000;26(4):351–356. 10873354
87. Frost, M.H., Schaid, D.J., Sellers, T.A., et al, Long-term satisfaction and psychological and social function following bilateral prophylactic mastectomy. JAMA. 2000;284(3):319–324. 10891963
88. Bresser, P.J., Seynaeve, C., Van Gool, A.R., et al, Satisfaction with prophylactic mastectomy and breast reconstruction in genetically predisposed women. Plast Reconstr Surg. 2006;117(6):1675–1682. 16651934
89. Garcia-Etienne, C.A., Barile, M., Gentilini, O.D., et al, Breast-conserving surgery in BRCA1/2 mutation carriers: are we approaching an answer? Ann Surg Oncol. 2009;16(12):3380–3387. 19649554
90. Tuttle, T., Habermann, E., Abraham, A., et al, Contralateral prophylactic mastectomy for patients with unilateral breast cancer. Expert Rev Anticancer Ther. 2007;7(8):1117–1122. 18028020
91. Eccles, D., Simmonds, P., Goddard, J., et al, Familial breast cancer: an investigation into the outcome of treatment for early stage disease. Fam Cancer. 2001;1(2):65–72. 14573999
92. Metcalfe, K., Lynch, H.T., Ghadirian, P., et al, Contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. J Clin Oncol. 2004;22(12):2328–2335. 15197194
93. Narod, S.A., Brunet, J.S., Ghadirian, P., et al, Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case–control study. Hereditary Breast Cancer Clinical Study Group. Lancet. 2000;356(9245):1876–1881. 11130383
94. Heemskerk-Gerritsen, B.A., Brekelmans, C.T., Menke-Pluymers, M.B., et al, Prophylactic mastectomy in BRCA1/2 mutation carriers and women at risk of hereditary breast cancer: long-term experiences at the Rotterdam Family Cancer Clinic. Ann Surg Oncol. 2007;14(12):3335–3344. 17541692
95. McDonnell, S.K., Schaid, D.J., Myers, J.L., et al, Efficacy of contralateral prophylactic mastectomy in women with a personal and family history of breast cancer. J Clin Oncol. 2001;19(19):3938–3943. 11579114
96. Herrinton, L.J., Barlow, W.E., Yu, O., et al, Efficacy of prophylactic mastectomy in women with unilateral breast cancer: a cancer research network project. J Clin Oncol. 2005;23(19):4275–4286. 15795415
97. Bedrosian, I., Hu, C.Y., Chang, G.J., Population-based study of contralateral prophylactic mastectomy and survival outcomes of breast cancer patients. J Natl Cancer Inst. 2010;102(6):401–409. 20185801
98. Boughey, J.C., Hoskin, T.L., Degnim, A.C., et al, Contralateral prophylactic mastectomy is associated with a survival advantage in high-risk women with a personal history of breast cancer. Ann Surg Oncol. 2010;17(10):2702–2709. 20853163
99. Rebbeck, T.R., Lynch, H.T., Neuhausen, S.L., et al, Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med. 2002;346(21):1616–1622. 12023993
100. Rebbeck, T.R., Friebel, T., Wagner, T., et al, Effect of short-term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol. 2005;23(31):7804–7810. 16219936
101. Armstrong, K., Schwartz, J.S., Randall, T., et al, Hormone replacement therapy and life expectancy after prophylactic oophorectomy in women with BRCA1/2 mutations: a decision analysis. J Clin Oncol. 2004;22(6):1045–1054. 14981106
102. Beral, V., Million Women Study Collaborators, Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362(9382):419–427. 12927427
103. Fisher, B., Costantino, J.P., Wickerham, D.L., et al, Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90(18):1371–1388. 9747868
104. Cuzick, J., Forbes, J., Edwards, R., et al, First results from the International Breast Cancer Intervention Study (IBIS-I): a randomised prevention trial. Lancet. 2002;360(9336):817–824. 12243915
105. Cuzick, J., Forbes, J.F., Sestak, I., et al, Long-term results of tamoxifen prophylaxis for breast cancer – 96-month follow-up of the randomized IBIS-I trial. J Natl Cancer Inst. 2007;99(4):272–282. 17312304
106. Vogel, V.G., The NSABP Study of Tamoxifen and Raloxifene (STAR) trial. Expert Rev Anticancer Ther. 2009;9(1):51–60. 19105706