[level-membership-for-physical-medicine-and-rehabilitation-category]
CHAPTER 15 The Diagnosis and Treatment of Anxiety Disorders in Chronic Spinal Pain
INTRODUCTION
ORIGIN AND DEFINITION OF THE MODERN CONCEPT OF ANXIETY
The concept of anxiety as an emotional experience distinct from fear is a relatively modern notion with its origins in the works of Sigmund Freud. It is interesting historical trivia that the German word for fear, ‘angst’ was inappropriately translated in some of Freud’s work to the subtly different word, ‘anxiety.’1 Although Freud made no distinction between fear and anxiety in his writings, this linguistic accident helped create a distinction between the concepts of fear and anxiety.1 This distinction, especially in relation to chronic anxiety disorders, has held up to scientific scrutiny. Fear, broadly defined, is a complex emotional and physical response to an actual threat (i.e. response to a physical attack.) Anxiety, by contrast, is a complex emotional and physical response to a perceived threat (i.e. believing an attack is imminent). Both fear and its analog, anxiety, are responses to stress that, when functioning properly, are adaptive and necessary for survival. It is when the physical and emotional sequelae of fear and anxiety are excessive in relation to their context, or when they lead to a state of chronic incapacity and loss of function, that they become a cause for clinical concern. As outlined above, chronic pain creates many situations that may provoke fear or anxiety.
Acute fear in a perceived dangerous situation has the effect of modulating pain sensation to enable successful fight-or-flight reactions. In the patient with chronic pain, the role that fear and chronic anxiety has in the maintenance and exacerbation of their symptoms cannot be underestimated. The common physical symptoms of anxiety, such as muscular tension, hyperarousal, insomnia, palpitations, and poorly localized pain, often confound a patient’s primary medical or surgical pain complaint. Anxiety, through activation of the noradrenergic system in the locus coeruleus, has both peripheral and central effects on pain perception in persons with nociceptive, neuropathic and visceral pain conditions, as listed in Table 15.1. Add to this the psychological symptoms associated with anxiety (worry, apprehension, irritability, poor concentration, overinterpretation of symptoms) along with the effects of anxiety on illness behavior, including presentation of pain complaints and compliance with treatment regimens, and the complexity of treating a patient with chronic pain with acute or chronic anxiety becomes obvious.
Table 15.1 Effects of Anxiety on Pain Conditions and Disorders
| Condition | Physiologic Effect of Anxiety | Consequences for Pain |
|---|---|---|
| Nociceptive pain | Sympathetic nervous system activation | Higher pain levels |
THE BIOLOGICAL BASIS OF FEAR AND ANXIETY
Figure 15.1 is a conceptual rendition of the basic neuroanatomical components of fear. The connections between these structures are much more complex than the pathways depicted in this illustration, but they provide a general idea of how these anatomical structures communicate and synthesize environmental, emotional, and learned stimuli. The spatial relationship of the brain regions depicted in this illustration is generally analogous to their location in the human brain.
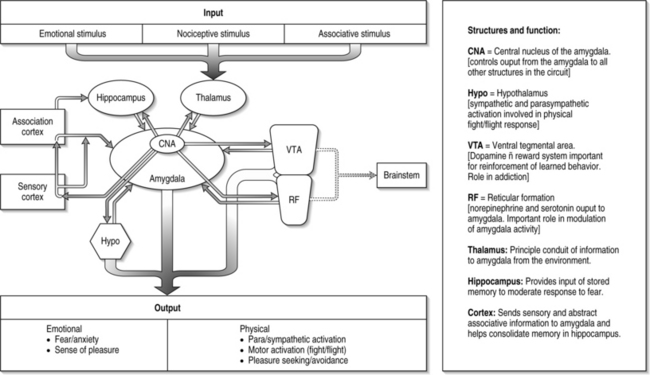
Figure 15.1 highlights the crucial role the amygdala plays in the fear response. It is essentially the central processor of fear. The amygdala receives and transmits information from the external world (via the thalamus, ventral tegmental area, and reticular formation). It merges this information with our memory, senses, executive functions, endocrine and musculoskeletal systems (via the hippocampus, sensory/motor cortex, association cortex, hypothalamus and brainstem) to elicit a complex emotional and behavioral response.
In humans, the amygdala is thought to be essential for the recognition of threatening environmental cues and modulation of the emotional response to them. Neuroimaging experiments of normal human brains show that the amygdala is important in our ability to recognize and respond to threatening stimuli in the form of disturbing faces, gestures, and scenes that evoke fear.2,3 This basic finding corresponds to the deficits observed in persons with surgical destruction of the amygdala and children with autism. In both cases, there is an enormous deficit in the individual’s ability to accurately identify and respond to fear-evoking stimuli.4,5
Other experiments have suggested the amygdala also plays an important role in the development of chronic anxiety and pain. One study has found that activation of the basolateral nucleus of the amygdala causes the emotional experience of anxiety without a corresponding increase in heart rate (which is controlled by the hypothalamus via a separate pathway).6 There is growing evidence that the amygdala plays a crucial role in the up- or downregulation of the emotional response to pain and that this ‘nociceptive amygdala’ can be influenced by a wide range of environmental and internal stimuli to modulate the subjective experience of pain.7
The hippocampus, a structure with robust connections to the amygdala, is a center for memory formation, storage, and retrieval. It provides information from detailed memories that are processed by the amygdala and given a particular emotional value. The emotional value the amygdala places on a particular memory is then fed back to the hippocampus, where it integrates this information and either strengthens or weakens the memory. This is why it is believed that events associated with high emotional content (a car accident, being wounded in battle, one’s first kiss) tend to be remembered in greater detail than those with little emotional significance (the drive to work every morning). In persons with bilateral destruction of the hippocampus secondary to herpes encephalitis, there is a preservation of memory related to processes (such as tying shoes, using a fork), but a deficit in memory related to particular events, persons, and experiences along with mood problems.8,9
In persons with post-traumatic stress disorder (PTSD), changes are seen in the amygdala, hippocampus, and other areas of the limbic system (the neural circuits of complex emotional experience). Neuroimaging studies have found consistent reductions in either total hippocampal volume or blood flow in men and women with PTSD.10–13 The primary function of the hypothalamus is to regulate the body’s homeostatic and endocrine systems. In relation to the fear response, the hypothalamus sends projections to the medulla which controls autonomic (fight-or-flight) functions such as heart rate, muscle tone, digestion, sweating, etc.14 With input from the amygdala, the hypothalamus contributes the hormonal and autonomic component of the fear response.
Research on other structures within this fear circuit has expanded our understanding of how the cortical structures influence the amygdala. Human brain imaging studies have found that the fusiform gyrus, prefrontal, and anterior cingulate gyrus are preferentially activated in response to fearful stimuli.3,15 The orbitofrontal cortex (OFC), which is involved in the evaluation of risk and reward and social norms, may also have a direct role in regulation of anxiety via its connection to the amygdala (Fig. 15.2).16 The cortex, then, plays an essential role in the categorization, appraisal, and attenuation of our reactions to fearful stimuli. The higher cortical connections to the more primitive fight, flight, and reward circuitry is what allows humans to have a degree of conscious recognition and control over these processes. These connections and their conditioning form the biological basis for the effects of behavioral training used widely in the treatment of pain, such as relaxation and biofeedback.
More recent functional brain chemistry research has provided neuroanatomical evidence for the overlap between the processing and perception of pain and anxiety. In an experiment comparing patients with chronic low back pain (CLBP) to normal controls, significant differences were found in two regions of the association cortex (orbitofrontal [OFC] and dorsolateral prefrontal cortex [DLPFC]), cingulate gyrus (part of the limbic system), and thalamus.17 The study showed that persons with CLBP have differences in regional brain chemistry in the OFC and DLPFC when comparing their perception of pain. Additionally, persons with CLBP and anxiety had changes in brain chemistry suggesting increased interaction between all four brain regions, whereas anxious controls only had changes observed in the OFC. Anxiety and pain, therefore, share common neurochemical pathways and can interact in a way that leads to the reorganization of normal perceptual pathways in the brain.
Role of neurotransmitters
Well over 300 different chemicals have been identified as ‘neurotransmitters.’ Endogenous neurotransmitters are broadly defined as chemicals synthesized in neurons, released in response to electrical impulses and acting on other neurons to cause changes in their electrochemical properties.17
Biogenic amines
Dopamine
The VTA sends dopaminergic projections throughout the brain. The VTA is primarily associated with reward and its connections with the nucleus accumbens are believed to be at the heart of the reinforcing effects of drugs of abuse.18 The VTA’s dopaminergic projections to the amygdala and hippocampus are the areas in which the reward circuit influences the emotional and memory-forming structures in the brain. The VTA is also regulated by the amino acid neurotransmitter GABA (γ-aminobutyric acid) and opioid peptide neurotransmitters (enkephalins, endorphins). This convergence of GABAergic and opioid peptide receptors with the dopaminergic neurons of the VTA may be an important pathway in the development of benzodiazepine and opiate addiction.
Serotonin and norepinephrine
There is a growing body of research that supports the role of 5-HT in modulating the amygdala and other limbic structures in persons with chronic anxiety disorders. Specifically, the SSRI drug paroxetine (Paxil) has been associated with a reduction of amygdala volume in persons with obsessive compulsive disorder (OCD), an anxiety disorder believed to be associated with a hyperactive amygdala.19 Conversely, in persons with PTSD, increases in hippocampal volume are positively associated with paroxetine treatment.20
A study examining the effects of tryptophan (the dietary amino acid precursor of 5-HT) depletion found decreases in the ability to recognize fear-related cues.21 Allelic variations in amygdalar 5-HT transporter proteins have also been found to correlate with individual responses to fear-related cues.22 Severe acute pain activates stress-related noradrenergic systems in the brain. Descending projections to the sympathetic nervous system may cause increased firing of pain neurons through several mechanisms as well as nociceptor activation and muscle tension and spasm. Ascending noradrenergic projections to the forebrain cause cognitive–emotional reactions, such as fear and anxiety, which to some degree are contextually determined. For example, pain in childbirth often does not evoke fear or anxiety, whereas pain in traumatic spinal and/or limb injury, with uncertain outcome, often does. The association of pain, anxiety, and depression may have a common neurochemical substrate in the serotonergic systems.
Amino acids
Glycine and GABA are amino acid neurotransmitters. Glycine is known as the primary excitatory neurotransmitter in the brain. GABA is the primary inhibitory neurotransmitter in the brain. Together, glycine and GABA are the most common endogenous neurotransmitters and 75–90% of all neurons in the CNS have glycine and GABA receptors.17 A thorough discussion of the biology of glycine and GABA is beyond the scope of this chapter. However, it is important to discuss the basic biology of GABA in the context of this chapter because of its central role in the action of benzodiazepines.
GABA
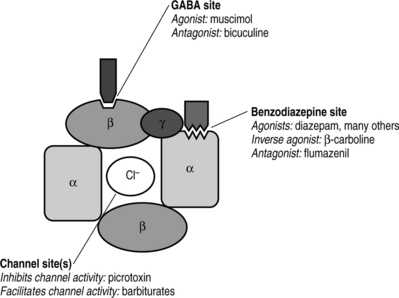
As mentioned previously, GABA is the primary inhibitory neurotransmitter in the CNS. It is estimated that 30% of all CNS synapses use GABA as a neurotransmitter and it is found in very high concentrations in CNS tissues (1000 to 1 000 000 times greater than concentrations of biogenic amines).23 The synaptic receptor for GABA is composed of two major receptor subtypes known as the GABAA and GABAB receptors. All benzodiazepines work at the GABAA receptor. Rather than occupying the entire GABA receptor as a competitive agonist (like morphine’s action at the opioid receptor), benzodiazepines bind to the GABAA subunit and actually facilitate endogenous GABA binding at the GABAA receptor. GABAA receptor activation opens chloride ion channels which hyperpolarizes neuronal membranes and inhibits firing (Fig. 15.3).23 This elegant mechanism is believed to be the primary pathway for the anxiolytic effects of benzodiazepines. Psychoactive compounds such as barbiturates and ethanol also act at the GABA receptor to produce similar anxiolytic and CNS depressant effects.
In an interesting study of the effects of benzodiazepines in acute pain, Di Piero and colleagues studied cerebral blood flow (CBF) by single photon emission computed tomography (SPECT), with and without diazepam.24 Diazepam, a benzodiazepine, binds to the benzodiazepine receptor on the GABAA receptor site and enhances GABA’s opening of the chloride channel, activating the GABA system and its anxiolytic actions. Diazepam, when given to healthy volunteers, following induction of pain by the cold pressor test (CPT), inhibited activation of the temporal regions, which was interpreted as part of the affective–emotional component of pain response. Diazepam-treated subjects tolerated the pain better and on SPECT this was associated with lack of temporal lobe activation of sensory–discriminative pain-related brain regions (contralateral hand region in the sensory motor cortex, pre-motor cortex and thalamus, and left anterior cingulate gyrus). This activity suggests that diazepam, which is useful in managing acute anxiety, interferes with affective–emotional components of pain perception and modifies temporal lobe activation patterns. The role of benzodiazepines in the treatment of acute and chronic anxiety will be discussed later in this chapter as will issues related to tolerance, dependence, and addiction to these medications.
DIAGNOSIS OF ANXIETY DISORDERS
Introduction
There are two major national surveys that estimated the prevalence of anxiety disorders in the United States. The Epidemiologic Catchment Area study estimated that 7% of people in the United States have a clinically significant anxiety disorder.25 The National Comorbidity Study (NCS) reported even higher prevalence rates for anxiety disorders. NCS estimated 12 month prevalence rates of 17.7% for at least one anxiety disorder and lifetime prevalence rates near 25%.1
The majority of persons with anxiety disorders suffer from simple phobias (i.e. needles, insects, heights). The NCS estimated the lifetime prevalence of generalized anxiety disorder (4.1–6.6%), PTSD (1.0–9.3%), panic disorder (2.3–2.7%), and social phobia (2.6–13.3%). The prevalence of any current psychiatric disorder in one survey of a large city spine clinic was estimated to be 59% and as high as 77% for lifetime prevalence.25 Of those with chronic anxiety disorders, 95% had evidence of a disorder before the onset of back pain.26 This strongly supports the need to carefully screen for anxiety disorders at the time of first intake to a spine clinic and to integrate treatment for anxiety disorders into the comprehensive treatment plan.
Clinical vignette 1: A case of untreated obsessive compulsive disorder
This case illustrates post-traumatic phobic anxiety complicating poorly managed acute postoperative pain as well as, the activation of a premorbid anxiety disorder, in this case OCD, with almost fatal consequences. Furthermore, her long history of undiagnosed and untreated ritualized hand washing and housecleaning caused by OCD certainly worsened cervical radiculopathy. Now, her residual phobic anxiety, resulting from poorly managed postoperative pain years ago, causes her to refuse consideration of needed spine surgery. This is a case in which appropriate postoperative pain pharmacotherapy and early screening and diagnosis for a chronic anxiety disorder might have prevented such an outcome.
Evaluation of clinical anxiety
Table 15.2 lists major anxiety disorders recognized in DSM IV-TR along with questions that can help a clinician screen for anxiety disorders. An affirmative answer to these questions should prompt a clinician to obtain further history, treat, or prompt a referral to a mental health professional.
| Panic disorder with and without agoraphobia |
In patients with pain disorders, the physician must be alert to several situations that commonly precipitate anxiety: increases in pain; threat of withdrawal of workers’ compensation benefits; threat of job loss; threat of marital discord or even spousal desertion caused by the stress of pain and disability; fear of surgery; fear of worsening disease, particularly when increasing or new pain may indicate cancer recurrence or spread; withdrawal from medications that are abruptly stopped, including opioids, antidepressants, anticonvulsants, sedatives and benzodiazepines; withdrawal from alcohol; stimulant intoxication; side effects of certain medications, such as SSRIs, tricyclics, and theophylline derivatives used for asthma; and drug–drug interactions, such as serotonin syndrome.27
INTRODUCTION AND GENERAL PRINCIPLES OF THE TREATMENT OF ANXIETY DISORDERS
General principles of biobehavioral medication use in chronic pain
To minimize the effects of anxiety on patients with chronic pain, physicians should routinely give patients behavioral tools for self-care and symptom management.28 Simple behavioral interventions, such as asking a patient to keep a diary and handing them a relaxation tape with instructions for daily use, take little time and may pay large benefits. Diaries give the physician important information about the pattern of pain and anxiety over each 24 hour period – factors such as activities that increase the pain or anxiety, or factors such as medication or activity avoidance that prevent escalations of anxiety and pain. The effects of new medications against baseline anxiety and pain scores can document effectiveness more robustly than questions such as, ‘How effective is the medication?,’ which may lead to information subject to recall bias, and does not provide the detail needed for effective treatment decisions. Relaxation tapes help patients restore control over emotions in response to pain or to stress that activates neuropathic pain and also helps them relax muscle groups.
When patients become disabled by chronic pain, to counter the impact of emotional factors, such as anxiety, and negative conditioning, comprehensive integrated, biobehavioral pain rehabilitation programs are more effective than both conventional medical treatment or ‘alternative’ therapies,29 for example, returning low back pain patients to work at a rate that is about 40% higher than conventional treatment.30 The ability to orchestrate constructively the complex network of supportive relationships necessary to rehabilitate these patients is essential. Skills in pain and stress management, such as pharmacology and behavioral pharmacology, group therapy and psychotherapy, greatly complement the skills of the rest of the team. Pain management training, usually taking place in behavioral educational groups, complemented as needed by individual psychotherapies, is effective in chronic pain patients and can reduce medical visits and costs.31 The sequence of tasks in such a group work in conjunction with physical rehabilitation and pharmacotherapy to enable helpless-feeling, functionally impaired patients to develop a sense of control over their pain, their fear of activity and their anxiety.28,32
Pharmacologic treatment of anxiety disorders
Anxiolytics for acute anxiety
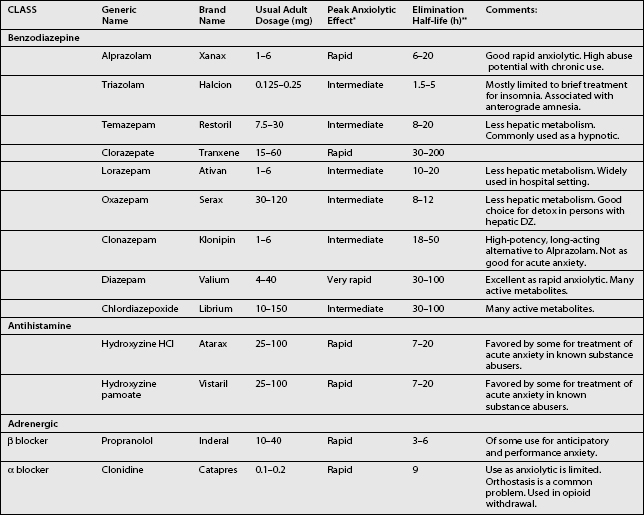
Table 15.3 provides a list of medications that are commonly used to relieve acute anxiety. When trying to find a medication that is well suited for the purpose of relieving acute anxiety, the time of onset of peak anxiolytic effect is most important, and sometimes not at all related to the elimination half-life of the drug. For example, diazepam has a very long elimination half-life, but a peak anxiolytic effect on par with, or even superior to, alprazolam.
Benzodiazepines
Since most benzodiazepines go through extensive hepatic metabolism and have many pharmacologically active metabolites, repetitive use of long-acting benzodiazepines in the elderly or in persons with hepatic disease should be avoided. Alprazolam, lorazepam and oxazepam undergo less hepatic metabolism and therefore are preferred in these patients. Overall, benzodiazepines have an excellent side effect profile compared to other anxiolytic medications. Sedation, ataxia, and cognitive changes are the principal adverse effects associated with benzodiazepine use. Sedation can occur with variable intensity in persons on the same dose of a benzodiazepine, so it is important to vary dosage according to individual tolerability. In the elderly, ataxia associated with benzodiazepine intoxication is a major clinical concern because of the risk of falls and fractures. Persons should also be warned about using benzodiazepines when driving or operating heavy machinery. The cognitive side effects of benzodiazepines include anterograde amnesia, cognitive slowing, and mental confusion. Again, elderly patients and patients early in the course of treatment are most at risk. Mental confusion is seen usually as the result of an overdose (either intentional or accidental) or from the build-up of long-acting metabolites (i.e. from chronic administration of diazepam or chlordiazepoxide). In the context of spine rehabilitation, chronic use of benzodiazepines may inhibit the learning of new coping skills.
Medications for chronic anxiety disorders
Antidepressants
Selective serotonin reuptake inhibitors (SSRIs) and serotonin norepinephrine reuptake inhibitors (SNRIs) have important roles in the treatment of chronic anxiety disorders. Only venlafaxine (Effexor) extended-release (XR) has been shown to possess unequivocal efficacy in generalized anxiety disorder (GAD).33,34 Pharmacotherapy of post-traumatic stress disorder includes several antidepressants.35–37 SSRIs protect against PTSD’s overwhelming intensity by modulating affect, memories, and impulses while loosening excessive inhibitions. Fluoxetine, fluvoxamine, sertraline, and paroxetine have all been FDA approved for the treatment of OCD. Higher doses may be necessary, such as fluoxetine 80 mg a day. The best outcomes are obtained when pharmacotherapy and behavior therapy are combined.38 Social phobia responds well to SSRIs and propanalol, a β blocker, can be used preemptively to quell anxiety prior to a performance. For panic disorder, generally, start with an SSRI, such as citalopram, sertraline, or fluoxetine.39 If a rapid control of anxiety symptoms is needed, a short acting benzodiazepine such as lorazepam should be used until the effects of SSRI are felt, keeping in mind the abuse potential and other potential negative effects of prolonged use of benzodiazepines. Once the SSRIs begin working, a benzodiazepine should be tapered and regular use avoided.
Medications approved for use in the treatment of chronic anxiety disorders are listed in Table 15.4, starting and maintenance doses of selected anxiolytics are listed in Table 15.5, and side effects of SSRIs are listed in Table 15.6.
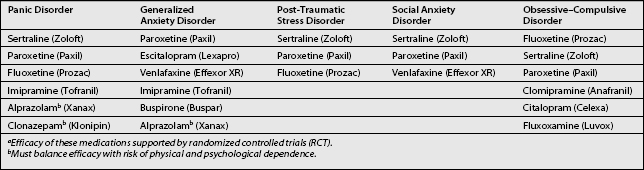
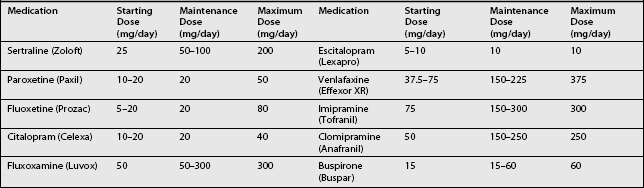
Table 15.6 Side Effects of SSRIs, Venlafaxine, and Duloxetine
| Sexual inhibition | |
| Diminished libido | Dose related. |
| Inhibited orgasm | 50–80% incidence. |
| Gastrointestinal | |
| Nausea/vomiting | Most frequent with sertraline, fluvoxamine and citalopram. |
| Diarrhea | |
| Anorexia | Usually transient symptoms that resolve within 4 weeks. |
| Dyspepsia | |
| Weight gain | |
| Up to 30% of patients | Most common with paroxetine. |
| Headache | |
| Fluoxetine most common | Usually subsides in 2–4 weeks. |
| Anxiety | |
| Fluoxetine most common | Insomnia greatest with fluoxetine. |
| Insomnia/sedation | |
| Helped by taking medication qam or qhs | Somnolence greatest with citalopram and paroxetine. |
| Dreams/nightmares | |
| Seizures | Low incidence (0.1–0.2%). |
| Increased risk with high doses. | |
| Extrapyramidal symptoms | Mainly limited to fine tremor. |
| Rarely causes dystonia or pseudoparkinsonism. | |
| Anticholinergic | |
| Dry mouth | Dose related. Most often with paroxetine. Much lower than with tricyclics. |
| Constipation | |
| Sedation | |
| Hypertension | Mild increases with higher doses (>150 mg) venlafaxine. |
| Withdrawal/discontinuation syndrome | |
| Flu-like illness, dizziness, anxiety, paresthesias, migraine, insomnia | Abrupt withdrawal of short acting medication. Most common with paroxetine and fluvoxemine. |
| Serotonin syndrome | |
| Diarrhea, restlessness, agitation, hyperreflexia, autonomic instability, myoclonus, seizure, delerium, coma | Potentially fatal. Maximum risk with MAO + SSRI combined. In patients with migraine headaches, caution when prescribing SSRIs and SNRIs with serotonin (5-HT 1b/1d) agonists, the so-called tryptans (e.g., sumatriptan and its successors). |
Buspirone
Buspirone (Buspar) is a unique anxiolytic medication indicated for the treatment of GAD and anxiety related to major depression. It has a mechanism of action different from traditional antidepressants and benzodiazepines. Buspirone works as a partial agonist at two serotonin receptor subtypes (5-HT1A and 5-HT2) and as a combined agonist/antagonist at the dopamine D2 receptor.39
Benzodiazepines
This chapter advocates for the use of benzodiazepines to be limited to the treatment of anxiety associated with acute and/or anticipated stressors. Certain benzodiazepines have clinical trial data supporting their efficacy for the treatment of GAD and panic disorder. Nevertheless, it is important to weigh the risks of using benzodiazepines with the potential benefits, especially when using the medication for more than 4 weeks.
Tolerance, withdrawal, and addiction
Perhaps the greatest clinical concern regarding the use of benzodiazepines for chronic anxiety or as a hypnotic is the potential for abuse and dependence. Although the vast majority of persons who are prescribed benzodiazepines do not go on to develop a clinically significant substance use disorder, regular use of benzodiazepines invariably causes physiologic tolerance (requiring larger amounts of a drug to achieve the same therapeutic effect) and withdrawal (a sign of physical dependence) if the medication is abruptly discontinued.40,41
Clinical vignette 3: A patient with benzodiazepine dependence
The addiction specialist does a thorough psychiatric evaluation and the patient reports that he uses cocaine recreationally about 2–4 times a year and has never met criteria for cocaine dependence. The patient also has a significant history of GAD. The patient, however, has been on three benzodiazepines for the last 4 years, ‘for my anxiety and to help me sleep.’ The patient, who saw his regular psychiatrist about 3 times a year, was getting clonazepam (Klonipin) for GAD and lorazepam (Ativan) for ‘breakthrough’ anxiety. The patient was getting temazepam (Restoril) from his PCP when he complained of trouble sleeping.
A patient who has reached this level of compulsive use clearly needs the help of professionals trained in the management of addiction. However, ‘treatment’ for a benzodiazepine addiction can start when a clinician first recognizes it. Simple, nonjudgmental recognition of a compulsive pattern of drug use is the first step, followed by education about the risks associated with benzodiazepine abuse. Very often, such a brief intervention can accurately assess a patient’s motivation to come off benzodiazepines and lead to a plan of action for discontinuation or dose reduction of the medication.
Although there is no strict consensus on the best way to successfully taper and discontinue benzodiazepines, Table 15.7 provides some general principles to apply with both inpatient and outpatient populations.40,41 Before initiating a benzodiazepine taper it is important to consider the following factors, which are associated with greater severity of withdrawal symptoms:
Table 15.7 General Recommendations for Tapering Benzodiazepines32,33
| Try to switch the patient to an equivalent cross-tolerant dose of longer-acting benzodiazepine (i.e. 4 mg of lorazepam to 100 mg of chlordiazepoxide). |
1 Brenner P, Wolf B, Rechlin T. Benzodiazepine dependence: detoxification under standardized conditions. Drug Alcoh Depend 1991; 29:195–204.
2 Rickels K, DeMartinins N, Rynn M. Pharmacologic strategies for discontinuing benzodiazepine treatment. J Clin Psychopharmacol 1999; 19(6 Suppl 2):12S–16S.
Table 15.8 provides a list of equivalent dose ranges for commonly prescribed benzodiazepines that is useful for conversion to a long- or short-acting medication when tapering or optimizing a benzodiazepine regimen.
Table 15.8 Equivalent Benzodiazepine Dosing Chart (Based on Lorazepam Equivalents)
| Benzodiazepine | Dosage | Half-Lifea |
|---|---|---|
| Lorazepam | 1.0 mg | Short |
| Alprazolam | 0.25 mg | Short |
| Triazolam | 0.125 mg | Short |
| Temazepam | 15.0 mg | Short |
| Oxazepam | 30 mg | Short |
| Clonazepam | 0.25 mg | Long |
| Clorazepate | 3.75 mg | Long |
| Diazepam | 5.0 mg | Long |
| Chlordiazepoxide | 25.0 mg | Long |
This is meant to be a general guide to pick equivalent doses for cross-tapering benzodiazepines.
a Half-life is defined as ‘short’ if ≤ 24 hours and ‘long’ of ≥ 24 hours.
SUMMARY
Anxiety is common and carries with it the potential for significant morbidity in patients suffering from spine pain. Patients may have a specific premorbid anxiety disorder with an exacerbation of symptoms as a result of reacting to the stress and pain of their spinal condition, or may develop distressing, even disabling, anxiety following spinal injury and prolonged pain with no evidence of a premorbid condition. Acute anxiety in reaction to pain, stress, and fear may be effectively treated by providing information about the condition and its treatment and by reassuring the patient that they will not be abandoned to agonizing pain. Anxiety is contagious, and insecure physicians can transmit uncertainty to their patients. The opposite happens as well; insecure patients and their resulting behavioral strategies, over which they may have little control, such as becoming demanding or refusal to participate in necessary aspects of care (e.g., further procedures), can exasperate physicians. A kindly, explanatory bedside manner calms the patient and, when necessary, benzodiazepines are situationally very effective, with the caveat that their use for more than 4 weeks carries an added risk of addiction. If anxiety persists, or if screening questions suggest a specific chronic anxiety disorder, patients should be treated by a clinician who can manage the psychopharmacology of each disorder. Patients with specific anxiety disorders respond well to antianxiety medication regimens that are specific for that particular disorder, as well as to the short-term use of benzodiazepines. Selectively and skillfully integrating anxiolytic treatment with psychotherapeutic treatments, such as coping skills training, including cognitive behavioral therapy and relaxation, usually confers the greatest benefit. Creating attitudes of self-help through knowledge (www.painconnection.org) and behavioral pain management training are complementary to the selective use of anxiolytics. A prevailing challenge to spine medicine is the mobilization of the needed resources to coordinate effective treatment for anxiety, with the treatment of spinal pain. The advent of evidence-based medicine, with reimbursement increasingly determined by outcomes, rather than procedural credentialing, encourages physicians to address more effectively the anxiety associated with spinal pain.
1 Saddock B, Saddock A. Anxiety disorders. In: Sadock B, Sadock A, editors. Synopsis of psychiatry. Philadelphia: Lippincott Williams and Wilkins; 2003:591-642.
2 Hariri AR, et al. The amygdala response to emotional stimuli: a comparison of faces and scenes. Neuroimage. 2002:317-323.
3 Hadjikhani N, de Gelder B. Seeing fearful body expressions activates the fusiform cortex and amygdala. Current Biology. 2003:2201-2205.
4 Amaral DG, Corbett BA. The amygdala, autism and anxiety. Novartis Found Symp. 2003:177-187. discussion 187–197, 281–297
5 Brierley B, et al. Emotional memory and perception in temporal lobectomy patients with amygdala damage. Journal of Neurology, Neurosurgery and Psychiatry. 2004:593-599.
6 Shekhar A, et al. The amygdala, panic disorder, and cardiovascular responses. Ann NY Acad Sci. 2003:308-325.
7 Neugebauer V, et al. The amygdala and persistent pain. Neuroscientist. 2004:221-234.
8 Bannerman DM, et al. Regional dissociations within the hippocampus – memory and anxiety. Neurosci Biobehav Rev. 2004:273-283.
9 Kapur N, et al. Herpes simplex encephalitis: long-term magnetic resonance imaging and neuropsychological profile. J Neurol Neurosurg Psychiatry. 1994:1334-1342.
10 Bremner JD, et al. MRI and PET study of deficits in hippocampal structure and function in women with childhood sexual abuse and posttraumatic stress disorder. Am J Psychiatry. 2003:924-932.
11 Mohanakrishnan Menon P, et al. Single-voxel proton MR spectroscopy of right versus left hippocampi in PTSD. Psychiatry Res. 2003;123:101-108. [Erratum appears in 2004; 30;130(3):313]
12 Lindauer RJ, et al. Smaller hippocampal volume in Dutch police officers with posttraumatic stress disorder. Biol Psychiatry. 2004:356-363.
13 Hedges DW, et al. Reduced hippocampal volume in alcohol and substance naive Vietnam combat veterans with posttraumatic stress disorder. Cogn Behav Neurol. 2003:219-224.
14 Kupfermann I. Hypothalamus and limbic system: peptidergic neurons, homeostasis, and emotional behavior. In: Kandel E, Schwartz J, Jessell T, editors. Principles of neural science. 3rd ed. New York: Elsevier; 1991:1135.
15 Hariri AR, et al. Neocortical modulation of the amygdala response to fearful stimuli. Biol Psychiatry. 2003:494-501.
16 Morris JS, Dolan RJ. Dissociable amygdala and orbitofrontal responses during reversal fear conditioning. Neuroimage. 2004:372-380.
17 Hyman S, Nestler E. The molecular foundations of psychiatry – overview of synaptic neurotransmission. Washington DC: American Psychiatric Press, Inc, 1993;1-239.
18 Childress AR, et al. Limbic activation during cue-induced cocaine craving. Am J Psychiatry. 1999:11-18.
19 Szeszko PR, et al. Amygdala volume reductions in pediatric patients with obsessive–compulsive disorder treated with paroxetine: preliminary findings. Neuropsychopharmacology. 2004:826-832.
20 Vermetten E, et al. Long-term treatment with paroxetine increases verbal declarative memory and hippocampal volume in posttraumatic stress disorder. Biol Psychiatry. 2003:693-702.
21 Harmer CJ, et al. Tryptophan depletion decreases the recognition of fear in female volunteers. Psychopharmacology (Berl). 2003:411-417.
22 Hariri AR, et al. Serotonin transporter genetic variation and the response of the human amygdala [see comment]. Science. 2002:400-403.
23 Hyman S, Nestler E. Molecular foundations of psychiatry–overview of neuropsychopharmacology. Washington DC: American Psychiatric Press, Inc, 1993;1-239.
24 Di Piero V, Feracutti S, Sabatini U, et al. Diazepam effects on the cerebral responses to tonic pain: A SPECT study. Psychopharmacology. 2001;158(3):252-258.
25 Regier DA, Narrow WE, Rae DS. The epidemiology of anxiety disorders: the Epidemiologic Catchment Area (ECA) experience. Br J Psychiatry Suppl. 1990:3-14.
26 Polatin PB, et al. Psychiatric illness and chronic low-back pain. The mind and the spine – which goes first? Spine. 1993:66-71.
27 Ener RA, Meglathery SB, Van Decker WA, et al. Serotonin syndrome and other serotonergic disorders. Pain Medicine. 2003;4(1):63-74.
28 Gallagher RM, Verma S. Treatment and rehabilitation of chronic orthopedic pain syndromes. Ch. 15. In: Stoudemire A, Fogel B, Greenblatt D, editors. Psychiatric care of the medical patient. New York: Oxford University Press; 2000:227-252.
29 Gallagher RM. Integrating medical and behavioral treatment in chronic pain management. Med Clin N Am. 1999;83(5):823-849.
30 Hazard RG, Fenwich JW, Kalish SM, et al. Functional restoration with behavioral support: A one year prospective study of patients with chronic low back pain. Spine. 1989;14:157-161.
31 Caudill M, Schnable R, Zuttermeister P, et al. Decreased clinic utilization in chronic pain patients: Response to behavioral medicine intervention. Clin J Pain. 1991;7:305-310.
32 Caudill MA. Managing pain before it manages you. New York: Guilford Press, 1995.
33 Davidson JR, DuPont RL, Hedges D, et al. Efficacy, safety, and tolerability of venlafaxine extended release and buspirone in outpatients with generalized anxiety disorder. J Clin Psychiatr. 1999;60:528-535.
34 Rickels K, Pollack MH, Sheehan DV, et al. Efficacy of extended-release venlafaxine in nondepressed outpatients with generalized anxiety disorder. Am J Psychiatr. 2000;157(6):968-974.
35 Brady KT, Pearlstein T, Asnis GM, et al. Efficacy and safety of sertraline treatment of posttraumatic stress disorder: a randomized controlled trial. JAMA. 2000;283:1837-1844.
36 Connor KM, Sutherland SM, Tupler LA, et al. Fluoxetine in post-traumatic stress disorder: randomized double-blind study. Br J Psychiatr. 1999;175:17-22.
37 Davidson JRT, Weisler RH, Malik ML, et al. Fluvoxamine in civilians with posttraumatic stress disorder. J Clin Psychopharmacol. 1998;18:93-95.
38 Jenike MA. Obsessive-compulsive disorder. N Engl J Med. 2004;350(3):259-265.
39 Stahl SM. Essential psychopharmacology. New York: Cambridge University Press, 2000;297-334.
40 Brenner P, Wolf B, Rechlin T. Benzodiazepine dependence: detoxification under standardized conditions. Drug Alcoh Depend. 1991;29:195-204.
41 Rickels K, DeMartinins N, Rynn M. Pharmacologic strategies for discontinuing benzodiazepine treatment. J Clin Psychopharmacol. 1999;19(6 Suppl 2):12S-16S.
[/level-membership-for-physical-medicine-and-rehabilitation-category][not-level-membership-for-physical-medicine-and-rehabilitation-category]
CHAPTER 15 The Diagnosis and Treatment of Anxiety Disorders in Chronic Spinal Pain
INTRODUCTION
ORIGIN AND DEFINITION OF THE MODERN CONCEPT OF ANXIETY
The concept of anxiety as an emotional experience distinct from fear is a relatively modern notion with its origins in the works of Sigmund Freud. It is interesting historical trivia that the German word for fear, ‘angst’ was inappropriately translated in some of Freud’s work to the subtly different word, ‘anxiety.’1 Although Freud made no distinction between fear and anxiety in his writings, this linguistic accident helped create a distinction between the concepts of fear and anxiety.1 This distinction, especially in relation to chronic anxiety disorders, has held up to scientific scrutiny. Fear, broadly defined, is a complex emotional and physical response to an actual threat (i.e. response to a physical attack.) Anxiety, by contrast, is a complex emotional and physical response to a perceived threat (i.e. believing an attack is imminent). Both fear and its analog, anxiety, are responses to stress that, when functioning properly, are adaptive and necessary for survival. It is when the physical and emotional sequelae of fear and anxiety are excessive in relation to their context, or when they lead to a state of chronic incapacity and loss of function, that they become a cause for clinical concern. As outlined above, chronic pain creates many situations that may provoke fear or anxiety.
Acute fear in a perceived dangerous situation has the effect of modulating pain sensation to enable successful fight-or-flight reactions. In the patient with chronic pain, the role that fear and chronic anxiety has in the maintenance and exacerbation of their symptoms cannot be underestimated. The common physical symptoms of anxiety, such as muscular tension, hyperarousal, insomnia, palpitations, and poorly localized pain, often confound a patient’s primary medical or surgical pain complaint. Anxiety, through activation of the noradrenergic system in the locus coeruleus, has both peripheral and central effects on pain perception in persons with nociceptive, neuropathic and visceral pain conditions, as listed in Table 15.1. Add to this the psychological symptoms associated with anxiety (worry, apprehension, irritability, poor concentration, overinterpretation of symptoms) along with the effects of anxiety on illness behavior, including presentation of pain complaints and compliance with treatment regimens, and the complexity of treating a patient with chronic pain with acute or chronic anxiety becomes obvious.
Table 15.1 Effects of Anxiety on Pain Conditions and Disorders
| Condition | Physiologic Effect of Anxiety | Consequences for Pain |
|---|---|---|
| Nociceptive pain | Sympathetic nervous system activation | Higher pain levels |
THE BIOLOGICAL BASIS OF FEAR AND ANXIETY
Figure 15.1 is a conceptual rendition of the basic neuroanatomical components of fear. The connections between these structures are much more complex than the pathways depicted in this illustration, but they provide a general idea of how these anatomical structures communicate and synthesize environmental, emotional, and learned stimuli. The spatial relationship of the brain regions depicted in this illustration is generally analogous to their location in the human brain.
Figure 15.1 highlights the crucial role the amygdala plays in the fear response. It is essentially the central processor of fear. The amygdala receives and transmits information from the external world (via the thalamus, ventral tegmental area, and reticular formation). It merges this information with our memory, senses, executive functions, endocrine and musculoskeletal systems (via the hippocampus, sensory/motor cortex, association cortex, hypothalamus and brainstem) to elicit a complex emotional and behavioral response.
In humans, the amygdala is thought to be essential for the recognition of threatening environmental cues and modulation of the emotional response to them. Neuroimaging experiments of normal human brains show that the amygdala is important in our ability to recognize and respond to threatening stimuli in the form of disturbing faces, gestures, and scenes that evoke fear.2,3 This basic finding corresponds to the deficits observed in persons with surgical destruction of the amygdala and children with autism. In both cases, there is an enormous deficit in the individual’s ability to accurately identify and respond to fear-evoking stimuli.4,5
Other experiments have suggested the amygdala also plays an important role in the development of chronic anxiety and pain. One study has found that activation of the basolateral nucleus of the amygdala causes the emotional experience of anxiety without a corresponding increase in heart rate (which is controlled by the hypothalamus via a separate pathway).6 There is growing evidence that the amygdala plays a crucial role in the up- or downregulation of the emotional response to pain and that this ‘nociceptive amygdala’ can be influenced by a wide range of environmental and internal stimuli to modulate the subjective experience of pain.7
The hippocampus, a structure with robust connections to the amygdala, is a center for memory formation, storage, and retrieval. It provides information from detailed memories that are processed by the amygdala and given a particular emotional value. The emotional value the amygdala places on a particular memory is then fed back to the hippocampus, where it integrates this information and either strengthens or weakens the memory. This is why it is believed that events associated with high emotional content (a car accident, being wounded in battle, one’s first kiss) tend to be remembered in greater detail than those with little emotional significance (the drive to work every morning). In persons with bilateral destruction of the hippocampus secondary to herpes encephalitis, there is a preservation of memory related to processes (such as tying shoes, using a fork), but a deficit in memory related to particular events, persons, and experiences along with mood problems.8,9
In persons with post-traumatic stress disorder (PTSD), changes are seen in the amygdala, hippocampus, and other areas of the limbic system (the neural circuits of complex emotional experience). Neuroimaging studies have found consistent reductions in either total hippocampal volume or blood flow in men and women with PTSD.10–13 The primary function of the hypothalamus is to regulate the body’s homeostatic and endocrine systems. In relation to the fear response, the hypothalamus sends projections to the medulla which controls autonomic (fight-or-flight) functions such as heart rate, muscle tone, digestion, sweating, etc.14 With input from the amygdala, the hypothalamus contributes the hormonal and autonomic component of the fear response.
Research on other structures within this fear circuit has expanded our understanding of how the cortical structures influence the amygdala. Human brain imaging studies have found that the fusiform gyrus, prefrontal, and anterior cingulate gyrus are preferentially activated in response to fearful stimuli.3,15 The orbitofrontal cortex (OFC), which is involved in the evaluation of risk and reward and social norms, may also have a direct role in regulation of anxiety via its connection to the amygdala (Fig. 15.2).16 The cortex, then, plays an essential role in the categorization, appraisal, and attenuation of our reactions to fearful stimuli. The higher cortical connections to the more primitive fight, flight, and reward circuitry is what allows humans to have a degree of conscious recognition and control over these processes. These connections and their conditioning form the biological basis for the effects of behavioral training used widely in the treatment of pain, such as relaxation and biofeedback.
More recent functional brain chemistry research has provided neuroanatomical evidence for the overlap between the processing and perception of pain and anxiety. In an experiment comparing patients with chronic low back pain (CLBP) to normal controls, significant differences were found in two regions of the association cortex (orbitofrontal [OFC] and dorsolateral prefrontal cortex [DLPFC]), cingulate gyrus (part of the limbic system), and thalamus.17 The study showed that persons with CLBP have differences in regional brain chemistry in the OFC and DLPFC when comparing their perception of pain. Additionally, persons with CLBP and anxiety had changes in brain chemistry suggesting increased interaction between all four brain regions, whereas anxious controls only had changes observed in the OFC. Anxiety and pain, therefore, share common neurochemical pathways and can interact in a way that leads to the reorganization of normal perceptual pathways in the brain.
Role of neurotransmitters
Well over 300 different chemicals have been identified as ‘neurotransmitters.’ Endogenous neurotransmitters are broadly defined as chemicals synthesized in neurons, released in response to electrical impulses and acting on other neurons to cause changes in their electrochemical properties.17
Biogenic amines
Dopamine
The VTA sends dopaminergic projections throughout the brain. The VTA is primarily associated with reward and its connections with the nucleus accumbens are believed to be at the heart of the reinforcing effects of drugs of abuse.18 The VTA’s dopaminergic projections to the amygdala and hippocampus are the areas in which the reward circuit influences the emotional and memory-forming structures in the brain. The VTA is also regulated by the amino acid neurotransmitter GABA (γ-aminobutyric acid) and opioid peptide neurotransmitters (enkephalins, endorphins). This convergence of GABAergic and opioid peptide receptors with the dopaminergic neurons of the VTA may be an important pathway in the development of benzodiazepine and opiate addiction.
Serotonin and norepinephrine
There is a growing body of research that supports the role of 5-HT in modulating the amygdala and other limbic structures in persons with chronic anxiety disorders. Specifically, the SSRI drug paroxetine (Paxil) has been associated with a reduction of amygdala volume in persons with obsessive compulsive disorder (OCD), an anxiety disorder believed to be associated with a hyperactive amygdala.19 Conversely, in persons with PTSD, increases in hippocampal volume are positively associated with paroxetine treatment.20
A study examining the effects of tryptophan (the dietary amino acid precursor of 5-HT) depletion found decreases in the ability to recognize fear-related cues.21 Allelic variations in amygdalar 5-HT transporter proteins have also been found to correlate with individual responses to fear-related cues.22 Severe acute pain activates stress-related noradrenergic systems in the brain. Descending projections to the sympathetic nervous system may cause increased firing of pain neurons through several mechanisms as well as nociceptor activation and muscle tension and spasm. Ascending noradrenergic projections to the forebrain cause cognitive–emotional reactions, such as fear and anxiety, which to some degree are contextually determined. For example, pain in childbirth often does not evoke fear or anxiety, whereas pain in traumatic spinal and/or limb injury, with uncertain outcome, often does. The association of pain, anxiety, and depression may have a common neurochemical substrate in the serotonergic systems.
Amino acids
Glycine and GABA are amino acid neurotransmitters. Glycine is known as the primary excitatory neurotransmitter in the brain. GABA is the primary inhibitory neurotransmitter in the brain. Together, glycine and GABA are the most common endogenous neurotransmitters and 75–90% of all neurons in the CNS have glycine and GABA receptors.17 A thorough discussion of the biology of glycine and GABA is beyond the scope of this chapter. However, it is important to discuss the basic biology of GABA in the context of this chapter because of its central role in the action of benzodiazepines.
GABA
As mentioned previously, GABA is the primary inhibitory neurotransmitter in the CNS. It is estimated that 30% of all CNS synapses use GABA as a neurotransmitter and it is found in very high concentrations in CNS tissues (1000 to 1 000 000 times greater than concentrations of biogenic amines).23 The synaptic receptor for GABA is composed of two major receptor subtypes known as the GABAA and GABAB receptors. All benzodiazepines work at the GABAA receptor. Rather than occupying the entire GABA receptor as a competitive agonist (like morphine’s action at the opioid receptor), benzodiazepines bind to the GABAA subunit and actually facilitate endogenous GABA binding at the GABAA receptor. GABAA receptor activation opens chloride ion channels which hyperpolarizes neuronal membranes and inhibits firing (Fig. 15.3).23 This elegant mechanism is believed to be the primary pathway for the anxiolytic effects of benzodiazepines. Psychoactive compounds such as barbiturates and ethanol also act at the GABA receptor to produce similar anxiolytic and CNS depressant effects.
In an interesting study of the effects of benzodiazepines in acute pain, Di Piero and colleagues studied cerebral blood flow (CBF) by single photon emission computed tomography (SPECT), with and without diazepam.24 Diazepam, a benzodiazepine, binds to the benzodiazepine receptor on the GABAA receptor site and enhances GABA’s opening of the chloride channel, activating the GABA system and its anxiolytic actions. Diazepam, when given to healthy volunteers, following induction of pain by the cold pressor test (CPT), inhibited activation of the temporal regions, which was interpreted as part of the affective–emotional component of pain response. Diazepam-treated subjects tolerated the pain better and on SPECT this was associated with lack of temporal lobe activation of sensory–discriminative pain-related brain regions (contralateral hand region in the sensory motor cortex, pre-motor cortex and thalamus, and left anterior cingulate gyrus). This activity suggests that diazepam, which is useful in managing acute anxiety, interferes with affective–emotional components of pain perception and modifies temporal lobe activation patterns. The role of benzodiazepines in the treatment of acute and chronic anxiety will be discussed later in this chapter as will issues related to tolerance, dependence, and addiction to these medications.
DIAGNOSIS OF ANXIETY DISORDERS
Introduction
There are two major national surveys that estimated the prevalence of anxiety disorders in the United States. The Epidemiologic Catchment Area study estimated that 7% of people in the United States have a clinically significant anxiety disorder.25 The National Comorbidity Study (NCS) reported even higher prevalence rates for anxiety disorders. NCS estimated 12 month prevalence rates of 17.7% for at least one anxiety disorder and lifetime prevalence rates near 25%.1
[/not-level-membership-for-physical-medicine-and-rehabilitation-category]
