[level-membership-for-opthalmology-category]
6 The Cornea
THE NORMAL CORNEA
The cornea has three primary functions. These are:
• the refraction of light—the main refractive interface being between air and the precorneal tear film
• transmission of light with a minimum of distortion, scatter and absorption
• the structural support and protection of the globe without compromising the optical performance.

Fig. 6.1 The cornea has five distinct histological layers. The epithelium supports the precorneal tear film and consists of a stratified squamous cellular layer attached to an underlying basement membrane. Bowman’s layer is an acellular condensation of superficial stroma approximately 10–20 μm thick that lies immediately beneath the epithelial basement membrane. The stroma forms over 90 per cent of the corneal thickness and consists of regularly spaced collagen lamellae (layers) in a proteoglycan matrix interspersed with keratocytes. Descemet’s membrane is composed of a lattice of collagen fibrils that is 3 μm thick at birth and increases in thickness with age. The endothelium is a monolayer of hexagonal cells.
By courtesy of Professor J Marshall.

Fig. 6.2 The precorneal tear film must be smooth and stable for regular refraction. The epithelial surface is thrown into multiple folds (microvilli and microplicae) which produce a glycocalyx (a branching mucoprotein layer) that renders the surface hydrophilic. The precorneal tear film is about 40 μm thick and is composed of mucus derived from the conjunctival epithelial cells, conjunctival goblet cells and the lacrimal glands. The aqueous component is secreted by the lacrimal glands and the superficial lipid layer from the meibomian glands. Atmospheric oxygen, metabolites and antimicrobial agents (e.g. IgA, lysosyme, lactoferrin) are dissolved in the tear film.
By courtesy of Professor J Marshall.
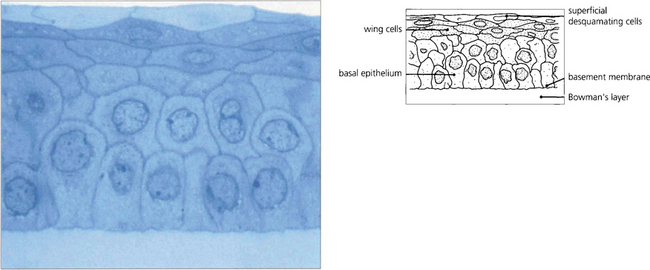
Fig. 6.3 The epithelium is about five cells thick and separated from Bowman’s layer by the epithelial basement membrane. The morphology of the basal cells changes as they migrate anteriorly to become intermediate wing cells and then elongated superficial cells with flattened nuclei that finally desquamate from the surface into the tear film. Macrophages (dendritic cells) are found in the epithelium having migrated from the limbus although they may be absent in the central zone. These have an antigen-presenting function. The epithelium has zonula occludens junctions between cells which make the healthy epithelium a virtually impermeable barrier.
By courtesy of Professor J Marshall.

Fig. 6.4 Cells from the corneal limbus continuously replace cells lost from the corneal epithelium. A population of stem cells is thought to be located in the basal epithelial cells of the folds of the palisades of Vogt. The stem cells divide to form ‘transient amplifying cells’ that move toward the centre of the cornea. Postmitotic daughter cells then migrate anteriorly from the basal layer to become terminally differentiated cells that are eventually lost from the anterior surface of the epithelium into the tear film.
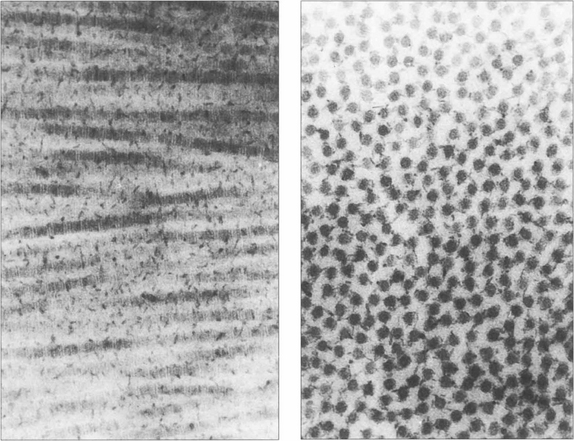
Fig. 6.5 The mechanical strength of the cornea is provided by the stroma, which is formed predominantly of collagen fibrils (mainly type 1) maintained in a proteoglycan matrix. The stromal fibrils are continuous from limbus to limbus and are arranged into about 200 layers or ‘lamellae’ with a small degree of interdigitation. The superficial Bowman’s layer is acellular and the collagen fibrils are finer and more densely packed. Transmission of light depends on the collagen fibrils regular size and spacing with small changes of refractive index. A relative dehydration of the stromal proteoglycans is required which is achieved because the epithelium is impermeable and by the endothelial pump which removes water from the stroma. Light transmission is maximal at 700 nm (98 per cent) and decreases to 80 per cent at 400 nm. Ultraviolet light with a wavelength below 310 nm is strongly absorbed by the stroma. By contrast, the cornea transmits infrared radiation up to 2400 nm.
By courtesy of Professor K Meek.
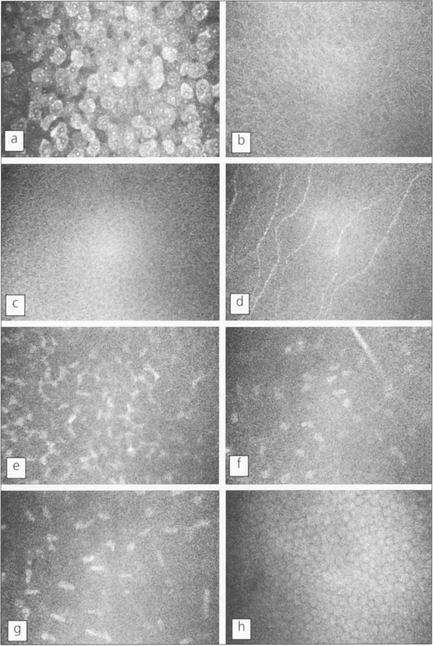
Fig. 6.6 Using in vivo confocal microscopy high-resolution images can be obtained in real-time at different depths within the intact living cornea without the need for staining or processing, giving a direct view of living cells. Shown here is the cellular morphology of a normal human cornea. Each image represents an optical volume with approximately 450 × 340 × 9 μm. a, Superficial epithelial cells; b, wing epithelial cells; c, basal epithelial cells; d, subepithelial nerves (on Bowman’s layer); e, first layer of keratocyte nuclei; f, nerve branch and keratocytes in the mid-stroma; g, keratocytes in front of Descemet’s membrane; h, endothelial cells.
By courtesy of Dr T Moller-Pedersen.
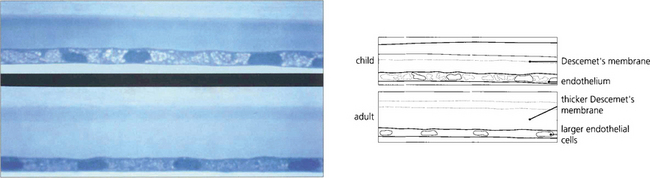
Fig. 6.7 Descemet’s membrane, which consists of type IV collagen, is secreted by the endothelium. It is composed of a lattice of collagen fibrils that is 3 μm thick at birth (top), when the entire layer appears striated or ‘banded’. A posterior ‘nonbanded’ layer is continuously laid down throughout life so that Descemet’s membrane increases in thickness with age to reach 30–40 μm in the elderly (bottom). Notice the decrease in endothelial cell population.
By courtesy of Professor J Marshall.
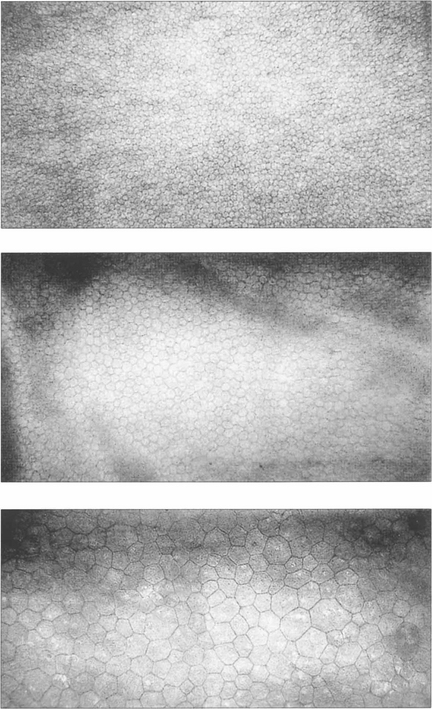
Fig. 6.8 The endothelium can be examined by specular or confocal microscopy. The endothelium does not replicate after birth and cell counts reduce from 3500–4000/mm2 at birth to approximately 2000–2500/mm2 in the adult cornea. Corneal decompensation is likely with cell counts of less than 500/mm2. Cell density is a good guide to function, which can be complemented by other parameters such as variation in cell size and morphology (polymegathism and polymorphism). These specular photographs show the endothelium of an 18-month-old infant (top) and a normal 74-year-old man (middle). Notice the decreasing cell count and larger cell size with age. Larger cells with variation in morphology are seen in the endothelium of a patient after traumatic cataract surgery (bottom).
NORMAL CORNEAL TOPOGRAPHY
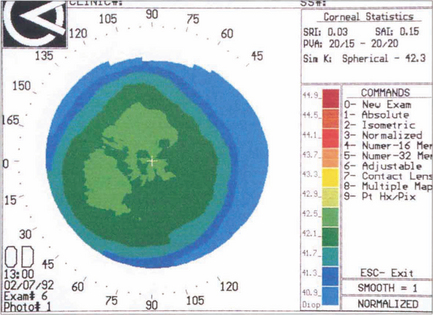
Fig. 6.9 With topography (see Ch. 1) a series of concentric rings is projected on to the surface of the corneal tear film. A difference in the relative distances between these rings compared to a calibrated spherical surface allows the corneal curvature to be measured from the visual axis to the periphery; this is then converted to dioptric power and displayed as a colour-coded topographical map in which colours towards the red end of the spectrum represent increasingly steep dioptric powers. A small range of dioptric powers can be seen on this normal cornea with flattening toward the periphery.
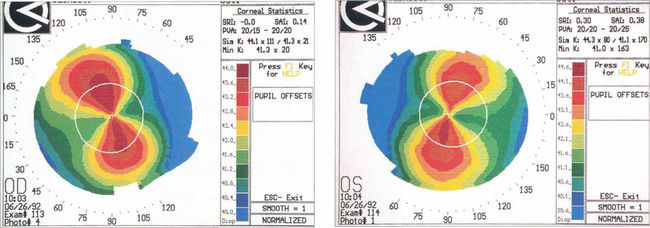
Fig. 6.10 Astigmatism is seen on topography as distortion of the circular projections into oval reflections. Topography of regular astigmatism appears as a ‘bow tie’ due to progressive peripheral flattening with axes at 90° to each other with mirror image symmetry between the two eyes. Irregular astigmatism, as in keratoconus, appears as steepening below the visual axis (see Fig. 6.33).
AGE-RELATED DEGENERATION
Involutional changes as a result of ageing must be distinguished from pathological changes.
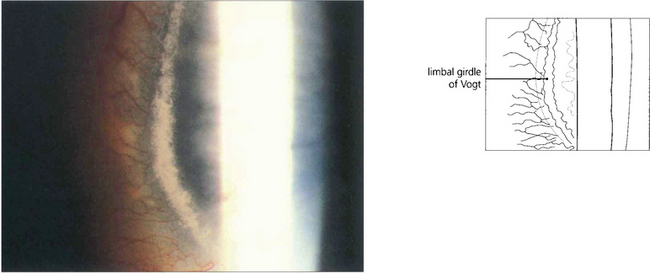
Fig. 6.11 The white limbal girdle of Vogt is a common ageing change at the interpalpebral limbus which appears as a semilunar opacity with a clear zone of separation from the limbus; it is best seen by sclerotic scatter. The nasal cornea is affected nearly twice as often as the temporal side. There may be clear patches in the opacity, which may then resemble a mild form of band keratopathy. Histological examination shows subepithelial hyaline degeneration at the level of Bowman’s layer.
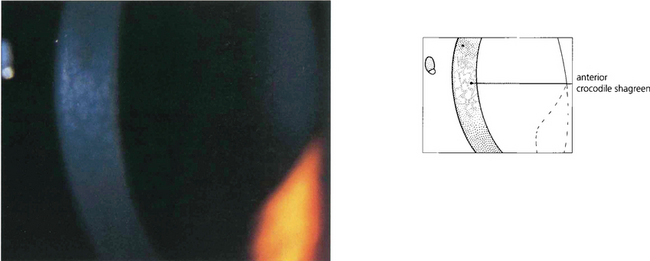
Fig. 6.12 Anterior crocodile shagreen is a pattern of polygonal opacities with intervening clear zones at the level of Bowman’s layer. It is best seen by wide-slit oblique illumination. Histological examination shows folding of Bowman’s layer. Although the superficial variant is an ageing change, a pre-Descemet’s form may be familial and related to the central cloudy corneal dystrophy (of François). Neither produces visual symptoms.
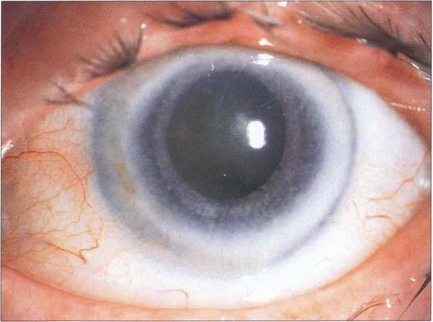
Fig. 6.13 Corneal arcus is due to the deposition of cholesterol and other lipids in the peripheral cornea, particularly adjacent to Bowman’s layer and Descemet’s membrane. It is a common ageing phenomenon and is almost universal by the eighth decade. A sharp clear zone lies between the limbus and the hazy inner border of the arcus. The condition is usually of no significance unless seen in patients under 40 years of age in whom it may be familial (arcus juvenilis); these patients require investigation for hyperlipidaemia. A lucent subepithelial zone with mild thinning may appear within the arcus in elderly people (senile furrow degeneration).
CONGENITAL CORNEAL ANOMALIES
Anterior segment dysgenesis produces a spectrum of anomalies. The current clinical classification does not reflect the underlying genetic defect. Both the Axenfeld–Rieger and Peter’s anomalies can be caused by abnormalities of at least four different genes. Posterior embryotoxon, the mildest expression, represents a centrally displaced Schwalbe ring. It is commonly a normal variant and not associated with glaucoma. The Axenfeld–Rieger syndrome (see Ch. 8) consists of posterior embryotoxon with anterior iris adhesions, corectopia and iris hypoplasia, probably due to arrest of neural crest development. Peter’s anomaly does not usually have posterior embryotoxon or peripheral anterior iris adhesions but there is a central iris to cornea adhesion with a defect of the endothelium and posterior stroma. The significance of the Axenfeld–Rieger syndrome and Peter’s anomaly lies in their association with childhood glaucoma, buphthalmos, corneal oedema and blindness. They may also be associated with systemic defects such as dental and cranial anomalies and malformations of the upper limbs and spine. Posterior keratoconus causes thinning of the posterior stroma with overlying haze and may be congenital; it has therefore been classified as a dysgenesis although many cases are thought to result from trauma. Congenital absence of the limbus is often associated with flattening of the cornea, as is seen in sclerocornea and cornea plana.
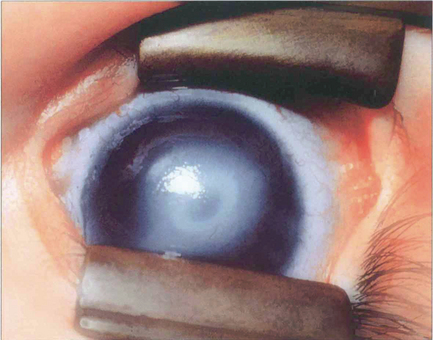
Fig. 6.14 Peter’s anomaly is characterized by a central corneal opacity with defects in the posterior stroma, Descemet’s membrane and endothelium and adhesions of the iris collarette to the posterior cornea, making it difficult to see the lens. There may be an anterior cataract or adhesions between the lens and cornea. Secondary glaucoma is common. Associated cardiac defects, cleft palate, craniofacial dysplasia and skeletal abnormalities may also occur (Peter’s-plus syndrome). The defects can be explained by abnormal separation of lens vesicle from surface ectoderm, and the heterogeneity by organs being affected that differentiate at same gestational age. The condition is usually bilateral with a sporadic incidence (80 per cent), although inherited cases occur.
By courtesy of Professor P Khaw.
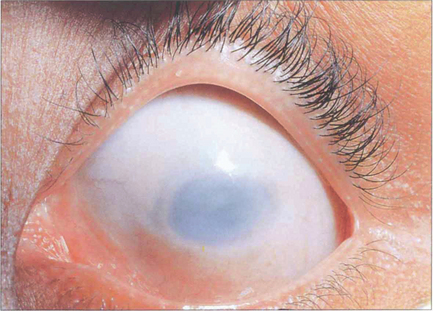
Fig. 6.15 Sclerocornea is a congenital nonprogressive scleralization of the cornea. Either the peripheral cornea or the entire cornea may be involved; the limbus cannot be identified and the radius of curvature is flattened. There is overgrowth of conjunctival and episcleral vessels on to the cornea. Opacification of the cornea is due to a similar organization of the corneal stromal collagen fibrils to those of normal sclera. The condition is usually bilateral with an equal sex incidence and is usually sporadic, although dominantly and recessively inherited forms have been described. Cornea plana is an inherited abnormality most commonly described in Finnish populations; the cornea is flat with a radius of curvature similar to that of the sclera. The cornea is not opaque unless associated with sclerocornea. Extreme hyperopia, abnormalities of the angle and secondary glaucoma are common.
CORNEAL DYSTROPHIES
The corneal dystrophies are inherited corneal diseases that exhibit a remarkable degree of phenotypic and genetic heterogeneity (Table 6.1). Molecular biology has given new insights into the pathogenesis of these diseases. While at present they are classified clinically or histopathologically, their future classification is likely to be genotypic. It is evident, however, that the distinction between some dystrophies is not as clear as had been thought as apparently different clinical appearances can be caused by mutations in the same gene. Conversely, clinically similar dystrophies can result from different genes on separate chromosomes determining different protein products.
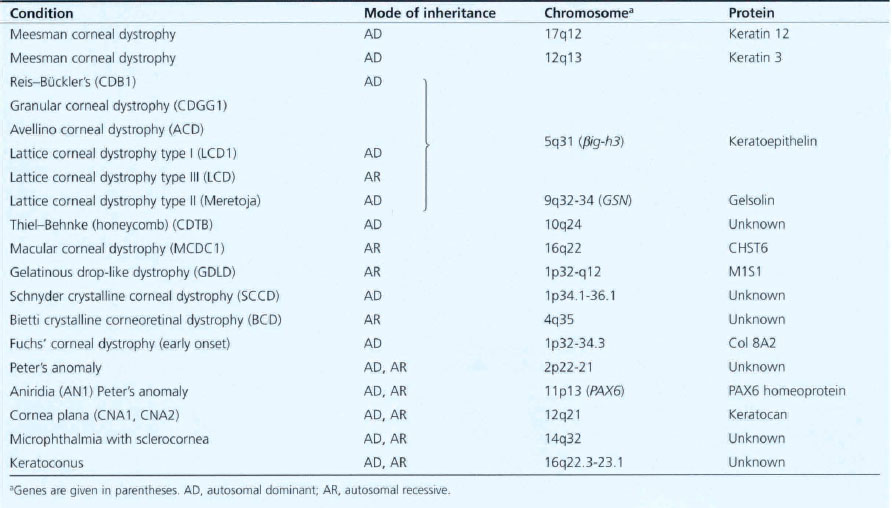
Table 6.1 Inheritance of corneal dystrophies
EPITHELIAL DYSTROPHIES
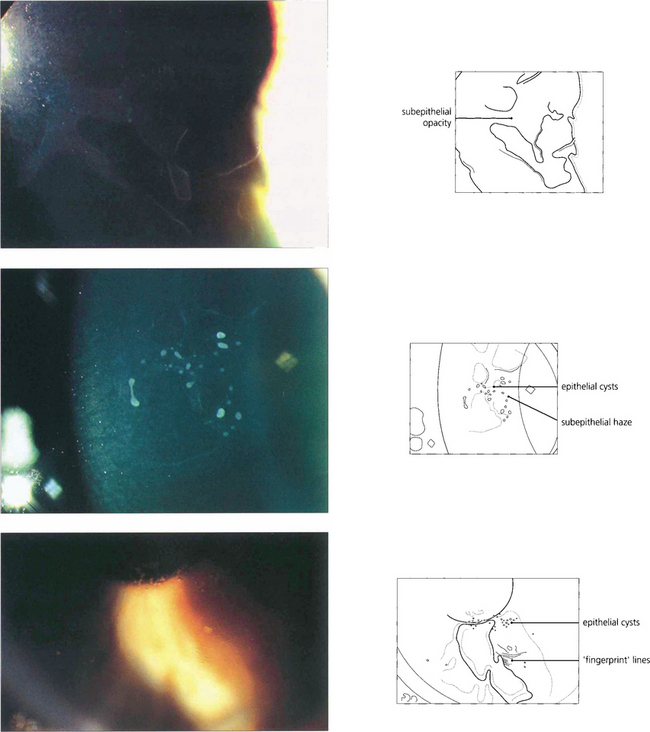
Fig. 6.16 The commonest corneal dystrophy is epithelial basement membrane dystrophy. This is bilateral, more frequent in women and its severity increases toward middle age. The clinical signs are variable and there may be grey subepithelial patches (top), dots or microcysts (middle), and concentric fine whorls of fingerprint lines best seen with retroillumination or broad-beam oblique illumination (bottom). The term Cogan’s dystrophy can be used if large grey dots are the prominent feature. Visual symptoms are uncommon but intensely painful recurrent erosions may occur following minor trauma. Symptoms are especially common on waking and can be relieved by using artificial tears during the day and lubricating ointment just before sleep. Severe cases may be helped by debridement of the epithelium, puncture of Bowman’s membrane with a needle or a bandage contact lens. Epithelial debridement followed by superficial excimer laser keratectomy can be useful in recurrent disease.
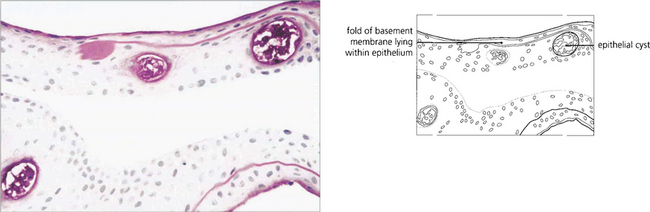
Fig. 6.17 Histological examination shows thickening and reduplication of the basement membrane and cyst formation.

Fig. 6.18 Meesman’s dystrophy is characterized by epithelial cysts spreading from limbus to limbus. These contain mucopolysaccharide.
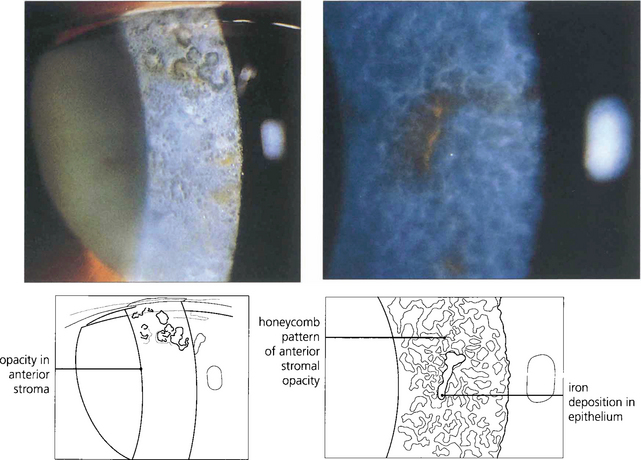
Fig. 6.19 Type I Bowman’s membrane dystrophy (Reis–Bückler’s) predominantly affects Bowman’s layer and the superficial stroma (left). It is dominantly inherited and has been linked to the same region of chromosome 5q31 as granular, lattice, and Avellino dystrophies. Patients usually present during the first or second decade of life with painful recurrent erosions; subsequent scarring and surface irregularity blur vision. Disease is bilateral and symmetrical with grey reticular opacities seen mainly in the central cornea.(Right) Type II Bowman’s membrane dystrophy (Thiel–Benhke dystrophy), a histologically distinct subgroup with a honeycomb pattern, has been linked to chromosome 10q24. Excimer laser superficial keratectomy or lamellar keratectomy is the initial treatment of choice but the disease usually recurs with time. Penetrating keratoplasty is rarely indicated.
STROMAL DYSTROPHIES
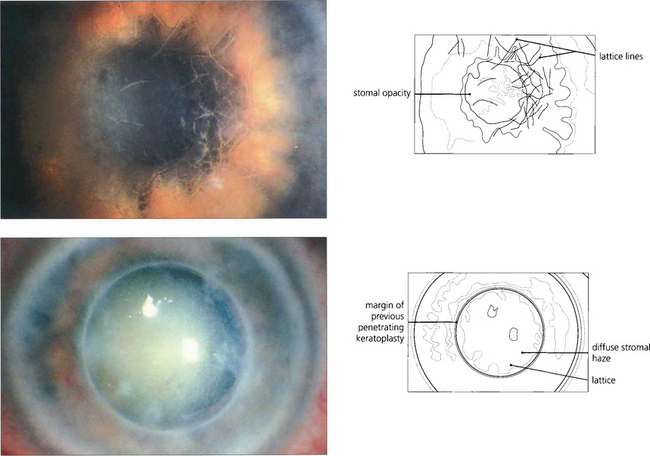
Fig. 6.20 Lattice dystrophy is a common, dominantly inherited, bilateral dystrophy with a varied appearance usually presenting in the first to second decade of life with blurred vision and recurrent epithelial erosions. The anterior stroma of the central cornea has a pattern of interlacing, clear, refractile lines and dots (top). Diffuse stromal haze is common in advanced disease. A bandage contact lens or laser phototherapeutic keratectomy can be used to manage symptoms of recurrent erosion, which are due to accumulated subepithelial amyloid impairing stromal–epithelial adhesion. Keratoplasty may be necessary by the fourth decade; lamellar keratoplasty is preferable to penetrating keratoplasty as disease commonly recurs in the transplant so that repeat keratoplasty may be needed (bottom). Changes similar to lattice dystrophy are seen in the corneal degeneration of climatic keratopathy.
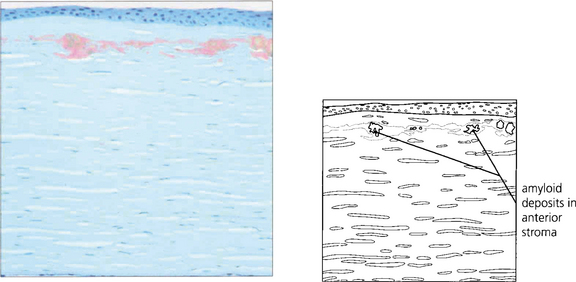
Fig. 6.21 Amyloid is deposited in the anterior stroma and beneath the epithelium. The lesions stain with Congo Red and exhibit dichroism and birefringence when viewed with crossed polarizing filters. The condition is considered a form of localized amyloid deposition but the gene defect (5q31) is different from that identified with systemic forms of amyloidosis. In Meretoja syndrome (type II lattice dystrophy) a defect of gelsolin production (9q34) is associated with systemic and mild corneal amyloidosis.
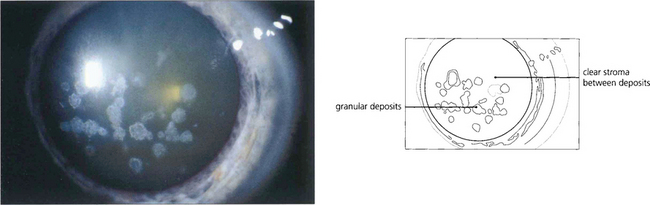
Fig. 6.22 Granular dystrophy is dominantly inherited. Different clinical subtypes have been described, although these are identical ultrastructurally. The condition presents in the first decade of life with white-grey breadcrumb-like or ring patterns in the central anterior stroma that become more widespread and posterior over time. The intervening stroma is usually clear and the lesions do not involve the peripheral cornea. Epithelial erosion is uncommon and symptoms are usually limited to glare from light scatter. Symptoms of erosion can be managed with either a bandage contact lens or laser superficial keratectomy. Rarely, either lamellar or penetrating keratoplasty is required for visual loss although disease can recur in the donor tissue.

Fig. 6.23 Histologically these amorphous hyaline lesions stain red with Masson trichrome stain. The origin of this material is uncertain. Patients with Avellino corneal dystrophy demonstrate clinical and pathological changes of both lattice and granular dystrophy.
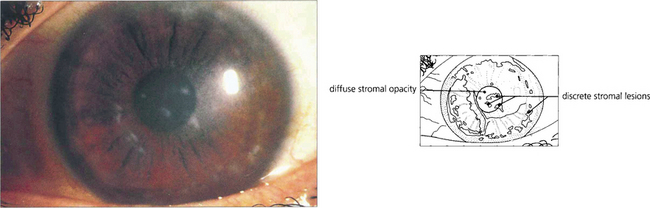
Fig. 6.24 Macular stromal dystrophy is uncommon and has an autosomal recessive inheritance. Poorly defined grey lesions appear in the superficial central corneal stroma during the first decade of life; these gradually spread deeper into the stroma and peripherally to the limbus. The intervening stroma becomes diffusely cloudy with whitish round lesions (macular spots) developing later in the deep stroma. There is associated corneal thinning. Vision is impaired early and recurrent corneal erosions may occur. Keratoplasty is often required although the condition may recur in the transplant.
By courtesy of Mr M G Falcon.
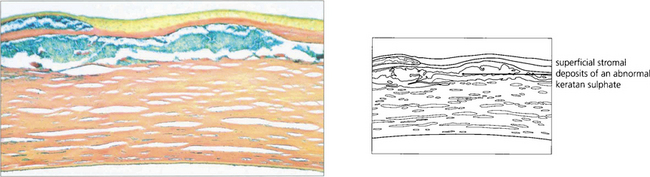
Fig. 6.25 An abnormal keratan sulphate accumulates intracellularly in keratocytes and extracellularly between stromal lamellae; these stain Prussian blue with colloidal iron.
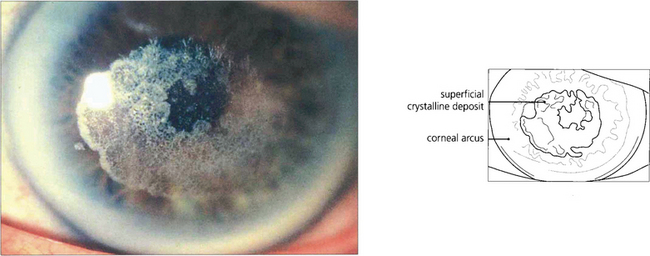
Fig. 6.26 Schnyder’s crystalline dystrophy is rare. It is dominantly inherited, bilateral, and often associated with a prominent arcus, as in this example. Central deposition of crystalline cholesterol esters occurs in the anterior corneal stroma and the condition appears to be a localized disorder of corneal lipid metabolism associated with hyperlipidaemia. Most cases can be treated by laser superficial keratectomy.
By courtesy of Mr J Kwartz.
ENDOTHELIAL DYSTROPHIES
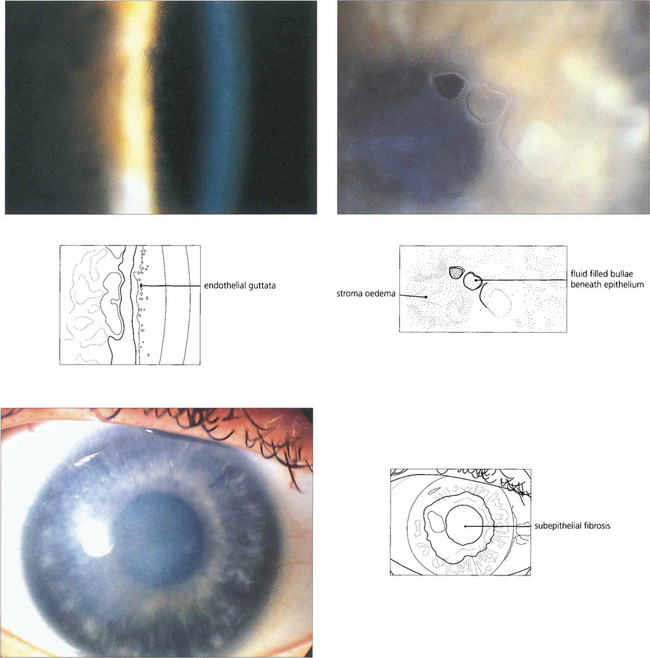
Fig. 6.27 Fuchs’ corneal endothelial dystrophy is common. It ranges from a few asymptomatic cornea guttata, which are localized excrescences of Descemet’s membrane in the central cornea best seen by retroillumination (top left), to confluent guttata associated with a reduced endothelial cell count and corneal oedema. The condition is more frequent and severe in women, and is usually inherited in an autosomal dominant manner. Its aetiology is unknown. Signs are rarely seen before the age of 50 years (top right). As the disease progresses, patients notice that vision is blurred on waking but clears during the day. In advanced disease there is pigment granule deposition on the endothelium, permanent stromal oedema, and epithelial microcysts and bullae. When cysts or bullae rupture they cause pain, watering and photophobia. Eventually subepithelial fibrosis develops and the eye may become more comfortable (bottom left). Penetrating keratoplasty may be indicated for pain or reduced vision. Cataract is a common association and the trauma of cataract surgery can precipitate permanent corneal decompensation. Combined keratoplasty and cataract extraction may be required.
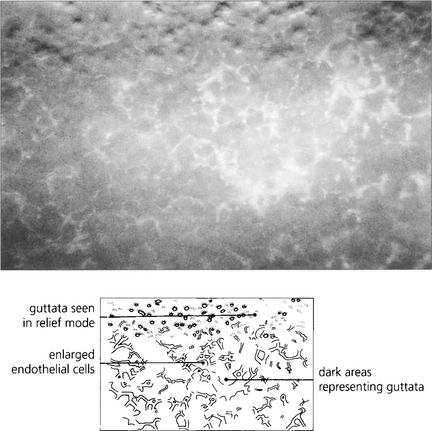
Fig. 6.28 Specular microscopy of the endothelium from a patient with moderately advanced disease shows numerous guttata that appear as dark areas due to loss of the specular reflection from the endothelial mosaic over the raised surface of each guttata (compare with Fig. 6.8).
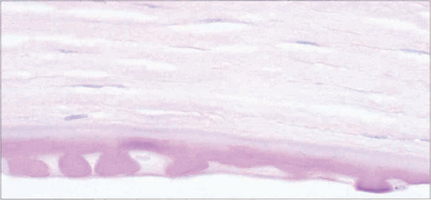
Fig. 6.29 Histological appearance of a keratoplasty specimen shows a thickened Descemet’s membrane with guttata formation and loss of endothelial cells.
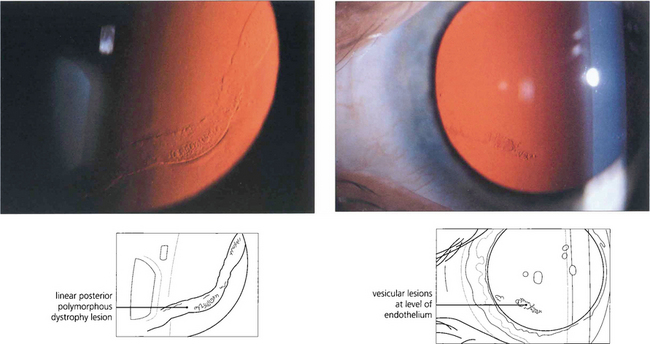
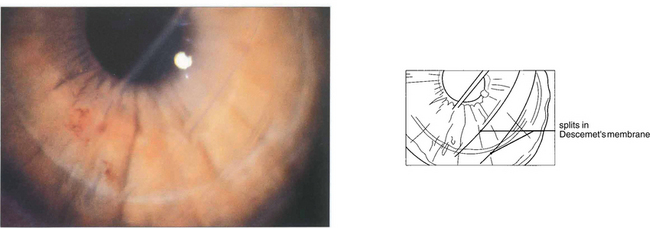
Fig. 6.30 Posterior polymorphous dystrophy develops early in life and the changes may be overlooked or noted only coincidentally. There are geographic ‘vesicular’ or linear lesions with grey scalloped edges at the level of Descemet’s membrane. These changes are only very slowly progressive and corneal oedema is rare. Filmy peripheral anterior synechiae may be present and secondary glaucoma may develop in gross cases. Specular microscopy shows decreased endothelial cell density and apparent splits in Descemet’s membrane.
CORNEAL ECTASIA
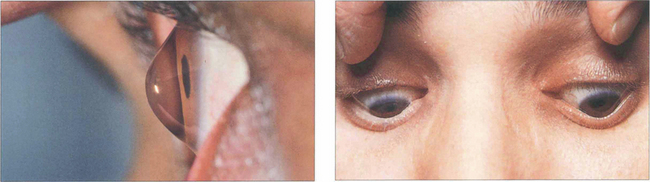
Fig. 6.32 The abnormally steep corneal apex of keratoconus can be seen on lateral view and may indent the lower lid on downgaze (Munsen’s sign).
By courtesy of Mr K Pullen.
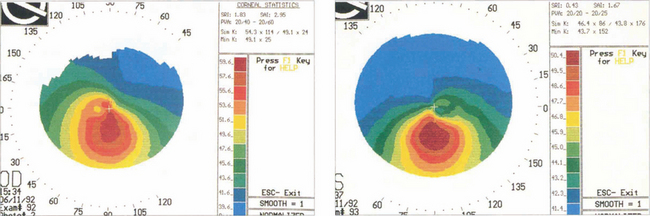
Fig. 6.33 The detection of early keratoconus is vital in patients considering corneal refractive surgery. In the absence of other clinical signs, pachymetry may show thinning below the visual axis and corneal topography demonstrates steepening below and temporal to the visual axis. Family members of patients with keratoconus may have subclinical disease detectable only by these methods.
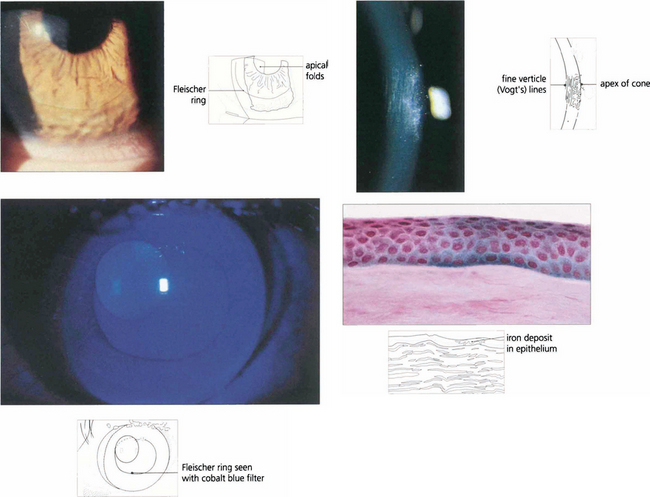
Fig. 6.34 Fine vertical folds in Descemet’s membrane occur near the apex of the cone (Vogt lines). Iron may be deposited in the deep epithelium at the base of the cone, best seen using the cobalt blue filter. This is known as a Fleischer ring; it delineates the area of corneal ectasia and may be a useful aid when planning surgery. Histological examination shows iron deposition in the epithelium, seen as Prussian blue with Perls’ stain.
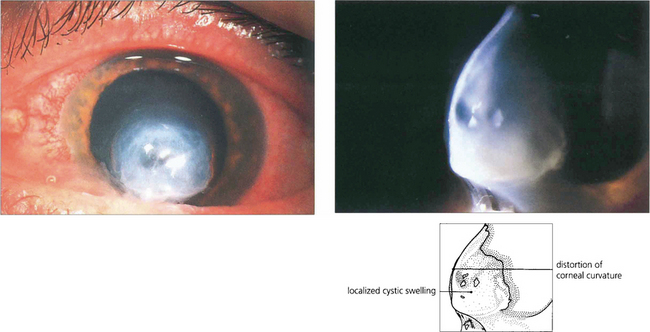
Fig. 6.35 Patients with keratoconus can suddenly develop blurred vision from an acute split in Descemet’s membrane results in a rapid influx of aqueous into the stroma (corneal hydrops). The condition is painful but resolves spontaneously over a few months as endothelial cells cover the defect. Although good vision may return, scarring often reduces visual acuity.
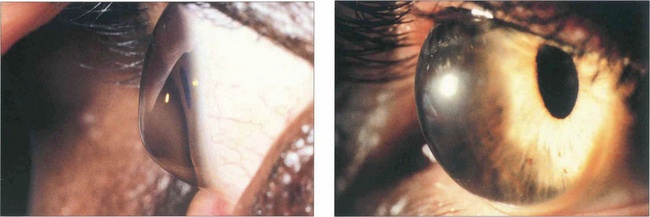
Fig. 6.36 Pellucid marginal degeneration (left) and keratoglobus (right) are rare variants of corneal ectasia. In pellucid marginal degeneration there is a zone of maximal thinning adjacent to the inferior limbus. In keratoglobus the entire cornea is ectatic. Management is more difficult than for keratoconus because of the very irregular shape. If corneal lenses fail, scleral contact lenses or lamellar graft surgery should be considered.
By courtesy of Mr K Pullen.
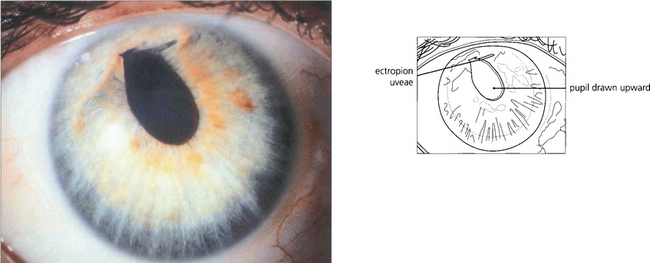
THE IRIDOCORNEAL ENDOTHELIAL SYNDROME
The iridocorneal endothelial (ICE) syndrome is characterized by abnormal endothelial cells that appear able to proliferate and migrate from the posterior corneal surface across the angle and on to the anterior surface of the iris. Characteristic changes of the corneal endothelium may be seen with specular microscopy; these may be focal or diffuse. The syndrome is more common in women and almost always unilateral. Iris atrophy, ectropion uveae, pseudo-nodules on the iris and broad-based peripheral anterior synechiae are common associated changes. Combinations of these changes have been named as three variants: essential iris atrophy, the Cogan–Reese syndrome and Chandler’s syndrome (see Ch. 8) although the underlying disease process is probably the same. Corneal oedema from endothelial failure or secondary glaucoma from outflow obstruction can cause visual loss. The aetiology is uncertain although herpes simplex antigen has been identified in the endothelium in some cases.

CORNEAL EPITHELIAL STEM CELL FAILURE
The corneal epithelium is continuously replaced from a population of stem cells located at the limbus (see Fig. 6.4). These cells divide to form daughter cells that move centrally to replace cells that are lost from the epithelium into the tear film. If these limbal cells are absent or destroyed the corneal epithelium is replaced by cells derived from conjunctiva. This may be accompanied by neovascularization, goblet cells in the epithelium, blurred vision and an unstable and irregular epithelial surface. Congenitally absent limbal stem cells are thought to underlie the keratopathy of aniridia. Acquired stem cell failure may follow alkali injury (see Ch. 3), Stevens–Johnson syndrome or chronic exposure to some contact lens care products, particularly thiomersal. In severe disease, limbal tissue can be transplanted either from the fellow eye or a donor eye to restore a stable corneal surface.
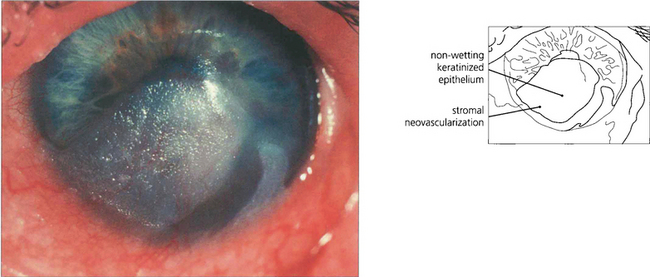
Fig. 6.39 Clinical stem cell failure is seen most frequently after Stevens–Johnson syndrome, alkali injury and, more rarely, after surgical excision of the limbus or chronic chemical injury. Pterygium is thought to be partly the result of focal stem cell failure following exposure to ultraviolet radiation. This eye shows an area of conjunctivalization with keratinization of the inferior cornea in Stevens–Johnson syndrome.
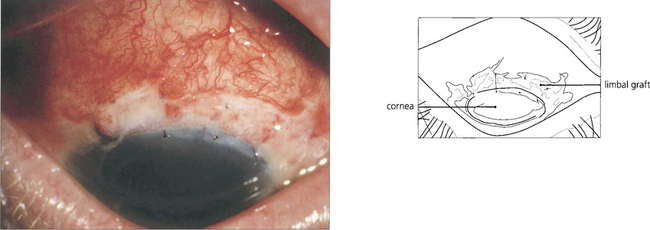
Fig. 6.40 Such eyes can be treated by removal of the abnormal epithelium and an allograft from the unaffected limbus of the same or fellow eye to restore normal corneal epithelium.
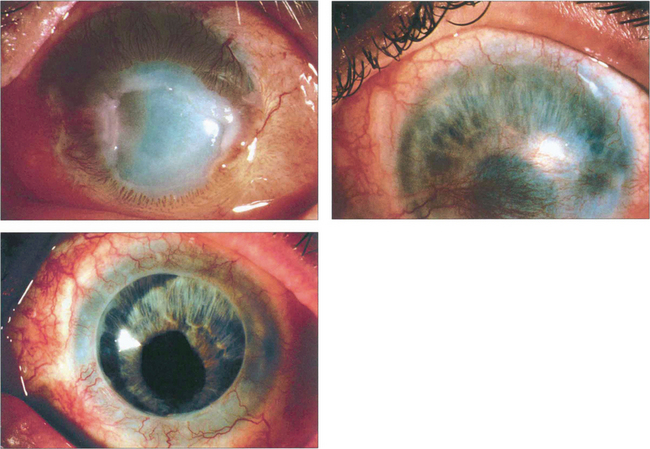
Fig. 6.41 This patient had a severe alkali burn with corneal vascularization and a persistent epithelial defect (top left). Following a limbal allograft from the fellow eye, the epithelial defect healed with residual corneal defect (top right). The patient went on to have a successful penetrating keratoplasty.
By courtesy of Professor Ed Holland.
CORNEAL POST-INFLAMMATORY CHANGES AND DEGENERATIONS
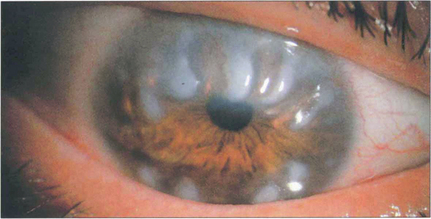
Fig. 6.42 In Salzmann’s nodular degeneration, elevated lesions resulting from the replacement of Bowman’s layer by hyaline material are typically distributed circumferentially around the mid peripheral cornea. The condition is more common in elderly women and is associated with chronic ocular surface diseases such as trachoma or severe blepharitis. Focal disease may occur over old scars. In symptomatic patients the nodules can be removed by surgical or phototherapeutic keratectomy.
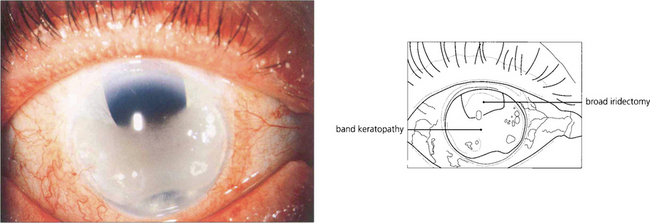
Fig. 6.43 Most cases of band keratopathy are idiopathic but the condition may be seen with chronic uveitis, especially in children (see Ch. 10), following the use of silicone oil for retinal detachment or in phthisis bulbi. A few cases are associated with hypercalcaemia or hyperphosphataemia with renal failure. Changes begin in the peripheral interpalpebral zone of the cornea as white deposits of calcium at the level of Bowman’s layer with a clear zone separating them from the limbus. Clear round holes can often be seen in the band representing the passage of nerves through the deposit. Large aggregated deposits may cause painful epithelial erosions. Treatment by surgical or laser keratectomy is indicated for patients with pain or impaired acuity.
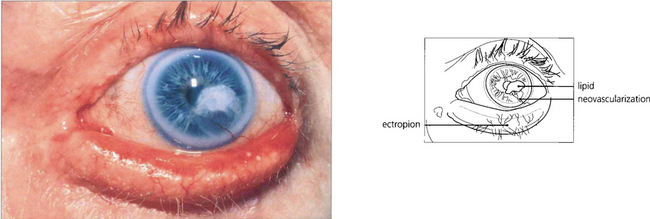
Fig. 6.44 Lipid keratopathy is due to yellow deposition of lipid and lipoproteins leaking from new vessels in the cornea. It is seen most commonly following chronic inflammation and vascularization, such as with herpes zoster keratitis (see Ch. 4).
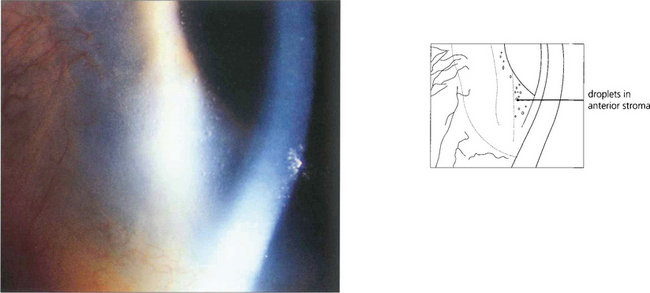
Fig. 6.45 Climatic droplet keratopathy (spheroidal degeneration, Labrador keratopathy) is characterized by translucent spheroidal deposits appearing in the superficial stroma. The deposits usually begin at the periphery in the interpalpebral zone. Lesions may appear adjacent to sites of chronic inflammation but more commonly they are the result of chronic exposure to sunlight.
CORNEAL TRAUMA
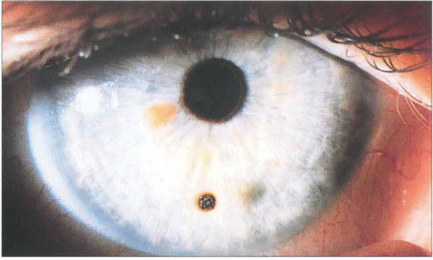
Fig. 6.46 An impacted metallic corneal foreign body is a common injury associated with drilling or grinding steel without protective goggles. These particles normally have insufficient energy to pass through the cornea and so lodge superficially. They become surrounded by a ring of rust within a few days; this should be lifted off with a sharp needle or dental burr under topical anaesthesia to prevent delayed healing.
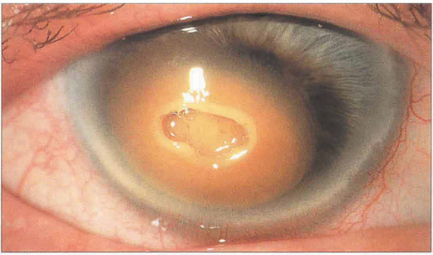
Fig. 6.47 Corneal blood staining is associated with large hyphemas, high intraocular pressure and endothelial injury. A dark brown discoloration from haemoglobin and haemosiderin develops in the posterior stroma that progresses to involve the full stromal thickness but leaves the peripheral cornea clear. Blood staining clears from the periphery and may take years to resolve completely. It can also occur following intrastromal bleeding from neovascularization. In this example, the overlying central stroma has also become necrotic.
By courtesy of Mr M G Falcon.
MICROBIAL KERATITIS
The majority of corneal infections in temperate regions are the result of bacterial infection. Infections due to fungi or acanthamoeba are rare in temperate regions but much more frequent in tropical countries. Most fungi will grow on blood agar but samples should be plated specifically on to Sabouraud agar if fungal infection is suspected. Samples for suspected acanthamoeba infection should be plated on to non-nutrient agar seeded secondarily with killed Escherichia coli. Viral keratitis is covered in Chapter 4.
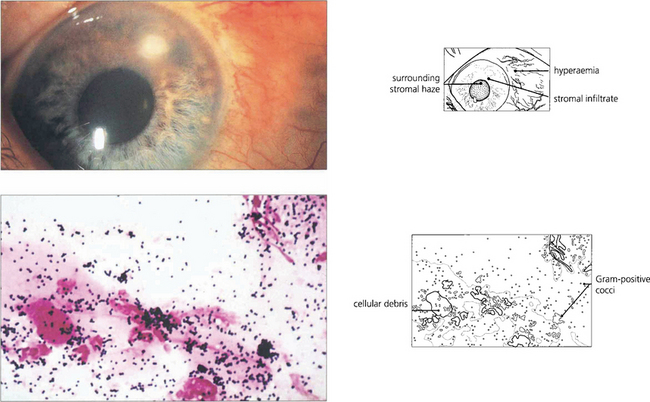
Fig. 6.49 Even small corneal infiltrates must be managed aggressively because they can typically progress. Lid and conjunctival commensals may act as pathogens if the natural defences of the cornea are breached. Gram-positive cocci (blue-black), as seen in this scrape, are therefore a common cause of microbial keratitis.
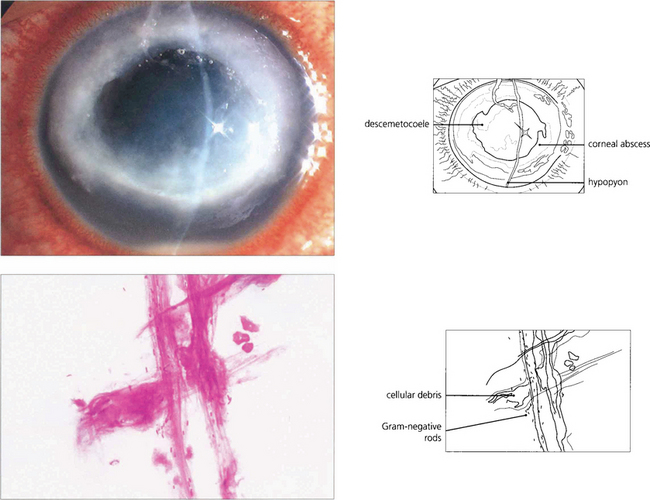
Fig. 6.50 Soft contact lenses (especially extended wear lenses) predispose the eye to Gram-negative microbial keratitis. Epithelial hypoxia or other trauma may enable Pseudomonas species to attach to and penetrate the corneal epithelium, rapidly destroy the corneal stroma, and cause perforation. Gram-negative rods (red) are seen in this corneal scrape.
By courtesy of M Matheson.
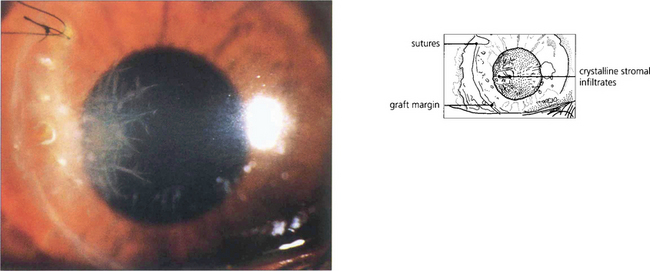
Fig. 6.51 Crystalline keratopathy is characteristic of a partially treated streptococcal keratitis in a patient whose inflammatory response has been suppressed by steroids. In this case the infection affects the margin of a corneal graft. The ‘crystals’ are proliferating bacteria in natural cleavage planes between stromal lamellae. It is important not to confuse the filamentary appearance with fungal keratitis (the filaments are much too large to be fungal hyphae).
By courtesy of Mr M G Falcon.
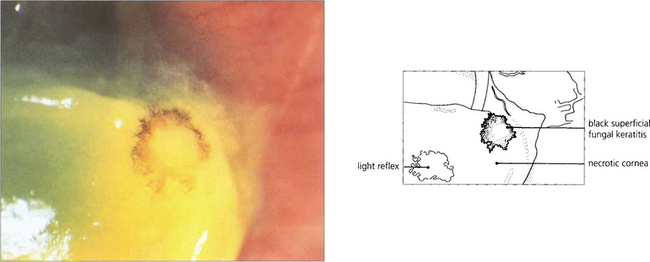
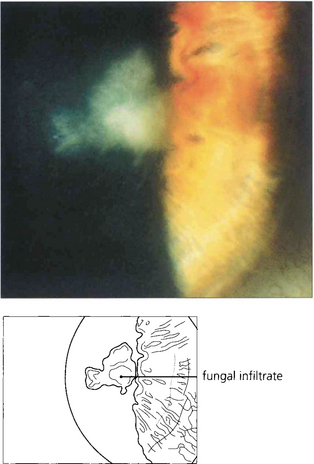
Fig. 6.52 Fungal keratitis is rare in temperate countries but important in tropical countries, often being caused by agricultural ocular injury. Fungal keratitis may also occur as a secondary infection in chronic ocular surface disease. Many different types of fungi have been reported to cause ocular infection; the most common are filamentous moulds such as Aspergillus and Fusarium or yeasts such as Candida. Treatment is with topical antifungal agents; severe infection may require oral treatment. Treatment is often ineffective owing to poor penetration and progressive disease may require wide surgical excision and keratoplasty. This patient has a pigmented stellate lesion (from Curvularia species) occurring on a grossly compromised cornea from exposure associated with proptosis from an orbital tumour.
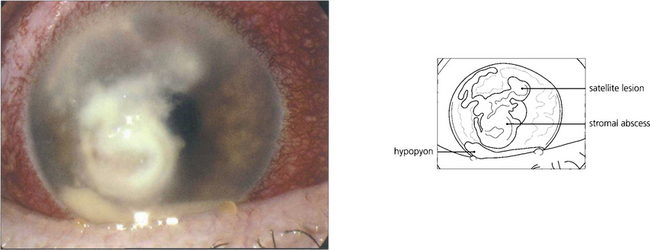
Fig. 6.53 This eye shows secondary infection of a stromal herpes simplex lesion with Fusarium. The satellite lesions superiorly are characteristic of fungal infection.
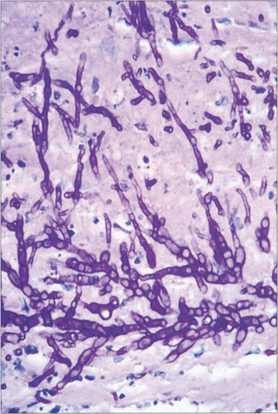
Fig. 6.54 Histology of the removed corneal button from another eye shows the filamentary hyphae of Fusarium throughout all levels with destruction of the stromal lamellae. The hyphae of filamentary fungi have a predilection to spread just anterior to Descemet’s membrane and may be missed at biopsy.
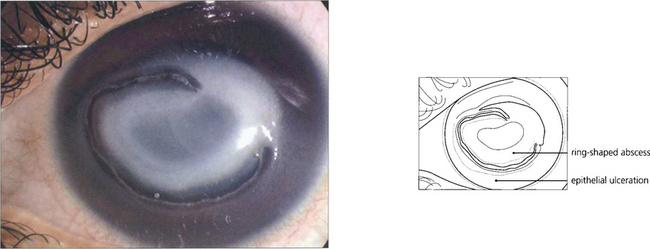
Fig. 6.55 Acanthamoebae are free-living organisms found in tap water; they can encyst when conditions are adverse. They are a rare cause of microbial keratitis which is usually associated with soft contact lens wear, although in tropical countries it is also commonly caused by agricultural injury. Early contact lens-associated disease may be limited to the epithelium and is readily curable. Neglected cases with stromal invasion may develop a ring-shaped abscess around the infected central stroma and hypopyon; amoebae can secrete powerful collagenases. Eradication is difficult as the amoebae form cysts that are resistant to treatment.
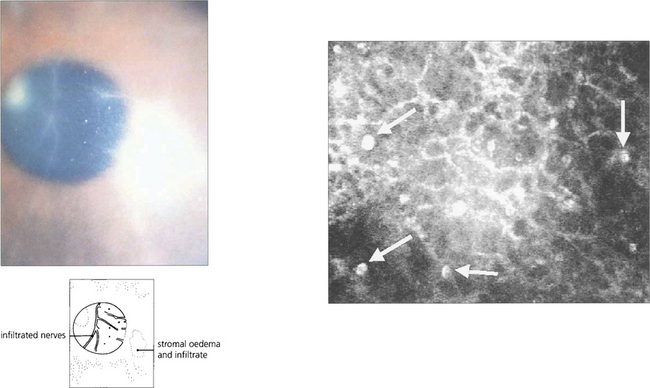
Fig. 6.56 Disproportionate pain is often a feature, and perineural infiltrates are probably pathognomonic (left). In vivo confocal microscopy can provide a quick and definite diagnosis in cases of suspected acanthamoebic keratitis (right). This is the anterior stroma of a contact lens wearer with a 3-week history of keratitis unresponsive to antibiotic treatment. Note the highly reflective, double-walled, ovoid structures (15–25 μm) consistent with acanthamoeba cysts (arrows).
By courtesy of Dr T Møller-Pedersen.
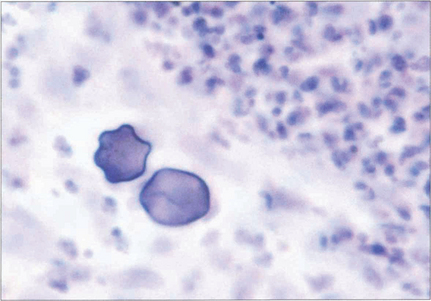
Fig. 6.57 Diagnosis is made by culturing the free-living form (trophozoites) on E. coli-seeded blood agar medium or by identifying the organism or its cyst by immunohistochemistry on an epithelial or stromal biopsy. This photograph demonstrates acanthamoebic cysts in a biopsy specimen.
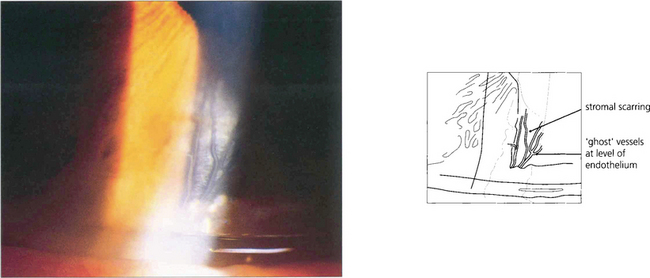
Fig. 6.58 Interstitial keratitis is believed to result from an immunological response to infectious organisms, and is usually thought of in the context of congenital syphilis. The active phase of corneal inflammation which is rarely seen in modern ophthalmology presents during the first two decades of life, with intense photophobia, deep peripheral vascularization (salmon patch) and an anterior uveitis. Treatment with intensive topical steroids should be started and serological investigation performed. Untreated, the cornea thickens and opacifies, resolving over several months to leave stromal scarring with the characteristic empty, deep, stromal ‘ghost’ vessels anterior to Descemet’s membrane.
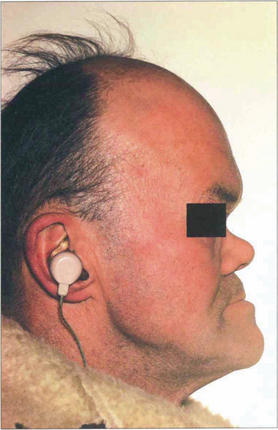
Fig. 6.59 Patients may have other signs of congenital syphilis such as neural deafness, Hutchinson’s teeth, a collapsed nasal bridge or intellectual impairment. Patients may also have a pigmentary retinopathy (see Ch. 10).
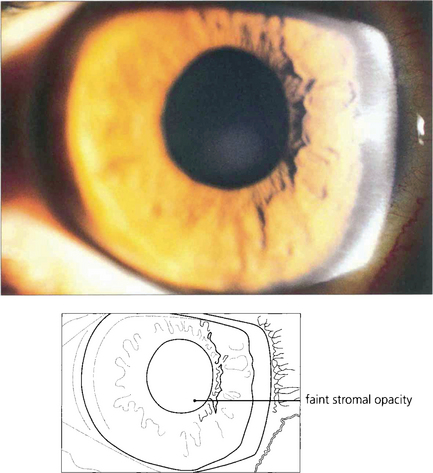
Fig. 6.60 Cogan’s syndrome is an autoimmune interstitial keratitis associated with vertigo and neural deafness that develops in young adults. It has no relationship to syphilis and the aetiology is unknown.
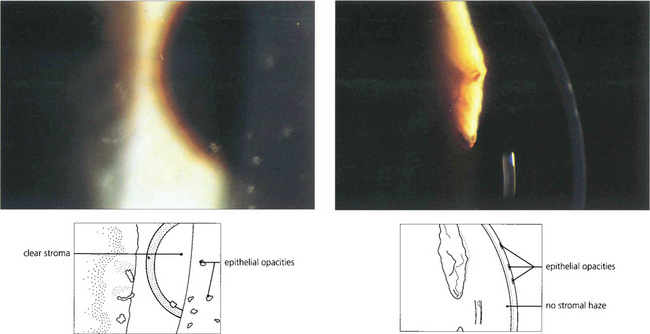
Fig. 6.61 Thygeson’s superficial punctate keratitis is an uncommon, noncontagious and usually bilateral disease with episodes of discomfort recurring over several years. The aetiology is unknown, but the rapid response to steroids suggests an immunological basis. Attempts to identify a viral agent have been unsuccessful. Discrete elevated grey opacities occur in the epithelium without stromal involvement. There is no associated conjunctival reaction or corneal neovascularization which helps to distinguish this condition from similar lesions from adenoviral infection or staphylococcal hypersensitivity. Discomfort is usually helped by a short course of topical steroids although it has been suggested that topical steroids may prolong the course of the disease.
CORNEAL THINNING AND MELTING DISORDERS
Corneal thinning results from uncontrolled lysis of the corneal stromal matrix. This is usually, but not always, associated with inflammation. The precise mechanism of stromal thinning is uncertain but it has been proposed that imbalance between the release of matrix metalloproteinases (MMPs) and their inhibitors (tissue inhibitors of metalloproteinases; TIMPs) may lead to lysis of collagen and the proteoglycan ground substance. Corneal thinning is also a feature of acne rosacea (see Ch. 4).
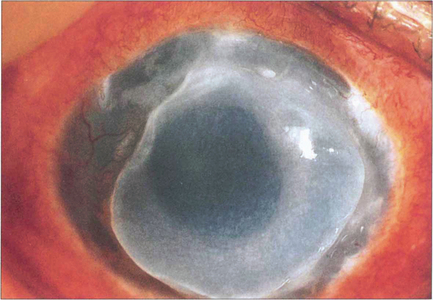
Fig. 6.62 Mooren’s ulcer is a progressive, painful, idiopathic ulceration of the peripheral corneal epithelium and stroma of uncertain aetiology. Recent studies suggest that autoimmunity against an antigen in corneal stroma is probably triggered by trauma in genetically susceptible individuals. In tropical regions a host–parasite interaction may incite autoimmunity. The disease may be unilateral or bilateral. Bilateral Mooren’s ulcer, irrespective of race or age of onset, is more aggressive; it is rapidly progressive and responds poorly to treatment. Its onset is heralded by pain, photophobia and blurred vision as a result of induced astigmatism and corneal oedema. A peripheral superficial stromal infiltrate progresses to epithelial ulceration that enlarges centrally and circumferentially with an undermined edge. Healing leaves an opaque, vascularized and thinned cornea but perforation, cataract and glaucoma may also occur. A variety of treatments has been used including topical and systemic immunosuppression or excision of the adjacent conjunctiva.
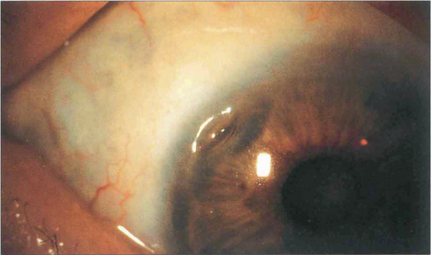
Fig. 6.63 Peripheral corneal ulceration is often associated with adjacent scleritis, most commonly due to seropositive rheumatoid arthritis but also other collagen vascular diseases such as systemic lupus erythematosus, polyarteritis nodosa, scleroderma, Wegener’s granulomatosis and relapsing polychondritis. Peripheral corneal melting with minimal overt inflammation may also occur and progress to perforation. The perforation can be sealed temporarily with tissue glue and covered with a soft bandage contact lens. Treatment of any associated systemic disease is essential. An oval tectonic graft can be performed electively (see also Ch. 5).
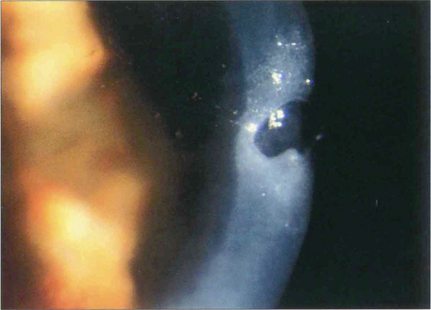
Fig. 6.64 Severe dry eye (keratoconjunctivitis sicca) is a feature of a minority of patients with rheumatoid arthritis. Such patients are often severely ill from their systemic disease. Epithelial breakdown may be followed by a central corneal melt (keratolysis) and descemetocele formation, with a risk of perforation. The possibility of coexisting infection must be excluded. The corneal disease is best managed medically if possible, using punctual occlusion, intensive lubrication and a soft bandage contact lens. Tissue glue may be used to close a small perforation. A lamellar corneal graft may be performed if required, once the epithelial surface has been stabilized.
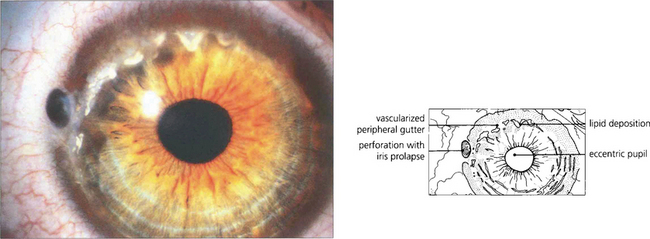
Fig. 6.65 Terrien’s marginal degeneration is seen in either sex and is usually bilateral, although often asymmetrical. It usually begins in the upper cornea as a faint peripheral stromal opacity with minimal superficial vascularization and slowly progresses to stromal thinning in an arcuate fashion. The thinned area, which may become ectatic, has a steeper central edge marked by lipid accumulation. Pain is unusual, although inflammatory episodes may occur. Vision may be affected by induced against the rule astigmatism from vertical flattening of the cornea in the affected area. Spontaneous perforation may occur, as in this patient, or may be induced by minor trauma. The aetiology is unknown.
CORNEAL CHANGES FROM METABOLIC DISORDERS AND MEDICATIONS
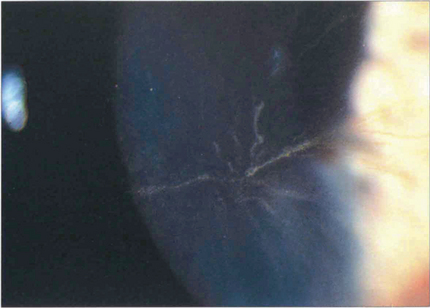
Fig. 6.66 Corneal verticillata are whorled epithelial deposits that delineate the pattern of epithelial cell migration. They are commonly seen in patients on treatment with amiodarone or chloroquine which are deposited in the epithelium. The deposits are asymptomatic and not an indication to stop therapy. Similar changes also occur in Fabry’s disease, an X-linked sphingolipidosis caused by a deficiency of the enzyme α-galactosidase associated with angiokeratomas in the skin.
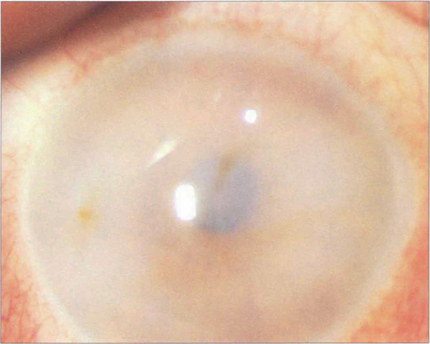
Fig. 6.67 The mucopolysaccharidoses are lysosomal storage disorders that are all recessively inherited (except Hunter’s syndrome, which is X-linked). A defect in degrading mucopolysaccharides (heparan, dermatan or keratan sulfate) leads to their accumulation and eventual excretion in urine. There are seven subtypes; corneal clouding is a feature of all except for the Hunter and Sanfilippo types. Pigmentary retinopathy and optic atrophy are other features. Corneal clouding is most marked in the Hurler–Sheie type. The example here is of Scheie syndrome in which the enzyme α-l-iduronidase is deficient (absence causes the more severe Hurler’s syndrome). Corneal clouding is usually present at birth and progresses slowly. Keratoplasty may be helpful if retinal and neurological function has been preserved but, unfortunately, the clouding tends to recur in the graft. Other features are claw hands with stiff joints and a characteristic facies, but the severe neurological and cardiological features of Hurler’s syndrome are absent. Corneal clouding reduces in some patients after successful bone marrow transplantation.
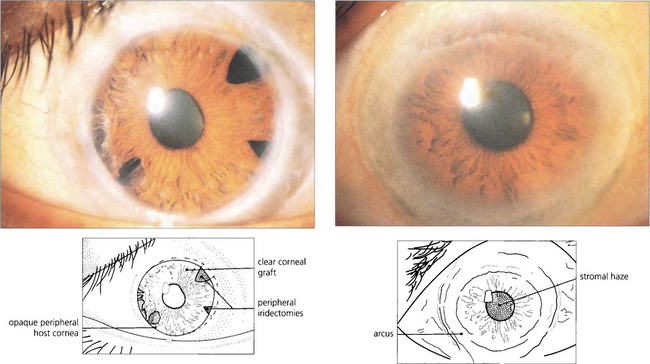
Fig. 6.68 Lecithin–cholesterol acyltransferase (LCAT) deficiency, Tangier disease and fish eye disease are autosomal recessive disorders of lipid metabolism (dyslipoproteinaemias). LCAT deficiency presents with early onset of a marked arcus and diffuse fine grey dots throughout the stroma that progress centrally from the periphery. Vision is usually preserved. This patient had acuity of 20/20 with the left eye; the right eye had been grafted 10 years previously and remained clear.
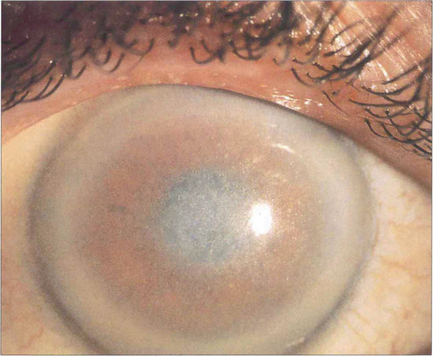
Fig. 6.69 Crystalline corneal deposits seen as grey or golden dots in the central cornea occur in cystinosis, oxalosis, gout and gold therapy for rheumatoid arthritis, as well as in Schnyder’s and Bietti’s dystrophies.
By courtesy of Mr M G Falcon.
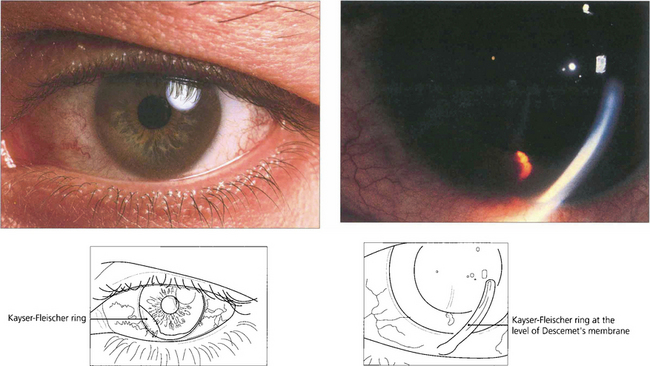
Fig. 6.70 Deposition of copper in Descemet’s membrane (Kayser–Fleischer ring) occurs in Wilson’s disease (hepatolenticular degeneration) from a deficiency of the transport protein caeruloplasmin in the blood with defective excretion of copper by the liver lysosomes. Patients develop liver failure and later neurological problems from deposition of copper in the basal ganglia. The Kayser–Fleischer ring is a peripheral orange, brown or green-brown discoloration at the level of Descemet’s membrane adjacent to the limbus. It is often better visualized with gonioscopy and is present in virtually all patients with neurological manifestations of Wilson’s disease, but may be absent in early cases.
By courtesy of Mr J Jagger.
REFRACTIVE SURGERY
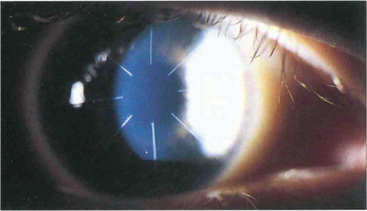
Fig. 6.72 Radial keratotomy relies on relaxing incisions that produce peripheral corneal ectasia to flatten the optical zone. Overall short-term results are good but there is a small risk of perforation or microbial keratitis following the procedure. Long-term complications include fluctuating myopia, haloes from incisions lying in the optical zone, a gradual hyperopic shift over several years, and increased risk of rupture from trauma from a weaker cornea. The procedure has generally been replaced by laser surgery.
By courtesy of Mr W Jory.
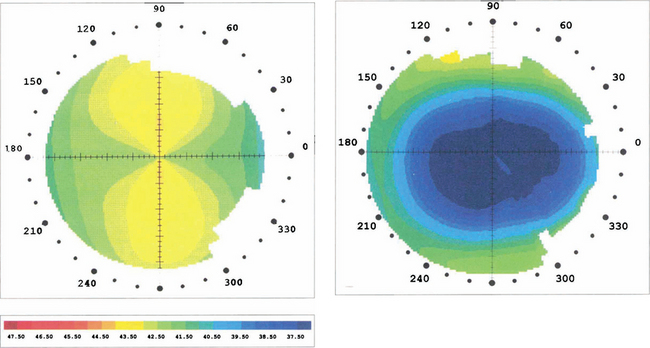
Fig. 6.73 Topography performed before and after laser refractive surgery for the correction of myopia and with the rule astigmatism shows that the radius of the anterior surface of the cornea has been flattened centrally and the astigmatism removed to leave a spherical anterior cornea surface.
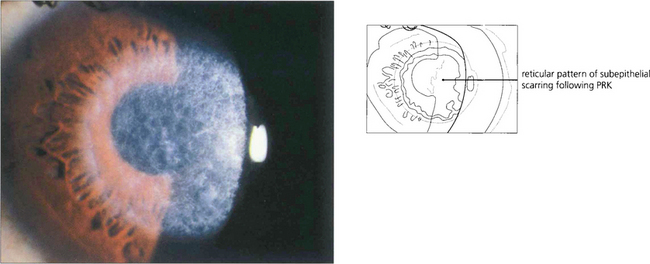
Fig. 6.74 Following photorefractive keratectomy (PRK) there is a risk of superficial stromal scarring that is seen as a central anterior stromal reticulated opacity on slit-lamp examination; this usually resolves over several months. Regression of myopia can occur following epithelial healing and regeneration. In general with PRK, the greater the refractive correction the greater the risk of stromal haze and the less predictable the refractive outcome.
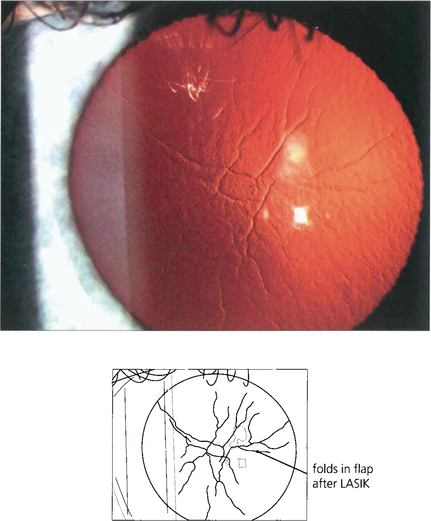
Fig. 6.75 Stromal scarring is not a feature after LASIK. Complications are due to incorrect positioning or calibration of the laser; flap-related problems (buttonholes, partial, free or irregular flaps) can result from defective microkeratomes or poor technique. Wrinkles or folds can sometimes be seen after the flap has been replaced; these may be visually significant.
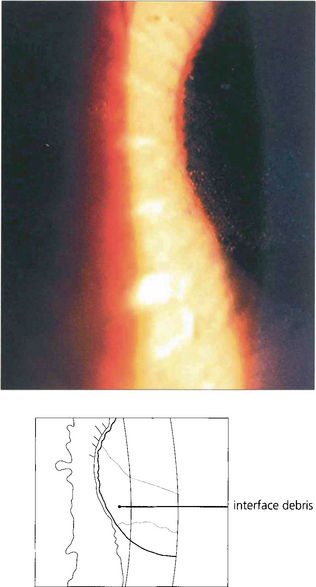
Fig. 6.76 A diffuse lamellar keratitis at the interface can be sometimes seen within days to weeks after treatment and is caused by debris from instruments or bacterial toxins. Treatment is with topical steroids; sometimes the flap requires lifting for irrigation and mechanical debridement.
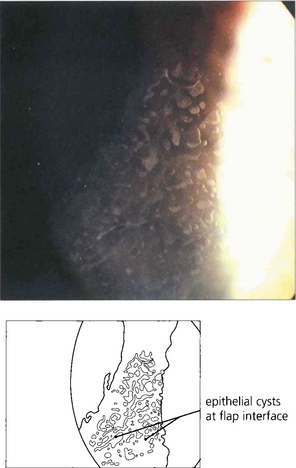
CORNEAL TRANSPLANTATION AND REJECTION
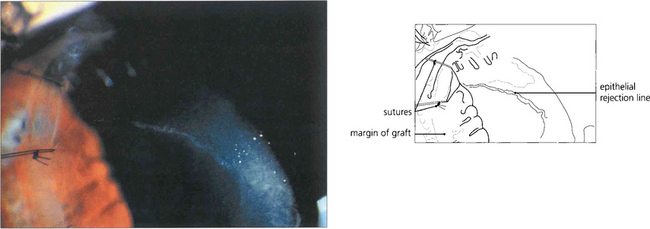
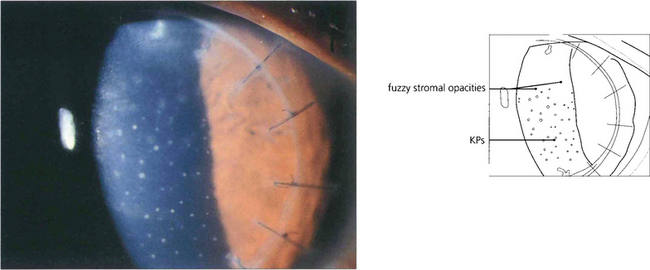
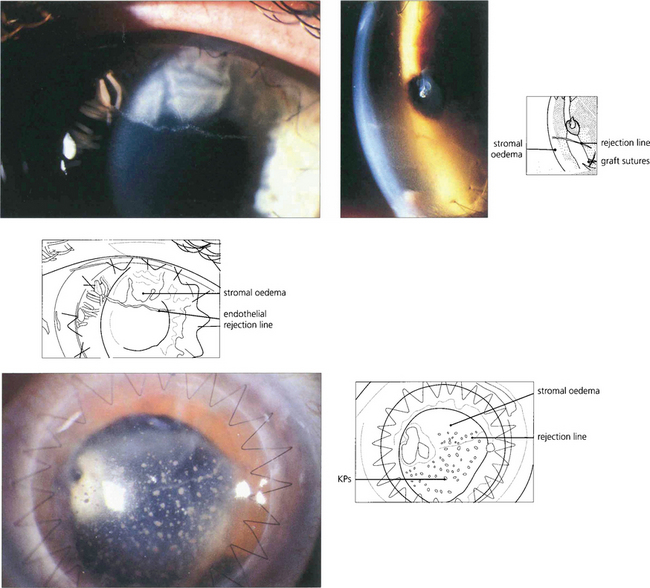
Fig. 6.79 Epithelial rejection appears as a line of raised epithelial and inflammatory cells that often originate from a focus of inflammation such as a loose suture. It is readily reversed with topical steroids and has no long-term effects.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
6 The Cornea
THE NORMAL CORNEA
The cornea has three primary functions. These are:
• the refraction of light—the main refractive interface being between air and the precorneal tear film
• transmission of light with a minimum of distortion, scatter and absorption
• the structural support and protection of the globe without compromising the optical performance.
Fig. 6.1 The cornea has five distinct histological layers. The epithelium supports the precorneal tear film and consists of a stratified squamous cellular layer attached to an underlying basement membrane. Bowman’s layer is an acellular condensation of superficial stroma approximately 10–20 μm thick that lies immediately beneath the epithelial basement membrane. The stroma forms over 90 per cent of the corneal thickness and consists of regularly spaced collagen lamellae (layers) in a proteoglycan matrix interspersed with keratocytes. Descemet’s membrane is composed of a lattice of collagen fibrils that is 3 μm thick at birth and increases in thickness with age. The endothelium is a monolayer of hexagonal cells.
By courtesy of Professor J Marshall.
Fig. 6.2 The precorneal tear film must be smooth and stable for regular refraction. The epithelial surface is thrown into multiple folds (microvilli and microplicae) which produce a glycocalyx (a branching mucoprotein layer) that renders the surface hydrophilic. The precorneal tear film is about 40 μm thick and is composed of mucus derived from the conjunctival epithelial cells, conjunctival goblet cells and the lacrimal glands. The aqueous component is secreted by the lacrimal glands and the superficial lipid layer from the meibomian glands. Atmospheric oxygen, metabolites and antimicrobial agents (e.g. IgA, lysosyme, lactoferrin) are dissolved in the tear film.
By courtesy of Professor J Marshall.
Fig. 6.3 The epithelium is about five cells thick and separated from Bowman’s layer by the epithelial basement membrane. The morphology of the basal cells changes as they migrate anteriorly to become intermediate wing cells and then elongated superficial cells with flattened nuclei that finally desquamate from the surface into the tear film. Macrophages (dendritic cells) are found in the epithelium having migrated from the limbus although they may be absent in the central zone. These have an antigen-presenting function. The epithelium has zonula occludens junctions between cells which make the healthy epithelium a virtually impermeable barrier.
By courtesy of Professor J Marshall.
Fig. 6.4 Cells from the corneal limbus continuously replace cells lost from the corneal epithelium. A population of stem cells is thought to be located in the basal epithelial cells of the folds of the palisades of Vogt. The stem cells divide to form ‘transient amplifying cells’ that move toward the centre of the cornea. Postmitotic daughter cells then migrate anteriorly from the basal layer to become terminally differentiated cells that are eventually lost from the anterior surface of the epithelium into the tear film.
Fig. 6.5 The mechanical strength of the cornea is provided by the stroma, which is formed predominantly of collagen fibrils (mainly type 1) maintained in a proteoglycan matrix. The stromal fibrils are continuous from limbus to limbus and are arranged into about 200 layers or ‘lamellae’ with a small degree of interdigitation. The superficial Bowman’s layer is acellular and the collagen fibrils are finer and more densely packed. Transmission of light depends on the collagen fibrils regular size and spacing with small changes of refractive index. A relative dehydration of the stromal proteoglycans is required which is achieved because the epithelium is impermeable and by the endothelial pump which removes water from the stroma. Light transmission is maximal at 700 nm (98 per cent) and decreases to 80 per cent at 400 nm. Ultraviolet light with a wavelength below 310 nm is strongly absorbed by the stroma. By contrast, the cornea transmits infrared radiation up to 2400 nm.
By courtesy of Professor K Meek.
Fig. 6.6 Using in vivo confocal microscopy high-resolution images can be obtained in real-time at different depths within the intact living cornea without the need for staining or processing, giving a direct view of living cells. Shown here is the cellular morphology of a normal human cornea. Each image represents an optical volume with approximately 450 × 340 × 9 μm. a, Superficial epithelial cells; b, wing epithelial cells; c, basal epithelial cells; d, subepithelial nerves (on Bowman’s layer); e, first layer of keratocyte nuclei; f, nerve branch and keratocytes in the mid-stroma; g, keratocytes in front of Descemet’s membrane; h, endothelial cells.
By courtesy of Dr T Moller-Pedersen.
Fig. 6.7 Descemet’s membrane, which consists of type IV collagen, is secreted by the endothelium. It is composed of a lattice of collagen fibrils that is 3 μm thick at birth (top), when the entire layer appears striated or ‘banded’. A posterior ‘nonbanded’ layer is continuously laid down throughout life so that Descemet’s membrane increases in thickness with age to reach 30–40 μm in the elderly (bottom). Notice the decrease in endothelial cell population.
By courtesy of Professor J Marshall.
Fig. 6.8 The endothelium can be examined by specular or confocal microscopy. The endothelium does not replicate after birth and cell counts reduce from 3500–4000/mm2 at birth to approximately 2000–2500/mm2 in the adult cornea. Corneal decompensation is likely with cell counts of less than 500/mm2. Cell density is a good guide to function, which can be complemented by other parameters such as variation in cell size and morphology (polymegathism and polymorphism). These specular photographs show the endothelium of an 18-month-old infant (top) and a normal 74-year-old man (middle). Notice the decreasing cell count and larger cell size with age. Larger cells with variation in morphology are seen in the endothelium of a patient after traumatic cataract surgery (bottom).
NORMAL CORNEAL TOPOGRAPHY
Fig. 6.9 With topography (see Ch. 1) a series of concentric rings is projected on to the surface of the corneal tear film. A difference in the relative distances between these rings compared to a calibrated spherical surface allows the corneal curvature to be measured from the visual axis to the periphery; this is then converted to dioptric power and displayed as a colour-coded topographical map in which colours towards the red end of the spectrum represent increasingly steep dioptric powers. A small range of dioptric powers can be seen on this normal cornea with flattening toward the periphery.
Fig. 6.10 Astigmatism is seen on topography as distortion of the circular projections into oval reflections. Topography of regular astigmatism appears as a ‘bow tie’ due to progressive peripheral flattening with axes at 90° to each other with mirror image symmetry between the two eyes. Irregular astigmatism, as in keratoconus, appears as steepening below the visual axis (see Fig. 6.33).
AGE-RELATED DEGENERATION
Involutional changes as a result of ageing must be distinguished from pathological changes.
Fig. 6.11 The white limbal girdle of Vogt is a common ageing change at the interpalpebral limbus which appears as a semilunar opacity with a clear zone of separation from the limbus; it is best seen by sclerotic scatter. The nasal cornea is affected nearly twice as often as the temporal side. There may be clear patches in the opacity, which may then resemble a mild form of band keratopathy. Histological examination shows subepithelial hyaline degeneration at the level of Bowman’s layer.
Fig. 6.12 Anterior crocodile shagreen is a pattern of polygonal opacities with intervening clear zones at the level of Bowman’s layer. It is best seen by wide-slit oblique illumination. Histological examination shows folding of Bowman’s layer. Although the superficial variant is an ageing change, a pre-Descemet’s form may be familial and related to the central cloudy corneal dystrophy (of François). Neither produces visual symptoms.
Fig. 6.13 Corneal arcus is due to the deposition of cholesterol and other lipids in the peripheral cornea, particularly adjacent to Bowman’s layer and Descemet’s membrane. It is a common ageing phenomenon and is almost universal by the eighth decade. A sharp clear zone lies between the limbus and the hazy inner border of the arcus. The condition is usually of no significance unless seen in patients under 40 years of age in whom it may be familial (arcus juvenilis); these patients require investigation for hyperlipidaemia. A lucent subepithelial zone with mild thinning may appear within the arcus in elderly people (senile furrow degeneration).
CONGENITAL CORNEAL ANOMALIES
Anterior segment dysgenesis produces a spectrum of anomalies. The current clinical classification does not reflect the underlying genetic defect. Both the Axenfeld–Rieger and Peter’s anomalies can be caused by abnormalities of at least four different genes. Posterior embryotoxon, the mildest expression, represents a centrally displaced Schwalbe ring. It is commonly a normal variant and not associated with glaucoma. The Axenfeld–Rieger syndrome (see Ch. 8) consists of posterior embryotoxon with anterior iris adhesions, corectopia and iris hypoplasia, probably due to arrest of neural crest development. Peter’s anomaly does not usually have posterior embryotoxon or peripheral anterior iris adhesions but there is a central iris to cornea adhesion with a defect of the endothelium and posterior stroma. The significance of the Axenfeld–Rieger syndrome and Peter’s anomaly lies in their association with childhood glaucoma, buphthalmos, corneal oedema and blindness. They may also be associated with systemic defects such as dental and cranial anomalies and malformations of the upper limbs and spine. Posterior keratoconus causes thinning of the posterior stroma with overlying haze and may be congenital; it has therefore been classified as a dysgenesis although many cases are thought to result from trauma. Congenital absence of the limbus is often associated with flattening of the cornea, as is seen in sclerocornea and cornea plana.
Fig. 6.14 Peter’s anomaly is characterized by a central corneal opacity with defects in the posterior stroma, Descemet’s membrane and endothelium and adhesions of the iris collarette to the posterior cornea, making it difficult to see the lens. There may be an anterior cataract or adhesions between the lens and cornea. Secondary glaucoma is common. Associated cardiac defects, cleft palate, craniofacial dysplasia and skeletal abnormalities may also occur (Peter’s-plus syndrome). The defects can be explained by abnormal separation of lens vesicle from surface ectoderm, and the heterogeneity by organs being affected that differentiate at same gestational age. The condition is usually bilateral with a sporadic incidence (80 per cent), although inherited cases occur.
By courtesy of Professor P Khaw.
Fig. 6.15 Sclerocornea is a congenital nonprogressive scleralization of the cornea. Either the peripheral cornea or the entire cornea may be involved; the limbus cannot be identified and the radius of curvature is flattened. There is overgrowth of conjunctival and episcleral vessels on to the cornea. Opacification of the cornea is due to a similar organization of the corneal stromal collagen fibrils to those of normal sclera. The condition is usually bilateral with an equal sex incidence and is usually sporadic, although dominantly and recessively inherited forms have been described. Cornea plana is an inherited abnormality most commonly described in Finnish populations; the cornea is flat with a radius of curvature similar to that of the sclera. The cornea is not opaque unless associated with sclerocornea. Extreme hyperopia, abnormalities of the angle and secondary glaucoma are common.
CORNEAL DYSTROPHIES
The corneal dystrophies are inherited corneal diseases that exhibit a remarkable degree of phenotypic and genetic heterogeneity (Table 6.1). Molecular biology has given new insights into the pathogenesis of these diseases. While at present they are classified clinically or histopathologically, their future classification is likely to be genotypic. It is evident, however, that the distinction between some dystrophies is not as clear as had been thought as apparently different clinical appearances can be caused by mutations in the same gene. Conversely, clinically similar dystrophies can result from different genes on separate chromosomes determining different protein products.
Table 6.1 Inheritance of corneal dystrophies
EPITHELIAL DYSTROPHIES
Fig. 6.16 The commonest corneal dystrophy is epithelial basement membrane dystrophy. This is bilateral, more frequent in women and its severity increases toward middle age. The clinical signs are variable and there may be grey subepithelial patches (top), dots or microcysts (middle), and concentric fine whorls of fingerprint lines best seen with retroillumination or broad-beam oblique illumination (bottom). The term Cogan’s dystrophy can be used if large grey dots are the prominent feature. Visual symptoms are uncommon but intensely painful recurrent erosions may occur following minor trauma. Symptoms are especially common on waking and can be relieved by using artificial tears during the day and lubricating ointment just before sleep. Severe cases may be helped by debridement of the epithelium, puncture of Bowman’s membrane with a needle or a bandage contact lens. Epithelial debridement followed by superficial excimer laser keratectomy can be useful in recurrent disease.
Fig. 6.17 Histological examination shows thickening and reduplication of the basement membrane and cyst formation.
Fig. 6.18 Meesman’s dystrophy is characterized by epithelial cysts spreading from limbus to limbus. These contain mucopolysaccharide.
Fig. 6.19 Type I Bowman’s membrane dystrophy (Reis–Bückler’s) predominantly affects Bowman’s layer and the superficial stroma (left). It is dominantly inherited and has been linked to the same region of chromosome 5q31 as granular, lattice, and Avellino dystrophies. Patients usually present during the first or second decade of life with painful recurrent erosions; subsequent scarring and surface irregularity blur vision. Disease is bilateral and symmetrical with grey reticular opacities seen mainly in the central cornea.(Right) Type II Bowman’s membrane dystrophy (Thiel–Benhke dystrophy), a histologically distinct subgroup with a honeycomb pattern, has been linked to chromosome 10q24. Excimer laser superficial keratectomy or lamellar keratectomy is the initial treatment of choice but the disease usually recurs with time. Penetrating keratoplasty is rarely indicated.
