Chapter 4
The Chemical Basis for Neuronal Communication
Fundamentals of Chemical Neurotransmission
Fast and Slow Synaptic Transmission
Information Flow Across Chemical Synapses
Synthesis, Storage, and Release of Chemical Messengers
Composition of Vesicle Membranes
Receptors and Receptor Subtypes
Regulation of Neuronal Excitability
Maintenance of the Synaptic Environment
Drug-Induced Parkinson Disease
Neurons in the human brain communicate primarily by the release of small quantities of chemical messengers, most of which are commonly called neurotransmitters. These chemicals alter the electrical activity of neurons after they interact with receptors on cell surfaces. Therapeutic alteration of brain function requires an understanding of the processes that regulate the synthesis and release of neurotransmitters and the means by which receptors alter neuronal electrical activity and biochemical function.
OVERVIEW
The brain contains approximately 100 billion (1011) neurons, each of which can make as many as 100,000 terminal contacts. Thus it has been estimated that the human brain contains approximately 1016 connections between neurons. Communication at most of these connections is mediated by chemical messengers. The transfer of information between neurons takes place at structurally and functionally specialized locations called synapses. Most synapses use chemical messengers that are released in discrete units (quanta) from presynaptic axonic or dendritic terminals in response to depolarization of the terminal.
Rapid diffusion of a chemical messenger across the synaptic cleft is followed by binding of this substance to receptors spanning the postsynaptic membrane. There is a resultant alteration in the electrical, biochemical, or genetic properties of that neuron. Less frequently, chemical messengers may also be released at sites without synaptic specializations. These messengers diffuse more widely than do neurotransmitters released at synaptic sites, and they influence receptors located at distant sites and on more than one neuron. Whether synaptic or nonsynaptic, chemical communication in the nervous system depends on (1) the nature of the presynaptically released chemical messenger, (2) the type of postsynaptic receptor to which it binds, and (3) the mechanism that couples receptors to effector systems in the target cell.
Western medicine is based fundamentally on modification of biologic function by drugs. Much of this alteration is focused on processes of chemical neurotransmission, whether in the central nervous system (CNS) or the periphery. The focus in this chapter is on basic elements of chemical neurotransmission. A model synapse, the noradrenergic synapse, is introduced, and a series of therapeutically important drugs is highlighted to illustrate potential medical targets in such a synapse.
FUNDAMENTALS OF CHEMICAL NEUROTRANSMISSION
Neurotransmitters
Specific criteria that define whether a chemical messenger can be identified as a neurotransmitter are listed in Table 4-1. Although a wide variety of putative neurotransmitters have been identified, these criteria have been met for only a few chemical substances. In general, these transmitters can be categorized as small-molecule messengers (having fewer than 10 carbon atoms) or larger neuropeptides (containing 10 or more carbon atoms).
Table 4-1 Criteria Necessary to Define a Substance as a Neurotransmitter

Small-molecule chemical messengers are classed as amino acids, biogenic amines, and nucleotides or nucleosides (Table 4-2). Amino acid neurotransmitters include γ-aminobutyric acid (GABA), glycine, aspartate, and glutamate. The vast majority of signaling within the nervous system is carried by amino acid neurotransmitters, specifically GABA and glutamate. For example, it has been estimated that roughly every fifth nerve cell and one of every six synaptic contacts uses GABA as a neurotransmitter. The biogenic amines include the familiar neurotransmitters acetylcholine, dopamine, norepinephrine, epinephrine, serotonin, and histamine. The nucleotide-nucleoside class includes adenosine and adenosine triphosphate (ATP). Nitric oxide, which functions as an endogenous nitrovasodilator in the cardiovascular system, has also been identified as a putative neurotransmitter.
Table 4-2 Substances Believed to Act as Chemical Messengers in the Central Nervous System
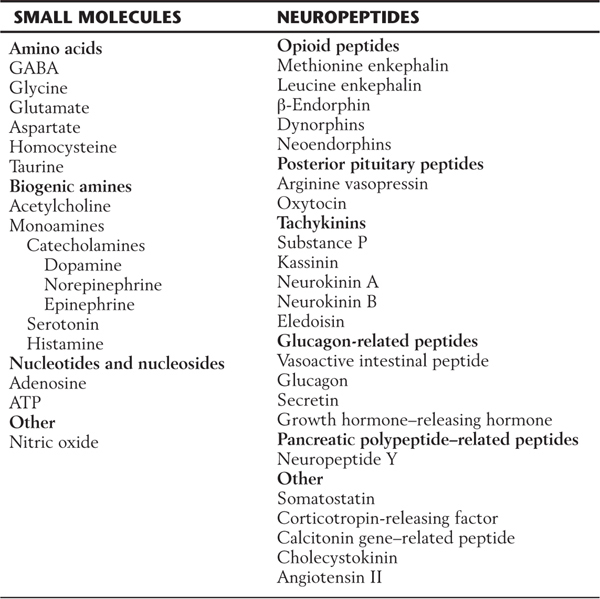
ATP, adenosine triphosphate; GABA, γ-aminobutyric acid.
More than 40 neuropeptides have been identified in brain tissue. These include methionine enkephalin (met-enkephalin) and leucine enkephalin (leu-enkephalin) as well as larger peptides, such as endorphins, calcitonin gene–related peptide (CGRP), arginine vasopressin, cholecystokinin, and many others (Table 4-2).
With a few exceptions, one being nitric oxide, the chemical messengers used by neurons are stored in secretory vesicles and released from them by exocytosis. In the case of neurotransmitters, these vesicles are found mainly in the presynaptic nerve terminals.
Fast and Slow Synaptic Transmission
The diffusion of a chemical message across the synaptic cleft can be quite rapid. At the neuromuscular junction, for example, it takes only about 50 microseconds for acetylcholine to reach the postsynaptic membrane. Total synaptic delay, the time from presynaptic release of neurotransmitter to the activation or inhibition of the postsynaptic neuron, is variable. This variability is influenced by the transduction mechanisms in the postsynaptic neuron.
Transduction mechanisms can be divided into fast and slow types. Fast chemical neurotransmission operates with a total synaptic delay of only a few milliseconds, whereas slow chemical neurotransmission usually requires hundreds of milliseconds. In both cases, the receptors on the postsynaptic membranes are glycoproteins that span the lipid bilayer membrane and transduce an extracellular chemical signal into a functional change in the target neuron. The difference relates to the complexity of the transduction mechanism.
In fast chemical neurotransmission, the postsynaptic receptor is itself an ion channel. This type of transmission is associated exclusively with small-molecule neurotransmitters. The binding of transmitter stimulates the channel to open, permitting a flux of ions across the membrane that alters the membrane potential. The process is fast because it is direct. Ion channels in this type of neurotransmission are called ligand-gated or receptor-gated ion channels; the ions normally involved are sodium, potassium, calcium, and chloride. Movement of these ions causes a change in the transmembrane electrical potential, which, if it exceeds threshold, may lead to generation of an action potential.
In slow chemical neurotransmission, the signal is transduced by a mechanism involving G protein–coupled receptors. These proteins and their action are discussed later in the chapter. Briefly, the binding of the transmitter (frequently a neuropeptide) causes the receptor to activate a G protein, which in turn binds to and influences an effector protein, which elicits the cellular effect. In some cases, the effector protein is an ion channel, which is induced to open or close. Transduction in these cases can be almost as rapid as in fast neurotransmission. More often, the effector is an enzyme that produces an intracellular second messenger, such as cyclic adenosine monophosphate (cAMP), whose cytoplasmic concentration is altered in response to the reception of a signal (binding of the transmitter) at the cell surface and that elicits intracellular responses to the signal. Second messengers can produce a plethora of cellular responses, ranging from the opening or closing of membrane ion channels to alterations in gene expression. These effects are mediated by complex sequences of chemical events, which is why they are relatively slow.
Information Flow Across Chemical Synapses
Transmission of information at a chemical synapse involves the following general sequence of events (Fig. 4-1): (1) secretory vesicle synthesis and transport to the synaptic terminal; (2) for small-molecule neurotransmitters, loading of the transmitter into the vesicle (for neuropeptides, this step accompanies vesicle synthesis); (3) depolarization of the presynaptic terminal; (4) vesicle docking with the presynaptic membrane, exocytosis of its contents, and trans-synaptic diffusion of the transmitter; (5) binding of transmitter to, and activation of, the postsynaptic receptor; (6) transduction of the signal, resulting in a postsynaptic response and one or two terminal steps; and (7) active reuptake of the transmitter by the presynaptic terminals or by glia or (8) enzymatic degradation of the transmitter in the synaptic cleft. These final events eliminate transmitter from the synaptic cleft and thereby terminate its action.
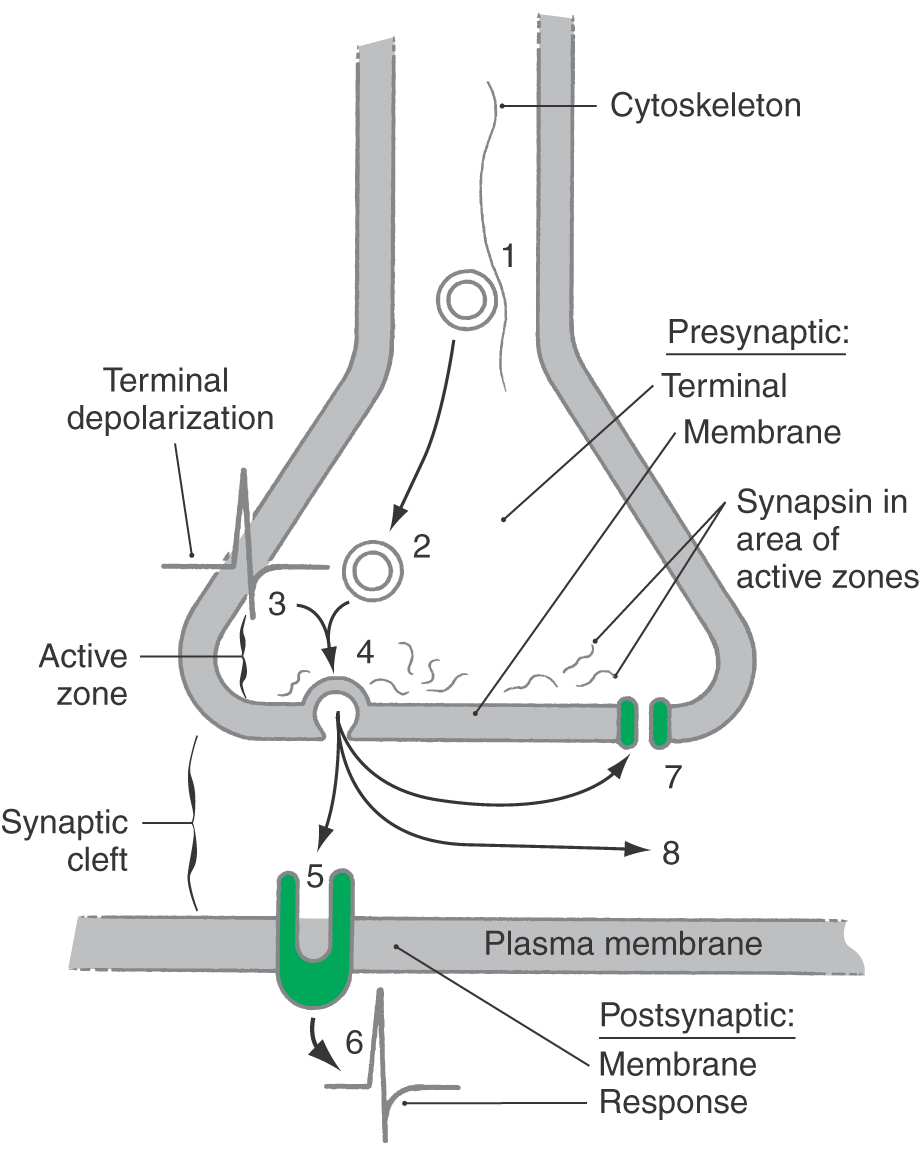
In many synapses, the amount of transmitter that a presynaptic terminal releases in response to an action potential can be regulated from outside the cell. Two regulatory mechanisms are (1) presynaptic receptor–mediated autoregulation and (2) retrograde transmission. In presynaptic receptor–mediated autoregulation, the neuron self-regulates the subsequent quantal release of its own chemical messenger. As a neurotransmitter enters the synaptic cleft, it stimulates not only postsynaptic receptors but also receptors located on the membranes of the terminal from which it was released. This constantly updates the presynaptic neuron concerning neurotransmitter synthesis and release and the efficiency of information transfer. In most cases, autoregulation is inhibitory. Loss or reduction of this input is interpreted as a reduction in signaling ability, and the presynaptic neuron increases the subsequent synthesis and release of stored neurotransmitter.
In retrograde transmission, the postsynaptic neuron responds to synaptic activation by releasing a second chemical messenger. This messenger diffuses back across the synapse and alters the function of the presynaptic terminal. Nitric oxide is currently the best example of a mediator of retrograde transmission.
SYNTHESIS, STORAGE, AND RELEASE OF CHEMICAL MESSENGERS
Neuronal chemical messengers are stored in two types of vesicles: small vesicles (also called synaptic vesicles) and large dense-cored vesicles. Synaptic small vesicles (~50 nm in diameter) appear clear and empty in electron micrographs and contain small-molecule chemical messengers such as GABA, glutamate, and acetylcholine. A subset of these small vesicles, with electron-dense cores, are found in both central and peripheral neurons. These vesicles contain the catecholamine family of biogenic amines (dopamine, norepinephrine, and epinephrine). Synaptic vesicles cluster near the exocytotic surface of a presynaptic nerve terminal in regions called active zones (Fig. 4-1).
Large dense-cored vesicles (~75 to 150 nm in diameter) are less numerous and appear in other intraneuronal locations as well as in the axon terminal. The electron-opaque, dense core is composed of soluble proteins that are mainly one or more neuropeptides. This core may also contain a small chemical messenger—often a biogenic amine, co-stored with a neuropeptide.
Neurons in certain hypothalamic nuclei contain a third type of vesicle called the neurosecretory vesicle. These vesicles are large (~150 to 200 nm in diameter), contain neurohormones, and are especially concentrated in axon terminals in the neurohypophysis (the posterior pituitary).
Composition of Vesicle Membranes
All vesicles are composed of a lipid bilayer membrane, spanned by a variety of proteins. Some proteins are common to both large dense-cored vesicles and synaptic vesicles, such as those that form calcium channels and the proteins synaptotagmin and SV2. Other proteins are found in high concentrations only in synaptic vesicles; these include synaptophysin and synaptobrevin. The differences in protein content reflect the different roles that large dense-cored vesicles and synaptic vesicles play in neurons.
Vesicles also contain proteins that act to accumulate small chemical messengers. These take the form of membrane pumps or transporters, most of which are coupled to the transport of protons. Synaptic vesicles contain at least four classes of proton-coupled transporters for chemical messengers, each specific for a different type of messenger. One class, the vesicular monamine transporter (VMAT), drives the accumulation of biogenic amines, including the catecholamines dopamine, norepinephrine, and epinephrine as well as the monoamine serotonin. Others are specific for acetylcholine, glutamate, and GABA or glycine. Large dense-cored vesicles can also accumulate small chemical messengers in addition to their neuropeptides. However, it is believed that the transporters involved are different from those used by synaptic vesicles.
Biosynthesis
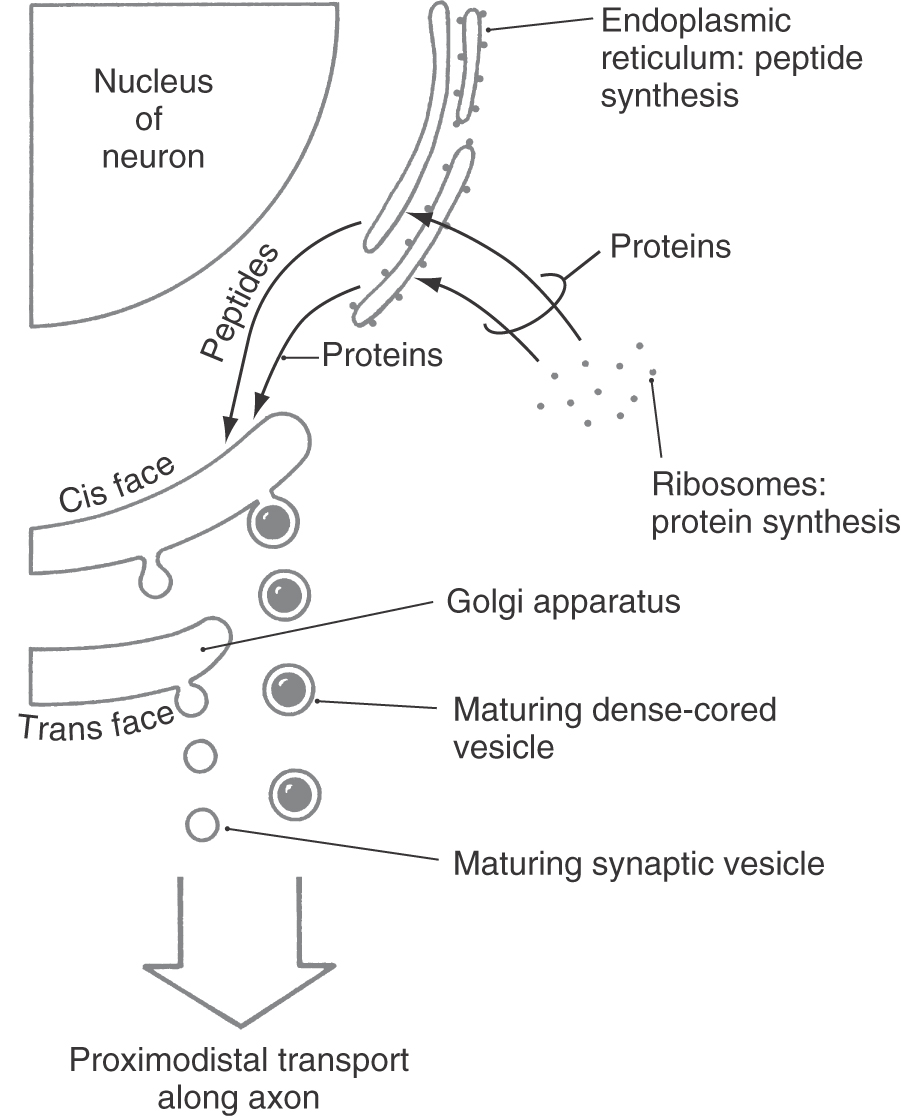
In terms of biosynthesis, an important difference between synaptic vesicles and large dense-cored vesicles is that the former can be recycled and refilled in the axon terminal, whereas the latter are both made and filled in the neuronal soma and are not recycled. This reflects the fact that small-molecule neurotransmitters can be synthesized in axon terminals, whereas neuropeptides, because they are synthesized on ribosomes and processed through the endoplasmic reticulum and Golgi complex, can be made only in the soma (Fig. 4-2). The cis face of the Golgi complex (also called the proximal or forming face) is prototypically concave toward the nucleus of the cell, whereas the trans face (distal or maturation face) is convex (Fig. 4-2). Peptides from the endoplasmic reticulum enter the cis face of the Golgi complex and are sorted and packaged into vesicles that bud from its trans face.
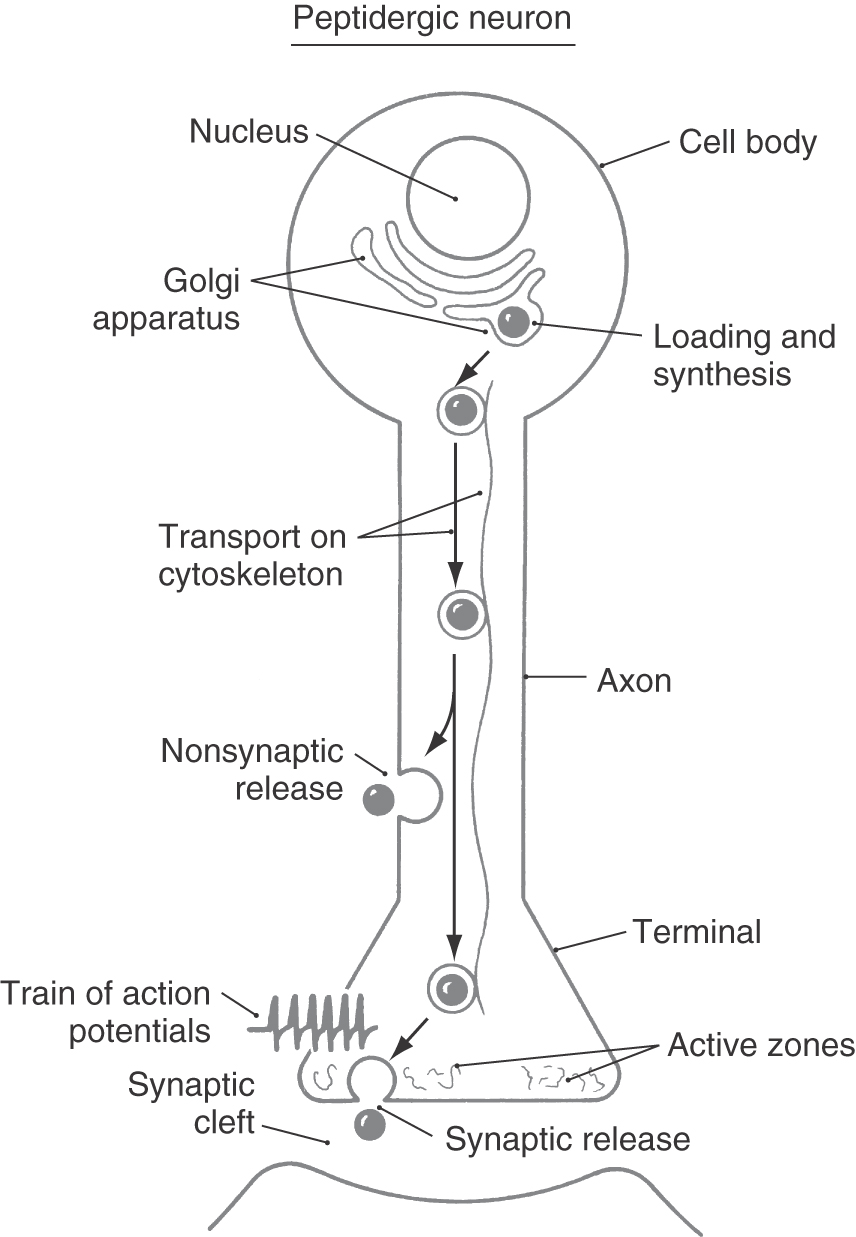
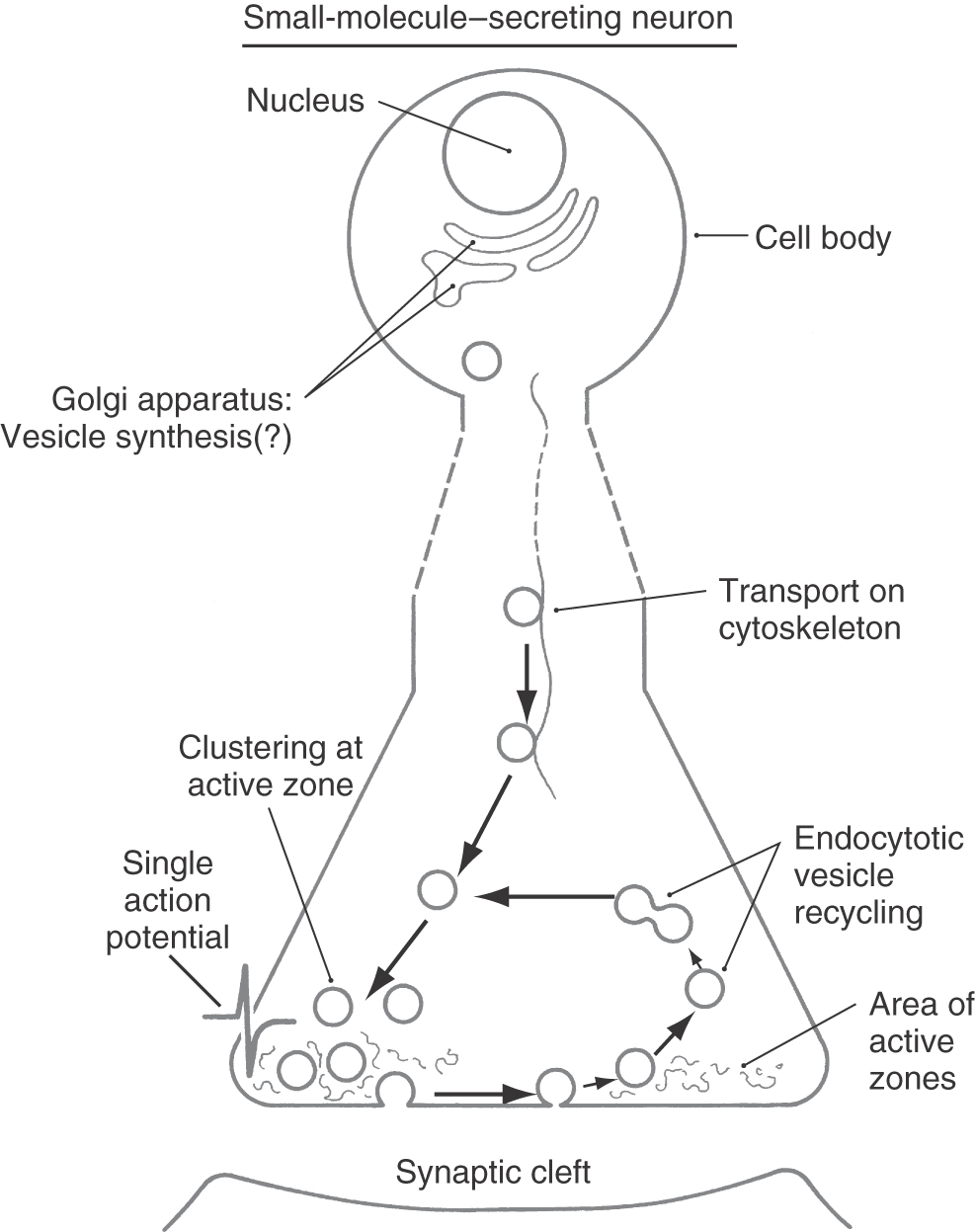
Large dense-cored vesicles contain neuropeptide messengers and are filled during the process of vesicle synthesis in the Golgi complex. These vesicles are translocated, by fast axonal transport (range, 4 to 17 mm/hr), from the cell body to axonal or dendritic release sites (Fig. 4-3). Frequently, neuropeptides are synthesized in the form of large precursor peptides that may be cleaved to yield more than one secreted bioactive neuropeptide. Maturation of neuropeptides can require covalent chemical modification of amino acid side chains, often with the addition of small chemical groups. Examples of the types of chemical modifications include the addition of methyl groups (methylation), sugar moieties (glycosylation), and sulfate groups (sulfation). This process of maturation can occur within the endoplasmic reticulum, during packaging of peptides into large dense-cored vesicles within the Golgi complex, or during axonal transport.
In general, synaptic vesicles are formed initially by budding from the Golgi apparatus within the cell body (Figs. 4-2 and 4-4). After transport to and release from the presynaptic terminal, however, the lipoprotein membrane components of the synaptic vesicles are recycled in a continuous process that occurs within nerve terminals (Fig. 4-4). Synthesis of the chemical messenger in a synaptic vesicle can occur while the vesicle is in the nerve terminal rather than in the cell body.
Some small-molecule neurotransmitters are synthesized in the cytosol of the axon and axon terminal and then transported into synaptic vesicles, whereas others are synthesized in the vesicle itself. The synthesis of acetylcholine is an example of the first of these mechanisms. The soluble enzyme choline acetyltransferase (CAT) catalyzes the acetylation of choline from acetyl coenzyme A (CoA) to yield the neurotransmitter acetylcholine. A high-affinity vesicular membrane transport protein concentrates this transmitter in cholinergic synaptic vesicles. Synthesis of the catecholamine norepinephrine is an example of the second mechanism. In the case of norepinephrine, synthesis occurs within the synaptic vesicle. The immediate precursor to norepinephrine, dopamine, is concentrated within the noradrenergic synaptic vesicle by a transporter specific for biogenic amines (the VMAT). Only then is dopamine converted to norepinephrine by the action of the enzyme dopamine β-hydroxylase, which is attached to the luminal border of the vesicular membrane.
Transporters for small chemical messengers concentrate compounds inside the vesicle to levels 10 to 1000 times higher than those found in the cytosol. The energy required for this transport is derived from an ATP-driven proton pump. The exchange of protons for the chemical messenger allows accumulation of the chemical messenger inside the vesicle.
Localization
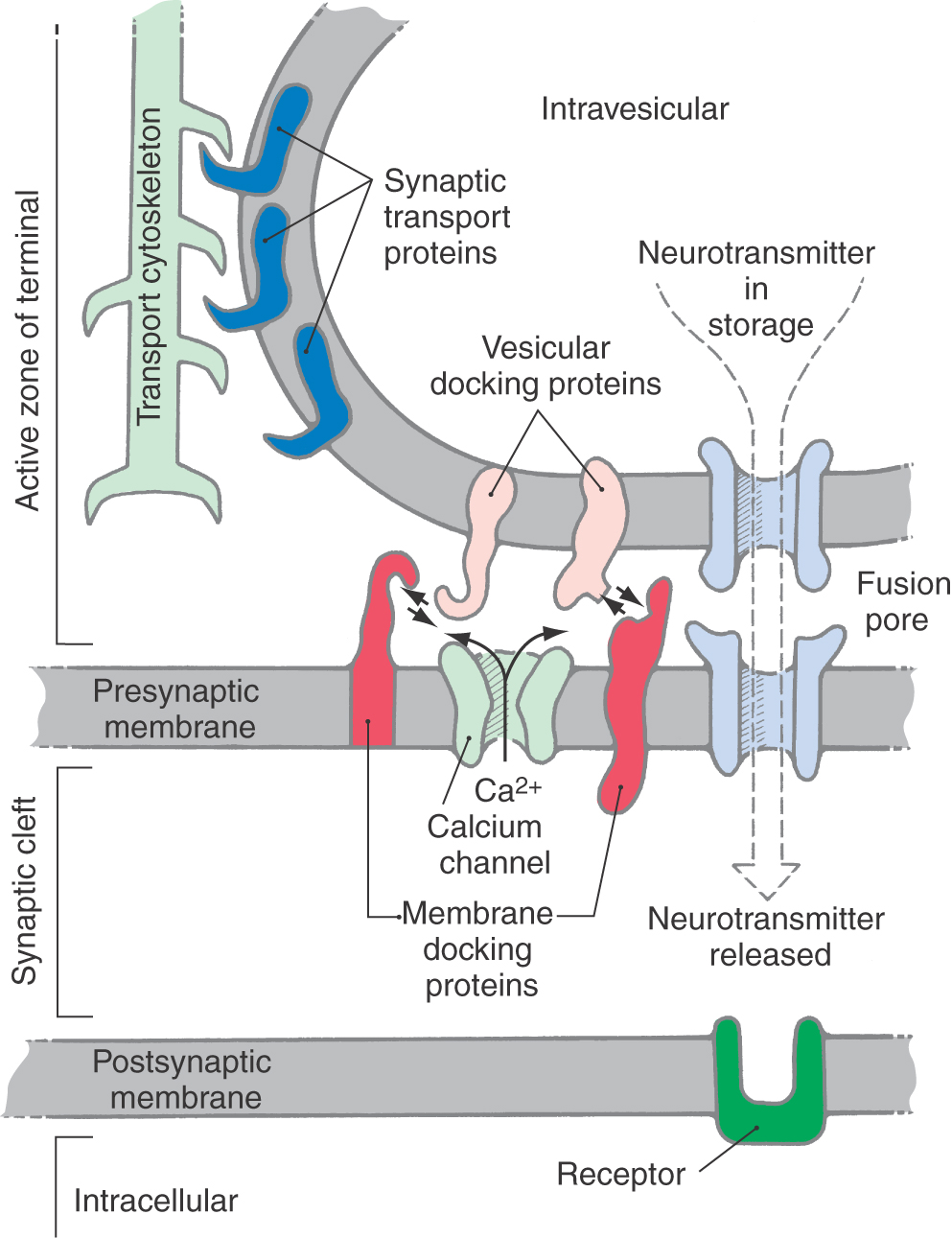
As mentioned earlier, synaptic vesicles are preferentially concentrated in active zones of the nerve terminal (Figs. 4-4 and 4-5). These zones are biochemically and anatomically specialized for neurotransmitter release. Large numbers of voltage-sensitive calcium channels are clustered in the plasma membrane of active zones. Consequently, depolarization of the axon terminal (or in special cases the dendrites) results in a high local concentration of calcium. This calcium causes synaptic vesicles to bind to the plasma membrane and stimulates exocytotic release of vesicle contents into the synaptic cleft. Active zones also contain high concentrations of the filamentous protein synapsin, which aids in the clustering of synaptic vesicles.
Although large dense-cored vesicles may accumulate in active zones, they also bind to the plasma membrane and release their contents from other sites in the terminal and axon that lack active zones (Fig. 4-3). As with synaptic vesicles, exocytosis depends on a local increase in calcium concentration. However, the release mechanisms for large dense-cored vesicles appear to be more sensitive to calcium than do those for synaptic vesicles. Therefore, sites of release do not require the high density of calcium channels found in active zones. Release sites outside active zones (such as those associated with large dense-cored vesicles) also do not have anchoring proteins such as synapsins.
Release
The essential structural elements critical for synaptic vesicle release are depicted in Figure 4-5. Proteins in the vesicle wall interact with cytoskeletal proteins to propel vesicles into the active zone. The surface of the synaptic vesicle contains two groups of proteins that are crucial for exocytotic release: docking proteins and elements of the fusion pore. A rise in intracellular calcium levels causes the vesicular docking proteins to interact with docking proteins on the cell membrane, creating a docking complex that brings the two membranes into apposition. Complementary proteins on both membranes then interact to form the fusion pore, and the lipid bilayers of the two membranes fuse at this site to form a rapidly expanding hole. Stored neurotransmitter in the vesicle begins to leak out through the fusion pore and exits in bulk (complete exocytosis) as the pore expands.
It is likely that large dense-cored vesicles use proteins and mechanisms for docking and exocytosis somewhat different from those used by synaptic vesicles. Also, whereas synaptic vesicles undergo exocytosis in response to single nerve impulses (Fig. 4-4), large dense-cored vesicles respond preferentially to high-frequency trains of impulses (Fig. 4-3). In experiments using peripheral nerves, a stimulation frequency of 10 Hz is often required to elicit neuropeptide release. Frequencies of that magnitude occur naturally in the autonomic nervous system under conditions of extreme behavioral or physiologic stress. Consequently, neuropeptides may play a role in stress responses.
SIGNAL TRANSDUCTION
Chemical messengers, once released from a presynaptic site, must interact with a postsynaptic neuron to transmit information. The postsynaptic membrane contains target molecules that exhibit an affinity for individual chemical messengers; these molecules are known as receptors. Most receptors are transmembrane glycoprotein chains. The binding of a messenger with its receptor precipitates a change in the architecture (conformation) of the glycoprotein chain that begins the process of information transfer. Some exceptions do exist. For example, there are intracellular receptors for testosterone. To be activated, drugs such as testosterone must first traverse the plasma membrane to gain access to the receptor.
Receptors and Receptor Subtypes
The receptor is capable of altering intracellular function in response to a change in the concentration of a specific chemical messenger in the environment. Thus, a receptor transduces a chemical signal (i.e., the concentration of a chemical messenger) into an intracellular event.
Receptors may be categorized by several means. One simplifying proposal identifies receptors into four general categories: (1) those termed ligand-gated channels (also called transmitter-gated channels), in which binding of a chemical messenger alters the probability of opening of transmembrane pores or channels; (2) those in which the receptor proteins are coupled to intracellular G proteins as transducing elements; (3) those consisting of single membrane–spanning protein units that have intrinsic enzyme activity (e.g., having tyrosine kinase activity); and (4) those termed ligand-dependent regulators of nuclear transcription (including receptors for corticosteroids such as testosterone).
On a more specific level, it is common for receptors to be grouped according to the type of native chemical messenger to which they respond. Thus all the receptors that respond to physiologically relevant concentrations of acetylcholine are called acetylcholine receptors (often termed cholinergic receptors). Similarly, adrenoceptors (often termed adrenergic receptors) respond to the catecholamine chemical messengers epinephrine (previously called adrenaline) and norepinephrine (noradrenaline). Traditionally (and functionally), receptors are identified by the response of a cell or tissue to a series of chemicals of different but closely allied molecular structures. Each compound in the series produces identical cell or tissue responses. However, each compound will exhibit a distinct potency (i.e., the concentration required to elicit the desired response) at each different receptor. The rank order of potency for a series of chemicals at each receptor then defines that unique receptor. It is common to rank potencies in terms of the concentration of an agent that produces 50% of the maximal biologic response in the test cell or tissue, or the effective concentration (EC50).
Agents that activate a receptor, whether they be native neurotransmitters or exogenous drugs, are termed receptor agonists. In contrast, receptor antagonists bind to a receptor but do not elicit any response. Rather, by preventing binding of an agonist to its receptor, the antagonist prevents any receptor-mediated signal from being produced. More recently, functional identification of a receptor has been complemented and amplified by molecular cloning techniques, which identify receptor similarities based on the primary amino acid sequence.
The receptors that respond to a given transmitter can often be divided into subtypes that elicit different biologic responses. For example, cholinergic receptors are divided into nicotinic and muscarinic subtypes. A cholinergic synapse with nicotinic receptors is commonly excitatory, whereas one with muscarinic receptors is commonly inhibitory. These receptor subtypes are named after plant compounds that stimulate them selectively and helped lead to their discovery. Nicotinic receptors are named after the nicotine of tobacco, and muscarinic receptors are named after muscarine, a substance found in the toxic mushroom Amanita muscaria.
The multiplicity of receptor subtypes can seem overwhelming at first. In the cholinergic system, both nicotinic and muscarinic receptors have subtypes of their own. For example, five different muscarinic receptor subtypes, termed M1 to M5, have been recognized. Adrenergic receptors (described in more detail elsewhere) are classified broadly into α-adrenoceptor and β-adrenoceptor subtypes, each of which is further delineated. Currently, six α-adrenoceptors (α1A, α1B, α1D, α2A, α2B, α1C) and four β-adrenoceptors (β1, β2, β3, and β4) are recognized. The serotonergic system is yet more complex, with 14 recognized receptor subtypes. Distinctions between subtypes are conferred by differences in coupling to intracellular second messenger systems, by changes in amino acid sequence of the receptor proteins, or by insertion of different protein subunits (in receptors in which the integral ion channel is oligomeric, i.e., constructed of multiple, distinct protein subunits). The GABAA receptor is a good example of this last type of modification. This receptor, like the nicotinic acetylcholine receptor, is a pentamer that forms a transmembrane ion channel. Molecular studies have identified 19 related GABAA receptor subunits in mammals. Regional differences in the subunit construction of the receptor, which are believed to confer subtle alterations in receptor function, are found in the brain and periphery.
Structure and Function
Transmembrane receptor proteins have a general structure that is based on glycoprotein chains that fold into the neural membrane in multiple loops (Fig. 4-6), although many of the receptors with intrinsic enzyme-associated activity consist of only a single transmembrane subunit. The protein is held in the membrane by several hydrophobic membrane-spanning segments (usually α helices but in specialized regions having a β-pleated sheet conformation), which are connected by loops that project into the aqueous environment on either side of the membrane. The N-terminal (NH2-terminal) and C-terminal (COOH-terminal) segments also project into the aqueous environment; they are typically relatively straight. In some transmembrane receptors, such as the β-adrenergic receptor, the N-terminal segment projects extracellularly and the C-terminal segment projects intracellularly (Fig. 4-6). In contrast, in voltage-gated ion channels, the N-terminal and C-terminal segments usually both project intracellularly. G protein–coupled receptors are formed from a single polypeptide chain, whereas most ligand-gated ion pores are multisubunit structures.
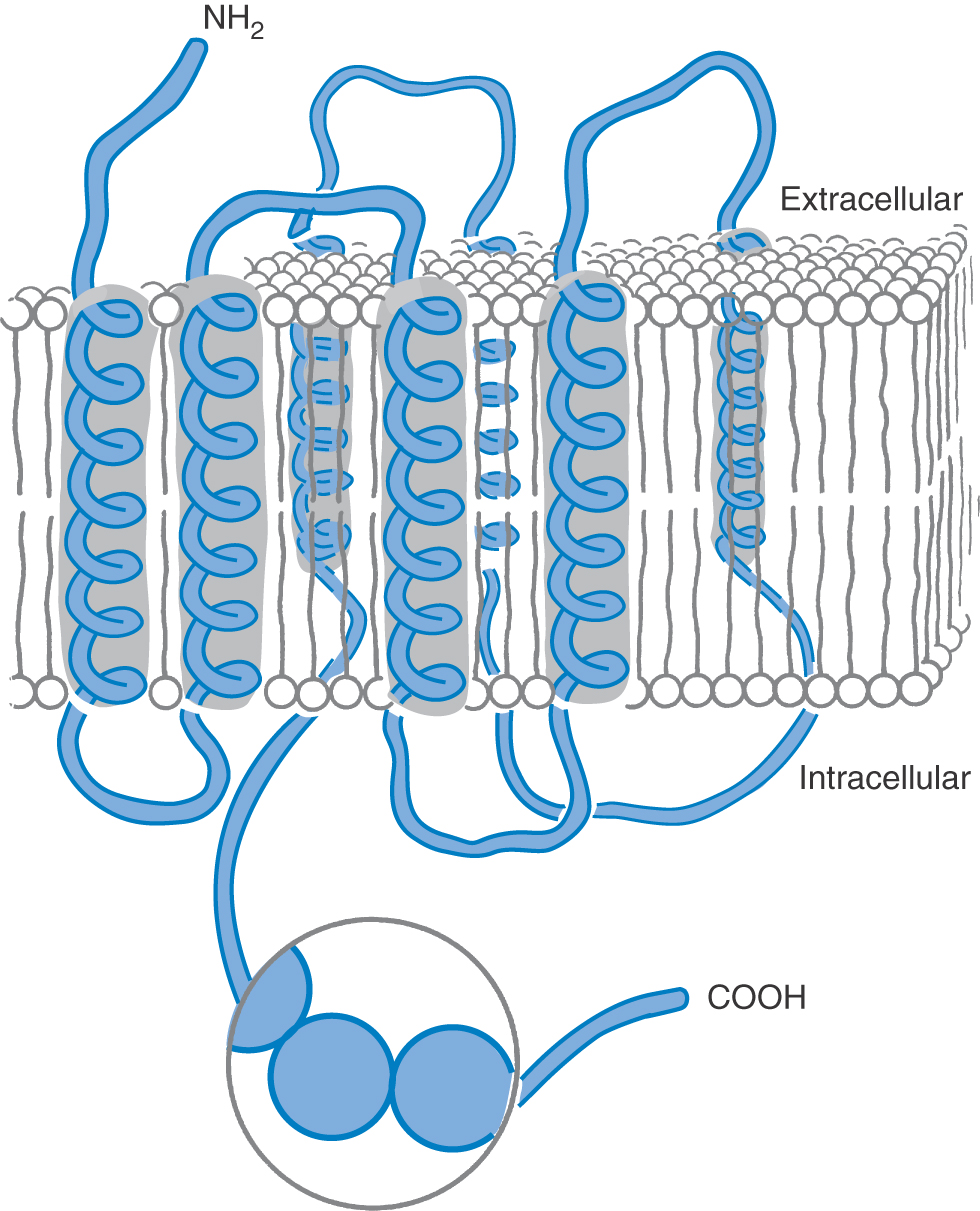
In G protein–coupled receptors, the transmembrane segments of the protein (usually seven in number) form a cluster that contains the binding site or sites for chemical messengers. The site is usually in a relatively hydrophobic pocket in the cluster, although it is sometimes on the extracellular surface of the protein. The receptor binds with its G protein transducer through multiple cationic sites on intracellular hydrophilic regions. The β-adrenergic receptors are the best characterized of the G protein–coupled receptors; their functioning is discussed later in the chapter.
Ligand-Gated Ion Channels
Ligand-gated ion channels are formed by several structurally distinct protein subunits called channel subunits (Fig. 4-7). Each channel subunit is a transmembrane glycoprotein (as described previously) with membrane-spanning segments connected by intracellular and extracellular loops. The channel subunits complex to form a roughly cylindrical structure that encloses a water-filled transmembrane channel. As exemplified by the nicotinic cholinergic receptor, the external face of the channel is enlarged and cup-like (Fig. 4-7). The channel narrows as it crosses the membrane, reducing the inner diameter such that it can selectively pass small cations (sodium, potassium, and calcium) or anions (chloride). The internal face of the channel widens again as it emerges from the lipid bilayer. The inner surface of the pore is blocked at rest by amino acid residues that project into the aqueous lumen of the pore and prevent the conductance of charged ions. This part of the channel is termed the gate. Binding sites, which most commonly occur at relatively hydrophobic regions within the transmembrane region of the channel, are specific for a chemical messenger. When a binding site is filled, conformational changes occur within the channel protein to open the gate and permit selective passage of ions across the membrane.
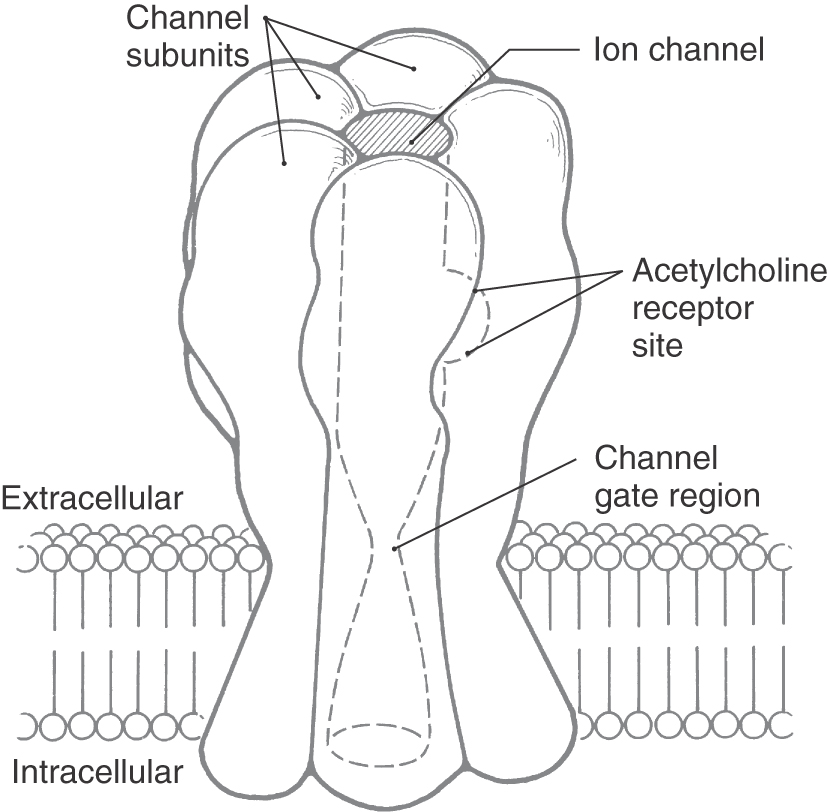
Two gene superfamilies of ligand-gated ion channels have been identified. One contains nicotinic cholinergic, serotonin (5-hydroxytryptamine), GABA, and glycine receptors; the other encodes the receptors for the excitatory neurotransmitter glutamate. The segregation of receptors into different superfamilies is based on the degree of homology of amino acid sequences. The subunits of the various receptors in a superfamily have 20% to 40% sequence homology with each other. The subunits of any given receptor generally have sequence homology of more than 40%.
G Protein–Coupled Receptors
The G protein receptors introduce another level of complexity to chemical transmission (Fig. 4-8). About three fourths of all chemical messengers transmit their information through G protein–coupled receptors. The basic elements of this system include a receptor, which must face the external surface of the membrane; the guanosine triphosphate (GTP)–binding protein, which consists of α, β, and γ subunits; and an effector protein, which may be an enzyme that alters the concentrations of intracellular second messengers (such as calcium, inositol 1,4,5-trisphosphate, diacylglycerol, or members of the eicosanoid family) or may be an ion channel (Fig. 4-8). The responses mediated by these receptors are generally slow (hundreds of milliseconds to minutes). The G protein–coupled receptor complex transduces an extremely wide range of chemical messages. About 100 different receptors have been identified that can link to a G protein, and at least 20 distinct G proteins have a similar number of effector proteins. The biogenic amines, bioactive peptides, eicosanoids, light (one of the first characterized G protein–coupled receptors was rhodopsin in the mammalian photoreceptor), and odorants all interact with G protein–coupled receptors.
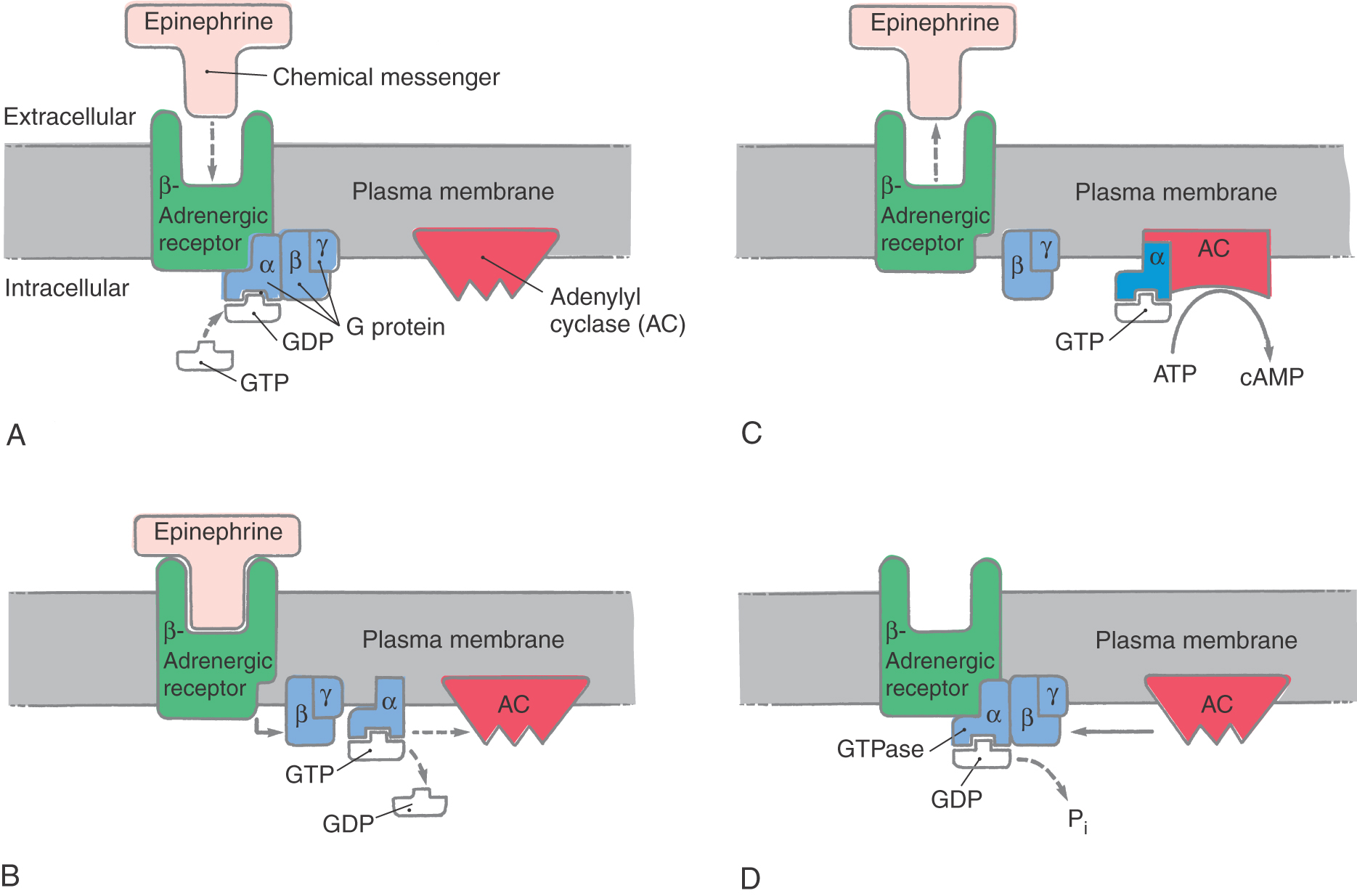
The G protein functions to amplify a signal received by a transmembrane receptor, transmitting that message to effector proteins within a neuron. Each G protein exists as a complex (a heterotrimer) formed by α, β, and γ subunits (Fig. 4-8). The αβγ heterotrimer maintains a loose association with the receptor glycoprotein but is not covalently bound to the receptor. Within the heterotrimer, the α subunit determines the nature of the G protein. It has the ability to bind GTP, to detach from the coupled βγ complex, and to alter the activity of an effector protein. The effector proteins whose activity can be modulated by α subunits are diverse and include the enzyme adenylyl cyclase as well as ion channels for calcium and potassium. In contrast, the βγ complex anchors α subunits to membrane sites and inhibits the GTP–guanosine diphosphate (GDP) exchange that activates the α subunit.
The resting form of a G protein exists as the heterotrimer, with GDP bound to the α subunit and the α subunit bound by the βγ complex (Fig. 4-8A). When it is activated, the α subunit exchanges GDP for GTP and dissociates from the βγ complex. The α subunit is then free to bind with and alter the activity of the effector protein; in this example, it is adenylyl cyclase (Fig. 4-8B, C). The α subunit has an integral slow GTPase activity, which eventually hydrolyzes the bound GTP to bound GDP (usually in 3 to 15 seconds). Reassociation of the α subunit–GDP complex with the βγ complex then completes the cycle (Fig. 4-8C, D).
The most completely characterized G protein–coupled receptor is the β2-adrenergic receptor. The endogenous ligand for this receptor is the catecholamine epinephrine. Epinephrine is a neurotransmitter that is confined to a very small number of cell groups within the brain and to chromaffin secretory cells in the adrenal gland. It is used clinically in cardiopulmonary resuscitation during cardiac asystole and to treat anaphylactic reactions. The β2-adrenergic receptor is coupled to a G protein that stimulates activity of adenylyl cyclase, catalyzing the formation of cAMP from intracellular ATP stores (Fig. 4-8). The stimulatory G protein associated with the β2-adrenergic receptor is called Gs.
Effector Proteins
As mentioned earlier, G proteins can interact with two main kinds of effector proteins: ion channels (called G protein–coupled ion channels) and enzymes that alter the level of intracellular second messenger compounds. The action of the G protein on its target may be positive or negative. That is, it may cause a channel to open or, more rarely, to close, or it may stimulate or inhibit a target enzyme. Most synaptic G protein responses are mediated through second messenger systems. Second messengers can elicit a variety of cellular responses, including the opening or closing of ion channels in the cell membrane (note that this indirect mechanism is in addition to the mechanism by which G proteins can interact directly with ion channels), the release or reuptake of calcium from intracellular storage sites, alterations in the activity of key cellular enzymes, and alterations in the expression of specific genes. Many of these effects are mediated by protein kinases, enzymes that regulate the activity of other proteins by phosphorylation. A given G protein–coupled receptor may activate more than one mechanism and produce multiple coordinated effects. G protein–coupled systems therefore have the capacity to mediate complex changes in neuronal function.
The best-known second messenger systems involve the enzymes adenylyl cyclase (Fig. 4-8) and guanylate cyclase. Adenylyl cyclase produces the second messenger cyclic guanosine monophosphate (cGMP) from cytoplasmic GTP. Another important second messenger system involves the enzyme phospholipase C, which hydrolyzes the membrane phospholipid phosphatidylinositol 4,5-bisphosphate to produce the second messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). G proteins can also interact with phospholipase A2, which stimulates the formation of members of the eicosanoid family, and with another enzyme called phospholipase D.
Receptor Regulation
The postsynaptic receptor is not static, either in the response to agonists or in the number of active receptors present on the membrane. The response of postsynaptic receptors to changes in the synaptic environment is a crucial element in neuronal communication. One of the most intensively studied postsynaptic receptor response systems is that of a G protein–coupled receptor, the β-adrenergic receptor—specifically, the β2-adrenergic receptor subtype. Continuous or repeated exposure of the β2-adrenergic receptor to an agonist will result in a diminution of the response of that agonist. The mechanisms by which this loss of response occurs are characterized by the time frame over which they occur. Exposure to an agonist for seconds to minutes will result in a reduction in the agonist-induced response through processes called desensitization. The loss of receptor responsiveness is mediated by agonist-induced changes in the receptor conformation that permit binding of additional intracellular proteins to the receptor. These proteins cause phosphorylation (i.e., addition of phosphate moieties) of the intracellular portions of the receptor. Phosphorylation changes the affinity of the receptors to other intracellular proteins that uncouple activated receptors from their effector proteins.
Homologous desensitization occurs when stimulation of the receptor by an agonist evokes the phosphorylation. Desensitization can also be produced without direct stimulation of the receptor in question, which is termed heterologous desensitization. In this case, intracellular phosphorylating enzymes are recruited by stimuli other than activation of the receptor. If the stimulus is maintained, the receptor protein may be subsequently sequestered into invaginations of the membrane that undergo internalization, effectively removing the receptor from the membrane surface. Once it is internalized, a receptor can be degraded or, in some circumstances, recycled back into the membrane as a fully sensitive, active receptor. Receptor downregulation, on the other hand, is caused by exposure to agonists for longer periods—hours to days—and is characterized by a reduction in the number of active receptors on the cell surface. Downregulation may be achieved by enhanced protein receptor degradation, decreased transcription of the messenger RNA (mRNA) for that receptor, or enhanced degradation of receptor mRNA.
REGULATION OF NEURONAL EXCITABILITY
As we have seen, neurotransmitters cause the opening or closing of ion channels in the postsynaptic membrane. If the transmitter signal is transduced by a G protein mechanism, it also may have other effects. The result will be a transient, local change in the polarization of the postsynaptic membrane, called a synaptic potential. This potential consists of either a depolarization or a hyperpolarization of the membrane relative to the resting potential. (Membrane potentials and the electrical properties of neuronal cell membranes are discussed in more detail in Chapter 3.) Synaptic potentials are graded in amplitude, reflecting the varying strengths of the incoming synaptic signals that elicit them. They generally do not exceed 20 mV. Local potentials of this type spread passively over the membrane of the postsynaptic cell, gradually losing amplitude and dying out. However, if they reach a trigger zone—a locus at which action potentials can be initiated—they may contribute to the production or suppression of action potentials. An action potential is triggered whenever the membrane is depolarized beyond a certain threshold potential. Therefore, depolarizing synaptic potentials tend to promote action potentials and are called excitatory postsynaptic potentials (EPSPs). Conversely, hyperpolarizing synaptic potentials inhibit the production of action potentials and are called inhibitory postsynaptic potentials (IPSPs).
In the CNS, a neuron is constantly bombarded by neurotransmitters, each of which can generate or modify a synaptic potential. Neurotransmitters that move the membrane toward depolarization (by reducing the −70 mV resting potential), with the resultant production of an action potential, are commonly called excitatory neurotransmitters. Neurotransmitters that move the membrane away from depolarization (by making the resting membrane more negative, the membrane is hyperpolarized) are frequently referred to as inhibitory neurotransmitters. Because the postsynaptic response is actually elicited by the receptor rather than by the transmitter, the postsynaptic receptor determines whether a given neurotransmitter will be excitatory or inhibitory. Some neurotransmitters can have either effect, depending on the type of postsynaptic receptor present.
Excitatory neurotransmitters act by promoting the opening of channels selective for cations (either sodium or calcium) that flux into the cell and depolarize the membrane. In some cases, as in that of certain glutamate receptor subtypes, the neurotransmitter binds directly to a stereospecific site on an associated ion channel. Glutamate receptor subtypes of this ilk are classified as ionotropic. Excitatory ionotropic glutamate receptors include the NMDA, AMPA, and kainate subtypes, each of which is named after a selective ligand for that subtype (NMDA, N-methyl-D-aspartate; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid). In other cases, opening of cation channels is accomplished indirectly, after alteration of an intracellular second messenger system or systems. Inhibitory neurotransmitters act by opening channels for potassium or chloride. Important examples of inhibitory neurotransmitters are the amino acids GABA and glycine.
A single chemical messenger can evoke either an EPSP or an IPSP, depending on the receptor to which it binds. A good example is the neurotransmitter norepinephrine. Like glutamate, norepinephrine binds to multiple receptor subtypes. In the CNS, receptors for norepinephrine fall into two categories: α-adrenergic and β-adrenergic receptors. Both types are G protein coupled. The G protein to which β-adrenergic receptors are coupled is of a type called Gs, which stimulates the activity of adenylyl cyclase and thus produces a rise in intracellular cAMP. This rise in cAMP leads to an EPSP. In contrast, the G protein to which the α2-adrenergic receptor subtype is coupled, called Gi, inhibits the activity of adenylyl cyclase. The resulting fall in intracellular cAMP leads to an IPSP. In both cases, cAMP acts through enzymes called cAMP-dependent protein kinases. In the pathway under discussion, the final targets are membrane ion channels, which open or close in response to the phosphorylation of sites on their cytoplasmic domains. Consequently, norepinephrine can elicit either an excitatory or inhibitory response, depending on the receptor.
MAINTENANCE OF THE SYNAPTIC ENVIRONMENT
The concentration of a chemical messenger in the synaptic cleft is crucial to information transfer. However, the time frame during which a chemical message is active must be limited if a temporally discrete signal is to be produced. This is particularly true when neurons fire at rates of more than several depolarizations per second. Simple diffusion out of the synaptic cleft is rarely adequate to effectively terminate the postsynaptic signal. Accordingly, active mechanisms exist to reduce or to eliminate chemical messengers in the synaptic cleft. The principal mechanisms are enzymatic degradation of transmitter in the cleft and transporter-mediated uptake across cell membranes.
Acetylcholine and the neuropeptides are examples of transmitters that are neutralized by enzymatic degradation in the cleft. Acetylcholine is cleaved by the enzyme acetylcholinesterase, which is synthesized by the neuron and inserted into the postsynaptic membrane near receptor sites. Neuropeptides are degraded through hydrolysis by the action of multiple peptidases, which are found in extracellular fluid.
The neurotransmitters whose action is terminated by uptake from the synaptic cleft include the monoamines (such as serotonin, histamine, and the catecholamines) and the amino acid neurotransmitters GABA, glycine, glutamate, and aspartate. This uptake is accomplished by the action of specific membrane-bound transport proteins. The monoamine class of biogenic amines (including the catecholamines, serotonin, and histamine) is avidly removed from the synaptic space by such transport proteins. Specificity for neurotransmitter uptake is provided by these transporters. Unique proteins have been identified that preferentially transport individual monoamines, the best identified of which are the norepinephrine (NET), dopamine (DAT), and serotonin (SERT) transporters.
In the case of norepinephrine, reuptake into the cytoplasm of the presynaptic terminal (a process known as uptake 1; subserved by the NET) is primarily responsible for terminating the action of the transmitter (Fig. 4-9). After reuptake, some norepinephrine is enzymatically degraded by the mitochondrial enzyme monoamine oxidase (MAO), whereas an additional fraction is retained in a cytoplasmic pool. The norepinephrine in this pool is an important target for drug action. Norepinephrine can also be removed through the action of a transporter on the postsynaptic membrane and in glia (uptake 2; subserved by the extraneuronal monoamine transport, EMT), although this process is usually less effective (Fig. 4-9). Norepinephrine transported into the postsynaptic neuron is degraded by the enzyme catechol-O-methyltransferase (COMT).
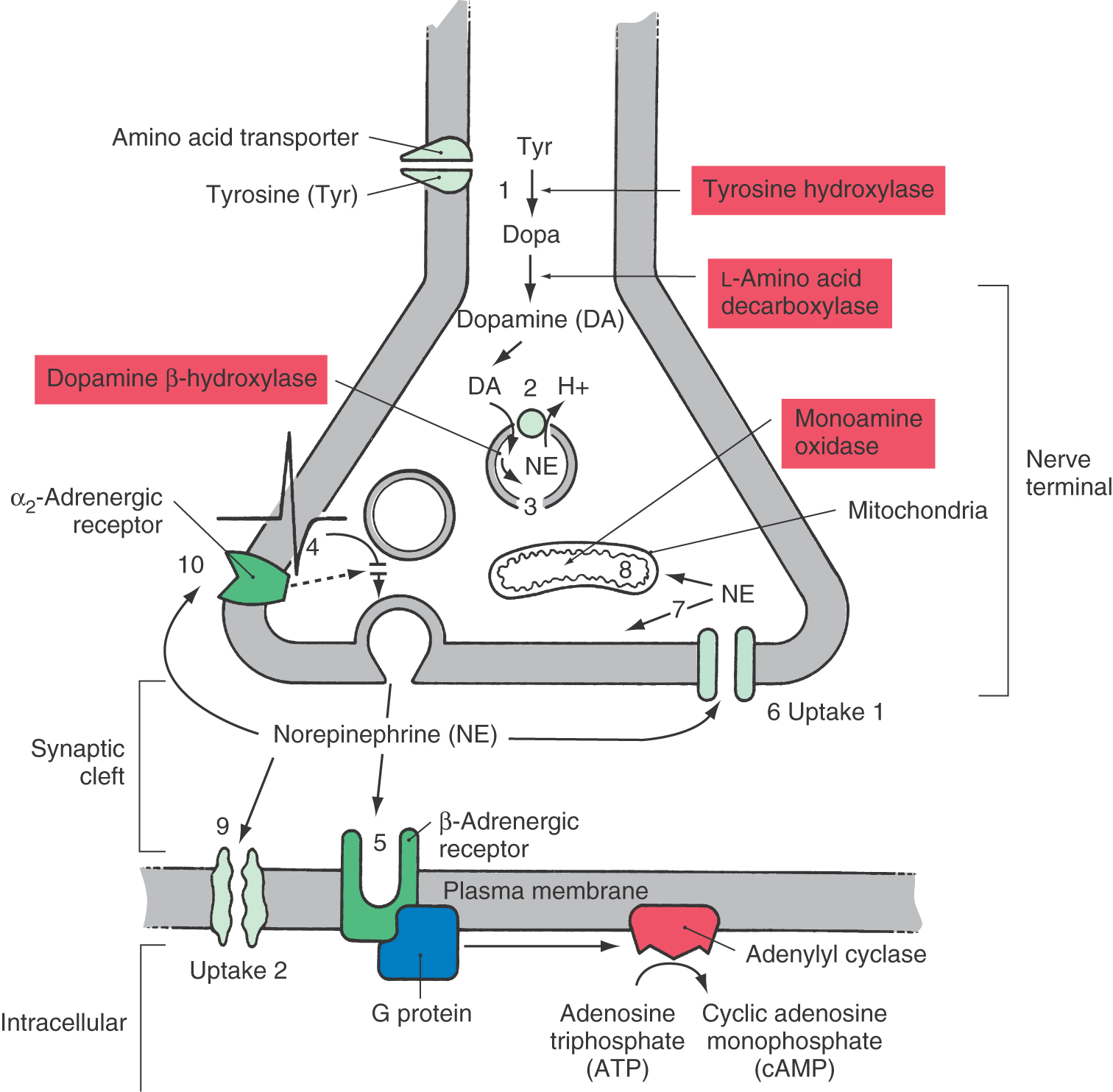
In the CNS, glial cells, primarily astrocytes, also express transporter proteins on their membranes and can remove transmitters from the synaptic cleft. The actions of the amino acids GABA, glycine, glutamate, and aspartate are terminated by active transport into neurons and glial cells. No active uptake mechanisms have been found that terminate the action of neuropeptides (see Chapter 2).
The mechanisms of termination of some other chemical messengers, such as adenosine, ATP, and nitric oxide, are less well understood. Nitric oxide is very labile; it undergoes redox reactions with membrane and cytoplasmic sulfhydryl moieties, reducing them and becoming oxidized itself. Specific ATPases may terminate the action of ATP functioning as a neurotransmitter.
DRUG-INDUCED PARKINSON DISEASE
A therapeutically relevant example of interaction with neurotransmitter transporters occurs in the substantia nigra, where the action of the neurotransmitter dopamine is terminated, in part, by uptake into glia by means of the EMT (uptake 2). Glia normally metabolize dopamine to inactive products by means of an MAO isoform, MAO-B, that is confined largely to the CNS. However, in the early 1980s in California, several cases of idiopathic paralysis that resembled severe Parkinson disease were noted in young persons. Through insightful clinical investigation, a young neurologist, W. Langston, recognized a connection between the clinical symptoms and the botched synthesis of an illicit analogue of meperidine (a potent opioid analgesic legally supplied under the trade name Demerol) that resulted in the formation of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). Subsequent study determined that MPTP is transported into glia and metabolized by MAO-B into a related neurotoxic compound, MPP+ (1-methyl-4-phenylpyridinium), that is released from glia and transported into nigral dopaminergic neurons by the DAT (uptake 1), selectively damaging them and producing a characteristic triphasic clinical syndrome that closely resembles Parkinson disease. The clinical hallmarks of this syndrome begin with intravenous injection of a contaminated meperidine analogue, which characteristically produces a burning sensation and a “high” that is more “spacey and giddy” than that usually experienced with heroin injection. Within 2 to 3 days, bradykinesia and rigidity of movement and awkward posture leading to inability to move begin to develop. These early responses may develop into a permanent parkinsonian syndrome with bradykinesia, rigidity, resting tremor, fixed stare, and loss of postural reflexes. The analysis of this tragedy has led to the use of MPTP as an extraordinarily helpful model of Parkinson disease.
PHARMACOLOGIC MODIFICATION OF SYNAPTIC TRANSMISSION
Drugs can alter virtually every level of neuronal and synaptic function. Therapeutic effects are most commonly achieved by actions of drugs on neurotransmitter synthesis, vesicular uptake and storage, depolarization-induced exocytosis, neurotransmitter-receptor binding, and termination of neurotransmitter action. Increasingly, drugs are being developed that modify neurotransmitter action through interaction with postsynaptic effector systems.
Noradrenergic Synapse
The noradrenergic synapse is used to illustrate the range of pharmacologic agents that can modify synaptic transmission (Fig. 4-9). Noradrenergic synapses use the neurotransmitter norepinephrine, and their postsynaptic receptors fall into the two classes introduced earlier, the α-adrenergic and β-adrenergic receptors. In the peripheral nervous system, norepinephrine is a critical neurotransmitter in the regulation of the sympathetic division of the autonomic nervous system. In the CNS, norepinephrine is synthesized in neurons concentrated in several discrete brainstem regions; these areas send noradrenergic axons throughout the brain and exert widespread effects. The greatest single source of norepinephrine in the mammalian CNS is the locus ceruleus, a paired structure in the floor of the pontine fourth ventricle. In humans, cerulear noradrenergic neurons number only about 12,500/side yet influence brain function from the spinal cord to the cerebral cortex.
Norepinephrine is synthesized from the amino acid tyrosine in a sequence of three enzymatic reactions (Fig. 4-9). The first two occur in the cytoplasm; the final reaction takes place within the synaptic vesicle. Tyrosine is accumulated in the terminal by a membrane-bound amino acid carrier and is converted to dopa by tyrosine hydroxylase (step 1 in Fig. 4-9). The rate of this conversion makes it the rate-limiting step in norepinephrine synthesis. Regulation of tyrosine hydroxylase is accomplished by phosphorylation of the enzyme by intracellular protein kinases, which increases the rate of enzymatic catalysis. The drug α-methyltyrosine can limit noradrenergic function by acting as a competitive inhibitor of tyrosine hydroxylase, thereby reducing the rate of norepinephrine synthesis. This drug, also called metyrosine (Demser), is used clinically in management of patients who have symptoms due to the excess production of catecholamines arising from tumors of adrenal chromaffin cell tissue, or pheochromocytomas.
Dopa is converted to dopamine (a neurotransmitter in its own right) by L-amino acid decarboxylase. Dopamine is transported into and concentrated within the synaptic vesicle by a monoamine-hydrogen transporter (the VMAT; step 2 in Fig. 4-9). The accumulation of dopamine (and ultimately norepinephrine) can be prevented by reserpine, a plant alkaloid that irreversibly inactivates the vesicular transporter (step 2 in Fig. 4-9). The inability to fill vesicles with neurotransmitter results in a progressive reduction in the level of transmitter in the axon terminal, which inhibits neurotransmission.
Reserpine, one of the earliest therapeutic agents available for the treatment of hypertensive cardiovascular disease, markedly reduces the ability of cardiac noradrenergic neurons to stimulate the heart to increase rate and contractility, thus lowering cardiac output. In addition, noradrenergic neurons that innervate arteriolar smooth muscle produce less norepinephrine, which results in less vasoconstriction and an overall reduction in blood pressure. Reserpine can precipitate parkinsonian symptoms that result from depletion of dopamine in nigrostriatal terminals as well as increased neurohypophyseal release of prolactin leading, to galactorrhea by deletion of tuberoinfundibular dopamine. Reserpine may also worsen clinical depression, a finding that contributed to the monoamine theory of depression. Today, reserpine is only rarely used to treat hypertension.
Within the vesicle, dopamine is converted to norepinephrine by dopamine β-hydroxylase. The accumulation of both dopamine and norepinephrine can also be reduced by administration of α-methyldopa (Aldomet). This dopa analogue is enzymatically converted in successive steps to α-methyldopamine and α-methylnorepinephrine, which take the place of the normal synthetic products, resulting in reduction of noradrenergic transmission (Fig. 4-9). Elevated sympathetic nerve activity contributes to hypertensive cardiovascular disease. Clinically, α-methyldopa is an effective antihypertensive drug and is one of the most commonly used drugs for management of hypertension during pregnancy.
The drug guanethidine interferes with the coupling between excitation of the nerve terminal and exocytotic release of norepinephrine, thereby reducing the amount of norepinephrine released. In addition, guanethidine acts like reserpine to inactivate vesicular transport. Unfortunately, guanethidine causes such a profound inhibition of noradrenergic neuron function that its use is associated with undesirable and unpleasant adverse effects, including excessively reduced heart rate, nasal congestion, and orthostatic hypotension (a decline in blood pressure that occurs on standing erect because of the effects of gravity, causing pooling of blood in the lower extremities) (see Chapter 29).
Once it is released into the synaptic cleft, norepinephrine can bind to two sets of receptors: (1) postsynaptic α-adrenergic or β-adrenergic receptors, which elicit the postsynaptic response; and (2) presynaptic receptors, partially but not exclusively of the α2-adrenergic subtype, which are involved in autoregulation. Eventually, the norepinephrine is removed from the synapse by reuptake into the presynaptic terminal or uptake into the postsynaptic cell. Propranolol (Inderal and others) is an example of a drug that interferes with the binding of norepinephrine to postsynaptic β-adrenergic receptors. This drug binds competitively to the receptor and prevents its activation, thereby blocking the postsynaptic response (in this case, the rise in intracellular cAMP mediated by Gs activation of adenylyl cyclase). Propranolol and related β-adrenergic receptor antagonists are extremely effective and widely used in cardiovascular medicine. Indications include but are not limited to management of hypertension, angina pectoris (chest pain due to cardiac ischemia), congestive heart failure, and myocardial infarction.
The autoregulatory presynaptic receptors for norepinephrine exert an inhibitory effect on the amount of norepinephrine released in response to an action potential. They influence both the synthesis of norepinephrine and its exocytotic release. The α2-adrenergic receptors that are responsible for these effects can be blocked by the drug yohimbine. The resulting loss of autoinhibition increases the amount of norepinephrine released and enhances noradrenergic function.
Blockade of α2-adrenergic receptors and reduction in norepinephrine release reduce postsynaptic effects of norepinephrine. In many peripheral tissues, the actions of the sympathetic (noradrenergic) component of the autonomic nervous system normally are finely balanced by those of the parasympathetic component (which, in most cases, releases acetylcholine) (see Chapter 29). Decline in the function of one component frequently leads to overexpression of the function of its opposing system. This is particularly true in the male urogenital tract, where erection has been linked both to activation of cholinergic nerves and to blockade of α2-adrenergic receptors. Yohimbine (Yocon, Aphrodyne, and others) is chemically similar to reserpine and obtained from botanical sources. Present in many herbal preparations and beverages, yohimbine has a lengthy folk history as an aphrodisiac. Therapeutic use has, in recent years, proved modestly effective in promoting erectile function in male patients with impotence of vascular or diabetic origin or of psychogenic origin.
Many psychomotor stimulant drugs, including cocaine and amphetamine, enhance motor performance, relieve fatigue, and exert positive reinforcing effects by enhancing synaptic concentrations of monoamines. Cocaine does so by reversible inhibition of membrane monoamine transporters. The order of potency for inhibition of NET, SERT, and DAT is SERT > DAT > NET. The action of amphetamine is more diverse. As a substrate for the monoamine transporters, amphetamine is said to “reverse” the normal process of membrane transport. Thus amphetamine promotes movement of cytoplasmic monoamines into the synapse. Amphetamine also enhances release of vesicular monoamine stores into the cytoplasmic pool and reduces intracellular degradation by inhibiting MAO. Monoamine oxidase has two isoforms, MAO-A and MAO-B. MAO-A is found in the periphery primarily and preferentially degrades norepinephrine and serotonin; MAO-B is largely confined to the brain. Both MAO-A and MAO-B have similar affinities for dopamine. A variety of MAO inhibitors are used clinically. Nonselective inhibitors that block both MAO-A and MAO-B, such as tranylcypromine (Parnate) and phenelzine (Nardil and others), have utility in management of major depressive disease but have frequent side effects. A selective inhibitor of MAO-B, selegiline (L-deprenyl; Eldepryl), preferentially blocks mitochondrial degradation of dopamine in the CNS and is useful in management of Parkinson disease. Agents such as tyramine displace norepinephrine from the cytoplasmic pool back into the synapse, also enhancing noradrenergic activity.
Sources and Additional Reading
Ballard PA, Tetrud JW, Langston JW. Permanent human parkinsonism due to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): Seven cases. Neurology. 1985;35:949–956.
Cowan WM, Sudhof TC, Stevens CF. Synapses. Baltimore: John Hopkins University Press; 2001.
Katzung BG. Basic and Clinical Pharmacology. New York: McGraw-Hill; 2004.
Kelly RB. Storage and release of neurotransmitters. Cell. 1993;72 (Suppl):43–53.