7 The Biologic Pacemaker
In October 1958, the first fully internalized pacemaker was implanted at the Karolinska Institute in Sweden. Within hours, the unit ceased to function, or as Senning wrote in a retrospective account, “At 2 am the pacemaker became silent.”1 In a Medline search with the keywords “implantable pacemaker,” the first five citations (starting in 1960) are case reports of various implants, and the sixth (1962) is entitled “Complications of an implantable cardiac pacemaker.”2 Since that initial implant, millions of pacemakers have been implanted, saving lives and reducing or even eliminating symptoms. Along with these benefits of electronic pacemakers, however, complications occur in 5% to 10% of patients undergoing implant procedures.3,4 Acute complications range from minor hematomas to more severe infections, pneumothoraces, and cardiac perforations. Long-term management of pacemaker patients is complicated by device and lead failures and a requirement to change out the device when the battery fails. These limitations of device therapy have motivated the search for alternatives. One widely publicized approach over the last few years is to create a “biologic pacemaker” by either gene transfer or cell transplantation methods (Table 7-1). This chapter reviews the literature on this approach.
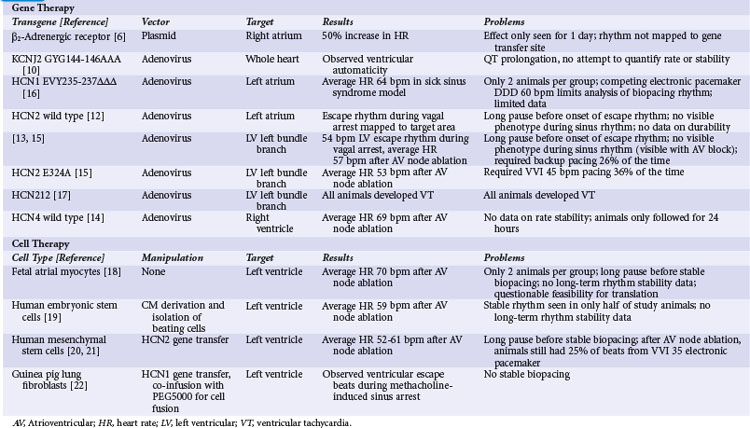
 Gene Therapy Approaches
Gene Therapy Approaches
Beta-2 Adrenergic Receptor Gene Transfer
In the first attempt at genetic modulation of heart rate, Edelberg et al.5 injected plasmids containing the beta-2 adrenergic receptor (β2-AR) into mouse right atria. They found expression of the vector in 81% of myocytes at the injection site and a heart rate of 550 beats per minute (bpm) for the active-treatment group, significantly higher than the 370 bpm rate in controls. In a subsequent pig study, these investigators did not quantify gene transfer but reported a heart rate of 163 bpm for the β2-AR–receiving pigs relative to 127 bpm for controls.6
These studies showed promising results but also raised questions. The investigators did not report the time course of heart rate increase for the mice, but in the pigs they found a statistically significant increase in heart rate only on the second day after injection. The usual experience with plasmid-mediated gene transfer is that the effect persists for several weeks or even months after gene transfer.7–9 The transient effect for the β2-AR experiments suggests that some toxic effect from expression of this gene may be a limiting factor. Additional concerns involve possible mechanisms for the increase in heart rate. The mouse study did not target the specialized conducting system, and under ordinary circumstances, atrial myocytes should not display automaticity. A potential mechanism for the heart rate increase from β2-AR expression in atrial myocytes is Gs-mediated activation of adenylate cyclase, causing an intracellular cascade that ultimately results in phosphorylation of the type 1 calcium channel and the ryanodine receptor. Increased activity of these calcium-handling proteins could cause automaticity by the calcium clock mechanism, but sustained increase in intracellular calcium would likely be toxic. The porcine study did target sinus node by injecting in the area electrically mapped as the earliest site of activation during sinus rhythm, but the extremely transient functional change in pigs suggests that toxicity may be a concern even in sinus nodal tissues.
Ik1 Knockout
In the next illustration of heart rate modulation, investigators used a dominant negative mutation of the gene encoding the IK1 channel (KCNJ2 GYG144-146AAA) to create automaticity.10 IK1 (also called the inward rectifier current) is the principal current responsible for maintaining the cardiac myocyte’s resting membrane potential. The logic behind this strategy was that differences in IK1 between atrial and sinus nodal myocytes account for the relative instability of sinus nodal membrane potential, so that a deficit of IK1 would increase the probability of phase 4 depolarization and automaticity. The investigators admitted that their primary interest was in the electrophysiologic effects of IK1 on the action potential, and that they did not intentionally create pacemaking nodes. Their delivery method essentially scattered the transgene around the ventricular myocardium. Nonetheless, they found evidence of ventricular automaticity in 40% of animals expressing the KCNJ2-AAA mutation. Further investigation found that at least 80% of endogenous IK1 needed to be eliminated for automaticity to occur.11 There have been no subsequent reports applying this strategy to other models.
No attempt was made to quantify rate or stability of the idioventricular rhythm in this report.10 Other concerns rising from the KCNJ2-AAA strategy include the repolarization delaying effects of gene expression and the possibility of atrioventricular (AV) nodal suppressing effects. The investigators reported some level of action potential prolongation and increased QT interval at all expression levels of the transgene, as opposed to the pacing phenotype that was only visible with 80% suppression of IK1. Inspection of the limited reported data also shows no apparent AV nodal conduction, even in areas where the atrial beat is timed far enough away from the proceeding ventricular beat that AV nodal conduction should reasonably be expected. This finding is extrapolated from extremely limited data and should be assessed with more care if IK1 knockout becomes part of a biopacing strategy. Overall, the findings with IK1 knockout suggest that this may be a component of an overall biopacing strategy (with the previous caveats), but that the limited rate in pigs suggests that it might need to be applied with other interventions to increase automaticity.
If Manipulations
Since the early attempts at pacemaker induction, almost all subsequent gene transfer approaches have involved manipulation of If, the putative pacemaker (funny) current. The first attempts consisted of intramyocardial injections of adenovirus vectors encoding wild-type HCN2. The initial report targeted the left atrium,12 and a subsequent report showed gene transfer to the left bundle branch (LBB).13 In both reports, transgene function was not apparent during normal sinus rhythm. During vagal stimulation–induced sinus arrest, the transgene-expressing animals had a significantly higher escape rhythm than controls, and this rhythm mapped to the target area. Overall, the escape rate was inadequate to be a viable pacemaker replacement, and the transgene-driven rhythm could not be seen outside of vagal stimulation, so the investigators could not comment on stability of this rhythm over time.
An additional report with the HCN4 isoform suggested that automaticity was not limited to HCN2. Cai et al.14 injected adenoviruses encoding either wild-type HCN4 with green fluorescent protein (GFP) or GFP alone into porcine ventricles.14 They performed AV nodal ablation 3 to 4 days after gene transfer and measured the residual ventricular rate. The HCN4-expressing animals had a ventricular escape rate of 69 bpm, whereas the rate was only 41 bpm in the GFP controls. Pace mapping of the rhythm in the HCN4-expressing animals showed that the rhythm originated from the target region. The heart rate measurement was essentially an acute study, so no data on rate stability were provided.
Bucchi et al.15 tested the hypothesis that mutations of the HCN channel could improve automaticity. They compared wild-type HCN2 to HCN2-E324A. This mutation was chosen because biophysical analysis suggested it would increase channel activation. The mutation alters the voltage dependence of activation so that the channel becomes active at less negative membrane potential, thus increasing the rate of channel opening during cellular repolarization. The authors hypothesized that this increase in If would lead to faster phase 4 depolarization and thus a faster heart rate. Events did not proceed as planned. The investigators injected adenoviruses encoding the wild-type HCN2, the HCN2-E324A mutant, or GFP into the LBB of dogs. They implanted electronic pacemakers, ablated the AV node, and watched. They saw no significant increase in heart rate when comparing the wild-type and mutant channels (both were better than GFP control), and they noted a greater need for the electronic pacemaker backup in animals expressing the HCN2 mutant than the wild-type HCN2. To investigate this discrepancy with expectations, the investigators found that the mutant channel did, indeed, have the predicted kinetics, but it also had a decrease in peak current caused by a decrease in expression compared with the wild-type channel. The investigators did not define the cause for the decreased expression, but presumably it was cellular toxicity to the mutant channel because the virus vector and DNA control elements were the same for the wild-type and mutant HCN2 genes.
In addition to finding that behavior of mutant channels involves factors other than the predicted biophysical profile, the AV node ablation allowed Bucchi et al.15 to comment on the long-term behavior of the biopacing node. Two important findings were the inadequacy of both pacing rate and rhythm stability after gene transfer. As noted, the average rate of animals receiving wild-type HCN2 was only 57 bpm, which is considerably lower than the normal canine heart rate. Even with this average heart rate of 57 bpm, the wild-type HCN2–expressing animals had frequent episodes when biopacemaking activity was lost, requiring the backup electronic pacemaker 26% of the time. The electronic pacemaker was programmed VVI 45 bpm, so drop-out of the biopacemaker was not caused by overdrive suppression from the electronic pacemaker. The cause was certainly failure of the biopacing node, but the exact mechanism was not reported; loss of automaticity and exit block were likely possibilities.
Tse et al.16 reported another strategy to improve pacing rate with a mutation of the HCN1 isoform. HCN1-EVY235-237ΔΔΔ was a deletion mutation that shortened the S3-4 linker of the channel, with a predicted biophysical effect of increased probability of channel opening. Using a porcine model, the investigators ablated enough of the sinus node to cause sick sinus syndrome, implanted an electronic dual-chamber pacemaker programmed to DDD mode with a lower rate limit of 60 bpm, and then injected either an adenovirus vector encoding the HCN1 mutant or saline into the left atrial appendage. There were only two animals in each group. After gene transfer, the active-treatment group had a spontaneous rate of 64 bpm but still required the electronic pacemaker 14% of the time, which is difficult to interpret given the programmed lower rate limit of 60 bpm and the observed average spontaneous rate of 64 bpm. There is little room for movement below the average rate, although the only 14% use of electronic pacing suggests that the heart rate could not have varied far from the average, or one would have expected a higher percentage of pacing. No rate or rhythm data from the two saline-injected animals were provided. Given the limitations of this report, it is difficult to interpret it in the overall context of biopacing strategies.
The danger of HCN mutation was illustrated by Plotnikov et al.,17 who reported ventricular tachycardia in dogs after injection of a chimeric HCN channel that had components of both HCN1 and HCN2 isoforms. One day after injection of adenoviruses encoding this chimeric channel into LBB, they noted ventricular tachycardia (VT) at rates in excess of 200 bpm in all animals. They further connected the arrhythmia with the gene transfer by showing VT elimination after exposure to ivabradine, an If-blocker with relative HCN1 specificity.
 Cell Therapy Strategies
Cell Therapy Strategies
The first report of cell transplantation for cardiac pacing used a strategy of first isolating canine fetal atrial myocytes, “including sinus node,” and then injecting these cells into the ventricles of adult dogs.18 The investigators did not attempt to identify or isolate a spontaneously beating subpopulation, choosing instead to inject the mixed population from cell isolation. Two dogs received the cardiomyocytes, and two control dogs received fibroblast injections. Three weeks after cell injections, the investigators performed AV node ablation and observed the ventricular rate. Initially after ablation, all animals had a nodal escape of approximately 35 bpm. After 5 to 30 minutes, the two dogs receiving atrial myocytes developed a ventricular escape rhythm at 70 bpm that mapped to the cell injection site. The two control animals had no alteration in rhythm beyond the initial 35-bpm nodal escape. Further study showed that the ventricular rhythm had an escape recovery time of 710 msec after 150-bpm burst pacing, which suggested some level of stability for the rhythm. As with many of the original gene therapy pacing studies, this phenotyping study was acute, and long-term rhythm stability was not assessed. Several practical problems limit translation of these findings, primarily the need to isolate cells from third-trimester fetuses and the need for information on long-term stability of the cells and the rhythm. Immunologic reaction to the cell transplant is another consideration. Overall, however, this report established the feasibility of cell transplant pacemakers.
Similar work showed viability of embryonic stem cell (ESC)–derived cardiomyocytes for pacing. Kehat et al.19 used methods frequently reported in the field to cultivate spontaneously beating cardiomyocytes from undifferentiated ESCs. They first allowed the human cells to grow with specialized culture medium overlying a “feeder layer” of neonatal mouse fibroblasts. The resulting cell population developed into “embryoid bodies,” with suspension culture again using specialized medium. Spontaneously beating cells were mechanically dissected away from the embryoid bodies and injected into pigs after AV node ablation. The 13 pigs that received cell therapy had backup electronic pacemakers in this uncontrolled study. Shortly after AV node ablation, the animals had only a nodal escape rhythm. At electrophysiologic study 1 to 3 weeks after cell injection/ablation, two of the animals still only had a nodal rhythm, five had isolated monomorphic premature ventricular contractions (PVCs) or short runs of ventricular rhythm, and six had sustained ventricular rhythm that mapped to the cell injection site. The average rate of the ventricular rhythm (59 bpm) was competitive with the nodal escape rhythm (61 bpm), so the investigators were unable to comment on rhythm stability or long-term viability of this approach. These issues are relevant when contemplating translation of the ESC results, in addition to ethical concerns voiced by some about the use of human ESCs, and the need for extensive manipulation of the cells, which would not be readily scalable.
Potapova et al.20 reported a novel hybrid cell and gene therapy approach. They transduced human mesenchymal stem cells (MSCs) with the HCN2 gene, then transplanted these cells into canine left ventricle. Their hypothesis was that the HCN2-expressing cell would couple with nearby ventricular myocytes and essentially act as a driver for phase 4 depolarization in the myocytes. With sufficient connectivity, the myocyte would transmit its membrane potential to the MSC during repolarization; this would activate If in the MSC, and that current would be transmitted across the gap junctions to the ventricular myocyte, causing phase 4 depolarization and thus automaticity in the two-cell unit. The investigators were motivated to use the cell therapy approach by concerns about dangers in long-term expression gene transfer vectors that would be needed to transduce the ventricular myocytes directly in vivo, although they admitted that these concerns had not yet been demonstrated in animal or human studies using adeno-associated virus. Potapova et al.20 compared four control animals to six animals receiving the HCN2-transduced MSCs. As in their initial HCN2 gene transfer studies, they saw no spontaneous rhythms, so they used strong vagal stimulation to induce sinus arrest. All animals had nodal or ventricular escape rhythms during the sinus arrest period. Of the four control animals, two had right ventricular (RV) escape rhythms and 2 had left ventricular (LV) escape rhythms. The average escape rate for the controls was 45 bpm. Five of the six HCN2-MSC animals had LV escape rhythms, and the average rate of 61 bpm was significantly higher than in the controls. The investigators mapped the escape to the target site in the HCN2-MSC animals.
In a subsequent study, Plotnikov et al.21 tested the dose-response for HCN2-transduced MSC injections.21 They evaluated animals receiving 155,000 to 850,000 cells. In this study the researchers performed AV nodal ablation with electronic pacemaker implantation (VVI 35 bpm) for long-term follow-up of the escape rhythm. In all animals, spontaneous rhythms became apparent after 2 to 3 days, but this rhythm remained unstable for at least 2 weeks. For animals receiving less than 600,000 cells, the rhythm never stabilized, and the average rate was 43 bpm. After 2 weeks the animals receiving 600,000 to 850,000 cells had a relatively stable rhythm at an average rate of 52 bpm for the 42-day duration of the study. With linear regression analysis, the investigators showed a correlation between rate and number of injected cells for the higher-dose animals. Even with this level of biopacing, however, 25% of observed beats originated from the VVI 35 bpm electronic pacemaker. A further concern was that only 70% of the ventricular beats pace-mapped to the injection site in the high-dose group, although this was an improvement over the 10% mapped to the target site in the low-dose group.
Cho et al.22 reported a variation on the HCN-transduced cell transplant approach. They transduced fibroblasts with the HCN1 isoform and then used polyethylene glycol 5000 (PEG5000) to induce fusion of the HCN1-expressing fibroblasts with ventricular myocytes. This largely in vitro study showed viability of the cell fusion concept and automaticity of the resulting fibroblast-myocyte complex. Very limited in vivo data in guinea pigs injected with the PEG5000/transduced fibroblast mixture showed occasional ventricular escape beats coming from the target region after methacholine-induced sinus arrest. The investigators hypothesized that an additional factor beyond the HCN1 expression was the increased cell surface area of the fibroblast/myocyte complex that diluted IK1 from the myocyte by approximately 20%.
An important in vitro investigation by Fahrenbach et al.23 showed improved pacing with suppression of connexin 43 (Cx43) expression. These investigators used ESCs isolated from Cx43 knockout transgenic mice. The cells still expressed connexins 40 and 45, so some cellular connectivity was possible, but the overall level of connectivity was reduced. The investigators cultured spontaneously beating ESC-derived cardiomyocytes from either wild type of Cx43−/− mice with HL-1 cells (an immortalized mouse atrial tumor cell line). The Cx43 knockout cells required a longer time to connect to the underlying HL-1 cells, but once connectivity was established, the Cx43−/− cells had a much higher probability of becoming the dominant pacing node for the culture dish. These results are consistent with the observations of reduced cell connectivity in histologic studies of sinus node tissues. The concept from sinus node studies is that limited cellular connectivity allows the rhythm to arise gradually in an isolated node, without suppression from the strong polarizing influence of the surrounding myocytes.
Based on the Fahrenbach et al.23 results, some rhythm stability problems in the previously noted gene and cell therapy examples could be caused by too much connectivity between the automatic cells, and the surrounding myocytes could be suppressing phase 4 depolarization in the biopacing node.
 Conclusion
Conclusion1 Senning A. Cardiac pacing in retrospect. Am J Surg. 1983;145:733-739.
2 Appelbaum I, Parsonnet V, Sonnet V, et al. Complications of an implantable cardiac pacemaker. J Newark Beth Isr Hosp. 1962;13:166-174.
3 Pakarinen S, Oikarinen L, Toivonen L. Short-term implantation-related complications of cardiac rhythm management device therapy: a retrospective single-centre 1-year survey. Europace. 2010;12:103-108.
4 Parsonnet V, Bernstein A, Lindsay B. Pacemaker-implantation complication rates: an analysis of some contributing factors. J Am Coll Cardiol. 1989;13:917-921.
5 Edelberg J, Aird W, Rosenberg R. Enhancement of murine cardiac chronotropy by the molecular transfer of the human beta-2 adrenergic receptor cDNA. J Clin Invest. 1998;101:337-343.
6 Edelberg J, Huang D, Josephson M, et al. Molecular enhancement of porcine cardiac chronotropy. Heart. 2001;86(5):559-562.
7 Perales JC, Ferkol T, Beegen H, et al. Gene transfer in vivo: sustained expression and regulation of genes introduced into the liver by receptor-targeted uptake. Proc Natl Acad Sci USA. 1994;91:4086-4090.
8 Muramatsu T, Arakawa S, Fukazawa K, et al. In vivo gene electroporation in skeletal muscle with special reference to the duration of gene expression. Int J Mol Med. 2001;7:37-42.
9 Ziady AG, Kim J, Colla J, et al. Defining strategies to extend duration of gene expression from targeted compacted DNA vectors. Gene Ther. 2004;11:1378-1390.
10 Miake J, Marban E, Nuss H. Biological pacemaker created by gene transfer. Nature. 2002;419:132-133.
11 Miake J, Marban E, Nuss H. Functional role of inward rectifier current in heart probed by Kir2.1 overexpression and dominant-negative suppression. J Clin Invest. 2003;111:1529-1536.
12 Qu J, Plotnikov A, Danilo PJ, et al. Expression and function of a biological pacemaker in canine heart. Circulation. 2003;107:1106-1109.
13 Plotnikov A, Sosunov E, Qu J, et al. Biological pacemaker implanted in canine left bundle branch provides ventricular escape rhythms that have physiologically acceptable rates. Circulation. 2004;109:r31-r37.
14 Cai J, Yi FF, Li YH, et al. Adenoviral gene transfer of HCN4 creates a genetic pacemaker in pigs with complete atrioventricular block. Life Sci. 2007;80:1746-1753.
15 Bucchi A, Plotnikov A, Shlapakova I, et al. Wild-type and mutant HCN channels in a tandem biological-electronic cardiac pacemaker. Circulation. 2006;114:992-999.
16 Tse H, Xue T, Lau C, et al. Bioartificial sinus node constructed via in vivo gene transfer of an engineered pacemaker HCN channel reduces the dependence on electronic pacemaker in a sick-sinus syndrome model. Circulation. 2006;114:1000-1011.
17 Plotnikov AN, Bucchi A, Shlapakova I, et al. HCN212-channel biological pacemakers manifesting ventricular tachyarrhythmias are responsive to treatment with I(f) blockade. Heart Rhythm. 2008;5:282-288.
18 Ruhparwar A, Tebbenjohanns J, Niehaus M, et al. Transplanted fetal cardiomyocytes as cardiac pacemaker. Eur J Cardiothorac Surg. 2002;21:853-857.
19 Kehat I, Khimovich L, Caspi O, et al. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat Biotechnol. 2004;22:1282-1289.
20 Potapova I, Plotnikov A, Lu Z, et al. Human mesenchymal stem cells as a gene delivery system to create cardiac pacemakers. Circ Res. 2004;94:952-959.
21 Plotnikov AN, Shlapakova I, Szabolcs MJ, et al. Xenografted adult human mesenchymal stem cells provide a platform for sustained biological pacemaker function in canine heart. Circulation. 2007;116:706-713.
22 Cho HC, Kashiwakura Y, Marban E. Creation of a biological pacemaker by cell fusion. Circ Res. 2007;100:1112-1115.
23 Fahrenbach JP, Ai X, Banach K. Decreased intercellular coupling improves the function of cardiac pacemakers derived from mouse embryonic stem cells. J Mol Cell Cardiol. 2008;45:642-649.

