[level-membership-for-basic-science-category]Chapter 26
The Basal Nuclei
Components of the Basal Nuclei
Parabrachial Pontine Reticular Formation
Direct and Indirect Pathways of Basal Nuclear Activity
Disinhibition as the Primary Mode of Basal Nuclear Function
Parallel Circuits of Information Flow Through the Basal Nuclei
Disease Implications of Topography of Basal Nuclear Circuits
Behavioral Functions of the Basal Nuclei
Integrated Function of the Basal Nuclei
Voluntary movement is essential to the well-being of living animals. Such behaviors are accomplished by signals that direct the actions of individual muscles. Although these signals originate in the cerebral cortex, they are modulated by a variety of subcortical structures. One such group of structures is the basal nuclei and their functionally associated cell groups. Classically, motor systems have been divided into pyramidal and extrapyramidal on the basis of whether the pathway is mediated by corticofugal neurons (pyramidal) or by the basal nuclei, cerebellum, or descending brainstem pathways (extrapyramidal). However, this distinction is overly simplistic if not inaccurate. Consequently, it is not used here. The basal nuclei are involved in a wide variety of motor and affective behaviors, in sensorimotor integration, and in cognitive functions.
OVERVIEW
These nuclei were traditionally called the basal ganglia rather than the basal nuclei, even though “ganglia” is usually reserved for groups of nerve cell bodies in the peripheral nervous system. The official term basal nuclei is used throughout this chapter, although the unofficial term basal ganglia is also commonly seen in the literature. For practical purposes, these terms may be considered interchangeable, although basal nuclei is the correct and preferred term.
The basal nuclei consist of cell groups embedded in the cerebral hemisphere. Although not classified as cell groups of the basal nuclei in a strict sense, the subthalamic nucleus, substantia nigra, and pedunculopontine tegmental nucleus are integral parts of the pathways passing through these forebrain cell groups. Collectively, the basal nuclei and their associated nuclei function primarily as components in a series of parallel circuits from the cerebral cortex through the basal nuclei to the thalamus and then back to the cerebral cortex.
Four fundamental concepts are crucial to understanding of the basal nuclei. First, damage to or disorders of the basal nuclei result in disruption of movements and may also cause significant deficits in other neural functions, such as cognition, perception, and mentation. Second, the basal nuclei are anatomically and functionally segregated into parallel circuits that process different types of behaviorally significant information. Third, the basal nuclei function primarily through disinhibition (release from inhibition). Fourth, diseases of the basal nuclei can be described as disruptions of the neurochemical interactions between elements of the basal nuclei. These neurochemical relationships rely not simply on the neurotransmitters involved but also on the characteristics of the transmitter receptors, on the locations of the synapses, and on other inputs received by these cells.
In summary, the basal nuclei integrate and modulate cortical information along multiple independent parallel channels. These channels affect behavior indirectly by feedback to the cerebral cortex and directly by providing information to subcortical centers that influence movements. Disruption of these channels by stroke or disease results in dysfunctions initially observed in the motor sphere with subsequent disruption in other behavioral domains.
COMPONENTS OF THE BASAL NUCLEI
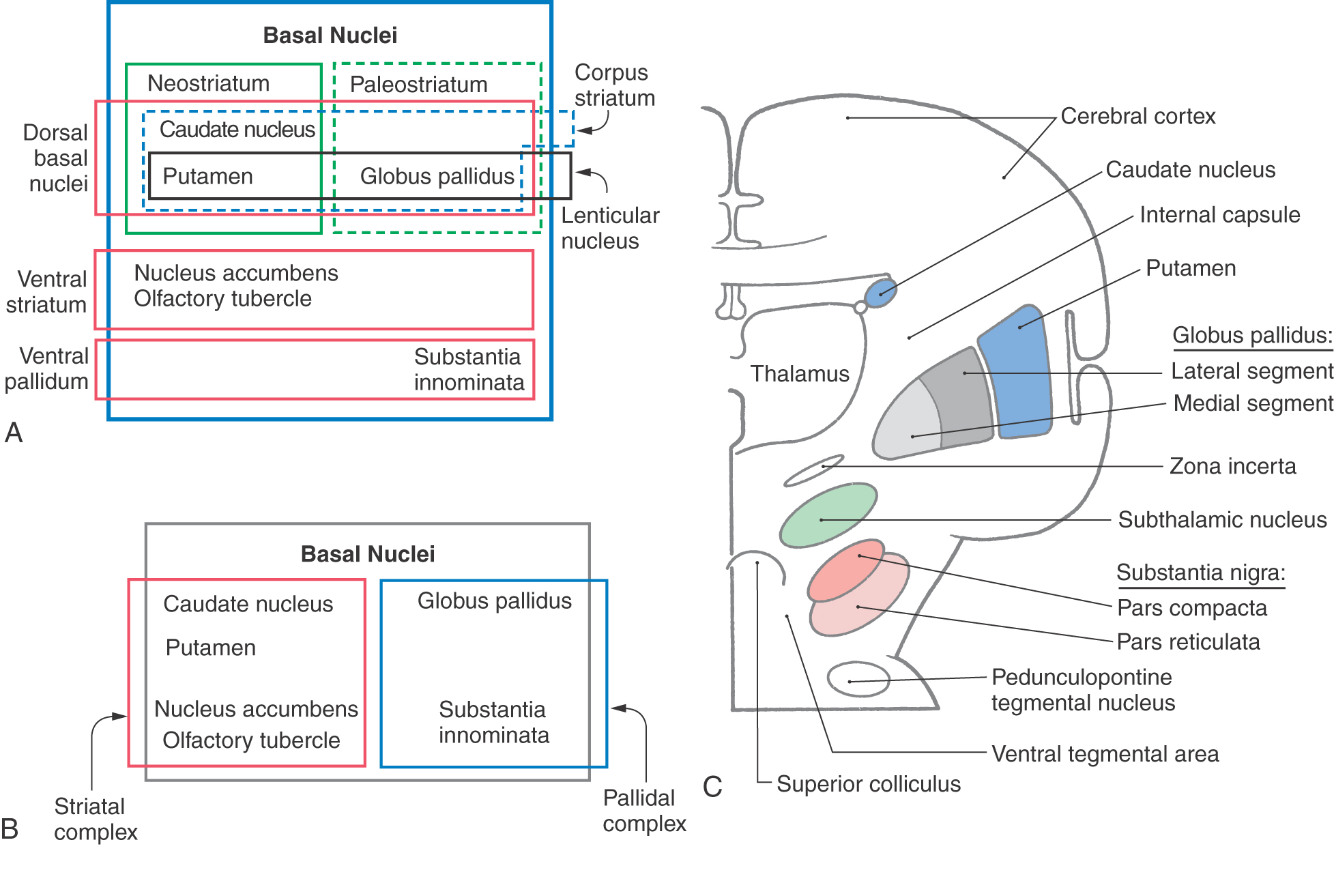
The basal nuclei are typically divided into dorsal and ventral divisions. The dorsal basal nuclei include the caudate and putamen (together constituting the neostriatum) and the globus pallidus (constituting the paleostriatum) (Fig. 26-1A). Associated with the dorsal basal nuclei, in a functional sense, are the substantia nigra, the subthalamic nucleus, and the parabrachial pontine reticular formation (containing the pedunculopontine tegmental nucleus). The ventral basal nuclei are located inferior to the anterior commissure and include the nucleus accumbens, substantia innominata, nucleus basalis of Meynert, and olfactory tubercle. This ventral region is intimately associated with portions of the amygdala and ventral tegmental area. For the purposes of this chapter, the basal nuclei are regarded as making up two complexes: the striatal complex and the pallidal complex (Fig. 26-1B).
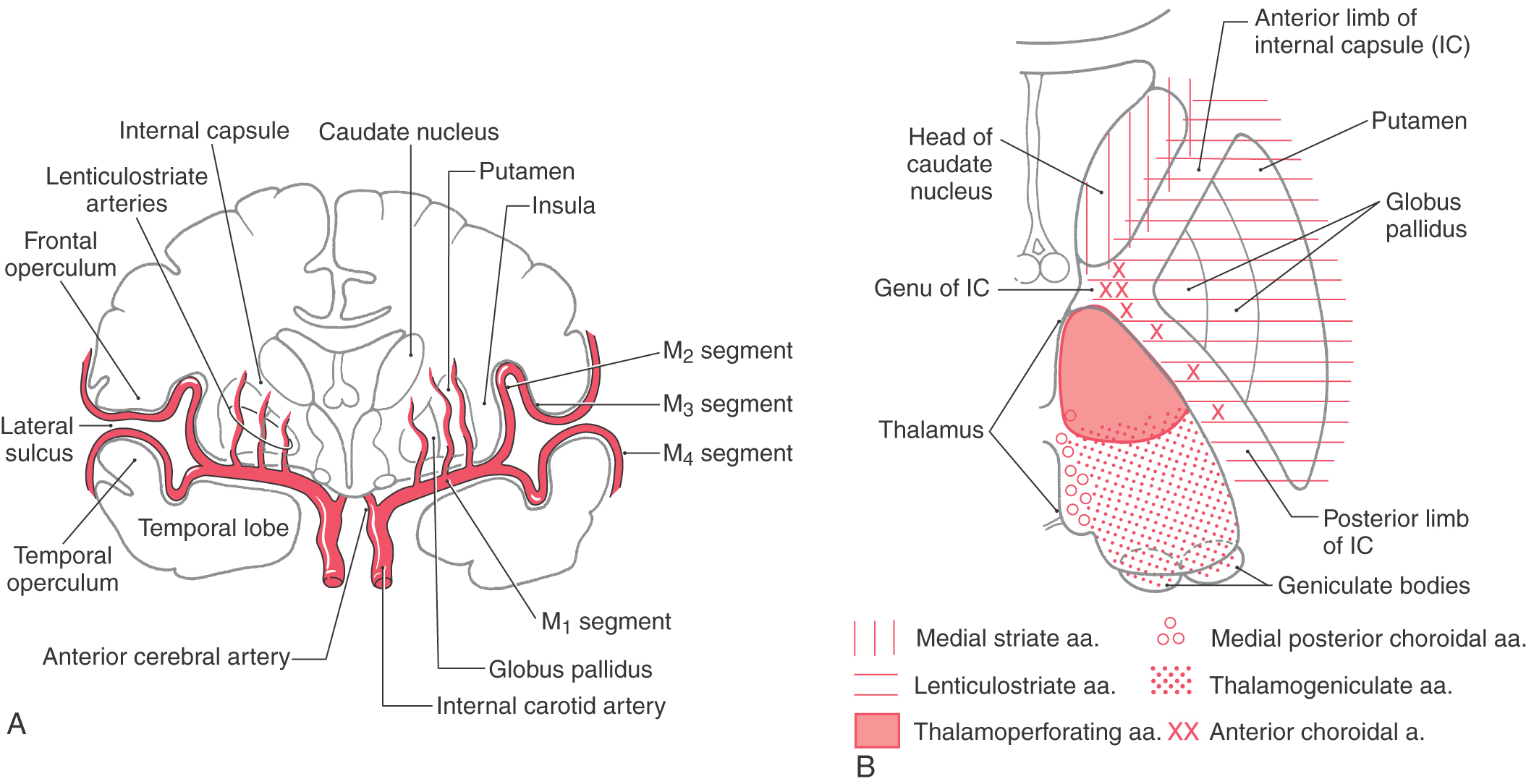
The telencephalic regions of the basal nuclei are supplied by the medial striate artery, the lenticulostriate branches of the M1 segment of the middle cerebral artery, and the anterior choroidal artery (Fig. 26-2). The diencephalic and mesencephalic regions are supplied by the posteromedial branches of the P1 segment of the posterior cerebral artery and branches of the posterior communicating artery. Diseases of these vessels may result in various behavioral or motor deficits, depending on which vessel and region are affected.
Striatal Complex
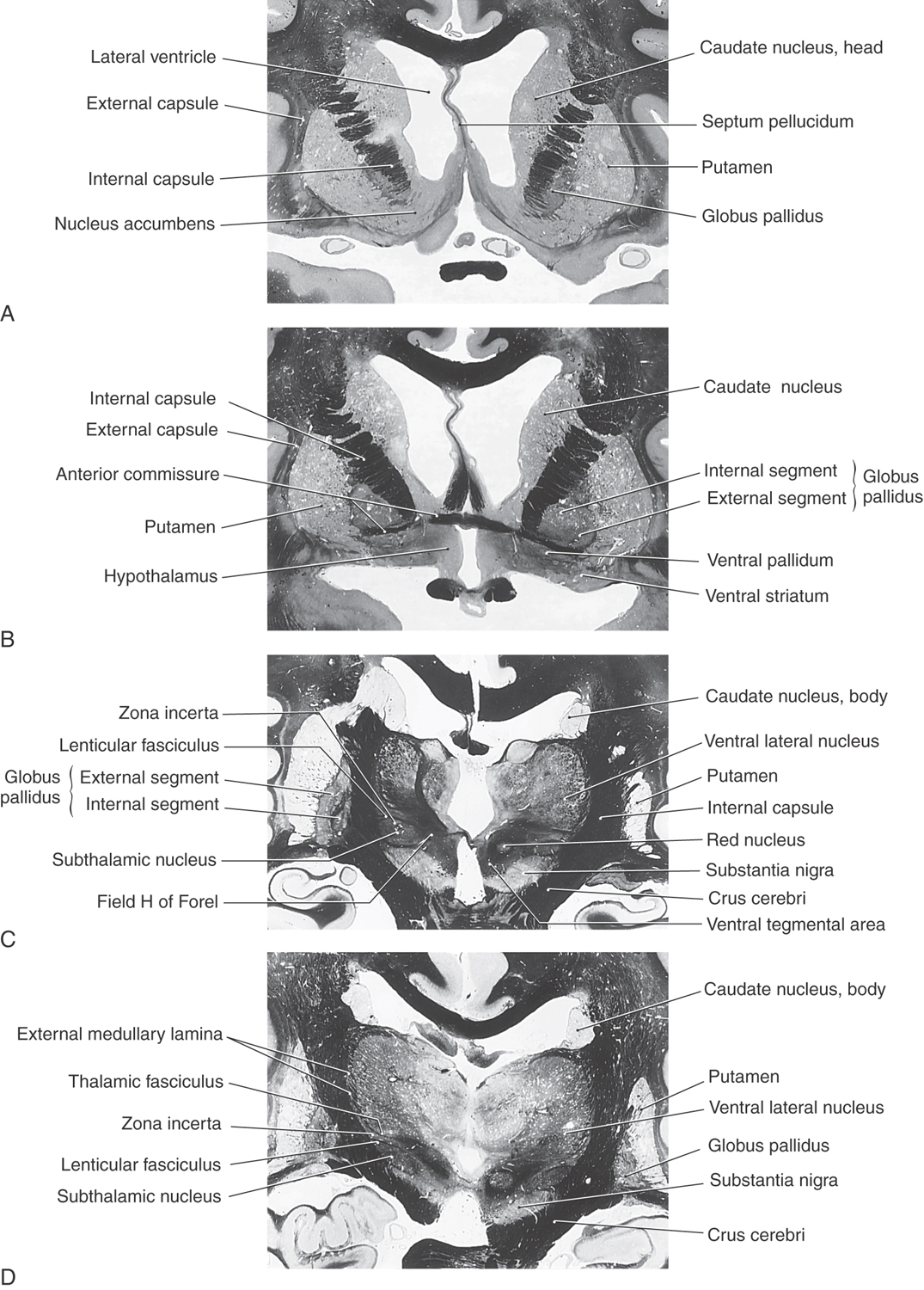
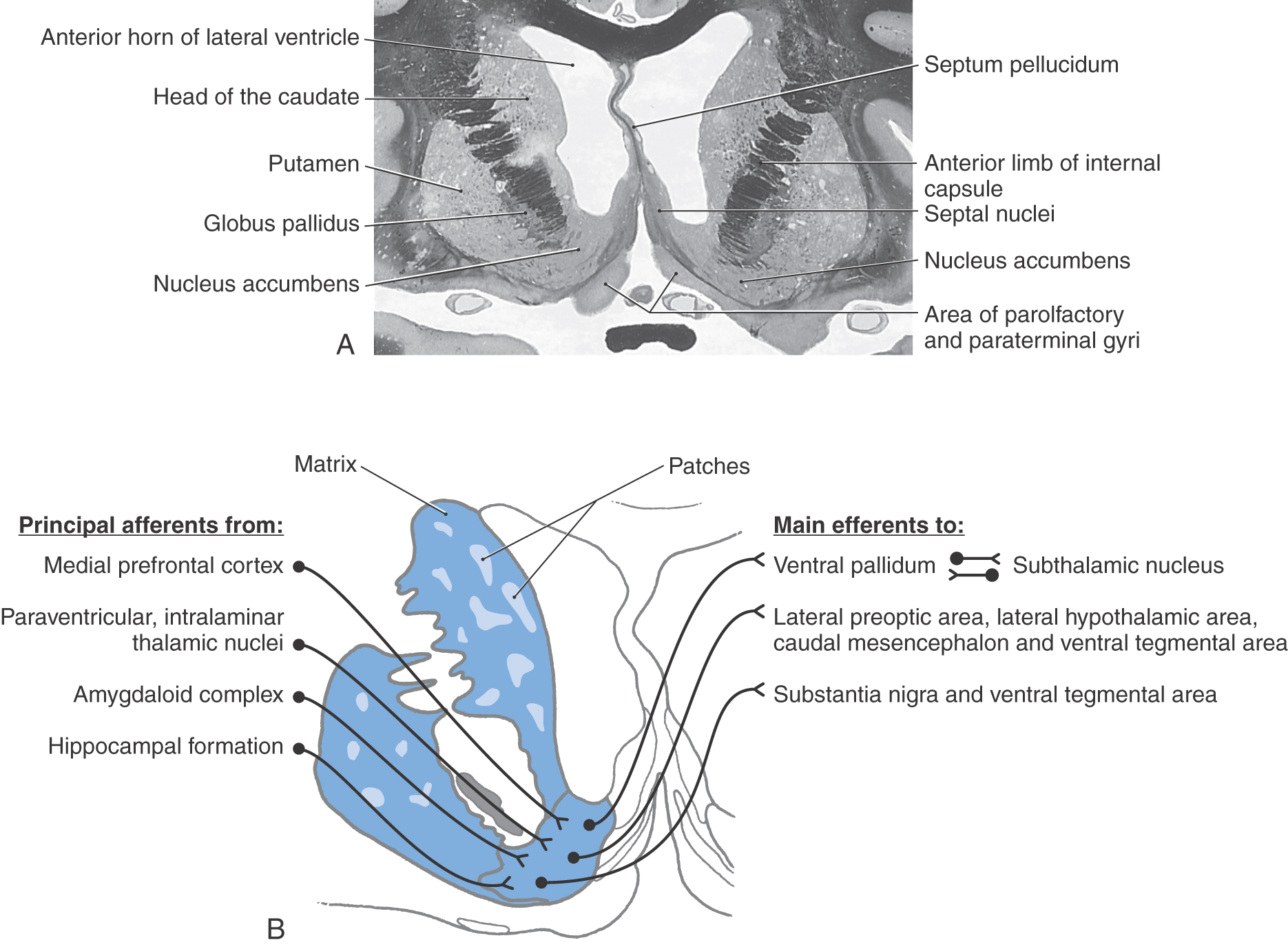
The striatal complex is a functional unit composed of the neostriatum and ventral striatum (Fig. 26-3A, B). The neostriatum consists of the caudate nucleus and putamen. These two nuclei have the same embryologic origin and similar connections. Although fused rostroventrally, they are separated throughout most of their extents by fibers of the internal capsule. The ventral striatum is composed of the nucleus accumbens and portions of the olfactory tubercle (Figs. 26-1A, B and 26-3A). The nucleus accumbens is located rostroventrally in the hemisphere, at the point where the putamen is continuous with the head of the caudate (Figs. 26-3A and 26-4A, B). It is internal to part of the anterior perforated substance. Portions of the olfactory tubercle are considered part of the ventral striatum because of functional, cytoarchitectural, and chemoarchitectural similarities. A defining characteristic of the striatal complex, patches (also called striosomes) are particularly prominent in the head of the caudate (Fig. 26-4B). Patches are acetylcholinesterase-poor regions within the striatal complex. They contain large amounts of one or more neuropeptides and one or more types of opiate receptors. Patches are surrounded by matrix (Fig. 26-4B), which contains high concentrations of acetylcholine and therefore stains darkly when tissue is histochemically reacted to reveal this enzyme. In addition to histochemical and receptor differences, these regions also receive projections from different cortical regions and project to different targets.
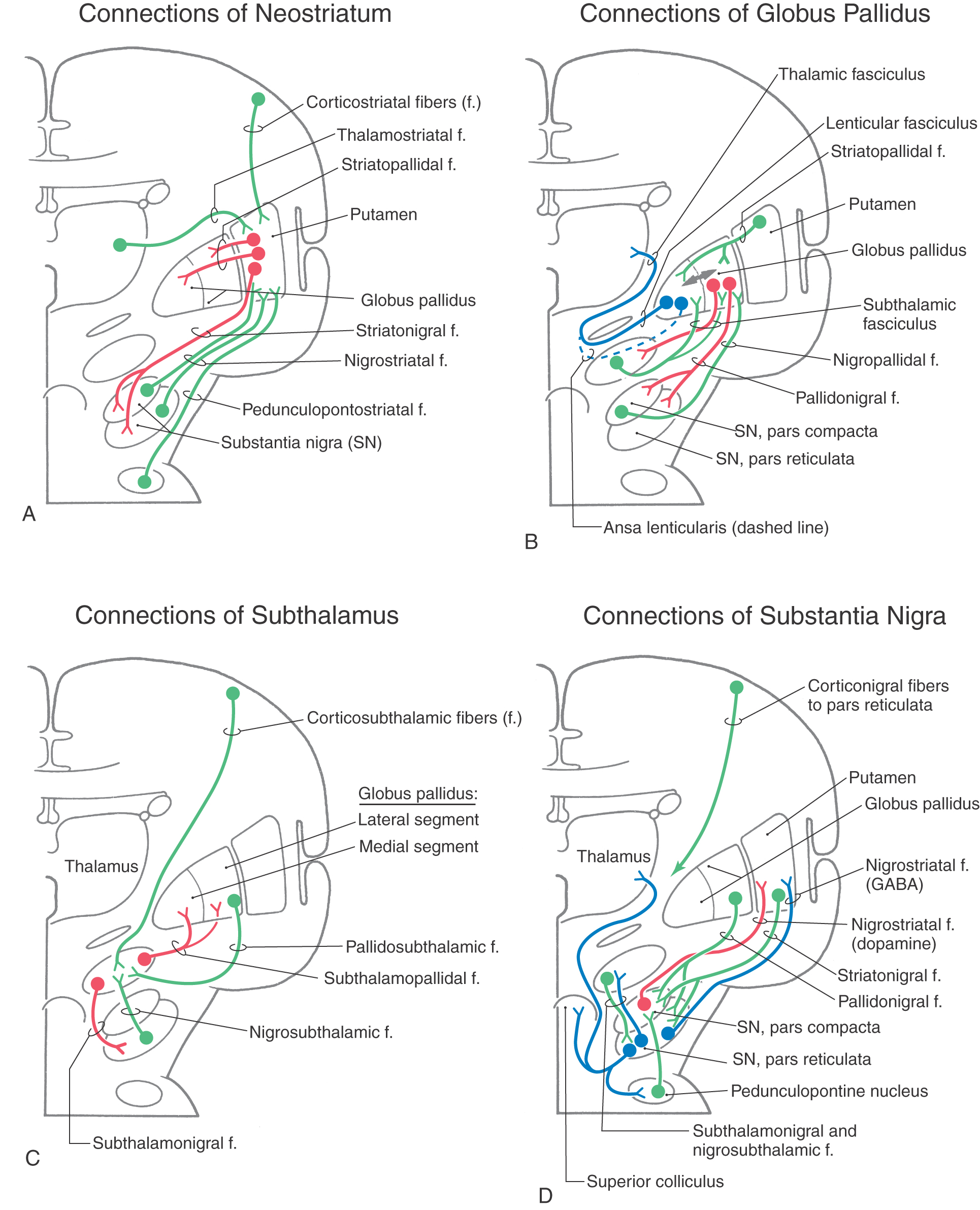
The largest afferent projections to the neostriatum are from the cerebral cortex (corticostriatal fibers) (Fig. 26-5A). Other afferents are from the thalamus (thalamostriatal fibers), substantia nigra (nigrostriatal fibers), and parabrachial pontine reticular formation (pedunculopontostriatal fibers) (Fig. 26-5A). The efferent projections of the striatum reach primarily the pallidum (striatopallidal fibers) and the nigral complex (striatonigral fibers) and to a small degree the subthalamic nucleus.
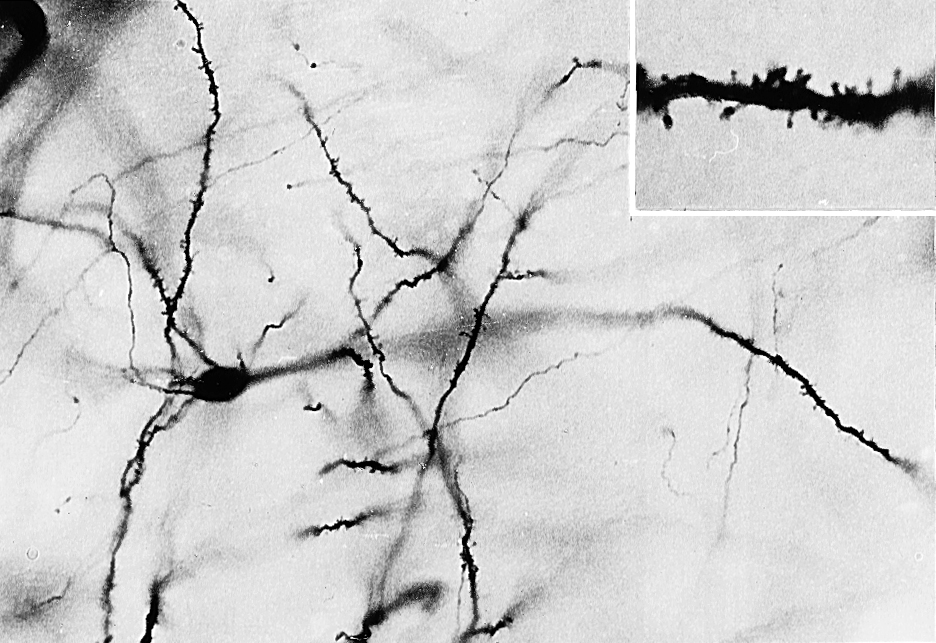
Most of the neurons in the neostriatum are called medium spiny neurons, so named because of their medium-sized cell bodies and the large numbers of spines on their dendrites (Fig. 26-6). Most medium spiny cells have dendritic fields that are restricted to the patch or matrix compartment in which the cell bodies are located.
The nucleus accumbens forms the majority of the ventral striatum (Fig. 26-4A). This nucleus is divided into a core region and a shell region. The core region is cytoarchitecturally and histochemically identical to the neostriatum. It has similar efferent and afferent connections, albeit from cortical regions and to different pallidal and midbrain nuclei. The shell is somewhat histochemically different and has a more diffuse set of connections. It receives projections primarily from allocortical regions and sends projections to the ventral pallidum, substantia nigra–ventral tegmental area, parabrachial nucleus, periaqueductal gray, lateral hypothalamus, and lateral preoptic area (Fig. 26-4B).
Medium spiny neurons fire few action potentials spontaneously and thus require activation by their afferent fibers. These cells use the inhibitory neurotransmitter γ-aminobutyric acid (GABA) and may also contain neuroactive peptides such as substance P and enkephalin. Thus when medium spiny neurons are activated, they subserve both direct inhibitory and neuromodulatory functions at their targets. The axons of medium spiny neurons are the efferent fibers of the neostriatum, collectively forming the striatopallidal fibers.
Also found in the neostriatum are large, acetylcholine-containing local circuit neurons that modulate local activity within the neostriatum. Huntington disease is characterized by progressive loss of medium spiny neurons and acetylcholine-containing neurons throughout the striatal complex.
Pallidal Complex
The pallidal complex is composed of the globus pallidus and the ventral pallidum. The latter is largely synonymous with the substantia innominata (Fig. 26-3B, C). The pallidal complex contains primarily GABAergic neurons with high rates of spontaneous activity. Consequently, these cells tonically inhibit their targets.
The globus pallidus is divided into medial (internal) and lateral (external) segments by a sheet of white matter (the medullary lamina) (Fig. 26-3C). The substantia innominata is located inferior to the anterior commissure and internal to the anterior perforated substance. One important cell group in the substantia innominata is the basal nucleus of Meynert. This nucleus has large acetylcholine-containing neurons, which are lost in Alzheimer disease. However, this disease is not considered a basal nuclear disorder because acetylcholine-containing cells in the cerebral cortex, hippocampus, and septum are also lost in patients with Alzheimer disease. This disease is further characterized by other biochemical and pathologic features, such as senile plaques.
The two divisions of the globus pallidus are reciprocally connected (pallidopallidal fibers) (Fig. 26-5B) but subserve different functions. The main afferent input to the pallidum is from the striatal complex. Medium spiny neurons from the striatum that project to the medial segment and substantia nigra use GABA and substance P; those that project to the lateral segment use GABA and enkephalin (Fig. 26-5B).
The medial division is composed of the medial segment of the globus pallidus. It subserves the direct basal nuclear pathway (described later) and projects primarily to the thalamus (pallidothalamic fibers) (Fig. 26-5B). These fibers exit the globus pallidus as two bundles: the ansa lenticularis and the lenticular fasciculus (Figs. 26-3C, D; see also Figs. 26-8 and 26-9). The ansa lenticularis originates from lateral portions of the medial segment and loops around the posterior limb of the internal capsule to enter the prerubral field (field H of Forel). The lenticular fasciculus (field H2 of Forel), on the other hand, originates in the posteromedial portion of the medial segment. These fibers traverse the internal capsule as small groups of axons, merge to form the lenticular fasciculus between the zona incerta and subthalamic nucleus, and then enter field H of Forel. In the Forel field, the ansa lenticularis and lenticular fasciculus join the thalamic fasciculus (field H1 of Forel), which courses immediately superior to the zona incerta (Fig. 26-3C, D; see also Figs. 26-8 and 26-9). These fibers ultimately terminate in ventral anterior, ventral lateral, and centromedian nuclei of the thalamus. The medial division of the pallidal complex is a principal efferent nucleus of the basal nuclei, the axons of these cells comprising the ansa lenticularis and the lenticular fasciculus.
The lateral division is composed of the external (or lateral) segment of the globus pallidus and the ventral pallidum. This division subserves the indirect basal nuclear pathway (see later). These nuclei receive a large input from the striatal complex (striatopallidal fibers) and small projections from the subthalamic nucleus (subthalamopallidal fibers) and the substantia nigra pars reticulata (nigropallidal fibers). They project strongly to the subthalamic nucleus (pallidosubthalamic fibers) and are also connected with the substantia nigra (pallidonigral fibers) (Fig. 26-5B).
Subthalamic Nucleus
The subthalamic nucleus is a lens-shaped cell group that makes up the largest part of the ventral thalamus. It is immediately inferior to the zona incerta and rostral to the substantia nigra (Fig. 26-3C, D). It receives projections from the lateral pallidal division (pallidosubthalamic fibers), cerebral cortex (corticosubthalamic fibers), nigral complex (nigrosubthalamic fibers), and parabrachial pontine reticular formation. The subthalamic nucleus projects to both pallidal divisions (subthalamopallidal fibers) and to the substantia nigra (subthalamonigral fibers) (Fig. 26-5C). These connections, especially the subthalamopallidal projections to the medial globus pallidus, are an essential part of the indirect pathway underlying basal nuclear function.
Subthalamic neurons use the excitatory neurotransmitter glutamate. Most of the time, subthalamic cells are inactive because of the constant inhibition by cells of the external pallidal segment. However, if this inhibition is removed, subthalamic neurons have a high level of activity resulting in a characteristic motor deficit described later in this chapter. This activity is mediated in part by a large corticosubthalamic projection.
Nigral Complex
The nigral complex is composed of the substantia nigra and the ventral (anterior) tegmental area (Fig. 26-3C, D). The substantia nigra is divided into a cell-dense portion (pars compacta) and a reticulated portion (referred to here as the pars reticulata, although it can be divided into a pars reticulata and a pars lateralis). The pars reticulata is located at and within the medial edge of the descending corticofugal fibers that form the crus cerebri. The pars compacta and the adjacent ventral tegmental area appear to subserve similar functions and to have a similar chemoarchitectural organization. The major afferents to the nigral complex are from the striatal and pallidal complexes. The nigral complex also receives cortical (corticonigral), subthalamic (subthalamonigral), and pedunculopontine fibers (Fig. 26-5D).
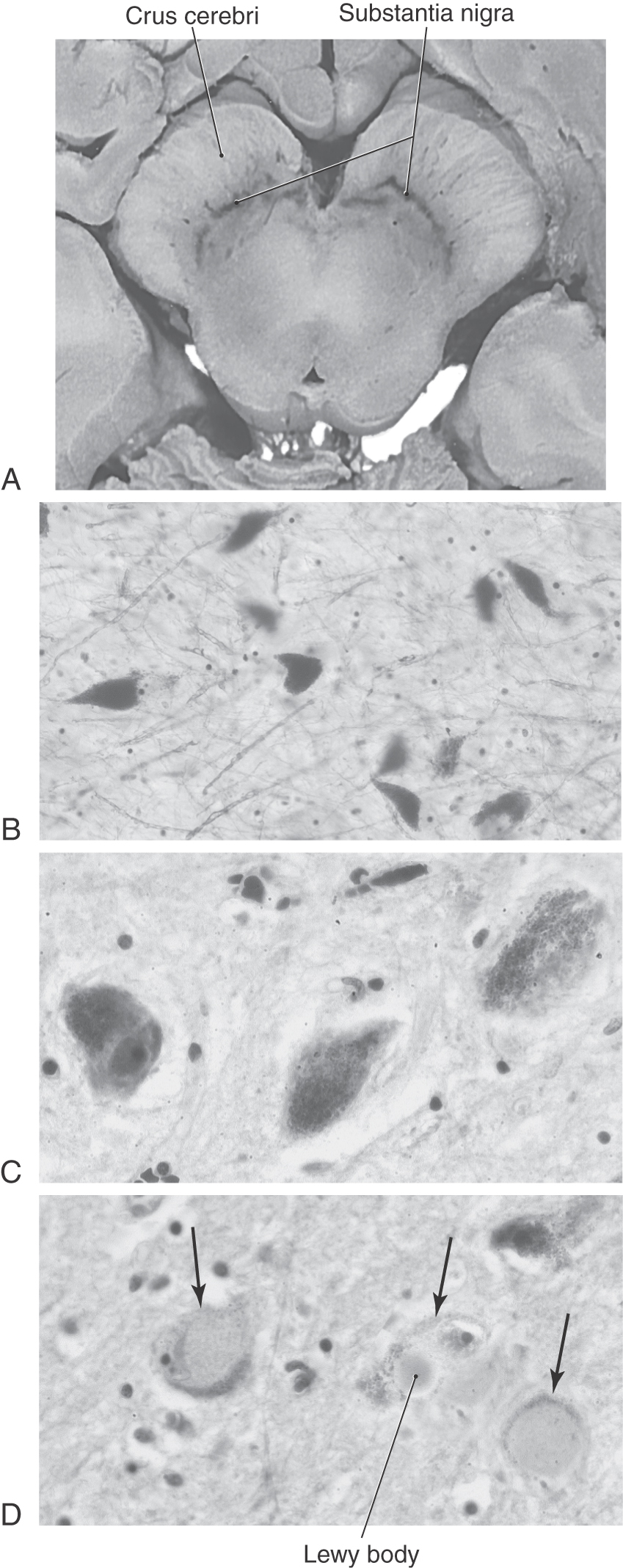
The pars compacta contains a large number of neuromelanin-containing cells, whose dark color gives the nucleus its name (substantia nigra, “black substance”). Cells of the pars compacta in healthy individuals appear characteristically dark in brain slices and are packed with small black granules in histologic sections (Fig. 26-7A–C). Neurons in the pars compacta use the neurotransmitter dopamine and project primarily to the neostriatum as nigrostriatal fibers. The dopamine released by these cells may excite or inhibit striatal neurons, depending on the type of receptor on the postsynaptic membrane.
The pars reticulata is formed by loose aggregations of medium-sized to large GABAergic neurons that are indistinguishable from those of the medial pallidum. Neurons in the pars reticulata have axons with an extensive system of collaterals; consequently, they may project to and inhibit one or more target structures. These targets include the neostriatum (nigrostriatal fibers), thalamus (nigrothalamic fibers), superior colliculus (nigrotectal fibers), and parabrachial pontine reticular formation. These cells have a high rate of discharge and tonically inhibit their targets. Projections of pars reticulata neurons represent an important pathway by which the basal nuclei influence other motor centers.
The pars compacta and the pars reticulata are interconnected. Dendrites of dopaminergic pars compacta neurons extend into the pars reticulata, where they release free dopamine (by a nonvesicular mechanism). The level of dopamine modulates the resting membrane potential of pars reticulata cells, making them either more or less likely to discharge, depending on the subtype of dopamine receptor they possess. In turn, pars reticulata neurons have axon collaterals that ramify extensively in the pars compacta and form GABAergic synapses. Collectively, these interactions form modulatory loops between neurons of the pars compacta and the pars reticulata.
These modulatory loops are influenced by the output of the striatal system. Neurons in the patches project predominantly onto the cells in the pars compacta, whereas the neurons in the matrix project predominantly onto the cells in the pars reticulata. Thus the striatonigral projection from patches directly inhibits the dopaminergic nigrostriatal neurons, and the projection from the matrix inhibits the GABAergic neurons in the pars reticulata. Consequently, inhibition of the pars reticulata is reduced and its targets, such as the pars compacta, are released from inhibition and may become more active.
The ventral tegmental area is located medial to the substantia nigra. It contains large numbers of dopaminergic neurons and forms connections with the ventral striatum, the amygdala, and other limbic system structures. Cells of the ventral tegmental area project to and terminate on striatal neurons that have postsynaptic D2 (dopamine) receptors. In schizophrenia, there is an increase in number and in sensitivity of these receptors. Neuroleptic drugs help control schizophrenia by blocking (downregulating) these D2 receptors.
Parabrachial Pontine Reticular Formation
Nuclei in the region of the parabrachial pontine reticular formation, primarily the pedunculopontine tegmental nucleus, are intimately connected with all portions of the basal nuclei and their associated nuclei. For example, GABAergic substantia nigra pars reticulata neurons project onto cells of the pedunculopontine tegmental nucleus. In turn, acetylcholine-containing pedunculopontine tegmental neurons project to the substantia nigra pars compacta. In addition, the pedunculopontine tegmental nucleus is reciprocally connected with the subthalamic nucleus. Moreover, the parabrachial pontine nuclei are connected with motor centers in the brainstem, which project to the spinal cord via the descending spinal pathways. Thus these nuclei serve as an efferent pathway for the basal nuclei. It has been suggested that damage to the connections between the pedunculopontine nucleus and the basal nuclei may partially account for motor deficits, such as tremor or chorea, in some patients.
Ventral Basal Nuclei
The ventral basal nuclear pathways are similar to those of the dorsal basal nuclei. The pathways originate in the allocortex (limbic-related cortical areas) and the orbital and medial prefrontal cortices. The corticostriatal pathway terminates in the ventral striatum (nucleus accumbens and portions of the olfactory tubercle). Striatopallidal neurons then project to the ventral pallidum (substantia innominata). The pallidothalamic neurons then project to the mediodorsal nucleus of the thalamus, whose neurons then project to the cerebral cortex. As with the dorsal basal nuclei, distinct regions of each nucleus in these pathways are connected with each other. Thus there is a clear loop through the ventral basal nuclei for information originating from different areas of the cortex, as is discussed later.
DIRECT AND INDIRECT PATHWAYS OF BASAL NUCLEAR ACTIVITY
Pathways through the basal nuclei consist of parallel circuits that share certain features. The basic circuit is divided into direct and indirect pathways that have opposing actions on targets of the basal nuclei (Figs. 26-8 to 26-10). As a general concept, the direct pathway facilitates a flow of information through the thalamus and the indirect pathway inhibits this flow. These pathways create a balance in the inhibitory outflow of the basal nuclei and function by modulating the extent of this inhibition on target nuclei.
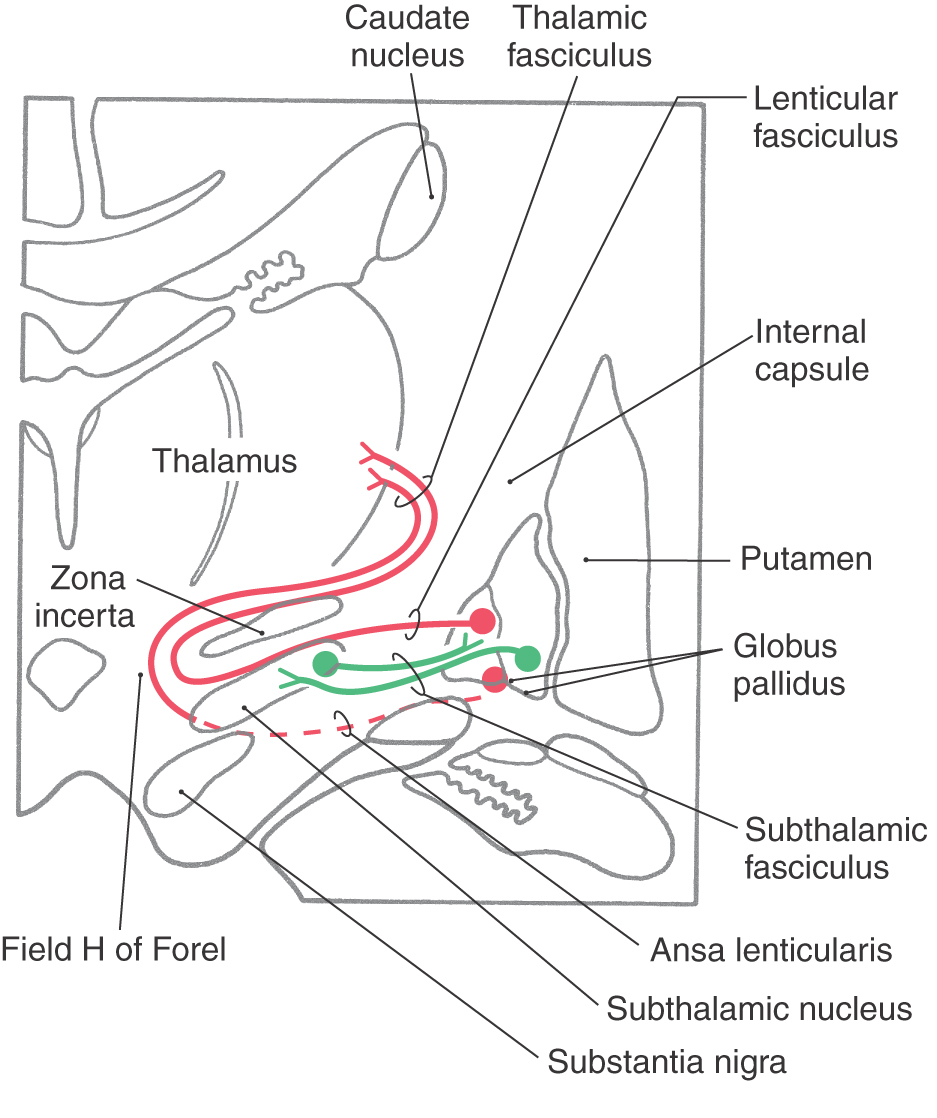
Figure 26-8. Schematic representation of pallidal connections to the thalamus and with the subthalamic nucleus.
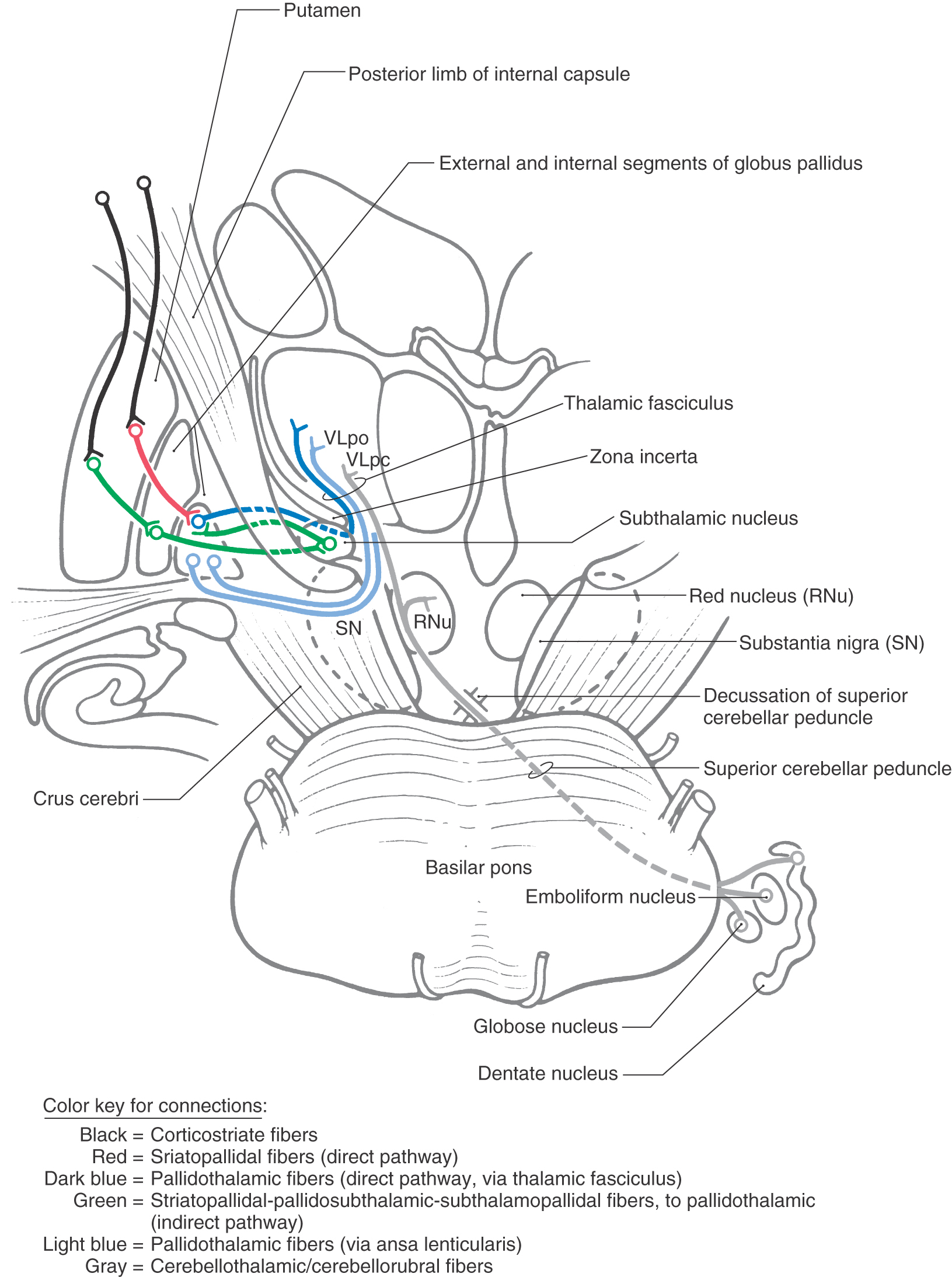
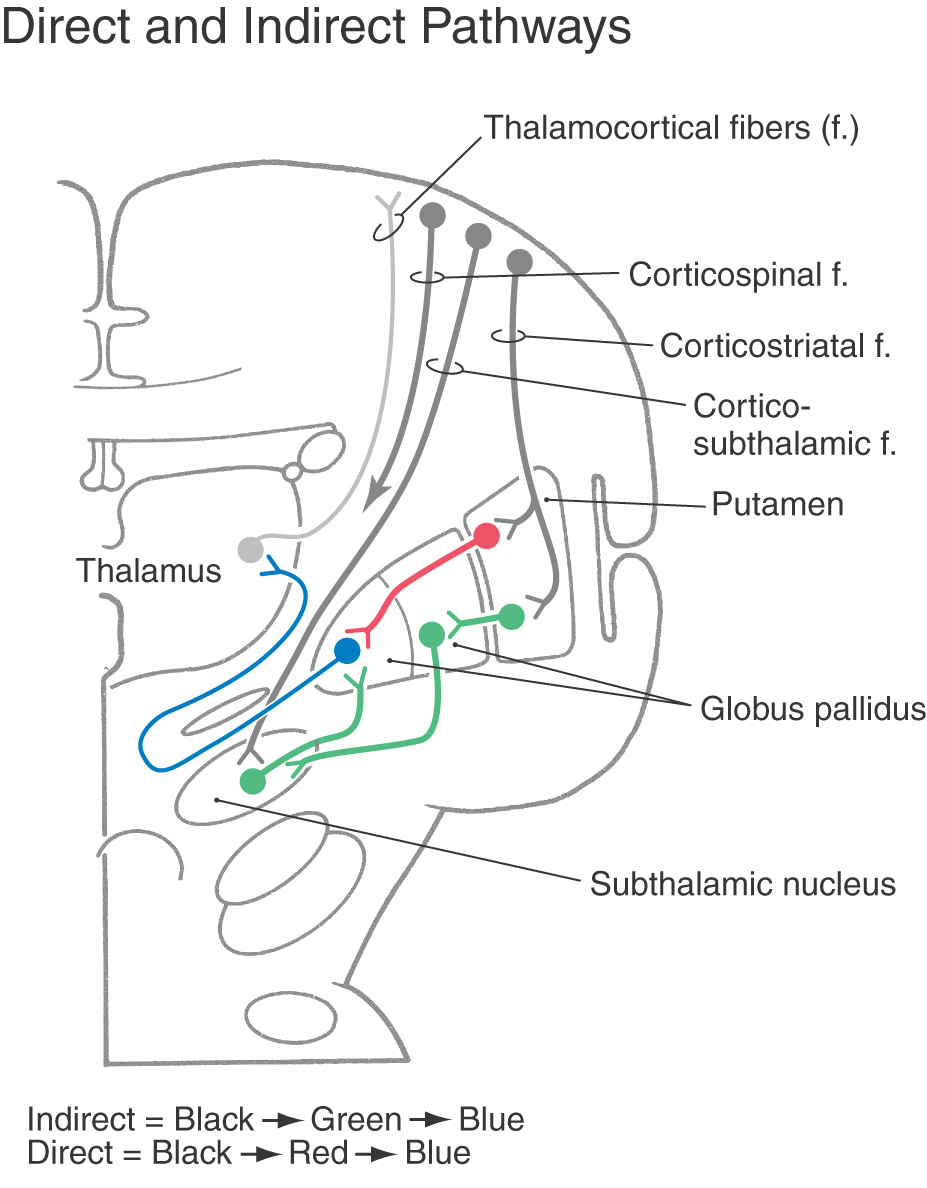
Figure 26-10. Schematic representation of direct and indirect pathways through the basal nuclei.
The direct pathway (Figs. 26-9, 26-10, and 26-11A) begins as an excitatory, glutamatergic projection from the cerebral cortex to the striatal complex. Striatal neurons inhibit cells in the internal (or medial) segment of the globus pallidus and in the substantia nigra pars reticulata. These striatopallidal and striatonigral fibers use GABA and substance P. Cells of the internal segment of the globus pallidus (as pallidothalamic fibers) and of the substantia nigra pars reticulata (as nigrothalamic fibers) project to thalamic neurons. These fibers have a high rate of spontaneous activity and thus tonically inhibit target thalamic neurons. Inhibition of these pallidal and nigral projections by striatal cells decreases the inhibitory inputs to thalamocortical neurons (thalamic disinhibition). The net effect of the direct pathway is to increase the activity of the thalamus and the consequent excitation of the cerebral cortex (Fig. 26-11B).
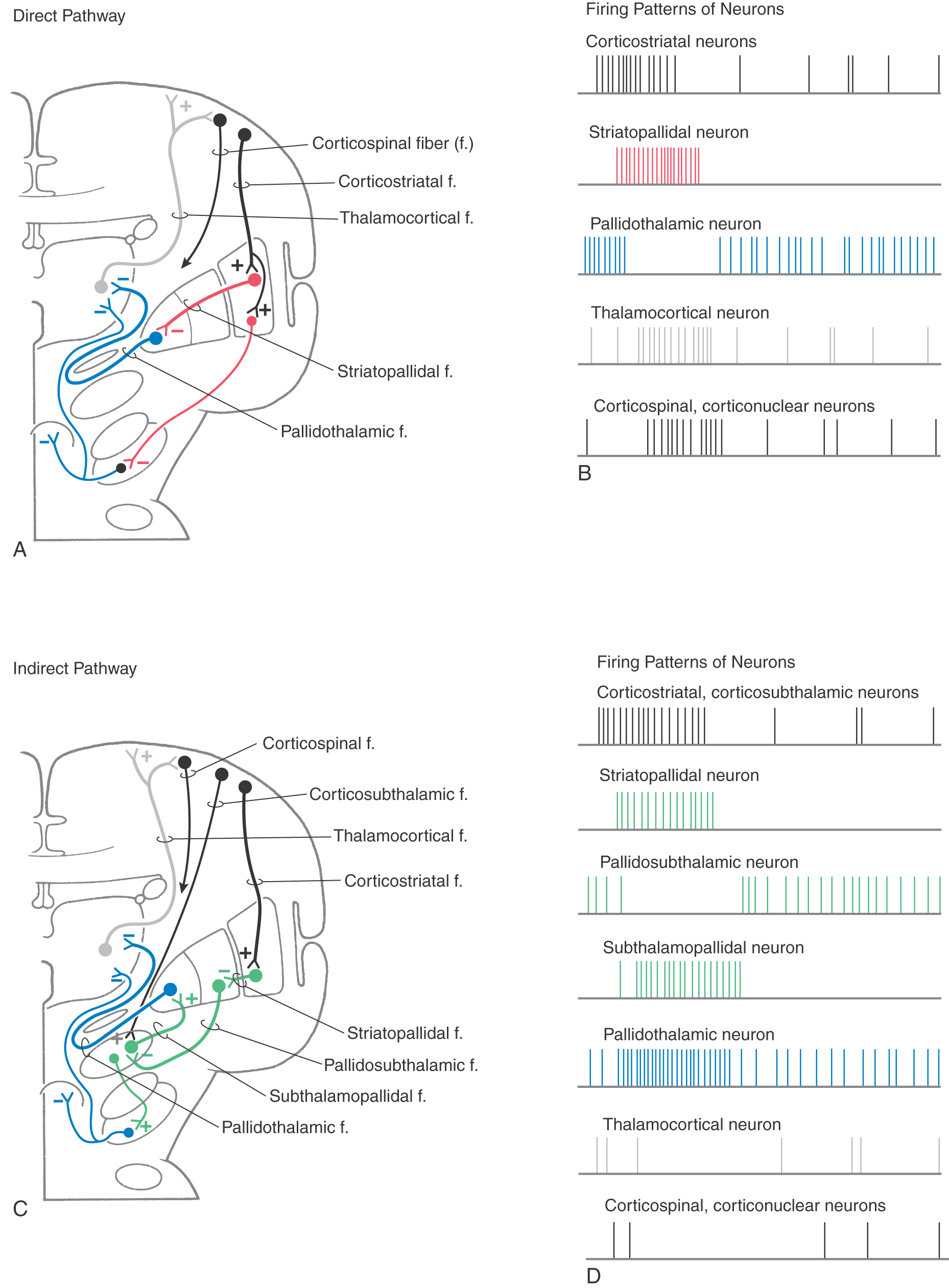
The indirect pathway includes a loop through the globus pallidus and subthalamic nucleus (Figs. 26-9, 26-10, and 26-11C). Striatopallidal neurons involved in this pathway contain GABA and enkephalin. They project into the lateral pallidal segment, which in turn sends pallidosubthalamic fibers into the subthalamic nucleus. These pallidosubthalamic fibers are GABAergic, have high spontaneous firing rates, and tonically inhibit subthalamic cells. Inhibition of these fibers by the neostriatum releases these subthalamic cells from their tonically inhibited state (subthalamic disinhibition). These subthalamic neurons have spontaneous activity and are also influenced by an excitatory corticosubthalamic projection. Together, these inputs increase the firing rates of glutamatergic subthalamopallidal fibers to the medial pallidal segment. As a consequence, the firing rate of inhibitory pallidothalamic fibers is increased, with a resultant decrease in the activity of thalamocortical neurons. The net effect of the indirect pathway is to decrease activity of the thalamus and consequently to decrease activity of the cerebral cortex (Fig. 26-11D).
Disinhibition as the Primary Mode of Basal Nuclear Function
The function of the direct pathway is to release the thalamus from its pallidal inhibition. This release is accomplished by striatopallidal inhibition of pallidothalamic neurons. In the indirect pathway, the subthalamic nucleus is released from inhibition by the lateral pallidal segment so that it can excite the inhibitory pallidothalamic cells. This mechanism that releases cells from inhibition is called disinhibition. Balance between thalamic disinhibition by the direct pathway and subthalamic disinhibition by the indirect pathway results in normal basal nuclear function. Behavioral deficits that accompany basal nuclear disorders can ultimately be traced to imbalances between the direct and indirect pathways.
Although the balance between the direct and indirect pathways determines the net outflow of the basal nuclei, other elements of the basal nuclei are also modulated by disinhibitory mechanisms. For example, in the earlier description of the suggested projections from the patches and matrix to the substantia nigra, the patch pathway to the pars compacta is a direct inhibitory pathway, whereas the pathway from the matrix, which inhibits the pars reticulata and releases the pars compacta nigrostriatal neurons, functions to disinhibit the pars compacta neurons.
PARALLEL CIRCUITS OF INFORMATION FLOW THROUGH THE BASAL NUCLEI
Information flow through the basal nuclei is separated into multiple distinct parallel circuits. These circuits originate from functionally distinct regions of the cerebral cortex, pass through distinct regions of each basal nuclear component, modulate different areas of the thalamus, and return to functionally distinct cortical regions. That is, each circuit can be considered an independent channel that processes information from one functional type of cortex by way of its own areas of the basal nuclei and thalamus and returns to the appropriate functionally related part of cortex. All these circuits have both direct and indirect pathways described previously. In addition, it is also clear that dopamine has a major role in each of these parallel circuits (see subsequent discussion).
A variety of anatomic and functional studies in humans and in other species have demonstrated that each loop projects to a restricted portion of each nucleus. Therefore, there is an anatomic as well as a functional separation of the basal nuclear circuits. This separation is called the closed component of the circuitry. However, at every stage of each circuit, the information is modulated and integrated with input from other centers by intrinsic basal nuclear connections. Thus, although the circuits are anatomically and functionally distinct, their activities are modulated by other modalities of the basal nuclei. This integration is called the open component of basal nuclear circuits.
Five classic circuits were described on the basis of their anatomic connections, and there is good evidence that there may be additional circuits that can be defined anatomically. More recently, there has been much focus on the function of these pathways. The major functional pathways are the motor, executive, motivational, and visuomotor loops. A brief description of each of these functional pathways is included here. These descriptions focus on the direct pathways, even though each of these loops has both direct and indirect pathways.
Motor Loop
The most obvious initial symptoms of basal nuclear disorders are those associated with the motor loop. This loop originates in the supplementary motor, primary motor, and premotor cortices. The corticostriatal projections terminate in the putamen (caudal striatum), which also receives projections from the somatosensory cortex. Efferents from the putamen terminate in the ventral lateral portions of the internal segment of the globus pallidus (GPi) and the caudolateral substantia nigra pars reticulata (SNr). These areas are topographically related to the lateral portions of the substantia nigra pars compacta (SNc). The GPi projects to the oral part of the ventral lateral nucleus (VLo) and the medial portions of the ventrolateral nucleus (Vlm) of the thalamus. These areas project back to the supplementary, premotor, and motor cortices. There are also direct projections to the superior colliculus and brainstem reticular formation. In this way, this basal nuclear pathway affects both cortical motor and brainstem motor centers. When these projections are disrupted by disease processes, a number of motor disturbances are observed. Depending on the progression of the disease, these motor signs may precede or may follow the onset of other signs characteristic of the other basal nuclear loops, such as cognitive loss.
Executive Loop
Executive functions are cognitive mechanisms that allow the individual to optimize function when multiple tasks need to be performed at the same time. These include functions such as learning of new tasks, selection between relevant and irrelevant information, and certain memory functions. Research into these functions suggests that this loop assists in recognition of a behavioral context and modulation of cortical activity appropriate for that context. This loop originates in the dorsolateral prefrontal and posterior parietal cortices that project to the head and dorsolateral portions of the caudate nucleus. This is related to the lateral dorsomedial portions of the globus pallidus internal segment (GPi) and the rostrolateral portions of the substantia nigra pars reticulata (SNr). These in turn project to the parvicellular region of the ventroanterior (VA) and parvicellular region of the dorsomedial (DM) nuclei of the thalamus. These thalamic nuclei in turn project back to the prefrontal and posterior parietal cortices. This circuit also has projections to brainstem motor centers. It is the disruption of these circuits that results in the cognitive loss observed in patients with basal nuclear diseases.
Motivational Loop
Motivational phenomena include such things as drive, pleasure, “wanting,” and emotions (affect). Research during the past two decades has emphasized the importance of the basal nuclei in these functions. In particular, the ventral basal nuclear dopamine pathway has been extensively studied, especially with regard to its relationship with diseases such as schizophrenia. This loop has a diverse set of telencephalic regions including lateral orbitofrontal cortex, anterior cingulate cortex, medial prefrontal cortex, amygdala, and hippocampus. These projections are to the ventromedial portions of the caudate nucleus and the ventral striatum (VS). In turn, these project to the rostromedial portions of the internal segment of the globus pallidus (GPi), nucleus accumbens (NA), ventral tegmental area, lateral preoptic area, and caudal mesencephalon. These areas in turn project to magnocellular portions of the ventroanterior nucleus (VA), magnocellular portions of the dorsomedial nucleus (DM), and posteromedial portions of the dorsomedial nucleus of the thalamus (DM). Treatments directed to the dopaminergic system within the motivational loop have resulted in reduction of schizophrenia and dementia and improvements in patients with affective and emotional disorders.
Visuomotor Loop
Certain visual functions and eye movements are modulated through the basal nuclei. This circuit begins in the visual portions of the temporal cortex and the frontal eye fields. They project onto the body and tail of the caudate nucleus. They project to the caudodorsomedial portions of the internal segment of the globus pallidus (GPi) and the ventrolateral portions of the substantia nigra pars reticulata (SNr). The thalamic areas related to this loop are the magnocellular portions of the ventroanterior nucleus (VA) and portions of the parvicellular region of the dorsomedial nucleus (DM) of the thalamus. The projections from this loop to the superior colliculus and the role of these connections in the control of eye movements have been particularly well studied.
Disease Implications of Topography of Basal Nuclear Circuits
As described, the loops are each primarily associated with different regions of the basal nuclear circuits. The key point is that generally speaking, these behavioral functions are segregated into anatomically distinct regions of the basal nuclei. This becomes important in understanding the progression of disturbances observed in various basal nuclear disorders.
In Parkinson disease, there is a loss of dopaminergic neurons in the substantia nigra. The loss typically progresses from lateral to medial within the substantia nigra. It affects the motor loop first and then progresses to the executive loop, followed by the visuomotor and motivational loops. Thus Parkinson patients first exhibit motor disturbances. As the disease progresses, cognitive and executive functions are increasingly affected. Last, dementia and other psychiatric symptoms become evident.
In Huntington disease, the medium spiny neurons in the striatum are lost. The progression of that disease typically moves dorsal to ventral, anterior to posterior, and medial to lateral. Thus the executive loop is affected first, and the motor loop is increasingly affected as the disease progresses. This results in the characteristic loss of cognitive and executive functions and progressive motor disturbances in Huntington disease. Because the motivational loop is affected last, dementia is usually the last set of symptoms observed in these patients.
BEHAVIORAL FUNCTIONS OF THE BASAL NUCLEI
The best-understood functions of the basal nuclei are associated with the motor systems, in particular the somatomotor (motor loop) and visuomotor (oculomotor loop) systems. Lesions in the basal nuclei resulting from stroke or other disease processes lead to significant changes in the motor behavior of the patient. Examination of the pathways damaged in these cases reveals how disruption of the basal nuclei can lead to seemingly opposite effects in different patients. For example, movements can be either reduced (hypokinetic disturbances) or increased (hyperkinetic disturbances).
Hypokinetic Disturbances
The two major types of hypokinetic disturbances seen in patients with basal nuclear disorders are akinesia and bradykinesia (see Fig. 26-16). Akinesia is an impairment in the initiation of movement; bradykinesia is a reduction in velocity and amplitude of movement. Both disturbances are characteristic in patients with Parkinson disease.
Akinesia, the impaired ability to initiate voluntary movements, may be due to disruption of the ability to plan a movement or to guide a movement to some desired position. Experimental studies in nonhuman primates reveal that many basal nuclear neurons are most active during the planning phase of a movement or when the subject is making an internally guided movement. The latter is a movement to a location at which no particular stimulus is present. Thus patients with akinesia have a generalized disruption of the role of the basal nuclei in planning and generating programmed movements.
Bradykinesia, the reduction in velocity and amplitude of movements, is due to disruption of the balance between the outflows of the direct and indirect pathways to the thalamus. The result is an increase in the activation of the antagonist muscles. Thus the observed abnormalities are due to an inappropriate activation of the antagonistic muscles and not necessarily to an overall decrease in muscle activity.
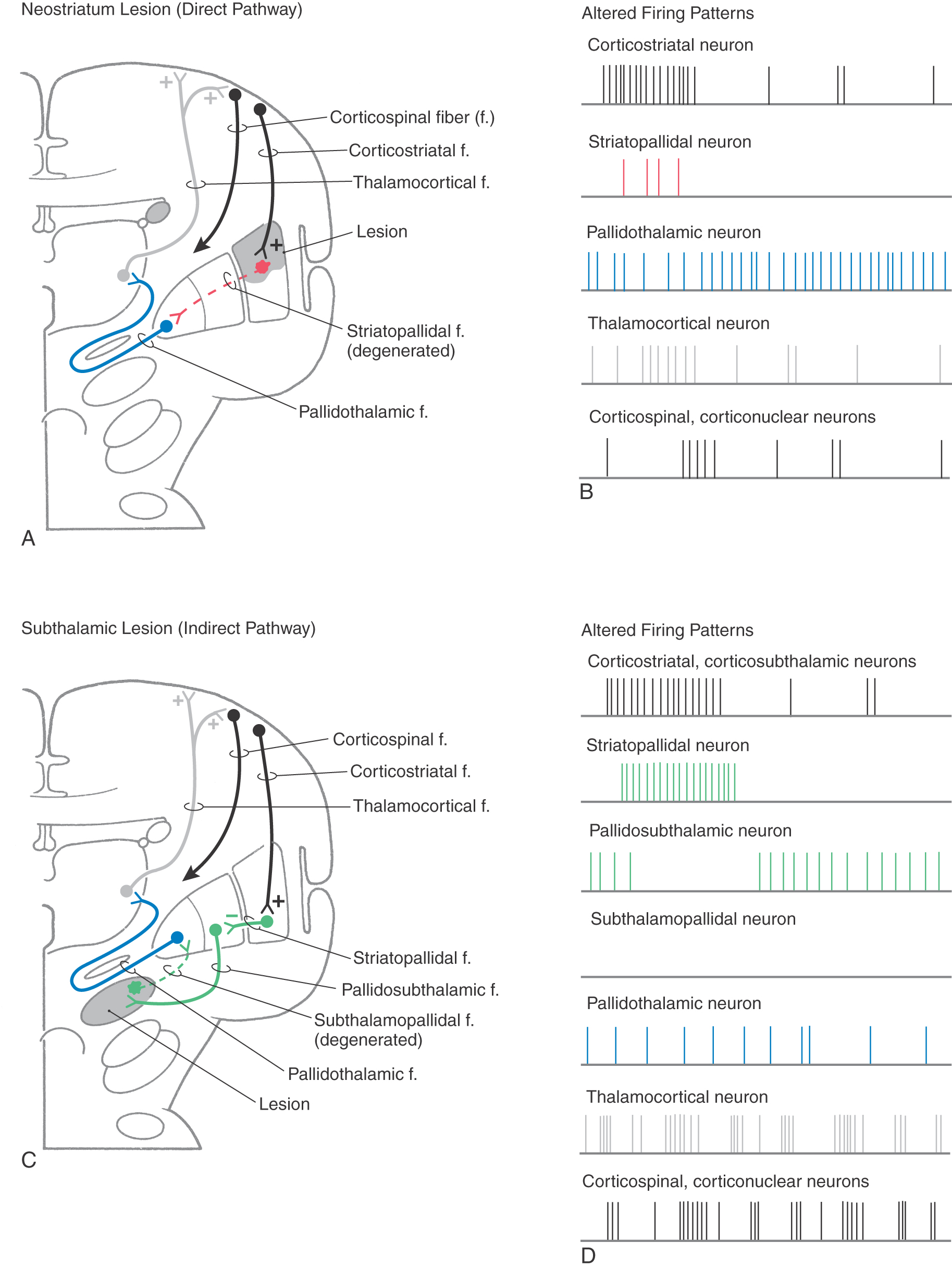
Hypokinetic disorders can be considered lesions of the neostriatum (Fig. 26-12A, B). These lesions result in the loss of inhibitory connections between the neostriatum and the internal segment of the globus pallidus. Thus tonically active pallidothalamic neurons continuously inhibit their thalamic targets. The thalamus is not disinhibited, so there is a decreased flow of information through the thalamus to the cerebral cortex. This decrease in turn causes a decrease in the activity of the appropriate corticospinal and other corticofugal neurons. In addition, most of the connections that subserve the indirect pathway remain intact. Therefore, when those connections are activated, as, for example, by a larger than normal burst in cortical neurons, subthalamopallidal cells excite pallidothalamic neurons, which results in increased inhibition of the thalamus. The combination of a lack of disinhibition of the thalamus by the direct pathway and an increased inhibition of the thalamus by the indirect pathway significantly decreases the level of appropriate activity in the cerebral cortex and increases the level of inappropriate cortical activity. Therefore, the patient becomes less able to execute the appropriate movements. Experiments in nonhuman primates have shown that bradykinetic animals have levels of neuronal activity that are consistent with this view.
Hyperkinetic Disturbances
Hyperkinetic disturbances take the form of dyskinesias. The three most common forms are ballismus, choreiform movements, and athetoid movements.
Ballismus is most typically seen as hemiballismus because it usually occurs on one side. It consists of uncontrolled flinging (ballistic) movements of an upper or lower extremity but is most characteristically seen in the upper extremity. This motor disorder is most commonly seen in patients with vascular lesions localized to the contralateral subthalamic nucleus (Fig. 26-12C, D).
Choreiform movements, which are present in Huntington disease and sometimes in treated Parkinson disease, are generalized irregular (and brisk) dance-like movements of the limbs. Similar movements may occur in oral and facial musculature. These patients may also have a decrease in muscle tone (hypotonia). In Huntington disease, there is an initial selective loss of the medium spiny cells in the striatum, which project to the lateral pallidum, and of acetylcholine-containing neurons in the striatal complex. It is thus likely that the neurons specifically associated with the indirect pathway are lost.
Finally, athetoid movements (athetosis) are a continuous writhing of distal portions of the extremity (Fig. 26-13). Athetosis is generally manifested as slow, sinuous, and writhing movements more obvious in the upper extremities and hands (Fig. 26-13) and face, although any muscle group may be affected. However, athetoid movements may also be seen as a range of abnormal movements. When they are more brisk and resemble chorea, the term choreoathetosis may be appropriate. When the movements are more intense and sustained, they may resemble dystonia, and the term athetotic dystonia may be used.
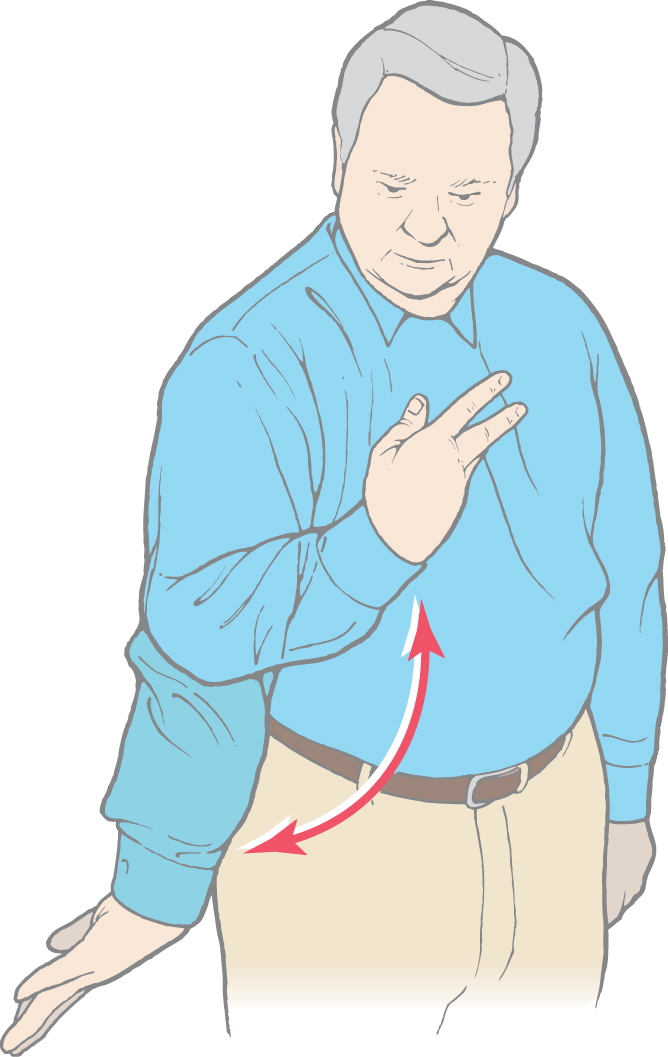
Figure 26-13. Athetoid movements (athetosis) of the upper extremity.
These hyperkinetic disturbances can most easily be explained by the disruption of the indirect pathway through the motor loop, resulting from the loss of excitatory subthalamopallidal neurons (Fig. 26-12C, D). The balance between excitation of pallidothalamic neurons (by the direct pathway) and their inhibition (by the indirect pathway) is skewed. The result is a decrease in the net amount of inhibition of thalamic cells, which results in more activity in the cerebral cortex.
Integrated Function of the Basal Nuclei
By combining the direct and indirect pathways, it is possible to gain a better appreciation of how the basal nuclei affect their targets through a balance between activation by the direct pathway and inactivation by the indirect pathway, as seen in Figure 26-14A, B. The activity in the cerebral cortex activates the striatopallidal neurons in both the direct (red) and indirect (green) pathways. Activation of the striatopallidal neurons in the direct pathway will inhibit pallidothalamic neurons (blue in Fig. 26-14B1). This pathway may be considered to be the initiator of the pause in pallidothalamic activity. At the same time, activation of the striatopallidal neurons in the indirect pathway inhibits the pallidosubthalamic neurons (Fig. 26-14B2). This inhibition releases the subthalamic neurons, which then excite the pallidothalamic neurons. This excitation by the subthalamopallidal neurons is what reactivates the pallidothalamic neurons that were inhibited through the direct pathway. Thus careful balance of the activity between the two pathways modulates the amount of time during which the thalamocortical neurons are activated.
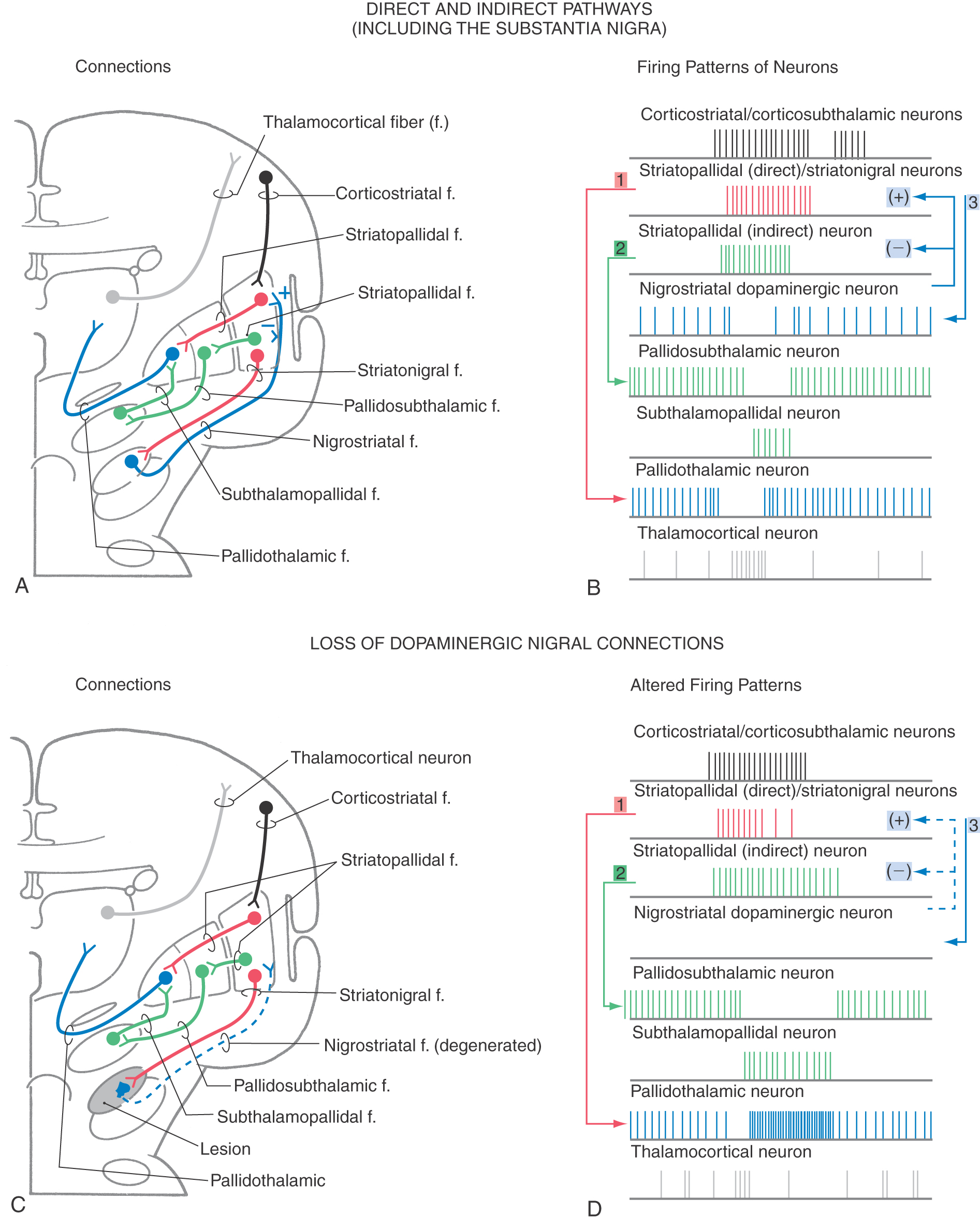
In addition to the activities of these pathways, the dopaminergic loop from the neostriatum to the substantia nigra and back to the neostriatum is also active. Activity of striatonigral projections originating in the striosomes will inhibit the dopaminergic nigrostriatal pathway (Fig. 26-14B3). Subsequent reactivation of the nigral neurons is due in part to a number of factors, including disinhibition of the pars compacta by means of the striatal projection originating from the matrix. The dopaminergic neurons project back onto striatal neurons. The effect of the dopamine depends on the receptor on the postsynaptic striatal neuron. D1 receptors are found on striatal neurons in the “direct” pathway. Stimulation of the D1 receptors causes excitation of the striatopallidal neurons in the direct pathway. This excitation accounts for the slightly longer train of action potentials seen in Figure 26-14B. The neurons belonging to the “indirect” pathway have D2 receptors. When dopamine binds to the D2 receptor, the neurons are inhibited. This inhibition accounts for the shorter train of action potentials for indirect striatopallidal neurons in the indirect pathway.
The modulation between the direct and indirect pathways by dopamine is significantly affected if dopamine is absent. For example, in Parkinson disease, the loss of melanin-containing dopaminergic neurons in the nigral complex has the net effect of decreasing thalamocortical neuronal activity (Fig. 26-14C, D). The striatopallidal activity of the direct pathway initiates the pause (Fig. 26-14D1). However, the activity is less sustained owing to the loss of excitation from the substantia nigra. The activity of the striatopallidal neurons in the indirect pathway is not inhibited by the dopamine and thus is of a somewhat longer duration. This longer duration of activity results in a longer lasting inhibition of the pallidosubthalamic neurons (Fig. 26-14D2), which increases the activity of the subthalamopallidal neurons and results in increased activation of the pallidothalamic neurons. Consequently, not only is the pause in pallidothalamic activity shortened but the inhibition of the thalamus by these neurons is enhanced. The net result is an increased inhibition of the thalamocortical neurons with decreased activity in the cerebral cortex.
This balance between the direct and indirect pathways, as mediated by GABA, glutamate, and dopamine, can be considered the critical component in changes seen in patients with movement disorders. Moreover, studies in animal models of aging have demonstrated that the change in the balance between these chemical systems, particularly in the nucleus accumbens and more ventral portions of the neostriatum, is likely to be responsible for reduction in spontaneous locomotion in older subjects. Furthermore, it appears to be an imbalance in dopamine between the more dorsal and ventral portions of the striatum that accounts for both cognitive enhancement and impairment in Parkinson patients performing different learning paradigms. Thus behavioral manifestations of basal nuclear disorders can be primarily attributed to changes in the balance between GABA, glutamate, and dopamine.
ETIOLOGY OF BASAL NUCLEAR–RELATED DISORDERS
The hallmark of basal nuclear disorders is a change in the neurochemical environment within the striatal complex. Careful examination of patients with basal nuclear syndromes reveals that the classic motor signs and symptoms constitute only one characteristic of these disorders and that associative memory and limbic dysfunctions also occur. Clinical and basic science studies of the chemistry of basal nuclear disorders have provided significant insight into their etiology and treatment.
Huntington Disease
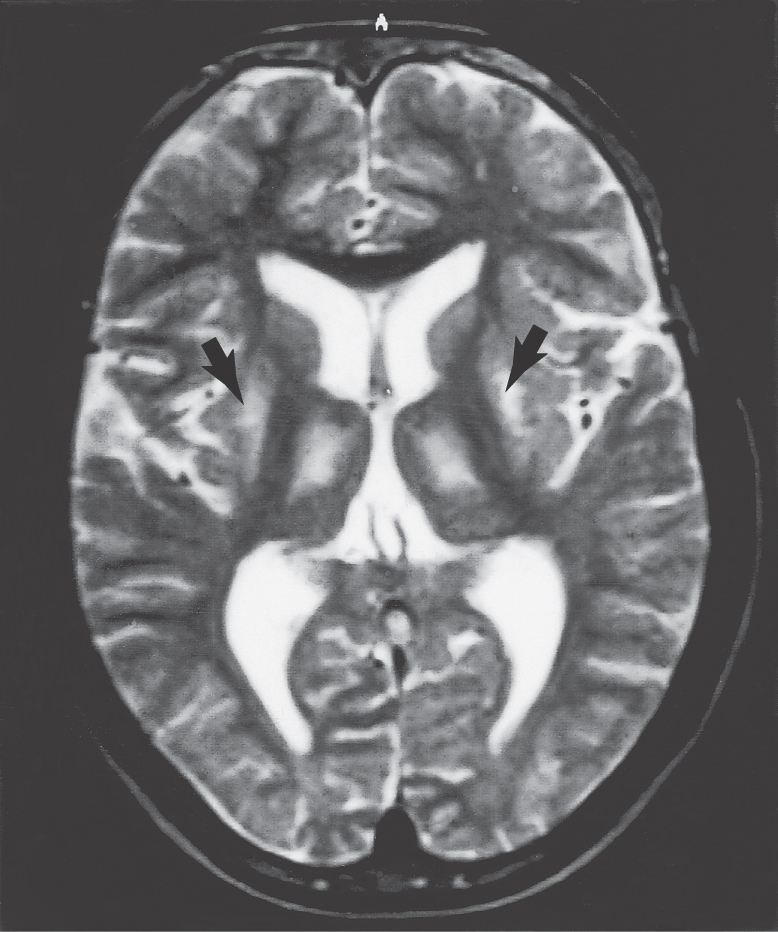
As originally described, Huntington disease is a progressive, untreatable disorder in which patients lose their ability to function and experience increasing dementia; death occurs 10 to 15 years after onset. The initial symptoms indicative of this disease generally begin to appear at 35 to 44 years of age. In the United States, the incidence of Huntington disease is approximately 1 in every 17,000 to 19,000 individuals. In the early stages, this disease is characterized by absentmindedness, irritability, depression, clumsiness, and sudden falls. Choreiform movements gradually increase until the patient is bedridden. Cognitive functions and speech progressively deteriorate. The later stages of this disease are characterized by severe dementia. One salient feature in the progressive memory loss is a difficulty in integrating newly acquired memory into useful information for planning of movements. In addition, a large proportion of these patients develop psychiatric disorders, such as major affective disorder, schizophrenia, and other behavioral disturbances.
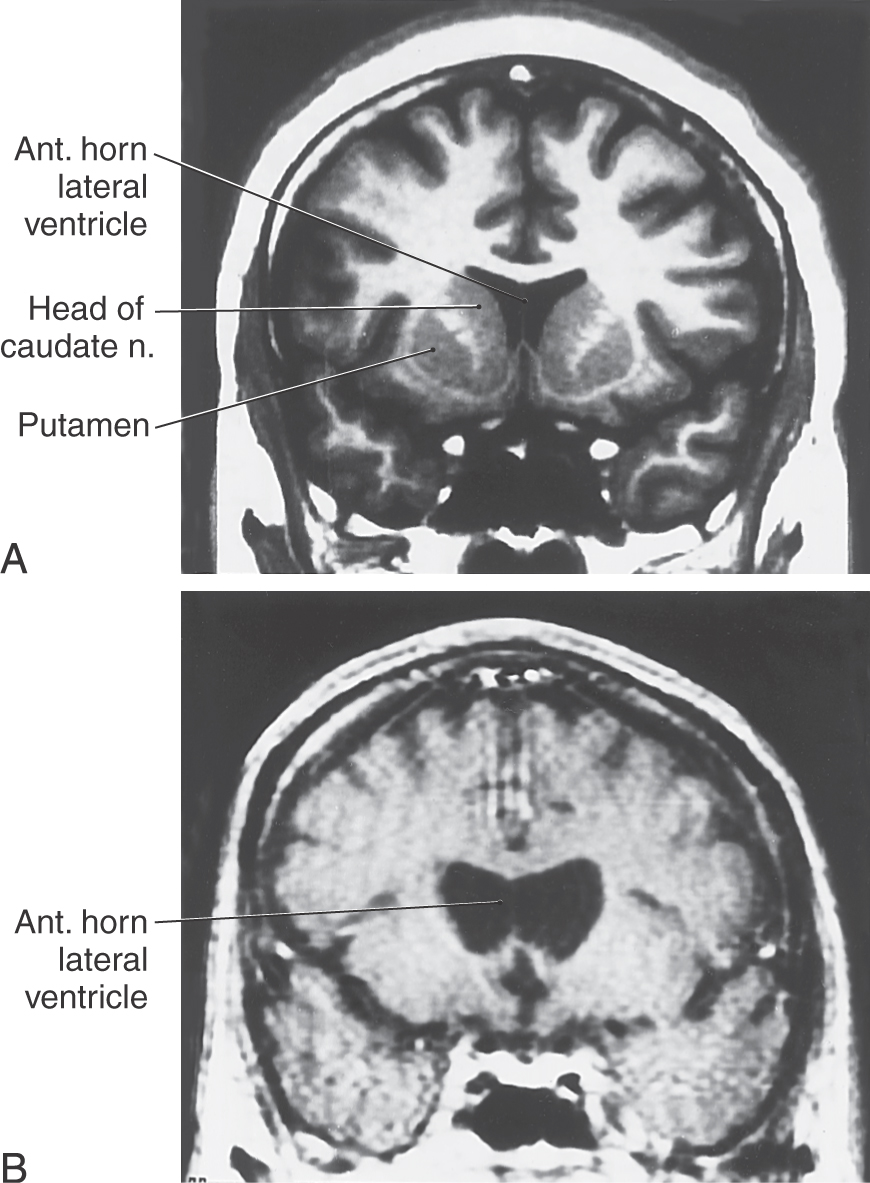
Postmortem examination reveals decrease in the size of the striatal complex caused by loss of about 90% of all striatal neurons and extensive astrocytosis. In particular, medium spiny cells, which project to the lateral pallidum, and the large acetylcholine-containing local circuit cells are lost. This disease can be demonstrated on magnetic resonance imaging as a flattening of the head of the caudate nucleus (Fig. 26-15).
Huntington disease is a genetic disorder inherited in autosomal dominant fashion, and any person who inherits the gene will develop the disease. It is most prevalent among persons of European descent. This disease is known to be due to a mutation on the short arm of chromosome 4. At the molecular level, the mutated gene has dozens of copies of the DNA sequence CAG (cytosine-adenine-guanine), which codes for glutamate. These CAG repeats result in the addition of glutamate residues inside the gene coding for the protein huntingtin. Patients with larger numbers of repeats have a more severe illness with an earlier onset. Thus there are now reliable markers for the disease.
Much effort has been devoted to understanding the mechanism underlying the death of striatal neurons in this disease. In the earliest stages of the disease, which may be months or years before the onset of clinical features, there is a diminution of glucose metabolism in the neostriatum of some patients. Glutamate excitotoxicity is thought to be primarily responsible for this process. Normally, cortical axons release glutamate as their neurotransmitter in the caudate and putamen. Glutamate binds with its receptor on the medium spiny neurons and opens the receptor channel to an influx of ions. This action depolarizes the membrane, resulting in an excitatory postsynaptic potential. As glutamate dissociates from the receptor, it is cleared from the extracellular space by uptake by astrocytes. In Huntington disease, an unknown mechanism causes glutamate to persist at one type of receptor, the N-methyl-D-aspartate (NMDA) receptor, which opens calcium ion channels. The resulting excessive influx of calcium causes an increase in intracellular calcium, which triggers a cascade that leads to cell death. Glutamate excitotoxicity is also thought to be the primary cause of localized neuronal death after acute brain injury, such as stroke (see Chapter 2).
Parkinson Disease
Parkinson disease is also a progressive, debilitating disorder. It affects more than 500,000 Americans, and there are about 50,000 new cases each year. Most cases of adult Parkinson disease appear initially as signs or symptoms in patients in the age range of 45 to 65 years (mean, 55 years). These initial signs may include a slight asymmetric gait or vague clumsiness of a hand, less blinking, and a reduction in or lack of arm swing when walking. In about 70% to 80% of cases, the initial onset of symptoms is a resting tremor that appears on one side but will become bilateral as the disease progresses. In addition, about the same percentage of nigral cells are lost before the onset of motor deficits.
In rare cases, signs or symptoms of Parkinson disease may be seen in patients younger than 20 years (juvenile Parkinson disease) or in patients in the age range of 20 to 40 years (young-onset Parkinson disease). This disease, in these younger patients, is more likely to have a genetic basis. Although equally rare, Parkinson disease may be seen resultant to repeated trauma, such as that experienced by boxers (pugilistic Parkinson disease). Some familial groups with Parkinson disease have been described.
Whereas the etiology of Parkinson disease is largely unknown, it is recognized that it may be inherited in about 2% or less of cases. On the other hand, in about 5% to 10% of patients with Parkinson disease, the history may reveal that other family members may have similar neurologic deficits or symptoms. In most Parkinson patients, however, the specific cause is enigmatic, and a wide range of possibilities, such as excitotoxicity, oxidative stress, malfunction of mitochondria, and recent or remote exposure to toxins, have been proposed.
This disorder is characterized by a progressive onset of movement and affective disturbances (Fig. 26-16). The movement disorders include tremor at rest, cogwheel (gamma) rigidity (increased muscle tone), akinesia, bradykinesia, disturbances of eye movements, and loss of postural reflexes. A classic picture of a patient with Parkinson disease is a person sitting or standing with pill-rolling tremor, a blank stare (reptilian or decreased blink), a flexed posture, and a paucity of movement (Fig. 26-16). When the patient starts to move, there is a shuffling start, as if the feet were stuck in place (also called a festinating gait), followed by nearly normal gait. A progressive decline in cognitive functions is seen in later stages of the disease. This may include slowness in responding to questions or instructions (bradyphrenia), memory dysfunction, and dementia.
At autopsy, the nigral complex has two characteristic features. First, compared with a healthy individual, the neurons of the pars compacta have largely or completely lost their dark melanin granules with a concurrent loss of dopamine in the nigrostriatal pathway (Fig. 26-7B, C, D). Clinical symptoms usually appear after approximately 70% to 80% of nigral neurons and their corresponding granules and dopamine are lost. Second, in surviving neurons that have lost most or all of their granules and dopamine, intracytoplasmic inclusion bodies are seen. These are Lewy bodies, and they appear as round eosinophilic structures surrounded by a light halo (Fig. 26-7D). Whereas Lewy bodies are a distinguishing feature of Parkinson disease, they may also be present in other neurologic disorders. Although there is degeneration of serotoninergic and noradrenergic pathways, it is this specific loss of dopamine that results in the observed symptoms. These motor and cognitive symptoms become progressively worse over time and presumably are related to loss of the dopaminergic cells over time. Dementia observed in the later stages of the disease is attributed to progressive chemical imbalances within the basal nuclei.
The standard treatment aims to replace the lost dopamine. Because dopamine itself will not cross the blood-brain barrier, patients are given L-3,4-hydroxyphenylalanine (L-dopa), which will. This agent is now combined with a second drug, carbidopa, which does not cross the blood-brain barrier but has the effect of inhibiting peripheral uptake of L-dopa and thus increasing the amount of L-dopa available to brain tissue. Persons receiving this combination therapy, along with other drugs such as dopamine agonists and monoamine oxidase inhibitors, show significant reductions in signs and symptoms, although progression of the disease is not arrested. Why L-dopa works is not clear. The method by which L-dopa is converted to dopamine in the brain of Parkinson disease patients is not known; these persons have very little tyrosine hydroxylase, the enzyme that is necessary for this catabolism. Moreover, the dopamine is not localized to specific nerve terminals or to the basal nuclei. Thus it appears that dopamine simply needs to be in the neural environment of the striatal complex to reduce the signs and symptoms of the patients. Different areas of the striatal complex are affected by the balance of dopamine, GABA, and glutamate within those regions. Moreover, the changes in the balance between dopamine and GABA in the substantia nigra and the ventral tegmental area also add to the complexity of understanding the palliative effects of L-dopa. However, L-dopa therapy is the most effective form of treatment for Parkinson disease.
Insight into one possible mechanism of Parkinson disease was inadvertently realized by a group of illicit drug manufacturers whose heroin was contaminated by a compound called 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Persons who used this drug developed symptoms exactly like those of patients with Parkinson disease but at an age (early 20s) when this disease is normally never manifested. Autopsy revealed a profound loss of dopaminergic neurons in the substantia nigra pars compacta.
Studies on the mechanism of action of MPTP in animal models showed that MPTP is converted into an active form (MPP+) before it causes a loss of dopaminergic cells. This metabolic pathway requires monoamine oxidase (MAO). Thus it was hypothesized that the use of MAO inhibitors may affect the progression of Parkinson disease. In fact, clinical trials have now shown that the drug L-deprenyl, an MAO-B inhibitor, slows the progression of Parkinson disease and increases the levels of dopamine in the brain. The increase in dopamine may result both from protection of neurons against toxicity and from blocking of the degradation pathway for dopamine, which requires the MAO enzyme.
In recent years, there have been an increasing number of surgical treatments for Parkinson disease. The most common type of surgical intervention is ablative surgery, in which either the ventral intermediate nucleus of the thalamus (thalamotomy) or the posterolateral part of the internal segment of the globus pallidus (pallidotomy) is lesioned. Another surgical approach involves introduction of stimulation electrodes directed at the thalamus, globus pallidus, or subthalamic nuclei. Each of these surgical approaches appears to be effective for treatment of one set of symptoms. However, none of these approaches completely eliminates the disease or prevents its progression.
A controversial method of treatment for Parkinson disease patients involves the use of human embryonic or autologous transplants. Tissues that produce dopamine, such as substantia nigra (embryonic) and the adrenal cortex (autologous), are obtained and separated into cell suspensions. These are then injected into the lateral ventricles of the patient. The idea is that these cells will adhere to the walls of the ventricle and produce dopamine. The dopamine will then diffuse into the nearby cerebral cortex and basal nuclei. This procedure has now been performed in humans as well as in animals. In the clinical trials to date, only a small number of patients have benefited from this approach.
Wilson Disease
Wilson disease, also known as hepatolenticular degeneration, is also related to the basal nuclei. This genetic disorder, inherited as an autosomal recessive trait, is due to a mutation on the long arm of chromosome 13 (13q14.3) and has a particularly high incidence in eastern European Jewish and southern Italian population groups or descendants of these groups.
Onset of the disorder is typically between 11 and 25 years of age. Wilson disease is a disorder of copper metabolism that results in accumulation of the metal in the liver, resulting in small necrotic lesions leading to cirrhotic nodules and progressive liver damage, and damage to various areas of the brain, most particularly the lenticular nucleus. Signs of liver damage may precede the onset of neurologic abnormalities by several years. Another metabolic feature, probably due to damage of the tubules in the kidney, is aminoaciduria, excessive amounts of amino acids in the urine. In the eye, copper accumulates in the periphery of the cornea; these deposits are responsible for the Kayser-Fleischer ring (present in about 75% of symptomatic patients; Fig. 26-17), which may appear yellow to green to brown. Degeneration of the putamen, often forming small cavities, is the predominant pathoanatomic feature in the brain (Fig. 26-18). However, other regions of the brain, including the thalamus, the head of the caudate, and the frontal and cerebellar cortices, may also show similar changes. This degeneration is due to a loss of neurons, axonal degeneration, and increasing numbers of protoplasmic astrocytes.
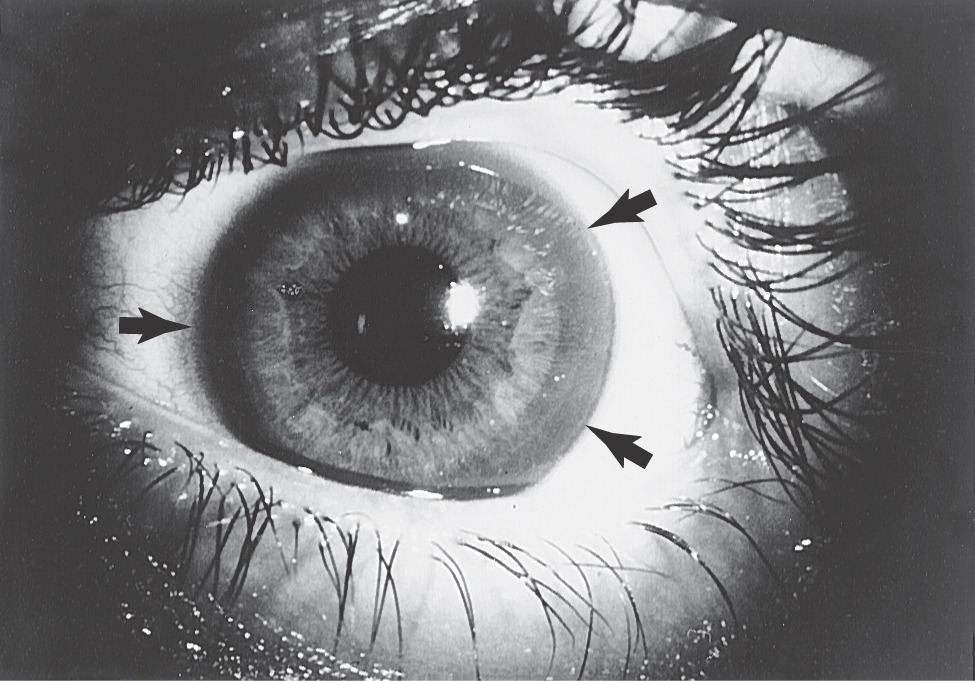
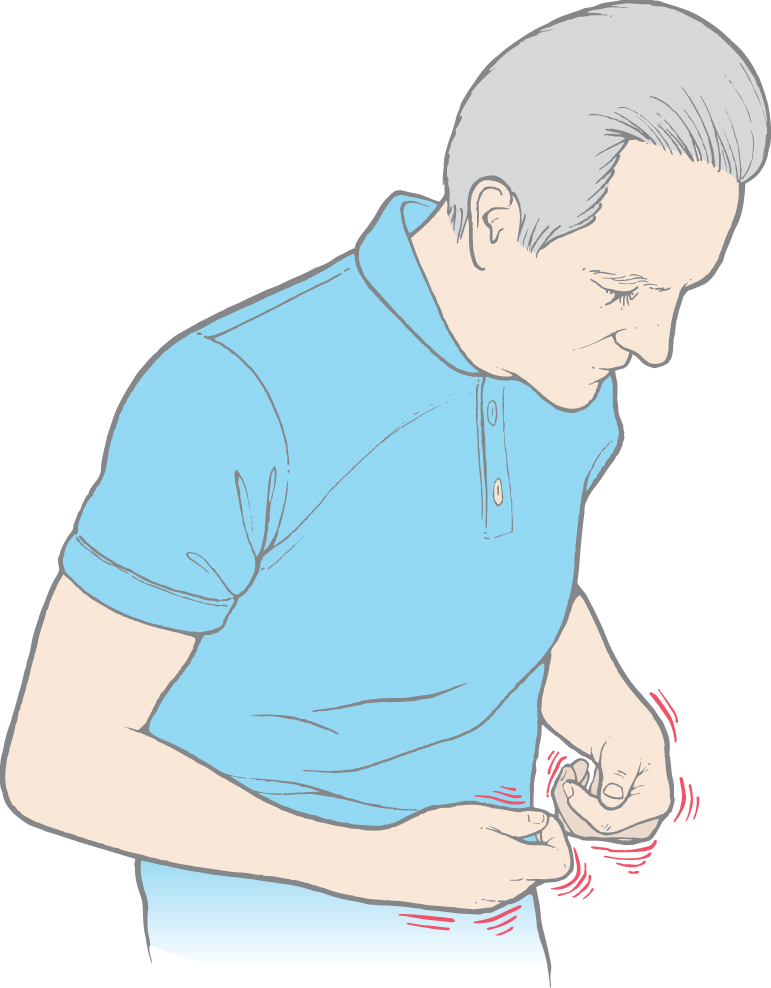
As in other basal nuclear disorders, many patients with Wilson disease will develop psychiatric symptoms, such as changes in personality, argumentative behavior, or emotional lability. However, the motor disturbances are often the most evident signs and include tremor, dysarthria, diminished dexterity, unsteady gait, and rigidity. The most common form of movement disorder in this disease is known as a wing-beating tremor; these movements are clearly different from resting and intention tremors. Affected patients do not have a tremor at rest. However, after the arms are extended, a beating movement develops that can be restricted to the wrists or can result in the arms being thrown up and down in a wide arc. These patients may also present with a mask-like facial expression, gaping mouth, and what can be called a vacuous smile.
Treatment is essential, and the goal is to decrease the amount of copper within the body, thus limiting its toxic effects, and to reduce dietary copper intake. Chelating agents such as TETA (triethylenetetramine dihydrochloride) and penicillamine may be appropriate for some patients. Others may require other agents, but all may have some side effects. Prognosis for patients who have completed the first few years of treatment is very good, with diminution of neurologic signs within 5 to 6 months from the onset of therapy.
Sydenham Chorea
This is a childhood autoimmune disease that is infrequently seen. This disease typically affects children between the ages of 5 and 15 years, and girls are affected more than boys by a ratio of about 2:1. It is a consequence of rheumatic fever, which is caused by infection with group A β-hemolytic streptococci, and the onset may be delayed by weeks or even months after the resolution of the precipitating infection. The disease is self-limited and is rarely fatal; fatalities are usually attributed to consequences of rheumatic fever. The chorea may not appear until 6 months or longer after infection and typically lasts 3 to 6 weeks. Patients present with rapid, irregular, aimless movements of the limbs, face, and trunk. These movements are more flowing and “restless” than those in Huntington disease patients. In addition, patients with Sydenham chorea have some muscle weakness and hypotonia. Other signs and symptoms may include irritability, emotional lability, obsessive-compulsive behaviors, attention deficit, and anxiety. Fortunately, this is a benign disease, and most patients experience complete recovery from the symptoms. However, about one third of patients may have recurrences of signs and symptoms after several months or even years. A minority of patients (2% or less) may have severe complications or death, due mainly to cardiac problems.
Tardive Dyskinesia
Tardive dyskinesia is a basal nuclear disorder that is iatrogenic in nature, that is, caused by medical intervention for another disease. This condition is caused by chronic treatment with neuroleptic medications, such as the phenothiazines (e.g., chlorpromazine, thioridazine) and butyrophenones (e.g., haloperidol). The manifestation of this condition is uncontrolled involuntary movements, particularly of the face, mouth, and tongue, and cogwheel rigidity. These abnormalities may be temporary or permanent. The action of these neuroleptic drugs is to block dopaminergic transmission throughout the brain. The primary target cells are those in the ventral tegmental area that form the mesolimbic dopaminergic pathway. Prolonged treatment with neuroleptic drugs may lead to blockage of the D2 dopamine receptor, which causes an imbalance in the nigrostriatal influence on the basal nuclear motor loop and ultimately results in movement disorders.
Treatment of tardive dyskinesia is complicated by the fact that withdrawal of the causative medication may result in exacerbation of the underlying psychotic state. Medications that may cause tardive dyskinesia should be used with full knowledge of their potential complications and when other treatments or medications may not be appropriate.
Sources and Additional Reading
Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381.
Caplan LR. Caplan’s Stroke: A Clinical Approach. 3rd ed. Boston: Butterworth Heinemann; 2000.
Goldman-Rakic PS, ed. Basal ganglia research (Special Issue). In: Trends Neurosci.13. 1990:241–308.
Hankey GJ, Wardlaw JM. Clinical Neurology. New York: Demos Medical Publishing; 2002.
Pryse-Phillips W. Companion to Clinical Neurology. 2nd ed. New York: Oxford University Press; 2003.
Rowland LP, ed. Merritt’s Neurology. 10th ed Philadelphia: Lippincott Williams & Wilkins; 2000.
Shultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]Chapter 26
The Basal Nuclei
Components of the Basal Nuclei
Parabrachial Pontine Reticular Formation
Direct and Indirect Pathways of Basal Nuclear Activity
Disinhibition as the Primary Mode of Basal Nuclear Function
Parallel Circuits of Information Flow Through the Basal Nuclei
Disease Implications of Topography of Basal Nuclear Circuits
Behavioral Functions of the Basal Nuclei
Integrated Function of the Basal Nuclei
Voluntary movement is essential to the well-being of living animals. Such behaviors are accomplished by signals that direct the actions of individual muscles. Although these signals originate in the cerebral cortex, they are modulated by a variety of subcortical structures. One such group of structures is the basal nuclei and their functionally associated cell groups. Classically, motor systems have been divided into pyramidal and extrapyramidal on the basis of whether the pathway is mediated by corticofugal neurons (pyramidal) or by the basal nuclei, cerebellum, or descending brainstem pathways (extrapyramidal). However, this distinction is overly simplistic if not inaccurate. Consequently, it is not used here. The basal nuclei are involved in a wide variety of motor and affective behaviors, in sensorimotor integration, and in cognitive functions.
OVERVIEW
These nuclei were traditionally called the basal ganglia rather than the basal nuclei, even though “ganglia” is usually reserved for groups of nerve cell bodies in the peripheral nervous system. The official term basal nuclei is used throughout this chapter, although the unofficial term basal ganglia is also commonly seen in the literature. For practical purposes, these terms may be considered interchangeable, although basal nuclei is the correct and preferred term.
The basal nuclei consist of cell groups embedded in the cerebral hemisphere. Although not classified as cell groups of the basal nuclei in a strict sense, the subthalamic nucleus, substantia nigra, and pedunculopontine tegmental nucleus are integral parts of the pathways passing through these forebrain cell groups. Collectively, the basal nuclei and their associated nuclei function primarily as components in a series of parallel circuits from the cerebral cortex through the basal nuclei to the thalamus and then back to the cerebral cortex.
Four fundamental concepts are crucial to understanding of the basal nuclei. First, damage to or disorders of the basal nuclei result in disruption of movements and may also cause significant deficits in other neural functions, such as cognition, perception, and mentation. Second, the basal nuclei are anatomically and functionally segregated into parallel circuits that process different types of behaviorally significant information. Third, the basal nuclei function primarily through disinhibition (release from inhibition). Fourth, diseases of the basal nuclei can be described as disruptions of the neurochemical interactions between elements of the basal nuclei. These neurochemical relationships rely not simply on the neurotransmitters involved but also on the characteristics of the transmitter receptors, on the locations of the synapses, and on other inputs received by these cells.
In summary, the basal nuclei integrate and modulate cortical information along multiple independent parallel channels. These channels affect behavior indirectly by feedback to the cerebral cortex and directly by providing information to subcortical centers that influence movements. Disruption of these channels by stroke or disease results in dysfunctions initially observed in the motor sphere with subsequent disruption in other behavioral domains.
COMPONENTS OF THE BASAL NUCLEI
The basal nuclei are typically divided into dorsal and ventral divisions. The dorsal basal nuclei include the caudate and putamen (together constituting the neostriatum) and the globus pallidus (constituting the paleostriatum) (Fig. 26-1A). Associated with the dorsal basal nuclei, in a functional sense, are the substantia nigra, the subthalamic nucleus, and the parabrachial pontine reticular formation (containing the pedunculopontine tegmental nucleus). The ventral basal nuclei are located inferior to the anterior commissure and include the nucleus accumbens, substantia innominata, nucleus basalis of Meynert, and olfactory tubercle. This ventral region is intimately associated with portions of the amygdala and ventral tegmental area. For the purposes of this chapter, the basal nuclei are regarded as making up two complexes: the striatal complex and the pallidal complex (Fig. 26-1B).
The telencephalic regions of the basal nuclei are supplied by the medial striate artery, the lenticulostriate branches of the M1 segment of the middle cerebral artery, and the anterior choroidal artery (Fig. 26-2). The diencephalic and mesencephalic regions are supplied by the posteromedial branches of the P1 segment of the posterior cerebral artery and branches of the posterior communicating artery. Diseases of these vessels may result in various behavioral or motor deficits, depending on which vessel and region are affected.
Striatal Complex
The striatal complex is a functional unit composed of the neostriatum and ventral striatum (Fig. 26-3A, B). The neostriatum consists of the caudate nucleus and putamen. These two nuclei have the same embryologic origin and similar connections. Although fused rostroventrally, they are separated throughout most of their extents by fibers of the internal capsule. The ventral striatum is composed of the nucleus accumbens and portions of the olfactory tubercle (Figs. 26-1A, B and 26-3A). The nucleus accumbens is located rostroventrally in the hemisphere, at the point where the putamen is continuous with the head of the caudate (Figs. 26-3A and 26-4A, B). It is internal to part of the anterior perforated substance. Portions of the olfactory tubercle are considered part of the ventral striatum because of functional, cytoarchitectural, and chemoarchitectural similarities. A defining characteristic of the striatal complex, patches (also called striosomes) are particularly prominent in the head of the caudate (Fig. 26-4B). Patches are acetylcholinesterase-poor regions within the striatal complex. They contain large amounts of one or more neuropeptides and one or more types of opiate receptors. Patches are surrounded by matrix (Fig. 26-4B), which contains high concentrations of acetylcholine and therefore stains darkly when tissue is histochemically reacted to reveal this enzyme. In addition to histochemical and receptor differences, these regions also receive projections from different cortical regions and project to different targets.
The largest afferent projections to the neostriatum are from the cerebral cortex (corticostriatal fibers) (Fig. 26-5A). Other afferents are from the thalamus (thalamostriatal fibers), substantia nigra (nigrostriatal fibers), and parabrachial pontine reticular formation (pedunculopontostriatal fibers) (Fig. 26-5A). The efferent projections of the striatum reach primarily the pallidum (striatopallidal fibers) and the nigral complex (striatonigral fibers) and to a small degree the subthalamic nucleus.
Most of the neurons in the neostriatum are called medium spiny neurons, so named because of their medium-sized cell bodies and the large numbers of spines on their dendrites (Fig. 26-6). Most medium spiny cells have dendritic fields that are restricted to the patch or matrix compartment in which the cell bodies are located.
The nucleus accumbens forms the majority of the ventral striatum (Fig. 26-4A). This nucleus is divided into a core region and a shell region. The core region is cytoarchitecturally and histochemically identical to the neostriatum. It has similar efferent and afferent connections, albeit from cortical regions and to different pallidal and midbrain nuclei. The shell is somewhat histochemically different and has a more diffuse set of connections. It receives projections primarily from allocortical regions and sends projections to the ventral pallidum, substantia nigra–ventral tegmental area, parabrachial nucleus, periaqueductal gray, lateral hypothalamus, and lateral preoptic area (Fig. 26-4B).
Medium spiny neurons fire few action potentials spontaneously and thus require activation by their afferent fibers. These cells use the inhibitory neurotransmitter γ-aminobutyric acid (GABA) and may also contain neuroactive peptides such as substance P and enkephalin. Thus when medium spiny neurons are activated, they subserve both direct inhibitory and neuromodulatory functions at their targets. The axons of medium spiny neurons are the efferent fibers of the neostriatum, collectively forming the striatopallidal fibers.
Also found in the neostriatum are large, acetylcholine-containing local circuit neurons that modulate local activity within the neostriatum. Huntington disease is characterized by progressive loss of medium spiny neurons and acetylcholine-containing neurons throughout the striatal complex.
Pallidal Complex
The pallidal complex is composed of the globus pallidus and the ventral pallidum. The latter is largely synonymous with the substantia innominata (Fig. 26-3B, C). The pallidal complex contains primarily GABAergic neurons with high rates of spontaneous activity. Consequently, these cells tonically inhibit their targets.
The globus pallidus is divided into medial (internal) and lateral (external) segments by a sheet of white matter (the medullary lamina) (Fig. 26-3C). The substantia innominata is located inferior to the anterior commissure and internal to the anterior perforated substance. One important cell group in the substantia innominata is the basal nucleus of Meynert. This nucleus has large acetylcholine-containing neurons, which are lost in Alzheimer disease. However, this disease is not considered a basal nuclear disorder because acetylcholine-containing cells in the cerebral cortex, hippocampus, and septum are also lost in patients with Alzheimer disease. This disease is further characterized by other biochemical and pathologic features, such as senile plaques.
The two divisions of the globus pallidus are reciprocally connected (pallidopallidal fibers) (Fig. 26-5B) but subserve different functions. The main afferent input to the pallidum is from the striatal complex. Medium spiny neurons from the striatum that project to the medial segment and substantia nigra use GABA and substance P; those that project to the lateral segment use GABA and enkephalin (Fig. 26-5B).
The medial division is composed of the medial segment of the globus pallidus. It subserves the direct basal nuclear pathway (described later) and projects primarily to the thalamus (pallidothalamic fibers) (Fig. 26-5B). These fibers exit the globus pallidus as two bundles: the ansa lenticularis and the lenticular fasciculus (Figs. 26-3C, D; see also Figs. 26-8 and 26-9). The ansa lenticularis originates from lateral portions of the medial segment and loops around the posterior limb of the internal capsule to enter the prerubral field (field H of Forel). The lenticular fasciculus (field H2 of Forel), on the other hand, originates in the posteromedial portion of the medial segment. These fibers traverse the internal capsule as small groups of axons, merge to form the lenticular fasciculus between the zona incerta and subthalamic nucleus, and then enter field H of Forel. In the Forel field, the ansa lenticularis and lenticular fasciculus join the thalamic fasciculus (field H1 of Forel), which courses immediately superior to the zona incerta (Fig. 26-3C, D; see also Figs. 26-8 and 26-9). These fibers ultimately terminate in ventral anterior, ventral lateral, and centromedian nuclei of the thalamus. The medial division of the pallidal complex is a principal efferent nucleus of the basal nuclei, the axons of these cells comprising the ansa lenticularis and the lenticular fasciculus.
The lateral division is composed of the external (or lateral) segment of the globus pallidus and the ventral pallidum. This division subserves the indirect basal nuclear pathway (see later). These nuclei receive a large input from the striatal complex (striatopallidal fibers) and small projections from the subthalamic nucleus (subthalamopallidal fibers) and the substantia nigra pars reticulata (nigropallidal fibers). They project strongly to the subthalamic nucleus (pallidosubthalamic fibers) and are also connected with the substantia nigra (pallidonigral fibers) (Fig. 26-5B).
Subthalamic Nucleus
The subthalamic nucleus is a lens-shaped cell group that makes up the largest part of the ventral thalamus. It is immediately inferior to the zona incerta and rostral to the substantia nigra (Fig. 26-3C, D). It receives projections from the lateral pallidal division (pallidosubthalamic fibers), cerebral cortex (corticosubthalamic fibers), nigral complex (nigrosubthalamic fibers), and parabrachial pontine reticular formation. The subthalamic nucleus projects to both pallidal divisions (subthalamopallidal fibers) and to the substantia nigra (subthalamonigral fibers) (Fig. 26-5C). These connections, especially the subthalamopallidal projections to the medial globus pallidus, are an essential part of the indirect pathway underlying basal nuclear function.
Subthalamic neurons use the excitatory neurotransmitter glutamate. Most of the time, subthalamic cells are inactive because of the constant inhibition by cells of the external pallidal segment. However, if this inhibition is removed, subthalamic neurons have a high level of activity resulting in a characteristic motor deficit described later in this chapter. This activity is mediated in part by a large corticosubthalamic projection.
Nigral Complex
The nigral complex is composed of the substantia nigra and the ventral (anterior) tegmental area (Fig. 26-3C, D). The substantia nigra is divided into a cell-dense portion (pars compacta) and a reticulated portion (referred to here as the pars reticulata, although it can be divided into a pars reticulata and a pars lateralis). The pars reticulata is located at and within the medial edge of the descending corticofugal fibers that form the crus cerebri. The pars compacta and the adjacent ventral tegmental area appear to subserve similar functions and to have a similar chemoarchitectural organization. The major afferents to the nigral complex are from the striatal and pallidal complexes. The nigral complex also receives cortical (corticonigral), subthalamic (subthalamonigral), and pedunculopontine fibers (Fig. 26-5D).
The pars compacta contains a large number of neuromelanin-containing cells, whose dark color gives the nucleus its name (substantia nigra, “black substance”). Cells of the pars compacta in healthy individuals appear characteristically dark in brain slices and are packed with small black granules in histologic sections (Fig. 26-7A–C). Neurons in the pars compacta use the neurotransmitter dopamine and project primarily to the neostriatum as nigrostriatal fibers. The dopamine released by these cells may excite or inhibit striatal neurons, depending on the type of receptor on the postsynaptic membrane.
The pars reticulata is formed by loose aggregations of medium-sized to large GABAergic neurons that are indistinguishable from those of the medial pallidum. Neurons in the pars reticulata have axons with an extensive system of collaterals; consequently, they may project to and inhibit one or more target structures. These targets include the neostriatum (nigrostriatal fibers), thalamus (nigrothalamic fibers), superior colliculus (nigrotectal fibers), and parabrachial pontine reticular formation. These cells have a high rate of discharge and tonically inhibit their targets. Projections of pars reticulata neurons represent an important pathway by which the basal nuclei influence other motor centers.
The pars compacta and the pars reticulata are interconnected. Dendrites of dopaminergic pars compacta neurons extend into the pars reticulata, where they release free dopamine (by a nonvesicular mechanism). The level of dopamine modulates the resting membrane potential of pars reticulata cells, making them either more or less likely to discharge, depending on the subtype of dopamine receptor they possess. In turn, pars reticulata neurons have axon collaterals that ramify extensively in the pars compacta and form GABAergic synapses. Collectively, these interactions form modulatory loops between neurons of the pars compacta and the pars reticulata.
These modulatory loops are influenced by the output of the striatal system. Neurons in the patches project predominantly onto the cells in the pars compacta, whereas the neurons in the matrix project predominantly onto the cells in the pars reticulata. Thus the striatonigral projection from patches directly inhibits the dopaminergic nigrostriatal neurons, and the projection from the matrix inhibits the GABAergic neurons in the pars reticulata. Consequently, inhibition of the pars reticulata is reduced and its targets, such as the pars compacta, are released from inhibition and may become more active.
The ventral tegmental area is located medial to the substantia nigra. It contains large numbers of dopaminergic neurons and forms connections with the ventral striatum, the amygdala, and other limbic system structures. Cells of the ventral tegmental area project to and terminate on striatal neurons that have postsynaptic D2 (dopamine) receptors. In schizophrenia, there is an increase in number and in sensitivity of these receptors. Neuroleptic drugs help control schizophrenia by blocking (downregulating) these D2 receptors.
Parabrachial Pontine Reticular Formation
[/not-level-membership-for-basic-science-category]
