[level-membership-for-cardiovascular-category]
CHAPTER 47 Tetralogy of Fallot
PREVALENCE AND EPIDEMIOLOGY
Tetralogy of Fallot is the most common congenital heart disease manifesting with cyanosis. The incidence is 0.06% of live births, and it constitutes 5% to 7% of all congenital heart disease.1,2 Genetic syndromes are associated with tetralogy of Fallot in 20% of cases with 22q11 deletion and trisomy 21 being the most common.3 Complete atrioventricular septal defects are present in 2% of patients with tetralogy of Fallot, especially patients with trisomy 21.4 Alagille syndrome, VACTERL syndrome (vertebral anomalies, anal atresia, cardiovascular anomalies, tracheoesophageal fistula, esophageal atresia, renal anomalies, limb anomalies), and CHARGE syndrome (coloboma of the eye, heart defects, atresia of the choanae, retardation of growth and development, genitourinary abnormalities; ear abnormalities and deafness) are also associated with tetralogy of Fallot. Atrial septal defect is present in one third of patients with tetralogy of Fallot. Currently, 85% of children with tetralogy of Fallot survive to adulthood, increasing the prevalence of tetralogy of Fallot in adults.
Aberrant subclavian arteries are associated in 5% to 8% of patients with tetralogy of Fallot. The prevalence of tetralogy of Fallot and anomalous subclavian artery is greater if associated with a right aortic arch or pulmonary atresia. Anomalous subclavian artery occurs in 24% of patients with tetralogy of Fallot, right aortic arch, and pulmonary atresia.5
Coronary artery anomalies are common, occurring in 5% to 12% of children with tetralogy of Fallot.6,7 Coronary artery anomalies that result in major branches coursing anterior to the right ventricular outflow tract (RVOT) interfere with transannular patch angioplasty of the outflow tract.
MANIFESTATIONS OF DISEASE
Clinical Presentation
A child with a palliative shunt has a continuous murmur similar to that heard in a child with a patent ductus arteriosus. After corrective surgery, symptoms may reflect continued right-sided obstruction or pulmonary regurgitation. Corrective surgery for tetralogy of Fallot requires enlargement of the RVOT and frequently a transannular patch, which results in pulmonary regurgitation.8–10 Pulmonary regurgitation is initially well tolerated, but later is associated with fatigue. The right ventricle and its outflow tract progressively dilate. Right ventricular enlargement leads to distortion of the tricuspid valve annulus and tricuspid regurgitation, compromising right heart function further. ECG abnormalities with lengthening of the QRS complex can lead to arrhythmia that may result in sudden death. Diastolic dysfunction can progress to irreversible systolic dysfunction.8 Revision of the RVOT with a valved conduit causes right ventricular remodeling and improved cardiac function. Pseudoaneurysms of the RVOT can follow corrective surgery with a transannular patch or pulmonary conduit.
Aortic root dilation and regurgitation are common in adults with tetralogy of Fallot and can lead to arterial medial abnormalities. Risk factors for aortopathy include patients with pulmonary atresia, right aortic arch, history of aorticopulmonary shunts, male sex, and chromosome 22q11 deletions.11
Imaging Technique and Findings
Radiography
Radiography plays a secondary role to echocardiography. The cardiac silhouette is frequently normal in size and configuration (Fig. 47-1). The classic radiographic appearance is a boot-shaped heart caused by elevation of the cardiac apex from right ventricular hypertrophy with a diminished or unapparent pulmonary artery segment (Fig. 47-2). The appearance of a boot-shaped heart can be mimicked if the central x-ray beam is caudal to the heart causing cephalic projection of the more anterior portion of the heart (Fig. 47-3). The pulmonary vascularity is diminished, but may be undetectable on a conventional radiograph if right ventricular obstruction is mild. A right aortic arch is visible in 25% of patients with tetralogy of Fallot. Direct visualization of the aortic arch and tracheal shift can be difficult to detect in infants and young children. The side of the aortic arch is reliably determined, however, by noting the increased density of the pedicles on the side of the aortic arch. Patients with tetralogy of Fallot and absent pulmonary artery have enlargement of the central pulmonary arteries (Fig. 47-4).
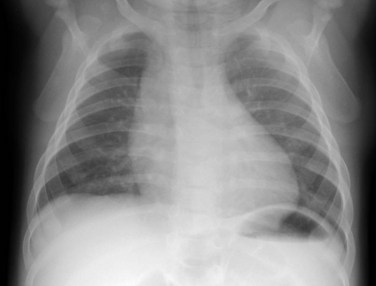
 FIGURE 47-1
FIGURE 47-1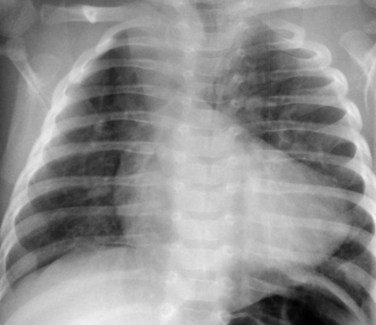
 FIGURE 47-2
FIGURE 47-2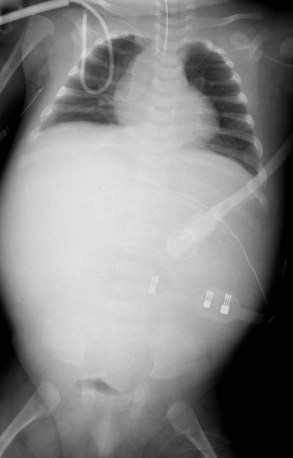
 FIGURE 47-3
FIGURE 47-3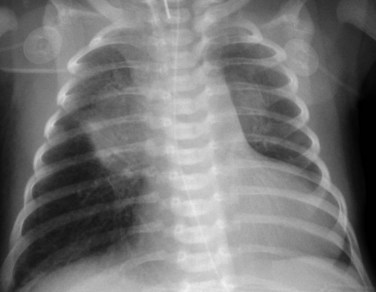
 FIGURE 47-4
FIGURE 47-4Ultrasonography
Echocardiography is definitive for the prenatal and postnatal diagnosis of tetralogy of Fallot.12 A large VSD is identified in utero on the four-chamber view, but evaluation of the outflow tracts is necessary for the diagnosis because other conotruncal abnormalities can have a VSD and an overriding aorta. A large aorta overriding the VSD is present. The aorta is larger than the pulmonary artery. Doppler interrogation of the fetal RVOT shows elevated velocity. A left-to-right shunt through the ductus arteriosus suggests severe RVOT obstruction.13
Postnatal echocardiography characterizes all intracardiac features of tetralogy of Fallot. The VSD and aorta override are identified on the parasternal long-axis view (Fig. 47-5). The outflow tract must also be evaluated (Fig. 47-6) because truncus arteriosus and VSD with pulmonary atresia can have the same appearance. In contrast to double outlet right ventricle, the aortic override to the right ventricle is less than 50%, and aortic valve fibrous continuity with the mitral valve is preserved. The RVOT including the infundibulum, pulmonary valve, and pulmonary artery is measured for stenosis. The measured infundibular volume is small, and owing to reduced growth compared with somatic growth becomes progressively more stenotic.14 Infants who require earlier surgical intervention have a greater reduction in infundibular volume. The pulmonary annulus can be hypoplastic and bicuspid. Hypoplasia with a z score less than 2 is an indication for a transannular patch. Branch pulmonary artery continuity and size is also determined. The position of the coronary arteries must be established. A coronary anomaly with a major coronary branch coursing anterior to the RVOT must be excluded before repair. CT or MRI may be necessary if the coronary arteries or pulmonary arteries are inadequately visualized.
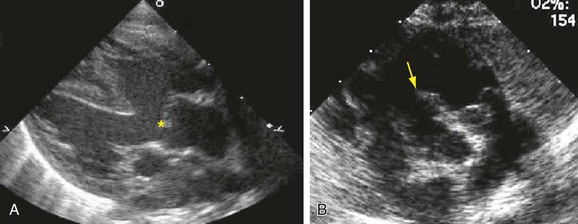
 FIGURE 47-5
FIGURE 47-5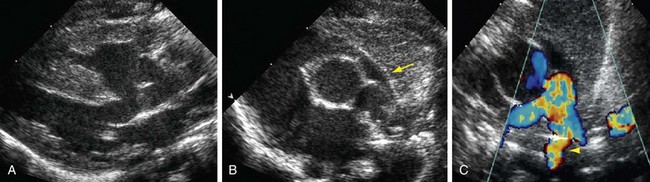
 FIGURE 47-6
FIGURE 47-6Computed Tomography
CT is a complementary imaging modality. Echocardiography is the primary imaging modality, and MRI is the preferred complementary imaging modality. The high diagnostic dose of ionizing radiation and the inability to detect flow patterns with CT make MRI the preferred complementary modality. Multidetector CT has an increasing role in the evaluation of tetralogy of Fallot in patients with contraindications to MRI, such as pacemakers or implantable cardioverter defibrillators. Multidetector CT is better than echocardiography and MRI for defining coronary artery abnormalities (Fig. 47-7) and can be used if echocardiography is inconclusive.15,16 High spatial resolution allows for excellent demonstration of the RVOT and pulmonary arteries, allowing for noninvasive measurements of hypoplasia, focal stenosis,17 or aneurysms (Fig. 47-8). CT is preferred to MRI to evaluate vascular stents (Figs. 47-9 and 47-10). Gated studies to measure cardiac function in patients with contraindications for MRI can be performed, but they require higher radiation dose gated techniques.18
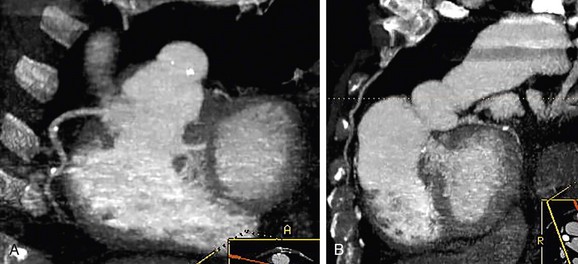
 FIGURE 47-7
FIGURE 47-7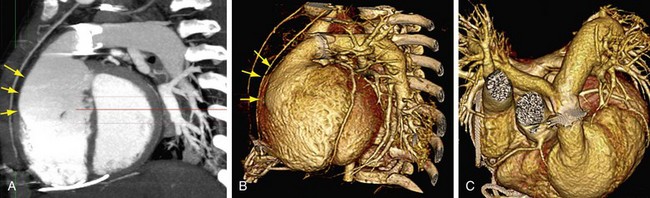
 FIGURE 47-8
FIGURE 47-8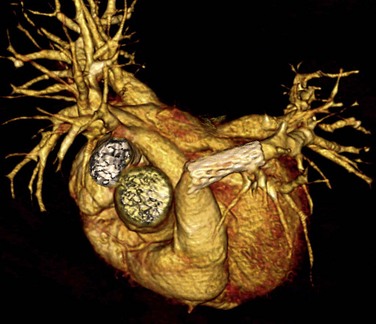
 FIGURE 47-9
FIGURE 47-9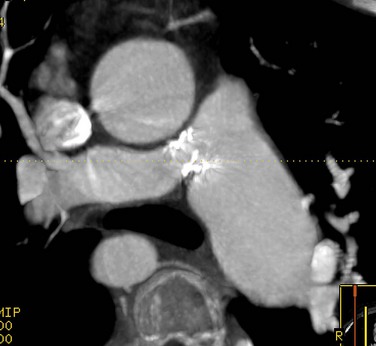
 FIGURE 47-10
FIGURE 47-10Magnetic Resonance Imaging
MRI is the primary complementary imaging modality after echocardiography. The primary diagnostic findings of tetralogy of Fallot can be detected by MRI, but are adequately imaged by echocardiography. Although echocardiography is adequate for evaluation of intracardiac abnormalities, the branch pulmonary arteries and collateral arteries are better characterized by MRI (Fig. 47-11). MRI, notably using MR angiography, can characterize branch pulmonary confluence, absence, or stenosis, and the number and location of major aorticopulmonary collateral arteries. Palliative shunts are better visualized by MR angiography than echocardiography (Fig. 47-12). Associated aortic arch anomalies that should be identified by echocardiography are more completely visualized by MR angiography (Fig. 47-13).16
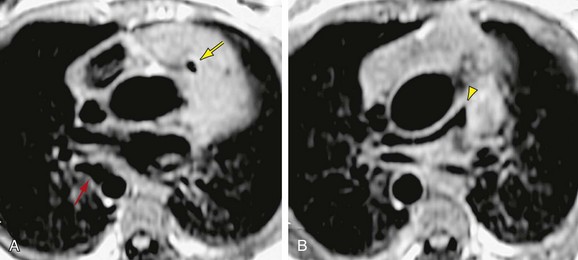
 FIGURE 47-11
FIGURE 47-11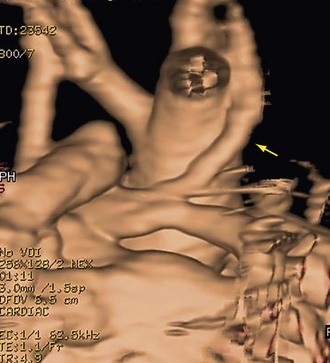
 FIGURE 47-12 MR angiography volume rendering posterior view shows wide patency of a modified Blalock-Taussig shunt (arrow).
FIGURE 47-12 MR angiography volume rendering posterior view shows wide patency of a modified Blalock-Taussig shunt (arrow).
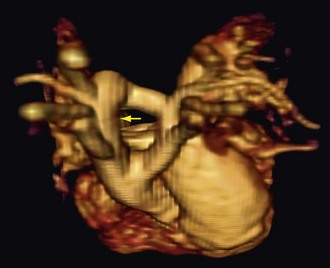
 FIGURE 47-13
FIGURE 47-13MRI has a more dominant role in postoperative patients. Echocardiography windows become progressively more restricted and are particularly limited for evaluation of the right ventricle and branch pulmonary arteries. Complications of palliations, such as branch pulmonary artery stenosis (Fig. 47-14), distortion, or occlusion resulting from prior systemic pulmonary artery shunts, are well visualized by MR angiography. Complications of corrective surgery, including residual RVOT stenosis, pseudoaneurysm (Fig. 47-15), or dilation from pulmonary regurgitation, are well imaged by a combination of black and white blood imaging techniques or MR angiography.
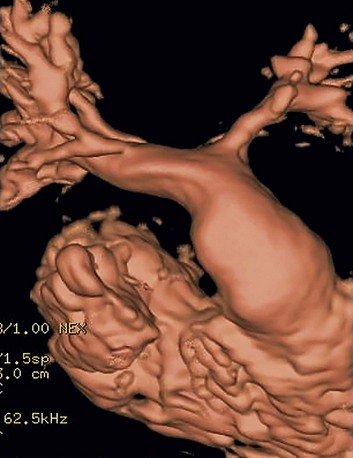
 FIGURE 47-14
FIGURE 47-14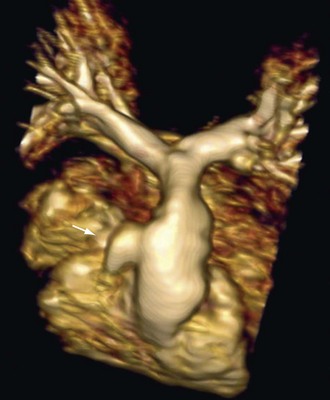
 FIGURE 47-15
FIGURE 47-15Accurate right ventricular size quantification cannot be obtained by two-dimensional echocardiography because of the right ventricular shape and the poor available windows. MRI biventricular volume measurements using short-axis cine steady-state free precession white blood images to quantify right and left ventricular systolic function and the right ventricular regurgitant fraction are the gold standard (Fig. 47-16). Right ventricular enlargement and an elevated right ventricular regurgitant fraction are among the criteria used to determine the timing for further reoperation to place a valved pulmonary conduit. Criteria for determining the appropriate time for valved conduit surgery are evolving.9
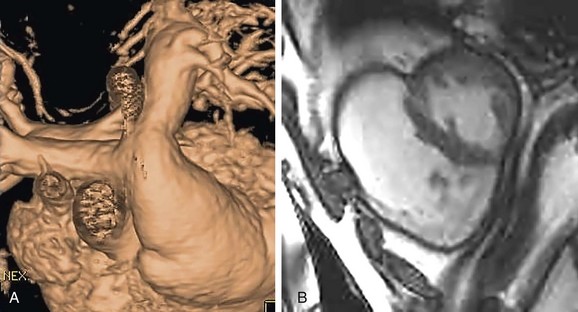
 FIGURE 47-16
FIGURE 47-16Pulmonary regurgitation and late diastolic forward flow consistent with right ventricular diastolic dysfunction can be quantified using phase contrast MRI flow quantification (also known as velocity-encoded MRI) (Fig. 47-17).19 A reduction in the normal E:A maximum flow amplitude ratio across the tricuspid valve detected by phase contrast MRI flow quantification across the tricuspid valve is also consistent with diastolic dysfunction and correlates with right ventriculomegaly (Fig. 47-18).20,21 Dobutamine stress MRI may unmask diastolic dysfunction not appreciated on rest imaging.22 Delayed hyperenhancement imaging can show fibrosis in the RVOT and is associated with outflow tract dilation.23
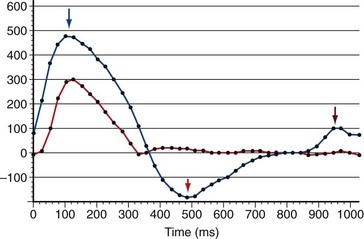
 FIGURE 47-17
FIGURE 47-17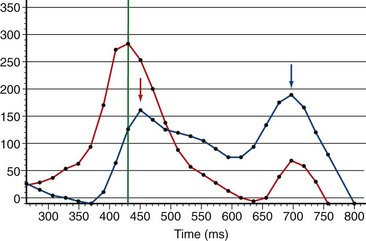
 FIGURE 47-18
FIGURE 47-18Late aortopathy associated with tetralogy of Fallot can be evaluated by MR angiography (Fig. 47-19). Associated aortic valve regurgitation is quantified by phase contrast MRI flow quantification.
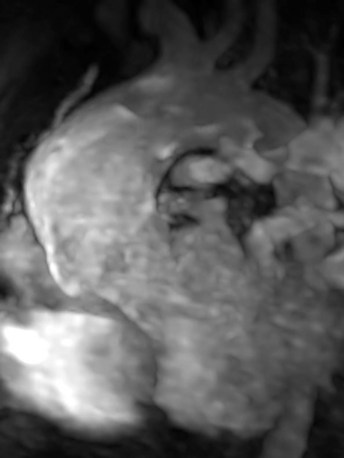
 FIGURE 47-19
FIGURE 47-19Classic Signs
The classic imaging sign of tetralogy of Fallot is the wooden shoe (coeur en sabot) or boot-shaped heart characterized on radiographs by elevation of the cardiac apex and pulmonary artery segment concavity. Right ventricular hypertrophy causes elevation of the cardiac apex. The greater the right ventricular hypertrophy, the more elevated the cardiac apex.24
DIFFERENTIAL DIAGNOSIS
Imaging Findings
Echocardiography is diagnostic. The parasternal long-axis view shows the VSD and overriding aorta (see Fig. 47-5), which is common to other conotruncal anomalies such as truncus arteriosus. Imaging of the RVOT detects stenosis, determining the diagnosis. The chest radiograph may show a boot-shaped heart that is normal in size with decreased pulmonary vascularity. Admixture lesions typically have large hearts and overcirculation. Patients with D-transposition of the great arteries clinically present early, and classic manifestations of cardiomegaly and increased pulmonary vascularity are absent. The chest radiograph in infants with D-transposition is initially normal with progressive enlargement and increased vascularity manifesting during the first week of life. Patients with truncus arteriosus frequently have a right aortic arch, but the cardiac silhouette is large and the pulmonary vascularity is increased compared with infants with tetralogy of Fallot. Patients with tricuspid atresia usually have normal-sized hearts and decreased pulmonary vascularity, but no elevation of the cardiac apex is present because the right ventricle is absent.
TREATMENT OPTIONS
Surgical
Primary repair bypassing initial palliation is growing in popularity. Repair includes VSD closure and RVOT enlargement. RVOT reconstruction frequently requires a transannular pulmonary patch that creates pulmonary valve incompetence. Symptomatic infants with pulmonary hypoplasia can still be treated with initial palliation. Increased morbidity is associated with primary repair in the first 3 months of life, but survival is not adversely affected.25 The ideal age for corrective surgery is 4 to 11 months in asymptomatic infants, but earlier repair is advocated in symptomatic infants.2,26
Long-term pulmonary regurgitation is associated with right ventricle, pulmonary artery, and branch pulmonary artery progressive dilation. Right heart dilation with associated complications can be prevented or reversed by placement of a valved conduit in the RVOT.27 No criteria are universally accepted to determine the timing for valved conduit placement. A combination of clinical, ECG, and imaging criteria is used to determine the timing of valve replacement. Clinical criteria include exercise intolerance and signs of heart failure. ECG abnormalities include prolonged tachycardia. A QRS length greater than 180 ms is associated with sudden death.28 MRI criteria noted by Geva8 include pulmonary regurgitant fraction of 25% or greater combined with increased right ventricular size or decreased right ventricular systolic function.
Right ventricular remodeling occurs in children after placement of a valved conduit with a reduction in right ventricular size when the right ventricular end-diastolic volume index was ≥150 mL/m2.29 Right ventricular remodeling also occurs in adults, but Oosterhof and colleagues30 found that right ventricular normalization did not occur if the right ventricular end-diastolic volume index was ≥160 mL/m2 or the right ventricular end-systolic volume index was ≥82 mL/m2. Studies in children and adults suggest that an optimal time exists for reoperation beyond which right ventricular remodeling is incomplete.
The aneurysmal size of the pulmonary artery and central branch pulmonary arteries in infants with tetralogy of Fallot and absent pulmonary valve requires additional surgical considerations. Surgical angioplasty to reduce the size of pulmonary arteries is essential to prevent airway compression.31 A Lecompte maneuver can be helpful (Fig. 47-20).
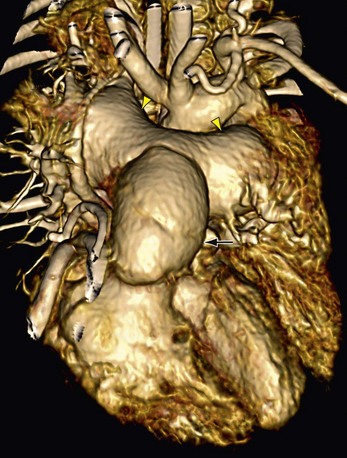
 FIGURE 47-20
FIGURE 47-20INFORMATION FOR THE REFERRING PHYSICIAN
After palliative procedures, additional findings include the following:
After corrective surgery, additional findings include the following:



Anderson RH, Weinberg PM. The clinical anatomy of tetralogy of Fallot. Cardiol Young Suppl. 2005;1:38-47.
Bashore TM. Adult congenital heart disease: right ventricular outflow tract lesions. Circulation. 2007;115:1933-1947.
Boechat MI, Ratib O, Williams PL, et al. Cardiac MR angiography for assessment of complex tetralogy of Fallot and pulmonary atresia. RadioGraphics. 2005;25:1535-1546.
Boshoff D, Gewillig M. A review of the options for treatment of major aortopulmonary collateral arteries in the setting of tetralogy of Fallot with pulmonary atresia. Cardiol Young. 2006;16:212-220.
Dorfman AL, Geva T. Magnetic resonance imaging evaluation of congenital heart disease: conotruncal anomalies. J Cardiovasc Magn Reson. 2006;8:645-659.
Gaca AM, Jaggers JJ, Dudley LT, et al. Repair of congenital heart disease: a primer—part 2. Radiology. 2008;248:44-60.
Gregg D, Foster E. Pulmonary insufficiency is the nexus of late complications in tetralogy of Fallot. Curr Cardiol Rep. 2007;9:315-322.
Norton KI, Tong C, Glass RBJ, et al. Cardiac MR imaging assessment following tetralogy of Fallot repair. RadioGraphics. 2006;26:197-211.
Oosterhof T, Mulder BJM, Vliegen HW, et al. Cardiovascular magnetic resonance in the follow-up of patients with corrected tetralogy of Fallot: a review. Am Heart J. 2006;151:265-272.
Restivo A, Placidi GPS, Saffirio C, et al. Cardiac outflow tract: a review of some embryogenetic aspects of the conotruncal region of the heart. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:936-943.
1 Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2008;39:1890-1900.
2 Derby CD, Pizarro C. Routine primary repair of tetralogy of Fallot in the neonate. Expert Tev Cardiovasc Ther. 2005;3:857-863.
3 Michielon G, Marino B, Formigari R, et al. Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Ann Thorac Surg. 2006;81:968-975.
4 Uretzky G, Puga FJ, Danielson GK, et al. Complete atrioventricular canal associated with tetralogy of Fallot: morphologic and surgical considerations. J Thorac Cardiovasc Surg. 1984;87:756-766.
5 Ramaswamy P, Lytrivi ID, Thanjan MR, et al. Frequency of aberrant subclavian artery, arch laterality and associated intracardiac anomalies detected by echocardiography. Am J Cardiol. 2008;101:677-682.
6 Need LR, Powell AJ, del Nido P, et al. Coronary echocardiography in tetralogy of Fallot: diagnostic accuracy, resource utilization and surgical implications over 13 years. J Am Coll Cardiol. 2000;36:1371-1377.
7 Li J, Soukaias ND, Carvalho JS, et al. Coronary arterial anatomy in tetralogy of Fallot: morphological and clinical correlations. Heart. 1998;80:174-183.
8 Geva T. Indications and timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2006;9:11-22.
9 Kadner A, Tulevski II, Bauerfeld U, et al. Chronic pulmonary valve insufficiency after repaired tetralogy of Fallot: diagnostics, reoperations and reconstruction possibilities. Expert Rev Cardiovasc Ther. 2007;5:221-230.
10 de Ruijter FTH, Weenink I, Hitchcock FJ, et al. Right ventricular dysfunction and pulmonary valve replacement after correction of tetralogy of Fallot. Ann Thorac Surg. 2002;73:1794-1800.
11 Niwa K. Aortic root dilatation in tetralogy of Fallot long-term after repair—histology of the aorta in tetralogy of Fallot: evidence of intrinsic aortopathy. Int J Cardiol. 2005;103:117-119.
12 Sivanandam S, Glickstein JS, Printz BF, et al. Prenatal diagnosis of conotruncal malformations: diagnostic accuracy, outcome, chromosomal abnormalities, and extracardiac anomalies. Am J Perinatol. 2006;23:241-245.
13 Shinebourne EA, Babu-Narayan SV, Carvalho JS. Tetralogy of Fallot: from fetus to adult. Heart. 2006;92:1353-1359.
14 Geva T, Ayres NA, Pac FA, et al. Quantitative morphometric analysis of progressive infundibular obstruction in tetralogy of Fallot. Circulation. 1995;92:886-892.
15 Wang XM, Wu LB, Sun C, et al. Clinical application of 64-slice spiral CT in the diagnosis of tetralogy of Fallot. Eur J Radiol. 2007;64:296-301.
16 Dogan OF, Karcaaltincaba M, Yorgancioglu C, et al. Demonstration of coronary arteries and major cardiac vascular structures in congenital heart disease by cardiac multidetector angiography. Heart Surg Forum. 2007;10:E90-E94.
17 Hayabuchi Y, Mori K, Kitagawa T, et al. Accurate quantification of pulmonary artery diameter in patients with cyanotic congenital heart disease using multidetector-row computed tomography. Am Heart J. 2007;154:783-788.
18 Leschka S, Oechslin E, Husmann L, et al. Pre- and postoperative evaluation of congenital heart disease in children and adults with 64-section CT. RadioGraphics. 2007;27:829-846.
19 Helbing WA, Niezen RA, Le Cessie S, et al. Right ventricular diastolic function in children with pulmonary regurgitation after repair of tetralogy of Fallot: volumetric evaluation by magnetic resonance velocity mapping. J Am Coll Cardiol. 1996;28:1827-1835.
20 Greenberg SB, Shah CC, Bhutta ST. Tricuspid valve magnetic resonance imaging phase contrast velocity-encoded flow quantification for follow up of tetralogy of Fallot. Int J Cardiovasc Imaging. 2008;24:861-865.
21 Paelinck BP, Lamb HJ, Bax JJ, et al. Assessment of diastolic function by cardiovascular magnetic resonance. Am Heart J. 2002;144:198-2005.
22 van den Berg J, Wielopolski PA, Meijboom FJ, et al. Diastolic function in repaired tetralogy of Fallot at rest and during stress: assessment with MR imaging. Radiology. 2007;243:212-219.
23 Oosterhof T, Mulder BJ, Vliegen HW, et al. Corrected tetralogy of Fallot: delayed enhancement in right ventricular outflow tract. Radiology. 2005;237:868-871.
24 Ferguson EC, Krishnamurthy R, Oldham SAA. Classic imaging signs of congenital cardiovascular abnormalities. RadioGraphics. 2007;27:1323-1334.
25 Vohra HA, Adamson L, Haw MP. Is early primary repair for correction of tetralogy of Fallot comparable to surgery after 6 months of age? Interact Cardiovasc Thorac Surg. 2008;7:698-701.
26 Kantorova A, Zbieranek K, Sauer H, et al. Primary early correction of tetralogy of Fallot irrespective of age. Cardiol Young. 2008;18:153-157.
27 Gengaskul A, Harris L, Bradley TJ, et al. The impact of pulmonary valve replacement after tetralogy of Fallot repair: a matched comparison. Eur J Cardiothorac Surg. 2007;32:462-468.
28 Knauth AL, Gauvreau K, Powell AJ, et al. Ventricular size and function assessed by cardiac MRI predict major adverse clinical outcomes late after tetralogy of Fallot repair. Heart. 2008;94:211-216.
29 Buechel ER, Dave HH, Kellenberger CJ, et al. Remodeling of the right ventricle after early pulmonary valve replacement in children with repaired tetralogy of Fallot: assessment by cardiovascular magnetic resonance. Eur Heart J. 2005;26:2614-2615.
30 Oosterhof T, van Straten A, Vliegen HW, et al. Preoperative thresholds for pulmonary valve replacement in patients with corrected tetralogy of Fallot using cardiovascular magnetic resonance. Circulation. 2007;116:545-551.
31 Hraska V. Repair of tetralogy of Fallot with absent pulmonary valve using a new approach. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2005:132-134.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
CHAPTER 47 Tetralogy of Fallot
PREVALENCE AND EPIDEMIOLOGY
Tetralogy of Fallot is the most common congenital heart disease manifesting with cyanosis. The incidence is 0.06% of live births, and it constitutes 5% to 7% of all congenital heart disease.1,2 Genetic syndromes are associated with tetralogy of Fallot in 20% of cases with 22q11 deletion and trisomy 21 being the most common.3 Complete atrioventricular septal defects are present in 2% of patients with tetralogy of Fallot, especially patients with trisomy 21.4 Alagille syndrome, VACTERL syndrome (vertebral anomalies, anal atresia, cardiovascular anomalies, tracheoesophageal fistula, esophageal atresia, renal anomalies, limb anomalies), and CHARGE syndrome (coloboma of the eye, heart defects, atresia of the choanae, retardation of growth and development, genitourinary abnormalities; ear abnormalities and deafness) are also associated with tetralogy of Fallot. Atrial septal defect is present in one third of patients with tetralogy of Fallot. Currently, 85% of children with tetralogy of Fallot survive to adulthood, increasing the prevalence of tetralogy of Fallot in adults.
Aberrant subclavian arteries are associated in 5% to 8% of patients with tetralogy of Fallot. The prevalence of tetralogy of Fallot and anomalous subclavian artery is greater if associated with a right aortic arch or pulmonary atresia. Anomalous subclavian artery occurs in 24% of patients with tetralogy of Fallot, right aortic arch, and pulmonary atresia.5
Coronary artery anomalies are common, occurring in 5% to 12% of children with tetralogy of Fallot.6,7 Coronary artery anomalies that result in major branches coursing anterior to the right ventricular outflow tract (RVOT) interfere with transannular patch angioplasty of the outflow tract.
MANIFESTATIONS OF DISEASE
Clinical Presentation
A child with a palliative shunt has a continuous murmur similar to that heard in a child with a patent ductus arteriosus. After corrective surgery, symptoms may reflect continued right-sided obstruction or pulmonary regurgitation. Corrective surgery for tetralogy of Fallot requires enlargement of the RVOT and frequently a transannular patch, which results in pulmonary regurgitation.8–10 Pulmonary regurgitation is initially well tolerated, but later is associated with fatigue. The right ventricle and its outflow tract progressively dilate. Right ventricular enlargement leads to distortion of the tricuspid valve annulus and tricuspid regurgitation, compromising right heart function further. ECG abnormalities with lengthening of the QRS complex can lead to arrhythmia that may result in sudden death. Diastolic dysfunction can progress to irreversible systolic dysfunction.8 Revision of the RVOT with a valved conduit causes right ventricular remodeling and improved cardiac function. Pseudoaneurysms of the RVOT can follow corrective surgery with a transannular patch or pulmonary conduit.
Aortic root dilation and regurgitation are common in adults with tetralogy of Fallot and can lead to arterial medial abnormalities. Risk factors for aortopathy include patients with pulmonary atresia, right aortic arch, history of aorticopulmonary shunts, male sex, and chromosome 22q11 deletions.11
Imaging Technique and Findings
Radiography
Radiography plays a secondary role to echocardiography. The cardiac silhouette is frequently normal in size and configuration (Fig. 47-1). The classic radiographic appearance is a boot-shaped heart caused by elevation of the cardiac apex from right ventricular hypertrophy with a diminished or unapparent pulmonary artery segment (Fig. 47-2). The appearance of a boot-shaped heart can be mimicked if the central x-ray beam is caudal to the heart causing cephalic projection of the more anterior portion of the heart (Fig. 47-3). The pulmonary vascularity is diminished, but may be undetectable on a conventional radiograph if right ventricular obstruction is mild. A right aortic arch is visible in 25% of patients with tetralogy of Fallot. Direct visualization of the aortic arch and tracheal shift can be difficult to detect in infants and young children. The side of the aortic arch is reliably determined, however, by noting the increased density of the pedicles on the side of the aortic arch. Patients with tetralogy of Fallot and absent pulmonary artery have enlargement of the central pulmonary arteries (Fig. 47-4).
Ultrasonography
Echocardiography is definitive for the prenatal and postnatal diagnosis of tetralogy of Fallot.12 A large VSD is identified in utero on the four-chamber view, but evaluation of the outflow tracts is necessary for the diagnosis because other conotruncal abnormalities can have a VSD and an overriding aorta. A large aorta overriding the VSD is present. The aorta is larger than the pulmonary artery. Doppler interrogation of the fetal RVOT shows elevated velocity. A left-to-right shunt through the ductus arteriosus suggests severe RVOT obstruction.13
Postnatal echocardiography characterizes all intracardiac features of tetralogy of Fallot. The VSD and aorta override are identified on the parasternal long-axis view (Fig. 47-5). The outflow tract must also be evaluated (Fig. 47-6) because truncus arteriosus and VSD with pulmonary atresia can have the same appearance. In contrast to double outlet right ventricle, the aortic override to the right ventricle is less than 50%, and aortic valve fibrous continuity with the mitral valve is preserved. The RVOT including the infundibulum, pulmonary valve, and pulmonary artery is measured for stenosis. The measured infundibular volume is small, and owing to reduced growth compared with somatic growth becomes progressively more stenotic.14 Infants who require earlier surgical intervention have a greater reduction in infundibular volume. The pulmonary annulus can be hypoplastic and bicuspid. Hypoplasia with a z score less than 2 is an indication for a transannular patch. Branch pulmonary artery continuity and size is also determined. The position of the coronary arteries must be established. A coronary anomaly with a major coronary branch coursing anterior to the RVOT must be excluded before repair. CT or MRI may be necessary if the coronary arteries or pulmonary arteries are inadequately visualized.
