FIGURE 173-2 Scarlet fever exanthem. Finely punctate erythema has become confluent (scarlatiniform); petechiae can occur and have a linear configuration within the exanthem in body folds (Pastia’s lines). (From Fitzpatrick, Johnson, Wolff: Color Atlas and Synopsis of Clinical Dermatology, 4th ed, New York, McGraw-Hill, 2001, with permission.)
Skin and Soft Tissue Infections GAS—and occasionally other streptococcal species—can cause a variety of infections involving the skin, subcutaneous tissues, muscles, and fascia. While several clinical syndromes offer a useful means for classification of these infections, not all cases fit exactly into one category. The classic syndromes are general guides to predicting the level of tissue involvement in a particular patient, the probable clinical course, and the likelihood that surgical intervention or aggressive life support will be required.
IMPETIGO (PYODERMA) Impetigo, a superficial infection of the skin, is caused primarily by GAS and occasionally by other streptococci or Staphylococcus aureus. Impetigo is seen most often in young children, tends to occur during warmer months, and is more common in semitropical or tropical climates than in cooler regions. Infection is more common among children living under conditions of poor hygiene. Prospective studies have shown that colonization of unbroken skin with GAS precedes clinical infection. Minor trauma, such as a scratch or an insect bite, may then serve to inoculate organisms into the skin. Impetigo is best prevented, therefore, by attention to adequate hygiene. The usual sites of involvement are the face (particularly around the nose and mouth) and the legs, although lesions may occur at other locations. Individual lesions begin as red papules, which evolve quickly into vesicular and then pustular lesions that break down and coalesce to form characteristic honeycomb-like crusts (Fig. 173-3). Lesions generally are not painful, and patients do not appear ill. Fever is not a feature of impetigo and, if present, suggests either infection extending to deeper tissues or another diagnosis. The classic presentation of impetigo usually poses little diagnostic difficulty. Cultures of impetiginous lesions often yield S. aureus as well as GAS. In almost all cases, streptococci are isolated initially and staphylococci appear later, presumably as secondary colonizing flora. In the past, penicillin was nearly always effective against these infections. However, an increasing frequency of penicillin treatment failure suggests that S. aureus may have become more prominent as a cause of impetigo. Bullous impetigo due to S. aureus is distinguished from typical streptococcal infection by more extensive, bullous lesions that break down and leave thin paper-like crusts instead of the thick amber crusts of streptococcal impetigo. Other skin lesions that may be confused with impetigo include herpetic lesions—either those of orolabial herpes simplex or those of chickenpox or zoster. Herpetic lesions can generally be distinguished by their appearance as more discrete, grouped vesicles and by a positive Tzanck test. In difficult cases, cultures of vesicular fluid should yield GAS in impetigo and the responsible virus in herpesvirus infections.
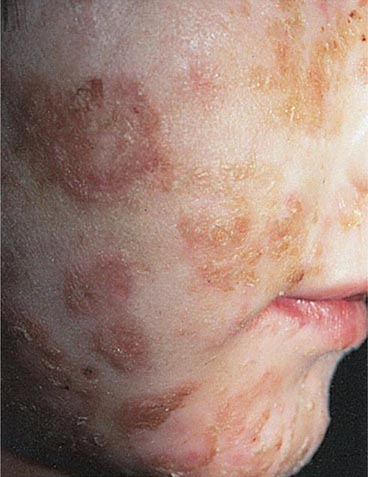
FIGURE 173-3 Impetigo contagiosa is a superficial streptococcal or Staphylococcus aureus infection consisting of honey-colored crusts and erythematous weeping erosions. Occasionally, bullous lesions may be seen. (Courtesy of Mary Spraker, MD; with permission.)
|
TREATMENT |
STREPTOCOCCAL IMPETIGO |
Treatment of streptococcal impetigo is the same as that for streptococcal pharyngitis. In view of evidence that S. aureus has become a relatively frequent cause of impetigo, empirical regimens should cover both streptococci and S. aureus. For example, either dicloxacillin or cephalexin can be given at a dose of 250 mg four times daily for 10 days. Topical mupirocin ointment is also effective. Culture may be indicated to rule out methicillin-resistant S. aureus, especially if the response to empirical treatment is unsatisfactory. ARF is not a sequela to streptococcal skin infections, although PSGN may follow either skin or throat infection. The reason for this difference is not known. One hypothesis is that the immune response necessary for development of ARF occurs only after infection of the pharyngeal mucosa. In addition, the strains of GAS that cause pharyngitis are generally of different M protein types than those associated with skin infections; thus the strains that cause pharyngitis may have rheumatogenic potential, while the skin-infecting strains may not.
CELLULITIS Inoculation of organisms into the skin may lead to cellulitis: infection involving the skin and subcutaneous tissues. The portal of entry may be a traumatic or surgical wound, an insect bite, or any other break in skin integrity. Often, no entry site is apparent. One form of streptococcal cellulitis, erysipelas, is characterized by a bright red appearance of the involved skin, which forms a plateau sharply demarcated from surrounding normal skin (Fig. 173-4). The lesion is warm to the touch, may be tender, and appears shiny and swollen. The skin often has a peau d’orange texture, which is thought to reflect involvement of superficial lymphatics; superficial blebs or bullae may form, usually 2–3 days after onset. The lesion typically develops over a few hours and is associated with fever and chills. Erysipelas tends to occur on the malar area of the face (often with extension over the bridge of the nose to the contralateral malar region) and the lower extremities. After one episode, recurrence at the same site—sometimes years later—is not uncommon. Classic cases of erysipelas, with typical features, are almost always due to β-hemolytic streptococci, usually GAS and occasionally group C or G. Often, however, the appearance of streptococcal cellulitis is not sufficiently distinctive to permit a specific diagnosis on clinical grounds. The area involved may not be typical for erysipelas, the lesion may be less intensely red than usual and may fade into surrounding skin, and/or the patient may appear only mildly ill. In such cases, it is prudent to broaden the spectrum of empirical antimicrobial therapy to include other pathogens, particularly S. aureus, that can produce cellulitis with the same appearance. Staphylococcal infection should be suspected if cellulitis develops around a wound or an ulcer.
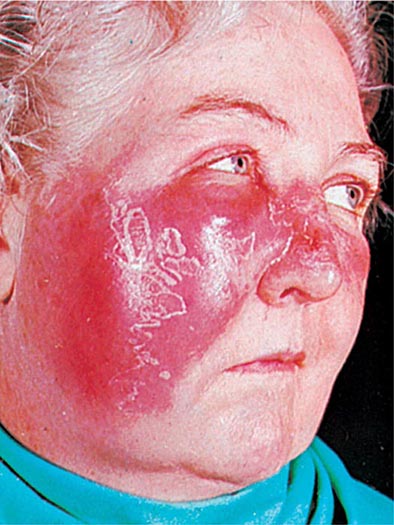
FIGURE 173-4 Erysipelas is a streptococcal infection of the superficial dermis and consists of well-demarcated, erythematous, edematous, warm plaques.
Streptococcal cellulitis tends to develop at anatomic sites in which normal lymphatic drainage has been disrupted, such as sites of prior cellulitis, the arm ipsilateral to a mastectomy and axillary lymph node dissection, a lower extremity previously involved in deep venous thrombosis or chronic lymphedema, or the leg from which a saphenous vein has been harvested for coronary artery bypass grafting. The organism may enter via a dermal breach some distance from the eventual site of clinical cellulitis. For example, some patients with recurrent leg cellulitis following saphenous vein removal stop having recurrent episodes only after treatment of tinea pedis on the affected extremity. Fissures in the skin presumably serve as a portal of entry for streptococci, which then produce infection more proximally in the leg at the site of previous injury. Streptococcal cellulitis may also involve recent surgical wounds. GAS is among the few bacterial pathogens that typically produce signs of wound infection and surrounding cellulitis within the first 24 h after surgery. These wound infections are usually associated with a thin exudate and may spread rapidly, either as cellulitis in the skin and subcutaneous tissue or as a deeper tissue infection (see below). Streptococcal wound infection or localized cellulitis may also be associated with lymphangitis, manifested by red streaks extending proximally along superficial lymphatics from the infection site.
|
TREATMENT |
STREPTOCOCCAL CELLULITIS |
See Table 173-3 and Chap. 156.
DEEP SOFT-TISSUE INFECTIONS Necrotizing fasciitis (hemolytic streptococcal gangrene) involves the superficial and/or deep fascia investing the muscles of an extremity or the trunk. The source of the infection is either the skin, with organisms introduced into tissue through trauma (sometimes trivial), or the bowel flora, with organisms released during abdominal surgery or from an occult enteric source, such as a diverticular or appendiceal abscess. The inoculation site may be inapparent and is often some distance from the site of clinical involvement; e.g., the introduction of organisms via minor trauma to the hand may be associated with clinical infection of the tissues overlying the shoulder or chest. Cases associated with the bowel flora are usually polymicrobial, involving a mixture of anaerobic bacteria (such as Bacteroides fragilis or anaerobic streptococci) and facultative organisms (usually gram-negative bacilli). Cases unrelated to contamination from bowel organisms are most commonly caused by GAS alone or in combination with other organisms (most often S. aureus). Overall, GAS is implicated in ~60% of cases of necrotizing fasciitis. The onset of symptoms is usually quite acute and is marked by severe pain at the site of involvement, malaise, fever, chills, and a toxic appearance. The physical findings, particularly early on, may not be striking, with only minimal erythema of the overlying skin. Pain and tenderness are usually severe. In contrast, in more superficial cellulitis, the skin appearance is more abnormal, but pain and tenderness are only mild or moderate. As the infection progresses (often over several hours), the severity and extent of symptoms worsen, and skin changes become more evident, with the appearance of dusky or mottled erythema and edema. The marked tenderness of the involved area may evolve into anesthesia as the spreading inflammatory process produces infarction of cutaneous nerves.
Although myositis is more commonly due to S. aureus infection, GAS occasionally produces abscesses in skeletal muscles (streptococcal myositis), with little or no involvement of the surrounding fascia or overlying skin. The presentation is usually subacute, but a fulminant form has been described in association with severe systemic toxicity, bacteremia, and a high mortality rate. The fulminant form may reflect the same basic disease process seen in necrotizing fasciitis, but with the necrotizing inflammatory process extending into the muscles themselves rather than remaining limited to the fascial layers.
|
TREATMENT |
DEEP SOFT-TISSUE INFECTIONS |
Once necrotizing fasciitis is suspected, early surgical exploration is both diagnostically and therapeutically indicated. Surgery reveals necrosis and inflammatory fluid tracking along the fascial planes above and between muscle groups, without involvement of the muscles themselves. The process usually extends beyond the area of clinical involvement, and extensive debridement is required. Drainage and debridement are central to the management of necrotizing fasciitis; antibiotic treatment is a useful adjunct (Table 173-3), but surgery is life-saving. Treatment for streptococcal myositis consists of surgical drainage—usually by an open procedure that permits evaluation of the extent of infection and ensures adequate debridement of involved tissues—and high-dose penicillin (Table 173-3).
Pneumonia and Empyema GAS is an occasional cause of pneumonia, generally in previously healthy individuals. The onset of symptoms may be abrupt or gradual. Pleuritic chest pain, fever, chills, and dyspnea are the characteristic manifestations. Cough is usually present but may not be prominent. Approximately one-half of patients with GAS pneumonia have an accompanying pleural effusion. In contrast to the sterile parapneumonic effusions typical of pneumococcal pneumonia, those complicating streptococcal pneumonia are almost always infected. The empyema fluid is usually visible by chest radiography on initial presentation, and its volume may increase rapidly. These pleural collections should be drained early, as they tend to become loculated rapidly, resulting in a chronic fibrotic reaction that may require thoracotomy for removal.
Bacteremia, Puerperal Sepsis, and Streptococcal Toxic Shock Syndrome GAS bacteremia is usually associated with an identifiable local infection. Bacteremia occurs rarely with otherwise uncomplicated pharyngitis, occasionally with cellulitis or pneumonia, and relatively frequently with necrotizing fasciitis. Bacteremia without an identified source raises the possibility of endocarditis, an occult abscess, or osteomyelitis. A variety of focal infections may arise secondarily from streptococcal bacteremia, including endocarditis, meningitis, septic arthritis, osteomyelitis, peritonitis, and visceral abscesses. GAS is occasionally implicated in infectious complications of childbirth, usually endometritis and associated bacteremia. In the preantibiotic era, puerperal sepsis was commonly caused by GAS; currently, it is more often caused by GBS. Several nosocomial outbreaks of puerperal GAS infection have been traced to an asymptomatic carrier, usually someone present at delivery. The site of carriage may be the skin, throat, anus, or vagina.
Beginning in the late 1980s, several reports described patients with GAS infections associated with shock and multisystem organ failure. This syndrome was called streptococcal TSS because it shares certain features with staphylococcal TSS. In 1993, a case definition for streptococcal TSS was formulated (Table 173-4). The general features of the illness include fever, hypotension, renal impairment, and respiratory distress syndrome. Various types of rash have been described, but rash usually does not develop. Laboratory abnormalities include a marked shift to the left in the white blood cell differential, with many immature granulocytes; hypocalcemia; hypoalbuminemia; and thrombocytopenia, which usually becomes more pronounced on the second or third day of illness. In contrast to patients with staphylococcal TSS, the majority with streptococcal TSS are bacteremic. The most common associated infection is a soft tissue infection—necrotizing fasciitis, myositis, or cellulitis—although a variety of other associated local infections have been described, including pneumonia, peritonitis, osteomyelitis, and myometritis. Streptococcal TSS is associated with a mortality rate of ≥30%, with most deaths secondary to shock and respiratory failure. Because of its rapidly progressive and lethal course, early recognition of the syndrome is essential. Patients should receive aggressive supportive care (fluid resuscitation, pressors, and mechanical ventilation) in addition to antimicrobial therapy and, in cases associated with necrotizing fasciitis, surgical debridement. Exactly why certain patients develop this fulminant syndrome is not known. Early studies of the streptococcal strains isolated from these patients demonstrated a strong association with the production of pyrogenic exotoxin A. This association has been inconsistent in subsequent case series. Pyrogenic exotoxin A and several other streptococcal exotoxins act as superantigens to trigger release of inflammatory cytokines from T lymphocytes. Fever, shock, and organ dysfunction in streptococcal TSS may reflect, in part, the systemic effects of superantigen-mediated cytokine release.
|
PROPOSED CASE DEFINITION FOR THE STREPTOCOCCAL TOXIC SHOCK SYNDROMEa |
aAn illness fulfilling criteria IA, IIA, and IIB is defined as a definite case. An illness fulfilling criteria IB, IIA, and IIB is defined as a probable case if no other etiology for the illness is identified.
Source: Modified from Working Group on Severe Streptococcal Infections: JAMA 269:390, 1993.
|
TREATMENT |
STREPTOCOCCAL TOXIC SHOCK SYNDROME |
In light of the possible role of pyrogenic exotoxins or other streptococcal toxins in streptococcal TSS, treatment with clindamycin has been advocated by some authorities (Table 173-3), who argue that, through its direct action on protein synthesis, clindamycin is more effective in rapidly terminating toxin production than is penicillin—a cell-wall agent. Support for this view comes from studies of an experimental model of streptococcal myositis, in which mice given clindamycin had a higher rate of survival than those given penicillin. Comparable data on the treatment of human infections are not available, although retrospective analysis has suggested a better outcome when patients with invasive soft-tissue infection are treated with clindamycin rather than with cell wall-active antibiotics. Although clindamycin resistance in GAS is uncommon (<2% among U.S. isolates), it has been documented. Thus, if clindamycin is used for initial treatment of a critically ill patient, penicillin should be given as well until the antibiotic susceptibility of the streptococcal isolate is known. IV immunoglobulin has been used as adjunctive therapy for streptococcal TSS (Table 173-3). Pooled immunoglobulin preparations contain antibodies capable of neutralizing the effects of streptococcal toxins. Anecdotal reports and case series have suggested favorable clinical responses to IV immunoglobulin, but no adequately powered, prospective, controlled trials have been reported.
PREVENTION
No vaccine against GAS is commercially available. A formulation that consists of recombinant peptides containing epitopes of 26 M-protein types has undergone phase 1 and 2 testing in volunteers. Early results indicate that the vaccine is well tolerated and elicits type-specific antibody responses. Vaccines based on a conserved region of M protein or on a mixture of other conserved GAS protein antigens are in earlier stages of development.
Household contacts of individuals with invasive GAS infection (e.g., bacteremia, necrotizing fasciitis, or streptococcal TSS) are at greater risk of invasive infection than the general population. Asymptomatic pharyngeal colonization with GAS has been detected in up to 25% of persons with >4 h/d of same-room exposure to an index case. However, antibiotic prophylaxis is not routinely recommended for contacts of patients with invasive disease because such an approach (if effective) would require treatment of hundreds of contacts to prevent a single case.
STREPTOCOCCI OF GROUPS C AND G
Group C and group G streptococci are β-hemolytic bacteria that occasionally cause human infections similar to those caused by GAS. Strains that form small colonies on blood agar (<0.5 mm) are generally members of the Streptococcus milleri (Streptococcus intermedius, Streptococcus anginosus) group (see “Viridans Streptococci,” below). Large-colony group C and G streptococci of human origin are now considered a single species, Streptococcus dysgalactiae subspecies equisimilis. These organisms have been associated with pharyngitis, cellulitis and soft tissue infections, pneumonia, bacteremia, endocarditis, and septic arthritis. Puerperal sepsis, meningitis, epidural abscess, intraabdominal abscess, urinary tract infection, and neonatal sepsis have also been reported. Group C or G streptococcal bacteremia most often affects elderly or chronically ill patients and, in the absence of obvious local infection, is likely to reflect endocarditis. Septic arthritis, sometimes involving multiple joints, may complicate endocarditis or develop in its absence. Distinct streptococcal species of Lancefield group C cause infections in domesticated animals, especially horses and cattle; some human infections are acquired through contact with animals or consumption of unpasteurized milk. These zoonotic organisms include Streptococcus equi subspecies zooepidemicus and S. equi subspecies equi.
|
TREATMENT |
GROUP C OR G STREPTOCOCCAL INFECTION |
Penicillin is the drug of choice for treatment of group C or G streptococcal infections. Antibiotic treatment is the same as for similar syndromes due to GAS (Table 173-3). Patients with bacteremia or septic arthritis should receive IV penicillin (2–4 mU every 4 h). All group C and G streptococci are sensitive to penicillin; nearly all are inhibited in vitro by concentrations of ≤0.03 μg/mL. Occasional isolates exhibit tolerance: although inhibited by low concentrations of penicillin, they are killed only by significantly higher concentrations. The clinical significance of tolerance is unknown. Because of the poor clinical response of some patients to penicillin alone, the addition of gentamicin (1 mg/kg every 8 h for patients with normal renal function) is recommended by some authorities for treatment of endocarditis or septic arthritis due to group C or G streptococci; however, combination therapy has not been shown to be superior to penicillin treatment alone. Patients with joint infections often require repeated aspiration or open drainage and debridement for cure; the response to treatment may be slow, particularly in debilitated patients and those with involvement of multiple joints. Infection of prosthetic joints almost always requires prosthesis removal in addition to antibiotic therapy.
GROUP B STREPTOCOCCI
Identified first as a cause of mastitis in cows, streptococci belonging to Lancefield’s group B have since been recognized as a major cause of sepsis and meningitis in human neonates. GBS is also a frequent cause of peripartum fever in women and an occasional cause of serious infection in nonpregnant adults. Since the widespread institution of prenatal screening for GBS in the 1990s, the incidence of neonatal infection per 1000 live births has fallen from ~2–3 cases to ~0.6 case. During the same period, GBS infection in adults with underlying chronic illnesses has become more common; adults now account for a larger proportion of invasive GBS infections than do newborns. Lancefield group B consists of a single species, S. agalactiae, which is definitively identified with specific antiserum to the group B cell wall–associated carbohydrate antigen. A streptococcal isolate can be classified presumptively as GBS on the basis of biochemical tests, including hydrolysis of sodium hippurate (in which 99% of isolates are positive), hydrolysis of bile esculin (in which 99–100% are negative), bacitracin susceptibility (in which 92% are resistant), and production of CAMP factor (in which 98–100% are positive). CAMP factor is a phospholipase produced by GBS that causes synergistic hemolysis with β lysin produced by certain strains of S. aureus. Its presence can be demonstrated by cross-streaking of the test isolate and an appropriate staphylococcal strain on a blood agar plate. GBS organisms causing human infections are encapsulated by one of ten antigenically distinct polysaccharides. The capsular polysaccharide is an important virulence factor. Antibodies to the capsular polysaccharide afford protection against GBS of the same (but not of a different) capsular type.
INFECTION IN NEONATES
Two general types of GBS infection in infants are defined by the age of the patient at presentation. Early-onset infections occur within the first week of life, with a median age of 20 h at onset. Approximately half of these infants have signs of GBS disease at birth. The infection is acquired during or shortly before birth from the colonized maternal genital tract. Surveillance studies have shown that 5–40% of women are vaginal or rectal carriers of GBS. Approximately 50% of infants delivered vaginally by carrier mothers become colonized, although only 1–2% develop clinically evident infection. Prematurity, prolonged labor, obstetric complications, and maternal fever are risk factors for early-onset infection. The presentation of early-onset infection is the same as that of other forms of neonatal sepsis. Typical findings include respiratory distress, lethargy, and hypotension. Essentially all infants with early-onset disease are bacteremic, one-third to one-half have pneumonia and/or respiratory distress syndrome, and one-third have meningitis.
Late-onset infections occur in infants 1 week to 3 months old and, in rare instances, in older infants (mean age at onset, 3–4 weeks). The infecting organism may be acquired during delivery (as in early-onset cases) or during later contact with a colonized mother, nursery personnel, or another source. Meningitis is the most common manifestation of late-onset infection and in most cases is associated with a strain of capsular type III. Infants present with fever, lethargy or irritability, poor feeding, and seizures. The various other types of late-onset infection include bacteremia without an identified source, osteomyelitis, septic arthritis, and facial cellulitis associated with submandibular or preauricular adenitis.
|
TREATMENT |
GROUP B STREPTOCOCCAL INFECTION IN NEONATES |
Penicillin is the agent of choice for all GBS infections. Empirical broad-spectrum therapy for suspected bacterial sepsis, consisting of ampicillin and gentamicin, is generally administered until culture results become available. If cultures yield GBS, many pediatricians continue to administer gentamicin, along with ampicillin or penicillin, for a few days until clinical improvement becomes evident. Infants with bacteremia or soft tissue infection should receive penicillin at a dosage of 200,000 units/kg per day in divided doses. For meningitis, infants ≤7 days of age should receive 250,000–450,000 units/kg per day in three divided doses; infants >7 days of age should receive 450,000–500,000 units/kg per day in four divided doses. Meningitis should be treated for at least 14 days because of the risk of relapse with shorter courses.
PREVENTION
The incidence of GBS infection is unusually high among infants of women with risk factors: preterm delivery, early rupture of membranes (>24 h before delivery), prolonged labor, fever, or chorioamnionitis. Because the usual source of the organisms infecting a neonate is the mother’s birth canal, efforts have been made to prevent GBS infections by the identification of high-risk carrier mothers and their treatment with various forms of antibiotic prophylaxis or immunoprophylaxis. Prophylactic administration of ampicillin or penicillin to such patients during delivery reduces the risk of infection in the newborn. This approach has been hampered by logistical difficulties in identifying colonized women before delivery; the results of vaginal cultures early in pregnancy are poor predictors of carrier status at delivery. The CDC recommends screening for anogenital colonization at 35–37 weeks of pregnancy by a swab culture of the lower vagina and anorectum; intrapartum chemoprophylaxis is recommended for culture-positive women and for women who, regardless of culture status, have previously given birth to an infant with GBS infection or have a history of GBS bacteriuria during pregnancy. Women whose culture status is unknown and who develop premature labor (<37 weeks), prolonged rupture of membranes (>18 h), or intrapartum fever or who have a positive intrapartum nucleic acid amplification test for GBS should also receive intrapartum chemoprophylaxis. The recommended regimen for chemoprophylaxis is a loading dose of 5 million units of penicillin G followed by 2.5 million units every 4 h until delivery. Cefazolin is an alternative for women with a history of penicillin allergy who are thought not to be at high risk for anaphylaxis. For women with a history of immediate hypersensitivity, clindamycin may be substituted, but only if the colonizing isolate has been demonstrated to be susceptible. If susceptibility testing results are not available or indicate resistance, vancomycin should be used in this situation.
Treatment of all pregnant women who are colonized or have risk factors for neonatal infection will result in exposure of up to one-third of pregnant women and newborns to antibiotics, with the attendant risks of allergic reactions and selection for resistant organisms. Although still in the developmental stages, a GBS vaccine may ultimately offer a better solution to prevention. Because transplacental passage of maternal antibodies produces protective antibody levels in newborns, efforts are under way to develop a vaccine against GBS that can be given to childbearing-age women before or during pregnancy. Results of phase 1 clinical trials of GBS capsular polysaccharide–protein conjugate vaccines suggest that a multivalent conjugate vaccine would be safe and highly immunogenic.
INFECTION IN ADULTS
The majority of GBS infections in otherwise healthy adults are related to pregnancy and parturition. Peripartum fever, the most common manifestation, is sometimes accompanied by symptoms and signs of endometritis or chorioamnionitis (abdominal distention and uterine or adnexal tenderness). Blood and vaginal swab cultures are often positive. Bacteremia is usually transitory but occasionally results in meningitis or endocarditis. Infections in adults that are not associated with the peripartum period generally involve individuals who are elderly or have an underlying chronic illness, such as diabetes mellitus or a malignancy. Among the infections that develop with some frequency in adults are cellulitis and soft tissue infection (including infected diabetic skin ulcers), urinary tract infection, pneumonia, endocarditis, and septic arthritis. Other reported infections include meningitis, osteomyelitis, and intraabdominal or pelvic abscesses. Relapse or recurrence of invasive infection weeks to months after a first episode is documented in ~4% of cases.
|
TREATMENT |
GROUP B STREPTOCOCCAL INFECTION IN ADULTS |
GBS is less sensitive to penicillin than GAS, requiring somewhat higher doses. Adults with serious localized infections (pneumonia, pyelonephritis, abscess) should receive doses of ~12 million units of penicillin G daily; patients with endocarditis or meningitis should receive 18–24 million units per day in divided doses. Vancomycin is an acceptable alternative for penicillin-allergic patients.
NONENTEROCOCCAL GROUP D STREPTOCOCCI
The main nonenterococcal group D streptococci that cause human infections were previously considered a single species, Streptococcus bovis. The organisms encompassed by S. bovis have been reclassified into two species, each of which has two subspecies: Streptococcus gallolyticus subspecies gallolyticus, S. gallolyticus subspecies pasteurianus, Streptococcus infantarius subspecies infantarius, and S. infantarius subspecies coli. Endocarditis caused by these organisms is often associated with neoplasms of the gastrointestinal tract—most frequently, a colon carcinoma or polyp—but is also reported in association with other bowel lesions. When occult gastrointestinal lesions are carefully sought, abnormalities are found in >60% of patients with endocarditis due to S. gallolyticus or S. infantarius. In contrast to the enterococci, nonenterococcal group D streptococci like these organisms are reliably killed by penicillin as a single agent, and penicillin is the agent of choice for the infections they cause.
VIRIDANS AND OTHER STREPTOCOCCI
VIRIDANS STREPTOCOCCI
Consisting of multiple species of α-hemolytic streptococci, the viridans streptococci are a heterogeneous group of organisms that are important agents of bacterial endocarditis (Chap. 155). Several species of viridans streptococci, including Streptococcus salivarius, Streptococcus mitis, Streptococcus sanguis, and Streptococcus mutans, are part of the normal flora of the mouth, where they live in close association with the teeth and gingiva. Some species contribute to the development of dental caries.
Previously known as Streptococcus morbillorum, Gemella morbillorum has been placed in a separate genus, along with Gemella haemolysans, on the basis of genetic-relatedness studies. These species resemble viridans streptococci with respect to habitat in the human host and associated infections.
The transient viridans streptococcal bacteremia induced by eating, toothbrushing, flossing, and other sources of minor trauma, together with adherence to biologic surfaces, is thought to account for the predilection of these organisms to cause endocarditis (see Fig. 155-1). Viridans streptococci are also isolated, often as part of a mixed flora, from sites of sinusitis, brain abscess, and liver abscess.
Viridans streptococcal bacteremia occurs relatively frequently in neutropenic patients, particularly after bone marrow transplantation or high-dose chemotherapy for cancer. Some of these patients develop a sepsis syndrome with high fever and shock. Risk factors for viridans streptococcal bacteremia include chemotherapy with high-dose cytosine arabinoside, prior treatment with trimethoprim-sulfamethoxazole or a fluoroquinolone, treatment with antacids or histamine antagonists, mucositis, and profound neutropenia.
The S. milleri group (also referred to as the S. intermedius or S. anginosus group) includes three species that cause human disease: S. intermedius, S. anginosus, and Streptococcus constellatus. These organisms are often considered viridans streptococci, although they differ somewhat from other viridans streptococci in both their hemolytic pattern (they may be α-, β-, or nonhemolytic) and the disease syndromes they cause. This group commonly produces suppurative infections, particularly abscesses of brain and abdominal viscera, and infections related to the oral cavity or respiratory tract, such as peritonsillar abscess, lung abscess, and empyema.
|
TREATMENT |
INFECTION WITH VIRIDANS STREPTOCOCCI |
Isolates from neutropenic patients with bacteremia are often resistant to penicillin; thus these patients should be treated presumptively with vancomycin until the results of susceptibility testing become available. Viridans streptococci isolated in other clinical settings usually are sensitive to penicillin.
ABIOTROPHIA AND GRANULICATELLA SPECIES (NUTRITIONALLY VARIANT STREPTOCOCCI)
Occasional isolates cultured from the blood of patients with endocarditis fail to grow when subcultured on solid media. These nutritionally variant streptococci require supplemental thiol compounds or active forms of vitamin B6 (pyridoxal or pyridoxamine) for growth in the laboratory. The nutritionally variant streptococci are generally grouped with the viridans streptococci because they cause similar types of infections. However, they have been reclassified on the basis of 16S ribosomal RNA sequence comparisons into two separate genera: Abiotrophia, with a single species (Abiotrophia defectiva), and Granulicatella, with three species associated with human infection (Granulicatella adiacens, Granulicatella para-adiacens, and Granulicatella elegans).
|
TREATMENT |
INFECTION WITH NUTRITIONALLY VARIANT STREPTOCOCCI |
Treatment failure and relapse appear to be more common in cases of endocarditis due to nutritionally variant streptococci than in those due to the usual viridans streptococci. Thus the addition of gentamicin (1 mg/kg every 8 h for patients with normal renal function) to the penicillin regimen is recommended for endocarditis due to the nutritionally variant organisms.
OTHER STREPTOCOCCI
Streptococcus suis is an important pathogen in swine and has been reported to cause meningitis in humans, usually in individuals with occupational exposure to pigs. Strains of S. suis associated with human infections have generally reacted with Lancefield group R typing serum and sometimes with group D typing serum as well. Isolates may be α- or β-hemolytic and are sensitive to penicillin. Streptococcus iniae, a pathogen of fish, has been associated with infections in humans who have handled live or freshly killed fish. Cellulitis of the hand is the most common form of human infection, although bacteremia and endocarditis have been reported. Anaerobic streptococci, or peptostreptococci, are part of the normal flora of the oral cavity, bowel, and vagina. Infections caused by the anaerobic streptococci are discussed in Chap. 201.
174 |
Enterococcal Infections |
Enterococci have been recognized as potential human pathogens for more than a century, but only in recent years have these organisms acquired prominence as important causes of nosocomial infections. The ability of enterococci to survive and/or disseminate in the hospital environment and to acquire antibiotic resistance determinants makes the treatment of some enterococcal infections in critically ill patients a difficult challenge. Enterococci were first mentioned in the French literature in 1899; the “entérocoque” was found in the human gastrointestinal tract and was noted to have the potential to produce significant disease. Indeed, the first pathologic description of an enterococcal infection dates to the same year. A clinical isolate from a patient who died as a consequence of endocarditis was initially designated Micrococcus zymogenes, was later named Streptococcus faecalis subspecies zymogenes, and would now be classified as Enterococcus faecalis. The ability of this isolate to cause severe disease in both rabbits and mice illustrated its potential lethality in the appropriate settings.
ETIOLOGY
Enterococci are gram-positive organisms. In clinical specimens, they are usually observed as single cells, diplococci, or short chains (Fig. 174-1), although long chains are noted with some strains. Enterococci were originally classified as streptococci because organisms of the two genera share many morphologic and phenotypic characteristics, including a generally negative catalase reaction. Only DNA hybridization studies and later 16S rRNA sequencing clearly demonstrated that enterococci should be grouped as a genus distinct from the streptococci. Nonetheless, unlike the majority of streptococci, enterococci hydrolyze esculin in the presence of 40% bile salts and grow at high salt concentrations (e.g., 6.5%) and at high temperatures (46°C). Enterococci are usually reported by the clinical laboratory to be nonhemolytic on the basis of their inability to lyse the ovine or bovine red blood cells (RBCs) commonly used in agar plates; however, some strains of E. faecalis do lyse RBCs from humans, horses, and rabbits. The majority of clinically relevant enterococcal species hydrolyze pyrrolidonyl-β-naphthylamide (PYR); this characteristic is helpful in differentiating enterococci from organisms of the Streptococcus gallolyticus group (formerly known as S. bovis), which includes S. gallolyticus, Streptococcus pasteurianus, and Streptococcus infantarius, and from Leuconostoc species. Although at least 18 species of enterococci have been isolated from human infections, the overwhelming majority of cases are caused by two species, E. faecalis and Enterococcus faecium. Less frequently isolated species include Enterococcus gallinarum, Enterococcus durans, Enterococcus hirae, and Enterococcus avium.
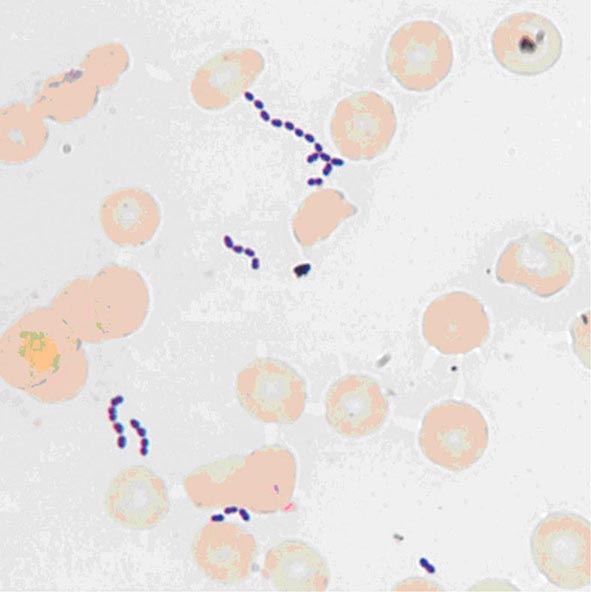
FIGURE 174-1 Gram’s stain of cultured blood from a patient with enterococcal bacteremia. Oval gram-positive bacterial cells are arranged as diplococci and short chains. (Courtesy of Audrey Wanger, PhD.)
PATHOGENESIS
Enterococci are normal inhabitants of the large bowel of human adults, although they usually make up <1% of the culturable intestinal microflora. In the healthy human gastrointestinal tract, enterococci are typical symbionts that coexist with other gastrointestinal bacteria; in fact, the utility of certain enterococcal strains as probiotics in the treatment of diarrhea suggests their possible role in maintaining the homeostatic equilibrium of the bowel. Enterococci are intrinsically resistant to a variety of commonly used antibacterial drugs. One of the most important factors that disrupts this equilibrium and promotes increased gastrointestinal colonization by enterococci is the administration of antimicrobial agents. In particular, antibiotics that are excreted in the bile and have broad-spectrum activity (e.g., certain cephalosporins that target anaerobes and gram-negative bacteria) are usually associated with the recovery of higher numbers of enterococci from feces. This increased colonization appears to be due not only to the simple enterococcal replacement in a “biologic niche” after the eradication of competing components of the flora, but also (at least in mice) to the suppression—upon reduction of the gram-negative microflora by antibiotics—of important immunologic signals (e.g., by the lectin RegIIIγ) that help keep enterococcal counts low in the normal human bowel. Several studies have shown that higher levels of gastrointestinal colonization are a critical factor in the pathogenesis of enterococcal infections. However, the mechanisms by which enterococci successfully colonize the bowel and gain access to the lymphatics and/or bloodstream remain incompletely understood.
Several vertebrate, worm, and insect models have been developed to study the role of possible pathogenic determinants in both E. faecalis and E. faecium. Three main groups of virulence factors may increase the ability of enterococci to colonize the gastrointestinal tract and/or cause disease. The first group, enterococcal secreted factors, are molecules released outside the bacterial cell that contribute to the process of infection. The best-studied of these molecules include enterococcal hemolysin/cytolysin and two enterococcal proteases (gelatinase and serine protease). Enterococcal cytolysin is a heterodimeric toxin produced by some strains of E. faecalis that is capable of lysing human RBCs as well as polymorphonuclear leukocytes and macrophages. E. faecalis gelatinase and serine protease are thought to mediate virulence by several mechanisms, including the degradation of host tissues and the modification of critical components of the immune system. Mutants lacking the genes corresponding to these proteins are highly attenuated in experimental peritonitis, endocarditis, and endophthalmitis.
A second group of virulence factors, enterococcal surface components, are thought to contribute to bacterial attachment to extracellular matrix molecules in the human host. Several molecules on the surface of enterococci have been characterized and shown to play a role in the pathogenesis of enterococcal infections. Among the characterized adhesins is aggregation substance of E. faecalis, which mediates the attachment of bacterial cells to each other, thereby facilitating conjugative plasmid exchange. Several lines of evidence indicate that aggregation substance and enterococcal cytolysin act synergistically to increase the virulence potential of E. faecalis strains in experimental endocarditis. The surface protein adhesin of collagen of E. faecalis (Ace) and its E. faecium homologue (Acm) recognize adhesive matrix molecules (MSCRAMMs) involved in bacterial attachment to host proteins such as collagen, fibronectin, and fibrinogen; both Ace and Acm are important in the pathogenesis of experimental endocarditis. Pili of gram-positive bacteria have been shown to be important mediators of attachment to and invasion of host tissues and are considered potential targets for immunotherapy. Both E. faecalis and E. faecium have surface pili. Mutants of E. faecalis lacking pili are attenuated in biofilm production, experimental endocarditis, and urinary tract infections (UTIs). Other surface proteins that share structural homology with MSCRAMMs and appear to play a role in enterococcal attachment to the host and in virulence include the E. faecalis surface protein Esp and its E. faecium homologue Espfm, the second collagen adhesin of E. faecium (Scm), the surface proteins of E. faecium (Fms), SgrA (which binds to components of the basal lamina), and EcbA (which binds to collagen type V). Additional surface components apparently associated with pathogenicity include the Elr protein (a protein from the WxL family) and polysaccharides, which are thought to interfere with phagocytosis of the organism by host immune cells. Some E. faecalis strains appear to harbor at least three distinct classes of capsular polysaccharide; some of these polysaccharides play a role in virulence and are potential targets for immunotherapy.
The third group of virulence factors has not been well characterized but consists of the E. faecalis stress protein Gls24, which has been associated with enterococcal resistance to bile salts and appears to be important in the pathogenesis of endocarditis, and the hylEfm-containing plasmids of E. faecium, which are transferable between strains and increase gastrointestinal colonization by E. faecium. In mouse peritonitis, acquisition of these plasmids increased the lethality of a commensal strain of E. faecium. Recently, a gene encoding a regulator of oxidative stress (AsrR) has been identified as an important virulence factor of E. faecium.
![]() The ability to sequence bacterial genomes has increased our understanding of bacterial diversity, evolution, pathogenesis, and mechanisms of antibiotic resistance. The genome sequences of more than 560 enterococcal strains are currently available, and some have been entirely closed and annotated. Sequence analysis has shown that the genetic diversity of enterococci is related in large part to the acquisition of exogenous DNA and the mobilization of large chromosomal regions, resulting in recombination of the “core” genomes. In addition, analyses indicate that E. faecium harbors a malleable accessory genome incorporating a substantial content of exogenous elements, including DNA from phages. Indeed, a hospital-associated E. faecium clade that contains most clinical and outbreak-associated strains is the predominant genetic lineage circulating in hospitals around the world. This clade appears to be evolving rapidly, and genomic comparisons suggest that this lineage emerged 75 years ago—a time point that coincides with the introduction of antimicrobial drugs—and evolved from animal strains, not from human commensal isolates. An initial genomic separation within E. faecium appears to have occurred ~3000 years ago, simultaneous with urbanization and domestication of animals. This genomic information provides new clues with regard to the evolution of enterococci from commensal organisms to important nosocomial pathogens.
The ability to sequence bacterial genomes has increased our understanding of bacterial diversity, evolution, pathogenesis, and mechanisms of antibiotic resistance. The genome sequences of more than 560 enterococcal strains are currently available, and some have been entirely closed and annotated. Sequence analysis has shown that the genetic diversity of enterococci is related in large part to the acquisition of exogenous DNA and the mobilization of large chromosomal regions, resulting in recombination of the “core” genomes. In addition, analyses indicate that E. faecium harbors a malleable accessory genome incorporating a substantial content of exogenous elements, including DNA from phages. Indeed, a hospital-associated E. faecium clade that contains most clinical and outbreak-associated strains is the predominant genetic lineage circulating in hospitals around the world. This clade appears to be evolving rapidly, and genomic comparisons suggest that this lineage emerged 75 years ago—a time point that coincides with the introduction of antimicrobial drugs—and evolved from animal strains, not from human commensal isolates. An initial genomic separation within E. faecium appears to have occurred ~3000 years ago, simultaneous with urbanization and domestication of animals. This genomic information provides new clues with regard to the evolution of enterococci from commensal organisms to important nosocomial pathogens.
EPIDEMIOLOGY
According to the National Healthcare Safety Network of the Centers for Disease Control and Prevention, enterococci are the second most common organisms (after staphylococci) isolated from hospital-associated infections in the United States. Although E. faecalis remains the predominant species recovered from nosocomial infections, the isolation of E. faecium has increased substantially in the past 20 years. In fact, E. faecium is now almost as common as E. faecalis as an etiologic agent of hospital-associated infections. This point is important, because E. faecium is by far the most resistant and challenging enterococcal species to treat; indeed, more than 80% of E. faecium isolates recovered in U.S. hospitals are resistant to vancomycin, and more than 90% are resistant to ampicillin (historically the most effective β-lactam agent against enterococci). Resistance to vancomycin and ampicillin in E. faecalis isolates is much less common (~7% and ~4%, respectively).
The dynamics of enterococcal transmission and dissemination in the hospital environment have been extensively studied, with a focus on vancomycin-resistant enterococci (VRE). These studies have revealed that VRE colonization of the gastrointestinal tract is a critical step in the natural history of enterococcal disease and that a substantial proportion of patients colonized with VRE remain colonized for prolonged periods (sometimes >1 year) and are more likely to develop an Enterococcus-related illness (e.g., bacteremia). The most important factors associated with VRE colonization and persistence in the gut include prolonged hospitalization; long courses of antibiotic therapy; hospitalization in long-term-care facilities, surgical units, and/or intensive care units; organ transplantation; renal failure (particularly in patients undergoing hemodialysis) and/or diabetes; high Acute Physiology and Chronic Health Evaluation (APACHE) scores; and physical proximity to patients infected or colonized with VRE or these patients’ rooms. Once a patient becomes colonized with VRE, several key factors are involved in the organisms’ dissemination in the hospital environment. VRE can survive exposure to heat and certain disinfectants and have been found on numerous inanimate objects in the hospital, including bed rails, medical equipment, doorknobs, gloves, telephones, and computer keyboards. Thus health care workers and the environment play pivotal roles in enterococcal transmission from patient to patient, and infection control measures are crucial in breaking the chain of transmission. Moreover, two meta-analyses have found that, independent of the patient’s clinical status, VRE infection increases the risk of death over that among individuals infected with a glycopeptide-susceptible enterococcal strain.
![]() The epidemiology of enterococcal disease and the emergence of VRE have followed slightly different trends in other parts of the world than in the United States. In Europe, the emergence of VRE in the mid-1980s was seen primarily in isolates recovered from animals and healthy humans rather than from hospitalized patients. The presence of VRE was associated with the use of the glycopeptide avoparcin as a growth promoter in animal feeds; this association prompted the European Union to ban the use of this compound in animal husbandry in 1996. However, after an initial decrease in the isolation of VRE from animals and humans, the prevalence of hospital-associated VRE infections has slowly increased in certain European countries, with important regional differences. For example, rates of vancomycin resistance among E. faecium clinical isolates in Europe are highest in Greece, the United Kingdom, and Portugal (10–30%), whereas rates in the Scandinavian countries and the Netherlands are <1%. These regional differences have been attributed in part to the implementation of aggressive “search-and-destroy” policies of infection control in countries such as the Netherlands; these policies have kept the frequency of nosocomial methicillin-resistant Staphylococcus aureus (MRSA) and VRE very low. Despite regional differences, rates of VRE continue to be much lower in Europe than in the United States. The reasons are not totally understood, although it has been postulated that this difference is related to the higher levels of human antibiotic use in the United States. Rates of enterococcal resistance to vancomycin in some Latin American countries are also lower (~4%) than those in the United States. Conversely, in Asia, rates of vancomycin resistance among enterococci appear to be similar to those in U.S. hospitals.
The epidemiology of enterococcal disease and the emergence of VRE have followed slightly different trends in other parts of the world than in the United States. In Europe, the emergence of VRE in the mid-1980s was seen primarily in isolates recovered from animals and healthy humans rather than from hospitalized patients. The presence of VRE was associated with the use of the glycopeptide avoparcin as a growth promoter in animal feeds; this association prompted the European Union to ban the use of this compound in animal husbandry in 1996. However, after an initial decrease in the isolation of VRE from animals and humans, the prevalence of hospital-associated VRE infections has slowly increased in certain European countries, with important regional differences. For example, rates of vancomycin resistance among E. faecium clinical isolates in Europe are highest in Greece, the United Kingdom, and Portugal (10–30%), whereas rates in the Scandinavian countries and the Netherlands are <1%. These regional differences have been attributed in part to the implementation of aggressive “search-and-destroy” policies of infection control in countries such as the Netherlands; these policies have kept the frequency of nosocomial methicillin-resistant Staphylococcus aureus (MRSA) and VRE very low. Despite regional differences, rates of VRE continue to be much lower in Europe than in the United States. The reasons are not totally understood, although it has been postulated that this difference is related to the higher levels of human antibiotic use in the United States. Rates of enterococcal resistance to vancomycin in some Latin American countries are also lower (~4%) than those in the United States. Conversely, in Asia, rates of vancomycin resistance among enterococci appear to be similar to those in U.S. hospitals.
![]() As mentioned above, genomic analyses of vancomycin-resistant E. faecium in different parts of the world suggest that the emergence and dissemination of these organisms in the hospital environment worldwide are due to the success of a unique hospital-associated genetic clade that acquired the genes responsible for vancomycin resistance as well as other antibiotic resistance determinants.
As mentioned above, genomic analyses of vancomycin-resistant E. faecium in different parts of the world suggest that the emergence and dissemination of these organisms in the hospital environment worldwide are due to the success of a unique hospital-associated genetic clade that acquired the genes responsible for vancomycin resistance as well as other antibiotic resistance determinants.
CLINICAL SYNDROMES
URINARY TRACT INFECTION AND PROSTATITIS
Enterococci are well-known causes of nosocomial UTI—the most common infection caused by these organisms (Chap. 162). Enterococcal UTIs are usually associated with indwelling catheters, instrumentation, or anatomic abnormalities of the genitourinary tract, and it is often challenging to differentiate between true infection and colonization (particularly in patients with chronic indwelling catheters). The presence of leukocytes in the urine in conjunction with systemic manifestations (e.g., fever) or local signs and symptoms of infection with no other explanation and a positive urine culture (≥105 colony-forming units [CFU]/mL) suggests the diagnosis. Moreover, enterococcal UTIs often occur in critically ill patients whose comorbidities may obscure the diagnosis. In many cases, removal of the indwelling catheter may suffice to eradicate the organism without specific antimicrobial therapy. In rare circumstances, UTIs caused by enterococci may run a complicated course, with the development of pyelonephritis and perinephric abscesses that may be a portal of entry for bloodstream infections (see below). Enterococci are also known causes of chronic prostatitis, particularly in patients whose urinary tract has been manipulated surgically or endoscopically. These infections can be difficult to treat because the agents most potent against enterococci (i.e., aminopenicillins and glycopeptides) penetrate prostatic tissue poorly. Chronic prostatic infection can be a source of recurrent enterococcal bacteremia.
BACTEREMIA AND ENDOCARDITIS
Bacteremia without endocarditis is one of the most common presentations of enterococcal disease. Intravascular catheters and other devices are commonly associated with these bacteremic episodes (Chap. 168). Other well-known sources of enterococcal bacteremia include the gastrointestinal and hepatobiliary tracts; pelvic and intraabdominal foci; and, less frequently, wound infections, UTIs, and bone infections. In the United States, enterococci are ranked second (after coagulase-negative staphylococci) as etiologic agents of central line–associated bacteremia. Patients with enterococcal bacteremia usually have comorbidities and have been in the hospital for prolonged periods; they commonly have received several courses of antibiotics. Several studies indicate that the isolation of E. faecium from the blood may lead to worse outcomes and higher mortality rates than when other enterococcal species are isolated; this finding may be related to the higher prevalence of vancomycin and ampicillin resistance in E. faecium than in other enterococcal species, with the consequent reduction of therapeutic options. In many cases (usually when the gastrointestinal tract is the source), enterococcal bacteremia may be polymicrobial, with gram-negative organisms isolated at the same time. In addition, several cases have now been documented in which enterococcal bacteremia was associated with Strongyloides stercoralis hyperinfection syndrome in immunocompromised patients.
Enterococci are important causes of community- and health care–associated endocarditis, ranking second after staphylococci in the latter infections. The presumed initial source of bacteremia leading to endocarditis is the gastrointestinal or genitourinary tract—e.g., in patients who have malignant and inflammatory conditions of the gut or have undergone procedures in which these tracts are manipulated. The affected patients tend to be male and elderly and to have other debilitating diseases and heart conditions. Both prosthetic and native valves can be involved; mitral and aortic valves are affected most often. Community-associated endocarditis (usually caused by E. faecalis) also occurs in patients with no apparent risk factors or cardiac abnormalities. Endocarditis in women of childbearing age has been well described. The typical presentation of enterococcal endocarditis is a subacute course of fever, weight loss, malaise, and cardiac murmur; typical stigmata of endocarditis (e.g., petechiae, Osler’s nodes, Roth’s spots) are found in only a minority of patients. Atypical manifestations include arthralgias and manifestations of metastatic disease (splenic abscesses, hiccups, pain in the left flank, pleural effusion, and spondylodiscitis). Embolic complications are variable and can affect the brain. Heart failure is a common complication of enterococcal endocarditis, and valve replacement may be critical in curing this infection, particularly when multidrug-resistant organisms or major complications are involved. The duration of therapy is usually 4–6 weeks, with more prolonged courses suggested for multidrug-resistant isolates in the absence of valvular replacement.
MENINGITIS
Enterococcal meningitis is an uncommon disease (accounting for only ~4% of meningitis cases) that is usually associated with neurosurgical interventions and conditions such as shunts, central nervous system (CNS) trauma, and cerebrospinal fluid (CSF) leakage. In some instances—usually in patients with a debilitating condition, such as cardiovascular or congenital heart disease, chronic renal failure, malignancy, receipt of immunosuppressive therapy, or HIV/AIDS—presumed hematogenous seeding of the meninges is seen in infections such as endocarditis or bacteremia. Fever and changes in mental status are common, whereas overt meningeal signs are less so. CSF findings are consistent with bacterial infection—i.e., pleocytosis with a predominance of polymorphonuclear leukocytes (average, ~500/μL), an elevated serum protein level (usually >100 mg/dL), and a decreased glucose concentration (average, 28 mg/dL). Gram’s staining yields a positive result in about half of cases, with a high rate of organism recovery from CSF cultures; the most common species isolated are E. faecalis and E. faecium. Complications include hydrocephalus, brain abscesses, and stroke. As mentioned before for bacteremia, an association with Strongyloides hyperinfection has also been documented.
INTRAABDOMINAL, PELVIC, AND SOFT TISSUE INFECTIONS
As mentioned earlier, enterococci are part of the commensal flora of the gastrointestinal tract and can produce spontaneous peritonitis in cirrhotic individuals and in patients undergoing chronic ambulatory peritoneal dialysis (Chap. 159). These organisms are commonly found (usually along with other bacteria, including enteric gram-negative species and anaerobes) in clinical samples from intraabdominal and pelvic collections. The presence of enterococci in intraabdominal infections is sometimes considered to be of little clinical relevance. Several studies have shown that the role of enterococci in intraabdominal infections originating in the community and involving previously healthy patients is minor, because surgery and broad-spectrum antimicrobial drugs that do not target enterococci are often sufficient to treat these infections successfully. In the last few decades, however, these organisms have become prominent as a cause of intraabdominal infections in hospitalized patients because of the emergence and spread of vancomycin resistance among enterococci and the increase in rates of nosocomial infections due to multidrug-resistant E. faecium isolates. In fact, several studies have now documented treatment failures due to enterococci, with consequently increased rates of postoperative complications and death among patients with intraabdominal infections. Thus, anti-enterococcal therapy is recommended for nosocomial peritonitis in immunocompromised and severely ill patients who have had a prolonged hospital stay, have undergone multiple procedures, have persistent abdominal sepsis and collections, or have risk factors for the development of endocarditis (e.g., prosthetic or damaged heart valves). Conversely, specific treatment for enterococci in the first episode of intraabdominal infections originating in the community and affecting previously healthy patients with no important cardiac risk factors for endocarditis does not appear to be beneficial.
Enterococci are commonly isolated from soft tissue infections (Chap. 156), particularly those involving surgical wounds (Chap. 168). In fact, these organisms rank third as agents of nosocomial surgical-site infections, with E. faecalis the most frequently isolated species. The clinical relevance of enterococci in some of these infections—as in intraabdominal infections—is a matter of debate; differentiating between colonization and true infection is sometimes challenging, although in some cases enterococci have been recovered from lung, liver, and skin abscesses. Diabetic foot and decubitus ulcers are often colonized with enterococci and may be the portal of entry for bone infections.
OTHER INFECTIONS
Enterococci are well-known causes of neonatal infections, including sepsis (mostly late-onset), bacteremia, meningitis, pneumonia, and UTI. Outbreaks of enterococcal sepsis in neonatal units have been well documented. Risk factors for enterococcal disease in newborns include prematurity, low birth weight, indwelling devices, and abdominal surgery. Enterococci have also been described as etiologic agents of bone and joint infections, including vertebral osteomyelitis, usually in patients with underlying conditions such as diabetes or endocarditis. Similarly, enterococci have been isolated from bone infections in patients who have undergone arthroplasty or reconstruction of fractures with the placement of hardware. Because enterococci can produce a biofilm that is likely to alter the efficacy of otherwise active anti-enterococcal agents, treatment of infections that involve foreign material is challenging, and removal of the hardware may be necessary to eradicate the infection. Rare cases of enterococcal pneumonia, lung abscess, and spontaneous empyema have been described.
|
TREATMENT |
ENTEROCOCCAL INFECTIONS |
GENERAL PRINCIPLES
Enterococci are intrinsically resistant and/or tolerant to several antimicrobial agents (with tolerance defined as lack of killing by drug concentrations 32 times higher than the minimal inhibitory concentration [MIC]). Monotherapy for endocarditis with a β-lactam antibiotic (to which many enterococci are tolerant) has produced disappointing results, with low cure rates at the end of therapy. However, the addition of an aminoglycoside to a cell wall–active agent (a β-lactam or a glycopeptide) increases cure rates and eradicates the organisms; moreover, this combination is synergistic and bactericidal in vitro. Therefore, for many decades, combination therapy with a cell wall–active agent and an aminoglycoside has been the standard of care for endovascular infections caused by enterococci. This synergistic effect can be explained, at least in part, by the increased penetration of the aminoglycoside into the bacterial cell, presumably as a result of cell wall alterations produced by the β-lactam (or glycopeptide). Nonetheless, attaining synergistic bactericidal activity in the treatment of severe enterococcal infections has become increasingly difficult because of the development of resistance to virtually all antibiotics available for this purpose.
The treatment of E. faecalis differs substantially from that of E. faecium (Tables 174-1 and 174-2), mainly because of differences in resistance profiles (see below). For example, resistance to ampicillin and vancomycin is rare in E. faecalis, whereas these antibiotics are only infrequently useful against current isolates of E. faecium. Moreover, as a consequence of the challenges and therapeutic limitations posed by the emergence of drug resistance in enterococci, valve replacement may need to be considered in the treatment of endocarditis caused by multidrug-resistant enterococci. Less severe infections are often related to indwelling intravascular catheters; removal of the catheter increases the likelihood of enterococcal eradication by a subsequent short course of appropriate antimicrobial therapy.
|
SUGGESTED REGIMENS FOR THE MANAGEMENT OF INFECTIONS CAUSED BY ENTEROCOCCUS FAECALIS |
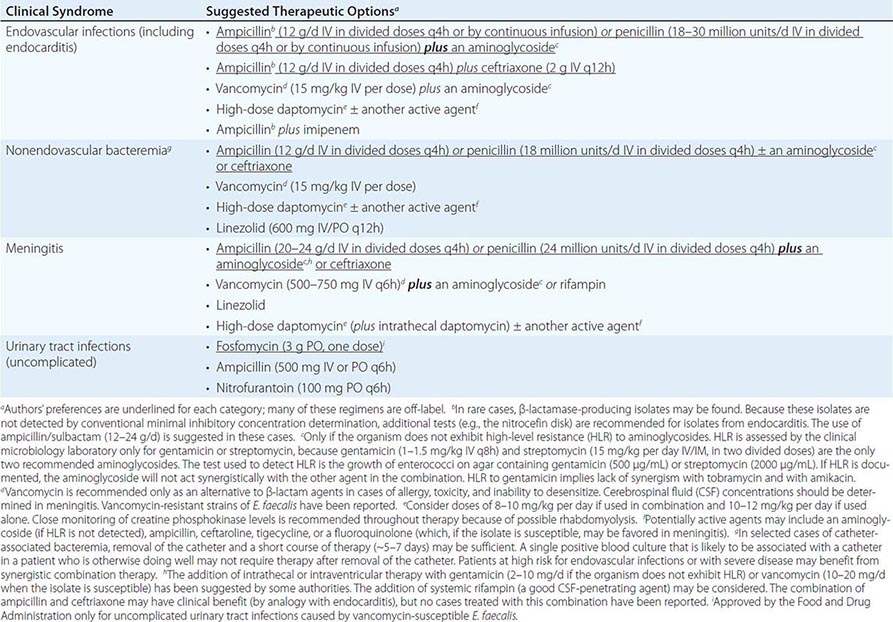
|
SUGGESTED REGIMENS FOR THE MANAGEMENT OF INFECTIONS CAUSED BY VANCOMYCIN- AND AMPICILLIN-RESISTANT ENTEROCOCCUS FAECIUM |
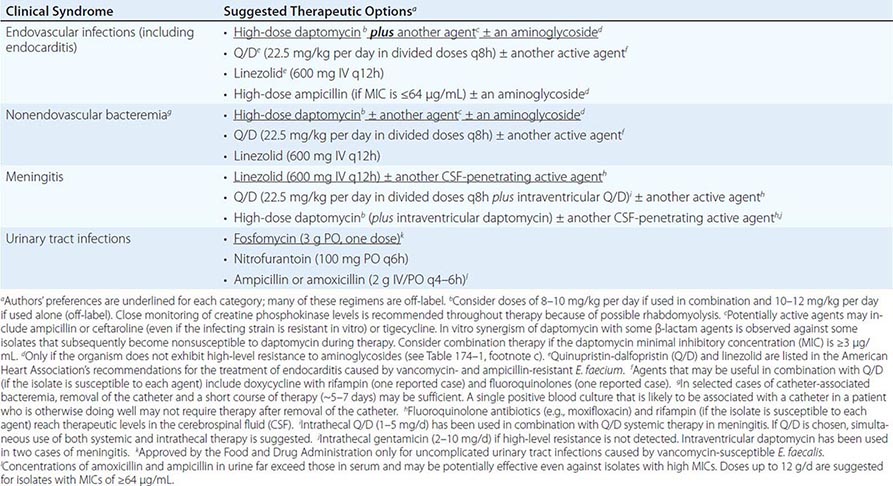
CHOICE OF ANTIMICROBIAL AGENTS
Among the β-lactams, the most active are the aminopenicillins (ampicillin, amoxicillin) and ureidopenicillins (i.e., piperacillin); next most active are penicillin G and imipenem. For E. faecium, a combination of high-dose ampicillin (up to 30 g/d) plus an aminoglycoside has been suggested—even for ampicillin-resistant strains if the MIC is ≤64 μg/mL—because a plasma ampicillin concentration of >100 μg/mL can be achieved at high doses. The only two aminoglycosides recommended for synergistic therapy in severe enterococcal infections are gentamicin and streptomycin. The use of amikacin is discouraged, tobramycin should never be used against E. faecium, and aminoglycoside monotherapy is not effective. Vancomycin is an alternative to β-lactam drugs for the treatment of E. faecalis infections but is less useful against E. faecium because resistance is common.
As mentioned above, use of the aminoglycoside–ampicillin combination for E. faecalis infections has become increasingly problematic because of toxicity in critically ill patients and increased rates of high-level resistance to aminoglycosides. A recent observational, nonrandomized, comparative study encompassing a multicenter cohort was conducted in 17 Spanish hospitals and 1 Italian hospital; this study found that the combination of ampicillin and ceftriaxone is as effective as ampicillin plus gentamicin in the treatment of E. faecalis endocarditis, with less risk of toxicity. Therefore, this regimen should be considered in patients at risk for aminoglycoside toxicity and could be considered for all patients.
Linezolid and quinupristin/dalfopristin (Q/D) are two agents approved by the U.S. Food and Drug Administration (FDA) for the treatment of some VRE infections (Table 174–2). Linezolid is not bactericidal, and its use in severe endovascular infections has produced mixed results; therefore, it is recommended only as an alternative to other agents. In addition, linezolid may cause significant toxicities (thrombocytopenia, peripheral neuropathy, and optic neuritis) when used in regimens given for >2 weeks. Nonetheless, linezolid may play a role in the treatment of enterococcal meningitis and other CNS infections, although clinical data are limited. Q/D is not active against most E. faecalis isolates, and its in vivo efficacy against E. faecium may often be compromised by resistance (see below). Adverse reactions to Q/D are common, including pain and inflammation at the infusion site and severe arthralgias and myalgias leading to discontinuation of treatment. Thus, Q/D should be used with caution and probably combined with other agents (Table 174–2).
The lipopeptide daptomycin is a bactericidal antibiotic with potent in vitro activity against all enterococci. Although daptomycin is not approved by the FDA for the treatment of VRE or E. faecium infections, it has been used alone (at high dosage) or in combination with other agents (ampicillin, ceftaroline, and tigecycline) with apparent success against multidrug-resistant enterococcal infections (Tables 174–1 and 174–2). The main adverse reactions to daptomycin are elevated creatine phosphokinase levels and eosinophilic pneumonitis (rare). Daptomycin is not useful against pulmonary infections because the pulmonary surfactant inhibits its antibacterial activity. Although the glycylcycline drug tigecycline is active in vitro against all enterococci (regardless of the isolates’ vancomycin susceptibility), its use as monotherapy for endovascular or severe enterococcal infections is not recommended because of low attainable blood levels. Telavancin, a lipoglycopeptide approved by the FDA for the treatment of skin and soft tissue infections as well as hospital-associated pneumonia, is active against vancomycin-susceptible enterococci but not VRE. Oritavancin, a compound of the same class that is active against VRE, has recently been approved by the FDA for the treatment of bacterial skin and soft tissue infections and may offer promise for the treatment of VRE in the future.
ANTIMICROBIAL RESISTANCE
As mentioned above, resistance to β-lactam agents continues to be observed only infrequently in E. faecalis, although rare outbreaks caused by β-lactamase-producing isolates have occurred in the United States and Argentina. However, ampicillin resistance is common in E. faecium. The mechanism of this resistance is related to a penicillin-binding protein (PBP) designated PBP5, which is the target of β-lactam antibiotics. PBP5 exhibits lower affinity for ampicillin and can synthesize cell wall in the presence of this antibiotic, even when other PBPs are inhibited. Two common mechanisms of high-level ampicillin resistance (MIC, >64 μg/mL) in clinical strains are (1) mutations in the PBP5-encoding gene that further decrease the protein’s affinity for ampicillin and (2) hyperproduction of PBP5. These factors preclude the use of all β-lactam agents in the treatment of E. faecium infections.
Vancomycin is a glycopeptide antibiotic that inhibits cell wall peptidoglycan synthesis in susceptible enterococci and has been widely used against enterococcal infections in clinical practice when the utility of β-lactams is limited by resistance, allergy, or adverse reactions. This effect is mediated by binding of the antibiotic to peptidoglycan precursors (UDP-MurNAc-pentapeptides) upon their exit from the bacterial cell cytoplasm. The interaction of vancomycin with the peptidoglycan is specific and involves the last two D-alanine residues of the precursor. The first isolates of VRE were documented in 1986, and vancomycin resistance (particularly in E. faecium) has since increased considerably around the world. The mechanism involves the replacement of the last D-alanine residue of peptidoglycan precursors with D-lactate or D-serine, with consequent high- and low-level resistance, respectively. There is significant heterogeneity among isolates, but either substitution substantially decreases the affinity of vancomycin for the peptidoglycan; with the D-lactate substitution, the MIC is increased by up to 1000-fold. Vancomycin-resistant organisms also produce enzymes that destroy the D-alanine-D-alanine ending precursors, ensuring that additional binding sites for vancomycin are not available.
High-level resistance to aminoglycosides (of which gentamicin and streptomycin are the only two tested by clinical laboratories) abolishes the synergism observed between cell wall–active agents and the aminoglycoside. This important phenotype is routinely sought in isolates from serious infections (Tables 174–1 and 174–2). The laboratory reports high-level resistance as gentamicin and streptomycin MICs of >500 μg/mL and >2000 μg/mL, respectively (agar dilution method) or as “SYN-R” (resistance to synergism). Genes encoding aminoglycoside-modifying enzymes are usually the cause of high-level resistance to these compounds and are widely disseminated among enterococci, decreasing the options for the treatment of severe enterococcal infections. The aforementioned enterococcal resistance to newer antibiotics such as linezolid (usually due to mutations in the 23S rRNA genes and the presence of an rRNA methylase), Q/D, daptomycin (involving major changes in cell membrane homeostasis), and tigecycline further reduces therapeutic alternatives.
175 |
Diphtheria and Other Corynebacterial Infections |
DIPHTHERIA
Diphtheria is a nasopharyngeal and skin infection caused by Corynebacterium diphtheriae. Toxigenic strains of C. diphtheriae produce a protein toxin that causes systemic toxicity, myocarditis, and polyneuropathy. The toxin is associated with the formation of pseudomembranes in the pharynx during respiratory diphtheria. While toxigenic strains most frequently cause pharyngeal diphtheria, nontoxigenic strains commonly cause cutaneous disease.
ETIOLOGY
C. diphtheriae is a gram-positive bacillus that is unencapsulated, nonmotile, and nonsporulating. The organism was first identified microscopically in 1883 by Klebs and a year later was isolated in pure culture by Löffler in Robert Koch’s laboratory. The bacteria have a characteristic club-shaped bacillary appearance and typically form clusters of parallel rays, or palisades, that are referred to as “Chinese characters.” The specific laboratory media recommended for the cultivation of C. diphtheriae rely upon tellurite, colistin, or nalidixic acid for the organism’s selective isolation from other autochthonous pharyngeal microbes. C. diphtheriae may be isolated from individuals with both nontoxigenic (tox–) and toxigenic (tox+) phenotypes. Uchida and Pappenheimer demonstrated that corynebacteriophage beta carries the structural gene tox, which encodes diphtheria toxin, and that a family of closely related corynebacteriophages are responsible for toxigenic conversion of tox– C. diphtheriae to the tox+ phenotype. Moreover, lysogenic conversion from a nontoxigenic to a toxigenic phenotype has been shown to occur in situ. Growth of toxigenic strains of C. diphtheriae under iron-limiting conditions leads to the optimal expression of diphtheria toxin and is believed to be a pathogenic mechanism during human infection.
EPIDEMIOLOGY
![]() While in many regions diphtheria has been controlled in recent years with effective vaccination, there have been sporadic outbreaks in the United States and Europe. Diphtheria is still common in the Caribbean, Latin America, and the Indian subcontinent, where mass immunization programs are not enforced. Large-scale epidemics of diphtheria have occurred in the post-Soviet independent states. Additional outbreaks have been reported in Algeria, China, and Ecuador.
While in many regions diphtheria has been controlled in recent years with effective vaccination, there have been sporadic outbreaks in the United States and Europe. Diphtheria is still common in the Caribbean, Latin America, and the Indian subcontinent, where mass immunization programs are not enforced. Large-scale epidemics of diphtheria have occurred in the post-Soviet independent states. Additional outbreaks have been reported in Algeria, China, and Ecuador.
C. diphtheriae is transmitted via the aerosol route, usually during close contact with an infected person. There are no significant reservoirs other than humans. The incubation period for respiratory diphtheria is 2–5 days, but disease onset has occurred as late as 10 days after exposure. Prior to the vaccination era, most individuals over the age of 10 were immune to C. diphtheriae; infants were protected by maternal IgG antibodies but became susceptible after ~6 months of age. Thus, the disease primarily affected children and nonimmune young adults. In temperate regions, respiratory diphtheria occurs year-round but is most common during winter months.
The development of diphtheria antitoxin in 1898 by von Behring and of the diphtheria toxoid vaccine in 1924 by Ramon led to the near-elimination of diphtheria in Western countries. The annual incidence rate in the United States peaked in 1921 at 191 cases per 100,000 population. In contrast, since 1980, the annual figure in the United States has been <5 cases per 100,000. Nevertheless, pockets of colonization persist in North America, particularly in South Dakota, Ontario, and recently the state of Washington. Immunity to diphtheria induced by childhood vaccination gradually decreases in adulthood. An estimated 30% of men 60–69 years old have antitoxin titers below the protective level. In addition to older age and lack of vaccination, risk factors for diphtheria outbreaks include alcoholism, low socioeconomic status, crowded living conditions, and Native American ethnic background. An outbreak of diphtheria in Seattle, Washington, between 1972 and 1982 comprised 1100 cases, most of which were cutaneous. During the 1990s in the states of the former Soviet Union, a much larger diphtheria epidemic included more than 150,000 cases and more than 5000 deaths. Clonally related toxigenic C. diphtheriae strains of the ET8 complex were associated with this outbreak. Given that the ET8 complex expressed a toxin against which the prevalent diphtheria toxoid vaccine was effective, the epidemic was attributed to failure of the public health infrastructure to effectively vaccinate the population. Beginning in 1998, this epidemic was controlled by mass vaccination programs. During the epidemic, the incidence rate was high among individuals between 16 and 50 years of age. Socioeconomic instability, migration, deteriorating public health programs, frequent vaccine shortages, delayed implementation of vaccination and treatment in response to cases, and lack of public education and awareness were contributing factors.
Significant outbreaks of diphtheria and diphtheria-related mortality continue to be reported from many developing countries, particularly in Africa and Asia. Statistics collected by the World Health Organization indicated the occurrence of ~7000 reported diphtheria cases in 2008 and ~5000 diphtheria deaths in 2004. Although ~82% of the global population has been adequately vaccinated, only 26% of countries have successfully vaccinated >80% of individuals in all districts.
Cutaneous diphtheria is usually a secondary infection that follows a primary skin lesion due to trauma, allergy, or autoimmunity. Most often, these isolates lack the tox gene and thus do not express diphtheria toxin. In tropical latitudes, cutaneous diphtheria is more common than respiratory diphtheria. In contrast to respiratory disease, cutaneous diphtheria is not reportable in the United States. Nontoxigenic strains of C. diphtheriae have also been associated with pharyngitis in Europe, causing outbreaks among men who have sex with men and persons who use illicit IV drugs.
PATHOGENESIS AND IMMUNOLOGY
Diphtheria toxin produced by tox+ strains of C. diphtheriae is the primary virulence factor in clinical disease. The toxin is synthesized in precursor form; is released as a 535-amino-acid, single-chain protein; and, in sensitive species (e.g., guinea pigs and humans, but not mice or rats), has a 50% lethal dose of ~100 ng/kg of body weight. The toxin is produced in the pseudomembranous lesion and is taken up in the bloodstream, from which it is distributed to all organ systems in the body. Once bound to its cell surface receptor (a heparin-binding epidermal growth factor–like precursor), the toxin is internalized by receptor-mediated endocytosis and enters the cytosol from an acidified early endosomal compartment. In vitro, the toxin may be separated into two chains by digestion with serine proteases: the N-terminal A fragment and the C-terminal B fragment. Delivery of the A fragment into the eukaryotic cell cytosol results in irreversible inhibition of protein synthesis by NAD+-dependent ADP-ribosylation of elongation factor 2. The eventual result is the death of the cell.
In 1926, Ramon at the Institut Pasteur found that formalinization of diphtheria toxin resulted in the production of a nontoxic but highly immunogenic diphtheria toxoid. Subsequent studies showed that immunization with diphtheria toxoid elicited antibodies that neutralized the toxin and prevented most disease manifestations. In the 1930s, mass immunization of children and susceptible adults with diphtheria toxoid commenced in the United States and Europe.
Individuals with a diphtheria antitoxin titer of >0.01 U/mL are at low risk of disease. In populations where a majority of individuals have protective antitoxin titers, the carrier rate for toxigenic strains of C. diphtheriae decreases and the overall risk of diphtheria among susceptible individuals is reduced. Nevertheless, individuals with nonprotective titers may contract diphtheria through either travel or exposure to individuals who have recently returned from regions where the disease is endemic.
Characteristic pathologic findings of diphtheria include mucosal ulcers with a pseudomembranous coating composed of an inner band of fibrin and a luminal band of neutrophils. Initially white and firmly adherent, in advanced diphtheria the pseudomembranes turn gray or even green or black as necrosis progresses. Mucosal ulcers result from toxin-induced necrosis of the epithelium accompanied by edema, hyperemia, and vascular congestion of the submucosal base. A significant fibrinosuppurative exudate from the ulcer develops into the pseudomembrane. Ulcers and pseudomembranes in severe respiratory diphtheria may extend from the pharynx into medium-sized bronchial airways. Expanding and sloughing membranes may result in fatal airway obstruction.
FIGURE 175-1 Respiratory diphtheria due to toxigenic C. diphtheriae producing exudative pharyngitis in a 47-year-old female patient displaying neck edema and a pseudomembrane extending from the uvula to the pharyngeal wall. The characteristic white pseudomembrane is caused by diphtheria toxin–mediated necrosis of the respiratory epithelial layer, producing a fibrinous coagulative exudate. Submucosal edema adds to airway narrowing. The pharyngitis is acute in onset, and respiratory obstruction from the pseudomembrane may occur in severe cases. Inoculation of pseudomembrane fragments or submembranous swabs onto Löffler’s or tellurite selective medium reveals C. diphtheriae. (Photograph by P. Strebel, MD, used by permission. From R. Kadirova et al: J Infect Dis 181:S110, 2000. With permission of Oxford University Press.)
CLINICAL MANIFESTATIONS
Respiratory Diphtheria The clinical diagnosis of diphtheria is based on the constellation of sore throat; adherent tonsillar, pharyngeal, or nasal pseudomembranous lesions; and low-grade fever. In addition, diagnosis requires the isolation of C. diphtheriae or histopathologic isolation of compatible gram-positive organisms. The Centers for Disease Control and Prevention (CDC) recognizes confirmed respiratory diphtheria (laboratory proven or epidemiologically linked to a culture-confirmed case) and probable respiratory diphtheria (clinically compatible but not laboratory proven or epidemiologically linked). Carriers are defined as individuals who have positive cultures for C. diphtheriae and who either are asymptomatic or have symptoms but lack pseudomembranes. Most patients seek medical care for sore throat and fever several days into the illness. Occasionally, weakness, dysphagia, headache, and voice change are the initial manifestations. Neck edema and difficulty breathing are evident in more advanced cases and carry a poor prognosis.
The systemic manifestations of diphtheria stem from the effects of diphtheria toxin and include weakness as a result of neurotoxicity and cardiac arrhythmias or congestive heart failure due to myocarditis. Most commonly, the pseudomembranous lesion is located in the tonsillopharyngeal region. Less commonly, the lesions are located in the larynx, nares, and trachea or bronchial passages. Large pseudomembranes are associated with severe disease and a poor prognosis. A few patients develop massive swelling of the tonsils and present with “bull-neck” diphtheria, which results from massive edema of the submandibular and paratracheal region and is further characterized by foul breath, thick speech, and stridorous breathing. The diphtheritic pseudomembrane is gray or whitish and sharply demarcated. Unlike the exudative lesion associated with streptococcal pharyngitis, the pseudomembrane in diphtheria is tightly adherent to the underlying tissues. Attempts to dislodge the membrane may cause bleeding. Hoarseness suggests laryngeal diphtheria, in which laryngoscopy may be diagnostically helpful.
Cutaneous Diphtheria This dermatosis is characterized by punched-out ulcerative lesions with necrotic sloughing or pseudomembrane formation (Figure 175-2). The diagnosis requires cultivation of C. diphtheriae from lesions, which most commonly occur on the lower and upper extremities, head, and trunk.
FIGURE 175-2 Cutaneous diphtheria due to nontoxigenic C. diphtheriae on the lower extremity. (From the Centers for Disease Control and Prevention.)
Infections Due to Non-diphtheriae Corynebacterium Species and Nontoxigenic C. diphtheriae Non-diphtheriae species of Corynebacterium and related genera (discussed below) as well as nontoxigenic strains of C. diphtheriae itself have been found in bloodstream and respiratory infections, often in individuals with immunosuppression or chronic respiratory disease. These organisms can cause disease manifestations and should not necessarily be dismissed as colonizers.
Other Clinical Manifestations C. diphtheriae causes rare cases of endocarditis and septic arthritis, most often in patients with preexisting risk factors, such as abnormal cardiac valves, injection drug use, or cirrhosis.
COMPLICATIONS
Airway obstruction poses a significant early risk in patients presenting with advanced diphtheria. Pseudomembranes may slough and obstruct the airway or may advance to the larynx or into the tracheobronchial tree. Children are particularly prone to obstruction because of their small airways.
![]() Polyneuropathy and myocarditis are late toxic manifestations of diphtheria. During a diphtheria outbreak in the Kyrgyz Republic in 1999, myocarditis was found in 22% and neuropathy in 5% of 656 hospitalized patients. The mortality rate was 7% among patients with myocarditis as opposed to 2% among those without myocardial manifestations. The median time to death in hospitalized patients was 4.5 days. Myocarditis is typically associated with dysrhythmia of the conduction tract and dilated cardiomyopathy.
Polyneuropathy and myocarditis are late toxic manifestations of diphtheria. During a diphtheria outbreak in the Kyrgyz Republic in 1999, myocarditis was found in 22% and neuropathy in 5% of 656 hospitalized patients. The mortality rate was 7% among patients with myocarditis as opposed to 2% among those without myocardial manifestations. The median time to death in hospitalized patients was 4.5 days. Myocarditis is typically associated with dysrhythmia of the conduction tract and dilated cardiomyopathy.
Polyneuropathy is seen 3–5 weeks after the onset of diphtheria and has a slow indolent course. However, patients may develop severe and prolonged neurologic abnormalities. The disorders typically occur in the mouth and neck, with lingual or facial numbness as well as dysphonia, dysphagia, and cranial nerve paresthesias. More ominous signs include weakness of respiratory and abdominal muscles and paresis of the extremities. Sensory manifestations and sensory ataxia also are observed. Cranial nerve dysfunction typically precedes disturbances of the trunk and extremities because of proximity to the site of infection. Autonomic dysfunction also is associated with polyneuropathy and can lead to hypotension. Polyneuropathy is typically reversible in patients who survive the acute phase.
Other complications of diphtheria include pneumonia, renal failure, encephalitis, cerebral infarction, pulmonary embolism, and serum sickness from antitoxin therapy.
DIAGNOSIS
The diagnosis of diphtheria is based on clinical signs and symptoms plus laboratory confirmation. Respiratory diphtheria should be considered in patients with sore throat, pharyngeal exudates, and fever. Other symptoms may include hoarseness, stridor, or palatal paralysis. The presence of a pseudomembrane should prompt strong consideration of diphtheria. Once a clinical diagnosis of diphtheria is made, diphtheria antitoxin should be obtained and administered as rapidly as possible.
Laboratory diagnosis of diphtheria is based either on cultivation of C. diphtheriae or toxigenic Corynebacterium ulcerans from the site of infection or on the demonstration of local lesions with characteristic histopathology. Corynebacterium pseudodiphtheriticum, a nontoxigenic organism, is a common component of the normal throat flora and does not pose a significant risk. Throat samples should be submitted to the laboratory for culture with the notation that diphtheria is being considered. This information should prompt cultivation on special selective medium and subsequent biochemical testing to differentiate C. diphtheriae from other nasopharyngeal commensal corynebacteria. All laboratory isolates of C. diphtheriae, including nontoxigenic strains, should be submitted to the CDC.
A diagnosis of cutaneous diphtheria requires laboratory confirmation since the lesions are not characteristic and are indistinguishable from other dermatoses. Diphtheritic ulcers occasionally—but not consistently—have a punched-out appearance (Fig. 175-2). Patients in whom cutaneous diphtheria is identified should have the nasopharynx cultured for C. diphtheriae. The laboratory medium for cutaneous diphtheria specimens is the same as that used for respiratory diphtheria: Löffler’s or Tinsdale’s selective medium in addition to nonselective medium such as blood agar. As has been mentioned, respiratory diphtheria remains a notifiable disease in the United States, whereas cutaneous diphtheria is not.
|
TREATMENT |
DIPHTHERIA |
DIPHTHERIA ANTITOXIN
Prompt administration of diphtheria antitoxin is critical in the management of respiratory diphtheria. Diphtheria antitoxin, a horse antiserum, is effective in reducing the extent of local disease as well as the risk of complications of myocarditis and neuropathy. Rapid institution of antitoxin therapy is also associated with a significant reduction in mortality risk. Because diphtheria antitoxin cannot neutralize cell-bound toxin, prompt initiation is important. This product, which is no longer commercially available in the United States, can be obtained from the CDC by calling the Bacterial Vaccine Preventable Disease Branch of the National Immunization Program at 404-639-8257 (8:00 A.M. to 4:30 P.M., U.S. Eastern time) or, at other hours, the Emergency Operations Center at 770-488-7100; the relevant website is www.cdc.gov/diphtheria/dat.html. The current protocol for the use of diphtheria antitoxin involves a test dose to rule out immediate hypersensitivity. Patients who demonstrate hypersensitivity require desensitization before a full therapeutic dose of antitoxin is administered.
ANTIMICROBIAL THERAPY
Antibiotics are used in the management of diphtheria primarily to prevent transmission to susceptible contacts. Antibiotics also prevent further toxin production and reduce the severity of local infection. Recommended treatment options for patients with respiratory diphtheria are as follows:
• Procaine penicillin G, 600,000 U IM q12h (for children: 12,500–25,000 U/kg IM q12h) until the patient can swallow comfortably; then oral penicillin V, 125–250 mg qid to complete a 14-day course
• Erythromycin, 500 mg IV q6h (for children: 40–50 mg/kg per day IV in two or four divided doses) until the patient can swallow comfortably; then 500 mg PO qid to complete a 14-day course
![]() A clinical study in Vietnam found that penicillin was associated with a more rapid resolution of fever and a lower rate of bacterial resistance than erythromycin; however, relapses were more common in the penicillin group. Erythromycin therapy targets protein synthesis and thus offers the presumed benefit of stopping toxin synthesis more quickly than a cell wall–active β-lactam agent. Alternative therapeutic agents for patients who are allergic to penicillin or cannot take erythromycin include rifampin and clindamycin. Eradication of C. diphtheriae should be documented after antimicrobial therapy is complete. A repeat throat culture 2 weeks later is recommended. For patients in whom the organism is not eradicated after a 14-day course of erythromycin or penicillin, an additional 10-day course followed by repeat culture is recommended. Drug-resistant strains of C. diphtheriae exist, and several reports have described multidrug-resistant strains, predominantly in Southeast Asia. Drug resistance should be considered when efforts at pathogen eradication fail.
A clinical study in Vietnam found that penicillin was associated with a more rapid resolution of fever and a lower rate of bacterial resistance than erythromycin; however, relapses were more common in the penicillin group. Erythromycin therapy targets protein synthesis and thus offers the presumed benefit of stopping toxin synthesis more quickly than a cell wall–active β-lactam agent. Alternative therapeutic agents for patients who are allergic to penicillin or cannot take erythromycin include rifampin and clindamycin. Eradication of C. diphtheriae should be documented after antimicrobial therapy is complete. A repeat throat culture 2 weeks later is recommended. For patients in whom the organism is not eradicated after a 14-day course of erythromycin or penicillin, an additional 10-day course followed by repeat culture is recommended. Drug-resistant strains of C. diphtheriae exist, and several reports have described multidrug-resistant strains, predominantly in Southeast Asia. Drug resistance should be considered when efforts at pathogen eradication fail.
Cutaneous diphtheria should be treated as described above for respiratory disease. Individuals infected with toxigenic strains should receive antitoxin. It is important to treat the underlying cause of the dermatoses in addition to the superinfection with C. diphtheriae.
Patients who recover from respiratory or cutaneous diphtheria should have antitoxin levels measured. If diphtheria antitoxin has been administered, this test should be performed 6 months later. Patients who recover from respiratory or cutaneous diphtheria should receive the appropriate vaccine to ensure the development of protective antibody titers.
MANAGEMENT STRATEGIES
Patients in whom diphtheria is suspected should be hospitalized in respiratory isolation rooms, with close monitoring of cardiac and respiratory function. A cardiac workup is recommended to assess the possibility of myocarditis. In patients with extensive pseudomembranes, an anesthesiology or an ear, nose, and throat consultation is recommended because of the possible need for tracheostomy or intubation. In some settings, pseudomembranes can be removed surgically. Treatment with glucocorticoids has not been shown to reduce the risk of myocarditis or polyneuropathy.
PROGNOSIS
Fatal pseudomembranous diphtheria typically occurs in patients with nonprotective antibody titers and in unimmunized patients. The pseudomembrane may actually increase in size from the time it is first noted. Risk factors for death include bullneck diphtheria; myocarditis with ventricular tachycardia; atrial fibrillation; complete heart block; an age of >60 years or <6 months; alcoholism; extensive pseudomembrane elongation; and laryngeal, tracheal, or bronchial involvement. Another important predictor of fatal outcome is the interval between the onset of local disease and the administration of antitoxin. Cutaneous diphtheria has a low mortality rate and is rarely associated with myocarditis or peripheral neuropathy.
PREVENTION
Vaccination Sustained campaigns for vaccination of children and adequate boosting vaccination of adults are responsible for the exceedingly low incidence of diphtheria in most developed nations. Currently, diphtheria toxoid vaccine is coadministered with tetanus vaccine (with or without acellular pertussis). DTaP (a full-level diphtheria and tetanus toxoids and acellular pertussis vaccine) is currently recommended for children up to the age of 7; DTaP replaced the earlier whole-cell pertussis vaccine DTP in 1997. Tdap is a tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine formulated for adolescents and adults. Tdap was licensed for use in the United States in 2005 and is the recommended booster vaccine for children 11–12 years old and the recommended catch-up vaccine for children 7–10 and 13–18 years of age. It is recommended that all adults (i.e., persons >19 years old) receive a single dose of Tdap if they have not received it previously, regardless of the interval since the last dose of Td (tetanus and reduced-dose diphtheria toxoids, adsorbed). Tdap vaccination is a priority for health care workers, pregnant women, adults anticipating contact with infants, and adults not previously vaccinated for pertussis. Adults who have received acellular pertussis vaccine should continue to receive decennial Td booster vaccinations. The vaccine schedule is detailed in Chap. 148.
Prophylaxis Administration to Contacts Close contacts of diphtheria patients should undergo throat culture to determine whether they are carriers. After samples for throat culture are obtained, antimicrobial prophylaxis should be considered for all contacts, even those whose cultures are negative. The options are 7–10 days of oral erythromycin or one dose of IM benzathine penicillin G (1.2 million units for persons ≥6 years of age or 600,000 units for children <6 years of age).
Contacts of diphtheria patients whose immunization status is uncertain should receive the appropriate diphtheria toxoid–containing vaccine. The Tdap vaccine (rather than Td) is now the booster vaccine of choice for adults who have not recently received an acellular pertussis–containing vaccine. Carriers of C. diphtheriae in the community should be treated and vaccinated when identified.
OTHER CORYNEBACTERIAL AND RHODOCOCCUS INFECTIONS
Nondiphtherial corynebacteria, referred to as diphtheroids or coryneforms, are frequently considered colonizers or contaminants; however, they have been associated with invasive disease, particularly in immunocompromised patients. These organisms have been isolated from the bloodstream, particularly in association with catheter infection, endocarditis, prosthetic valve infection, meningitis, neurosurgical shunt infection, brain abscess, and peritonitis and often in the setting of chronic ambulatory peritoneal dialysis, osteomyelitis, septic arthritis, urinary tract infection, empyema, and pneumonia, among other infections. Patients infected with these organisms usually have significant medical comorbidity or are immunosuppressed. The nondiphtherial coryneforms are a diverse collection of bacteria that are taxonomically grouped together in the genus Corynebacterium on the basis of their 16S rDNA signature nucleotides. Despite the shared rDNA signatures, these isolates are quite diverse. For example, their guanine-cytosine content ranges from 45% to 70%. Several nondiphtheroid corynebacteria, including Corynebacterium jeikeium and Corynebacterium urealyticum, are associated with resistance to multiple antibiotics. Rhodococcus equi is associated with necrotizing pneumonia and granulomatous infection, particularly in immunocompromised individuals.
MICROBIOLOGY AND LABORATORY DIAGNOSIS
These organisms are non-acid-fast, catalase-positive, aerobic or facultatively anaerobic rods. Their colonial morphologies vary widely; some species are small and α-hemolytic (similar to lactobacilli), whereas others form large white colonies (similar to yeasts). Many nondiphtherial coryneforms require special media, such as Löffler’s, Tinsdale’s, or telluride medium. These cultivation idiosyncrasies have led to a complex taxonomic categorization of the organisms.
EPIDEMIOLOGY
Humans are the natural reservoirs for several nondiphtherial coryneforms, including C. xerosis, C. pseudodiphtheriticum, C. striatum, C. minutissimum, C. jeikeium, C. urealyticum, and Arcanobacterium haemolyticum. Animal reservoirs are responsible for carriage of Arcanobacterium pyogenes, C. ulcerans, and C. pseudotuberculosis. Soil is the natural reservoir for R. equi.
C. pseudodiphtheriticum is a component of the normal flora of the human pharynx and skin. C. xerosis is found on the skin, nasopharynx, and conjunctiva; C. auris in the external auditory canal; and C. striatum in the anterior nares and on the skin. C. jeikeium and C. urealyticum are found in the axilla, groin, and perineum, particularly in hospitalized patients. Infections with C. ulcerans and C. pseudotuberculosis have been associated with the consumption of raw milk from infected cattle.
C. ulcerans This organism causes a diphtheria-like illness and produces both diphtheria toxin and a dermonecrotic toxin. The organism is a commensal in horses and cattle and has been isolated from cow’s milk. C. ulcerans causes exudative pharyngitis, primarily during summer months, in rural areas, and among individuals exposed to cattle. In contrast to diphtheria, this infection is considered a zoonosis whose person-to-person transmission has not been documented. Nevertheless, treatment with antitoxin and antibiotics should be initiated when respiratory C. ulcerans is identified, and a contact investigation (including throat cultures to determine the need for antimicrobial prophylaxis and, in unimmunized contacts, administration of the appropriate diphtheria toxoid–containing vaccine) should be conducted. The organism grows on Löffler’s, Tinsdale’s, and telluride agars as well as blood agar. In addition to exudative pharyngitis, cutaneous disease due to C. ulcerans has been reported. C. ulcerans is susceptible to a wide panel of antibiotics. Erythromycin and macrolides appear to be the first-line agents.
C. pseudotuberculosis (ovis) Infection caused by C. pseudotuberculosis is rare and is reported almost exclusively from Australia. C. pseudotuberculosis causes suppurative granulomatous lymphadenitis and an eosinophilic pneumonia syndrome among individuals who handle horses, cattle, goats, and deer or who drink raw milk. The organism is an important veterinary pathogen, causing suppurative lymphadenitis, abscesses, and pneumonia, but is rarely a human pathogen. Successful treatment with erythromycin or tetracycline has been reported, with surgery also performed when indicated.
C. jeikeium (Group JK) Originally described in American hospitals, C. jeikeium infection was subsequently reported in Europe. After a 1976 survey of diseases caused by nondiphtherial corynebacteria, CDC group JK emerged as an important opportunistic pathogen among neutropenic and HIV-infected patients. The organism has now been designated a separate species. C. jeikeium forms small, gray to white, glistening, nonhemolytic colonies on blood agar. It lacks urease and nitrate reductase and does not ferment most carbohydrates. The predominant syndrome associated with C. jeikeium is sepsis with pneumonia, endocarditis, meningitis, osteomyelitis, and epidural abscess. Risk factors for C. jeikeium infection include hematologic malignancy, neutropenia from comorbid conditions, prolonged hospitalization, exposure to multiple antibiotics, and skin disruption. There is evidence that C. jeikeium is part of the inguinal, axillary, genital, and perirectal flora of hospitalized patients.
Broad-spectrum antimicrobial therapy appears to select for colonization. Gram’s staining shows gram-positive coccobacillary forms slightly resembling streptococci. Moreover, C. jeikeium is resistant to all antibiotics tested except vancomycin. Effective therapy involves removal of the infectious source, whether a catheter, prosthetic joint, or prosthetic valve. Efforts have been made to prevent C. jeikeium infection by improving hygienic conditions for high-risk patients in intensive care settings with antibacterial soap.
C. urealyticum (Group D2) Identified as a urease-positive nondiphtherial Corynebacterium in 1972, C. urealyticum is an opportunistic pathogen causing sepsis and urinary tract infection. C. urealyticum appears to be the etiologic agent of a severe urinary tract syndrome known as alkaline-encrusted cystitis, a chronic inflammatory bladder infection associated with deposition of ammonium magnesium phosphate on the surface and walls of ulcerating lesions in the bladder. In addition, C. urealyticum has been associated with pneumonia, peritonitis, endocarditis, osteomyelitis, and wound infection. It is similar to C. jeikeium in its resistance to most antibiotics except vancomycin. Vancomycin therapy has been used successfully in severe infections.
C. minutissimum (Erythrasma) Erythrasma is a cutaneous infection producing reddish-brown, macular, scaly, pruritic intertriginous patches. The dermatologic presentation under the Wood’s lamp is of coral red fluorescence. C. minutissimum appears to be a common cause of erythrasma, although there is evidence for a polymicrobial etiology in certain settings. This microbe has also been associated with bacteremia in patients with hematologic malignancy. Erythrasma responds to topical erythromycin, clarithromycin, clindamycin, or fusidic acid, although more severe infections may require oral macrolide therapy.
Other Nondiphtherial Corynebacterial Infections C. xerosis is a human commensal found in the conjunctiva, nasopharynx, and skin. This nontoxigenic organism is occasionally identified as a source of invasive infection in immunocompromised or postoperative patients and prosthetic joint recipients. C. striatum is found in the anterior nares, skin, face, and upper torso of healthy individuals. Also nontoxigenic, this organism has been associated with invasive opportunistic infections in severely ill or immunocompromised patients. C. amycolatum is isolated from human skin and is identified on the basis of a unique 16S ribosomal RNA sequence associated with opportunistic infection. C. glucuronolyticum is a nonlipophilic species that causes male genitourinary tract infections such as prostatitis and urethritis. These infections may be successfully treated with a wide variety of antibacterial agents, including β-lactams, rifampin, aminoglycosides, or vancomycin; however, the organism appears to be resistant to fluoroquinolones, macrolides, and tetracyclines. C. imitans has been identified in eastern Europe as a nontoxigenic cause of pharyngitis. C. auris has been identified in children with otitis media; it is susceptible to fluoroquinolones, rifampin, tetracycline, and vancomycin but resistant to penicillin G and variably susceptible to macrolides. C. pseudodiphtheriticum (C. hoffmanii) is a nontoxigenic species that is part of the normal human flora. Human infections—particularly endocarditis of either prosthetic or natural valves and invasive pneumonia—have been reported only rarely. Although C. pseudodiphtheriticum may be isolated from the nasopharynx of patients with suspected diphtheria, it is part of the normal flora and does not produce diphtheria toxin. C. propinquum, a close relative of C. pseudodiphtheriticum, is part of CDC group ANF-3 and is isolated from the human respiratory tract and blood. C. afermentans subspecies lipophilum belongs to CDC group ANF-1 and has been isolated from human blood and abscesses. C. accolens has been isolated from wound drainage, throat swabs, and sputum and is typically identified as a satellite of staphylococcal organisms; this species has been associated with endocarditis. C. bovis is a veterinary commensal that has not been clearly associated with human disease. C. aquaticum is a water-dwelling organism that is occasionally isolated from patients using medical devices (e.g., for chronic ambulatory peritoneal dialysis or venous access).
Rhodococcus Rhodococcus species are phylogenetically related to the corynebacteria. These gram-positive coccobacilli have been associated with tuberculosis-like infections in humans with granulomatous pathology. While R. equi is best known, other species have been identified, including R. (Gordonia) bronchialis, R. (Tsukamurella) aurantiacus, R. luteus, R. erythropolis, R. rhodochrous, and R. rubropertinctus.
R. equi has been recognized as a cause of pneumonia in horses since the 1920s and as a cause of related infections in cattle, sheep, and swine. It is found in soil as an environmental microbe. The organisms vary in length; appear as spherical to long, curved, clubbed rods; and produce large irregular mucoid colonies. R. equi cannot ferment carbohydrates or liquefy gelatin and is often acid fast. An intracellular pathogen of macrophages, R. equi can cause granulomatous necrosis and caseation. This organism has most commonly been identified in pulmonary infection, but infections of brain, bone, and skin also have been reported. Most commonly, R. equi disease manifests as nodular cavitary pneumonia of the upper lobe—a picture similar to that seen in tuberculosis or nocardiosis. Most patients are immunocompromised, often by HIV infection. Subcutaneous nodular lesions have also been identified. The involvement of R. equi should be considered when any patient presents with a tuberculosis-like syndrome.
Infection due to R. equi has been treated successfully with antibiotics that penetrate intracellularly, including macrolides, clindamycin, rifampin, and trimethoprim-sulfamethoxazole. β-Lactam antibiotics have not been useful. The organism is routinely susceptible to vancomycin, which is considered the drug of choice.
Other Related Species • ACTINOMYCES PYOGENES This organism, a well-known pathogen of cattle, sheep, goats, and pigs, causes seasonal leg ulcers in rural Thailand. A few human cases of sepsis, endocarditis, septic arthritis, pneumonia, meningitis, and empyema have been reported. This species is susceptible to β-lactams, tetracyclines, aminoglycosides, and fluoroquinolones.
ARCANOBACTERIUM HAEMOLYTICUM A. haemolyticum was identified as an agent of wound infections in U.S. soldiers in the South Pacific during World War II. It appears to be a human commensal of the nasopharynx and skin, but has also been implicated in pharyngitis and chronic skin ulcers. In contrast to the much more common pharyngitis caused by Streptococcus pyogenes, A. haemolyticum pharyngitis is associated with a scarlatiniform rash on the trunk and proximal extremities in about half of cases; this illness is occasionally confused with toxic shock syndrome. Because A. haemolyticum pharyngitis primarily affects teenagers, it has been postulated that the rash-pharyngitis syndrome may represent copathogenicity, synergy, or opportunistic secondary infection with Epstein-Barr virus. A. haemolyticum has also been reported as a cause of bacteremia, soft tissue infections, osteomyelitis, and cavitary pneumonia, predominantly in the setting of underlying diabetes mellitus. The organism is susceptible to β-lactams, macrolides, fluoroquinolones, clindamycin, vancomycin, and doxycycline. Penicillin resistance has been reported.
176 |
Listeria monocytogenes Infections |
Listeria monocytogenes is a food-borne pathogen that can cause serious infections, particularly in pregnant women and immunocompromised individuals. A ubiquitous saprophytic environmental bacterium, L. monocytogenes is also a facultative intracellular pathogen with a broad host range. Humans are probably accidental hosts for this microorganism. L. monocytogenes is of interest not only to clinicians but also to basic scientists as a model intracellular pathogen that is used to study basic mechanisms of microbial pathogenesis and host immunity.
MICROBIOLOGY
L. monocytogenes is a facultatively anaerobic, nonsporulating, gram-positive rod that grows over a broad temperature range, including refrigeration temperatures. This organism is motile during growth at low temperatures but much less so at 37°C. The vast majority of cases of human listerial disease can be traced to serotypes 1/2a, 1/2b, and 4. L. monocytogenes is weakly β-hemolytic on blood agar, and (as detailed below) its β-hemolysin is an essential determinant of its pathogenicity.
PATHOGENESIS
Infections with L. monocytogenes follow ingestion of contaminated food that contains the bacteria at high concentrations. The conversion from environmental saprophyte to pathogen involves the coordinate regulation of bacterial determinants of pathogenesis that mediate entry into cells, intracellular growth, and cell-to-cell spread. Many of the organism’s pathogenic strategies can be examined experimentally in tissue culture models of infection (Fig. 176-1). Like other enteric pathogens, L. monocytogenes induces its own internalization by cells that are not normally phagocytic. Its entry into cells is mediated by host surface proteins classified as internalins. Internalin-mediated entry is important in the crossing of intestinal, blood-brain, and fetoplacental barriers, although how L. monocytogenes traffics from the intestine to the brain or fetus is only beginning to be investigated. In a pregnant guinea pig model of infection, L. monocytogenes was shown to traffic from maternal organs to the placenta; surprisingly, however, it also trafficked from the placenta back to maternal organs. These data are consistent with a model in which miscarriage can be viewed as a host defense strategy to eliminate a nidus of infection.
FIGURE 176-1 Stages in the intracellular life cycle of Listeria monocytogenes. The central diagram depicts cell entry, escape from a vacuole, actin nucleation, actin-based motility, and cell-to-cell spread. Surrounding the diagram are representative electron micrographs from which it was derived. ActA, surface protein mediating nucleation of host actin filaments to propel bacteria intra- and intercellularly; LLO, listeriolysin O; PLCs, phospholipases C; Inl, internalin. See text for further details. (Adapted with permission from LG Tilney, DA Portnoy: J Cell Biol 109:1597, 1989. © Rockefeller University Press.)
An essential determinant of the pathogenesis of L. monocytogenes is its β-hemolysin, listeriolysin O (LLO). LLO is a pore-forming, cholesterol-dependent cytolysin. (Related cytolysins include streptolysin O, pneumolysin, and perfringolysin O, all of which are produced by extracellular pathogens.) LLO is largely responsible for mediating the rupture of the phagosomal membrane that forms after phagocytosis of L. monocytogenes. LLO probably acts by insertion into an acidifying phagosome, which prevents the vesicle’s maturation. In addition, LLO acts as a translocation pore for one or both of the L. monocytogenes phospholipases that also contribute to vacuolar lysis. LLO synthesis and activity are controlled at multiple levels to ensure that its lytic activity is limited to acidic vacuoles and does not affect the cytosol. Mutations in LLO that influence its synthesis, cytosolic half-life, or pH optimum cause premature toxicity to infected cells. There is an inverse relationship between toxicity and virulence—i.e., the more cytotoxic the strain, the less virulent it is in animals. This relationship may seem paradoxical, but, as an intracellular pathogen, L. monocytogenes benefits from leaving its host cell unharmed.
Shortly after exposure to the mammalian-cell cytosol, L. monocytogenes expresses a surface protein, ActA, that mediates the nucleation of host actin filaments to propel the bacteria intra- and intercellularly. ActA mimics host proteins of the Wiskott-Aldrich syndrome protein (WASP) family by promoting the actin nucleation properties of the Arp2/3 complex. Thus, L. monocytogenes can enter the cytosol of almost any eukaryotic cell or cell extract and can exploit a conserved and essential actin-based motility system. Other pathogens as diverse as certain Shigella, Mycobacterium, Rickettsia, and Burkholderia species use a related pathogenic strategy that allows cell-to-cell spread without exposure to the extracellular milieu.
IMMUNE RESPONSE
The innate and acquired immune responses to L. monocytogenes have been studied extensively in mice. Shortly after IV injection, most bacteria are found in Kupffer cells in the liver, with some organisms in splenic dendritic cells and macrophages. Listeriae that survive the bactericidal activity of initially infected macrophages grow in the cytosol and spread from cell to cell. L. monocytogenes triggers three innate immune pathways: a MyD88-dependent pathway leading to inflammatory cytokines, a STING/IRF3 pathway leading to a type I interferon response; and low-level inflammasome activation. Neutrophils are crucial to host defense during the first 24 h of infection, whereas an influx of activated macrophages from the bone marrow is critical subsequently. Mice that survive sublethal infection clear the organisms within a week, with consequent sterile immunity. Studies with knockout mice have been instrumental in dissecting the roles played by chemokines and cytokines during infection. For example, interferon γ, tumor necrosis factor, and CCR2 are essential in controlling infection. While innate immunity is sufficient to control infection, the acquired immune response is required for sterile immunity. Immunity is cell mediated; antibody plays no measurable role. The critical effector cells are cytotoxic (CD8+) T cells that recognize and lyse infected cells; the resulting extracellular bacteria are killed by circulating activated phagocytes.
A hallmark of the L. monocytogenes model is that killed vaccines do not provide protective immunity. The explanation for this fundamental observation is multifactorial, involving the generation of appropriate cytokines and the compartmentalization of bacterial proteins for antigen processing and presentation. Because the organism has the capacity to induce a robust cell-mediated immune response, attenuated strains have been engineered to express foreign antigens and are undergoing clinical studies as therapeutic vaccines for cancer.
EPIDEMIOLOGY
L. monocytogenes usually enters the body via the gastrointestinal tract in foods. Listeriosis is most often sporadic, although outbreaks do occur. No epidemiologic or clinical evidence supports person-to-person transmission (other than vertical transmission from mother to fetus) or waterborne infection. In line with its survival and multiplication at refrigeration temperatures, L. monocytogenes is commonly found in processed and unprocessed foods of animal and plant origin, especially soft cheeses, delicatessen meats, hot dogs, milk, and cold salads; fresh fruits and vegetables can also transmit the organism. Because food supplies are increasingly centralized and normal hosts tolerate the organism well, outbreaks may not be immediately apparent. The U.S. Food and Drug Administration has a zero-tolerance policy for L. monocytogenes in ready-to-eat foods.
DIAGNOSIS
Symptoms of listerial infection overlap greatly with those of other infectious diseases. Timely diagnosis requires that the illness be considered in groups at risk: pregnant women; elderly persons; neonates; individuals immunocompromised by organ transplantation, cancer, or treatment with tumor necrosis factor antagonists or glucocorticoids; and patients with a variety of chronic medical conditions, including alcoholism, diabetes, renal disease, and rheumatologic and hepatic illnesses. Meningitis in older adults (especially with parenchymal brain involvement or subcortical brain abscess) should trigger consideration of L. monocytogenes infection and treatment. Listeriosis occasionally affects healthy, young, nonpregnant individuals. HIV-infected patients are at risk; however, listeriosis seems to be prevented by trimethoprim-sulfamethoxazole (TMP-SMX) prophylaxis targeting other AIDS-related infections. The diagnosis is typically made by culture of blood, cerebrospinal fluid (CSF), or amniotic fluid. L. monocytogenes may be confused with “diphtheroids” or pneumococci in Gram-stained CSF or may be gram-variable and confused with Haemophilus species. Polymerase chain reaction diagnostics have been described but are not widely available, and serology is not clinically useful.
CLINICAL MANIFESTATIONS
Listerial infections present as several clinical syndromes, of which meningitis and septicemia are most common. Monocytosis is seen in infected rabbits but is not a hallmark of human infection.
Gastroenteritis Appreciated only since the outbreaks of the late 1980s, listerial gastroenteritis typically develops within 48 h of ingestion of a large inoculum of bacteria in contaminated foods. Attack rates are high (50–100%). L. monocytogenes is neither sought nor found in routine fecal cultures, but its involvement should be considered in outbreaks when cultures for other likely pathogens are negative. Sporadic intestinal illness appears to be uncommon. Manifestations include fever, diarrhea, headache, and constitutional symptoms. The largest reported outbreak occurred in an Italian school system and included 1566 individuals; ~20% of patients were hospitalized, but only one person had a positive blood culture. Isolated gastrointestinal illness does not require antibiotic treatment. Surveillance studies show that 0.1–5% of healthy asymptomatic adults may have stool cultures positive for the organism.
Bacteremia L. monocytogenes septicemia presents with fever, chills, and myalgias/arthralgias and cannot be differentiated from septicemia involving other organisms. Meningeal symptoms, focal neurologic findings, or mental status changes may suggest the diagnosis. Bacteremia is documented in 70–90% of cancer patients with listeriosis. A nonspecific flulike illness with fever is a common presentation in pregnant women. Endocarditis of prosthetic and native valves is an uncommon complication, with reported fatality rates of 35–50% in case series. A lumbar puncture is often prudent, although not necessary, in pregnant women without central nervous system (CNS) symptoms.
Meningitis L. monocytogenes causes ~5–10% of all cases of community-acquired bacterial meningitis in adults in the United States. Case-fatality rates are reported to be 15–26% and do not appear to have changed over time. This diagnosis should be considered in all older or chronically ill adults with “aseptic” meningitis. The presentation is more frequently subacute (with illness developing over several days) than in meningitis of other bacterial etiologies, and nuchal rigidity and meningeal signs are less common. Photophobia is infrequent. Focal findings and seizures are common in some but not all series. The CSF profile in listerial meningitis most often shows white blood cell counts in the range of 100–5000/μL (rarely higher); 75% of patients have counts below 1000/μL, usually with a neutrophil predominance more modest than that in other bacterial meningitides. Low glucose levels and positive results on Gram’s staining are found ~30–40% of the time. Hydrocephalus can occur.
Meningoencephalitis and Focal CNS Infection L. monocytogenes can directly invade the brain parenchyma, producing either cerebritis or focal abscess. Approximately 10% of cases of CNS infection are macroscopic abscesses resulting from bacteremic seeding; the affected patients often have positive blood cultures. Concurrent meningitis can exist, but the CSF may appear normal. Abscesses can be misdiagnosed as metastatic or primary tumors and, in rare instances, occur in the cerebellum and the spinal cord. Invasion of the brainstem results in a characteristic severe rhombencephalitis, usually in otherwise healthy older adults (although there are numerous other infectious and noninfectious causes of this syndrome). The presentation may be biphasic, with a prodrome of fever and headache followed by asymmetric cranial nerve deficits, cerebellar signs, and hemiparetic and hemisensory deficits. Respiratory failure can occur. The subacute course and the often minimally abnormal CSF findings may delay the diagnosis, which may be suggested by MRI showing ring-enhancing lesions after gadolinium contrast and hyperintense lesions on diffusion-weighted imaging. MRI is superior to CT for the diagnosis of these infections.
Infection in Pregnant Women and Neonates Listeriosis in pregnancy is a severe and important infection. The usual presentation is a nonspecific acute or subacute febrile illness with myalgias, arthralgias, backache, and headache. Pregnant women with listeriosis are usually bacteremic. This syndrome should prompt blood cultures, especially if there is no other reasonable explanation. Involvement of the CNS is rare in the absence of other risk factors. Preterm delivery is a common complication, and the diagnosis may be made only post-partum. As many as 70–90% of fetuses from infected women can become infected. Prepartum treatment of bacteremic women enhances the chances of delivery of a healthy infant. Women usually do well after delivery: maternal deaths are very rare, even when the diagnosis is made late in pregnancy or post-partum. Overall mortality rates for fetuses infected in utero approach 50% in some series; among live-born neonates treated with antibiotics, mortality rates are much lower (~20%). Granulomatosis infantiseptica is an overwhelming listerial fetal infection with miliary microabscesses and granulomas, most often in the skin, liver, and spleen. Less severe neonatal infection acquired in utero presents at birth. “Late-onset” neonatal illness typically develops ~10–30 days post-partum. Mothers of infants with late-onset disease are not ill.
|
TREATMENT |
INFECTIONS CAUSED BY LISTERIA MONOCYTOGENES |
ANTIBIOTICS
No clinical trials have compared antimicrobial agents for the treatment of L. monocytogenes infections. Data from studies conducted in vitro and in animals as well as observational clinical data indicate that ampicillin is the drug of choice, although penicillin also is highly active. Adults should receive IV ampicillin at high doses (2 g every 6 h). Many experts recommend the addition of gentamicin for synergy (1.0–1.7 mg/kg every 8 h); retrospective uncontrolled trials are not conclusive, but one study suggests that gentamicin may not help. TMP-SMX, given IV, is the best alternative for the penicillin-allergic patient (15–20 mg of TMP/kg per day in divided doses every 6–8 h). The dosages recommended cover CNS infection and bacteremia (see below for duration); dosages must be reduced for patients with renal insufficiency. One small nonrandomized study supports a combination of ampicillin and TMP-SMX. Case reports document success with vancomycin, imipenem, meropenem, linezolid, tetracycline, and macrolides, although there are also reports of clinical failure or disease development with some of these agents. Acquired resistance to antimicrobial agents has been sought but not found in large strain collections. Cephalosporins are not effective and should not be used. Neonates should receive ampicillin and gentamicin at doses based on weight.
DURATION
The duration of therapy depends on the syndrome: 2 weeks for bacteremia, 3 weeks for meningitis, 6–8 weeks for brain abscess/encephalitis, and 4–6 weeks for endocarditis in both neonates and adults. Early-onset neonatal disease may be more severe and should be treated for >2 weeks.
COMPLICATIONS AND PROGNOSIS
Many individuals who are promptly diagnosed and treated recover fully, but permanent neurologic sequelae are common in patients with brain abscess or rhombencephalitis. Focal infections of visceral organs; the eye; the pleural, peritoneal, and pericardial spaces; the bones; and both native and prosthetic joints have all been reported. Of 100 live-born, treated neonates in one series, 60% recovered fully, 24% died, and 13% had long-term neurologic or other complications.
PREVENTION
Healthy persons should take standard precautions to prevent food-borne illness: fully cooking meats, washing fresh vegetables, carefully cleaning utensils, and avoiding unpasteurized dairy products. In addition, persons at risk for listeriosis, including pregnant women, should avoid soft cheeses (hard cheeses and yogurt are not problematic) and should avoid or thoroughly reheat ready-to-eat and delicatessen foods.
177 |
Tetanus |
![]() Tetanus is an acute disease manifested by skeletal muscle spasm and autonomic nervous system disturbance. It is caused by a powerful neurotoxin produced by the bacterium Clostridium tetani and is completely preventable by vaccination. C. tetani is found throughout the world, and tetanus commonly occurs where the vaccination coverage rate is low. In developed countries, the disease is seen occasionally in individuals who are incompletely vaccinated. In any setting, established tetanus is a severe disease with a high mortality rate.
Tetanus is an acute disease manifested by skeletal muscle spasm and autonomic nervous system disturbance. It is caused by a powerful neurotoxin produced by the bacterium Clostridium tetani and is completely preventable by vaccination. C. tetani is found throughout the world, and tetanus commonly occurs where the vaccination coverage rate is low. In developed countries, the disease is seen occasionally in individuals who are incompletely vaccinated. In any setting, established tetanus is a severe disease with a high mortality rate.
DEFINITION
Tetanus is diagnosed on clinical grounds (sometimes with supportive laboratory confirmation of the presence of C. tetani; see “Diagnosis,” below), and case definitions are often used to facilitate clinical and epidemiologic assessments. The Centers for Disease Control and Prevention (CDC) defines tetanus as “the acute onset of hypertonia or … painful muscular contractions (usually of the muscles of the jaw and neck) and generalized muscle spasms without other apparent medical cause.” Neonatal tetanus is defined by the World Health Organization (WHO) as “an illness occurring in a child who has the normal ability to suck and cry in the first 2 days of life but who loses this ability between days 3 and 28 of life and becomes rigid and has spasms.” Given the unique presentation of neonatal tetanus, the history generally permits accurate classification of the illness with a high degree of probability. Maternal tetanus is defined by the WHO as tetanus occurring during pregnancy or within 6 weeks after the conclusion of pregnancy (whether with birth, miscarriage, or abortion).
ETIOLOGY
C. tetani is an anaerobic, gram-positive, spore-forming rod whose spores are highly resilient and can survive readily in the environment throughout the world. Spores resist boiling and many disinfectants. In addition, C. tetani spores and bacilli survive in the intestinal systems of many animals, and fecal carriage is common. The spores or bacteria enter the body through abrasions, wounds, or (in the case of neonates) the umbilical stump. Once in a suitable anaerobic environment, the organisms grow, multiply, and release tetanus toxin, an exotoxin that enters the nervous system and causes disease. Very low concentrations of this highly potent toxin can result in tetanus (minimum lethal human dose, 2.5 ng/kg).
In 20–30% of cases of tetanus, no puncture entry wound is found. Superficial abrasions to the limbs are the commonest infection sites in adults. Deeper infections (e.g., attributable to open fracture, abortion, or drug injection) are associated with more severe disease and worse outcomes. In neonates, infection of the umbilical stump can result from inadequate umbilical cord care; in some cultures, for example, the cord is cut with grass or animal dung is applied to the stump. Circumcision or ear-piercing also can result in neonatal tetanus.
EPIDEMIOLOGY
Tetanus is a rare disease in the developed world. Two cases of neonatal tetanus have occurred in the United States since 1989. Between 2001 and 2008, a total of 231 cases of tetanus were reported to the U.S. national surveillance system. Most cases occur in incompletely vaccinated or unvaccinated individuals. Vaccination status is known in 50% of cases reported in the United States between 1972 and 2009; among these cases, only 16% of patients had had three or more doses of tetanus toxoid.
Persons >60 years of age are at greater risk of tetanus because antibody levels decrease over time. One-third of recent cases in the United States were in persons >65 years old. Injection drug users—particularly those injecting heroin subcutaneously (“skin-popping”)—are increasingly recognized as a high-risk group (15% of all cases in 2001–2008). In 2004, an outbreak of tetanus occurred in the United Kingdom, which had previously reported low rates among drug users. The reasons for this outbreak remain unclear but are thought to involve a combination of heroin contamination, skin-popping, and incomplete vaccination. Since then, only seven sporadic cases have been reported in the United Kingdom.
PATHOGENESIS
Genome sequencing of C. tetani has allowed identification of several exotoxins and virulence factors. Only those bacteria producing tetanus toxin (tetanospasmin) can cause tetanus. Although closely related to the botulinum toxins in structure and mode of action, tetanus toxin undergoes retrograde transport into the central nervous system and thus produces clinical effects different from those caused by the botulinum toxins, which remain at the neuromuscular junction.
Toxin is transported by intra-axonal transport to motor nuclei of the cranial nerves or ventral horns of the spinal cord. Tetanus toxin is produced as a single 150-kDa protein that is cleaved to produce heavy (100-kDa) and light (50-kDa) chains linked by a disulfide bond and noncovalent forces. The carboxy terminal of the heavy chain binds to specific membrane components in presynaptic α-motor nerve terminals; evidence suggests binding to both polysialogangliosides and membrane proteins. This binding results in toxin internalization and uptake into the nerves. Once inside the neuron, the toxin enters a retrograde transport pathway, whereby it is transported proximally to the motor neuron body in what appears to be a highly specific process. Unlike other components of the endosomal contents, which undergo acidification following internalization, tetanus toxin is transported in a carefully regulated pH-neutral environment that prevents an acid-induced conformational change that would result in light-chain expulsion into the surrounding cytosol.
The next stage in toxin trafficking is less clearly understood but involves tetanus toxin’s escaping normal lysosomal degradation processes and undergoing translocation across the synapse to the GABA-ergic presynaptic inhibitory interneuron terminals. Here the light chain, which is a zinc-dependent endopeptidase, cleaves vesicle-associated membrane protein 2 (VAMP2, also known as synaptobrevin). This molecule is necessary for presynaptic binding and release of neurotransmitter; thus tetanus toxin prevents transmitter release and effectively blocks inhibitory interneuron discharge. The result is unregulated activity in the motor nervous system. Similar activity in the autonomic system accounts for the characteristic features of skeletal muscle spasm and autonomic system disturbance. The increased circulating catecholamine levels in severe tetanus are associated with cardiovascular complications.
Relatively little is known about the processes of recovery from tetanus. Recovery can take several weeks. Peripheral nerve sprouting is involved in recovery from botulism, and similar central nervous system sprouting may occur in tetanus. Other evidence suggests toxin degradation as a mechanism of recovery.
CLINICAL MANIFESTATIONS
Tetanus produces a wide spectrum of clinical features that are broadly divided into generalized (including neonatal) and local. In the usually mild form of local tetanus, only isolated areas of the body are affected and only small areas of local muscle spasm may be apparent. If the cranial nerves are involved in localized cephalic tetanus, the pharyngeal or laryngeal muscles may spasm, with consequent aspiration or airway obstruction, and the prognosis may be poor. In the typical progression of generalized tetanus (Fig. 177-1), muscles of the face and jaw often are affected first, presumably because of the shorter distances toxin must travel up motor nerves to reach presynaptic terminals. Neonates typically present with inability to suck.
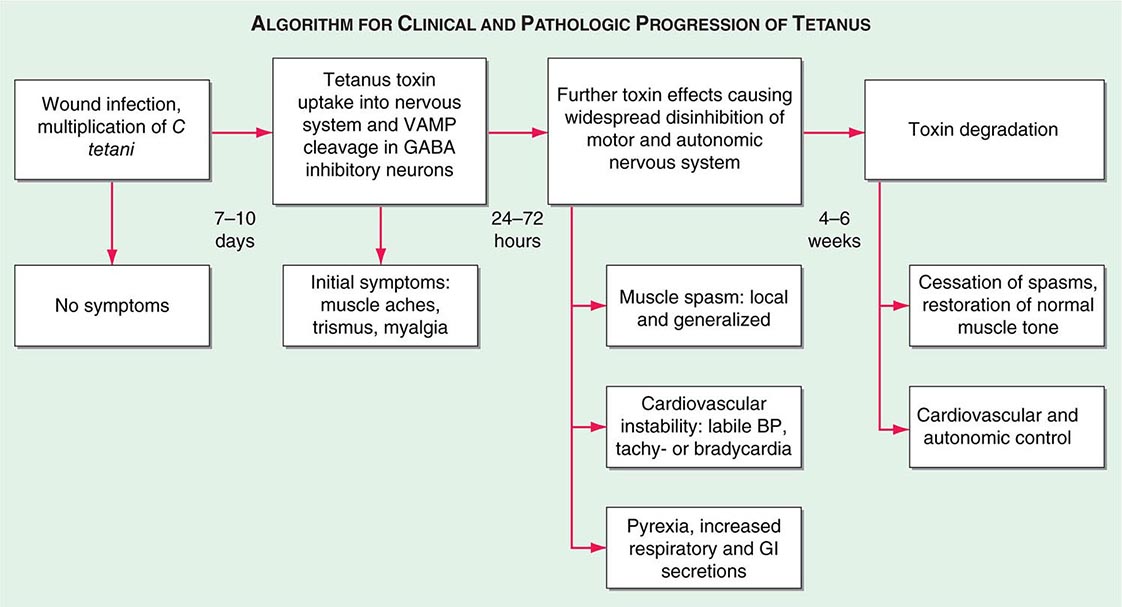
FIGURE 177-1 Clinical and pathologic progression of tetanus. BP, blood pressure; GABA, γ-aminobutyric acid; GI, gastrointestinal; VAMP, vesicle-associated membrane protein (synaptobrevin).
In assessing prognosis, the speed at which tetanus develops is important. The incubation period (time from wound to first symptom) and the period of onset (time from first symptom to first generalized spasm) are of particular significance; shorter times are associated with worse outcome. In neonatal tetanus, the younger the infant is when symptoms occur, the worse the prognosis.
The commonest initial symptoms are trismus (lockjaw), muscle pain and stiffness, back pain, and difficulty swallowing. In neonates, difficulty in feeding is the usual presentation. As the disease progresses, muscle spasm develops. Generalized muscle spasm can be very painful. Commonly, the laryngeal muscles are involved early or even in isolation. This is a life-threatening event as complete airway obstruction may ensue. Spasm of the respiratory muscles results in respiratory failure. Without ventilatory support, respiratory failure is the commonest cause of death in tetanus. Spasms strong enough to produce tendon avulsions and crush fractures have been reported, but this outcome is rare.
Autonomic disturbance is maximal during the second week of severe tetanus, and death due to cardiovascular events becomes the major risk. Blood pressure is usually labile, with rapid fluctuations from high to low accompanied by tachycardia. Episodes of bradycardia and heart block can also occur. Autonomic involvement is evidenced by gastrointestinal stasis, sweating, increased tracheal secretions, and acute (often high-output) renal failure.
DIAGNOSIS
The diagnosis of tetanus is based on clinical findings. As stated above, treatment should not be delayed while laboratory tests are conducted. Culture of C. tetani from a wound provides supportive evidence. Serum anti-tetanus immunoglobulin G may also be measured in a sample taken before the administration of antitoxin or immunoglobulin. Serum levels >0.1 IU/mL are deemed protective and do not support the diagnosis of tetanus. If levels are below this threshold, a bioassay for serum tetanus toxin may be helpful, but a negative result does not exclude the diagnosis. Polymerase chain reaction also has been used for detection of tetanus toxin, but its sensitivity is unknown.
The few conditions that mimic generalized tetanus include strychnine poisoning and dystonic reactions to antidopaminergic drugs. Abdominal muscle rigidity is characteristically continuous in tetanus but is episodic in the latter two conditions. Cephalic tetanus can be confused with other causes of trismus, such as oropharyngeal infection. Hypocalcemia and meningoencephalitis are included in the differential diagnosis of neonatal tetanus.
|
TREATMENT |
TETANUS |
If possible, the entry wound should be identified, cleaned, and debrided of necrotic material in order to remove anaerobic foci of infection and prevent further toxin production. Metronidazole (400 mg rectally or 500 mg IV every 6 h for 7 days) is the preferred antibiotic. An alternative is penicillin (100,000–200,000 IU/kg per day), although this drug theoretically may exacerbate spasms. Failure to remove pockets of ongoing infection may result in recurrent or prolonged tetanus.
Antitoxin should be given early in an attempt to deactivate any circulating tetanus toxin and prevent its uptake into the nervous system. Two preparations are available: human tetanus immune globulin (TIG) and equine antitoxin. TIG is the preparation of choice, as it is less likely to be associated with anaphylactoid reactions. Recommended therapy is 3000–5000 IU of TIG as a single IM dose, a portion of which should be injected around the wound. Equine-derived antitoxin is available widely and is used in low-income countries at a dosage of 10,000–20,000 U administered IM as a single dose or as divided doses after testing for hypersensitivity. Some evidence indicates that intrathecal administration of TIG inhibits disease progression and leads to a better outcome. The results of relevant studies have been supported by a meta-analysis of trials involving both adults and neonates, with TIG doses of 50–1500 IU administered intrathecally.
Spasms are controlled by heavy sedation with benzodiazepines. Chlorpromazine and phenobarbital are commonly used worldwide, and IV magnesium sulfate has been used as a muscle relaxant. A significant problem with all these treatments is that the doses necessary to control spasms also cause respiratory depression; thus, in resource-limited settings without mechanical ventilators, controlling spasms while maintaining adequate ventilation is problematic, and respiratory failure is a common cause of death. In locations with ventilation equipment, severe spasms are best controlled with a combination of sedatives or magnesium and relatively short-acting, cardiovascularly inert, nondepolarizing neuromuscular blocking agents that allow titration against spasm intensity. Infusions of propofol have also been used successfully to control spasms and provide sedation.
It is important to establish a secure airway early in severe tetanus. Ideally, patients should be nursed in calm, quiet environments because light and noise can trigger spasms. Tracheal secretions are increased in tetanus, and dysphagia due to pharyngeal involvement combined with hyperactivity of laryngeal muscles makes endotracheal intubation difficult. Patients may need ventilator support for several weeks. Thus tracheostomy is the usual method of securing the airway in severe tetanus.
Cardiovascular instability in severe tetanus is notoriously difficult to treat. Rapid fluctuations in blood pressure and heart rate can occur. Cardiovascular stability is improved by increasing sedation with IV magnesium sulfate (plasma concentration, 2–4 mmol/L or titrated against disappearance of the patella reflex), morphine, or other sedatives. In addition, drugs acting specifically on the cardiovascular system (e.g., esmolol, calcium antagonists, and inotropes) may be required. Short-acting drugs that allow rapid titration are preferred; particular care should be taken when longer-acting β antagonists are administered, as their use has been associated with hypotensive cardiac arrest.
Complications arising from treatment are common and include thrombophlebitis associated with diazepam injection, ventilator-associated pneumonia, central-line infections, and septicemia. In some centers, prophylaxis against deep-vein thrombosis and thromboembolism is routine.
Recovery from tetanus may take 4–6 weeks. Patients must be given a full primary course of immunization, as tetanus toxin is poorly immunogenic and the immune response following natural infection is inadequate.
PROGNOSIS
Rapid development of tetanus is associated with more severe disease and poorer outcome; it is important to note time of onset and length of incubation period. More sophisticated modeling has revealed other important predictors of prognosis (Table 177-1). Few studies have formally addressed long-term outcomes of tetanus. However, it is generally accepted that recovery is typically complete unless periods of hypoventilation have been prolonged or other complications have ensued. Studies of children and neonates have suggested a higher incidence of neurologic sequelae. Neonates may be at increased risk of learning disabilities, behavioral problems, cerebral palsy, and deafness.
|
FACTORS ASSOCIATED WITH A POOR PROGNOSIS IN TETANUS |

PREVENTION
Tetanus is prevented by good wound care and immunization (Chap. 148). In neonates, use of safe, clean delivery and cord-care practices as well as maternal vaccination are essential. The WHO guidelines for tetanus vaccination consist of a primary course of three doses in infancy, boosters at 4–7 and 12–15 years of age, and one booster in adulthood. In the United States, the CDC suggests an additional dose at 14–16 months and boosters every 10 years. “Catch-up” schedules recommend a three-dose primary course for unimmunized adolescents followed by two further doses. For persons who have received a complete primary course in childhood but no further boosters, two doses at least 4 weeks apart are recommended.
Standard WHO recommendations for prevention of maternal and neonatal tetanus call for administration of two doses of tetanus toxoid at least 4 weeks apart to previously unimmunized pregnant women. However, in high-risk areas, a more intensive approach has been successful, with all women of childbearing age receiving a primary course along with education on safe delivery and postnatal practices.
Individuals sustaining tetanus-prone wounds should be immunized if their vaccination status is incomplete or unknown or if their last booster was given >10 years earlier. Patients sustaining wounds not classified as clean or minor should also undergo passive immunization with TIG. It is recommended that tetanus toxoid be given in conjunction with diphtheria toxoid in a preparation with or without acellular pertussis: DTaP for children <7 years old, Td for 7- to 9-year olds, and Tdap for children >9 years old and adults.
![]() In the early 1980s, tetanus caused more than 1 million deaths a year, accounting for an estimated 5% of maternal deaths and 14% of all neonatal deaths. In 1989, the World Health Assembly adopted a resolution to eliminate neonatal tetanus by the year 2000; elimination was defined as <1 case/1000 live births in every district in every country. By 1999, elimination was still to be achieved in 57 countries and the deadline was extended until 2005, with the additional target of eliminating maternal tetanus (tetanus occurring during pregnancy or within 6 weeks of its end). Ratification of the Millennium Development Goals, in particular goal 4 (achieving a two-thirds reduction in the mortality rate among children under 5 by 2015), has further focused attention on reducing deaths from vaccine-preventable disease, particularly in the first 4 weeks of life.
In the early 1980s, tetanus caused more than 1 million deaths a year, accounting for an estimated 5% of maternal deaths and 14% of all neonatal deaths. In 1989, the World Health Assembly adopted a resolution to eliminate neonatal tetanus by the year 2000; elimination was defined as <1 case/1000 live births in every district in every country. By 1999, elimination was still to be achieved in 57 countries and the deadline was extended until 2005, with the additional target of eliminating maternal tetanus (tetanus occurring during pregnancy or within 6 weeks of its end). Ratification of the Millennium Development Goals, in particular goal 4 (achieving a two-thirds reduction in the mortality rate among children under 5 by 2015), has further focused attention on reducing deaths from vaccine-preventable disease, particularly in the first 4 weeks of life.
Because vaccination reduces the incidence of neonatal tetanus by an estimated 94%, immunization of pregnant women with two doses of tetanus toxoid at least 4 weeks apart has been the primary method of maternal and neonatal tetanus elimination. In some areas, all women of childbearing age have been targeted as a means of increasing vaccination coverage. In addition, educational programs have focused on improving hygiene during the birth process, an intervention that in itself is estimated to reduce neonatal tetanus deaths by up to 40%.
The latest available data show that 34 countries have yet to eliminate maternal and neonatal tetanus, although incidence has declined significantly. Worldwide, deaths from neonatal tetanus fell by 92% between 1990 and 2008; in the latter year, with 84% of newborns protected from the disease by maternal vaccination, there were an estimated 59,000 neonatal tetanus deaths. Despite this relative success, immunization programs need to be ongoing as there is no tetanus herd immunity effect and C. tetani contamination of soil and feces is widespread.
178 |
Botulism |
Botulism, recognized at least since the eighteenth century, is a neuroparalytic disease caused by botulinum toxin, one of the most toxic substances known. While initially thought to be caused only by the ingestion of botulinum toxin in contaminated food (food-borne botulism), three additional forms caused by in situ toxin production after germination of spores in either a wound or the intestine are now recognized worldwide: wound botulism, infant botulism, and adult intestinal colonization botulism. In addition to occurring in these recognized natural forms of the disease, botulism symptoms have been reported in patients receiving injections of botulinum toxin for cosmetic or therapeutic purposes (iatrogenic botulism). Moreover, botulism was reported after inhalation of botulinum toxin in a laboratory setting. All forms of botulism manifest as a relatively distinct clinical syndrome of symmetric cranial-nerve palsies followed by descending bilateral flaccid paralysis of voluntary muscles, which may progress to respiratory compromise and death. The mainstays of therapy are meticulous intensive care and treatment with antitoxin as soon as botulism is suspected and before other illnesses have been ruled out.
ETIOLOGY AND PATHOGENESIS
Seven serologically distinct confirmed serotypes of botulinum toxin (A through G) have been confirmed. Botulinum toxin is produced by four recognized species of clostridia: Clostridium botulinum, Clostridium argentinense, Clostridium baratii, and Clostridium butyricum. Certain strains may produce more than one serotype. All are anaerobic gram-positive organisms that form subterminal spores; C. botulinum and C. argentinense spores have been recovered from the environment. The spores survive environmental conditions and ordinary cooking procedures. Toxin production, however, requires a rare confluence of product storage conditions: an anaerobic environment, a pH of >4.6, low salt and sugar concentrations, and temperatures of >4°C. Although commonly ingested, spores do not normally germinate and produce toxin in the adult human intestine.
Food-borne botulism is caused by consumption of foods contaminated with botulinum toxin; no confirmed host-specific factors are involved in the disease. Wound botulism is caused by contamination of wounds with C. botulinum spores, subsequent spore germination, and toxin production in the anaerobic milieu of an abscess or a wound. Infant botulism results from absorption of toxin produced in situ by toxigenic clostridia colonizing the intestine of children ≤1 year of age. Colonization is thought to occur because the normal bowel microbiota is not yet fully established; this theory is supported by studies in animals. Adult intestinal colonization botulism, a very rare form that is poorly understood, has a pathology similar to that of infant botulism but occurs in adults; typically, patients have some anatomic or functional bowel abnormality or have recently used antibiotics that may help toxigenic clostridia compete more successfully against the normal bowel microbiota. Despite antitoxin treatment, relapse due to intermittent intraluminal production of toxin may be observed in patients with adult intestinal colonization botulism.
Regardless of how exposure occurs, botulinum neurotoxin enters the vascular system and is transported to peripheral cholinergic nerve terminals, including neuromuscular junctions, postganglionic parasympathetic nerve endings, and peripheral ganglia. Botulinum toxin is a zinc-endopeptidase protein of ~150 kDa, consisting of a 100-kDa heavy chain and a 50-kDa light chain. Steps in neurotoxin activity include (1) heavy-chain binding to nerve terminals, (2) internalization in endocytic vesicles, (3) translocation of the light chain to cytosol, and (4) light-chain serotype-specific cleavage of one of several proteins involved in the release of the neurotransmitter acetylcholine. Inhibition of acetylcholine release by any of the seven toxin serotypes results in characteristic flaccid paralysis. Recovery follows sprouting of new nerve terminals.
All botulinum toxin serotypes have been demonstrated to cause botulism in nonhuman primates. Human cases associated with serotypes A, B, E, and F are reported each year. Serotype A produces the most severe syndrome, with the greatest proportion of patients requiring mechanical ventilation. Serotype B appears to cause milder disease than type A in both food-borne and infant botulism. Serotype E, most often associated with foods of aquatic origin, produces a syndrome of variable severity. The rare cases of illness caused by toxin serotype F, whether in infants or adults, are characterized by rapid progression to quadriplegia and respiratory failure but also by relatively rapid recovery. Recent studies have shown that at least some serotypes can be differentiated into subtypes through neurotoxin gene sequencing; however, the impact of these subtype differences on clinical illness is not yet known.
EPIDEMIOLOGY
![]() Botulism occurs worldwide, but the number of cases reported varies among countries and regions. The variation may be due not only to actual differences in incidence but also to (1) availability of resources to identify botulism, a rare disease; (2) differences in reporting requirements; and (3) limited external access to data collections. There is no universal surveillance system to capture worldwide botulism incidence. However, 30 countries currently participate in voluntary reporting of botulism cases to the European Union through an established surveillance system that includes standardized case definitions similar to those used in the United States and Canada. Other countries (e.g., Argentina, China, Thailand, Japan) maintain independent botulism surveillance.
Botulism occurs worldwide, but the number of cases reported varies among countries and regions. The variation may be due not only to actual differences in incidence but also to (1) availability of resources to identify botulism, a rare disease; (2) differences in reporting requirements; and (3) limited external access to data collections. There is no universal surveillance system to capture worldwide botulism incidence. However, 30 countries currently participate in voluntary reporting of botulism cases to the European Union through an established surveillance system that includes standardized case definitions similar to those used in the United States and Canada. Other countries (e.g., Argentina, China, Thailand, Japan) maintain independent botulism surveillance.
Food-Borne Botulism From 1899 to 2011, 1225 food-borne botulism events (single cases or outbreaks) were reported in the United States; from 1990 to 2000, a median of 23 cases were reported annually. Most such events (~80%) involve vegetables or fish/aquatic animals, usually home-preserved (canned, jarred). Native communities in both the United States (Alaska) and Canada have a high incidence of food-borne botulism due to traditional food-preparation practices; 85% of all cases in Canada occur in Native communities. Outbreaks typically involve two or three cases; however, one restaurant-associated outbreak in 1977 affected 59 persons. Worldwide, the highest incidence rate is reported from Georgia and Armenia in the southern Caucasus region, where illness is also associated with home-canning practices. Outbreaks in Asia are attributable to consumption of home-preserved fish or vegetable products such as bean curd and bamboo shoots. In parts of Europe, including Poland, France, and Germany, illness is often associated with home-preserved meat such as ham or sausage. Since 1950, commercial products have rarely been implicated in botulism in the United States, and botulism from commercial products is most often attributed to consumer error in storage or cooking. However, manufacturer deficiencies do occur. In 2007, botulism developed in eight persons in the United States who consumed a commercially canned hot-dog chili sauce. Significant deficiencies discovered by regulatory authorities involved 91 different products and resulted in the recall of 111 million cans of food.
Wound Botulism This form of disease was first recognized in 1951 as a result of a review of the clinical records on an accidental injury in 1943. Between 1943 and 2011, 491 cases of wound botulism were reported in the United States; 97% of cases reported after 1990 were associated with injection drug use. The typical patient was a 30- to 50-year-old resident of the western United States with a long history of black-tar heroin injection. In the early 2000s, wound botulism associated with drug use emerged in Europe, and at least two case clusters have been reported.
Infant Botulism More than 3900 infant botulism cases have been reported worldwide (84% in the United States alone) since this form of the disease was first recognized in 1976; ~80–100 cases (commonly caused by serotypes A and B) are reported annually in the United States.
Adult Intestinal Colonization Botulism This form of botulism is difficult to confirm because it is poorly understood and because no clear guidelines are available to help differentiate it from other adult botulism cases. Often these cases are caused by C. baratii type F, but the involvement of both C. botulinum type A and C. butyricum type E has been reported.
Iatrogenic Botulism Paralysis of variable severity has followed injection of licensed botulinum toxin products for treatment of conditions involving hypertonicity of large muscle groups. The U.S. Food and Drug Administration received 658 reports of adverse events related to botulinum toxin use—some very serious—between 1997 and 2006. Although some patients had symptoms consistent with botulism, no cases were laboratory confirmed. Injection of approved doses of licensed products for cosmetic purposes has not been associated with botulism. However, four cases of laboratory-confirmed botulism resulted from illegal injection of research-grade toxin for cosmetic purposes in a U.S. medical facility in 2004.
Inhalational Botulism Inhalational botulism does not occur naturally. One report from Germany has described botulism resulting from possible inhalational exposure to botulinum toxin in a laboratory incident.
Intentional Botulism Botulinum toxin has been “weaponized” by governments and terrorist organizations. An attack might use aerosolization of toxin or contamination of foods or beverages ranging in scope from small-scale tampering to contamination of a widely distributed food item. An unnatural event may be suggested by unusual relationships between patients (e.g., a visit to the same building), atypical exposure vehicles, or atypical toxin serotypes.
CLINICAL MANIFESTATIONS
The distinctive clinical syndrome of botulism consists of symmetric cranial-nerve palsies followed by bilateral descending flaccid paralysis that may progress to respiratory failure and death. The incubation period from ingestion of contaminated food to onset of symptoms in food-borne botulism is usually 8–36 h but can be as long as 10 days and is dose dependent. Incubation periods of 4–17 days have been documented in wound botulism associated with accidental injury. However, estimation is difficult in cases involving injection drug users because most of these patients inject drugs several times daily. Similarly, the incubation period for infant botulism has not been established, but the fact that the illness has affected infants <3 days old suggests that this interval may be very short.
Cranial nerve deficits may include some of the following: diplopia, dysarthria, dysphonia, ptosis, ophthalmoplegia, facial paralysis, and impaired gag reflex. Pupillary reflexes may be depressed, and fixed or dilated pupils are sometimes noted. Autonomic symptoms such as dizziness, dry mouth, and very dry, occasionally sore throat are common. Constipation due to paralytic ileus is nearly universal, and urinary retention is also common. Patients are afebrile and remain alert and oriented. Respiratory failure may occur due to either paralysis of the diaphragm and accessory breathing muscles or pharyngeal collapse secondary to cranial nerve paralysis. Weakness descends, often rapidly, from the head to involve the neck, arms, thorax, and legs; occasionally, weakness is asymmetric. Deep tendon reflexes may be normal or may progressively disappear. Paresthesias, while rare, have been reported and may represent secondary nerve compression from immobility due to paralysis. Absence of cranial nerve palsies or their onset after the appearance of other true neurologic symptoms makes botulism highly unlikely. Nausea, vomiting, and abdominal pain may precede or follow the onset of paralysis in food-borne botulism. Infants with botulism typically present with reduced ability to suck and swallow, constipation, weakened voice, ptosis, sluggish pupils, hypotonia, and floppy neck; as in adults, illness can progress to generalized flaccidity and respiratory compromise.
Even when intubated, patients can respond to questions by moving their fingers or toes unless paralysis has affected the digits. In some instances, unfortunately, the severe ptosis, expressionless face, and weak phonation of patients with botulism have been interpreted as signs of mental status changes due to alcohol intoxication, drug overdose, encephalitis, or meningitis—a conclusion that delays an accurate diagnosis. Because of skeletal muscle paralysis, patients experiencing respiratory distress may appear placid and detached even as they near respiratory arrest. Death in untreated botulism is usually due to airway obstruction from pharyngeal muscle paralysis and inadequate tidal volume resulting from paralysis of diaphragmatic and accessory respiratory muscles. Death can also result from hospital-associated infections and other sequelae of long-term paralysis, hospitalization, and mechanical ventilation.
A history of preparing home-canned foods may assist with the diagnosis. Patients with wound botulism may or may not have a discernible wound or abscess. A history of injection drug use or the presence of track marks may raise suspicion of the diagnosis. Clinical improvement follows sprouting of new nerve terminals and may take weeks to months. Patients often require outpatient rehabilitation therapy and may experience residual deficits.
DIAGNOSIS
Botulism is diagnosed primarily on clinical grounds, with laboratory confirmation by specific tests that identify botulinum toxin in clinical and food samples. In the setting of an outbreak with multiple patients presenting to the same treatment facility, the diagnosis is apparent as long as physicians recognize that cases within a cluster may have varied signs and symptoms. The temporal occurrence of two or more cases with symptoms compatible with botulism is essentially pathognomonic because other illnesses that resemble botulism do not usually occur in clusters. In lone (sporadic) cases, the diagnosis is often missed. The rarity of this disease prevents many physicians from gaining experience with its clinical diagnosis, and some patients present with signs and symptoms that do not fit the classic pattern. Assessing clinical characteristics of other paralytic illnesses in single cases is sometimes critical to rule in or rule out the diagnosis of botulism.
In adults, a food history and the names of contacts who may have shared foods should be obtained before illness progresses to respiratory failure; specific questions should include information about the consumption of home-preserved and/or exotic foods and of products requiring refrigeration that have been left at room temperature in sealed plastic containers or bags. A history of recent consumption of home-canned food substantially enhances the probability of food-borne botulism.
Ascertainment of the patient’s behavioral history related to injection drug use is critical to the diagnosis of wound botulism unless an accidental wound is evident. Caretakers’ observations up to and including the onset of symptoms are vital to the diagnosis of infant botulism. A history of recent abdominal surgery or antibiotic use may be important in the diagnosis of adult intestinal colonization botulism.
Differential Diagnosis The illnesses most commonly considered in the differential diagnosis of adult botulism cases include Guillain-Barré syndrome (GBS), myasthenia gravis, stroke syndromes, Eaton-Lambert syndrome, and tick paralysis. Less likely possibilities are poisoning by tetrodotoxin, shellfish, or a host of rarer agents and antimicrobial drug–associated paralysis. A thorough history and a meticulous physical examination can effectively eliminate most alternative diagnoses, but a workup for other diagnoses should not delay treatment with botulinum antitoxin.
GBS, a rare autoimmune demyelinating polyneuropathy that often follows an acute infection, presents most often as an ascending paralysis. Case clusters are rare. Occasional GBS cases present as the Miller Fisher variant, whose characteristic triad of ophthalmoplegia, ataxia, and areflexia is easily mistaken for the early descending paralysis of botulism. Protein levels in cerebrospinal fluid (CSF) are elevated in GBS; because this increase may be delayed until several days after symptom onset, an early lumbar puncture with a negative result may need to be repeated. In contrast, CSF findings are generally normal in botulism, although marginally elevated CSF protein concentrations have been reported. In experienced hands, electromyography may differentiate GBS from botulism.
The edrophonium (Tensilon) test is sometimes of value in distinguishing botulism (usually a negative result) from myasthenia gravis (usually a positive result).
In most cerebrovascular accidents, physical examination reveals asymmetry of paralysis and upper motor neuron signs. Brain imaging can reveal the rare basilar stroke that produces symmetric bulbar palsies. Eaton-Lambert syndrome usually manifests as proximal limb weakness in a patient already debilitated by cancer. Tick paralysis is a rare flaccid condition closely resembling botulism and caused by neurotoxins of certain ticks.
Botulism-Specific Laboratory Tests Botulism is confirmed in the laboratory by demonstration of toxin in clinical specimens (e.g., serum, stool, gastric aspirate, and sterile-water enema samples) or in samples of ingested foods. Isolation of toxigenic clostridia from stool also provides evidence of botulism. Wound cultures yielding the organism are highly suggestive in symptomatic cases. The universally accepted method for confirmation of botulism is the mouse bioassay, which is available only in specialized laboratories. Specific guidance about what specimens to collect should be obtained from the testing laboratory because the requirements vary with the form of botulism suspected. Clinical specimens collected early in the hospital admission process should be submitted for testing; toxin results may be negative if specimens are collected >7 days after symptom onset. Because of the extreme potency of botulinum toxin, a test may yield a negative result even when a patient has botulism; thus, a negative result does not exclude this diagnosis. In suspected wound botulism, material from abscesses should be collected in anaerobic culture tubes. New laboratory tests for botulism are being developed but remain experimental. Nerve conduction studies showing reduced amplitude of motor potentials—with or without potentiation by rapid repetitive stimulation in weak muscles—and needle electromyography showing small-motor-unit action potentials are consistent with botulism; these results and those that make alternative diagnoses more likely may be useful. Standard blood work and radiologic studies are not useful in diagnosing botulism.
|
TREATMENT |
BOTULISM |
The cornerstones of treatment for botulism are meticulous intensive care and immediate administration of botulinum antitoxin. Because antitoxin is most beneficial early in the course of clinical illness, it should be administered as soon as botulism is suspected and before the time-consuming workup for other illnesses is complete. Persons of all ages (including infants) in whom botulism is suspected should be hospitalized immediately so that respiratory failure—the usual cause of death—can be detected and managed promptly. Vital capacity should be monitored frequently and mechanical ventilation provided as needed. Botulinum antitoxin can limit the progression of illness because it neutralizes toxin molecules in the circulation that have not yet bound to nerve endings. However, antitoxin does not reverse existing paralysis, which may take weeks to improve. In the United States, there are two licensed antitoxin products: Botulism Antitoxin Heptavalent® (BAT; Emergent Biosolutions, Rockville, MD), an equine-derived heptavalent (A through G) product enzymatically de-speciated for treatment of all forms of adult botulism and infant cases not involving serotypes A and B; and Botulism Immune Globulin Intravenous (BabyBIG®; California Department of Public Health, Sacramento, CA), a human-derived product for treating infant botulism caused by serotype A and/or B only. Antitoxin is also available in some other countries. Aminoglycosides and other medications that block the neuromuscular junction may potentiate botulism and should be avoided.
In wound botulism, suspect wounds and abscesses should be cleaned, debrided, and drained promptly. The role of penicillin and metronidazole in treatment and decolonization is unclear. It has been hypothesized that antimicrobial agents may increase circulating botulinum toxin from lysis of bacterial cells.
Person-to-person transmission of botulism does not occur. Universal precautions are the only infection-control measures required during inpatient care.
NOTIFICATION, EXPERT CONSULTATION, AND ANTITOXIN PROVISION
Every botulism case is a public health emergency. Antitoxin is not universally available. Some countries maintain stockpiles of antitoxin for immediate response, whereas others must access supplies from other nations when an outbreak occurs.
In the United States, clinicians must report every suspected case, regardless of form, on an emergency basis to their state health department. The state health department may put the physician in contact with the 24/7 botulism consultation service at the Centers for Disease Control and Prevention (CDC) through the CDC Emergency Operations Center (770-488-7100) or a locally available service. The botulism consultant will review the case and determine whether botulism is likely. If indicated, the consultant will coordinate laboratory confirmation at appropriate testing facilities and facilitate emergency shipment of antitoxin for all adult cases and for infant cases not involving serotypes A and B. In this country, botulinum antitoxin for noninfant cases is available exclusively from the CDC. Physicians who see suspected infant botulism cases should contact the California Department of Public Health Infant Botulism Treatment and Prevention Program (510-231-7600), which provides 24-h consultation and distributes antitoxin (BabyBIG) for the treatment of infant botulism nationwide. Except in cases involving infants who reside in California, laboratory testing requests must still be authorized by the state health department where the infant is located or by the CDC.
PREVENTION
No prophylaxis or licensed vaccine is available against botulism. Home-canning instructions and equipment have changed over the years. Up-to-date canning instructions from reliable sources (e.g., the U.S. Department of Agriculture or the U.S. Food and Drug Administration) should be followed to ensure food safety. Processed food should be stored properly and heated thoroughly prior to consumption. Because of the possible presence of spores, honey should not be given to infants (≤12 months of age). Injection of illicit drugs should be avoided. All wounds should be meticulously cleaned to eliminate possible contamination with bacterial spores. Clinicians should educate individuals or family members of at-risk individuals, including infants, illegal drug users, and preparers of home-preserved foods.
ACKNOWLEDGMENT
The authors thank Dr. Jeremy Sobel for his valued contributions to the previous version of this chapter.
179 |
Gas Gangrene and Other Clostridial Infections |
The genus Clostridium encompasses more than 60 species that may be commensals of the gut microflora or may cause a variety of infections in humans and animals through the production of a plethora of proteinaceous exotoxins. C. tetani and C. botulinum, for example, cause specific clinical disease by elaborating single but highly potent toxins. In contrast, C. perfringens and C. septicum cause aggressive necrotizing infections that are attributable to multiple toxins, including bacterial proteases, phospholipases, and cytotoxins.
ETIOLOGIC AGENT
Vegetative cells of Clostridium species are pleomorphic, rod-shaped, and arranged singly or in short chains (Fig. 179-1); the cells have rounded or sometimes pointed ends. Although clostridia stain gram-positive in the early stages of growth, they may appear to be gram-negative or gram-variable later in the growth cycle or in infected tissue specimens. Most strains are motile by means of peritrichous flagella; C. septicum swarms on solid media. Nonmotile species include C. perfringens, C. ramosum, and C. innocuum. Most species are obligately anaerobic, although clostridial tolerance to oxygen varies widely; some species (e.g., C. septicum, C. tertium) will grow but will not sporulate in air.
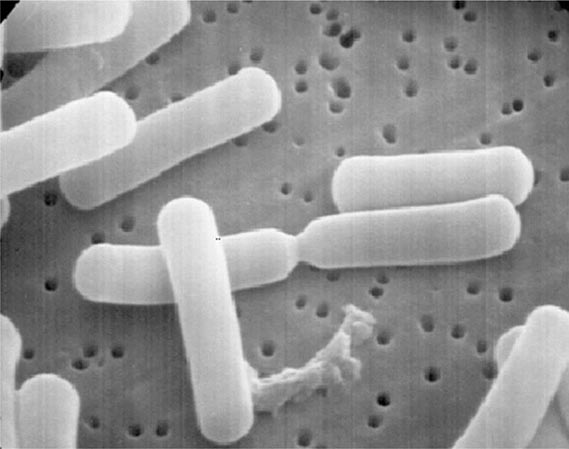
FIGURE 179-1 Scanning electron micrograph of C. perfringens.
Clostridia produce more protein toxins than any other bacterial genus, and more than 25 clostridial toxins lethal to mice have been identified. These proteins include neurotoxins, enterotoxins, cytotoxins, collagenases, permeases, necrotizing toxins, lipases, lecithinases, hemolysins, proteinases, hyaluronidases, DNases, ADP-ribosyltransferases, and neuraminidases. Botulinum and tetanus neurotoxins are the most potent toxins known, with lethal doses of 0.2–10 ng/kg for humans. Epsilon toxin, a 33-kDa protein produced by C. perfringens types B and D, causes edema and hemorrhage in the brain, heart, spinal cord, and kidneys of animals. It is among the most lethal of the clostridial toxins and is considered a potential agent of bioterrorism (Chap. 261e). The genomic sequences of some pathogenic clostridia are now available and are likely to facilitate a comprehensive approach to understanding the virulence factors involved in clostridial pathogenesis.
EPIDEMIOLOGY AND TRANSMISSION
Clostridium species are widespread in nature, forming endospores that are commonly found in soil, feces, sewage, and marine sediments. The ecology of C. perfringens in soil is greatly influenced by the degree and duration of animal husbandry in a given location and is relevant to the incidence of gas gangrene caused by contamination of war wounds with soil. For example, the incidence of clostridial gas gangrene is higher in agricultural regions of Europe than in the Sahara Desert of Africa. Similarly, the incidences of tetanus and food-borne botulism are clearly related to the presence of clostridial spores in soil, water, and many foods. Clostridia are present in large numbers in the indigenous microbiota of the intestinal tract of humans and animals, in the female genital tract, and on the oral mucosa. It should be noted that not all commensal clostridia are toxigenic.
![]() Clostridial infections remain a serious public health concern worldwide. In developing nations, food poisoning, necrotizing enterocolitis, and gas gangrene are common because large portions of the population are poor and have little or no immediate access to health care. These infections remain prevalent in developed countries as well. Gas gangrene commonly follows knife or gunshot wounds or vehicular accidents or develops as a complication of surgery or gastrointestinal carcinoma. Severe clostridial infections have emerged as a health threat to injection drug users and to women undergoing childbirth or abortion. Historically, clostridial gas gangrene has been the scourge of the battlefield. The global political situation portends another possible scenario involving mass casualties of war or terrorism, with extensive injuries conducive to gas gangrene. Thus there is an ongoing need to develop novel strategies to prevent or attenuate the course of clostridial infections in both civilians and military personnel. Vaccination against exotoxins important in pathogenesis would be of great benefit in developing nations and could also be used safely in at-risk populations such as the elderly, patients with diabetes who may require lower-limb surgery due to trauma or poor circulation, and those undergoing intestinal surgery. Moreover, a hyperimmune globulin would be a valuable tool for prophylaxis in victims of acute traumatic injury or for attenuation of the spread of infection in patients with established gas gangrene.
Clostridial infections remain a serious public health concern worldwide. In developing nations, food poisoning, necrotizing enterocolitis, and gas gangrene are common because large portions of the population are poor and have little or no immediate access to health care. These infections remain prevalent in developed countries as well. Gas gangrene commonly follows knife or gunshot wounds or vehicular accidents or develops as a complication of surgery or gastrointestinal carcinoma. Severe clostridial infections have emerged as a health threat to injection drug users and to women undergoing childbirth or abortion. Historically, clostridial gas gangrene has been the scourge of the battlefield. The global political situation portends another possible scenario involving mass casualties of war or terrorism, with extensive injuries conducive to gas gangrene. Thus there is an ongoing need to develop novel strategies to prevent or attenuate the course of clostridial infections in both civilians and military personnel. Vaccination against exotoxins important in pathogenesis would be of great benefit in developing nations and could also be used safely in at-risk populations such as the elderly, patients with diabetes who may require lower-limb surgery due to trauma or poor circulation, and those undergoing intestinal surgery. Moreover, a hyperimmune globulin would be a valuable tool for prophylaxis in victims of acute traumatic injury or for attenuation of the spread of infection in patients with established gas gangrene.
CLINICAL SYNDROMES
Life-threatening clostridial infections range from intoxications (e.g., food poisoning, tetanus) to necrotizing enteritis/colitis, bacteremia, myonecrosis, and toxic shock syndrome (TSS). Tetanus and botulism are discussed in Chaps. 177 and 178, respectively. Colitis due to C. difficile is discussed in Chap. 161.
CLOSTRIDIAL WOUND CONTAMINATION
Of open traumatic wounds, 30–80% reportedly are contaminated with clostridial species. In the absence of devitalized tissue, the presence of clostridia does not necessarily lead to infection. In traumatic injuries, clostridia are isolated with equal frequency from both suppurative and well-healing wounds. Thus, diagnosis and treatment of clostridial infection should be based on clinical signs and symptoms and not solely on bacteriologic findings.
POLYMICROBIAL INFECTIONS INVOLVING CLOSTRIDIA
Clostridial species may be found in polymicrobial infections also involving microbial components of the indigenous flora. In these infections, clostridia often appear in association with non-spore-forming anaerobes and facultative or aerobic organisms. Head and neck infections, conjunctivitis, brain abscess, sinusitis, otitis, aspiration pneumonia, lung abscess, pleural empyema, cholecystitis, septic arthritis, and bone infections all may involve clostridia. These conditions are often associated with severe local inflammation but may lack the characteristic systemic signs of toxicity and rapid progression seen in other clostridial infections. In addition, clostridia are isolated from ~66% of intraabdominal infections in which the mucosal integrity of the bowel or respiratory system has been compromised. In this setting, C. ramosum, C. perfringens, and C. bifermentans are the most commonly isolated species. Their presence does not invariably lead to a poor outcome. Clostridia have been isolated from suppurative infections of the female genital tract (e.g., ovarian or pelvic abscess) and from diseased gallbladders. Although the most frequently isolated species is C. perfringens, gangrene is not typically observed; however, gas formation in the biliary system can lead to emphysematous cholecystitis, especially in diabetic patients. C. perfringens in association with mixed aerobic and anaerobic microbes can cause aggressive life-threatening type I necrotizing fasciitis or Fournier’s gangrene.
The treatment of mixed aerobic/anaerobic infection of the abdomen, perineum, or gynecologic organs should be based on Gram’s staining, culture, and antibiotic sensitivity information. Reasonable empirical treatment consists of ampicillin or ampicillin/sulbactam combined with either clindamycin or metronidazole (Table 179-1). Broader gram-negative coverage may be necessary if the patient has recently been hospitalized or treated with antibiotics. Such coverage can be obtained by substituting ticarcillin/clavulanic acid, piperacillin/sulbactam, or a penem antibiotic for ampicillin or by adding a fluoroquinolone or an aminoglycoside to the regimen. Empirical treatment should be given for 10–14 days or until the patient’s clinical condition improves.
|
TREATMENT OF CLOSTRIDIAL INFECTIONS |
ENTERIC CLOSTRIDIAL INFECTIONS
C. perfringens type A is one of the most common bacterial causes of food-borne illness in the United States and Canada. The foods typically implicated include improperly cooked meat and meat products (e.g., gravy) in which residual spores germinate and proliferate during slow cooling or insufficient reheating. Illness results from the ingestion of food containing at least ~108 viable vegetative cells, which sporulate in the alkaline environment of the small intestine, producing C. perfringens enterotoxin in the process. The diarrhea that develops within 7–30 h of ingestion of contaminated food is generally mild and self-limiting; however, in the very young, the elderly, and the immunocompromised, symptoms are more severe and occasionally fatal. Enterotoxin-producing C. perfringens has been implicated as an etiologic agent of persistent diarrhea in elderly patients in nursing homes and tertiary-care institutions and has been considered to play a role in antibiotic-associated diarrhea without pseudomembranous colitis.
![]() C. perfringens strains associated with food poisoning possess the gene (cpe) coding for enterotoxin, which acts by forming pores in host cell membranes. C. perfringens strains isolated from non-food-borne diseases, such as antibiotic-associated and sporadic diarrhea, carry cpe on a plasmid that may be transmitted to other strains. Several methods have been described for the detection of C. perfringens enterotoxin in feces, including cell culture assay (Vero cells), enzyme-linked immunosorbent assay, reversed-phase latex agglutination, and polymerase chain reaction (PCR) amplification of cpe. Each method has its advantages and limitations.
C. perfringens strains associated with food poisoning possess the gene (cpe) coding for enterotoxin, which acts by forming pores in host cell membranes. C. perfringens strains isolated from non-food-borne diseases, such as antibiotic-associated and sporadic diarrhea, carry cpe on a plasmid that may be transmitted to other strains. Several methods have been described for the detection of C. perfringens enterotoxin in feces, including cell culture assay (Vero cells), enzyme-linked immunosorbent assay, reversed-phase latex agglutination, and polymerase chain reaction (PCR) amplification of cpe. Each method has its advantages and limitations.
![]() Enteritis necroticans (gas gangrene of the bowel) is a fulminating clinical illness characterized by extensive necrosis of the intestinal mucosa and wall. Cases can occur sporadically in adults or as epidemics in people of all ages. Enteritis necroticans is caused by α toxin– and β toxin–producing strains of C. perfringens type C; β toxin is located on a plasmid and is mainly responsible for pathogenesis. This life-threatening infection causes ischemic necrosis of the jejunum. In Papua New Guinea during the 1960s, enteritis necroticans (known in that locale as pigbel) was found to be the most common cause of death in childhood; it was associated with pig feasts and occurred both sporadically and in outbreaks. Intramuscular immunization against the β toxin resulted in a decreased incidence of the disease in Papua New Guinea, although the condition remains common. Enteritis necroticans has also been recognized in the United States, the United Kingdom, Germany (where it is known as darmbrand), and other developed nations; especially affected are adults who are malnourished or who have diabetes, alcoholic liver disease, or neutropenia.
Enteritis necroticans (gas gangrene of the bowel) is a fulminating clinical illness characterized by extensive necrosis of the intestinal mucosa and wall. Cases can occur sporadically in adults or as epidemics in people of all ages. Enteritis necroticans is caused by α toxin– and β toxin–producing strains of C. perfringens type C; β toxin is located on a plasmid and is mainly responsible for pathogenesis. This life-threatening infection causes ischemic necrosis of the jejunum. In Papua New Guinea during the 1960s, enteritis necroticans (known in that locale as pigbel) was found to be the most common cause of death in childhood; it was associated with pig feasts and occurred both sporadically and in outbreaks. Intramuscular immunization against the β toxin resulted in a decreased incidence of the disease in Papua New Guinea, although the condition remains common. Enteritis necroticans has also been recognized in the United States, the United Kingdom, Germany (where it is known as darmbrand), and other developed nations; especially affected are adults who are malnourished or who have diabetes, alcoholic liver disease, or neutropenia.
Necrotizing enterocolitis, a disease resembling enteritis necroticans but associated with C. perfringens type A, has been found in North America in previously healthy adults. It is also a serious gastrointestinal disease of low-birth-weight (premature) infants hospitalized in neonatal intensive care units. The etiology and pathogenesis of this disease have remained enigmatic for more than four decades. Pathologic similarities between necrotizing enterocolitis and enteritis necroticans include the pattern of small-bowel necrosis involving the submucosa, mucosa, and muscularis; the presence of gas dissecting the tissue planes; and the degree of inflammation. In contrast to enteritis necroticans, which most commonly involves the jejunum, necrotizing enterocolitis affects the ileum and frequently the ileocecal valve. Both diseases may manifest as intestinal gas cysts, although this feature is more common in necrotizing enterocolitis. The sources of the gas, which contains hydrogen, methane, and carbon dioxide, are probably the fermentative activities of intestinal bacteria, including clostridia. Epidemiologic data support an important role for C. perfringens or other gas-producing microorganisms (e.g., C. neonatale, certain other clostridia, or Klebsiella species) in the pathogenesis of necrotizing enterocolitis.
Patients with suspected clostridial enteric infection should undergo nasogastric suction and receive IV fluids. Pyrantel is given by mouth, and the bowel is rested by fasting. Benzylpenicillin (1 mU) is given IV every 4 h, and the patient is observed for complications requiring surgery. Patients with mild cases recover without surgical intervention. If surgical indications are present (gas in the peritoneal cavity, absent bowel sounds, rebound tenderness, abdominal rigidity), however, the mortality rate ranges from 35% to 100%; a fatal outcome is due in part to perforation of the intestine.
As pigbel continues to be a common disease in Papua New Guinea, consideration should be given to the use of a C. perfringens type C βtoxoid vaccine in local areas. Two doses given 3–4 months apart are preventive.
CLOSTRIDIAL BACTEREMIA
Clostridium species are important causes of bloodstream infections. Molecular epidemiologic studies of anaerobic bacteremia have identified C. perfringens and C. tertium as the two most frequently isolated species; these organisms cause up to 79% and 5%, respectively, of clostridial bacteremias. Occasionally, C. perfringens bacteremia occurs in the absence of an identifiable infection at another site. When associated with myonecrosis, bacteremia has a grave prognosis.
C. septicum is also commonly associated with bacteremia. This species is isolated only rarely from the feces of healthy individuals but may be found in the normal appendix. More than 50% of patients whose blood cultures are positive for this organism have some gastrointestinal anomaly (e.g., diverticular disease) or underlying malignancy (e.g., carcinoma of the colon). In addition, a clinically important association of C. septicum bacteremia with neutropenia of any origin—and, more specifically, with neutropenic enterocolitis involving the terminal ileum or cecum—has been observed. Patients with diabetes mellitus, severe atherosclerotic cardiovascular disease, or anaerobic myonecrosis (gas gangrene) also may develop C. septicum bacteremia. C. septicum has been recovered from the bloodstream of cirrhotic patients, as have C. perfringens, C. bifermentans, and other clostridia. Infections of the bloodstream by C. sordellii and C. perfringens have been associated with TSS.
Bloodstream infection by C. tertium, either alone or in combination with C. septicum or C. perfringens, can be found in patients with serious underlying disease such as malignancy or acute pancreatitis, with or without neutropenic enterocolitis; the frequency has not been systematically studied. C. tertium may present special problems in terms of both identification and treatment. This organism may stain gram-negative; is aerotolerant; and is resistant to metronidazole, clindamycin, and cephalosporins.
Other clostridia from the C. clostridioforme group (including C. clostridioforme, C. hathewayi, and C. bolteae) can cause bacteremia.
The clinical importance of recognizing clostridial bacteremia—especially that due to C. septicum—and starting appropriate treatment immediately (Table 179-1) cannot be overemphasized. Patients with this condition usually are gravely ill, and infection may metastasize to distant anatomic sites, resulting in spontaneous myonecrosis (see next section). Alternative methods to identify bacteremia-causing clostridial species, such as PCR or other rapid diagnostic tests, are not currently available. Anaerobic blood cultures and Gram’s stain interpretation remain the best diagnostic tests at this point.
CLOSTRIDIAL SKIN AND SOFT-TISSUE INFECTIONS
Histotoxic clostridial species such as C. perfringens, C. histolyticum, C. septicum, C. novyi, and C. sordellii cause aggressive necrotizing infections of the skin and soft tissues. These infections are attributable in part to the elaboration of bacterial proteases, phospholipases, and cytotoxins. Necrotizing clostridial soft-tissue infections are rapidly progressive and are characterized by marked tissue destruction, gas in the tissues, and shock; they frequently end in death. Severe pain, crepitus, brawny induration with rapid progression to skin sloughing, violaceous bullae, and marked tachycardia are characteristics found in the majority of patients.
Clostridial Myonecrosis (Gas Gangrene) • TRAUMATIC GAS GANGRENE C. perfringens myonecrosis (gas gangrene) is one of the most fulminant gram-positive bacterial infections of humans. Even with appropriate antibiotic therapy and management in an intensive care unit, tissue destruction can progress rapidly. Gas gangrene is accompanied by bacteremia, hypotension, and multiorgan failure and is invariably fatal if untreated. Gas gangrene is a true emergency and requires immediate surgical debridement.
The development of gas gangrene requires an anaerobic environment and contamination of a wound with spores or vegetative organisms. Devitalized tissue, foreign bodies, and ischemia reduce locally available oxygen levels and favor outgrowth of vegetative cells and spores. Thus conditions predisposing to traumatic gas gangrene include crush-type injury, laceration of large or medium-sized arteries, and open fractures of long bones that are contaminated with soil or bits of clothing containing the bacterial spores. Gas gangrene of the abdominal wall and flanks follows penetrating injuries such as knife or gunshot wounds that are sufficient to compromise intestinal integrity, with resultant leakage of the bowel contents into the soft tissues. Proximity to fecal sources of bacteria is a risk factor for cases following hip surgery, adrenaline injections into the buttocks, or amputation of the leg for ischemic vascular disease. In the last decade, cutaneous gas gangrene caused by C. perfringens, C. novyi, and C. sordellii has been described in the United States and northern Europe among persons injecting black-tar heroin subcutaneously.
The incubation period for traumatic gas gangrene can be as short as 6 h and is usually <4 days. The infection is characterized by the sudden onset of excruciating pain at the affected site and the rapid development of a foul-smelling wound containing a thin serosanguineous discharge and gas bubbles. Brawny edema and induration develop and give way to cutaneous blisters containing bluish to maroon-colored fluid. Such tissue later may become liquefied and slough. The margin between healthy and necrotic tissue often advances several inches per hour despite appropriate antibiotic therapy, and radical amputation remains the single best life-saving intervention. Shock and organ failure frequently accompany gas gangrene; when patients become bacteremic, the mortality rate exceeds 50%.
Diagnosis of traumatic gas gangrene is not difficult because the infection always begins at the site of significant trauma, is associated with gas in the tissue, and is rapidly progressive. Gram’s staining of drainage or tissue biopsy is usually definitive, demonstrating large gram-positive (or gram-variable) rods, an absence of inflammatory cells, and widespread soft-tissue necrosis.
SPONTANEOUS (NONTRAUMATIC) GAS GANGRENE Spontaneous gas gangrene generally occurs via hematogenous seeding of normal muscle with histotoxic clostridia—principally C. perfringens, C. septicum, and C. novyi and occasionally C. tertium—from a gastrointestinal tract portal of entry (as in colonic malignancy, inflammatory bowel disease, diverticulitis, necrotizing enterocolitis, cecitis, or distal ileitis or after gastrointestinal surgery). These gastrointestinal pathologies permit bacterial access to the bloodstream; consequently, aerotolerant C. septicum can proliferate in normal tissues. Patients surviving bacteremia or spontaneous gangrene due to C. septicum should undergo aggressive diagnostic studies to rule out gastrointestinal pathology.
Additional predisposing host factors include leukemia, lymphoproliferative disorders, cancer chemotherapy, radiation therapy, and AIDS. Cyclic, congenital, or acquired neutropenia also is strongly associated with an increased incidence of spontaneous gas gangrene due to C. septicum; in such cases, necrotizing enterocolitis, cecitis, or distal ileitis is common, particularly among children.
The first symptom of spontaneous gas gangrene may be confusion followed by the abrupt onset of excruciating pain in the absence of trauma. These findings, along with fever, should heighten suspicion of spontaneous gas gangrene. However, because of the lack of an obvious portal of entry, the correct diagnosis is frequently delayed or missed. The infection is characterized by rapid progression of tissue destruction with demonstrable gas in the tissue (Fig. 179-2). Swelling increases, and bullae filled with clear, cloudy, hemorrhagic, or purplish fluid appear. The surrounding skin has a purple hue, which may reflect vascular compromise resulting from the diffusion of bacterial toxins into surrounding tissues. Invasion of healthy tissue rapidly ensues, with quick progression to shock and multiple-organ failure. Mortality rates in this setting range from 67 to 100% among adults; among children, the mortality rate is 59%, with the majority of deaths occurring within 24 h of onset.
FIGURE 179-2 Radiograph of patient with spontaneous gas gangrene due to C. septicum, demonstrating gas in the affected arm and shoulder.
PATHOGENESIS OF GAS GANGRENE In traumatic gas gangrene, organisms are introduced into devitalized tissue. It is important to recognize that for C. perfringens and C. novyi, trauma must be sufficient to interrupt the blood supply and thereby to establish an optimal anaerobic environment for growth of these species. These conditions are not strictly required for the more aerotolerant species such as C. septicum and C. tertium, which can seed normal tissues from gastrointestinal lesions. Once introduced into an appropriate niche, the organisms proliferate locally and elaborate exotoxins.
The major C. perfringens extracellular toxins implicated in gas gangrene are α toxin and θ toxin. A lethal hemolysin that has both phospholipase C and sphingomyelinase activities, α toxin has been implicated as the major virulence factor of C. perfringens: immunization of mice with the C-terminal domain of α toxin provides protection against lethal challenge with C. perfringens, and isogenic α toxin–deficient mutant strains of C. perfringens are not lethal in a murine model of gas gangrene. It has been shown in experimental models that the severe pain, rapid progression, marked tissue destruction, and absence of neutrophils in C. perfringens gas gangrene are attributable in large part to α toxin–induced occlusion of blood vessels by heterotypic aggregates of platelets and neutrophils. The formation of these aggregates, which occurs within minutes, is largely mediated by α toxin’s ability to activate the platelet adhesion molecule gpIIb/IIIa (Fig. 179-3); the implication is that platelet glycoprotein inhibitors (e.g., eptifibatide, abciximab) may be therapeutic for maintaining tissue blood flow.
FIGURE 179-3 Schematic illustration of the molecular mechanisms of C. perfringens α toxin–induced platelet/neutrophil aggregates. Homotypic aggregates of platelets (not shown) and heterotypic aggregates of platelets and leukocytes are due to α toxin–induced activation of the platelet fibrinogen receptor gpIIb/IIIa and upregulation of leukocyte CD11b/CD18. Binding of fibrinogen (red) bridges the connection between these adhesion molecules on adjacent cells. An auxiliary role for α toxin–induced upregulation of platelet P-selectin and its binding to leukocyte P-selectin glycoprotein ligand 1 (PSGL-1) or other leukocyte surface carbohydrates also has been demonstrated.
C. perfringens θ toxin (perfringolysin) is a member of the thiol-activated cytolysin family known as cholesterol-dependent cytolysins, which includes streptolysin O from group A Streptococcus, pneumolysin from Streptococcus pneumoniae, and several other toxins. Cholesterol-dependent cytolysins bind as oligomers to cholesterol in host cell membranes. At high concentrations, these toxins form ringlike pores resulting in cell lysis. At sublytic concentrations, θ toxin hyperactivates phagocytes and vascular endothelial cells.
Cardiovascular collapse and end-organ failure occur late in the course of C. perfringens gas gangrene and are largely attributable to both direct and indirect effects of α and θ toxins. In experimental models, θ toxin causes markedly reduced systemic vascular resistance but increased cardiac output (i.e., “warm shock”), probably via induction of endogenous mediators (e.g., prostacyclin, platelet-activating factor) that cause vasodilation. This effect is similar to that observed in gram-negative sepsis. In sharp contrast, α toxin directly suppresses myocardial contractility; the consequence is profound hypotension due to a sudden reduction in cardiac output. The roles of other endogenous mediators, such as cytokines (e.g., tumor necrosis factor, interleukin 1, interleukin 6) and vasodilators (e.g., bradykinin) have not been fully elucidated.
C. septicum produces four main toxins—α toxin (lethal, hemolytic, necrotizing activity), β toxin (DNase), γ toxin (hyaluronidase), and Δ toxin (septicolysin, an oxygen-labile hemolysin)—as well as a protease and a neuraminidase. Unlike the α toxin of C. perfringens, that of C. septicum does not possess phospholipase activity. The mechanisms remain to be fully elucidated, but it is likely that each of these toxins contributes uniquely to C. septicum gas gangrene.
|
TREATMENT |
GAS GANGRENE |
Patients with suspected gas gangrene (either traumatic or spontaneous) should undergo prompt surgical inspection of the infected site. Direct examination of a Gram-stained smear of the involved tissues is of major importance. Characteristic histologic findings in clostridial gas gangrene include widespread tissue destruction, a paucity of leukocytes in infected tissues in conjunction with an accumulation of leukocytes in adjacent vessels (Fig. 179-4), and the presence of gram-positive rods (with or without spores). CT and MRI are invaluable for determining whether the infection is localized or is spreading along fascial planes, and needle aspiration or punch biopsy may provide an etiologic diagnosis in at least 20% of cases. However, these techniques should not replace surgical exploration, Gram’s staining, and histopathologic examination. When spontaneous gas gangrene is suspected, blood should be cultured since bacteremia usually precedes cutaneous manifestations by several hours.
FIGURE 179-4 Histopathology of experimental gas gangrene due to C. perfringens, demonstrating widespread muscle necrosis, a paucity of leukocytes in infected tissues, and accumulation of leukocytes in adjacent vessels (arrows). These features are due to the effects of α and θ toxins on muscle cells, platelets, leukocytes, and endothelial cells.
For patients with evidence of clostridial gas gangrene, thorough emergent surgical debridement is of extreme importance. All devitalized tissue should be widely resected back to healthy viable muscle and skin so as to remove conditions that allow anaerobic organisms to continue proliferating. Closure of traumatic wounds or compound fractures should be delayed for 5–6 days until it is certain that these sites are free of infection.
Antibiotic treatment of traumatic or spontaneous gas gangrene (Table 179-1) consists of the administration of penicillin and clindamycin for 10–14 days. Penicillin is recommended on the basis of in vitro sensitivity data; clindamycin is recommended because of its superior efficacy over penicillin in animal models of C. perfringens gas gangrene and in some clinical reports. Controlled clinical trials comparing the efficacy of these agents in humans have not been performed. In the penicillin-allergic patient, clindamycin may be used alone. The superior efficacy of clindamycin is probably due to its ability to inhibit bacterial protein toxin production, its insensitivity to the size of the bacterial load or the stage of bacterial growth, and its ability to modulate the host’s immune response.
C. tertium is resistant to penicillin, cephalosporins, and clindamycin. Appropriate antibiotic therapy for C. tertium infection is vancomycin (1 g every 12 h IV) or metronidazole (500 mg every 8 h IV).
The value of adjunctive treatment with hyperbaric oxygen (HBO) for gas gangrene remains controversial. Basic science studies suggest that HBO can inhibit the growth of C. perfringens but not that of the more aerotolerant C. septicum. In vitro, blood and macerated muscle inhibit the bactericidal potential of HBO. Numerous studies in animals demonstrate little efficacy of HBO alone, whereas antibiotics alone—especially those that inhibit bacterial protein synthesis—confer marked benefits. Addition of HBO to the therapeutic regimen provides some additional benefit, but only if surgery and antibiotic administration precede HBO treatment.
In conclusion, gas gangrene is a rapidly progressive infection whose outcome depends on prompt recognition, emergent surgery, and timely administration of antibiotics that inhibit toxin production. Gas gangrene associated with bacteremia probably represents a later stage of illness and is associated with the worst outcomes. Emergent surgical debridement is crucial to ensure survival, and ancillary procedures (e.g., CT or MRI) or transport to HBO units should not delay this intervention. Some trauma centers associated with HBO units may have special expertise in managing these aggressive infections, but proximity and speed of transfer must be carefully weighed against the need for haste.
PROGNOSIS OF GAS GANGRENE The prognosis for patients with gas gangrene is more favorable when the infection involves an extremity rather than the trunk or visceral organs, since debridement of the latter sites is more difficult. Gas gangrene is most likely to progress to shock and death in patients with associated bacteremia and intravascular hemolysis. Mortality rates are highest for patients in shock at the time of diagnosis. Mortality rates are relatively high among patients with spontaneous gas gangrene, especially that due to C. septicum. Survivors of gas gangrene may undergo multiple debridements and face long periods of hospitalization and rehabilitation.
PREVENTION OF GAS GANGRENE Initial aggressive debridement of devitalized tissue can reduce the risk of gas gangrene in contaminated deep wounds. Interventions to be avoided include prolonged application of tourniquets and surgical closure of traumatic wounds; patients with compound fractures are at significant risk for gas gangrene if the wound is closed surgically. Vaccination against α toxin is protective in experimental animal models of C. perfringens gas gangrene but has not been investigated in humans. In addition, as mentioned above, a hyperimmune globulin would represent a significant advance for prophylaxis in victims of acute traumatic injury or for attenuation of the spread of infection in patients with established gas gangrene.
Toxic Shock Syndrome Clostridial infection of the endometrium, particularly that due to C. sordellii, can develop after gynecologic procedures, childbirth, or abortion (spontaneous or elective, surgical or medical) and, once established, proceeds rapidly to TSS and death. Systemic manifestations, including edema, effusions, profound leukocytosis, and hemoconcentration, are followed by the rapid onset of hypotension and multiple-organ failure. Elevation of the hematocrit to 75–80% and leukocytosis of 50,000–200,000 cells/μL, with a left shift, are characteristic of C. sordellii infection. Pain may not be a prominent feature, and fever is typically absent. In one series, 18% of 45 cases of C. sordellii infection were associated with normal childbirth, 11% with medically induced abortion, and 0.4% with spontaneous abortion; the case-fatality rate was 100% in these groups. Of the infections in this series that were not related to gynecologic procedures or childbirth, 22% occurred in injection drug users, and 50% of these patients died. Other infections followed trauma or surgery (42%), mostly in healthy persons, and 53% of these patients died. Overall, the mortality rate was 69% (31 of 45 cases). Of patients who succumbed, 85% died within 2–6 days after infection onset or following procedures.
Early diagnosis of C. sordellii infections often proves difficult for several reasons. First, the prevalence of these infections is low. Second, the initial symptoms are nonspecific and frankly misleading. Early in the course, the illness resembles any number of infectious diseases, including viral syndromes. Given these vague symptoms and an absence of fever, physicians usually do not aggressively pursue additional diagnostic tests. The absence of local evidence of infection and the lack of fever make early diagnosis of C. sordellii infection particularly problematic in patients who develop deep-seated infection following childbirth, therapeutic abortion, gastrointestinal surgery, or trauma. Such patients are frequently evaluated for pulmonary embolization, gastrointestinal bleeding, pyelonephritis, or cholecystitis. Unfortunately, such delays in diagnosis increase the risk of death, and, as in most necrotizing soft-tissue infections, patients are hypotensive with evidence of organ dysfunction by the time local signs and symptoms become apparent. In contrast, infection is more readily suspected in injection drug users presenting with local swelling, pain, and redness at injection sites; early recognition probably contributes to the lower mortality rates in this group.
Physicians should suspect C. sordellii infection in patients who present within 2–7 days after injury, surgery, drug injection, childbirth, or abortion and who report pain, nausea, vomiting, and diarrhea but are afebrile. There is little information regarding appropriate treatment for C. sordellii infections. In fact, the interval between onset of symptoms and death is often so short that there is little time to initiate empirical antimicrobial therapy. Indeed, anaerobic cultures of blood and wound aspirates are time-consuming, and many hospital laboratories do not routinely perform antimicrobial sensitivity testing on anaerobes. Antibiotic susceptibility data from older studies suggest that C. sordellii, like most clostridia, is susceptible to β-lactam antibiotics, clindamycin, tetracycline, and chloramphenicol but is resistant to aminoglycosides and sulfonamides. Antibiotics that suppress toxin synthesis (e.g., clindamycin) may possibly prove useful as therapeutic adjuncts since they are effective in necrotizing infections due to other toxin-producing gram-positive organisms.
Other Clostridial Skin and Soft-Tissue Infections Crepitant cellulitis (also called anaerobic cellulitis) occurs principally in diabetic patients and characteristically involves subcutaneous tissues or retroperitoneal tissues, whereas the muscle and fascia are not involved. This infection can progress to fulminant systemic disease.
Cases of C. histolyticum infection with cellulitis, abscess formation, or endocarditis have also been documented in injection drug users. Endophthalmitis due to C. sordellii or C. perfringens has been described. C. ramosum is also isolated frequently from clinical specimens, including blood and both intraabdominal and soft tissues. This species may be resistant to clindamycin and multiple cephalosporins.
SECTION 6 |
DISEASES CAUSED BY GRAM-NEGATIVE BACTERIA |
180 |
Meningococcal Infections |
DEFINITION
Infection with Neisseria meningitidis most commonly manifests as asymptomatic colonization in the nasopharynx of healthy adolescents and adults. Invasive disease occurs rarely, usually presenting as either bacterial meningitis or meningococcal septicemia. Patients may also present with occult bacteremia, pneumonia, septic arthritis, conjunctivitis, and chronic meningococcemia.
ETIOLOGY AND MICROBIOLOGY
N. meningitidis is a gram-negative aerobic diplococcus that colonizes humans only and that causes disease after transmission to a susceptible individual. Several related organisms have been recognized, including the pathogen N. gonorrhoeae and the commensals N. lactamica, N. flavescens, N. mucosa, N. sicca, and N. subflava. N. meningitidis is a catalase- and oxidase-positive organism that utilizes glucose and maltose to produce acid.
Meningococci associated with invasive disease are usually encapsulated with polysaccharide, and the antigenic nature of the capsule determines an organism’s serogroup (Table 180-1). In total, 13 serogroups have been identified (A–D, X–Z, 29E, W, H–J, and L), but just 6 serogroups—A, B, C, X, Y, and W (formerly W135)—account for the majority of cases of invasive disease. Acapsular meningococci are commonly isolated from the nasopharynx in studies of carriage; the lack of capsule often is a result of phase variation of capsule expression, but as many as 16% of isolates lack the genes for capsule synthesis and assembly. These “capsule-null” meningococci and those that express capsules other than A, B, C, X, Y, and W are only rarely associated with invasive disease and are most commonly identified in the nasopharynx of asymptomatic carriers.
|
STRUCTURE OF THE POLYSACCHARIDE CAPSULE OF COMMON DISEASE-CAUSING MENINGOCOCCI |
Beneath the capsule, meningococci are surrounded by an outer phospholipid membrane containing lipopolysaccharide (LPS, endotoxin) and multiple outer-membrane proteins (Figs. 180-1 and 180-2). Antigenic variability in porins expressed in the outer membrane defines the serotype (PorB) and serosubtype (PorA) of the organism, and structural differences in LPS determine the immunotype. Serologic methods for typing of meningococci are restricted by the limited availability of serologic reagents that can distinguish among the organisms’ highly variable surface proteins. Where available, high-throughput antigen gene sequencing has superseded serology for meningococcal typing. A large database of antigen gene sequences for the outer-membrane proteins PorA, PorB, FetA, Opa, and factor H–binding protein is available online (www.neisseria.org). The number of specialized iron-regulated proteins found in the meningococcal outer membrane (e.g., FetA and transferrin-binding proteins) highlights the organisms’ dependence on iron from human sources. A thin peptidoglycan cell wall separates the outer membrane from the cytoplasmic membrane.
FIGURE 180-1 Electron micrograph of Neisseria meningitidis. Black dots are gold-labeled polyclonal antibodies binding surface opacity proteins. Blebs of outer membrane can be seen being released from the bacterial surface (arrow). (Photo courtesy of D. Ferguson, Oxford University.)
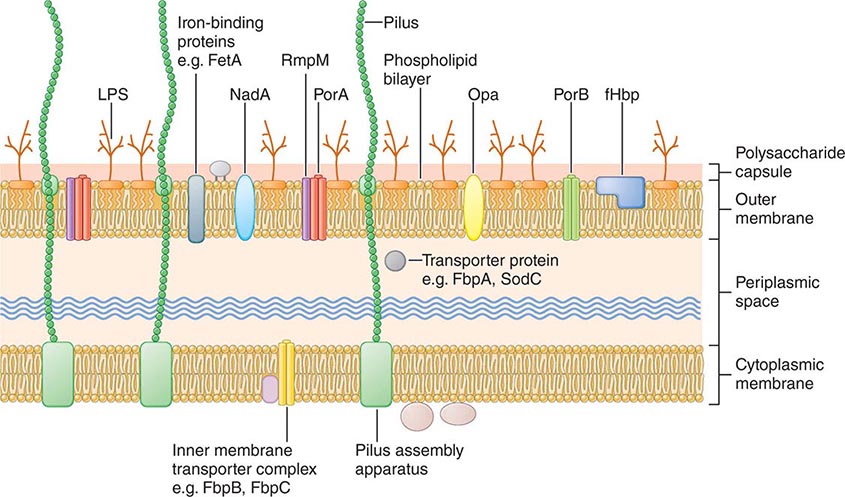
FIGURE 180-2 Cross-section through surface structures of Neisseria meningitidis. LPS, lipopolysaccharide. (Reprinted with permission from M Sadarangani, AJ Pollard: Lancet Infect Dis 10:112, 2010.)
![]() The structure of meningococcal populations involved in local and global spread has been studied with multilocus enzyme electrophoresis (MLEE), which characterizes isolates according to differences in the electrophoretic mobility of cytoplasmic enzymes. However, this technique has mostly been replaced by multilocus sequence typing (MLST), in which meningococci are characterized by sequence types assigned on the basis of sequences of internal fragments of seven housekeeping genes. The online MLST database currently includes more than 27,000 meningococcal isolates and 10,500 unique sequence types (pubmlst.org/neisseria/). A limited number of hyperinvasive lineages of N. meningitidis have been recognized and are responsible for the majority of cases of invasive meningococcal disease worldwide. The apparent genetic stability of these meningococcal clones over decades and during wide geographic spread indicates that they are well adapted to the nasopharyngeal environment of the host and to efficient transmission. While MLST has become established as the main method of genotyping meningococci in many reference laboratories over the past decade, whole-genome sequencing is set to replace this approach in the decade ahead, with almost 1000 genomes already available in the United Kingdom’s national library (www.meningitis.org/genome-library).
The structure of meningococcal populations involved in local and global spread has been studied with multilocus enzyme electrophoresis (MLEE), which characterizes isolates according to differences in the electrophoretic mobility of cytoplasmic enzymes. However, this technique has mostly been replaced by multilocus sequence typing (MLST), in which meningococci are characterized by sequence types assigned on the basis of sequences of internal fragments of seven housekeeping genes. The online MLST database currently includes more than 27,000 meningococcal isolates and 10,500 unique sequence types (pubmlst.org/neisseria/). A limited number of hyperinvasive lineages of N. meningitidis have been recognized and are responsible for the majority of cases of invasive meningococcal disease worldwide. The apparent genetic stability of these meningococcal clones over decades and during wide geographic spread indicates that they are well adapted to the nasopharyngeal environment of the host and to efficient transmission. While MLST has become established as the main method of genotyping meningococci in many reference laboratories over the past decade, whole-genome sequencing is set to replace this approach in the decade ahead, with almost 1000 genomes already available in the United Kingdom’s national library (www.meningitis.org/genome-library).
The group B meningococcal genome is >2 megabases in length and contains 2158 coding regions. Many genes undergo phase variation that makes it possible to control their expression; this capacity is likely to be important in meningococcal adaptation to the host environment and evasion of the immune response. Meningococci can obtain DNA from their environment and can acquire new genes—including the capsular operon—such that capsule switching from one serogroup to another can occur.
EPIDEMIOLOGY
![]() Patterns of Disease Up to 500,000 cases of meningococcal disease are thought to occur worldwide each year, and ~10% of the individuals affected die. There are several patterns of disease: epidemic, outbreak (small clusters of cases), hyperendemic, and sporadic or endemic.
Patterns of Disease Up to 500,000 cases of meningococcal disease are thought to occur worldwide each year, and ~10% of the individuals affected die. There are several patterns of disease: epidemic, outbreak (small clusters of cases), hyperendemic, and sporadic or endemic.
Epidemics have continued since the original descriptions of meningococcal disease, especially affecting the sub-Saharan meningitis belt of Africa, where tens to hundreds of thousands of cases (caused mainly by serogroup A but also by serogroups W and X) may be reported over a season and rates may be as high as 1000 cases per 100,000 population. Serogroup A epidemics took place in Europe and North America after the First and Second World Wars, and serogroup A outbreaks have been documented over the past 30 years in New Zealand, China, Nepal, Mongolia, India, Pakistan, Poland, and Russia.
Clusters of cases occur where there is an opportunity for increased transmission—i.e., in (semi-)closed communities such as schools, colleges, universities, military training centers, and refugee camps. Recently, such clusters have been especially strongly linked with a particular clone (sequence type 11) that is mainly associated with the serogroup C or W capsule but was first described in association with serogroup B. Wider and more prolonged community outbreaks (hyperendemic disease) due to single clones of serogroup B meningococci account for ≥10 cases per 100,000. Regions affected in the past decade include the U.S. Pacific Northwest, New Zealand (both islands), and the province of Normandy in France.
Most countries now experience predominantly sporadic cases (0.3–5 cases per 100,000 population), with many different disease-causing clones involved and usually no clear epidemiologic link between one case and another. The disease rate and the distribution of meningococcal strains vary in different regions of the world and also in any one location over time. For example, in the United States, the rate of meningococcal disease fell from 1.2 cases per 100,000 population in 1997 to <0.15 case per 100,000 in 2012 (Fig. 180-3). Meningococcal disease in the United States was previously dominated by serogroups B and C; however, serogroup Y emerged during the 1990s and became more common than serogroup C in 2007. In contrast, rates of disease in England and Wales rose to >5 cases per 100,000 during the 1990s because of an increase in cases caused by the ST11 serogroup C clone. As a result of a mass immunization program against serogroup C in 1999, almost all cases in the United Kingdom are now attributed to serogroup B (Fig. 180-4). Over the last decade, most industrialized nations have seen a general decrease in meningococcal disease; this decrease is linked to immunization against serogroup C meningococci in Europe, Canada, and Australia and to adolescent immunization programs for A, C, Y and W in the United States. However, other factors, including changes in population immunity (probably the explanation for the cyclic nature of meningococcal disease rates) as well as a reduction in smoking and passive exposure to tobacco smoke (driven by bans on smoking in buildings and public spaces) across wealthy countries are likely to have contributed to the fall in cases.
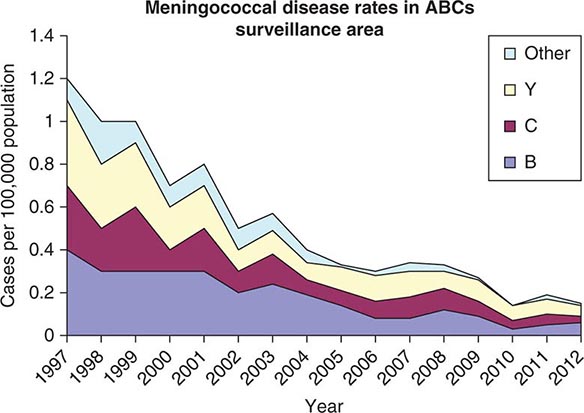
FIGURE 180-3 Meningococcal disease in the United States over time. ABCs, active bacterial cores. (Adapted from ABC Surveillance data, Centers for Disease Control and Prevention; www.cdc.gov.)
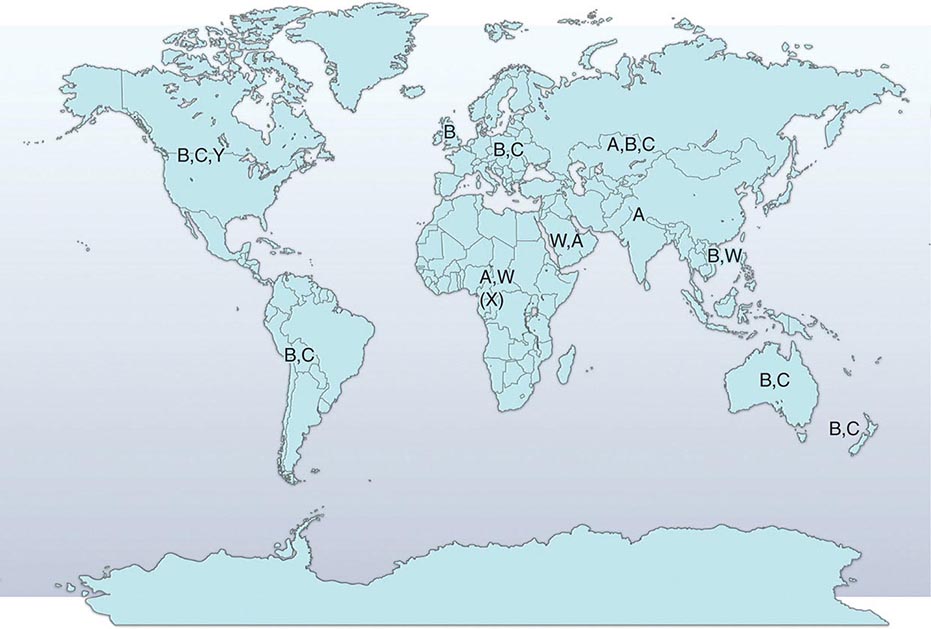
FIGURE 180-4 Global distribution of meningococcal serogroups, 1999–2009.
Factors Associated with Disease Risk and Susceptibility The principal determinant of disease susceptibility is age, with the peak incidence in the first year of life (Fig. 180-5). The susceptibility of the very young presumably results from an absence of specific adaptive immunity in combination with very close contact with colonized individuals, including parents. Compared with other age groups, infants appear to be particularly susceptible to serogroup B disease: >30% of serogroup B cases in the United States occur during the first year of life. In the early 1990s in North America, the median ages for patients with disease due to serogroups B, C, Y, and W were 6, 17, 24, and 33 years, respectively.
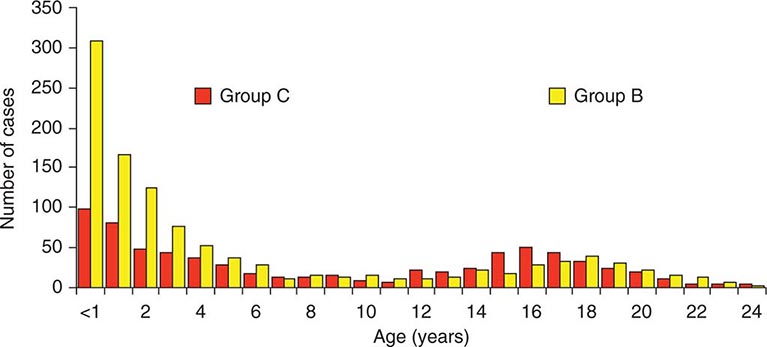
FIGURE 180-5 Age distribution of serogroups B and C meningococcal disease in England and Wales, 1998–1999. (Health Protection Agency, UK; www.hpa.org.uk.)
![]() After early childhood, a second peak of disease occurs among adolescents and young adults (15–25 years of age) in Europe and North America. It is thought that this peak relates to social behaviors and environmental exposures in this age group, as discussed below. Most cases of infection with N. meningitidis in developed countries today are sporadic, and the rarity of the disease suggests that individual susceptibility may be important. A number of factors probably contribute to individual susceptibility, including the host’s genetic constitution, environment, and contact with a carrier or a case.
After early childhood, a second peak of disease occurs among adolescents and young adults (15–25 years of age) in Europe and North America. It is thought that this peak relates to social behaviors and environmental exposures in this age group, as discussed below. Most cases of infection with N. meningitidis in developed countries today are sporadic, and the rarity of the disease suggests that individual susceptibility may be important. A number of factors probably contribute to individual susceptibility, including the host’s genetic constitution, environment, and contact with a carrier or a case.
The best-documented genetic association with meningococcal disease is complement deficiency, chiefly of the terminal complement components (C5–9), properdin, or factor D; such a deficiency increases the risk of disease by up to 600-fold and may result in recurrent attacks. Complement components are believed to be important for the bactericidal activity of serum, which is considered the principal mechanism of immunity against invasive meningococcal disease. However, when investigated, complement deficiency is found in only a very small proportion of individuals with meningococcal disease (0.3%). Conversely, 7–20% of persons whose disease is caused by the less common serogroups (W, X, Y, Z, 29E) have a complement deficiency. Complement deficiency appears to be associated with serogroup B disease only rarely. Individuals with recurrences of meningococcal disease, particularly those caused by non-B serogroups, should be assessed for complement deficiency by measurement of total hemolytic complement activity. There is also limited evidence that hyposplenism (through reduction in phagocytic capacity) and hypogammaglobulinemia (through absence of specific antibody) increase the risk of meningococcal disease. Genetic studies have revealed various associations with disease susceptibility, including complement and mannose-binding lectin deficiency, single-nucleotide polymorphisms in Toll-like receptor (TLR) 4 and complement factor H, and variants of Fc gamma receptors.
Factors that increase the chance of a susceptible individual’s acquiring N. meningitidis via the respiratory route also increase the risk of meningococcal disease. Acquisition occurs through close contact with carriers as a result of overcrowding (e.g., in poor socioeconomic settings, in refugee camps, during the Hajj pilgrimage to Mecca, and during freshman-year residence in college dormitories) and certain social behaviors (e.g., attendance at bars and nightclubs, kissing). Secondary cases may occur in close contacts of an index case (e.g., household members and persons kissing the infected individual); the risk to these contacts may be as high as 1000 times the background rate in the population. Factors that damage the nasopharyngeal epithelium also increase the risk of both colonization with N. meningitidis and invasive disease. The most important of these factors are cigarette smoking (odds ratio, 4.1) and passive exposure to cigarette smoke. In addition, recent viral respiratory tract infection, infection with Mycoplasma species, and winter or the dry season have been associated with meningococcal disease; all of these factors presumably either increase the expression of adhesion molecules in the nasopharynx, thus enhancing meningococcal adhesion, or facilitate meningococcal invasion of the bloodstream.
PATHOGENESIS
N. meningitidis has evolved as an effective colonizer of the human nasopharynx, with asymptomatic infection rates of >25% described in some series of adolescents and young adults and among residents of crowded communities. Point-prevalence studies reveal widely divergent rates of carriage for different types of meningococci. This variation suggests that some types may be adapted to a short duration of carriage with frequent transmission to maintain the population, while others may be less efficiently transmitted but may overcome this disadvantage by colonizing for a long period. Despite the high rates of carriage among adolescents and young adults, only ~10% of adults carry meningococci, and colonization is very rare in early childhood. Many of the same factors that increase the risk of meningococcal disease also increase the risk of carriage, including smoking, crowding, and respiratory viral infection. Colonization of the nasopharynx involves a series of interactions of meningococcal adhesins (e.g., Opa proteins and pili) with their ligands on the epithelial mucosa. N. meningitidis produces an IgA1 protease that is likely to reduce interruption of colonization by mucosal IgA.
Colonization should be considered the normal state of meningococcal infection, with an increased risk of invasion being the unfortunate consequence (for both host and organism) of adaptations of hyperinvasive meningococcal lineages. The meningococcal capsule is an important virulence factor: acapsular strains only very rarely cause invasive disease. The capsule provides resistance to phagocytosis and may be important in preventing desiccation during transmission between hosts. Antigenic diversity in surface structures and an ability to vary levels of their expression have probably evolved as important factors in maintaining meningococcal populations within and between individual hosts.
Invasion through the mucosa into the blood occurs rarely, usually within a few days of acquisition of an invasive strain by a susceptible individual. Only occasional cases of prolonged colonization prior to invasion have been documented. Once the organism is in the bloodstream, its growth may be limited if the individual is partially immune, although bacteremia may allow seeding of another site, such as the meninges or the joints. Alternatively, unchecked proliferation may continue, resulting in high bacterial counts in the circulation. During growth, meningococci release blebs of outer membrane (Fig. 180-1) containing outer-membrane proteins and LPS. Endotoxin binds cell-bound CD14 in association with TLR4 to initiate an inflammatory cascade with the release of high levels of various mediators, including tumor necrosis factor (TNF) α, soluble TNF receptor, interleukin (IL) 1, IL-1 receptor antagonist, IL-1β, IL-6, IL-8, IL-10, plasminogen-activator inhibitor 1 (PAI-1), and leukemia inhibitory factor. Soluble CD14-bound endotoxin acts as a mediator of endothelial activation. The severity of meningococcal disease is related both to the levels of endotoxin in the blood and to the magnitude of the inflammatory response. The latter is determined to some extent by polymorphisms in the inflammatory response genes (and their inhibitors), and the release of the inflammatory cascade heralds the development of meningococcal septicemia (meningococcemia). Endothelial injury is central to many clinical features of meningococcemia, including increased vascular permeability, pathologic changes in vascular tone, loss of thromboresistance, intravascular coagulation, and myocardial dysfunction. Endothelial injury leads to increased vascular permeability (attributed to loss of glycosaminoglycans and endothelial proteins), with subsequent gross proteinuria. Leakage of fluid and electrolytes into the tissues from capillaries (“capillary leak syndrome”) leads to hypovolemia, tissue edema, and pulmonary edema. Initial compensation results in vasoconstriction and tachycardia, although cardiac output eventually falls. While resuscitation fluids may restore circulating volume, tissue edema will continue to increase, and, in the lung, the consequence may be respiratory failure.
Intravascular thrombosis (caused by activation of procoagulant pathways in association with upregulation of tissue factor on the endothelium) occurs in some patients with meningococcal disease and results in purpura fulminans and infarction of areas of skin or even of whole limbs. At the same time, multiple anticoagulant pathways are downregulated through loss of endothelial thrombomodulin and protein C receptors and decreases in levels of antithrombin III, protein C, protein S, and tissue factor pathway inhibitor. Thrombolysis is also profoundly impaired in meningococcal sepsis through the release of high levels of PAI-1.
Shock in meningococcal septicemia appears to be attributable to a combination of factors, including hypovolemia, which results from the capillary leak syndrome secondary to endothelial injury, and myocardial depression, which is driven by hypovolemia, hypoxia, metabolic derangements (e.g., hypocalcemia), and cytokines (e.g., IL-6). Decreased perfusion of tissues as a result of intravascular thrombosis, vasoconstriction, tissue edema, and reduced cardiac output in meningococcal septicemia can cause widespread organ dysfunction, including renal impairment and—later in the disease—a decreased level of consciousness due to central nervous system involvement.
Bacteria that reach the meninges cause a local inflammatory response—with release of a spectrum of cytokines similar to that seen in septicemia—that presents clinically as meningitis and is thought to determine the severity of neuronal injury. Local endothelial injury may result in cerebral edema and rapid onset of raised intracranial pressure in some cases.
CLINICAL MANIFESTATIONS
As discussed above, the most common form of infection with N. meningitidis is asymptomatic carriage of the organism in the nasopharynx. Despite the location of infection in the upper airway, meningococcal pharyngitis is rarely reported; however, upper respiratory tract symptoms are common prior to presentation with invasive disease. It is not clear whether these symptoms relate to preceding viral infection (which may promote meningococcal acquisition) or to meningococcal acquisition itself. After acquiring the organism, susceptible individuals develop disease manifestations in 1–10 days (usually <4 days, although colonization for 11 weeks has been documented).
Along the spectrum of presentations of meningococcal disease, the most common clinical syndromes are meningitis and meningococcal septicemia. In fulminant cases, death may occur within hours of the first symptoms. Occult bacteremia is also recognized and, if untreated, progresses in two-thirds of cases to focal infection, including meningitis or septicemia. Meningococcal disease may also present as pneumonia, pyogenic arthritis or osteomyelitis, purulent pericarditis, endophthalmitis, conjunctivitis, primary peritonitis, or (rarely) urethritis. Perhaps because it is difficult to diagnose, pneumococcal pneumonia is not commonly reported but is associated with serogroups Y, W, and Z and appears most often to affect individuals >10 years of age.
Rash A nonblanching rash (petechial or purpuric) develops in >80% of cases of meningococcal disease; however, the rash is often absent early in the illness. Usually initially blanching in nature (macules, maculopapules, or urticaria) and indistinguishable from more common viral rashes, the rash of meningococcal infection becomes petechial or frankly purpuric over the hours after onset. In the most severe cases, large purpuric lesions develop (purpura fulminans). Some patients (including those with overwhelming sepsis) may have no rash. While petechial rash and fever are important signs of meningococcal disease, fewer than 10% of children (and, in some clinical settings, fewer than 1% of patients) with this presentation are found to have meningococcal disease. Most patients presenting with a petechial or purpuric rash have a viral infection (Table 180-2). The skin lesions exhibit widespread endothelial necrosis and occlusion of small vessels in the dermis and subcutaneous tissues, with a neutrophilic infiltrate.
|
COMMON CAUSES OF PETECHIAL OR PURPURIC RASHES |
Meningitis Meningococcal meningitis commonly presents as nonspecific manifestations, including fever, vomiting, and (especially in infants and young children) irritability, and is indistinguishable from other forms of bacterial meningitis unless there is an associated petechial or purpuric rash, which occurs in two-thirds of cases. Headache is rarely reported in early childhood but is more common in later childhood and adulthood. When headache is present, the following features, in association with fever or a history of fever, are suggestive of bacterial meningitis: neck stiffness, photophobia, decreased level of consciousness, seizures or status epilepticus, and focal neurologic signs. Classic signs of meningitis, such as neck stiffness and photophobia, are often absent in infants and young children with bacterial meningitis, who more usually present with fever and irritability and may have a bulging fontanelle.
While 30–50% of patients present with a meningitis syndrome alone, up to 40% of meningitis patients also present with some features of septicemia. Most deaths from meningococcal meningitis alone (i.e., without septicemia) are associated with raised intracranial pressure presenting as a reduced level of consciousness, relative bradycardia and hypertension, focal neurologic signs, abnormal posturing, and signs of brainstem involvement—e.g., unequal, dilated, or poorly reactive pupils; abnormal eye movement; and impaired corneal responses (Chap. 328).
Septicemia Meningococcal septicemia alone accounts for up to 20% of cases of meningococcal disease. The condition may progress from early nonspecific symptoms to death within hours. Mortality rates among children with this syndrome have been high (25–40%), but early aggressive management (as discussed below) may reduce the figure to <10%. Early symptoms are nonspecific and suggest an influenza-like illness with fever, headache, and myalgia accompanied by vomiting and abdominal pain. As discussed above, the rash, if present, may appear to be viral early in the course until petechiae or purpuric lesions develop. Purpura fulminans occurs in severe cases, with multiple large purpuric lesions and signs of peripheral ischemia. Surveys of patients have indicated that limb pain, pallor (including a mottled appearance and cyanosis), and cold hands and feet may be prominent. Shock is manifested by tachycardia, poor peripheral perfusion, tachypnea, and oliguria. Decreased cerebral perfusion leads to confusion, agitation, or decreased level of consciousness. With progressive shock, multiorgan failure ensues; hypotension is a late sign in children, who more commonly present with compensated shock (tachycardia, poor peripheral perfusion, and normal blood pressure). Poor outcome is associated with an absence of meningism, hypotension, young age, coma, relatively low temperature (<38°C), leukopenia, and thrombocytopenia. Spontaneous hemorrhage (pulmonary, gastric, or cerebral) may result from consumption of coagulation factors and thrombocytopenia.
Chronic Meningococcemia Chronic meningococcemia, which is rarely recognized, presents as repeated episodes of petechial rash associated with fever, joint pain, features of arthritis, and splenomegaly that may progress to acute meningococcal septicemia if untreated. During the relapsing course, bacteremia characteristically clears without treatment and then recurs. The differential diagnosis includes bacterial endocarditis, acute rheumatic fever, Henoch-Schönlein purpura, infectious mononucleosis, disseminated gonococcal infection, and immune-mediated vasculitis. This condition has been associated with complement deficiencies in some cases and with inadequate sulfonamide therapy in others.
![]() A study from the Netherlands found that half of isolates from patients with chronic meningococcemia had an underacylated lipid A (part of the surface LPS molecule) due to an lpxL1 gene mutation, which markedly reduces the inflammatory response to endotoxin.
A study from the Netherlands found that half of isolates from patients with chronic meningococcemia had an underacylated lipid A (part of the surface LPS molecule) due to an lpxL1 gene mutation, which markedly reduces the inflammatory response to endotoxin.
Postmeningococcal Reactive Disease In a small proportion of patients, an immune complex disease develops ~4–10 days after the onset of meningococcal disease, with manifestations that include a maculopapular or vasculitic rash (2% of cases), arthritis (up to 8% of cases), iritis (1%), pericarditis, and/or polyserositis associated with fever. The immune complexes involve meningococcal polysaccharide antigen and result in immunoglobulin and complement deposition with an inflammatory infiltrate. These features resolve spontaneously without sequelae. It is important to recognize this condition since a new onset of fever and rash can lead to concerns about relapse of meningococcal disease and unnecessarily prolonged antibiotic treatment.
DIAGNOSIS
Like other invasive bacterial infections, meningococcal disease may produce elevations of the white blood cell (WBC) count and of values for inflammatory markers (e.g., C-reactive protein and procalcitonin levels or the erythrocyte sedimentation rate). Values may be normal or low in rapidly progressive disease, and a lack of rise in these laboratory test values does not exclude the diagnosis. However, in the presence of fever and a petechial rash, these elevations are suggestive of meningococcal disease. In patients with severe meningococcal septicemia, common laboratory findings include hypoglycemia, acidosis, hypokalemia, hypocalcemia, hypomagnesemia, hypophosphatemia, anemia, and coagulopathy.
Although meningococcal disease is often diagnosed on clinical grounds, in suspected meningococcal meningitis or meningococcemia, blood should routinely be sent for culture to confirm the diagnosis and to facilitate public health investigations; blood cultures are positive in up to 75% of cases. Culture media containing sodium polyanethol sulfonate, which may inhibit meningococcal growth, should be avoided. Meningococcal viability is reduced if there is a delay in transport of the specimen to the microbiology laboratory for culture or in plating of cerebrospinal fluid (CSF) samples. In countries where treatment with antibiotics before hospitalization is recommended for meningococcal disease, the majority of clinically suspected cases are culture negative. Real-time polymerase chain reaction (PCR) analysis of whole-blood samples increases the diagnostic yield by >40%, and results obtained with this method may remain positive for several days after administration of antibiotics. Indeed, in the United Kingdom, more than half of clinically suspected cases are currently identified by PCR.
Unless contraindications exist (raised intracranial pressure, uncorrected shock, disordered coagulation, thrombocytopenia, respiratory insufficiency, local infection, ongoing convulsions), lumbar puncture should be undertaken to identify and confirm the etiology of suspected meningococcal meningitis, whose presentation cannot be distinguished from that of meningitis of other bacterial causes. Some authorities have recommended a CT brain scan prior to lumbar puncture because of the risk of cerebral herniation in patients with raised intracranial pressure. However, a normal CT scan is not uncommon in the presence of raised intracranial pressure in meningococcal meningitis, and the decision to perform a lumbar puncture should be made on clinical grounds. CSF features of meningococcal meningitis (elevated protein level and WBC count, decreased glucose level) are indistinguishable from those of other types of bacterial meningitis unless a gram-negative diplococcus is identified. (Gram’s staining is up to 80% sensitive for meningococcal meningitis.) CSF should be submitted for culture (sensitivity, 90%) and (where available) PCR analysis. CSF antigen testing with latex agglutination is insensitive and should be replaced by molecular diagnosis when possible.
Lumbar puncture should generally be avoided in meningococcal septicemia, as positioning for the procedure may critically compromise the patient’s circulation in the context of hypovolemic shock. Delayed lumbar puncture may still be useful when the diagnosis is uncertain, particularly if molecular diagnostic technology is available.
In other types of focal infection, culture and PCR analysis of normally sterile body fluids (e.g., synovial fluid) may aid in the diagnosis. Although some authorities have recommended cultures of scrapings or aspirates from skin lesions, this procedure adds little to the diagnostic yield when compared with a combination of blood culture and PCR analysis. Urinary antigen testing also is insensitive, and serologic testing for meningococcal infection has not been adequately studied. Because N. meningitidis is a component of the normal human nasopharyngeal flora, identification of the organism on throat swabs has no diagnostic value.
|
TREATMENT |
MENINGOCOCCAL INFECTIONS |
Death from meningococcal disease is associated most commonly with hypovolemic shock (meningococcemia) and occasionally with raised intracranial pressure (meningococcal meningitis). Therefore, management should focus on the treatment of these urgent clinical issues in addition to the administration of specific antibiotic therapy. Delayed recognition of meningococcal disease or its associated physiologic derangements, together with inadequate emergency management, is associated with poor outcome. Since the disease is rare, protocols for emergency management have been developed (see www.meningitis.org).
Airway patency may be compromised if the level of consciousness is depressed as a result of shock (impaired cerebral perfusion) or raised intracranial pressure; this situation may require intervention. In meningococcemia, pulmonary edema and pulmonary oligemia (presenting as hypoxia) require oxygen therapy or elective endotracheal intubation. In cases with shock, aggressive fluid resuscitation (with replacement of the circulating volume several times in severe cases) and inotropic support may be necessary to maintain cardiac output. If shock persists after volume resuscitation at 40 mL/kg, the risk of pulmonary edema is high, and elective intubation is recommended to improve oxygenation and decrease the work of breathing. Metabolic derangements, including hypoglycemia, acidosis, hypokalemia, hypocalcemia, hypomagnesemia, hypophosphatemia, anemia, and coagulopathy, should be anticipated and corrected. In the presence of raised intracranial pressure, management includes correction of coexistent shock and neurointensive care to maintain cerebral perfusion.
Empirical antibiotic therapy for suspected meningococcal disease consists of a third-generation cephalosporin such as ceftriaxone (75–100 mg/kg per day [maximum, 4 g/d] in one or two divided IV doses) or cefotaxime (200 mg/kg per day [maximum, 8 g/d] in four divided IV doses) to cover the various other (potentially penicillin-resistant) bacteria that may produce an indistinguishable clinical syndrome. Although unusual in most isolates, reduced meningococcal sensitivity to penicillin (a minimal inhibitory concentration of 0.12–1.0 μg/mL) has been reported widely.
Both meningococcal meningitis and meningococcal septicemia are conventionally treated for 7 days, although courses of 3–5 days may be equally effective. Furthermore, a single dose of ceftriaxone or an oily suspension of chloramphenicol has been used successfully in resource-poor settings. No data are available to guide the duration of treatment for meningococcal infection at other foci (e.g., pneumonia, arthritis); antimicrobial therapy is usually continued until clinical and laboratory evidence of infection has resolved. Cultures usually become sterile within 24 h of initiation of appropriate antibiotic chemotherapy.
The use of glucocorticoids for adjunctive treatment of meningococcal meningitis remains controversial since no relevant studies have had sufficient power to determine true efficacy. One large study in adults did indicate a trend toward benefit, and in clinical practice a decision to use glucocorticoids usually must precede a definite microbiologic diagnosis. Therapeutic doses of glucocorticoids are not recommended in meningococcal septicemia, but many intensivists recommend replacement glucocorticoid doses for patients who have refractory shock in association with impaired adrenal gland responsiveness.
Various other adjunctive therapies for meningococcal disease have been considered, but few have been subjected to clinical trials and none can currently be recommended. An antibody to LPS (HA1A) failed to confer a demonstrable benefit. Recombinant bactericidal/permeability-increasing protein (which is not currently available) was tested in a study that had inadequate power to show an effect on mortality rates; however, there were trends toward lower mortality rates among patients who received a complete infusion, and this group also had fewer amputations, fewer blood-product transfusions, and a significantly improved functional outcome. Given that protein C concentrations are reduced in meningococcal disease, the use of activated protein C has been considered since a survival benefit was demonstrated in adult sepsis trials; however, trials in pediatric sepsis (of particular relevance for meningococcal disease) found no benefit and indicated a potential risk of bleeding complications with use of activated protein C.
The postmeningococcal immune-complex inflammatory syndrome has been treated with nonsteroidal anti-inflammatory agents until spontaneous resolution occurs.
COMPLICATIONS
About 10% of patients with meningococcal disease die despite the availability of antimicrobial therapy and other intensive medical interventions. The most common complication of meningococcal disease (10% of cases) is scarring after necrosis of purpuric skin lesions, for which skin grafting may be necessary. The lower limbs are most often affected; next in frequency are the upper limbs, the trunk, and the face. On average, 13% of the skin surface area is involved. Amputations are necessary in 1–2% of survivors of meningococcal disease because of a loss of tissue viability after peripheral ischemia or compartment syndromes. Unless there is local infection, amputation should usually be delayed to allow the demarcation between viable and nonviable tissue to become apparent. Approximately 5% of patients with meningococcal disease suffer hearing loss, and 7% have neurologic complications. In one study pain was reported by 21% of survivors, and in a recent analysis of serogroup B meningococcal disease (the MOSAIC study) as many as one-quarter of survivors had psychological disorders. In some investigations, the rate of complications is higher for serogroup C disease (mostly associated with the ST11 clone) than for serogroup B disease. In patients with severe hypovolemic shock, renal perfusion may be impaired and prerenal failure is common, but permanent renal replacement therapy is rarely needed.
Several studies suggest adverse psychosocial outcomes after meningococcal disease, with reduced quality of life, lowered self-esteem, and poorer neurologic development, including increased rates of attention deficit/hyperactivity disorder and special educational needs. Other studies have not found evidence of such outcomes.
PROGNOSIS
Several prognostic scoring systems have been developed to identify patients with meningococcal disease who are least likely to survive. Factors associated with a poorer prognosis are shock; young age (infancy), old age, and adolescence; coma; purpura fulminans; disseminated intravascular coagulation; thrombocytopenia; leukopenia; absence of meningitis; metabolic acidosis; low plasma concentrations of antithrombin and proteins S and C; high blood levels of PAI-1; and a low erythrocyte sedimentation rate or C-reactive protein level. The Glasgow Meningococcal Septicaemia Prognostic Score (GMSPS) is probably the best-performing scoring system studied so far and may be clinically useful for severity assessment in meningococcal disease. However, scoring systems do not direct the clinician to specific interventions, and the priority in management should be recognition of compromised airways, breathing, or circulation and direct, urgent intervention. Most patients improve rapidly with appropriate antibiotics and supportive therapy. Fulminant meningococcemia is more likely to result in death or ischemic skin loss than is meningitis; optimal emergency management may reduce mortality rates among the most severely affected patients.
PREVENTION
Since mortality rates in meningococcal disease remain high despite improvements in intensive care management, immunization is the only rational approach to prevention at a population level. Secondary cases are common among household and “kissing” contacts of cases, and secondary prophylaxis with antibiotic therapy is widely recommended for these contacts (see below).
Polysaccharide Vaccines Purified meningococcal capsular polysaccharide has been used for immunization since the 1960s. Meningococcal polysaccharide vaccines are currently formulated as either bivalent (serogroups A and C) or quadrivalent (serogroups A, C, Y, and W), with 50 μg of each polysaccharide per dose. Local reactions (erythema, induration, and tenderness) may occur in up to 40% of vaccinees, but serious adverse events (including febrile convulsions in young children) are very rarely reported. In adults, the vaccines are immunogenic, but immunity appears to be relatively short-lived (with antibody levels above baseline for only 2–10 years), and booster doses do not induce a further rise in antibody concentration. Indeed, a state of immunologic hyporesponsiveness has been widely reported to follow booster doses of plain polysaccharide vaccines. The repeating units of these vaccines cross-link B cell receptors to drive specific memory B cells to become plasma cells and produce antibody. Because meningococcal polysaccharides are T cell–independent antigens, no memory B cells are produced after immunization, and the memory B-cell pool is depleted such that fewer polysaccharide-specific cells are available to respond to a subsequent dose of vaccine (Fig. 180-6). The clinical relevance of hyporesponsiveness is unknown. Plain polysaccharide vaccines generally are not immunogenic in early childhood, possibly because marginal-zone B cells are involved in polysaccharide responses and maturation of the splenic marginal zone is not complete until 18 months to 2 years of age. The efficacy of the meningococcal serogroup C component is >90% in young adults; no efficacy data are available for the serogroup Y and W polysaccharides in this age group.
FIGURE 180-6 A. Polysaccharides from the encapsulated bacteria that cause disease in early childhood stimulate B cells by cross-linking the BCR and driving the production of immunoglobulins. There is no production of memory B cells, and the B-cell pool may be depleted by this process such that subsequent immune responses are decreased. B. The carrier protein from protein-polysaccharide conjugate vaccines is processed by the polysaccharide-specific B cell, and peptides are presented to carrier peptide–specific T cells, with the consequent production of both plasma cells and memory B cells. BCR, B-cell receptor; MHC, major histocompatibility complex; TCR, T-cell receptor. (Reprinted from AJ Pollard et al: Nat Rev Immunol 9:213, 2009.)
![]() Group A meningococcal polysaccharides are exceptional in that they have been found to be effective in preventing disease at all ages. Two doses administered 2–3 months apart to children 3–18 months of age or a single dose administered to older children or adults has a protective efficacy rate of >95%. The vaccine has been widely used in the control of meningococcal disease in the African meningitis belt. The duration of protection appears to be only 3–5 years.
Group A meningococcal polysaccharides are exceptional in that they have been found to be effective in preventing disease at all ages. Two doses administered 2–3 months apart to children 3–18 months of age or a single dose administered to older children or adults has a protective efficacy rate of >95%. The vaccine has been widely used in the control of meningococcal disease in the African meningitis belt. The duration of protection appears to be only 3–5 years.
There is no meningococcal serogroup B plain polysaccharide vaccine because α-2,8-N-acetylneuraminic acid is expressed on the surface of neural cells in the fetus such that the B polysaccharide is perceived as “self” and therefore is not immunogenic in humans.
Conjugate Vaccines The poor immunogenicity of plain polysaccharide vaccines in infancy has been overcome by chemical conjugation of the polysaccharides to a carrier protein (CRM197, tetanus toxoid, or diphtheria toxoid). Conjugates that contain monovalent serogroup C polysaccharide and quadrivalent vaccines with A, C, Y, and W polysaccharides have been developed, as have vaccines including various other antigen combinations (e.g., tetanus conjugates with serogroup C and/or Y polysaccharide with Haemophilus influenzae type b polysaccharide). After immunization, peptides from the carrier protein are conventionally thought to be presented to peptide-specific T cells in association with major histocompatibility complex (MHC) class II molecules (some recent data suggesting that carrier protein peptide may actually be presented in association with an oligosaccharide and MHCII) by polysaccharide-specific B cells; the result is a T cell–dependent immune response that allows production of antibody and generation of an expanded B-cell memory pool. Unlike responses to booster doses of plain polysaccharides, responses to booster doses of conjugate vaccines have the characteristics of memory responses. Indeed, conjugate vaccines overcome the hyporesponsiveness induced by plain polysaccharides by replenishing the memory pool. The reactogenicity of conjugate vaccines is similar to that of plain polysaccharide vaccines.




