[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 53 Testicular Cancer
Testicular tumors are uncommon in the general population but are some of the most common and important malignant tumors in young men. Worldwide, 52,000 new cases and 9,000 deaths were estimated from this disease in 2008.1 The vast majority are primary germ cell tumors (GCTs); the others include lymphomas and sarcomas. The incidence of GCTs has doubled in the past 30 years. Although most patients (70% to 80%) present with early-stage, highly curable disease, the continued rise in incidence of these tumors presents a major challenge. Management is based on histologic type and disease extent. Advances in chemotherapy, imaging, and multidisciplinary care have led to improvements in outcomes over the past three decades. In this chapter, we will discuss the management principles for patients with primary GCTs of the testis, malignant extragonadal GCTs, and other rare testicular tumors.
Etiology and Epidemiology
Testicular cancer accounts for only 1% to 2 % of all cancers in men in most populations across the world.2 Germ cell testicular tumors are the most common solid malignant tumors in men between 20 and 35 years of age, and it was estimated that in the United States in 2010, there would be 8480 new cases and 350 deaths from testicular cancer.3 The cumulative lifetime risk of developing a GCT for a white man in the United States is 0.2%.4 Although the disease is rare, the observed rising incidence is a concern. Between 1973 and 2003 the incidence of GCTs rose by 61% in the United States, with the major rise occurring in seminomas rather than nonseminomas.5
Risk factors for testicular germ cell cancer (TGCC) include a family history of cancer, the presence of an undescended testis or gonadal dysgenesis, subfertility, and testicular microlithiasis. Men with a history of cryptorchidism have an approximately six-fold increased chance of developing testicular cancer.6 Orchiopexy prior to puberty appears to lower the risk of developing a subsequent tumor and may help preserve Leydig cell function and enhance fertility.7,8 Although most testicular cancers in men with a history of maldescent occur on the ipsilateral side, approximately 5% to 20% develop in the contralateral testicle.6 The mechanism by which cryptorchidism increases the risk of developing GCTs is unknown, but the effects of maldescent on the testis (i.e., increased temperature or increased risk of trauma if the testis is in the inguinal region) have been suggested as possible factors. The increase in the incidence of tumors in the contralateral testicle, however, suggests that maldescent and testicular cancer may result from the same prenatal etiologic process. Other genitourinary abnormalities associated with testicular cancers include hydrocele and hypospadias.9
There is considerable geographic and ethnic variation in the incidence of testicular tumors, with the highest incidence being reported from Denmark (8.4 per 100,000 men per year) and Switzerland (6.2 to 8.8 per 100,000 men per year).8 The incidence of testicular cancer is lower in non-Europeans as compared with Europeans. In the United States, white men (6.36 per 100,000 per year) are five to six times more likely to develop testicular cancers than black men (1.30 per 100,000 per year); low incidence rates are seen in other ethnic groups, such as Americans of Chinese and Japanese descent.5 A high incidence of testicular cancer is seen in some nonwhite populations such as the Maoris in New Zealand and Native Americans.10
Prior testicular cancer is a major risk factor for the development of a contralateral malignant tumor. In a large population-based follow-up study of 29,515 patients with unilateral testicular cancer, the cumulative risk (at 15 years after diagnosis of the primary tumor) of developing a contralateral malignant tumor was 1.9%.11
Heritability of Testicular Germ Cell Cancer
Given the strong familial association of TGCC, extended pedigrees with this disease are curiously rare. Most families with TGCC have two affected members, most commonly sibling pairs. The International Testicular Germ Cell Linkage Consortium has carried out two large-scale genome scans, neither of which provided convincing or replicable evidence for susceptibility gene(s).12 Candidate association studies have demonstrated that rare gr/gr deletions of the Y chromosome predispose to TGCC, but such deletions cannot explain more than a fraction of heritable cases of this disease.
More recently, two groups carried out association studies, based on genome-wide, single-nucleotide polymorphisms (SNPs), of patients with TGCC and controls.13,14 Both research groups identified strong evidence for two susceptibility loci for seminoma and nonseminoma; one locus is on chromosome 12p22, within the KITLG gene, encoding the ligand for the receptor tyrosine kinase KIT, and the second locus is on chromosome 5q31.3 near the SPRY4 gene, encoding sprouty 4, an inhibitor of the MAP kinase pathway. A third locus on chromosome 6 was identified by only one of the research groups and is not associated with a known gene. The relative risk was approximately 3 for the KITLG locus and about 1.4 for the other two loci. Both the KITLG gene and the SPTY4 gene are biologically feasible candidates because their products lie in the same signal transduction pathway. Moreover, activating mutations of the KIT gene have been identified in a subset of seminomas, while paradoxically, deletions of the KIT gene predispose to germ cell cancer in the SV/129 mouse strain. Of note, cases in the association studies included patients with and patients without a family history of TGCC, but none of these loci were enriched in the familial cases and neither were they associated with other abnormalities such as an undescended testis.
As mentioned above, the SV/129 mouse strain is particularly susceptible to the development of germ cell cancer, and this system has been exploited by Nadeau and colleagues14a to map several genes predisposing to the disease. Curiously, none of the corresponding loci in humans appear to have any role in TGCC development.
Environmental Factors and Testicular Germ Cell Cancer
Other factors linked to the development of TGCCs include a history of testicular trauma, an increased body mass index, immunosuppression following organ transplantation, and human immunodeficiency virus (HIV) infection.2,15–17 There is no evidence to suggest a causal relationship between testicular trauma and the development of a tumor, and the likely explanation is that testicular trauma leads to examination of the testes. Because of the age distribution of testicular cancer, exposure must occur early in life if there is an environmental contribution to the etiology of these tumors. Prenatal factors linked to the later development of testicular cancers include threatened miscarriage, excessive maternal nausea, and birth by caesarean delivery.2 To explain these associations it has been suggested that exposure of the germinal epithelium in utero to an elevated level of free unbound maternal estrogen could give rise to subsequent cryptorchidism and an elevated risk of developing a testicular tumor; the prenatal estrogen theory remains unproven, however.2
Prevention and Early Detection
The identification of carcinoma in situ (CIS) or testicular intraepithelial neoplasia (TIN) as a precursor of testicular GCTs has raised the possibility that the development of invasive testicular cancer could be prevented by treating CIS. In adults, CIS is found adjacent to GCTs in virtually 100% of cases, and it is thought that, with the exception of spermatocytes seminoma, CIS precedes the development of all invasive tumors.18 The natural history of testicular CIS is unknown, but the Danish experience suggests that all cases of adult CIS will ultimately progress to invasive cancer.18
The diagnosis of testicular CIS can only be made by testicular biopsy. Because the incidence of CIS in the general population is low (at most, 0.7%), screening biopsies are currently not recommended. They should be considered in high-risk patients, however, including those with presumed extragonadal germ cell cancer, intersex individuals, and select patients with contralateral GCT (age <40 years and testicular volumes of <12 mL).19
The management of patients with testicular CIS is controversial. Orchiectomy has been suggested for unilateral disease, and in cases diagnosed after orchiectomy for GCT, three options can be considered and discussed with the patient: orchiectomy, low-dose RT, or surveillance.19 Although both orchiectomy and RT offer definitive treatment for testicular CIS, they both destroy any residual fertility. Surveillance, with careful follow-up of the affected testis, is a reasonable option, especially given the excellent prognosis for metachronous testicular cancers.20
Biologic Characteristics and Molecular Biology
GCTs have a distinctive capacity for totipotential differentiation as demonstrated by the frequent finding of combinations of choriocarcinoma, embryonal carcinoma, and seminoma in a single tumor. They also can retain their ability to differentiate as displayed by the not infrequent identification of mature teratoma in residual post-treatment retroperitoneal masses. Cytogenetic analysis of GCTs has shown that chromosome numbers are more homogenous in seminomas than in nonseminomas.21–23 Triploid and tetraploid chromosomal patterns are common in seminomas, and hyperdiploid to hypertriploid counts are common in nonseminomas. A characteristic chromosome anomaly in GCTs of all histologic types is the presence of an isochromosome of the short arm of chromosome 12.24 This isochromosome, first reported by Atkin and Baker in 1982,21 consists essentially of two chromosome 12 short arms. It is present in more than 80% of cases, and GCTs without a 12p isochromosome have extra copies of 12p segments incorporated into other chromosomes. The 12p isochromosome is also found in testicular CIS.24 Isochromosome copies tend to be more numerous in nonseminomas than in seminomas.
It is not known how these chromosomal changes contribute to the development of the neoplastic phenotype. The frequent finding of the 12p isochromosome in both CIS and invasive tumors, however, indicates that this chromosome plays an important role in the biology of these tumors. The possibility of amplification of normal or modified genes, such as the DDX1 gene on the 12p isochromosome, is currently being investigated.25 Proto-oncogenes present on the short arm of chromosome 12 could be activated by point mutations, deletions, or translocations to become oncogenes, which could then act in a dominant manner.26 The KRAS gene is located on the short arm of chromosome 12, and amplification or enhanced expression of this gene has been reported to occur in testicular tumors and derived cell lines. Other candidate genes being studied include the c-KIT oncogene and its ligand KITGL or SCF, the c-MOS oncogene, and the CCND2 gene. In addition to chromosome 12, 17q is overrepresented in 50% of cases of GCT; this area contains a number of genes of interest, including the GRB7 gene and the plakoglobin gene.22
The study of the genetics and molecular biology of GCTs has enhanced our understanding of these tumors. To date, however, cytogenetics has had little impact in the clinic, with the possible exception of the use of various markers for the identification of non-TGCCs located elsewhere in the body.27–29
Pathology and Pathways of Spread
Pathology
Testicular cancers can arise from intratesticular and paratesticular cells (Table 53-1). The vast majority are of germ cell origin, and three major classification schemes have been in use worldwide30–32 (Table 53-2). The Dixon and Moore classification as modified by Mostofi has been adopted by the World Health Organization (WHO) and is the classification scheme most widely used in North America.33
TABLE 53-1 Histologic Classification of Testicular Neoplasms
TABLE 53-2 Three Classifications of Germ Cell Tumors
| Dixon and Moore, 195330 | WHO Classification33 | Pugh, 197632 |
|---|---|---|
For clinical purposes, GCTs are classified into two major groups: seminomas and nonseminomas (NSGCTs). It should be noted that patients with pure seminoma may have recurrence with pure NSGCT, and vice versa. Approximately 60% of GCTs are pure seminomas, 30% are NSGCTs, and 10% are mixed tumors (both seminomatous and NSGCT elements are present).34 Patients with mixed tumors are clinically considered to have NSGCT, the only exception being tumors with syncytiotrophoblastic cells in cases of seminoma.
Carcinoma in Situ
Intratubular germ cell neoplasia, or CIS, is felt to precede the development of all cases of seminoma and NSGCT in adults (with the exception of spermatocytic seminoma).35 On light microscopy, CIS cells closely resemble seminoma cells and in most cases are found within the seminiferous tubules. Cytologically, there is no difference between the CIS cells that develop into seminomas and those that develop into NSGCTs. In the general population, the incidence of CIS is very low (0.2%), but it is somewhat higher in men with impaired fertility (0.5%) and in those with cryptorchid testes (2% to 4%).18
Seminomas
Seminoma, the most common type of testicular GCT, is most often seen in the fourth decade of life. On gross examination, the tumors are usually well demarcated from the residual testicular tissue and rarely have foci of necrosis or hemorrhage. On microscopic examination, the classical or typical seminoma type is made up of large cells with abundant cytoplasm divided by connective tissue septae into sheets or cords.36 These cells typically have round, hyperchromatic or vesicular nuclei with prominent nucleoli. Frequently, there is a lymphocytic infiltrate, and macrophages, plasma cells, and multinucleated giant cells are often present. Syncytiotrophoblasts are present in 15% to 20% of cases, and their presence does not appear to alter the prognosis.
On immunohistochemical testing, virtually all seminomas express placental leukocyte alkaline phosphatase (PLAP) and do not express low-molecular-weight keratins, blood group antigens, or vimentin. Several histologic variants of seminoma have been identified, including anaplastic seminoma and spermatocytic seminoma. Anaplastic seminoma is diagnosed when there are three or more mitoses seen per high-power field.36 Spermatocytic seminoma is a rare subtype mainly seen in older men and is not associated with CIS or bilateral disease.35 Also, these tumors do not stain for PLAP on immunohistochemical testing and rarely, if ever, metastasize. An atypical variant of seminoma with some features similar to those of NSGCT on immunohistochemical examination has been reported, although its morphologic appearance is similar to that of classical seminoma.37 Usually, there is little or no lymphocytic infiltrate and the tumor cells have less cytoplasm than classical seminoma cells. In terms of prognosis, this atypical variant appears to have the same prognosis as classical seminoma.
Nonseminomatous Germ Cell Tumors
Nonseminomatous tumors make up 40% of TGCCs and occur most commonly in the third decade of life. In the WHO classification system, NSGCTs include embryonal carcinoma, teratoma (mature, immature, or with malignant differentiation), choriocarcinoma, yolk sac tumor, and mixed GCTs. Most tumors are mixed, with two or more cell types present. Although some tumors have a component of seminoma, the association of seminoma within a histologically confirmed NSGCT has no major impact on the clinical outcome.38 Patients with combined tumors present at an age (median, 33 years) intermediate between those with seminoma (median, 36 years) and those with nonseminoma (median, 27 years).38 On gross examination, there is usually a soft irregular mass poorly demarcated from the surrounding testicular tissue, and a considerable amount of necrosis and hemorrhage is often present. Immunohistochemical studies usually demonstrate cytoplasmic expression of low-molecular-weight keratins in embryonal carcinomas, and yolk sac elements, low-molecular-weight keratin, and/or vimentin expression in mature teratomas.
Pathways of Spread
Lymphatic spread is the most common route of metastatic spread. The lymphatic drainage of the testis is directly to the para-aortic lymph nodes. There are differences in the distribution of metastases from left or right testicular tumors. The left testicular vein drains to the left renal vein, and the lymphatic drainage is primarily to the lymph nodes in the para-aortic area, directly below the left renal hilum. On the right side, the testicular vein drains directly to the inferior vena cava below the level of the renal vein, and, therefore, paracaval and interaortocaval nodes are the first ones to be involved in right-sided tumors. Figure 53-1 shows the distribution of retroperitoneal lymph node metastases in NSGCT.39 Contralateral nodal involvement occurs in approximately 15% of cases and is rarely found in the absence of ipsilateral involvement. Supradiaphragmatic spread can occur through the thoracic duct, and although left supraclavicular nodal disease is infrequent at presentation, it is often seen at the time of relapse.
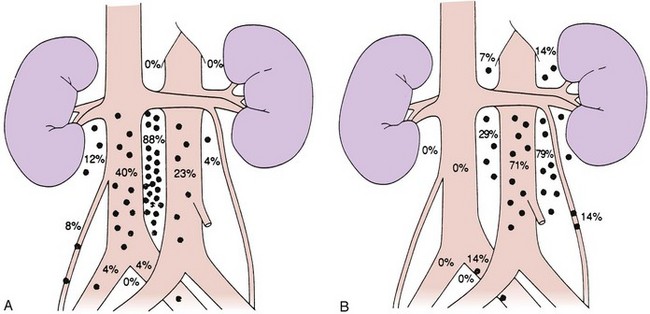
Pelvic and inguinal lymph node involvement is rare (<3%). Factors predisposing to inguinal lymph node involvement include prior scrotal or inguinal surgery, scrotal orchiectomy with incision of the tunica albuginea, tumor invasion of the tunica vaginalis or lower third of the epididymis, and cryptorchid testis.40,41 Disruption of lymphatic vessels in the spermatic cord during inguinal surgery has been shown to induce anastomoses between the testicular lymphatic vessels and the regional lymphatics destined for inguinal or pelvic lymph nodes. In an occasional patient, a connection with the contralateral inguinal lymph nodes may be established, but this is very uncommon. In a small proportion of patients with inguinal relapse, no predisposing factors may be apparent.40
In patients with NSGCTs, hematogenous spread occurs early in the course of the disease. The lung parenchyma is the commonest site of hematogenous spread, but liver, bone, brain, kidney, and gastrointestinal metastases are also seen. In a review of over 5000 patients with metastatic GCT, pulmonary metastases were present in 44% of cases and liver metastases in 6% of cases, with all other areas of hematogenous spread present in 1% or less of cases.42 Mediastinal and neck node involvement was present in 11% to 12% of cases.
Clinical Manifestations, Patient Evaluation, and Staging
Clinical Manifestations
Patients with testicular tumors most commonly present with a painless testicular mass. Up to 45% of patients may have testicular pain, with signs and symptoms suggestive of acute epididymitis in up to 25% of patients. Much less common presenting features include those related to the presence of metastases, for example, back pain, dyspnea, and gynecomastia (from malignant tumors that produce HCG). Up to 3.5% of patients—those with very high HCG levels—may develop hyperthyroidism because the alpha subunit of HCG is identical to that of thyroid-stimulating hormone (TSH) and may therefore stimulate the thyroid.43 The differential diagnosis of a testicular mass (in addition to tumor) includes torsion, hydrocele, varicocele, spermatocele, and epididymitis. A small percentage of tumors are associated with a hydrocele, so the presence of transillumination on examination does not rule out a diagnosis of malignancy.
Patient Evaluation
Routine staging investigations include chest x-ray, CT scanning of the abdomen and pelvis, CT scanning of the thorax, and tumor markers. CT scanning of the thorax does not add value in patients with seminoma with no evidence of retroperitoneal lymph node involvement. There is no role for bipedal lymphography in staging patients; however, magnetic resonance imaging (MRI) with lymphotrophic nanoparticles has shown promise in a number of studies but needs further evaluation before it can be adopted into clinical practice.44 In stage II and III seminomas, especially in patients with bulky retroperitoneal disease, a bone scan should also be performed. Abnormal serum marker levels should be monitored to document postorchiectomy decay according to their respective half-lives. Patients with extensive metastatic disease, nonpulmonary visceral metastases (NPVMs), or very high tumor marker levels are at risk for brain metastases, and CT or MRI of the brain should be performed.45 Baseline pulmonary and renal functions are assessed in patients who require chemotherapy.
Tumor Markers
Measurements of AFP, β-HCG, and LDH are essential in the diagnosis and management of patients with GCTs.46 HCG is a glycoprotein with a molecular weight of 45,000 daltons, composed of two subunits, of which the α-subunit is identical to that of luteinizing hormone (LH), follicle-stimulating hormone (FSH), and thyroid-stimulating hormone (TSH), and a distinct β-subunit. HCG is normally produced by the placenta. About 15% of patients with seminoma have elevated β-HCG levels. Low levels of β-HCG may be found in other neoplasms, including prostate, bladder, and renal tumors. The use of marijuana derivatives also may lead to elevated levels. Some cross-reactivity with LH does occur, and in cases where an elevated HCG level is thought to be due to this cross-reactivity, consideration should be given to having levels remeasured after treatment with testosterone.47 The half-life of β-HCG in blood is approximately 22 hours. In the subset of GCT patients with very high levels of HCG, a plateau of this tumor marker is often reached after the fourth cycle of chemotherapy that is considerably greater than normal. Such patients often remain in remission with slowly falling levels of HCG, possibly resulting from tissue binding of this tumor marker. Such persistent elevation of HCG levels following chemotherapy is not in itself an indication for salvage therapy.48
One or both of these markers are elevated in 85% of patients with nonseminomatous GCTs. Experience with surveillance of patients with early-stage disease has shown that normal markers do not exclude the presence of occult disease.49
LDH is another important marker in patients with GCTs. It is elevated in up to 60% of patients with nonseminomas and also in a high proportion of patients with advanced seminomas.42,50
PLAP is an isoenzyme of alkaline phosphatase and is normally expressed by placental syncytiotrophoblasts. It is also expressed by testicular tissue and has been investigated as a tumor marker in seminoma. Although PLAP levels are often elevated in patients with seminoma, this factor has proven to be of little value in clinical management.51
Staging
The 2009 American Joint Committee on Cancer/International Union Against Cancer (AJCC/UICC) TNM classification (Table 53-3) is the recommended staging system. In stage I disease, one of the most important determinants of outcome is the presence of vascular invasion in the primary tumor, and this differentiates a pT1 tumor from a pT2 tumor.52 Tumors with invasion of the spermatic cord are staged as pT3 cancers, and the rare tumors with scrotal invasion are classified as pT4 lesions.
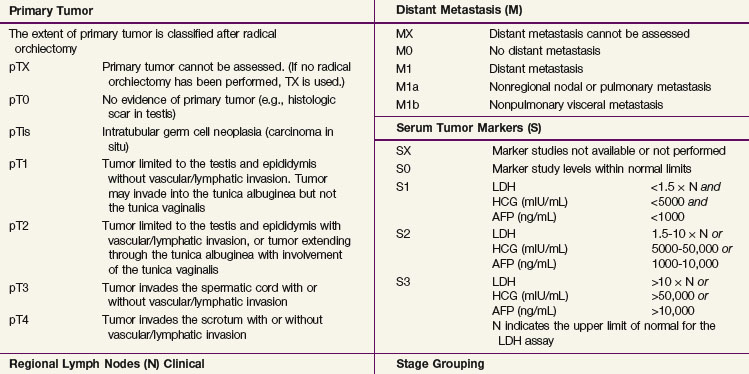
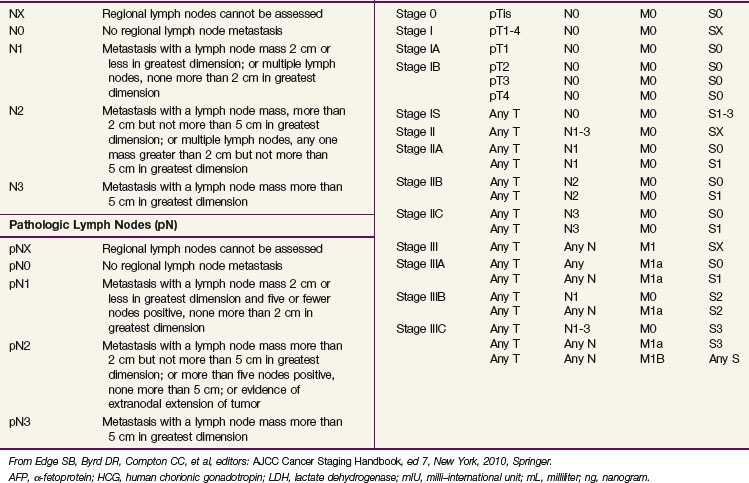
The International Germ Cell Cancer Collaborative Group (IGCCCG) analyzed data from 5168 patients with advanced disease.42 Independent prognostic variables identified by the IGCCCG were included: histologic type (nonseminoma vs. seminoma), site of the primary tumor (testis, retroperitoneal site, or other), presence or absence of NPVMs (brain, bone, or liver), and degree of marker elevation (AFP, β-HCG, and LDH). Based on the results of multivariate analysis, the IGCCCG recommended three prognostic groups for nonseminomatous tumors and two prognostic groups for seminomatous tumors. For nonseminomatous cancers, the 5-year overall survival (OS) for the good-prognosis group was 92%, for the intermediate-prognosis group, 80%, and for the poor-prognosis group, 48%. For seminoma, only two prognostic groups were identified: a good-prognosis group without NPVMs with a 5-year OS of 86% and an intermediate-prognosis group with NPVMs with a 5-year OS of 72%. This classification system has been validated by van Dijk and associates50 using Cox regression analysis and recursive partitioning in a cohort of 3048 NSGCT patients.
Primary Therapy
Surgery
Surgery involves a radical inguinal orchiectomy to allow high division of the spermatic cord. Orchiectomy is both diagnostic and therapeutic by providing adequate tissue to ascertain the diagnosis and offering cure in a high proportion (60% to 90%) of patients with stage I disease.19,53 Although not recommended, an accidental scrotal approach does not appear to compromise the outcome, and no additional therapy is required following scrotal violation, provided that the scrotal cavity has not been grossly contaminated by tumor. In patients with life-threatening metastatic disease and a clear-cut diagnosis of germ cell malignancy, initial management should be with chemotherapy and surgery should be postponed until completion of systemic treatment.19,53 The role of partial orchiectomy in patients with metachronous or synchronous bilateral tumors will be discussed later.
Seminoma
In stage I seminoma, postorchiectomy management options include surveillance, RT, or adjuvant chemotherapy. In the past, the standard management of patients with stage I seminoma has been adjuvant retroperitoneal RT. Although RT provided excellent long-term results, with local control in the abdomen and pelvis in virtually all patients, it is now known that over 85% of patients are cured with orchiectomy alone and that RT has been associated with an increased risk of late gonadal toxicity, development of secondary malignant tumors, and, in some cases, an increased risk of cardiovascular disease.54–58 Various approaches have been tried to reduce radiation morbidity, including a reduced radiation dose and treatment volume. In addition, such alternative management strategies as surveillance (including risk-adapted surveillance) and adjuvant chemotherapy have been investigated.59–63 Although adjuvant retroperitoneal RT constituted the standard of care for the past 50 to 60 years, surveillance is now the standard approach, minimizing the burden of treatment while maintaining the cure rate at 100%.64,65,66
In stage III disease, chemotherapy is the treatment of choice.19,53
Management of Stage I Seminoma
Surveillance
The data on seminoma surveillance are now mature, and relapse rates of 15% to 24% have been reported* (Table 53-4). The two largest reported series are from Copenhagen and Toronto.60,66 In the Copenhagen series of 394 patients, with a median follow-up of 60 months, the crude relapse rate was 17.5%. In the Princess Margaret Hospital, Toronto, series of 421 patients, with a median follow-up of 8.1 years, the actuarial 5-year relapse-free survival (RFS) was 85.5%. The other studies with adequate follow-up (>36 months) have reported similar relapse rates. The predominant site of relapse in all studies was the para-aortic lymph nodes (82% in the Danish Testicular Cancer Study Group (DATECA) study and 89% in the Princess Margaret Hospital series.60,66 The median time to relapse ranged from 12 to 18 months, but occasional late relapses (>4 years) have also been reported.70
Prognostic factors for relapse have been studied in a number of the surveillance studies, and age, tumor size, and small vessel invasion have been reported as predictive of relapse. To more accurately determine prognostic factors for relapse in patients with stage I testicular seminoma managed by surveillance, a pooled analysis of four large surveillance series was performed using individual patient data.71 In 638 patients, tumor size and rete testis invasion predicted for relapse on multivariable analysis. The effect of tumor size on the relapse rate is shown in Figure 53-2, and the hazard ratio for relapse with a tumor size of more than 4 cm was 2 (95% CI, 1.3 to 3.2) relative to baseline (tumor size <4 cm and no rete testis invasion). This model has not been validated in an independent dataset and does not have sufficient discrimination to be clinically useful, because patients in the high-risk group have only a 35% risk of relapse during surveillance. For these reasons, these prognostic factors should not dictate treatment strategy.
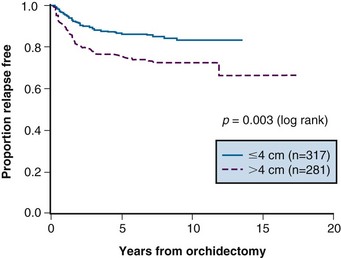
Figure 53-2 Relapse-free rate based on primary tumor size.
From Warde P, Specht L, Horwich A, et al: Prognostic factors for relapse in stage I seminoma managed by surveillance. A pooled analysis. J Clin Oncol 20:4448-4452, 2002. Copyright © American Society of Clinical Oncology.
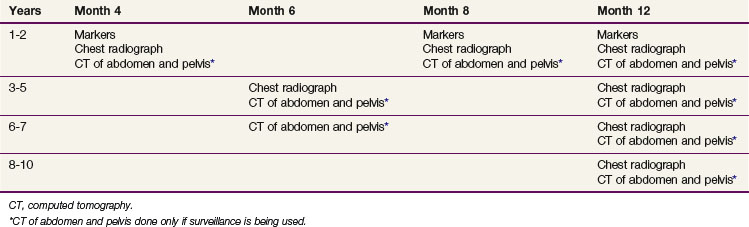
At relapse, most patients have been treated with retroperitoneal RT. The incidence of second relapse after irradiation is approximately 10%. The likelihood of detecting early progression in the retroperitoneal lymph nodes (nodes <5 cm) and, therefore, suitability of the patient for RT depends on the frequency of follow-up CT scans of the abdomen and pelvis. The current Princess Margaret Hospital follow-up policy is shown in Table 53-5. One of the concerns regarding the routine use of surveillance was the potential for an increased need for chemotherapy. In the Toronto experience, a similar percentage of patients managed with surveillance (4.6%) and adjuvant RT (3.9%) required chemotherapy as part of their management. Therefore, surveillance does not lead to an increased use of chemotherapy, with its ensuing toxicity.
Another concern regarding surveillance relates to the increased use of imaging studies. There are no prospective data available that quantitate the risk of radiation-induced cancer from CT scanning. A recent paper used long-term follow-up data from survivors of the Hiroshima and Nagasaki atomic bombings to estimate the risk of low-level exposure approximating that of one or more CT scans. This approach has been criticized for methodologic reasons. Nonetheless, the seminoma surveillance population is likely at some risk of induced cancer, given the young age of many patients and the use of repeated CT scans of the abdomen. It seems reasonable to attempt to decrease the frequency of imaging as much as possible until more data on the risk of such scans are available. In addition, the use of low-dose CT protocols is currently under study, and these may be used in standard surveillance protocols in the future.72
Radiation Therapy
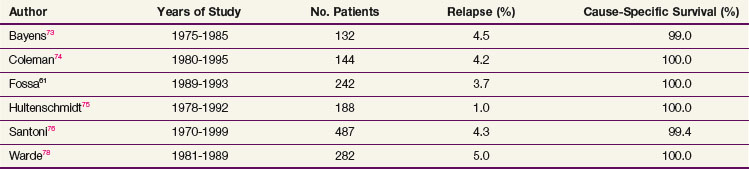
Overall survival (OS) ranges between 92% and 99% at 5 to 10 years, with the cause-specific survival (CSS) approaching 100%. Most deaths are due to intercurrent illness, but concern exists that premature deaths may be occurring from radiation-induced cancers or cardiac disease.54,58 With modern-era RT, the relapse rate has varied from 0.5% to 5%66,73–77 (Table 53-6). In-field relapse is rare, and when it is suspected, biopsy should be performed to rule out a nonseminomatous tumor or another malignant tumor. The commonest sites of relapse following adjuvant RT are the mediastinum, lungs, and left supraclavicular fossa. A small proportion of patients, usually with predisposing factors, develop relapse in the inguinal nodes. Uncommon sites of isolated metastases such as the brain and tonsils have been noted.45,77 For supradiaphragmatic relapse, chemotherapy is the treatment of choice and results in a cure rate of nearly 100%. Inguinal relapse can often be treated successfully with RT to the involved area.78
With such excellent results, the prognostic factors for relapse are difficult to identify. One of the potential adverse factors examined was the presence of anaplastic histologic findings. In the Princess Margaret Hospital experience, 8 of 55 patients with anaplastic seminoma relapsed compared with 2 of 116 patients with classical seminoma (14.5% vs. 1.7%).78 The WHO criteria for the diagnosis of anaplastic seminoma (three or more mitoses per high-power field) are not uniformly used, and other series indicate that the prognosis for patients with anaplastic seminoma is similar to that for patients with classical seminoma.79 Other factors associated with a higher risk of relapse include tumor invasion of the tunica albuginea, lymphovascular invasion, invasion of the epididymis, raised preoperative HCG levels, and spermatic cord involvement.73,78
Various strategies have been investigated to reduce the long-term complications of adjuvant RT in stage I seminoma. They include reducing the radiation dose and/or treatment volume (see the section on Techniques and Dose of Irradiation), deciding to use surveillance for more patients and reserve RT for patients who relapse, and using adjuvant chemotherapy rather than RT.
Adjuvant Chemotherapy
Because chemotherapy is very effective in seminoma, adjuvant carboplatin has been investigated as an alternative strategy to RT or surveillance in stage I disease. The use of one to two courses of carboplatin after orchiectomy was initially pioneered by Oliver,80 who treated 78 patients (53 patients with two courses and 25 patients with one course). With a median follow-up of 44 months, only one patient had relapsed. Subsequent phase II studies have reported relapse rates of between 0% and 8.6%; the variability in results noted may be related to the dosing formula used in the various reports.81
The Medical Research Council in the United Kingdom (MRC UK) has conducted a randomized phase III study comparing adjuvant RT and a single course of carboplatin in 1447 patients.82 The 2008 report showed similar relapse rates at 3 years in both arms of the study (4% RT vs. 5% carboplatin), with most of the relapses in the carboplatin arm occurring in the retroperitoneal lymph nodes.83 One possible benefit of adjuvant carboplatin noted in this study was a reduction in the incidence of second primary testicular germ cell tumors, although this observation has recently been questioned by Powles.63 In addition, this study reported that patients who received higher doses of carboplatin (using the area under the curve [AUC] formula) had lower relapse rates.
Data from other single-institution series indicate that if adjuvant carboplatin is given in this setting, then two courses of treatment are necessary.59,84 Even with two courses of carboplatin, a small but significant percentage of patients develop relapse in the retroperitoneum, and the usefulness of this approach is therefore questionable.59,84 The relapse pattern mandates that continued surveillance of the retroperitoneal lymph nodes is required (similar to the surveillance policy), and the reduction in relapse rates is only 10% (15% with surveillance and 5% with adjuvant chemotherapy). Eighty-five percent of patients receive unnecessary treatment, and the potential long-term toxicity of this strategy is unknown.
Overview of Management of Stage I Seminoma
Almost 100% of patients with stage I testicular seminoma are cured regardless of the choice of postorchiectomy management. The most attractive feature of surveillance is the ability to limit treatment to orchiectomy alone. Surveillance does not compromise the survival rate, and only one disease-related death was observed in the two largest surveillance series.60,66 Surveillance requires a commitment to close and prolonged follow-up from both patients and clinicians. Although isolated occurrences of second no testicular tumors following treatment of seminoma with irradiation have been reported for decades, there is now convincing data from long-term studies indicating that patients with seminoma treated with irradiation have an increased risk of developing cardiac disease and second malignant tumors, and these factors must be considered when deciding on postorchiectomy management.19,54,56–58
Although physicians may view one management approach as preferable, surveillance should be offered to all patients with stage I seminoma and individual patients’ preferences must be considered.85 These preferences may be based on many socioeconomic factors as well as the side effects associated with both approaches. Also, the identification of prognostic factors for occult disease may allow patients and clinicians to choose management based on a more accurate assessment of an individual patient’s risk of relapse.
Management of Stage II Seminoma
The most important prognostic factor in stage II seminoma is the bulk of retroperitoneal disease, usually defined by the transverse diameter of the largest lymph node mass visible on CT scanning. This was the only prognostic factor for relapse in a consecutive series of 95 patients with stage II seminoma treated with RT at the Princess Margaret Hospital between 1981 and 1999.86 The 5-year relapse-free rate in 79 patients with stage IIA or IIB disease was 91%, compared with 44% in 16 patients with stage IIC disease. Relapse occurred most commonly in mediastinal or supraclavicular lymph nodes, lung, or bone. These results are similar to other series in the literature (Table 53-7) and support the continued use of primary RT in stage IIA or IIB patients. In stage IIC disease, chemotherapy is recommended.
The use of carboplatin with RT in stage IIA or IIB seminoma has been suggested by Gilbert.87 In a series of 59 patients treated with one course of carboplatin 4 to 6 weeks prior to RT, the 5-year RFS was 98.3% versus 80.7% for patients treated in the 1980s with RT alone. This approach cannot be accepted as routine practice without further data, because stage migration could explain the better results in the modern cohort of patients.
The technique of irradiation in stage II seminoma is similar to that used in stage I disease. This is discussed in the section on Techniques and Dose of Irradiation.
Residual Retroperitoneal Mass
Following treatment, patients with stage II disease require follow-up imaging of the abdomen until complete regression of disease is observed. A stable persistent mass usually represents fibrosis or necrosis, and only the minority contain an active tumor. The possibility of a nonseminomatous component to explain the residual mass needs to be kept in mind, however, even in patients whose primary tumors show pure seminoma. In addition, surgical extirpation of retroperitoneal nodes in the setting of seminoma is technically challenging and is associated with considerable morbidity.88
Therapeutic options for patients with residual masses after treatment include observation, surgical removal, or, after chemotherapy, irradiation. The role of positron emission tomography (PET) imaging to detect residual tumor is controversial, and the decision to treat the residual mass should not be based exclusively on a positive PET image, because false-positive results occur.89–91
A number of centers have reviewed their experience with surgery for residual masses in the setting of seminoma. The Memorial Sloan-Kettering Cancer Center (MSKCC) group published their data on 55 of 104 patients who demonstrated residual masses following chemotherapy.92 Of these 55 patients, 32 (58%) had a formal RPLND and 23 (42%) had multiple intraoperative biopsies performed because the residual mass was deemed unresectable. Among patients with a mass of more than 3 cm (27 patients), 8 (30%) had residual viable tumor. Interestingly, two of the eight patients had teratoma and six had seminoma. No patients with tumors of less than 3 cm had a viable tumor when the final pathologic findings were obtained. Among the eight patients with preoperative tumor masses of more than 3 cm and positive pathologic findings, six remained relapse-free at 47 months of follow-up. Two patients died of disease; both had poorly defined masses on CT scanning. Given this high proportion of persistent malignant disease, MSKCC investigators have recommended resection or biopsy of masses of 3 cm or larger. In contrast, Culine and Droz from the Centre Léon-Bérard93 have suggested that as long as the retroperitoneal mass continues to decrease in size after treatment, continued observation is a reasonable strategy.
The use of RT in patients with masses following chemotherapy is often mentioned as a therapeutic option. Horwich and colleagues94 published their experience with both observation and RT for these masses and found that the relapse rate was similar whether RT or observation was performed. The MRC Testicular Tumor Working Party published a retrospective pooled analysis assessing the role of RT for postchemotherapy residual masses among men with seminoma.95 In 123 patients with a residual abdominal mass, 56% received consolidative RT. There was no significant difference in outcome among patients who did or did not receive RT. Given these data it was concluded that routine RT is not indicated for a postchemotherapy residual mass.
Management of Stage III Seminoma
This is an uncommon presentation seen in less than 5% of patients. Seminoma is exceptionally chemosensitive, and the use of cisplatin-based regimens results in high cure rates. Patients with stage III seminoma should be classified according to the IGCCCG system into either the good prognostic group or intermediate prognostic group based on the presence or absence of NPVMs.42 For patients in the good-prognosis group, three courses of 5-day BEP chemotherapy (bleomycin, etoposide, cisplatin) should be considered the standard of care.53,96 In patients unsuitable for bleomycin, four courses of etoposide-cisplatin (EP) can be given.53 For patients with intermediate-prognosis disease, four courses of 5-day BEP should be administered.53,96
Nonseminoma
Since the 1970s, when cisplatin, bleomycin, etoposide, and vinblastine were introduced, nonseminoma has become one of the most curable adult cancers. Before these drugs were available, nonseminomatous tumors were treated with a wide range of chemotherapeutic agents and there were occasional reports of long-term remissions with the use of single-agent mithramycin and actinomycin D.97 The modern era in treating advanced disease began at the M.D. Anderson Hospital with the use of continuous infusions of bleomycin and vinblastine; at MSKCC with the vincristine plus actinomycin D plus bleomycin (VAB) regimens; and, later, at Indiana University, with the cisplatin plus vinblastine plus bleomycin (PVB) regimen.98–100 With these regimens, complete responses to treatment became frequent and cures were seen in patients with metastatic disease. The current BEP protocol (bleomycin, etoposide, cisplatin) emerged in the 1990s and is now the standard for first-line chemotherapeutic treatment of metastatic seminoma and nonseminoma.
Management of Stage I Nonseminoma
Current treatment options include surveillance, a modified bilateral retroperitoneal lymph node dissection (RPLND), and adjuvant chemotherapy. The presence of vascular invasion or embryonal carcinoma, the absence of yolk sac elements, and the immunohistochemical proliferation rates, as assessed by the MIB-1 score in the primary tumor, are associated with a high risk of occult nodal disease.101–104 Vascular invasion is the most important factor predicting relapse, and the presence or absence of this factor has been used to divide patients into those with high-risk disease (a third of cases), who have an approximately 50% risk of relapse, and those with low-risk disease, who have an approximately 15% to 20% risk of relapse.52
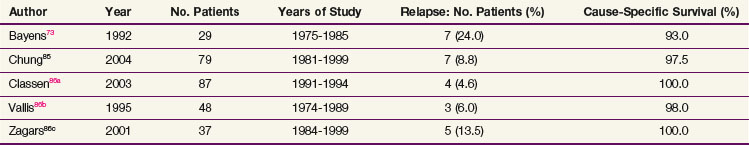
In studies of surveillance in 1768 patients with a follow-up range of 19.5 months to 76 months, the overall relapse rate was 21.4% (378 cases), with 14 deaths (0.79%) from testicular cancer and/or treatment.105 Up to 95% of patients who progress do so within 12 months of diagnosis; progression after 24 months from diagnosis is rare. Approximately 60% of patients progress in the retroperitoneal lymph nodes, with or without other evidence of disease. The other common presentations of disease progression are lung metastases or tumor marker elevation alone. At the time of progression, patients are usually treated with chemotherapy, although RPLND may be successfully used in selected cases with small retroperitoneal lymph nodes. Most patients are cured, and CSS in most series is greater than 98%* (Table 53-8).
The optimal follow-up protocol for nonseminoma surveillance has not been defined. In most centers, clinical examination, tumor marker measurements, chest radiographs, and physical examination are performed every 2 months, with CT scanning of the abdomen and pelvis every 4 months for 2 years and less frequent follow-up thereafter. It has been suggested that the number of CT scans could be reduced to two, at 3 and 12 months after diagnosis, but this approach has not been widely accepted.114 Given the concern that CT scanning may increase the risk of second malignant tumors, this approach should be explored further.
RPLND is a recognized alternative to surveillance in clinical stage I disease. A modified unilateral infrahilar RPLND is performed, with nerve-sparing techniques to preserve ejaculation. For right-sided tumors, the interaortocaval nodes and paracaval nodes are removed, with preservation of the left sympathetic chain, and for left-sided tumors, the para-aortic and interaortocaval nodes are removed and the right autonomic chain is preserved. If significant lymphadenopathy is revealed at surgery, a more extensive surgical resection is performed. The relapse rate following surgery is approximately 10% for patients with pathologic stage I disease.115 Most patients who relapse after RPLND are cured with subsequent chemotherapy.116
In some centers, patients with clinical stage I NSGCT at high risk of relapse on surveillance are offered adjuvant chemotherapy.117 In a recent meta-analysis of one randomized and seven nonrandomized studies of adjuvant chemotherapy, with a total of 873 evaluable patients, 23 relapses (2.6%) were observed.105 To reduce treatment-related toxicity, a number of studies have tested a single cycle of adjuvant BEP. In a recent randomized clinical trial comparing one course of BEP versus RPLND alone, the relapse rate was only 1% (2 of 191 patients) with BEP, and this has sparked renewed interest in this approach.118
The OS rate in stage I nonseminoma approaches 100% in most series irrespective of the treatment strategy used.105 A risk-adapted approach with surveillance for low-risk disease and treatment (either adjuvant chemotherapy or RPLND for patients at a moderate to high risk of occult metastatic disease) has been advocated by some. A surveillance policy, however, is associated with excellent outcomes, and overall, 75% of patients avoid any treatment after orchidectomy.105,119 International consensus statements have concluded that all three approaches should be discussed with the patient.19,53,96
Management of Clinical Stage II Nonseminoma
The cure rate for stage II nonseminoma is approximately 98%. Patients with stage IIA disease with marker elevations (AFP, β-HCG, or LDH) or stage IIB disease regardless of marker status should be treated with chemotherapy according to the algorithms for patients with advanced disease.53,96 Patients with stage IIA disease with no marker have several options. The moderately enlarged nodes may be benign or they may contain teratoma, pure embryonal carcinoma, or mixed tumor elements. If the primary tumor histologic type is pure embryonal carcinoma, either short-term observation to see if the nodes regress or immediate treatment with chemotherapy is a reasonable option. If the primary tumor is of a mixed histologic type or a teratoma, either short-term observation or immediate RPLND is the preferred option. After RPLND, surveillance or adjuvant chemotherapy may be employed. For patients with pathologic stage IIA or IIB disease, the risk of relapse with no adjuvant treatment after surgery is 30% to 50%.96,120 Relapses occur almost exclusively outside the retroperitoneum. Adjuvant chemotherapy with two cycles of BEP in all pathologic stage IIA or IIB patients after RPLND reduces this risk of relapse to 0% to 7%.121 However, adjuvant chemotherapy represents overtreatment in 50% to 70% of pathologic stage IIA or IIB patients, with resulting treatment-related toxicity and possible late sequelae.
Management of Stage III Disease
Chemotherapy with or without Retroperitoneal Lymph Node Dissection
The standard first-line chemotherapy for all patients is bleomycin, etoposide, and cisplatin (BEP) using a 5-day schedule.53,96 Modifications in BEP, such as substitution of cisplatin with carboplatin to reduce toxicity or improve convenience, should be avoided because they may reduce efficacy.53 In patients with IGCCCG good-prognosis disease, three cycles of BEP should be given, and if there is a contraindication to bleomycin, four cycles of etoposide and cisplatin (EP) can be given. This strategy, however, has been associated with a nonstatistically significant but higher death rate in one randomized clinical trial, possibly because of cisplatin toxicity.122
In patients with IGCCCG intermediate- or poor-prognosis disease, four cycles of BEP should be considered the standard therapy.53,96 The VIP regimen (etoposide, ifosfamide, and cisplatin) has been compared with BEP in this patient population and shows similar outcomes but with more toxicity. The VIP regimen represents an alternative to BEP for patients with a contraindication to bleomycin or who develop pulmonary compromise while receiving BEP.123 For intermediate- or poor-prognosis patients, there is no evidence to date that the use of high-dose chemotherapy with autologous stem cell transplantation is superior to the standard four cycles of BEP.124,125 Chemotherapy should be given, preferably without dose reductions, even in the setting of a critically ill patient, at 21-day intervals with no delays between cycles. In patients with life-threatening disease, orchiectomy should not delay the initiation of curative therapy and can be performed at the end of therapy.53 If possible, these patients should be treated in centers with expertise in the management of germ cell tumors.
Residual Retroperitoneal Mass
In many patients who complete chemotherapy and have normalized tumor markers, residual masses are seen on imaging. If technically feasible, these masses should be resected 4 to 6 weeks after chemotherapy. On histologic testing. approximately 50% will show necrosis, 35% mature teratoma, and 15% malignant disease.53 In some centers, RPLND is performed in all cases, even in patients with no residual retroperitoneal disease on imaging. However, there is increasing evidence that observation in patients with complete response after chemotherapy, reserving surgery for those who relapse, gives excellent results and spares many patients unnecessary surgery.126
Treatment Complications
Radiation Therapy Complications
With the low-dose RT used in seminoma, most patients tolerate treatment well.127 Although mild nausea and vomiting occur, only a small proportion of patients require regular antiemetics and are unable to complete daily tasks. Diarrhea develops in a minority of patients. Severe late radiation complications are not expected, unless the patient has an underlying medical problem or a technical error occurs.
Late Gonadal Toxicity
Testicular germinal epithelium is exquisitely sensitive to ionizing radiation. Although the contralateral testis is not located directly in the radiation field, a scatter radiation dose may cause profound depression of spermatogenesis and compromise fertility. In a series of 451 consecutive patients treated for testicular cancer, RT had a significantly greater deleterious effect on fertility than chemotherapy.128 A radiation dose between 20 and 50 cGy may produce temporary aspermia, and doses greater than 50 cGy may preclude recovery of spermatogenesis.129 The use of scrotal shielding reduces the scattered radiation dose to the testis but cannot ensure protection of spermatogenesis in all patients. In men who recover spermatogenesis after RT for seminoma, there is no evidence of an increased incidence of genetic abnormalities among offspring.130 Limiting the target volume to the para-aortic and common iliac area does not eliminate concerns regarding irradiation-induced fertility. In the MRC randomized trial of para-aortic RT alone versus para-aortic and pelvic RT, the median time to a normal post-treatment sperm count was 13 months in patients treated to the para-aortic lymph nodes alone.61 This was significantly better than the time for patients treated to the para-aortic and pelvic lymph nodes (20 months). However, at 3 years of follow-up, there was no significant difference in sperm counts between the two groups. Testicular shielding should be used in all patients who wish to retain fertility after treatment.131
Cardiovascular Toxicity
Since mediastinal RT has been abandoned, the risk of cardiovascular disease in long-term survivors has decreased. Data from the M.D. Anderson Cancer Center and the Royal Marsden Hospital, however, suggest that long-term survivors of testicular seminoma treated with RT following orchiectomy are at significant excess risk of death as a result of cardiac disease.54,58 The etiology of this effect is currently unclear. Although the relevance of these data to the modern practice of RT has been questioned by some, patients should be informed of this increased risk of cardiac disease when deciding whether to undergo RT in the adjuvant setting.132
Second Malignant Tumor
In discussing second malignant tumors after treatment of testicular cancer, a distinction must be made between second germ cell tumors of the testis, which reflect a common risk factor for this disease, and unrelated malignant tumors, which may be treatment induced. An increased risk of second cancers has been documented in a number of studies, and because this increased risk is expressed more than 10 to 15 years following RT, it is often not apparent in series with shorter follow-up times.133 The largest study of second cancers in long-term survivors of testicular cancer was conducted by Travis and co-workers56 at the National Cancer Institute. This report combined 14 population-based registries including 10,534 patients with seminoma treated with RT. The overall relative risk of a second nontesticular malignant tumor was 2 (95% CI, 1.8 to 2.2). For a 35-year-old patient with seminoma, the cumulative 40-year risk of a second malignant tumor was 36%, compared with 23% in the normal population. The increased risk of second malignant tumors after treatment with RT was also shown in a Dutch population-based study of more than 2700 long-term survivors where the risk of a second malignant tumor with subdiaphragmatic RT was 2.6-fold increased as compared with surgery alone.57 The increased risk associated with irradiation was similar to the increased cancer risk seen with smoking. Limitation of the RT field to the infradiaphragmatic region results in a lower risk of second malignant tumors than more extensive fields.
Psychological Toxicity
Testicular cancer survivors have a higher level of anxiety and a higher prevalence of anxiety disorders than the normal population, and screening using a simple tool such as the Hospital Anxiety and Depression Scale (HADS) should be part of routine follow-up care.127,134 Appropriate social support should be offered to patients with testicular tumors. Although highly curable, testicular cancer is still a life-threatening condition in young patients with a concomitant threat of infertility and impaired sexual function as a result of the disease or its treatment.135
Chemotherapy Complications
The complications of chemotherapy in testicular GCTs are related to the drugs employed.127 Nausea and vomiting occur with all the drugs used but are controlled effectively in most patients with 5-hydroxytryptamine receptor-3 (5-HT3) antagonists and steroids. Myelosuppression is frequent. Febrile neutropenia is seen in about 10% to 15% of patients receiving EP, and such patients may benefit from hematopoietic growth factors. Nephrotoxicity, primarily a reduction in the glomerular filtration rate, occurs with the use of cisplatin and ifosfamide. Renal tubule function is normal in most patients, but hypomagnesemia is seen in patients after repeated use of cisplatin. Raynaud phenomenon has been reported in 23% to 49% of patients, although resolution of symptoms is common.136 Chronic peripheral neuropathy (often asymptomatic) is well recognized after treatment with cisplatin, vinblastine, and etoposide. This symptom may require up to 12 to 18 months to resolve or may be permanent. Cisplatin-induced hearing loss is common, particularly at higher frequencies. Many patients report a loss of discrimination (cocktail party effect); tinnitus is another common complaint. Risk factors for hearing loss include a family history of deafness and exposure to other ototoxic drugs. Pulmonary toxicity is seen with the use of bleomycin and can be fatal in a small number of cases.137 Impaired fertility, which may precede the use of chemotherapy, may also continue after its use. Although chemotherapy also produces infertility, recovery of sperm counts at least to levels compatible with fertility has been observed in the majority of patients.138 Approximately 75% of patients who attempt paternity after chemotherapy are successful.139
Non–Germ Cell Testicular Tumors
Leydig Cell Tumors
Leydig cell, or interstitial cell, tumors account for less than 2% of testicular tumors.140 Twenty-five percent occur in children and can present with signs of prepubertal virilization. Seventy-five percent are in patients presenting between the age of 20 and 50 years, usually with a painless testicular mass. Most Leydig cell tumors are benign, and there are no definite histologic criteria for malignancy.140 The regional lymph nodes are the commonest site of metastatic disease, but lung, bone, and liver metastases have also been reported. The initial management is similar to that for GCTs, with a radical inguinal orchiectomy followed by staging assessment with a chest radiograph and CT scanning of the abdomen and pelvis.141 Testis-sparing surgery can be considered in experienced hands. Urinary and serum steroids should also be assessed. RPLND can be considered in selected cases. There is no role for adjunctive or definitive irradiation or chemotherapy.
Sertoli Cell Tumors
Sertoli cell tumors account for less than 1% of all testicular tumors.140 They have been classified into three subtypes: classic, large cell calcifying, and sclerosing.142 Most present with a painless testicular mass, and initial management is with a radical inguinal orchiectomy. Both precocious puberty and feminization have been reported to be associated with Sertoli cell tumors in children. Most Sertoli cell tumors are benign, and orchiectomy is nearly always curative.
Extragonadal Germ Cell Tumors
Extragonadal germ cell tumors (EGCTs) have histologic findings similar to those of testicular GCTs but are found in other parts of the body, in the absence of a testicular mass. They account for 1% to 5% of all GCTs and like testicular GCTs tend to occur in young men, although the median age of presentation is 5 to 10 years older than with testicular GCTs.143 Ten percent of adult EGCTs occur in women, usually as ovarian dysgerminomas, and these will be discussed elsewhere (see Ovarian Cancer, Chapter 59). In infants, EGCTs are more common than testicular primary tumors (usually, sacrococcygeal teratomas).143
Overall, patients with EGCTs (especially NSGCTs) have a worse prognosis than patients with testicular primary tumors.144 An increased incidence of EGCTs is seen in Klinefelter syndrome. A number of patients present with poorly differentiated carcinomas (predominantly in midline locations), with a cytogenetic pattern similar to those with typical EGCTs, and do respond to cisplatin-based chemotherapy regimens.145
The results of an international analysis of 635 consecutive patients with EGCT treated in 11 European and U.S. centers have shown that patients with pure seminomatous histologic typing had a long-term cure rate of 89%, irrespective of the primary site (retroperitoneum or mediastinum).144 In patients with nonseminomatous tumors, however, a mediastinal primary site was an adverse feature, with only 45% of these patients alive at 5 years as compared with 63% of patients with retroperitoneal presentations.144 Prognostic variables for response and outcome have been identified, and four prognostic risk groups have been identified based on histologic findings, the presence of liver, lung or CNS metastases, elevation of β-HCG levels, and number of metastatic sites involved with disease.146 The best prognostic group has an 89% 5-year survival rate and encompasses all patients with seminoma. The other groups all have nonseminomatous histologic findings and 5-year survival rates of 69%, 55%, and 17%, respectively. The excellent treatment results with chemotherapy alone of mediastinal seminomatous tumors would suggest that there is no routine role for RT in their management. Platinum-based chemotherapy regimens are recommended. Although a 5-year survival rate of 73% has been reported with the use of cisplatin, vincristine, methotrexate, bleomycin, actinomycin D, cyclophosphamide, and etoposide (POMB/ACE), there is no evidence that this regimen is superior to BEP.147 In contrast, POMB/ACE is considerably more toxic, and its routine use cannot be recommended.
Special Treatment Situations
Germ Cell Tumors in Patients with Horseshoe Kidney
Horseshoe kidney (pelvic kidney) occurs in approximately 1 in 400 persons in the general population.148 There are two main problems in the management of GCTs associated with horseshoe kidney. The first is related to the technical problem of delivery of RT in patients with seminoma. In a number of cases of horseshoe kidney, a large part of the renal parenchyma directly overlies the regional lymph nodes and lies within the standard radiation volume. The delivery of a standard radiation dose would be associated with an unacceptable risk of radiation nephritis. The second problem is related to the possible abnormalities in lymphatic drainage of the testis and, therefore, the possibility of relapse when the standard radiation fields are used. Unusual patterns of relapse have been observed in patients managed by surveillance, confirming concerns regarding abnormal lymphatic pathways.149 For these reasons, in patients with seminoma and NSGCTs, postorchiectomy surveillance in stage I and chemotherapy in stage II have usually been recommended. However, an RPLND is another option for patients unwilling to follow the surveillance program. Reports in the literature suggest that RPLND was both safe and effective in the selected cases where it was performed.149
Testicular Tumors Developing in Immunosuppressed Patients
It is well established that the risks of developing Kaposi’s sarcoma and non-Hodgkin lymphoma are markedly increased in patients with human immunodeficiency virus (HIV) infection.150 Initial data suggested that HIV-infected men have an increased incidence of testicular cancer, but more recent studies have indicated that this may not be true.17,150 Organ transplant patients may also have an increased incidence of testicular tumors.151 The clinical course of immunosuppressed patients with testicular tumors is similar to that of nonimmunosuppressed patients, and these patients should be offered standard oncologic therapy. Both chemotherapy and RT reduce the CD4 count even when antiretroviral therapy is used; therefore, wherever possible, surveillance should be used in patients with stage I disease.17 Caution must be exercised because benign retroperitoneal adenopathy related to acquired immunodeficiency syndrome (AIDS) may be mistaken for metastasis from a testicular primary tumor. For those receiving chemotherapy, consideration should be given to concomitant prophylaxis for opportunistic infection. Most patients can be cured of their tumors, and the outcome of patients with HIV-related GCTs is similar to that of non-HIV patients.
Bilateral Testicular Germ Cell Tumors or Tumors Arising in a Single Testicle
Patients with cancer in one testicle are significantly more likely to develop a testicular tumor in the contralateral testicle, with rates of metachronous or synchronous development of second testis cancers of 1% to 5% reported.152 Bilateral orchiectomy is recommended as standard management, with resulting infertility, lifelong dependence on androgen substitution, and psychological morbidity due to castration at a young age.153
To preserve endogenous hormonal function, organ-sparing surgery followed by low-dose RT (16 to 20 Gy in 2-Gy daily fractions using a direct field with electrons) to the remaining testis to eradicate residual CIS is a viable therapeutic option with a high likelihood of cure in a highly select group of patients.19,154 Adjuvant irradiation after surgery may be postponed if fertility is an issue provided that follow-up is rigorous.154 Patients with large tumors (>30% of the testis volume) or clinical evidence of metastatic disease should not be considered for this approach.154 The recent European Consensus on the diagnosis and treatment of testis cancer has emphasized the need for organ-sparing approaches to be offered only in centers with experience in this area.19
Central Nervous System Metastases
Approximately 2% to 3 % of patients with metastatic GCT will present with brain metastases, and up to 40% of patients who die of progressive disease will have brain metastases at autopsy. Patients who present at the time of diagnosis with brain metastases can achieve approximately 50% 5-year CSS with aggressive treatment.53 The optimal sequence of chemotherapy, RT, and surgery has not yet been fully established. Systemic chemotherapy is required in all cases, and in one study the addition of cranial RT improved OS.155 It is unclear, however, if RT is necessary if a complete response is obtained with chemotherapy. Surgery can also be considered in resectable disease. If RT is given, a dose of 40 to 45 Gy should be given to gross disease, preferably using a stereotactic approach. The role of total brain irradiation is unclear, but if this treatment is given, the dose should not exceed 40 Gy. Patients presenting with late relapse with central nervous system (CNS) metastases should be treated aggressively because long-term disease-free survival times are possible.156 Patients who develop brain metastases during chemotherapy have a very poor prognosis.156
Spermatocytic Seminoma
Spermatocytic seminoma is a rare testicular tumor accounting for only 1% to 2% of all seminomas.157 It usually occurs at age 50 to 60 years but is seen in younger patients.157 It is distinct in its histologic characteristics, usually having three different cell sizes, spherical nuclei, a lack of cytoplasmic glycogen, and sparse or absent lymphocytic infiltrate when compared with classical seminoma.158 Unlike classical seminoma, it does not appear to arise from CIS and it occurs solely in the testis and has no ovarian equivalent. All patients present with stage I disease, and subsequent metastatic disease is extremely rare. In the past, the treatment approach was similar to seminoma with adjuvant RT. Current recommendations call for surveillance in all patients.159
Irradiation Techniques and Dose
Stage I Seminoma
Reduction in Radiation Treatment Volume
The low incidence of pelvic lymph node involvement in stage I seminoma and a desire to reduce radiation-induced morbidity led to the investigation of RT directed to the para-aortic lymph nodes alone. The advantages of such an approach include decreased scatter to the contralateral testicle and a reduction in the integral irradiation dose with a presumed decrease in the risk of second malignant tumors. Reports from phase II trials and institutional experiences have shown excellent results with few pelvic failures.79,86a In the MRC Testicular Study Group randomized study of 478 patients comparing para-aortic plus pelvic RT and para-aortic RT alone, patients treated with para-aortic RT alone had a 4% relapse rate as compared with a 3.4% relapse rate with para-aortic and pelvic irradiation.155 All patients who received para-aortic and pelvic RT relapsed in supradiaphragmatic sites, but four patients (1.6%) treated to the para-aortic lymph nodes alone failed in the pelvis. This trial demonstrated that a small risk of pelvic failure is present if para-aortic RT alone is given. It highlighted that regular imaging with CT of the pelvic lymph nodes must be performed to detect pelvic relapse. If no routine evaluation of the pelvis is carried out after para-aortic RT alone, patients fail with bulky disease (median, 5 cm). Recent data from the German Testicular Cancer Study Group indicate that follow-up imaging may be confined to the first 3 years after irradiation.160 The advantages of para-aortic RT alone are not clear, when surveillance is an excellent option.
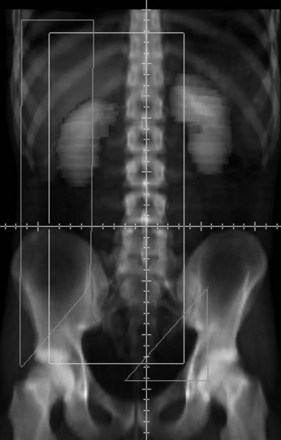
A compromise between traditional irradiation fields and para-aortic irradiation alone is to treat the para-aortic and ipsilateral common iliac lymph nodes by positioning the inferior border of the radiation fields at midpelvis161 (Fig. 53-3). This encompasses the lymph nodes that are typically removed at lymphadenectomy in patients with nonseminomatous tumors and also covers the vast majority of pelvic nodal relapses in patients treated with para-aortic irradiation alone.86a
Radiation Dose
The minimum dose of radiation required to control occult seminomatous tumor has not been clearly defined; however, there are ample data to suggest that a dose of 20 to 25 Gy is sufficient. At Princess Margaret Hospital in Toronto, a dose of 25 Gy prescribed at midline and delivered in 20 daily fractions of 1.25 Gy has been used for over 25 years and no in-field recurrences have been observed.66 In the MRC TE18 trial, 625 patients were randomized to 30 Gy in 15 fractions over 3 weeks and 20 Gy in 10 fractions over 2 weeks.62 The median follow-up was 61 months. A total of 10 and 11 relapses, respectively, occurred in the 30-Gy and 20-Gy groups (hazard ratio [HR], 1.11; 90% CI, 0.54 to 2.28). The 5-year relapse rates were 3% and 3.6%, respectively, for the 30-Gy and 20-Gy groups. This trial was updated at the 2008 American Society of Clinical Oncology (ASCO) meeting, and the original findings were confirmed with longer follow-up.83
Stage II Seminoma
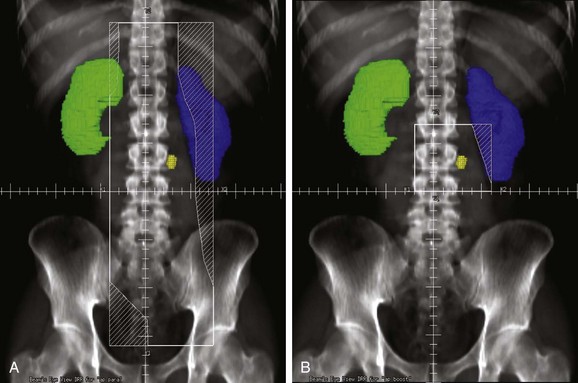
The RT technique in stage II seminoma is similar to that used in stage I disease. The penis is always positioned out of the field, and the contralateral testis is placed in a scrotal shield. The treatment volume includes the gross tumor as well as the para-aortic and ipsilateral common and external iliac lymph nodes (Fig. 53-4A). The radiation dose is typically 25 Gy in 20 daily fractions plus a boost of a further 10 Gy to the gross lymphadenopathy (see Fig. 53-4B). At Princess Margaret Hospital, this boost is given concurrently with the large-field treatment. The contralateral iliac lymph nodes may also be treated in cases where lymphadenopathy in the low para-aortic area is deemed to increase the risk of these nodes being involved by tumor. This is probably of most concern, however, in patients with bulky retroperitoneal lymphadenopathy who are better treated with primary chemotherapy as discussed previously. Adjuvant RT of the supraclavicular lymph nodes in patients with stage II disease has been suggested by some, although it is not justified on a routine basis in view of the low risk of isolated supraclavicular relapse (2 of 79 patients with stage IIA or IIB disease in the Princess Margaret Hospital series).86c,162
Treatment Algorithms, Conclusions, and Future Possibilities
The recommended treatment algorithms for seminomas and nonseminomas based on disease extent are outlined in Figures 53-5 and 53-6. Although the overall results are excellent, there are ongoing controversies regarding the optimal management of GCTs.
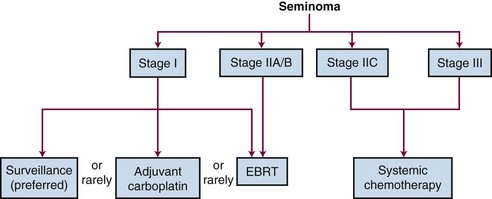
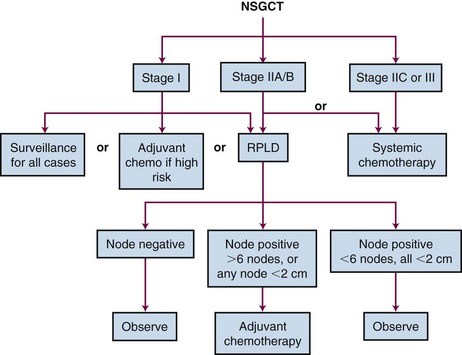
In seminoma, the major area of controversy involves the management of stage I disease. The mature data from surveillance and adjuvant RT series suggest that almost 100% of patients with stage I testicular seminoma are cured no matter which approach is chosen as postorchiectomy management. Surveillance should be considered the management option of choice. Change is adopted slowly, however, and it is disturbing to note that a considerable proportion of urologists and radiation oncologists do not discuss the option of surveillance with their patients.85
Acknowledgments
The authors would like to thank Mrs. Eleni Sachinidis for her help in chapter preparation.
2 Manecksha RP, Fitzpatrick JM. Epidemiology of testicular cancer. BJU Int. 2009;104(9 Pt B):1329-1333.
3 Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2010;60:277-300.
19 Krege S, Beyer J, Souchon R, et al. European consensus conference on diagnosis and treatment of germ cell cancer. A report of the second meeting of the European Germ Cell Cancer Consensus Group (EGCCCG). Part I. Eur Urol. 2008;53(3):478-496.
42 International Germ Cell Consensus Classification. A prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15(2):594-603.
44 Sohaib SA, Koh DM, Husband JE. The role of imaging in the diagnosis, staging, and management of testicular cancer. AJR Am J Roentgenol. 2008;191(2):387-395.
52 Read G, Stenning SP, Cullen MH, et al. Medical Research Council prospective study of surveillance for stage I testicular teratoma. Medical Research Council Testicular Tumors Working Party. J Clin Oncol. 1992;10(11):1762-1768.
53 Krege S, Beyer J, Souchon R, et al. European consensus conference on diagnosis and treatment of germ cell cancer. A report of the second meeting of the European Germ Cell Cancer Consensus Group (EGCCCG). Part II. Eur Urol. 2008;53(3):497-513.
54 Huddart RA, Norman A, Shahidi M, et al. Cardiovascular disease as a long-term complication of treatment for testicular cancer. J Clin Oncol. 2003;21(8):1513-1523.
55 Robinson D, Moller H, Horwich A. Mortality and incidence of second cancers following treatment for testicular cancer. Br J Cancer. 2007;96(3):529-533.
56 Travis LB, Fossa SD, Schonfeld SJ, et al. Second cancers among 40,576 testicular cancer patients. Focus on long-term survivors. J Natl Cancer Inst. 2005;97(18):1354-1365.
57 van den Belt-Dusebout AW, de Wit R, Gietema JA, et al. Treatment-specific risks of second malignancies and cardiovascular disease in 5-year survivors of testicular cancer. J Clin Oncol. 2007;25(28):4370-4378.
58 Zagars GK, Ballo MT, Lee AK, Strom SS. Mortality after cure of testicular seminoma. J Clin Oncol. 2004;22(4):640-647.
59 Aparicio J, Germa JR, Garcia Del Muro XG, et al. Risk-adapted management for patients with clinical stage I seminoma. The Second Spanish Germ Cell Cancer Cooperative Group Study. J Clin Oncol. 2005;23:8717-8723.
60 Daugaard G, Petersen PM, Rorth M. Surveillance in stage I testicular cancer. APMIS. 2003;111(1):76-83. discussion APMIS 111(1):83-75, 2003
61 Fossa SD, Horwich A, Russell JM, et al. Optimal planning target volume for stage I testicular seminoma. A Medical Research Council randomized trial. Medical Research Council Testicular Tumor Working Group [see comment]. J Clin Oncol. 1999;17(4):1146.
62 Jones WG, Fossa SD, Mead GM, et al. Randomized trial of 30 versus 20 Gy in the adjuvant treatment of stage I testicular seminoma. A report on Medical Research Council Trial TE18, European Organisation for the Research and Treatment of Cancer Trial 30942 (ISRCTN18525328). J Clin Oncol. 2005;23(6):1200-1208.
63 Powles T, Robinson D, Shamash J, et al. The long-term risks of adjuvant carboplatin treatment for stage I seminoma of the testis. Ann Oncol. 2008;19(3):443-447.
66 Warde PR, Chung P, Sturgeon J, et al. Should surveillance be considered the standard of care in stage I seminoma? J Clin Oncol (Meeting Abstracts). 2005;23(16_suppl):4520.
71 Warde P, Specht L, Horwich A, et al. Prognostic factors for relapse in stage I seminoma managed by surveillance. A pooled analysis. J Clin Oncol. 2002;20(22):4448-4452.
72 O’Malley ME, Chung P, Haider M, et al. Comparison of low dose with standard dose abdominal/pelvic multidetector CT in patients with stage 1 testicular cancer under surveillance. Eur Radiol. 2010;20(7):1624-1630.
79 Logue JP, Harris MA, Livsey JE, et al. Short course para-aortic radiation for stage I seminoma of the testis. Int J Radiat Oncol Biol Phys. 2003;57(5):1304-1309.
83 Mead GM, Fossa SD, Oliver RT, et al. Randomized trials in 2466 patients with stage I seminoma: patterns of relapse and follow-up. J Natl Cancer Inst. 2011;103(3):241-249.
84 Steiner H, Holtl L, Wirtenberger W, et al. Long-term experience with carboplatin monotherapy for clinical stage I seminoma. A retrospective single-center study. Urology. 2002;60(2):324-328.
85 Choo R, Sandler H, Warde P, et al. Survey of radiation oncologists. Practice patterns of the management of stage I seminoma of testis in Canada and a selected group in the United States. Can J Urol. 2002;9(2):1479-1485.
86c Zagars GK, Pollack A. Radiotherapy for stage II testicular seminoma. Int J Radiat Oncol Biol Phys. 2001;51(3):643-649.
87 Gilbert DC, Vanas NJ, Beesley S, et al. Treating IIA/B seminoma with combination carboplatin and radiotherapy. J Clin Oncol. 2009;27(12):2101-2102. author reply J Clin Oncol 27(12):2102-2103, 2009
88 Mosharafa AA, Foster RS, Leibovich BC, et al. Is post-chemotherapy resection of seminomatous elements associated with higher acute morbidity? J Urol. 2003;169(6):2126-2128.
89 Becherer A, De Santis M, Karanikas G, et al. FDG PET is superior to CT in the prediction of viable tumour in post-chemotherapy seminoma residuals. Eur J Radiol. 2005;54(2):284-288.
90 Ganjoo KN, Chan RJ, Sharma M, Einhorn LH. Positron emission tomography scans in the evaluation of postchemotherapy residual masses in patients with seminoma. J Clin Oncol. 1999;17(11):3457-3460.
91 Hinz S, Schrader M, Kempkensteffen C, et al. The role of positron emission tomography in the evaluation of residual masses after chemotherapy for advanced stage seminoma. J Urol. 2008;179(3):936-940. discussion J Urol 179(3):940, 2008
92 Herr HW, Sheinfeld J, Puc HS, et al. Surgery for a post-chemotherapy residual mass in seminoma. J Urol. 1997;157(3):860-862.
93 Culine S, Droz JP. Optimal management of residual mass after chemotherapy in advanced seminoma. There is time for everything. J Clin Oncol. 1996;14(10):2884-2885.
94 Horwich A, Paluchowska B, Norman A, et al. Residual mass following chemotherapy of seminoma. Ann Oncol. 1997;8(1):37-40.
96 Wood L, Kollmannsberger C, Jewett M, et al. Canadian consensus guidelines for the management of testicular germ cell cancer. Can Urol Assoc J. 2010;4(2):e19-38.
97 Kennedy BJ. Mithramycin therapy in advanced testicular neoplasms. Cancer. 1970;26(4):755-766.
116 Stephenson AJ, Klein EA. Surgical management of low-stage nonseminomatous germ cell testicular cancer. BJU Int. 2009;104(9 Pt B):1362-1368.
120 Pizzocaro G, Zanoni F, Milani A, et al. Retroperitoneal lymphadenectomy and aggressive chemotherapy in nonbulky clinical Stage II nonseminomatous germinal testis tumors. Cancer. 1984;53(6):1363-1368.
128 Huyghe E, Matsuda T, Daudin M, et al. Fertility after testicular cancer treatments. Results of a large multicenter study. Cancer. 2004;100(4):732-737.
129 Fossa SD, Abyholm T, Normann N, Jetne V. Post-treatment fertility in patients with testicular cancer. III. Influence of radiotherapy in seminoma patients. Br J Urol. 1986;58(3):315-319.
132 Horwich A. Radiotherapy in stage I seminoma of the testis. J Clin Oncol. 2004;22(4):585-588.
146 Hartmann JT, Nichols CR, Droz JP, et al. Prognostic variables for response and outcome in patients with extragonadal germ-cell tumors. Ann Oncol. 2002;13(7):1017-1028.
152 Hentrich M, Weber N, Bergsdorf T, et al. Management and outcome of bilateral testicular germ cell tumors. Twenty-five year experience in Munich. Acta Oncol. 2005;44(6):529-536.
153 Heidenreich A, Weissbach L, Holtl W, et al. Organ sparing surgery for malignant germ cell tumor of the testis. J Urol. 2001;166(6):2161-2165.
154 Giannarini G, Dieckmann KP, Albers P, et al. Organ-sparing surgery for adult testicular tumours. A systematic review of the literature. Eur Urol. 2010;57:780-790.
156 Bokemeyer C, Nowak P, Haupt A, et al. Treatment of brain metastases in patients with testicular cancer. J Clin Oncol. 1997;15(4):1449-1454.
158 Aggarwal N, Parwani AV. Spermatocytic seminoma. Arch Pathol Lab Med. 2009;133(12):1985-1988.
160 Classen J, Souchon R, Hehr T, et al. Posttreatment surveillance after paraaortic radiotherapy for stage I seminoma. A systematic analysis. J Cancer Res Clin Oncol. 2009;136(2):227-232.
161 Thomas GM. Is “optimal” radiation for stage I seminoma yet defined? J Clin Oncol. 1999;17(9):3004-3005.
162 Chung PW, Warde PR, Panzarella T, et al. Appropriate radiation volume for stage IIA/B testicular seminoma. Int J Radiat Oncol Biol Phys. 2003;56(3):746-748.
1 Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOLBOCAN 2008. Int J Cancer. 2010;127(12):2893-2917.
2 Manecksha RP, Fitzpatrick JM. Epidemiology of testicular cancer. BJU Int. 2009;104(9 Pt B):1329-1333.
3 Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2010;60:277-300.
4 Sokoloff MH, Joyce GF, Wise M. Testis cancer. J Urol. 2007;177(6):2030-2041.
5 Shah MN, Devesa SS, Zhu K, McGlynn KA. Trends in testicular germ cell tumours by ethnic group in the United States. Int J Androl. 2007;30(4):206-213. discussion Int J Androl 30(4):213-204, 2007
6 Akre O, Pettersson A, Richiardi L. Risk of contralateral testicular cancer among men with unilaterally undescended testis. A meta analysis. Int J Cancer. 2009;124(3):687-689.
7 Pettersson A, Richiardi L, Nordenskjold A, et al. Age at surgery for undescended testis and risk of testicular cancer. N Engl J Med. 2007;356(18):1835-1841.
8 Walsh TJ, Dall’Era MA, Croughan MS, et al. Prepubertal orchiopexy for cryptorchidism may be associated with lower risk of testicular cancer. J Urol. 2007;178(4 Pt 1):1440-1446. discussion J Urol 178(4 Pt 1): 1446, 2007
9 Virtanen HE, Rajpert-De Meyts E, Main KM, et al. Testicular dysgenesis syndrome and the development and occurrence of male reproductive disorders. Toxicol Appl Pharmacol. 2005;207(2 Suppl):501-505.
10 Parkin D, Muir C, Whelan S, et al. Cancer Incidence in Five Continents: Comparability and Quality of Data. IARC Scientific Publication 120, vol. VI. Lyon, France: IARC Scientific Publications; 1992.
11 Fossa SD, Chen J, Schonfeld SJ, et al. Risk of contralateral testicular cancer. A population-based study of 29,515 U.S. men. J Natl Cancer Inst. 2005;97(14):1056-1066.
12 Mai PL, Friedlander M, Tucker K, et al. The International Testicular Cancer Linkage Consortium. A clinicopathologic descriptive analysis of 461 familial malignant testicular germ cell tumor kindred. Urol Oncol. 2010;28:492-499.
13 Rapley EA, Turnbull C, Al Olama AA, et al. A genome-wide association study of testicular germ cell tumor. Nat Genet. 2009;41(7):807-810.
14 Cook MB, Zhang Y, Graubard BI, et al. Risk of testicular germ-cell tumours in relation to childhood physical activity. Br J Cancer. 2008;98(1):174-178.
14a Anderson PD, Nelson VR, Tesar PJ, Nadeau JH. Genetic factors on mouse chromosome 18 affecting susceptibility to testicular germ cell tumors and permissiveness to embryonic stem cell derivation. Cancer Res. 2009;69(23):9112-9117.
15 Dieckmann KP, Hartmann JT, Classen J, et al. Is increased body mass index associated with the incidence of testicular germ cell cancer? J Cancer Res Clin Oncol. 2009;135(5):731-738.
16 Kasiske BL, Snyder JJ, Gilbertson DT, Wang C. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4(6):905-913.
17 Powles T, Nelson M, Bower M. HIV-related testicular cancer. Int J STD AIDS. 2003;14(1):24-27.
18 Rorth M, Rajpert-De Meyts E, Andersson L, et al. Carcinoma in situ in the testis. Scand J Urol Nephrol Suppl. 2000;205:166-186.
19 Krege S, Beyer J, Souchon R, et al. European consensus conference on diagnosis and treatment of germ cell cancer. A report of the second meeting of the European Germ Cell Cancer Consensus Group (EGCCCG). Part I. Eur Urol. 2008;53(3):478-496.
20 Heidenreich A. Contralateral testicular biopsy in testis cancer. Current concepts and controversies. BJU Int. 2009;104(9 Pt B):1346-1350.
21 Atkin N, Baker M. High chromosome numbers of testicular germ cell tumors. An update. Cancer Genet Cytogenet. 1995;4:88-90.
22 Goddard NC, McIntyre A, Summersgill B, et al. KIT and RAS signalling pathways in testicular germ cell tumours. New data and a review of the literature. Int J Androl. 2007;30(4):337-348. discussion Int J Androl 30(4):349, 2007
23 Sandberg AA, Meloni AM, Suijkerbuijk RF. Reviews of chromosome studies in urological tumors. III. Cytogenetics and genes in testicular tumors. J Urol. 1996;155(5):1531-1556.
24 Oosterhuis JW, Looijenga LH. Current views on the pathogenesis of testicular germ cell tumours and perspectives for future research. Highlights of the 5th Copenhagen Workshop on Carcinoma in situ and Cancer of the Testis. APMIS. 2003;111(1):280-289.
25 Tanaka K, Okamoto S, Ishikawa Y, et al. DDX1 is required for testicular tumorigenesis, partially through the transcriptional activation of 12p stem cell genes. Oncogene. 2009;28(21):2142-2151.
26 Shuin T, Misaki H, Kubota Y, et al. Differential expression of protooncogenes in human germ cell tumors of the testis. Cancer. 1994;73:1721-1727.
27 Donadio AC, Motzer RJ, Bajorin DF, et al. Chemotherapy for teratoma with malignant transformation. J Clin Oncol. 2003;21(23):4285-4291.
28 Ilson DH, Motzer RJ, Rodriguez E, et al. Genetic analysis in the diagnosis of neoplasms of unknown primary tumor site. Semin Oncol. 1993;20(3):229-237.
29 Motzer RJ, Amsterdam A, Prieto V, et al. Teratoma with malignant transformation. Diverse malignant histologies arising in men with germ cell tumors. J Urol. 1998;159(1):133-138.
30 Dixon F, Moore R. Tumors of the male sex organs. In: Atlas of Tumor Pathology. Washington, DC: Armed Forces Institute of Pathology; 1953:31-32.
31 Mostofi F, Price EJ. Tumors of the male genital system. In: Hartmann W, Cowan W, editors. Atlas of Tumor Pathology. ed 66. Washington, DC: Armed Forces Institute of Pathology; 1973:85.
32 Pugh R. Pathology of Testis. Oxford: Blackwell; 1976.
33 Ebele J, Sauter G, Epstein J, Sesterhenn I. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Lyon, France: IARC Press; 2004.
34 McGlynn KA, Devesa SS, Sigurdson AJ, et al. Trends in the incidence of testicular germ cell tumors in the United States. Cancer. 2003;97(1):63-70.
35 Grigor KM. A new classification of germ cell tumours of the testis. Eur Urol. 1993;23(1):93-100. discussion Eur Urol 23(1):101-103, 1993.
36 Ulbright TM. Germ cell neoplasms of the testis. Am J Surg Pathol. 1993;17(11):1075-1091.
37 Tickoo SK, Hutchinson B, Bacik J, et al. Testicular seminoma. A clinicopathologic and immunohistochemical study of 105 cases with special reference to seminomas with atypical features. Int J Surg Pathol. 2002;10(1):23-32.
38 Thomas R, Dearnaley D, Nicholls J, et al. An analysis of surveillance for stage I combined teratoma—seminoma of the testis. Br J Cancer. 1996;74(1):59-62.
39 Donohue J, Zachary J, Maynard B. Distribution of nodal metastases in nonseminomatous testis cancer. J Urol. 1982;128:315-320.
40 Klein FA, Whitmore W, Sogani PC, et al. Inguinal lymph node metastases from germ cell testicular tumors. J Urol. 1984;131(3):497-500.
41 Mason MD, Featherstone T, Olliff J, Horwich A. Inguinal and iliac lymph node involvement in germ cell tumours of the testis. Implications for radiological investigation and for therapy. Clin Oncol. 1991;3(3):147-150.
42 International Germ Cell Consensus Classification. A prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15(2):594-603.
43 Oosting SF, de Haas EC, Links TP, et al. Prevalence of paraneoplastic hyperthyroidism in patients with metastatic non-seminomatous germ-cell tumors. Ann Oncol. 2010;21(1):104-108.
44 Sohaib SA, Koh DM, Husband JE. The role of imaging in the diagnosis, staging, and management of testicular cancer. AJR Am J Roentgenol. 2008;191(2):387-395.
45 Raina V, Singh SP, Kamble N, et al. Brain metastasis as the site of relapse in germ cell tumor of testis. Cancer. 1993;72(7):2182-2185.
46 Bosl GJ, Motzer RJ. Testicular germ-cell cancer. N Engl J Med. 1997;337(4):242-253.
47 Catalona WJ, Vaitukaitis JL, Fair WR. Falsely positive specific human chorionic gonadotropin assays in patients with testicular tumors. Conversion to negative with testosterone administration. J Urol. 1979;122(1):126-128.
48 Zon RT, Nichols C, Einhorn LH. Management strategies and outcomes of germ cell tumor patients with very high human chorionic gonadotropin levels. J Clin Oncol. 1998;16(4):1294-1297.
49 Sturgeon JF, Jewett MA, Alison RE, et al. Surveillance after orchiectomy for patients with clinical stage I nonseminomatous testis tumors. J Clin Oncol. 1992;10(4):564-568.
50 van Dijk MR, Steyerberg EW, Stenning SP, Habbema JD. Identifying subgroups among poor prognosis patients with nonseminomatous germ cell cancer by tree modeling. A validation study. Ann Oncol. 2004;15(9):1400-1405.
51 Nielsen OS, Munro AJ, Duncan W, et al. Is placental alkaline phosphatase (PLAP) a useful marker for seminoma? Eur J Cancer. 1990;26(10):1049-1054.
52 Read G, Stenning SP, Cullen MH, et al. Medical Research Council prospective study of surveillance for stage I testicular teratoma. Medical Research Council Testicular Tumors Working Party. J Clin Oncol. 1992;10(11):1762-1768.
53 Krege S, Beyer J, Souchon R, et al. European consensus conference on diagnosis and treatment of germ cell cancer. A report of the second meeting of the European Germ Cell Cancer Consensus Group (EGCCCG). Part II. Eur Urol. 2008;53(3):497-513.
54 Huddart RA, Norman A, Shahidi M, et al. Cardiovascular disease as a long-term complication of treatment for testicular cancer. J Clin Oncol. 2003;21(8):1513-1523.
55 Robinson D, Moller H, Horwich A. Mortality and incidence of second cancers following treatment for testicular cancer. Br J Cancer. 2007;96(3):529-533.
56 Travis LB, Fossa SD, Schonfeld SJ, et al. Second cancers among 40,576 testicular cancer patients. Focus on long-term survivors. J Natl Cancer Inst. 2005;97(18):1354-1365.
57 van den Belt-Dusebout AW, de Wit R, Gietema JA, et al. Treatment-specific risks of second malignancies and cardiovascular disease in 5-year survivors of testicular cancer. J Clin Oncol. 2007;25(28):4370-4378.
58 Zagars GK, Ballo MT, Lee AK, Strom SS. Mortality after cure of testicular seminoma. J Clin Oncol. 2004;22(4):640-647.
59 Aparicio J, Germa JR, Garcia Del Muro XG, et al. Risk-adapted management for patients with clinical stage I seminoma. The Second Spanish Germ Cell Cancer Cooperative Group Study. J Clin Oncol. 2005;23:8717-8723.
60 Daugaard G, Petersen PM, Rorth M. Surveillance in stage I testicular cancer. Apmis. 2003;111(1):76-83. discussion APMIS 111(1):83-75, 2003
61 Fossa SD, Horwich A, Russell JM, et al. Optimal planning target volume for stage I testicular seminoma. A Medical Research Council randomized trial. Medical Research Council Testicular Tumor Working Group [see comment]. J Clin Oncol. 1999;17(4):1146.
62 Jones WG, Fossa SD, Mead GM, et al. Randomized trial of 30 versus 20 Gy in the adjuvant treatment of stage I testicular seminoma. A report on Medical Research Council Trial TE18, European Organisation for the Research and Treatment of Cancer Trial 30942 (ISRCTN18525328). J Clin Oncol. 2005;23(6):1200-1208.
63 Powles T, Robinson D, Shamash J, et al. The long-term risks of adjuvant carboplatin treatment for stage I seminoma of the testis. Ann Oncol. 2008;19(3):443-447.
64 Horwich A, Alsanjari N, A’Hern R, et al. Surveillance following orchiectomy for stage I testicular seminoma. Br J Cancer. 1992;65(5):775-778.
65 von der Maase H, Specht L, Jacobsen GK, et al. Surveillance following orchiectomy for stage I seminoma of the testis [see comment]. Eur J Cancer. 1993;29A(14):1931-1934.
66 Warde PR, Chung P, Sturgeon J, et al. Should surveillance be considered the standard of care in stage I seminoma? J Clin Oncol (Meeting Abstracts). 2005;23(16_suppl):4520.
67 Germa-Lluch JR, Garcia del Muro X, Maroto P, et al. Clinical pattern and therapeutic results achieved in 1490 patients with germ-cell tumours of the testis. The experience of the Spanish Germ-Cell Cancer Group (GG). Eur Urol. 2002;42(6):553-562. discussion Eur Urol 42(6):562-553, 2002
68 Oliver RT, Lore S, Ong J. Alternatives to radiotherapy in the management of seminoma. Br J Urol. 1990;65(1):61-67.
69 Tyldesley S, Voduc D, McKenzie M, et al. Surveillance of stage I testicular seminoma. British Columbia Cancer Agency Experience 1992 to 2002. Urology. 2006;67(3):594-598.
70 Chung P, Parker C, Panzarella T, et al. Surveillance in stage I testicular seminoma—risk of late relapse. Can J Urol. 2002;9(5):1637-1640.
71 Warde P, Specht L, Horwich A, et al. Prognostic factors for relapse in stage I seminoma managed by surveillance. A pooled analysis. J Clin Oncol. 2002;20(22):4448-4452.
72 O’Malley ME, Chung P, Haider M, et al. Comparison of low dose with standard dose abdominal/pelvic multidetector CT in patients with stage 1 testicular cancer under surveillance. Eur Radiol. 2010;20(7):1624-1630.
73 Bayens YC, Helle PA, Van PW, Mali SP. Orchiectomy followed by radiotherapy in 176 stage I and II testicular seminoma patients. Benefits of a 10-year follow-up study. Radiother Oncol. 1992;25(2):97-102.
74 Coleman JM, Coleman RE, Turner AR, et al. The management and clinical course of testicular seminoma. 15 years’ experience at a single institution. Clin Oncol (R Coll Radiol). 1998;10(4):237-241.
75 Hultenschmidt B, Budach V, Genters K, Sack H. Results of radiotherapy for 230 patients with stage I-II seminomas. Strahlenther Onkol. 1996;172(4):186-192.
76 Santoni R, Barbera F, Bertoni F, et al. Stage I seminoma of the testis. A bi-institutional retrospective analysis of patients treated with radiation therapy only. BJU Int. 2003;92(1):47-52. discussion BJU Int 92(1):52, 2003
77 Rathmell AJ, Mapstone NP, Jones WG. Testicular seminoma metastasizing to palatine tonsil. Clin Oncol. 1993;5(3):185-186.
78 Warde P, Gospodarowicz MK, Panzarella T, et al. Stage I testicular seminoma. Results of adjuvant irradiation and surveillance. J Clin Oncol. 1995;13(9):2255-2262.
79 Logue JP, Harris MA, Livsey JE, et al. Short course para-aortic radiation for stage I seminoma of the testis. Int J Radiat Oncol Biol Phys. 2003;57(5):1304-1309.
80 Oliver RT, Edmonds PM, Ong JY, et al. Pilot studies of 2 and 1 course carboplatin as adjuvant for stage I seminoma. Should it be tested in a randomized trial against radiotherapy? Int J Radiat Oncol Biol Phys. 1994;29(1):3-8.
81 Calvert AH. Dose optimisation of carboplatin in adults. Anticancer Res. 1994;14(6A):2273-2278.
82 Oliver RT, Mason MD, Mead GM, et al. Radiotherapy versus single-dose carboplatin in adjuvant treatment of stage I seminoma. A randomised trial. Lancet. 2005;366(9482):293-300.
83 Mead GM, Fossa SD, Oliver RT, et al. Randomized trials in 2466 patients with stage I seminoma: patterns of relapse and follow-up. J Natl Cancer Inst. 2011;103(3):241-249.
84 Steiner H, Holtl L, Wirtenberger W, et al. Long-term experience with carboplatin monotherapy for clinical stage I seminoma. A retrospective single-center study. Urology. 2002;60(2):324-328.
85 Choo R, Sandler H, Warde P, et al. Survey of radiation oncologists. Practice patterns of the management of stage I seminoma of testis in Canada and a selected group in the United States. Can J Urol. 2002;9(2):1479-1485.
86 Chung PW, Gospodarowicz MK, Panzarella T, et al. Stage II testicular seminoma. Patterns of recurrence and outcome of treatment. Eur Urol. 2004;45(6):754-759. discussion Eur Urol 45(6):759-760, 2004
86a Classen J, Schmidberger H, Meisner C, et al. Para-aortic irradiation for stage I testicular seminoma. Results of a prospective study in 675 patients. A trial of the German Testicular Cancer Study Group (GTCSG). Br J Cancer. 2004;90(12):2305-2311.
86b Vallis KA, Howard GC, Duncan W, et al. Radiotherapy for stages I and II testicular seminoma. Results and morbidity in 238 patients. Br J Radiol. 1995;68:400-405.
86c Zagars GK, Pollack A. Radiotherapy for stage II testicular seminoma. Int J Radiat Oncol Biol Phys. 2001;51(3):643-649.
87 Gilbert DC, Vanas NJ, Beesley S, et al. Treating IIA/B seminoma with combination carboplatin and radiotherapy. J Clin Oncol. 2009;27(12):2101-2102. author reply J Clin Oncol 27(12):2102-2103, 2009
88 Mosharafa AA, Foster RS, Leibovich BC, et al. Is post-chemotherapy resection of seminomatous elements associated with higher acute morbidity? J Urol. 2003;169(6):2126-2128.
89 Becherer A, De Santis M, Karanikas G, et al. FDG PET is superior to CT in the prediction of viable tumour in post-chemotherapy seminoma residuals. Eur J Radiol. 2005;54(2):284-288.
90 Ganjoo KN, Chan RJ, Sharma M, Einhorn LH. Positron emission tomography scans in the evaluation of postchemotherapy residual masses in patients with seminoma. J Clin Oncol. 1999;17(11):3457-3460.
91 Hinz S, Schrader M, Kempkensteffen C, et al. The role of positron emission tomography in the evaluation of residual masses after chemotherapy for advanced stage seminoma. J Urol. 2008;179(3):936-940. discussion J Urol 179(3):940, 2008
92 Herr HW, Sheinfeld J, Puc HS, et al. Surgery for a post-chemotherapy residual mass in seminoma. J Urol. 1997;157(3):860-862.
93 Culine S, Droz JP. Optimal management of residual mass after chemotherapy in advanced seminoma. There is time for everything. J Clin Oncol. 1996;14(10):2884-2885.
94 Horwich A, Paluchowska B, Norman A, et al. Residual mass following chemotherapy of seminoma. Ann Oncol. 1997;8(1):37-40.
95 Duchesne GM, Stenning SP, Aass N, et al. Radiotherapy after chemotherapy for metastatic seminoma—a diminishing role. MRC Testicular Tumour Working Party. Eur J Cancer. 1997;33(6):829-835.
96 Wood L, Kollmannsberger C, Jewett M, et al. Canadian consensus guidelines for the management of testicular germ cell cancer. Can Urol Assoc J. 2010;4(2):e19-38.
97 Kennedy BJ. Mithramycin therapy in advanced testicular neoplasms. Cancer. 1970;26(4):755-766.
98 Einhorn L, Donohue J. Cis-diamminedichloroplatinum, vinblastine and bleomycin combination chemotherapy in disseminated testicular cancer. Ann Intern Med. 1977;187:293-298.
99 Samuels M, Johnson E, Holoye P. Continuous intravenous bleomycin therapy with vinblastine in stage III testicular neoplasms. Cancer Chemother Rep. 1975;59:563-570.
100 Vugrin D, Herr H, Whitmore WJ, et al. VAB-6 combination chemotherapy in disseminated cancer of the testis. Ann Intern Med. 1981;95(1):59-61.
101 Albers P, Siener R, Kliesch S, et al. Risk factors for relapse in clinical stage I nonseminomatous testicular germ cell tumors. Results of the German Testicular Cancer Study Group Trial. J Clin Oncol. 2003;21(8):1505-1512.
102 Freedman LS, Parkinson MC, Jones WG, et al. Histopathology in the prediction of relapse of patients with stage I testicular teratoma treated by orchiectomy alone. Lancet. 1987;2(8554):294-298.
103 Jacobsen GK, Rorth M, Osterlind K, et al. Histopathological features in stage I non-seminomatous testicular germ cell tumours correlated to relapse. Danish Testicular Cancer Study Group. APMIS. 1990;98(4):377-382.
104 Klepp O, Olsson AM, Henrikson H, et al. Prognostic factors in clinical stage I nonseminomatous germ cell tumors of the testis. Multivariate analysis of a prospective multicenter study. Swedish-Norwegian Testicular Cancer Group. J Clin Oncol. 1990;8(3):509-518.
105 Hotte SJ, Mayhew LA, Jewett M, et al. Management of stage I non-seminomatous testicular cancer. A systematic review and meta-analysis. Clin Oncol (R Coll Radiol). 2010;22(1):17-26.
106 Freiha F, Torti F. Orchiectomy only for clinical stage I nonseminomatous germ cell testis tumors. Comparison with pathologic stage I disease. Urology. 1989;34(6):347-348.
107 Peckham MJ, Brada M. Surveillance following orchiectomy for stage I testicular cancer. Int J Androl. 1987;10(1):247-254.
108 Pizzocaro G, Nicolai N, Salvioni R, et al. Comparison between clinical and pathological staging in low stage nonseminomatous germ cell testicular tumors. J Urol. 1992;148(1):76-79.
109 Sharir S, Jewett M, Sturgeon J, et al. Progression detection of stage I nonseminomatous testis cancer on surveillance. Implication for the follow-up protocol. J Urol. 1999;161:472-476.
110 Sogani PC, Perrotti M, Herr HW, et al. Clinical stage I testis cancer. Long-term outcome of patients on surveillance. J Urol. 1998;159(3):855-858.
111 Thompson PI, Nixon J, Harvey VJ. Disease relapse in patients with stage I nonseminomatous germ cell tumor of the testis on active surveillance. J Clin Oncol. 1988;6(10):1597-1603.
112 Wishnow KI, Johnson DE, Swanson DA, et al. Identifying patients with low-risk clinical stage I nonseminomatous testicular tumors who should be treated by surveillance. Urology. 1989;34(6):339-343.
113 Raghavan D, Colls B, Levi J, et al. Surveillance for stage I non-seminomatous germ cell tumours of the testis. The optimal protocol has not yet been defined. Br J Urol. 1988;61(6):522-526.
114 Rustin GJ, Mead GM, Stenning SP, et al. Randomized trial of two or five computed tomography scans in the surveillance of patients with stage I nonseminomatous germ cell tumors of the testis. Medical Research Council Trial TE08, ISRCTN56475197—the National Cancer Research Institute Testis Cancer Clinical Studies Group. J Clin Oncol. 2007;25(11):1310-1315.
115 Stephenson AJ, Bosl GJ, Motzer RJ, et al. Retroperitoneal lymph node dissection for nonseminomatous germ cell testicular cancer. Impact of patient selection factors on outcome. J Clin Oncol. 2005;23(12):2781-2788.
116 Stephenson AJ, Klein EA. Surgical management of low-stage nonseminomatous germ cell testicular cancer. BJU Int. 2009;104(9 Pt B):1362-1368.
117 Vaughn DJ. Chemotherapy for clinical stage I nonseminomatous germ cell tumours. BJU Int. 2009;104(9 Pt B):1381-1386.
118 Albers P, Siener R, Krege S, et al. Randomized phase III trial comparing retroperitoneal lymph node dissection with one course of bleomycin and etoposide plus cisplatin chemotherapy in the adjuvant treatment of clinical stage I nonseminomatous testicular germ cell tumors. AUO trial AH 01/94 by the German Testicular Cancer Study Group. J Clin Oncol. 2008;26(18):2966-2972.
119 Kollmannsberger C, Moore C, Chi KN, et al. Non-risk-adapted surveillance for patients with stage I nonseminomatous testicular germ-cell tumors. Diminishing treatment-related morbidity while maintaining efficacy. Ann Oncol. 2010;21:1296-1301.
120 Pizzocaro G, Zanoni F, Milani A, et al. Retroperitoneal lymphadenectomy and aggressive chemotherapy in nonbulky clinical Stage II nonseminomatous germinal testis tumors. Cancer. 1984;53(6):1363-1368.
121 Kondagunta GV, Sheinfeld J, Mazumdar M, et al. Relapse-free and overall survival in patients with pathologic stage II nonseminomatous germ cell cancer treated with etoposide and cisplatin adjuvant chemotherapy. J Clin Oncol. 2004;22(3):464-467.
122 Culine S, Kerbrat P, Kramar A, et al. Refining the optimal chemotherapy regimen for good-risk metastatic nonseminomatous germ-cell tumors. A randomized trial of the Genito-Urinary Group of the French Federation of Cancer Centers (GETUG T93BP). Ann Oncol. 2007;18(5):917-924.
123 de Wit R, Stoter G, Sleijfer DT, et al. Four cycles of BEP vs four cycles of VIP in patients with intermediate-prognosis metastatic testicular non-seminoma. A randomized study of the EORTC Genitourinary Tract Cancer Cooperative Group. European Organization for Research and Treatment of Cancer. Br J Cancer. 1998;78(6):828-832.
124 Droz JP, Kramar A, Biron P, et al. Failure of high-dose cyclophosphamide and etoposide combined with double-dose cisplatin and bone marrow support in patients with high-volume metastatic nonseminomatous germ-cell tumours. Mature results of a randomised trial. Eur Urol. 2007;51(3):739-746. discussion Eur Urol 51(3):747-738, 2007
125 Motzer RJ, Nichols CJ, Margolin KA, et al. Phase III randomized trial of conventional-dose chemotherapy with or without high-dose chemotherapy and autologous hematopoietic stem-cell rescue as first-line treatment for patients with poor-prognosis metastatic germ cell tumors. J Clin Oncol. 2007;25(3):247-256.
126 Kollmannsberger C, Daneshmand S, So A, et al. Management of disseminated nonseminomatous germ cell tumors with risk-based chemotherapy followed by response-guided postchemotherapy surgery. J Clin Oncol. 2010;28(4):537-542.
127 Fossa SD, Oldenburg J, Dahl AA. Short- and long-term morbidity after treatment for testicular cancer. BJU Int. 2009;104(9 Pt B):1418-1422.
128 Huyghe E, Matsuda T, Daudin M, et al. Fertility after testicular cancer treatments. Results of a large multicenter study. Cancer. 2004;100(4):732-737.
129 Fossa SD, Abyholm T, Normann N, Jetne V. Post-treatment fertility in patients with testicular cancer. III. Influence of radiotherapy in seminoma patients. Br J Urol. 1986;58(3):315-319.
130 Senturia YD, Peckham CS, Peckham MJ. Children fathered by men treated for testicular cancer. Lancet. 1985;2(8458):766-769.
131 Bieri S, Rouzaud M, Miralbell R. Seminoma of the testis. Is scrotal shielding necessary when radiotherapy is limited to the para-aortic nodes? Radiother Oncol. 1999;50(3):349-353.
132 Horwich A. Radiotherapy in stage I seminoma of the testis. J Clin Oncol. 2004;22(4):585-588.
133 Moller H, Mellemgaard A, Jacobsen GK, et al. Incidence of second primary cancer following testicular cancer. Eur J Cancer. 1993;29A(5):672-676.
134 Dahl AA, Haaland CF, Mykletun A, et al. Study of anxiety disorder and depression in long-term survivors of testicular cancer. J Clin Oncol. 2005;23(10):2389-2395.
135 Barrass BJ, Jones R, Graham JD, Persad RA. Practical management issues in bilateral testicular cancer. BJU Int. 2004;93(9):1183-1187.
136 Vogelzang NJ, Bosl GJ, Johnson K, Kennedy BJ. Raynaud’s phenomenon. A common toxicity after combination chemotherapy for testicular cancer. Ann Intern Med. 1981;95(3):288-292.
137 Keijzer A, Kuenen B. Fatal pulmonary toxicity in testis cancer with bleomycin-containing chemotherapy. J Clin Oncol. 2007;25(23):3543-3544.
138 Lampe H, Horwich A, Norman A, et al. Fertility after chemotherapy for testicular germ cell cancers. J Clin Oncol. 1997;15(1):239-245.
139 Brydoy M, Fossa SD, Klepp O, et al. Paternity following treatment for testicular cancer. J Natl Cancer Inst. 2005;97(21):1580-1588.
140 Young RH. Testicular tumors—some new and a few perennial problems. Arch Pathol Lab Med. 2008;132(4):548-564.
141 Loeser A, Vergho DC, Katzenberger T, et al. Testis-sparing surgery versus radical orchiectomy in patients with Leydig cell tumors. Urology. 2009;74(2):370-372.
142 Giglio M, Medica M, De Rose AF, et al. Testicular Sertoli cell tumours and relative sub-types. Analysis of clinical and prognostic features. Urol Int. 2003;70(3):205-210.
143 McKenney JK, Heerema-McKenney A, Rouse RV. Extragonadal germ cell tumors. A review with emphasis on pathologic features, clinical prognostic variables, and differential diagnostic considerations. Adv Anat Pathol. 2007;14(2):69-92.
144 Bokemeyer C, Nichols CR, Droz JP, et al. Extragonadal germ cell tumors of the mediastinum and retroperitoneum. Results from an international analysis. J Clin Oncol. 2002;20(7):1864-1873.
145 Motzer RJ, Rodriguez E, Reuter VE, et al. Genetic analysis as an aid in diagnosis for patients with midline carcinomas of uncertain histologies. J Natl Cancer Inst. 1991;83(5):341-346.
146 Hartmann JT, Nichols CR, Droz JP, et al. Prognostic variables for response and outcome in patients with extragonadal germ-cell tumors. Ann Oncol. 2002;13(7):1017-1028.
147 Bower M, Brock C, Holden L, et al. POMB/ACE chemotherapy for mediastinal germ cell tumours. Eur J Cancer. 1997;33(6):838-842.
148 Bauer SB, Perlmutter AD, Retik AB. Anomalies of the upper urinary tract. In: Walsh PC, Retik AB, Stamey TA, et al, editors. Campbell’s Urology. ed 6. Philadelphia: WB Saunders; 1992:1357-1442.
149 Evans CP, Tunuguntla HS, Saffarian AR, Wood CG. Does retroperitoneal lymphadenectomy for testicular germ cell tumor require a different approach in the presence of horseshoe kidney? J Urol. 2003;169(2):503-506.
150 Engels EA, Biggar RJ, Hall HI, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187-194.
151 Leibovitch I, Baniel J, Rowland RG, et al. Malignant testicular neoplasms in immunosuppressed patients. J Urol. 1996;155(6):1938-1942.
152 Hentrich M, Weber N, Bergsdorf T, et al. Management and outcome of bilateral testicular germ cell tumors. Twenty-five year experience in Munich. Acta Oncol. 2005;44(6):529-536.
153 Heidenreich A, Weissbach L, Holtl W, et al. Organ sparing surgery for malignant germ cell tumor of the testis. J Urol. 2001;166(6):2161-2165.
154 Giannarini G, Dieckmann KP, Albers P, et al. Organ-sparing surgery for adult testicular tumours. A systematic review of the literature. Eur Urol. 2010;57:780-790.
155 Fossa SD, Bokemeyer C, Gerl A, et al. Treatment outcome of patients with brain metastases from malignant germ cell tumors. Cancer. 1999;85(4):988-997.
156 Bokemeyer C, Nowak P, Haupt A, et al. Treatment of brain metastases in patients with testicular cancer. J Clin Oncol. 1997;15(4):1449-1454.
157 Carriere P, Baade P, Fritschi L. Population based incidence and age distribution of spermatocytic seminoma. J Urol. 2007;178(1):125-128.
158 Aggarwal N, Parwani AV. Spermatocytic seminoma. Arch Pathol Lab Med. 2009;133(12):1985-1988.
159 Chung PW, Bayley AJ, Sweet J, et al. Spermatocytic seminoma. A review. Eur Urol. 2004;45(4):495-498.
160 Classen J, Souchon R, Hehr T, et al. Posttreatment surveillance after paraaortic radiotherapy for stage I seminoma. A systematic analysis. J Cancer Res Clin Oncol. 2009;136(2):227-232.
161 Thomas GM. Is “optimal” radiation for stage I seminoma yet defined? J Clin Oncol. 1999;17(9):3004-3005.
162 Chung PW, Warde PR, Panzarella T, et al. Appropriate radiation volume for stage IIA/B testicular seminoma. Int J Radiat Oncol Biol Phys. 2003;56(3):746-748.
163 Nicolai N, Pizzocaro G. A surveillance study of clinical stage I nonseminomatous germ cell tumors of the testis. 10-year follow-up. J Urol. 1995;154(3):1045-1049.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 53 Testicular Cancer
Testicular tumors are uncommon in the general population but are some of the most common and important malignant tumors in young men. Worldwide, 52,000 new cases and 9,000 deaths were estimated from this disease in 2008.1 The vast majority are primary germ cell tumors (GCTs); the others include lymphomas and sarcomas. The incidence of GCTs has doubled in the past 30 years. Although most patients (70% to 80%) present with early-stage, highly curable disease, the continued rise in incidence of these tumors presents a major challenge. Management is based on histologic type and disease extent. Advances in chemotherapy, imaging, and multidisciplinary care have led to improvements in outcomes over the past three decades. In this chapter, we will discuss the management principles for patients with primary GCTs of the testis, malignant extragonadal GCTs, and other rare testicular tumors.
Etiology and Epidemiology
Testicular cancer accounts for only 1% to 2 % of all cancers in men in most populations across the world.2 Germ cell testicular tumors are the most common solid malignant tumors in men between 20 and 35 years of age, and it was estimated that in the United States in 2010, there would be 8480 new cases and 350 deaths from testicular cancer.3 The cumulative lifetime risk of developing a GCT for a white man in the United States is 0.2%.4 Although the disease is rare, the observed rising incidence is a concern. Between 1973 and 2003 the incidence of GCTs rose by 61% in the United States, with the major rise occurring in seminomas rather than nonseminomas.5
Risk factors for testicular germ cell cancer (TGCC) include a family history of cancer, the presence of an undescended testis or gonadal dysgenesis, subfertility, and testicular microlithiasis. Men with a history of cryptorchidism have an approximately six-fold increased chance of developing testicular cancer.6 Orchiopexy prior to puberty appears to lower the risk of developing a subsequent tumor and may help preserve Leydig cell function and enhance fertility.7,8 Although most testicular cancers in men with a history of maldescent occur on the ipsilateral side, approximately 5% to 20% develop in the contralateral testicle.6 The mechanism by which cryptorchidism increases the risk of developing GCTs is unknown, but the effects of maldescent on the testis (i.e., increased temperature or increased risk of trauma if the testis is in the inguinal region) have been suggested as possible factors. The increase in the incidence of tumors in the contralateral testicle, however, suggests that maldescent and testicular cancer may result from the same prenatal etiologic process. Other genitourinary abnormalities associated with testicular cancers include hydrocele and hypospadias.9
There is considerable geographic and ethnic variation in the incidence of testicular tumors, with the highest incidence being reported from Denmark (8.4 per 100,000 men per year) and Switzerland (6.2 to 8.8 per 100,000 men per year).8 The incidence of testicular cancer is lower in non-Europeans as compared with Europeans. In the United States, white men (6.36 per 100,000 per year) are five to six times more likely to develop testicular cancers than black men (1.30 per 100,000 per year); low incidence rates are seen in other ethnic groups, such as Americans of Chinese and Japanese descent.5 A high incidence of testicular cancer is seen in some nonwhite populations such as the Maoris in New Zealand and Native Americans.10
Prior testicular cancer is a major risk factor for the development of a contralateral malignant tumor. In a large population-based follow-up study of 29,515 patients with unilateral testicular cancer, the cumulative risk (at 15 years after diagnosis of the primary tumor) of developing a contralateral malignant tumor was 1.9%.11
Heritability of Testicular Germ Cell Cancer
Given the strong familial association of TGCC, extended pedigrees with this disease are curiously rare. Most families with TGCC have two affected members, most commonly sibling pairs. The International Testicular Germ Cell Linkage Consortium has carried out two large-scale genome scans, neither of which provided convincing or replicable evidence for susceptibility gene(s).12 Candidate association studies have demonstrated that rare gr/gr deletions of the Y chromosome predispose to TGCC, but such deletions cannot explain more than a fraction of heritable cases of this disease.
More recently, two groups carried out association studies, based on genome-wide, single-nucleotide polymorphisms (SNPs), of patients with TGCC and controls.13,14 Both research groups identified strong evidence for two susceptibility loci for seminoma and nonseminoma; one locus is on chromosome 12p22, within the KITLG gene, encoding the ligand for the receptor tyrosine kinase KIT, and the second locus is on chromosome 5q31.3 near the SPRY4 gene, encoding sprouty 4, an inhibitor of the MAP kinase pathway. A third locus on chromosome 6 was identified by only one of the research groups and is not associated with a known gene. The relative risk was approximately 3 for the KITLG locus and about 1.4 for the other two loci. Both the KITLG gene and the SPTY4 gene are biologically feasible candidates because their products lie in the same signal transduction pathway. Moreover, activating mutations of the KIT gene have been identified in a subset of seminomas, while paradoxically, deletions of the KIT gene predispose to germ cell cancer in the SV/129 mouse strain. Of note, cases in the association studies included patients with and patients without a family history of TGCC, but none of these loci were enriched in the familial cases and neither were they associated with other abnormalities such as an undescended testis.
As mentioned above, the SV/129 mouse strain is particularly susceptible to the development of germ cell cancer, and this system has been exploited by Nadeau and colleagues14a to map several genes predisposing to the disease. Curiously, none of the corresponding loci in humans appear to have any role in TGCC development.
Environmental Factors and Testicular Germ Cell Cancer
Other factors linked to the development of TGCCs include a history of testicular trauma, an increased body mass index, immunosuppression following organ transplantation, and human immunodeficiency virus (HIV) infection.2,15–17 There is no evidence to suggest a causal relationship between testicular trauma and the development of a tumor, and the likely explanation is that testicular trauma leads to examination of the testes. Because of the age distribution of testicular cancer, exposure must occur early in life if there is an environmental contribution to the etiology of these tumors. Prenatal factors linked to the later development of testicular cancers include threatened miscarriage, excessive maternal nausea, and birth by caesarean delivery.2 To explain these associations it has been suggested that exposure of the germinal epithelium in utero to an elevated level of free unbound maternal estrogen could give rise to subsequent cryptorchidism and an elevated risk of developing a testicular tumor; the prenatal estrogen theory remains unproven, however.2
Prevention and Early Detection
The identification of carcinoma in situ (CIS) or testicular intraepithelial neoplasia (TIN) as a precursor of testicular GCTs has raised the possibility that the development of invasive testicular cancer could be prevented by treating CIS. In adults, CIS is found adjacent to GCTs in virtually 100% of cases, and it is thought that, with the exception of spermatocytes seminoma, CIS precedes the development of all invasive tumors.18 The natural history of testicular CIS is unknown, but the Danish experience suggests that all cases of adult CIS will ultimately progress to invasive cancer.18
The diagnosis of testicular CIS can only be made by testicular biopsy. Because the incidence of CIS in the general population is low (at most, 0.7%), screening biopsies are currently not recommended. They should be considered in high-risk patients, however, including those with presumed extragonadal germ cell cancer, intersex individuals, and select patients with contralateral GCT (age <40 years and testicular volumes of <12 mL).19
The management of patients with testicular CIS is controversial. Orchiectomy has been suggested for unilateral disease, and in cases diagnosed after orchiectomy for GCT, three options can be considered and discussed with the patient: orchiectomy, low-dose RT, or surveillance.19 Although both orchiectomy and RT offer definitive treatment for testicular CIS, they both destroy any residual fertility. Surveillance, with careful follow-up of the affected testis, is a reasonable option, especially given the excellent prognosis for metachronous testicular cancers.20
Biologic Characteristics and Molecular Biology
GCTs have a distinctive capacity for totipotential differentiation as demonstrated by the frequent finding of combinations of choriocarcinoma, embryonal carcinoma, and seminoma in a single tumor. They also can retain their ability to differentiate as displayed by the not infrequent identification of mature teratoma in residual post-treatment retroperitoneal masses. Cytogenetic analysis of GCTs has shown that chromosome numbers are more homogenous in seminomas than in nonseminomas.21–23 Triploid and tetraploid chromosomal patterns are common in seminomas, and hyperdiploid to hypertriploid counts are common in nonseminomas. A characteristic chromosome anomaly in GCTs of all histologic types is the presence of an isochromosome of the short arm of chromosome 12.24 This isochromosome, first reported by Atkin and Baker in 1982,21 consists essentially of two chromosome 12 short arms. It is present in more than 80% of cases, and GCTs without a 12p isochromosome have extra copies of 12p segments incorporated into other chromosomes. The 12p isochromosome is also found in testicular CIS.24 Isochromosome copies tend to be more numerous in nonseminomas than in seminomas.
It is not known how these chromosomal changes contribute to the development of the neoplastic phenotype. The frequent finding of the 12p isochromosome in both CIS and invasive tumors, however, indicates that this chromosome plays an important role in the biology of these tumors. The possibility of amplification of normal or modified genes, such as the DDX1 gene on the 12p isochromosome, is currently being investigated.25 Proto-oncogenes present on the short arm of chromosome 12 could be activated by point mutations, deletions, or translocations to become oncogenes, which could then act in a dominant manner.26 The KRAS gene is located on the short arm of chromosome 12, and amplification or enhanced expression of this gene has been reported to occur in testicular tumors and derived cell lines. Other candidate genes being studied include the c-KIT oncogene and its ligand KITGL or SCF, the c-MOS oncogene, and the CCND2 gene. In addition to chromosome 12, 17q is overrepresented in 50% of cases of GCT; this area contains a number of genes of interest, including the GRB7 gene and the plakoglobin gene.22
The study of the genetics and molecular biology of GCTs has enhanced our understanding of these tumors. To date, however, cytogenetics has had little impact in the clinic, with the possible exception of the use of various markers for the identification of non-TGCCs located elsewhere in the body.27–29
Pathology and Pathways of Spread
Pathology
Testicular cancers can arise from intratesticular and paratesticular cells (Table 53-1). The vast majority are of germ cell origin, and three major classification schemes have been in use worldwide30–32 (Table 53-2). The Dixon and Moore classification as modified by Mostofi has been adopted by the World Health Organization (WHO) and is the classification scheme most widely used in North America.33
TABLE 53-1 Histologic Classification of Testicular Neoplasms
TABLE 53-2 Three Classifications of Germ Cell Tumors
| Dixon and Moore, 195330 | WHO Classification33 | Pugh, 197632 |
|---|---|---|
For clinical purposes, GCTs are classified into two major groups: seminomas and nonseminomas (NSGCTs). It should be noted that patients with pure seminoma may have recurrence with pure NSGCT, and vice versa. Approximately 60% of GCTs are pure seminomas, 30% are NSGCTs, and 10% are mixed tumors (both seminomatous and NSGCT elements are present).34 Patients with mixed tumors are clinically considered to have NSGCT, the only exception being tumors with syncytiotrophoblastic cells in cases of seminoma.
Carcinoma in Situ
Intratubular germ cell neoplasia, or CIS, is felt to precede the development of all cases of seminoma and NSGCT in adults (with the exception of spermatocytic seminoma).35 On light microscopy, CIS cells closely resemble seminoma cells and in most cases are found within the seminiferous tubules. Cytologically, there is no difference between the CIS cells that develop into seminomas and those that develop into NSGCTs. In the general population, the incidence of CIS is very low (0.2%), but it is somewhat higher in men with impaired fertility (0.5%) and in those with cryptorchid testes (2% to 4%).18
Seminomas
Seminoma, the most common type of testicular GCT, is most often seen in the fourth decade of life. On gross examination, the tumors are usually well demarcated from the residual testicular tissue and rarely have foci of necrosis or hemorrhage. On microscopic examination, the classical or typical seminoma type is made up of large cells with abundant cytoplasm divided by connective tissue septae into sheets or cords.36 These cells typically have round, hyperchromatic or vesicular nuclei with prominent nucleoli. Frequently, there is a lymphocytic infiltrate, and macrophages, plasma cells, and multinucleated giant cells are often present. Syncytiotrophoblasts are present in 15% to 20% of cases, and their presence does not appear to alter the prognosis.
On immunohistochemical testing, virtually all seminomas express placental leukocyte alkaline phosphatase (PLAP) and do not express low-molecular-weight keratins, blood group antigens, or vimentin. Several histologic variants of seminoma have been identified, including anaplastic seminoma and spermatocytic seminoma. Anaplastic seminoma is diagnosed when there are three or more mitoses seen per high-power field.36 Spermatocytic seminoma is a rare subtype mainly seen in older men and is not associated with CIS or bilateral disease.35 Also, these tumors do not stain for PLAP on immunohistochemical testing and rarely, if ever, metastasize. An atypical variant of seminoma with some features similar to those of NSGCT on immunohistochemical examination has been reported, although its morphologic appearance is similar to that of classical seminoma.37 Usually, there is little or no lymphocytic infiltrate and the tumor cells have less cytoplasm than classical seminoma cells. In terms of prognosis, this atypical variant appears to have the same prognosis as classical seminoma.
Nonseminomatous Germ Cell Tumors
Nonseminomatous tumors make up 40% of TGCCs and occur most commonly in the third decade of life. In the WHO classification system, NSGCTs include embryonal carcinoma, teratoma (mature, immature, or with malignant differentiation), choriocarcinoma, yolk sac tumor, and mixed GCTs. Most tumors are mixed, with two or more cell types present. Although some tumors have a component of seminoma, the association of seminoma within a histologically confirmed NSGCT has no major impact on the clinical outcome.38 Patients with combined tumors present at an age (median, 33 years) intermediate between those with seminoma (median, 36 years) and those with nonseminoma (median, 27 years).38 On gross examination, there is usually a soft irregular mass poorly demarcated from the surrounding testicular tissue, and a considerable amount of necrosis and hemorrhage is often present. Immunohistochemical studies usually demonstrate cytoplasmic expression of low-molecular-weight keratins in embryonal carcinomas, and yolk sac elements, low-molecular-weight keratin, and/or vimentin expression in mature teratomas.
Pathways of Spread
Lymphatic spread is the most common route of metastatic spread. The lymphatic drainage of the testis is directly to the para-aortic lymph nodes. There are differences in the distribution of metastases from left or right testicular tumors. The left testicular vein drains to the left renal vein, and the lymphatic drainage is primarily to the lymph nodes in the para-aortic area, directly below the left renal hilum. On the right side, the testicular vein drains directly to the inferior vena cava below the level of the renal vein, and, therefore, paracaval and interaortocaval nodes are the first ones to be involved in right-sided tumors. Figure 53-1 shows the distribution of retroperitoneal lymph node metastases in NSGCT.39 Contralateral nodal involvement occurs in approximately 15% of cases and is rarely found in the absence of ipsilateral involvement. Supradiaphragmatic spread can occur through the thoracic duct, and although left supraclavicular nodal disease is infrequent at presentation, it is often seen at the time of relapse.
Pelvic and inguinal lymph node involvement is rare (<3%). Factors predisposing to inguinal lymph node involvement include prior scrotal or inguinal surgery, scrotal orchiectomy with incision of the tunica albuginea, tumor invasion of the tunica vaginalis or lower third of the epididymis, and cryptorchid testis.40,41 Disruption of lymphatic vessels in the spermatic cord during inguinal surgery has been shown to induce anastomoses between the testicular lymphatic vessels and the regional lymphatics destined for inguinal or pelvic lymph nodes. In an occasional patient, a connection with the contralateral inguinal lymph nodes may be established, but this is very uncommon. In a small proportion of patients with inguinal relapse, no predisposing factors may be apparent.40
In patients with NSGCTs, hematogenous spread occurs early in the course of the disease. The lung parenchyma is the commonest site of hematogenous spread, but liver, bone, brain, kidney, and gastrointestinal metastases are also seen. In a review of over 5000 patients with metastatic GCT, pulmonary metastases were present in 44% of cases and liver metastases in 6% of cases, with all other areas of hematogenous spread present in 1% or less of cases.42 Mediastinal and neck node involvement was present in 11% to 12% of cases.
Clinical Manifestations, Patient Evaluation, and Staging
Clinical Manifestations
Patients with testicular tumors most commonly present with a painless testicular mass. Up to 45% of patients may have testicular pain, with signs and symptoms suggestive of acute epididymitis in up to 25% of patients. Much less common presenting features include those related to the presence of metastases, for example, back pain, dyspnea, and gynecomastia (from malignant tumors that produce HCG). Up to 3.5% of patients—those with very high HCG levels—may develop hyperthyroidism because the alpha subunit of HCG is identical to that of thyroid-stimulating hormone (TSH) and may therefore stimulate the thyroid.43 The differential diagnosis of a testicular mass (in addition to tumor) includes torsion, hydrocele, varicocele, spermatocele, and epididymitis. A small percentage of tumors are associated with a hydrocele, so the presence of transillumination on examination does not rule out a diagnosis of malignancy.
Patient Evaluation
Routine staging investigations include chest x-ray, CT scanning of the abdomen and pelvis, CT scanning of the thorax, and tumor markers. CT scanning of the thorax does not add value in patients with seminoma with no evidence of retroperitoneal lymph node involvement. There is no role for bipedal lymphography in staging patients; however, magnetic resonance imaging (MRI) with lymphotrophic nanoparticles has shown promise in a number of studies but needs further evaluation before it can be adopted into clinical practice.44 In stage II and III seminomas, especially in patients with bulky retroperitoneal disease, a bone scan should also be performed. Abnormal serum marker levels should be monitored to document postorchiectomy decay according to their respective half-lives. Patients with extensive metastatic disease, nonpulmonary visceral metastases (NPVMs), or very high tumor marker levels are at risk for brain metastases, and CT or MRI of the brain should be performed.45 Baseline pulmonary and renal functions are assessed in patients who require chemotherapy.
Tumor Markers
Measurements of AFP, β-HCG, and LDH are essential in the diagnosis and management of patients with GCTs.46 HCG is a glycoprotein with a molecular weight of 45,000 daltons, composed of two subunits, of which the α-subunit is identical to that of luteinizing hormone (LH), follicle-stimulating hormone (FSH), and thyroid-stimulating hormone (TSH), and a distinct β-subunit. HCG is normally produced by the placenta. About 15% of patients with seminoma have elevated β-HCG levels. Low levels of β-HCG may be found in other neoplasms, including prostate, bladder, and renal tumors. The use of marijuana derivatives also may lead to elevated levels. Some cross-reactivity with LH does occur, and in cases where an elevated HCG level is thought to be due to this cross-reactivity, consideration should be given to having levels remeasured after treatment with testosterone.47 The half-life of β-HCG in blood is approximately 22 hours. In the subset of GCT patients with very high levels of HCG, a plateau of this tumor marker is often reached after the fourth cycle of chemotherapy that is considerably greater than normal. Such patients often remain in remission with slowly falling levels of HCG, possibly resulting from tissue binding of this tumor marker. Such persistent elevation of HCG levels following chemotherapy is not in itself an indication for salvage therapy.48
One or both of these markers are elevated in 85% of patients with nonseminomatous GCTs. Experience with surveillance of patients with early-stage disease has shown that normal markers do not exclude the presence of occult disease.49
LDH is another important marker in patients with GCTs. It is elevated in up to 60% of patients with nonseminomas and also in a high proportion of patients with advanced seminomas.42,50
PLAP is an isoenzyme of alkaline phosphatase and is normally expressed by placental syncytiotrophoblasts. It is also expressed by testicular tissue and has been investigated as a tumor marker in seminoma. Although PLAP levels are often elevated in patients with seminoma, this factor has proven to be of little value in clinical management.51
Staging
The 2009 American Joint Committee on Cancer/International Union Against Cancer (AJCC/UICC) TNM classification (Table 53-3) is the recommended staging system. In stage I disease, one of the most important determinants of outcome is the presence of vascular invasion in the primary tumor, and this differentiates a pT1 tumor from a pT2 tumor.52 Tumors with invasion of the spermatic cord are staged as pT3 cancers, and the rare tumors with scrotal invasion are classified as pT4 lesions.
The International Germ Cell Cancer Collaborative Group (IGCCCG) analyzed data from 5168 patients with advanced disease.42 Independent prognostic variables identified by the IGCCCG were included: histologic type (nonseminoma vs. seminoma), site of the primary tumor (testis, retroperitoneal site, or other), presence or absence of NPVMs (brain, bone, or liver), and degree of marker elevation (AFP, β-HCG, and LDH). Based on the results of multivariate analysis, the IGCCCG recommended three prognostic groups for nonseminomatous tumors and two prognostic groups for seminomatous tumors. For nonseminomatous cancers, the 5-year overall survival (OS) for the good-prognosis group was 92%, for the intermediate-prognosis group, 80%, and for the poor-prognosis group, 48%. For seminoma, only two prognostic groups were identified: a good-prognosis group without NPVMs with a 5-year OS of 86% and an intermediate-prognosis group with NPVMs with a 5-year OS of 72%. This classification system has been validated by van Dijk and associates50 using Cox regression analysis and recursive partitioning in a cohort of 3048 NSGCT patients.
Primary Therapy
Surgery
Surgery involves a radical inguinal orchiectomy to allow high division of the spermatic cord. Orchiectomy is both diagnostic and therapeutic by providing adequate tissue to ascertain the diagnosis and offering cure in a high proportion (60% to 90%) of patients with stage I disease.19,53 Although not recommended, an accidental scrotal approach does not appear to compromise the outcome, and no additional therapy is required following scrotal violation, provided that the scrotal cavity has not been grossly contaminated by tumor. In patients with life-threatening metastatic disease and a clear-cut diagnosis of germ cell malignancy, initial management should be with chemotherapy and surgery should be postponed until completion of systemic treatment.19,53 The role of partial orchiectomy in patients with metachronous or synchronous bilateral tumors will be discussed later.
Seminoma
In stage I seminoma, postorchiectomy management options include surveillance, RT, or adjuvant chemotherapy. In the past, the standard management of patients with stage I seminoma has been adjuvant retroperitoneal RT. Although RT provided excellent long-term results, with local control in the abdomen and pelvis in virtually all patients, it is now known that over 85% of patients are cured with orchiectomy alone and that RT has been associated with an increased risk of late gonadal toxicity, development of secondary malignant tumors, and, in some cases, an increased risk of cardiovascular disease.54–58 Various approaches have been tried to reduce radiation morbidity, including a reduced radiation dose and treatment volume. In addition, such alternative management strategies as surveillance (including risk-adapted surveillance) and adjuvant chemotherapy have been investigated.59–63 Although adjuvant retroperitoneal RT constituted the standard of care for the past 50 to 60 years, surveillance is now the standard approach, minimizing the burden of treatment while maintaining the cure rate at 100%.64,65,66
In stage III disease, chemotherapy is the treatment of choice.19,53
Management of Stage I Seminoma
Surveillance
The data on seminoma surveillance are now mature, and relapse rates of 15% to 24% have been reported* (Table 53-4). The two largest reported series are from Copenhagen and Toronto.60,66 In the Copenhagen series of 394 patients, with a median follow-up of 60 months, the crude relapse rate was 17.5%. In the Princess Margaret Hospital, Toronto, series of 421 patients, with a median follow-up of 8.1 years, the actuarial 5-year relapse-free survival (RFS) was 85.5%. The other studies with adequate follow-up (>36 months) have reported similar relapse rates. The predominant site of relapse in all studies was the para-aortic lymph nodes (82% in the Danish Testicular Cancer Study Group (DATECA) study and 89% in the Princess Margaret Hospital series.60,66 The median time to relapse ranged from 12 to 18 months, but occasional late relapses (>4 years) have also been reported.70
Prognostic factors for relapse have been studied in a number of the surveillance studies, and age, tumor size, and small vessel invasion have been reported as predictive of relapse. To more accurately determine prognostic factors for relapse in patients with stage I testicular seminoma managed by surveillance, a pooled analysis of four large surveillance series was performed using individual patient data.71 In 638 patients, tumor size and rete testis invasion predicted for relapse on multivariable analysis. The effect of tumor size on the relapse rate is shown in Figure 53-2, and the hazard ratio for relapse with a tumor size of more than 4 cm was 2 (95% CI, 1.3 to 3.2) relative to baseline (tumor size <4 cm and no rete testis invasion). This model has not been validated in an independent dataset and does not have sufficient discrimination to be clinically useful, because patients in the high-risk group have only a 35% risk of relapse during surveillance. For these reasons, these prognostic factors should not dictate treatment strategy.
Figure 53-2 Relapse-free rate based on primary tumor size.
From Warde P, Specht L, Horwich A, et al: Prognostic factors for relapse in stage I seminoma managed by surveillance. A pooled analysis. J Clin Oncol 20:4448-4452, 2002. Copyright © American Society of Clinical Oncology.
At relapse, most patients have been treated with retroperitoneal RT. The incidence of second relapse after irradiation is approximately 10%. The likelihood of detecting early progression in the retroperitoneal lymph nodes (nodes <5 cm) and, therefore, suitability of the patient for RT depends on the frequency of follow-up CT scans of the abdomen and pelvis. The current Princess Margaret Hospital follow-up policy is shown in Table 53-5. One of the concerns regarding the routine use of surveillance was the potential for an increased need for chemotherapy. In the Toronto experience, a similar percentage of patients managed with surveillance (4.6%) and adjuvant RT (3.9%) required chemotherapy as part of their management. Therefore, surveillance does not lead to an increased use of chemotherapy, with its ensuing toxicity.
Another concern regarding surveillance relates to the increased use of imaging studies. There are no prospective data available that quantitate the risk of radiation-induced cancer from CT scanning. A recent paper used long-term follow-up data from survivors of the Hiroshima and Nagasaki atomic bombings to estimate the risk of low-level exposure approximating that of one or more CT scans. This approach has been criticized for methodologic reasons. Nonetheless, the seminoma surveillance population is likely at some risk of induced cancer, given the young age of many patients and the use of repeated CT scans of the abdomen. It seems reasonable to attempt to decrease the frequency of imaging as much as possible until more data on the risk of such scans are available. In addition, the use of low-dose CT protocols is currently under study, and these may be used in standard surveillance protocols in the future.72
Radiation Therapy
Overall survival (OS) ranges between 92% and 99% at 5 to 10 years, with the cause-specific survival (CSS) approaching 100%. Most deaths are due to intercurrent illness, but concern exists that premature deaths may be occurring from radiation-induced cancers or cardiac disease.54,58 With modern-era RT, the relapse rate has varied from 0.5% to 5%66,73–77 (Table 53-6). In-field relapse is rare, and when it is suspected, biopsy should be performed to rule out a nonseminomatous tumor or another malignant tumor. The commonest sites of relapse following adjuvant RT are the mediastinum, lungs, and left supraclavicular fossa. A small proportion of patients, usually with predisposing factors, develop relapse in the inguinal nodes. Uncommon sites of isolated metastases such as the brain and tonsils have been noted.45,77 For supradiaphragmatic relapse, chemotherapy is the treatment of choice and results in a cure rate of nearly 100%. Inguinal relapse can often be treated successfully with RT to the involved area.78
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
